

Abstract
More than 15 million people have been affected by coronavirus disease 2019 (COVID‐19) and it has caused 640 016 deaths as of July 26, 2020. Currently, no effective treatment option is available for COVID‐19 patients. Though many drugs have been proposed, none of them has shown particular efficacy in clinical trials. In this article, the relationship between the Adrenergic system and the renin‐angiotensin‐aldosterone system (RAAS) is focused in COVID‐19 and a vicious circle consisting of the Adrenergic system‐RAAS‐Angiotensin converting enzyme 2 (ACE2)‐severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) (which is referred to as the “ARAS loop”) is proposed. Hyperactivation of the ARAS loop may be the underlying pathophysiological mechanism in COVID‐19, and beta‐adrenergic blockers are proposed as a potential treatment option. Beta‐adrenergic blockers may decrease the SARS‐CoV‐2 cellular entry by decreasing ACE2 receptors expression and cluster of differentiation 147 (CD147) in various cells in the body. Beta‐adrenergic blockers may decrease the morbidity and mortality in COVID‐19 patients by preventing or reducing acute respiratory distress syndrome (ARDS) and other complications. Retrospective and prospective clinical trials should be conducted to check the validity of the hypothesis. Also see the video abstract here https://youtu.be/uLoy7do5ROo.
Keywords: ACE2, ARDS, beta‐adrenergic blockers, COVID‐19, pulmonary embolism, SARS‐CoV‐2, septic shock
The underlying pathophysiology of COVID‐19 is unknown and no effective treatment is available at present. This paper suggests the vicious cycle formed by adrenergic system, RAAS, ACE2, and SARS‐CoV‐2 in COVID‐19, which facilitates SARS‐CoV‐2 entry and produce complications. Beta adrenergic blockers treatment may decrease the SARS‐CoV‐2 cellular entry and reduce the complications in COVID‐19.
1. Introduction
Coronavirus disease 2019 (COVID‐19) has affected people's lives across the world, and as of 26 July 2020, 15 785 641 people are affected globally and caused 640 016 deaths.[ 1 ] Severe acute respiratory syndrome coronavirus 2 (SARS‐COV‐2), a positive‐sense single‐stranded RNA virus, belongs to the beta coronavirus genus. It is known that the SARS‐CoV‐2 spike protein binds to human angiotensin‐converting enzyme 2 (ACE2), which acts as a receptor, and binding to ACE2 is crucial for the cellular entry of SARS‐COV‐2.[ 2 , 3 ] It has been proposed recently that cluster of differentiation 147 (CD147) also known as Basigin, maybe another receptor in the host cells that play a role in the SARS‐CoV‐2 entry into the cell. SARS‐CoV‐2 invades host cells via a novel route: CD147‐spike protein.[ 4 , 5 ] In this paper, I develop a novel proposal for treatment of COVID‐19 infection based on the following components: (i) Adrenergic system by its action on Renin‐angiotensin‐aldosterone system (RAAS), ACE2, and SARS‐CoV‐2 might play a crucial role in COVID‐19. (ii) Hyperactivation of the vicious ARAS loop may be the mechanism behind COVID‐19. (iii) Beta‐adrenergic blockers should be used as a potential treatment option in COVID‐19 patients.
1.1. Pathophysiological Features in COVID‐19
1.1.1. Clinical and Laboratory Features of COVID‐19
Clinical course of COVID‐19 patients are variable—ranging from being asymptomatic to having mild symptoms like fever, cough, sore throat, dyspnea, muscle pain, tiredness, loss of taste or smell sensation, etc.[ 6 ] Around 15% of COVID‐19 patients exhibit pneumonia and ≈5% progress to acute respiratory distress syndrome (ARDS) wherein patients have tachypnoea, decreased oxygen saturation, decreased partial pressure of oxygen in arterial blood, and lung infiltrates in the X‐ray and CT chest imaging.[ 7 ] Severe COVID‐19 patients with complications like ARDS, respiratory failure, and septic shock have a high mortality rate.[ 8 ] Laboratory investigations in moderately or severely affected COVID‐19 patients showed increased Interleukin 6 (IL‐6), D‐dimer (a degradation product of fibrin), C‐reactive protein (CRP), and lactate dehydrogenase (LDH).[ 6 ] Severely affected COVID‐19 patients had lymphopenia and increased IL‐6, Interleukin‐1β (IL‐1β), tumor necrosis factor alpha (TNFα) cytokines levels.[ 7 ]
1.1.2. IL‐6 Increase in COVID‐19 Produce Complications
Increased IL‐6 is an indicator of poor outcome in patients with ARDS and Tocilizumab can be used to block the action of IL‐6.[ 7 ] It has been shown that IL‐6 is increased in COVID‐19 nonsurvivors.[ 9 ] Increased mucus aggregation has been noticed in the distal airways and alveoli in the postmortem study of COVID‐19 patients.[ 10 ] It is known that IL‐6 plays a key role in the activation of MUC5AC and MUC5B gene expression and increases the mucus secretion.[ 11 ] It has been suggested that Tocilizumab by the inhibition of IL‐6 receptor may help decrease the mucus secretion in COVID‐19.[ 12 ] Postmortem study has shown the presence of hyperplasia of type II alveolar epithelial cells, diffuse alveolar damage, macrophage and neutrophil infiltration, fibrosis, hyaline membrane formation.[ 13 ] It is known that cytokine storm occurs in COVID‐19, as the levels of IL‐1β, IL‐6, TNFα are increased and this leads to multiple organ dysfunction and septic shock.[ 7 ]
1.1.3. Pulmonary Complications in COVID‐19 Patients
Inflammatory responses like macrophage and neutrophil infiltration may lead to alveolar damage, pulmonary capillary endothelial dysfunction, and the pulmonary capillary leak that may lead to pulmonary edema. Since type II pulmonary alveolar epithelial cells are the main cells that express ACE2 receptors, their damage might cause problems in surfactant secretion by these cells. Decreased surfactant leads to increased surface tension in the alveoli, which along with the damage in the alveolar epithelium and pulmonary capillary endothelium, may lead to pulmonary edema. Respiratory dysfunction as a result of this leads to decreased partial pressure of oxygen in arterial blood (PaO2), decreased oxygen saturation, and severe cases warrant mechanical ventilation. If refractory hypoxemia develops despite mechanical ventilation, then extracorporeal membrane oxygenation (ECMO) can be tried as per World Health Organization's (WHO) interim guidance.[ 14 ]
1.1.4. Severely Affected Patients Exhibit Complications Like ARDS, Pulmonary Embolism, Septic Shock
Around 30% of the severely affected COVID‐19 patients developed pulmonary embolism and D‐dimer level is found higher in these patients.[ 15 , 16 , 17 ] Around 5% of them had severe complications like ARDS, septic shock.[ 18 ] Septic shock occurs in severely affected COVID‐19 patients wherein the mean arterial pressure is low (<65 mmHg) irrespective of fluid management and plasma lactate concentration will be >2 × 10−3 m L−1. Septic shock state carries a high mortality rate and the primary drug used in the treatment is norepinephrine.[ 19 ]
1.1.5. Cytokine Storm, Nucleotide‐binding domain (NOD)‐like Receptor Protein 3 (NLRP3) Activation and Hyperinflammation Occurs in COVID‐19
Cytokine storm and hyperinflammation occurs in COVID‐19 irrespective of lymphopenia.[ 20 , 21 ] Considering the cytokine storm and potential activation of the NLRP3 inflammasome, it is likely that immune hyperactivation occurs in COVID‐19. Nucleocapsid protein of SARS‐CoV has been shown to increase IL‐6 via the activation of Nuclear factor kappa B (NF‐κB).[ 22 ] Since the nucleocapsid protein of SARS‐CoV‐2 shares 89.6% homology with SARS‐CoV nucleocapsid,[ 23 ] it is likely that the increase in IL‐6 found in severely affected COVID‐19 patients may also have a similar underlying mechanism. IL‐6 level has been shown to positively correlate with the SARS‐CoV‐2 viral load.[ 24 ] NLRP3 inflammasome has been suggested to be activated by SARS‐CoV‐2. It is known that colchicine inhibits NLRP3 inflammasome, and clinical trials have already been registered to test whether colchicine treatment in COVID‐19 improves the clinical condition by inhibiting NLRP3.[ 25 ] It is known that the SARS‐COV orf3a protein activates the NLRP3.[ 26 ] SARS‐CoV viroporin 3a has also been shown to activate NLRP3.[ 27 ] SARS‐CoV‐2 may also activate NLRP3 by a similar mechanism. The downstream effectors of NLRP3 are caspase 1, IL‐1β, and IL‐18.[ 28 ] It is known that the decrease in surfactant protein D in ARDS patients is associated with mortality[ 29 ] and interestingly surfactant protein D has been shown to inhibit NLRP3.[ 30 ] Since the type 2 alveolar epithelial cells which secrete the surfactant are one of the main cells affected by SARS‐CoV‐2, surfactant secretion may likely be decreased. Reduction in surfactant D may result in loss of its inhibitory role on NLRP3 and the resultant activation of the NLRP3 inflammasome pathway may produce an inflammatory response in COVID‐19.
1.1.6. Hypercoagulation State in COVID‐19 Patients
As mentioned earlier, severely affected COVID‐19 patients developed pulmonary embolism, and D‐dimer levels were shown to be high.[ 15 , 16 , 17 ] But so far, it is not known what causes pulmonary embolism or deep vein thrombosis (DVT) in COVID‐19. I hypothesize that the hypercoagulation state in COVID‐19 may be due to a sympathetic storm. Increased catecholamine levels may hyperactivate the vicious ARAS loop, which will be explained in a later section. It has been known that beta2‐adrenergic agonist nebulization increases the plasma catecholamine levels.[ 31 ] Sympathetic hyperactivation has been shown to be related to hypercoagulation state.[ 32 ] Inhalation of beta2‐adrenergic agonists has been shown to increase coagulation factor VIII, von Willebrand factor (VWF), D‐dimer, and lead to hypercoagulation state.[ 33 ] I hypothesize that use of catecholamines in COVID‐19 patients may lead to hypercoagulation state, and nebulization with beta2‐adrenergic agonists for respiratory symptoms and intravenous administration of norepinephrine for treating septic shock should be avoided in COVID‐19, as it may lead to hypercoagulation state and may induce complications like deep vein thrombosis and pulmonary embolism.
Other organs damage in COVID‐19 is not discussed in this article due to space constraints.
1.2. Current Treatment Recommendations are Largely Ineffective and Controversial
At present, there is no effective drug available against the COVID‐19 pandemic.[ 6 , 34 ] Many potential treatments are proposed and some are on clinical trials. Many are focusing on drugs that interrupt the binding of spike protein with the ACE2 receptor. Remdesivir, hydroxychloroquine, azithromycin, tocilizumab are used in COVID‐19 condition all over the world, though there is no evidence so far, for their efficacy. Remdesivir is presumed to work, by its inhibitory action on RNA dependent RNA polymerase restricting the viral replication but so far, its efficacy has not been shown in clinical trials.[ 34 ] Hydroxychloroquine may restrict the SARS‐CoV‐2 cellular entry but its efficacy has not been shown in clinical trials. Tocilizumab by its action on the IL‐6 receptor blocks the inflammatory actions due to IL‐6, but its efficacy has not been proven in clinical trials. As mentioned earlier, Tocilizumab may help decrease the mucus secretion in COVID‐19.[ 12 ] Recently National Institutes of Health (NIH) had recommended Remdesivir for COVID‐19 patients who are on supplemental oxygen but not for those who are on mechanical ventilation. NIH has also recommended dexamethasone due to their immunomodulatory role for the COVID‐19 patients who are on mechanical ventilation. NIH has not recommended hydroxychloroquine, azithromycin, tocilizumab treatment in COVID‐19.[ 35 ]
Another treatment option on the trial is convalescent plasma therapy. Supportive measures are used depending on the clinical condition such as the use of mechanical ventilation, ECMO, etc. As mentioned earlier, WHO suggests that if refractory hypoxemia persists despite mechanical ventilation, then ECMO can be opted for in selected cases. Many have raised concerns about the use of ECMO in COVID‐19, as some studies showed increased mortality in patients who suffered from ARDS and were on ECMO support.[ 9 , 36 , 37 ]
In summary, the underlying pathophysiology of the COVID‐19 is not known and no effective treatment is available at present. In this article, I speculate that the dangerous relationship between the adrenergic system, RAAS, ACE2, and SARS‐CoV‐2 may be the underlying pathological mechanism in COVID‐19. I propose the existence of a vicious Adrenergic system‐RAAS‐ACE2‐SARS‐CoV‐2 (ARAS) loop in COVID‐19 condition. Any drugs like norepinephrine, beta2 agonist that increase the activity of ARAS loop, may worsen the condition in COVID‐19 patients. Adrenergic blockers may inhibit this vicious ARAS loop and provide beneficial effect in COVID‐19 patients. I propose beta‐adrenergic blockers as a potential drug option for the treatment of COVID‐19.
1.3. The Adrenergic System‐RAAS‐ACE2‐SARS‐CoV‐2 (ARAS) Loop May be the Underlying Pathophysiological Mechanism in COVID‐19
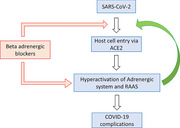
It is important to study whether sympathetic storm, i.e., increased catecholamine level in the body occurs in COVID‐19. Considering all the above evidences and clinical features of pulmonary edema, pulmonary embolism, deep vein thrombosis, and septic shock it appears that the critical illness in COVID‐19 may exhibit sympathetic storm in the body and maybe the basis for all the complications. Increased catecholamine level in the COVID‐19 patients may trigger a hypothetical ARAS loop. Adrenergic system regulation of RAAS is crucial in the COVID‐19. Increased plasma catecholamine level will activate the adrenergic system leading to the activation of RAAS and increase in ACE2, as a result of which the entry of SARS‐CoV‐2 increases, producing complications in COVID‐19 patients; in response to the critical illness, endogenous catecholamines will be increased and the vicious cycle goes on. I would like to call this as adrenergic system‐RAAS‐ACE2‐SARS‐CoV‐2 (ARAS) loop (Figure 1 ).
Figure 1.
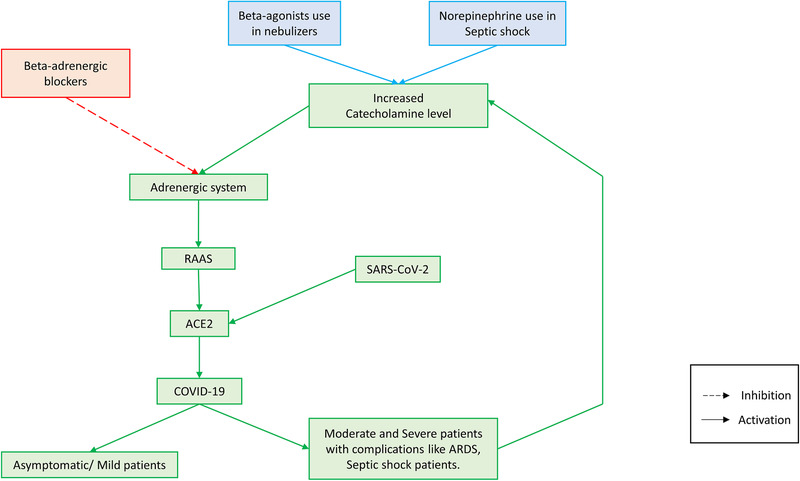
The adrenergic system‐RAAS‐ACE2‐SARS‐CoV‐2 (ARAS) loop: A hypothetical vicious circle consisting of the adrenergic system‐RAAS‐ACE2‐SARS‐CoV‐2 (ARAS) is predicted to emerge in COVID‐19 pathology. Severely affected COVID‐19 patients may have “sympathetic storm” (increased catecholamine levels in the body), which may hyperactivate this loop, and increase ACE2 which in turn may increase the viral entry into cells. Administration of catecholamines exogenously via application of beta2‐adrenergic agonists in a nebulizer solution, or by intravenous administration of norepinephrine for treating the septic shock condition in COVID‐19 patients, will serve as an agonist of the ARAS loop, and worsen the condition. Beta‐adrenergic blockers will inhibit this ARAS loop and may produce beneficial effects in COVID‐19 patients by preventing or reducing complications such as pulmonary embolism, ARDS and septic shock.
It is known that critical illnesses will increase the plasma catecholamine level.[ 38 ] It is interesting to note that adenoviral respiratory illness causes increased plasma catecholamine level. SARS‐CoV‐2 infection may also lead to increased catecholamine levels.[ 39 ] I speculate that in the severely affected COVID‐19 patients with complications like ARDS and septic shock, it is likely that endogenous catecholamine levels may be high and it may trigger the ARAS loop. Apart from the increased endogenous catecholamine level in these complicated COVID‐19 patients, giving exogenous catecholamines like beta‐agonists in the nebulizer solution for treating dyspnea or norepinephrine given intravenously for the patients having septic shock, may further increase the catecholamine level and hyperactivate the vicious ARAS loop and worsen the condition. I speculate that any drug that augments the activation of the ARAS loop will worsen the COVID‐19 condition and increase the mortality. The potential dangers of using norepinephrine, beta2‐agonists, and angiotensin receptor‐blockers (ARBs) in COVID‐19 are discussed in a later section.
1.4. RAAS Inhibitors Role in COVID‐19 Patients is not Clear
It is known that the RAAS plays a crucial role in the regulation of blood pressure, sodium level, extracellular fluid volume, etc. RAAS system has two opposing arms, the classic Renin‐angiotensinI‐angiotensin converting enzyme (ACE)–angiotensin II(ATII)‐aldosterone and Renin‐ATII‐angiotensin‐converting‐enzyme2(ACE2)‐angiotensin‐(1‐7)‐Mas receptor (MasR). Along with the classic pathway which produces effects like vasoconstriction and inflammation, one more pathway with opposing actions to classic pathway exist which produces effects like vasorelaxation and is anti‐inflammatory. Unlike the classic RAAS pathway which uses ACE, the vasorelaxation arm uses ACE2.[ 40 ] Angiotensin II receptor‐blockers (ARBs) and angiotensin‐converting‐enzyme inhibitors (ACEIs) are common drugs used in hypertension patients. They have inhibitory action on RAAS pathway and produce the antihypertensive effect. Recently concerns were raised regarding the use of angiotensin receptor blocker (ARB) drugs as it may increase the ACE2 expression, which in turn may increase the SARS‐CoV‐2 entry.[ 41 , 42 ] It has been suggested that other antihypertensive drugs like beta‐adrenergic blockers and calcium channel blockers can be used in COVID‐19 cases with hypertension instead of ARBs.[ 41 ] On the contrary, many opined that in the absence of sufficient evidence there is no need to change the drugs like ARBs in the COVID‐19 patients who are already taking them.[ 43 ] Withdrawal of ARBs and ACEIs in COVID‐19 patients with hypertension was suggested not to be done without sufficient evidence.[ 44 ] I speculate that beta‐adrenergic blockers decrease the renin level by their inhibitory action on the sympathetic system and decrease ATII. Unlike ARBs and ACEIs, beta‐adrenergic blockers have an advantage by their action on upstream protein renin which may decrease the activity of both arms. Therefore, beta‐adrenergic blockers not only decrease ATII levels but also reduce the ACE2 receptors, which will be useful in regulating the blood pressure and at the same time decrease the SARS‐CoV‐2 cellular entry. I suggest that beta‐adrenergic blockers can be considered as an alternative to ARBs in the COVID‐19 patients having hypertension and ARBs should be avoided in COVID‐19 patients. Since ARBs, by blocking the angiotensin II receptor increase the ATII level, they may trigger the ARAS loop and complicate the COVID‐19 condition. It has been already shown that ATII induces pulmonary edema in the animal model study,[ 45 ] which supports the view that increased ATII may be detrimental in COVID‐19.
1.5. Why Beta2‐adrenergic Agonist in Nebulizer Form Likely Worsens Symptoms?
Clinicians treat dyspneic patients routinely with beta2‐adrenergic agonists like salbutamol, anticholinergics, and corticosteroids either alone or in combinations. Beta2‐adrenergic agonists are one of the primary drugs used in nebulizers. Considering their role in sympathetic activation and renin release, beta2‐adrenergic agonists may increase ACE2 expression in the alveolar epithelial cells which may help the SARS‐CoV‐2 and worsen the condition. It has been already suggested that beta‐2 agonists should be avoided in ARDS, as they worsen the condition.[ 46 ] As mentioned earlier, beta2‐adrenergic agonists like salbutamol used in the nebulizer solutions, increase the plasma catecholamine level, and produce hypercoagulable state.[ 31 , 32 , 33 ] Considering the vicious ARAS loop, I suggest beta2‐adrenergic agonists should be avoided in the nebulizer solutions, as they may worsen the clinical condition in COVID‐19 patients and may produce complications like pulmonary embolism.
1.6. Why Norepinephrine Given for Septic Shock Complication in COVID‐19 Patients May Worsen the Condition?
Adrenergic system hyperactivation occurs in critical conditions, which increases the catecholamine level. Critically ill COVID‐19 patients may also have sympathetic storm, i.e., increased catecholamine level in the body. So far whether increased catecholamine level occurs in COVID‐19 patients is not known. As any critical condition may activate the adrenergic system, it is likely that in severe COVID‐19 patients catecholamine levels may increase. Increased catecholamine level will lead to increase renin release, which increases the activity of both its arms including an increase in ACE2 expression facilitating the SARS‐Cov‐2 cellular entry and worsening the condition. Some of the severe COVID‐19 patients may end up in septic shock. The primary drug used in septic shock is norepinephrine.[ 19 ] As mentioned earlier, in the critical COVID‐19 patients, endogenous catecholamine levels may be increased, and giving norepinephrine exogenously will further worsen the condition. Many have raised concerns about the use of norepinephrine in septic shock, as norepinephrine may worsen the condition and increase the mortality rate in septic shock.[ 47 , 48 , 49 , 50 ] Not only increased catecholamine but also increased RAAS activation is detrimental in sepsis patients. It has been shown that increased ATII and plasma renin activity worsen the sepsis condition.[ 51 ] Recent studies showed that beta‐adrenergic blockers may be beneficial in septic shock.[ 52 , 53 ] I suggest that norepinephrine should not be used in the treatment of COVID‐19 patients with septic shock, instead beta‐adrenergic blockers should be used.
2. Beta‐Adrenergic Blocker Treatment in COVID‐19: How They Work, and Why They Hold Promise in Treating COVID‐19?
The aforementioned considerations suggest that beta‐adrenergic blockers like Propranolol should be used in COVID‐19 patients, as per the schematic reasoning presented in Figure 2 . Here, I focus mainly on the beta‐adrenergic blocker's role in COVID‐19. Which subtype of beta‐adrenergic receptor blocker plays a crucial role in COVID‐19 remains to be seen. It is possible that alpha1 adrenergic blockers such as Prazosin or combination of beta‐ and alpha‐adrenergic blockers may also be beneficial in COVID‐19. I discuss the advantages of using beta‐adrenergic blockers in COVID‐19 in the following section.
Figure 2.
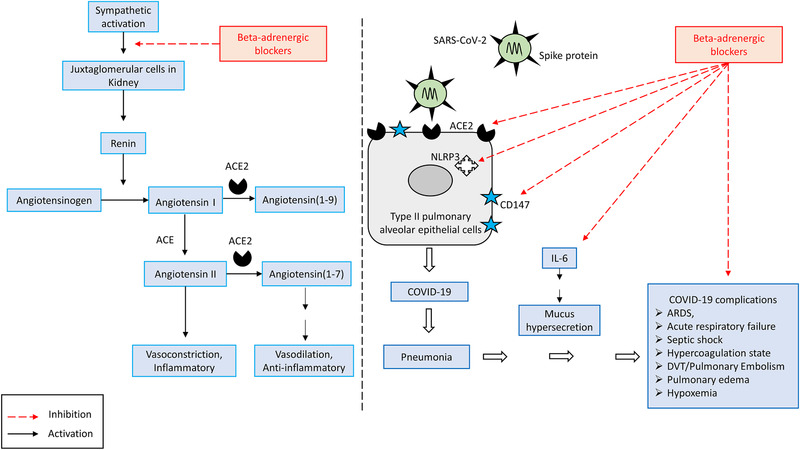
The effect of beta‐adrenergic blockers on RAAS and COVID‐19: A) Beta‐adrenergic blockers by their inhibitory action on the sympathetic nervous system decrease renin release by juxtaglomerular (JG) cells in the Kidney. A decrease in renin may reduce activity in both arms of the RAAS, and may decrease ACE2 receptor expression on cells. B) Beta‐adrenergic blockers decrease ACE2 receptor abundance, hence reducing the opportunities for SARS‐CoV‐2 cellular entry and thereby reducing viral infectivity. Beta‐adrenergic blockers reduce mortality in cases of ARDS, septic shock and respiratory failure. Beta‐adrenergic blockers may inhibit the NLRP3 inflammasome, reduce IL‐6 level, decrease mucus secretion, reduce the pulmonary edema, pulmonary embolism, and refractory hypoxemia complications.
2.1. Beta‐Adrenergic Blockers Reduce the SARS‐COV‐2 Host Cell Entry
Beta‐blockers by its negative regulation on the juxtaglomerular cells in the kidney reduce the activity of both arms of the RAAS pathway, thereby it may decrease the ACE2 level. As ACE2 is the receptor for SARS‐CoV‐2, beta‐adrenergic blockers may decrease the cellular entry of SARS‐CoV‐2. It is already known that beta‐adrenergic blocker propranolol down‐regulates CD147.[ 54 ] Therefore, beta‐adrenergic blockers treatment in COVID‐19 patients may reduce the cellular entry of SARS‐COV‐2 via downregulation of the ACE2 receptor as well as CD147.
2.2. Beta Adrenergic Blockers Reduce IL‐6
It is known that activation of beta‐adrenergic receptors plays a role in the IL‐6 secretion.[ 55 ] It is also known that beta‐adrenergic blockers decrease IL‐6 in cardiac disorder patients.[ 56 , 57 ] I suggest beta‐adrenergic blockers will reduce the IL‐6 level in COVID‐19 patients and reduce the inflammatory complications associated with it.
2.3. Beta Adrenergic Blockers Decrease Proinflammatory Cytokines and Inhibit Cytokine Storm
Beta‐adrenergic blockers have been shown to decrease a variety of proinflammatory cytokines expression including IL‐1β, IL‐6, TNFα, IFNγ.[ 56 , 57 , 58 , 59 , 60 ] The use of beta‐adrenergic blockers in COVID‐19 patients may reduce the cytokine storm by decreasing the expression of the proinflammatory cytokines and the inflammation associated with it.
2.4. Beta‐Adrenergic Blockers Might be Beneficial in ARDS and Respiratory Failure Condition
It is known that beta‐adrenergic agonists counterintuitively worsen the ARDS condition and beta‐adrenergic blockers have been shown to reduce the mortality in ARDS.[ 46 ] Based on the BASEL II ICU study Noveanu et al had shown that beta‐adrenergic blockers reduce mortality in respiratory failure cases.[ 61 ] A recent study has shown that beta‐adrenergic blockers reduce mortality in ARDS.[ 62 ]
2.5. Beta‐Adrenergic Blockers are Beneficial in Septic Shock
Recent trends suggest that beta‐adrenergic blockers are beneficial in septic shock and reduce the mortality rate.[ 52 , 53 ] As mentioned earlier, many have raised concerns about the use of norepinephrine in septic shock.[ 47 , 48 , 49 , 50 ] I suggest that norepinephrine for treating septic shock in the COVID‐19 condition should be avoided. Instead beta‐adrenergic blockers should be used for treating septic shock in COVID‐19 patients.
2.6. Beta‐Adrenergic Blockers May Reduce the Pulmonary Edema
As mentioned earlier, increased catecholamine levels may trigger the ARAS loop and worsen the COVID‐19. It is well known that increased catecholamine induces pulmonary edema in the animal model studies. Studies have shown that both alpha‐ and beta‐adrenergic receptors may be involved in this. Both alpha1 and beta‐adrenergic blockers have shown to relieve pulmonary edema in many animal models studies. Due to adrenergic receptor involvement in the etiology of pulmonary edema, it has been suggested that beta‐2 agonists should not be used routinely in the mechanically ventilated patients.[ 63 ] Increased catecholamine level induced pulmonary edema can be prevented by adrenergic receptor blockers. I suggest that beta‐adrenergic blockers like Propranolol or alpha1 adrenergic blockers like Prazosin should be used to reduce or prevent pulmonary edema in COVID‐19 patients.
2.7. Beta Adrenergic Blockers May Reduce the Hypercoagulation State and Prevent Pulmonary Embolism in COVID‐19 Patients
Severely affected COVID‐19 patients developed pulmonary embolism in COVID‐19 and D‐dimer level was shown to be high.[ 15 , 16 , 17 ] This indicates that the hypercoagulation state occurs in COVID‐19. Adrenergic hyperactivation is known to be associated with the hypercoagulation state. Beta‐adrenergic blockers have been shown to reduce the coagulation parameters and prevent complications like venous thrombosis in both animal models and patients.[ 64 , 65 ] I suggest that beta‐adrenergic blockers should be used in COVID‐19 patients to prevent or reduce the hypercoagulation state complications like pulmonary embolism and deep vein thrombosis.
2.8. Improvement of Oxygenation level by Beta Adrenergic Blockers
Beta‐adrenergic blockers have been shown to improve the oxygenation level in the patients who were on ECMO.[ 66 ] This suggests the possibility that the use of beta‐adrenergic blockers in COVID‐19 patients who are on ECMO may improve the oxygenation level. It has been shown that beta‐adrenergic blockers improved the oxygenation level in the hypoxic condition induced in an animal study.[ 67 ] It is known that beta‐adrenergic blockers improve the PaO2 level in critically ill patients and increase the oxygenation.[ 68 , 69 ] Considering the above evidences, I hypothesize that beta‐adrenergic blockers may improve the oxygenation level and reduce the hypoxemia in COVID‐19 patients.
2.9. Beta‐Adrenergic Blockers May Reduce the Mucus Hypersecretion in COVID‐19
Increased mucus aggregation has been noticed in the distal airways and alveoli in the postmortem study of COVID‐19 patients.[ 10 ] It is known that IL‐6 plays a key role in the activation of MUC5AC and MUC5B gene expression and increases the mucus secretion.[ 11 ] It has been suggested that Tocilizumab by the inhibition of IL‐6 receptor may help decrease the mucus secretion in COVID‐19.[ 12 ] As mentioned earlier, beta‐adrenergic blockers decrease IL‐6. It is interesting to note that chronic propranolol administration has been shown to decrease MUC5AC expression and mucus secretion.[ 70 ] I hypothesize that the use of beta‐adrenergic blockers like propranolol may decrease the mucus secretion in COVID‐19 patients.
2.10. Computational Studies Suggest the Potential use of Beta‐Adrenergic Blocker in COVID‐19
Wu et al. using the computational method has shown some potential molecules that could be used for the treatment in COVID‐19. In the article, beta‐adrenergic blocker Oxprenolol has been suggested to potentially inhibit SARS‐CoV‐2 papain‐like protease (PLpro) and Carvedilol has been suggested to potentially inhibit SARS‐CoV‐2 3‐chymotrypsin like protease.[ 71 ] Using a network‐based approach Zhou et al shown 16 potential drugs that can be used in the treatment of COVID‐19 and one of the drugs mentioned was beta‐adrenergic blocker Carvedilol.[ 23 ] The above two recent computational studies support the hypothesis that beta‐adrenergic blockers may be useful in the treatment of COVID‐19 patients.
2.11. Beta‐Adrenergic Blockers Inhibit NLRP3 Inflammasome
As mentioned earlier, NLRP3 has been suggested to be activated in COVID‐19 and a clinical trial targeting the inhibition of NLRP3 using Colchicine is already registered.[ 25 ] It is interesting to note that beta‐adrenergic blocker carvedilol has been shown to inhibit NLRP3 inflammasome.[ 72 ] Thus, one of the many uses of beta‐adrenergic blockers in COVID‐19 condition may be the inhibition of NLRP3 inflammasome and thereby reducing the inflammation by decreasing the NLRP3 downstream effectors IL‐1 and IL‐8.
2.12. Use of Beta‐Adrenergic Blockers are Safe in Respiratory Illness Condition
Traditionally clinicians are hesitant in using beta‐blockers in patients with respiratory illness. But recent studies are changing this viewpoint, for example, beta‐adrenergic blockers have been shown beneficial effects in chronic obstructive pulmonary disease (COPD) patients.[ 73 ] I suggest that clinicians should not hesitate to use beta‐adrenergic blockers in COVID‐19 patients having respiratory dysfunction.
Considering the above evidence beta‐adrenergic blockers like Propranolol may be a good candidate for treating COVID‐19 patients. As mentioned earlier which subtype of beta‐adrenergic receptor blocker works better in COVID‐19 needs to be clarified in the future. It is possible that alpha1‐adrenergic blockers like Prazosin or combination of beta and alpha‐adrenergic blockers may also be beneficial in COVID‐19.
3. Testing of the Hypothesis
3.1. Testing the Existence of ARAS Loop and Sympathetic Storm in COVID‐19 Patients
To check the presence of the hypothetical ARAS loop, I suggest measuring the following in COVID‐19 patients—plasma catecholamine levels especially plasma norepinephrine levels, AT II, ACE, ACE2, Renin levels. These parameters need to be measured in all the COVID‐19 patients along with the viral load measurement. If the plasma concentration of above said ARAS loop molecules correlates with the viral load in the COVID‐19, it would validate the existence of the ARAS loop in COVID‐19. The plasma catecholamine levels in COVID‐19 patients have to be measured in all the COVID‐19 patients which would clarify whether a sympathetic storm occurs in this condition.
3.2. Retrospective and Prospective Clinical Trials Would Test the Effect of Beta‐Adrenergic Blockers in COVID‐19 Patients
I suggest conducting retrospective and prospective clinical trials to clarify the following questions;
Do beta‐adrenergic blockers reduce the morbidity and mortality in COVID‐19 patients?
Do beta‐adrenergic blockers reduce the SARS‐CoV‐2 host cell entry and viral load?
4. Conclusion
The interrelationship between the adrenergic system, RAAS, ACE2, and SARS‐CoV‐2 plays a crucial role in COVID‐19 pathology. I propose a hypothetical ARAS loop, hyperactivation of which may be the pathophysiological mechanism behind COVID‐19. Beta‐adrenergic blockers may produce beneficial effects in COVID‐19 patients in many ways ranging from decreasing the SARS‐CoV‐2 virus entry, inhibiting NLRP3 inflammasome, reduction of IL‐6 to decreasing the complications such as pulmonary embolism, ARDS, and septic shock. It is important now to act quickly and do retrospective clinical studies on the COVID‐19 patients who were already on beta‐adrenergic blockers for pre‐existing cardiovascular illness, and assess their effect on mortality. The proposed retrospective studies will test the validity of the hypothesis that I develop in this paper. If the retrospective study result transpires as expected, it would help millions of people all over the world, because beta‐adrenergic blockers are well‐known drugs with longstanding safety profiles. Furthermore, they are relatively inexpensive, can be taken orally and intravenously, and are readily available in all parts of the world. It is imperative to do the proposed retrospective and prospective clinical trial studies as soon as possible, with the aim of clarifying the role of beta‐adrenergic blockers in COVID‐19 patients.
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgements
The author would like to thank Dr. Raja for his help in editing the manuscript.
Vasanthakumar N., Beta‐Adrenergic Blockers as a Potential Treatment for COVID‐19 Patients. BioEssays 2020, 42, 2000094. 10.1002/bies.202000094
References
- 1. https://www.who.int/docs/default-source/coronaviruse/situation-reports/20200726-covid-19-sitrep-188.pdf?sfvrsn=f177c3fa_2 (accessed: July 2020).
- 2. Hoffmann M., Kleine‐Weber H., Schroeder S., Krüger N., Herrler T., Erichsen S., Schiergens T. S., Herrler G., Wu N. H., Nitsche A., Müller M. A., Drosten C., Pöhlmann S., Cell 2020, 181, 271.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zhang H., Penninger J. M., Li Y., Zhong N., Slutsky A. S., Intensive Care Med. 2020, 46, 586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ulrich H., Pillat M. M., Stem Cell Rev. Rep. 2020, 16, 434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wang K., Chen W., Zhou Y. S., Lian J. Q., Zhang Z., Du P., Gong L., Zhang Y., Cui H. Y., Geng J. J., Wang B.. SARS‐CoV‐2 invades host cells via a novel route: CD147‐spike protein. BioRxiv. 2020 Jan 1 (accessed: July 2020).
- 6. Gandhi R. T., Lynch J. B., Del Rio C., N. Engl. J. Med. 2020. [Google Scholar]
- 7. Cao X., Nat. Rev. Immunol. 2020, 20, 269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhou F., Yu T., Du R., Fan G., Liu Y., Liu Z., Xiang J., Wang Y., Song B., Gu X., Guan L., Wei Y., Li H., Wu X., Xu J., Tu S., Zhang Y., Chen H., Cao B., Lancet 2020, 395, 1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Henry B. M., Lancet Respir. Med. 2020, 8, e24.32178774 [Google Scholar]
- 10. Wang C., Xie J., Zhao L., Fei X., Zhang H., Tan Y., Nie X., Zhou L., Liu Z., Ren Y., Yuan L., Zhang Y.u, Zhang J., Liang L., Chen X., Liu X., Wang P., Han X., Weng X., Chen Y., Yu T., Zhang X., Cai J., Chen R., Shi Z.‐L.i, Bian X.‐W.u, EBioMedicine 2020, 57, 102833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chen Y., Thai P., Zhao Y.u‐H., Ho Y.e‐S., Desouza M. M., Wu R., J. Biol. Chem. 2003, 278, 17036. [DOI] [PubMed] [Google Scholar]
- 12. Zhu Z., Cai T., Fan L., Lou K., Hua X., Huang Z., Gao G., Int. J. Infect. Dis. 2020, 95, 332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhang H., Zhou P., Wei Y., Yue H., Wang Y.i, Hu M., Zhang S., Cao T., Yang C., Li M., Guo G., Chen X., Chen Y., Lei M., Liu H., Zhao J., Peng P., Wang C.‐Y.i, Du R., Ann. Intern. Med. 2020, 172, 629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. https://www.who.int/publications/i/item/clinical-management-of-severe-acute-respiratory-infection-when-novel-coronavirus-(ncov)-infection-is-suspected (accessed: July 2020).
- 15. Leonard‐Lorant I., Delabranche X., Severac F., Helms J., Pauzet C., Collange O., Schneider F., Labani A., Bilbault P., Moliere S., Leyendecker P., Roy C., Ohana M., Radiology 2020, 201561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Poissy J., Goutay J., Caplan M., Parmentier E., Duburcq T., Lassalle F., Jeanpierre E., Rauch A., Labreuche J., Susen S., Circulation 2020, 142, 184. [DOI] [PubMed] [Google Scholar]
- 17. Grillet F., Behr J., Calame P., Aubry S., Delabrousse E., Radiology 2020, 201544, 296, E186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wu Z., Mcgoogan J. M., JAMA 2020, 323, 1239. [DOI] [PubMed] [Google Scholar]
- 19. Rhodes A., Evans L. E., Alhazzani W., Levy M. M., Antonelli M., Ferrer R., Kumar A., Sevransky J. E., Sprung C. L., Nunnally M. E., Rochwerg B., Rubenfeld G. D., Angus D. C., Annane D., Beale R. J., Bellinghan G. J., Bernard G. R., Chiche J. D., Coopersmith C., De Backer D. P., French C. J., Fujishima S., Gerlach H., Hidalgo J. L., Hollenberg S. M., Jones A. E., Karnad D. R., Kleinpell R. M., Koh Y., Lisboa T. C., Machado F. R., Marini J. J., Marshall J. C., Mazuski J. E., Mcintyre L. A., Mclean A. S., Mehta S., Moreno R. P., Myburgh J., Navalesi P., Nishida O., Osborn T. M., Perner A., Plunkett C. M., Ranieri M., Schorr C. A., Seckel M. A., Seymour C. W., Shieh L., Shukri K. A., Simpson S. Q., Singer M., Thompson B. T., Townsend S. R., Van Der Poll T., Vincent J. L., Wiersinga W. J., Zimmerman J. L., Dellinger R. P., Intensive Care Med. 2017, 43, 304.28101605 [Google Scholar]
- 20. Pedersen S. F., Ho Y. A. C., J. Clin. Invest. 2020, 130, 2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mehta P., Mcauley D. F., Brown M., Sanchez E., Tattersall R. S., Manson J. J., Lancet 2020, 395, 1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhang X., Wu K., Wang D. I., Yue X., Song D., Zhu Y., Wu J., Virology 2007, 365, 324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhou Y., Hou Y., Shen J., Huang Y., Martin W., Cheng F., Cell Discov. 2020, 6, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chen X., Zhao B., Qu Y., Xiong J., Feng Y., Men D., Huang Q., Liu Y., Yang B., Ding J., Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Deftereos S. G., Siasos G., Giannopoulos G., Vrachatis D. A., Angelidis C., Giotaki S. G., Gargalianos P., Giamarellou H., Gogos C., Daikos G., Lazanas M., Lagiou P., Saroglou G., Sipsas N., Tsiodras S., Chatzigeorgiou D., Moussas N., Kotanidou A., Koulouris N., Oikonomou E., Kaoukis A., Kossyvakis C., Raisakis K., Fountoulaki K., Comis M., Tsiachris D., Sarri E., Theodorakis A., Martinez‐Dolz L., Sanz‐Sanchez J., Reimers B., Stefanini G. G., Cleman M., Filippou D., Olympios C. D., Pyrgakis V. N., Goudevenos J., Hahalis G., Kolettis T. M., Iliodromitis E., Tousoulis D., Stefanadis C., Hell. J. Cardiol. 2020, 61, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Siu K. L., Yuen K. S., Castano‐Rodriguez C., Ye Z. W., Yeung M. L., Fung S. Y., Yuan S., Chan C. P., Yuen K. Y., Enjuanes L., Jin D. Y., FASEB J. 2019, 33, 8865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chen I. Y., Moriyama M., Chang M. F., Ichinohe T., Front. Microbiol. 2019, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yang Y., Wang H., Kouadir M., Song H., Shi F., Cell Death Dis. 2019, 10, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Greene K. E., Wright J. R., Steinberg K. P., Ruzinski J. T., Caldwell E., Wong W. B., Hull W., Whitsett J. A., Akino T., Kuroki Y., Nagae H., Hudson L. D., Martin T. R., Am J. Respir. Crit. Care Med. 1999, 160, 1843. [DOI] [PubMed] [Google Scholar]
- 30. Yu J., Ni L., Zhang X., Zhang J., Abdel‐Razek O., Wang G., Shock 2019, 51, 557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Snyder E. M., Wong E. C., Foxx‐Lupo W. T., Wheatley C. M., Cassuto N. A., Patanwala A. E., Pharmacother.: J. Hum. Pharmacol. Drug Ther. 2011, 31, 748. [DOI] [PubMed] [Google Scholar]
- 32. Von Kanel R., Dimsdale J. E., Eur. J. Haematol. 2000, 65, 357. [DOI] [PubMed] [Google Scholar]
- 33. Ali‐Saleh M., Sarig G., Ablin J. N., Brenner B., Jacob G., PLoS One 2016, 11, e0158652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sanders J. M., Monogue M. L., Jodlowski T. Z., Cutrell J. B., JAMA 2020, 323, 1824. [DOI] [PubMed] [Google Scholar]
- 35. https://www.covid19treatmentguidelines.nih.gov/whats-new/ (accessed: July 2020).
- 36. Henry B. M., Lippi G., J. Crit. Care 2020, 58, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ñamendys‐Silva S. A., Heart Lung J. Crit. Care 2020, 49, 348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Groves A. C., Griffiths J., Leunc F., Meek R. N., Ann. Surg. 1973, 178, 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mason J. W., Buescher E. L., Belfer M. L., Artenstein M. S., Mougey E. H., J. Hum. Stress 1979, 5, 18. [DOI] [PubMed] [Google Scholar]
- 40. Rabelo L. A., Alenina N., Bader M., Hypertens. Res. Off. J. Jpn. Soc. Hypertens. 2011, 34, 154. [DOI] [PubMed] [Google Scholar]
- 41. Esler M., Esler D., J. Hypertens. 2020, 38, 781. [DOI] [PubMed] [Google Scholar]
- 42. Fang L., Karakiulakis G., Roth M., Lancet Respir. Med. 2020, 8, e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kuster G. M., Pfister O., Burkard T., Zhou Q., Twerenbold R., Haaf P., Widmer A. F., Osswald S., Eur. Heart J. 2020, 41, 1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Vaduganathan M., Vardeny O., Michel T., Mcmurray J. J. V., Pfeffer M. A., Solomon S. D., N. Engl. J. Med. 2020, 382, 1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yamamoto T., Wang L.i‐M., Shimakura K., Sanaka M., Koike Y., Mineshita S., Jpn. J. Pharmacol. 1997, 73, 33. [DOI] [PubMed] [Google Scholar]
- 46. Sweeney R. M., McAuley D. F., Lancet 2016, 388, 2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Stolk R. F., Van Der Poll T., Angus D. C., Van Der Hoeven J. G., Pickkers P., Kox M., Am J. Respir. Crit. Care Med. 2016, 194, 550. [DOI] [PubMed] [Google Scholar]
- 48. Martin C., Medam S., Antonini F., Alingrin J., Haddam M., Hammad E., Meyssignac B., Vigne C., Zieleskiewicz L., Leone M., Shock 2015, 44, 305. [DOI] [PubMed] [Google Scholar]
- 49. Natesan V., Int. J. Clin. Exp. Physiol. 2018, 5, 3. [Google Scholar]
- 50. Singer M., Lancet 2007, 370, 636. [DOI] [PubMed] [Google Scholar]
- 51. Doerschug K. C., Delsing A. S., Schmidt G. A., Ashare A., Crit. Care 2010, 14, R24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Coppola S., Froio S., Chiumello D., Crit. Care 2015, 19, 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Tan K., Harazim M., Tang B., Mclean A., Nalos M., Crit. Care. 2019, 23, 298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Xie W., Xie H., Liu F., Li W., Dan J., Liu L., Dan L., Xiao X., Li J., Chen X., Br. J. Dermatol. 2013, 168, 739. [DOI] [PubMed] [Google Scholar]
- 55. Chen C., Du J., Feng W., Song Y., Lu Z., Xu M., Li Z., Zhang Y., Br. J. Pharmacol. 2012, 166, 676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Doo Y. C., Kim D. M., Oh D. J., Ryu K. H., Rhim C. Y., Lee Y., Am. J. Cardiol. 2001, 88, 422. [DOI] [PubMed] [Google Scholar]
- 57. Matsumura T., Tsushima K., Ohtaki E., Misu K., Tohbaru T., Asano R., Nagayama M., Kitahara K., Umemura J., Sumiyoshi T., Hosoda S., J. Cardiol. 2002, 39, 253. [PubMed] [Google Scholar]
- 58. Deten A., Volz H. C., Hölzl A., Briest W., Zimmer H. G., Mol. Cell. Biochem. 2003, 251, 127. [PubMed] [Google Scholar]
- 59. Hajighasemi F., Mirshafiey A., Int. J. Hematol.‐Oncol. Stem Cell Res. 2016, 10, 99. [PMC free article] [PubMed] [Google Scholar]
- 60. Woiciechowsky C., Schöning B., Stoltenburg‐Didinger G., Stockhammer F., Stockhammer H. D., Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2004, 10, BR325. [PubMed] [Google Scholar]
- 61. Noveanu M., Breidthardt T., Reichlin T., Gayat E., Potocki M., Pargger H., Heise A., Meissner J., Twerenbold R., Muravitskaya N., Mebazaa A., Mueller C., Crit. Care. 2010, 14, R198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Al‐Qadi M. O., Kashyap R., Am J. Respir. Crit. Care Med. 2015, 191, p. A1602. [Google Scholar]
- 63. Rassler B., Scientifica 2012, 2012, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Gruszecki M., Rółkowski R., Pawlak R., Buczko W., Pol. J. Pharmacol. 2001, 53, 5. [PubMed] [Google Scholar]
- 65. Hoppener M. R., Kraaijenhagen R. A., Hutten B. A., Buller H. R., Peters R. J. G., Levi M., J. Thromb. Haemost. JTH 2004, 2, 1316. [DOI] [PubMed] [Google Scholar]
- 66. Bunge J. J. H., Diaby S., Valle A. L., Bakker J., Gommers D., Vincent J. L., Creteur J., Taccone F. S., Reis Miranda D., J. Crit. Care 2019, 53, 248. [DOI] [PubMed] [Google Scholar]
- 67. Khambatta H. J., Stone J. G., Askanazi J., Khan E., Br. J. Anaesth. 1987, 59, 1171. [DOI] [PubMed] [Google Scholar]
- 68. Vincent J. L., Lignian H., Gillet J. B., Berre J., Contu E., Chest 1985, 88, 558. [DOI] [PubMed] [Google Scholar]
- 69. Wood G., Crit. Care Med. 1997, 25, 1807. [DOI] [PubMed] [Google Scholar]
- 70. Zhou Y., Zhang Y., Guo Y., Zhang Y., Xu M., He B., PLoS One 2014, 9, e97788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Wu C., Liu Y., Yang Y., Zhang P., Zhong W.u, Wang Y., Wang Q., Xu Y., Li M., Li X., Zheng M., Chen L., Li H., Acta Pharm. Sin. B 2014, 10, 766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Wong W. T., Li L. H., Rao Y. K., Yang S. P., Cheng S. M., Lin W. Y., Cheng C. C., Chen A., Hua K. F., Front. Immunol. 2018, 9, 1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Nielsen A. O., Pedersen L., Sode B. F., Dahl M., EClinicalMedicine 2019, 7, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]