

Summary
Understanding Covid‐19 pathophysiology is crucial for a better understanding of the disease and development of more effective treatments. Alpha‐1‐antitrypsin (A1AT) is a constitutive tissue protector with antiviral and anti‐inflammatory properties. A1AT inhibits SARS‐CoV‐2 infection and two of the most important proteases in the pathophysiology of Covid‐19: the transmembrane serine protease 2 (TMPRSS2) and the disintegrin and metalloproteinase 17 (ADAM17). It also inhibits the activity of inflammatory molecules, such as IL‐8, TNF‐α, and neutrophil elastase (NE). TMPRSS2 is essential for SARS‐CoV‐2‐S protein priming and viral infection. ADAM17 mediates ACE2, IL‐6R, and TNF‐α shedding. ACE2 is the SARS‐CoV‐2 entry receptor and a key component for the balance of the renin‐angiotensin system, inflammation, vascular permeability, and pulmonary homeostasis. In addition, clinical findings indicate that A1AT levels might be important in defining Covid‐19 outcomes, potentially partially explaining associations with air pollution and with diabetes. In this review, we focused on the interplay between A1AT with TMPRSS2, ADAM17 and immune molecules, and the role of A1AT in the pathophysiology of Covid‐19, opening new avenues for investigating effective treatments.
Keywords: A1AT, ADAM17, Covid‐19, SARS‐CoV‐2, TMPRSS2
Abbreviations
- A1AT
Alfa‐1‐antitrypsin
- A549
pulmonary type II‐like epithelial cells
- ACE2
angiotensin I converting enzyme II
- ADAM17
disintegrin and metalloproteinase 17
- Ang‐
angiotensin
- AREG
amphiregulin
- AT1R
protein G receptor for angiotensin
- BEAS‐2B
normal human pulmonary epithelial cell line
- CD19
cluster of differentiation 19
- CD32a
glycoprotein surface receptor that belongs to the Ig gene superfamily
- CD40
cluster of differentiation 40
- COPD
chronic obstructive pulmonary disease
- COVID‐19
Corona Virus disease 2019
- CR3
complement Receptor 3
- CXCR1
chemokine receptor type 1
- delC
nucleotide deletion
- DIC
disseminated intravascular coagulation
- EGFR
epithelial growth factor receptor
- ENaC‐α
epithelial sodium channel
- eQTL
expression quantitative trait locus
- FcyRIIIB
type IIIFcγ receptor
- FGF7
fibroblast growth factor 7
- fMLP
N‐formyl peptide receptor
- HRV
human rhinoviruses
- IL
interleukin
- IRF1/2
regulatory factor of interferon 1/2
- Ki‐67+B
B cell that express the nuclear protein Ki‐67
- LPS
lipopolysaccharide
- MAS
cell membrane protein, part of RAS system
- Met358
metihonine residue 358
- MMP‐12
matrix metallopeptidase 12
- NCI‐H292
airway epithelium like cell line
- NE
neutrophil elastase
- NPS
neuropeptide S
- P2
purine 2
- PAR‐1
protease‐activated receptor I
- PKC
protein kinase C
- PS
phosphatidylserine
- RAS
renin angiotensin system
- RRARSVAS
New Corona virus exclusive S1/S2 cleavage site
- S
virus spike protein
- SARS‐CoV‐2‐
Severe Acute Respiratory Syndrome Corona Virus 2
- STAT3
signal transducer and activator of transcription III
- T735
threonine residua 735
- TMPRSS2
protease transmembrane protease, serine 2
- VIRIP
virus inhibitory peptide
1. INTRODUCTION
In December 2019, the severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) emerged in Wuhan, Hubei province, China to cause a pandemic. 1 Improved understanding of Covid‐19 pathophysiology, the role of host defense molecules and mechanisms against viral infection are crucial for better understanding the disease, and developing more effective treatment strategies.
Alpha‐1‐antitrypsin (A1AT) is a constitutive tissue protector, as well as an antiviral and anti‐inflammatory molecule. 2 , 3 A1AT is an inhibitor of SARS‐CoV‐2 infection and two of the most important proteases in the pathophysiology of Covid‐19: the transmembrane serine protease 2 (TMPRSS2) and the disintegrin and metalloproteinase 17 (ADAM17), as was well as an inhibitor of inflammatory molecules, such as IL‐8, TNF‐α, and neutrophil elastase. 4 , 5 , 6 Moreover, recent data indicate that lower IL‐6:A1AT levels are related to worse prognosis in Covid‐19 patients. 7
This review addresses the interplay between A1AT, TMPRSS2, ADAM17, and inflammatory molecules during SARS‐CoV‐2 infection with the aim of identifying new avenues for effective treatments against Covid‐19.
2. SARS‐COV‐2 INFECTION
SARS‐CoV can use a non‐endosomal or an endosomal pathway to enter cells. 8 The non‐endosomal pathway requires the virus spike (S) protein to bind host angiotensin II converting enzyme receptor (ACE2). Immediately after, the proteolytic action of TMPRSS2 processes S into S1 and S2 subunits. 9 The endosomal pathway is a less efficient and non‐essential pathway; it depends on cathepsin L, a pH‐dependent host‐cell protease, to fuse the S protein in lysosomes. 8 , 10
A recent study demonstrated that blockage of both TMPRSS2 and cathepsin L by camostat mesylate inhibits, but does not abrogate, SARS‐CoV‐2 infection of Calu‐3 cells, inferring the possible involvement of other proteases; furin is a strong candidate. 9 Sequence‐based studies found four SARS‐CoV‐2‐redundant furin cleavage sites that are absent from SARS‐CoV. 11 In addition, the new coronavirus presents an exclusive S1/S2 cleavage site (RRARSVAS) that mimics a host furin‐cleavable peptide in the epithelial sodium channel α‐subunit (ENaC‐α). 12 This potential furin‐mediated cleavage was thought to explain the higher infectivity of SARS‐CoV‐2 compared to SARS‐CoV. However, Xia et al recently demonstrated, in an in vitro assay using 293T cells, that furin cleavage sites might not be very relevant for SARS‐CoV‐2 infections in the human airway. 13 Moreover, an elastase cleavage site near the S1‐S2 protein was recently identified in the A2a SARS‐CoV subtype (D614G mutation), suggesting an important role of neutrophil elastase in this infection. 14 Therefore, the possible participation of other proteases in viral entry requires further investigation.
3. ACE2/TMPRSS2/ADAM17 INTERPLAY AND PATHOPHYSIOLOGY OF COVID‐19
The ACE2 receptor is a type I transmembrane protein mainly expressed in the testicles, cardiovascular system, intestine, brain, oral mucosa, and lungs. 15 Besides being the entry receptor for SARS‐CoV‐2, ACE2 is a component of the renin‐angiotensin system (RAS). It is responsible for converting angiotensin (Ang) I into Ang 1‐9 and Ang II into Ang 1‐7, which act on the MAS‐receptor. 16 , 17 The ACE2/Ang1‐7, Ang1‐9/MAS axis counterbalances the ACE1/AngII/AT1R axis, enabling protection against fibrosis, pulmonary injury, diabetic cardiovascular complications, myocardial infarction, and disseminated intravascular coagulation (DIC), all of which are associated with severe Covid‐19 outcomes. 18 , 19 , 20 Therefore, decreased tissue ACE2 might be a Covid‐19 risk factor. 21
Indeed, ACE2 plasma levels are low in healthy individuals and increase during male ageing process. 22 , 23 In addition, Sama et al observed higher ACE2 plasma levels in men with heart failure than in women with the same condition. 21 Circulating levels of ACE2 were increased in diabetes, chronic obstructive pulmonary disease (COPD) and nasal and bronchial airways of smokers compared with non‐smokers, some Covid‐19 comorbidities. 24 , 25 These findings, along with the observation of higher SARS‐CoV‐2 viral load and high soluble ACE2 plasma levels in elderly men, support the relevance of ACE2 tissue levels in Covid‐19 severity. 22 , 26
Tissue ACE2 is down‐modulated by SARS‐CoV‐2 endocytosis and cleavage by ADAM17 and TMPRSS2. 27 , 28 Analysis of the differential fate of the ACE2 cleavage products and amino acids required for shedding suggests that ADAM17 and TMPRSS2 cleave different sites, resulting in distinct biological consequences. While a stable ACE2 ectodomain was found in culture supernatants previously treated with ADAM17, it was absent from TMPRSS2‐treated culture supernatants. 29
The protease ADAM17 is a type I sheddase transmembrane protein expressed in muscle, lungs, the placenta, ovaries, testicles, pancreas, kidneys, small intestine, thymus, and heart. 30 The mechanisms by which ADAM17 is regulated remain poorly understood. However, its membrane sheddase function was induced by apoptosis, Ca2+ ionophores, fibroblast growth factor 7 (FGF7), protein kinase C (PKC) activators, and purine 2 (P2) receptor agonists. 31 The activation of ADAM17 also involves the translocation of phosphatidylserine (PS) to the cell membrane outer leaflet. 31 Moreover, in vitro assays revealed that IL‐1β can also activate ADAM17 through threonine residue 735 (T735) phosphorylation in a cytoplasmic domain non‐dependent manner. 32 In addition, the uptake of SARS‐S‐ACE2 also promotes ADAM17 activation. 33 Although the mechanism is unclear, ADAM17 siRNA prevented SARS‐CoV infection, indicating an important role of ADAM17 in viral infection. 33
ADAM17 is involved in several physiological and pathophysiological processes, such as the activation of TNF‐α and IL6R cleavage in pulmonary inflammation. 34 The pro‐inflammatory cytokine TNF‐α is related to the “cytokine storm,” a Covid‐19 phenomenon that increases the risk of vascular hyper‐permeability, multiorgan failure, and death. 35 A study described high tissue levels of ADAM17 and TNF‐α in an animal model of COPD. 36 Patients with COPD present elevated serum ACE2 levels and severe Covid‐19 outcomes, suggesting that ADAM17 might be a risk factor for this new disease. 37
Levels of soluble TNF‐α were elevated in patients with type 2 diabetes. Patients with diabetic low‐density lipoprotein (LDL) have shown higher gene expressions of ADAM17. 38 Since, ADAM17 plays an important role in TNF‐α shedding, this suggests that ADAM17 activity is increased in a high sugar environment, explaining why free TNF‐α levels are elevated in diabetic patients.
TNF‐α induces IL‐8 release from pulmonary type II‐like epithelial cells (A549) and IL‐8 secretion in airway epithelium‐like NCI‐H292 cells. 39 , 40 IL‐8 is an important neutrophil chemotactic factor produced by lung fibroblasts and type II epithelial cells, plays an essential role in variety pulmonary acute inflammations and is highly expressed in Covid‐19 patients. 41 , 42 , 43 The administration of ADAM17 inhibitors in acute allergic lung inflammation models diminished neutrophil and eosinophil migration, decreasing inflammation and damage, suggesting a contribution of ADAM17 to inflammatory processes. 44 Additionally, a neutrophil ex vivo assay showed that the activity of ADAM17 is upregulated under immune complex stimuli, leading to FcγRIIIb shedding. FcγRIIIb can then interact with CR3, CD32a, or the fMLP receptor, enabling cell migration during inflammatory processes. 4
IL‐6R, which is expressed in the airway epithelial cells, can be cleaved by ADAM17. The resulting soluble IL‐6R/IL‐6 complex can promote autocrine or paracrine activation by binding to gp130, activating STAT3 signaling in lung fibroblasts, myofibroblasts, as well as smooth muscle cells and inflammation. 45 In addition, ADAM17 also sheds an airway epithelial cell EGFR ligand amphiregulin (AREG). 46 AREG binding to EGFR can also lead to STAT3 signaling paracrine activation. 47 Thus, Stolarczyk and Scholte suggested that the trans‐signaling of AREG and IL‐6R could be relevant in lung fibrosis and COPD. 47
TMPRSS2 is a type II transmembrane serine protease. 48 It contains a trypsin‐like substrate domain, binding sites for calcium and LDL. 49 It is expressed in the pancreas, prostate, colon, kidneys, liver, and lungs. 50 Pulmonary expression occurs through type I and II alveolar cells, macrophages, and bronchial epithelial cells. 51 Despite being androgen‐induced in the prostate, TMPRSS2 lung levels show no significant difference between men and women (below 60 years old). 52 TMPRSS2 is crucial in SARS‐CoV‐2 infection and seems to have no relevant known physiological function. 53 Therefore, TMPRSS2 modulation has become a promising therapeutic approach against Covid‐19. 54
Recently, Bhattacharyya et al described a nucleotide deletion (delC) in the lung‐specific expression quantitative trait locus (eQTL) rs35074065, that regulates the expression of TMPRSS2 and MX1 genes. The rs35074065‐delC variant, common in Europeans and North Americans, does not hold the binding site for the repressor IRF2, only for the IRF1 transcription factor. Thus, the rs35074065‐delC variant may promote an increased expression of TMPRSS2 and MX1 genes, increasing the cleavage of SARS‐CoV‐2S protein in these populations. The expression of MX1 leads to IFN type‐I activation and neutrophil infiltration, possibly increasing the release of neutrophil elastase (NE). 14
In summary, the pathophysiological relevance of ACE2 down‐regulation on Covid‐19 is controversial. Since ACE2 is a SARS‐CoV‐2 entry receptor, one might consider that ACE2, virus entry and disease severity are directly proportional. 55 Moreover, the ADAM17‐mediated release of the ACE2 ectodomain in the blood could lead to virus neutralization. 56 However, the loss of cell membrane active ACE2 due to ADAM17, TMPRSS2, and virus uptake, might lead to the increased production of Ang II and activation of AT1R, resulting in breakdown of homeostasis of the RAS system and detrimental effects, such as inflammation, increased vascular permeability, pulmonary injury, and DIC in Covid‐19 patients. 56 , 57
Considering the deleterious effects of TMPRSS2, ADAM17, and inflammatory cytokines involved in Covid‐19, the identification of cellular inhibitors for these molecules is crucial. In this context, the host protein A1AT emerges as an important candidate against SARS‐CoV‐2 infection and Covid‐19, as discussed below.
4. ALPHA‐1‐ANTITRYPSIN‐A1AT
The serine protease inhibitor alpha‐1‐antitrypsin (A1AT) is a Mr 52 000 protein with a half life of 3 to 5 days. A1AT is synthesized in the endoplasmic reticulum and released by the Golgi apparatus. It is expressed in hepatocytes, neutrophils, macrophages, pulmonary alveolar, intestine and corneal cells. Daily, under normal conditions, the liver synthesizes and secretes into circulation approximately 34 mg of A1AT per kilogram of body mass, resulting in a normal plasma level of 0.9 to 2.23 mg per milliliter. During inflammatory acute‐phase response, the A1AT levels can increase more than fourfold in “normal variants” individuals. 2 , 6
4.1. A1AT deficiency
Encoded by SERPINA1, A1AT presents more than 100 recognized allelic variations associated with distinct A1AT serum levels. 58 Determined by codominant expression, the “normal variants” are M1‐M6, related to serum levels ≥0.9 mg/mL 2 , 59 ; meanwhile, homozygous or heterozygous S or Z are associated with lower serum levels (<0.9 mg/mL), resulting in different magnitudes of A1AT deficiency, lung, and liver diseases. 60 Intravenous augmentation therapy is usually prescribed for severe A1AT deficiency. 61
Null mutations are not common; while mild A1AT deficiency is caused by the S allele, which encodes the Glu264Val substitution, severe cases of A1AT deficiency are due to the Z allele. 60 About 70% of the altered A1AT molecules are degraded by hepatocytes. 62 The rest of them form a polymerized conformation, generating cell stress and an inflammatory response that can lead to chronic liver injury, fibrosis, and cirrhosis. 63 Exposure to oxidising agents can lead to polymerization, nitrosylation, and the oxidation of Met‐358, modifying A1AT function in individuals with “normal variants.” 64
Serres and Blanco investigated the worldwide distribution of PI*S and PI*Z. They concluded that there are approximately 190 million deficiency genotypes in the 97 analyzed countries, with 75% being the MS slightly deficient genotype; 24% being MZ and SS, presenting a potential risk of A1AT deficiency; 0.7% being SZ, with an increased risk of deficiency; and being 0.1% ZZ, which is the high risk genotype. 65 Moreover, 1 in 25 European descendants presents the Z allele and 1 in 2000 are homozygous. In the Iberian peninsula, 1 in 4 individuals present the S allele. 66
Genotyping of A1AT performed in 3751 Italians from different regions showed a higher prevalence of the deficiency‐related phenotypes SZ, MZ, ZZ in northern Italy, the region that was most affected by Covid‐19. 67 , 68 More recent data collected by Italian Registry for Alpha‐1‐Antitrypsin Deficiency confirmed that the northern region of Italy had a higher prevalence of A1AT deficiency in comparison to the southern region, with 47% of the total cases registered only in Lombardia. 69
Another study analyzed A1AT serum levels of 125 individuals aged from 20 to 76 years. It was observed that middle‐aged men showed a decrease in A1AT levels, while women did not show any variation. Altogether, women presented higher levels than men and no significant variation was observed in older groups. 70
4.2. A1AT: A multitask protein
The main function of A1AT is inactivating proteolytic enzymes, which are constantly released in pulmonary tissue due to the frequent exposure to pathogens and high cellular immune activity. The inhibition of NE, which is very harmful to lung tissue, is the most evident role of A1AT; therefore, it can be inferred that changes in A1AT expression may be linked to pulmonary pathologies, such as fibrosis. 71 The A1AT protein is formed of three β‐sheets (A‐C) and a reactive loop. NE binds to the reactive loop, which approaches β‐sheet A, forming an irreversible complex and promoting NE inhibition. 72 This new conformation might be recognized by hepatic receptors and removed from the circulation. 73
Furthermore, A1AT appears to participate in several other important biological processes and can increase four to six times during inflammation. 74 In a cohort of 51 individuals with fever stress, 76.4% presented elevated A1AT levels, showing the adaptive capacity of A1AT during the acute‐phase response. 75 Moreover, A1AT levels in women using oral contraceptives proved to be significantly higher, suggesting responsiveness to estrogen. 76 In addition, Larsson et al showed that women have increased levels of A1AT during pregnancy, with a peak at 34 to 38 weeks and a decrease in the postpartum period. 77 Since low levels of A1AT were related to spontaneous abortion, it was suggested that A1AT reduces embryotoxicity and contributes to successful pregnancy. 78
Evidence supports the fact that A1AT molecules also play anti‐inflammatory roles. High concentrations of A1AT were able to neutralize airway plasmin and thrombin in vitro in smoke‐conditioned medium. Both plasmin and thrombin activate PAR‐1 receptors, resulting in macrophage MMP‐12 release, which in turn is a TNF‐α converting enzyme. 79 High levels of pro‐inflammatory cytokines were observed in A1AT‐deficient individuals 80 ; this could be explained by in vitro A1AT‐mediated NF‐κB inhibition. 81 Also, A1AT can inhibit caspase‐3, ‐6, and ‐7 in a dose‐response fashion. 82 A1AT reduced mice hepatocyte apoptosis induced by TNF‐α, in vitro normal human pulmonary epithelial cell line (BEAS‐2B) apoptosis in an inflammatory response model and alveolar cell apoptosis by directly binding to caspase‐3 in a mouse model of apoptosis emphysema. 83 , 84 , 85
Furthermore, an in vitro assay showed that A1AT can bind to TNFR1 and TNFR2 receptors, diminishing TNF‐α‐TNFR1 by 35% and TNF‐α‐TNFR2 by 50%. Neutrophils pre‐exposed to physiological levels of A1AT and treated with exogenous TNF‐α presented 55% less bound TNF‐α when compared to neutrophils cultivated in non‐A1AT culture. 86 Also, biophysical experiments showed that A1AT can bind to retinoic acid, which is related to inflammatory disorders. 87 , 88
Moreover, Janciauskiene et al observed that A1AT promotes the release of IL‐10 in human monocytes by increasing cAMP and activating cAMP‐dependent protein kinase A in response to LPS, contributing to an anti‐inflammatory response. 89 Besides, A1AT increases IL‐1Ra expression without promoting expression of the NF‐κB gene. 74 , 90
Mizrahi et al described A1AT as a B‐lymphocyte modulator. 91 In cell culture under LPS stimuli, A1AT reduced B‐lymphocyte activation, Ki‐67+ B‐cell population size, IgM release and expression of the activation markers CD40 and CD19. Also, the authors observed a low expression of CD80 and CD86 by CD40 stimulation, suggesting that A1AT interferes with T‐cell‐dependent B‐cell activation. 91
Recently, Azouz et al demonstrated in vitro assays that A1AT can reverse inflammatory conditions caused by the loss of SPINK7, that might be related to cytokine release and eosinophil infiltration. Therefore, the authors propose A1AT as a therapeutic approach against eosinophilic esophagitis and Netherton syndrome. 92
Finally, A1AT appears to be involved in diabetes. Despite the increased A1AT levels in diabetic patients, its inhibitory capacity is decreased. Hashemi et al suggested that the non‐enzymatic glycosylation of A1AT lysine is responsible for the low activity. 93
4.3. A1AT and pulmonary protection
COPD emphysema is strongly related to an imbalance in A1AT activity or a deficiency of this protein. As the majority of people who develop the disease have normal levels of A1AT, environmental changes in A1AT, promoted by cigarette exposure and pollution, appear to be more relevant risk factors than genetics. 94 , 95 , 96
The protective effects of A1AT in COPD are mainly involved in neutrophil regulation. Ordinarily, A1AT reaches the lung by passive diffusion endocytosis irreversibly inhibiting NE and can be taken up by lung endothelial cells through clathrin‐dependent endocytosis. 97 , 98 Additionally, A1AT has been described as a neutrophil chemotaxis modulator because it can bind to IL‐8, preventing interaction with CXCR1. 4 A1AT can inhibit ADAM17, which is activated by soluble immune complexes and is responsible for the release of FcyRIIIb from neutrophils. Thus, the inhibition of ADAM17 reduces FcγRIIIb release and diminishes the migration of cells during inflammation. 4
In summary, also considering the previously described anti‐apoptotic and TNF‐α modulation performed by A1AT, its protective role in the lung during inflammation appears to have substantial physiological relevance. 79 , 84 , 85
4.4. A1AT and viral infections
One of the first reports of the protective role of A1AT against viral infection was written by Shapiro et al. in 2001. 81 In this study, the proteinase inhibitor attenuated HIV replication in peripheral blood mononuclear cells. Afterward, a small C‐terminus of 20aa residue of A1AT called “virus‐inhibitory peptide (VIRIP)” can bind to the HIV envelope glycoprotein gp41, disabling viral entry into host cells. 3 , 99 VIRIP also has anti‐HIV activity in in vivo assays. 3 In addition, A1AT interacts with cytosolic IkBα of HIV infected CD4+ T cells, modifying its ubiquitinylation pattern, resulting in the inhibition of NF‐kB activation. 100
It was observed that low levels of A1AT in serum might be a risk factor for HIV infection and disease progression. 101 In addition, the presence of emphysema in HIV positive patients was related to increased levels of oxidized A1AT in bronchoalveolar lavage fluid (but not in serum) and NPS imbalance. 102
A protective role for A1AT has been described for several other viral infections. A1AT affects human rhinovirus (HRV) infection in airway epithelial cells. 103 Cells exposed to cigarette smoke exhibited an increased viral load. However, the addition of A1AT after exposure resulted in a 29‐fold decrease in viral replication. Plus, bronchial epithelial cells treated with A1AT and infected with HRV showed reduced ICAM‐1 expression, which normally acts on vascular permeability and immune responses, and is used by HRV as an entry receptor. 103
Finally, A1AT deficiency genotypes in Iranian, Egyptian, and Brazilian individuals were linked to HIV, hepatitis B, and hepatitis C infection and complications, such as emphysema, liver, and hepatic disease. Therefore, A1AT levels may be relevant to the development of these viral diseases. 104 , 105 , 106
5. A1AT AND COVID‐19
Covid‐19 is associated with comorbidities, such as cardiovascular diseases, pulmonary diseases, hypertension, diabetes, and liver and kidney diseases. 20 , 107 However, the patient's prognosis varies widely and may respond to several intrinsic and environmental factors. 108 A1AT is an innate immune system regulator that plays a protective role during viral infections, as well as in lung homeostasis, and might be relevant in the RAS system imbalance (Figure 1). 71 In this context, determination of the role of A1AT in SARS‐CoV‐2 infection and how it interferes with the progress of Covid‐19 may provide a better understanding of the pathophysiology of the disease and would open up an avenue for new therapeutic approaches.
FIGURE 1.
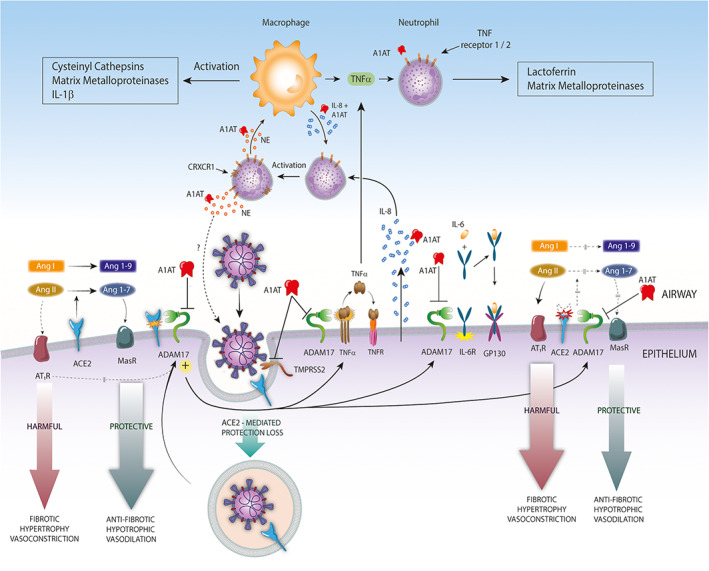
Possible protective role of A1AT against Covid‐19. Alpha‐1‐antitrypsin (A1AT) inhibits transmembrane serine protease 2‐mediated SARS‐CoV2 infection and ADAM17 protease. A1AT‐mediated ADAM17 inhibition can modulate ACE2 cleavage, protecting pulmonary, and cardiac tissue from deleterious effects of RAS imbalance. In addition, A1AT‐ADAM17 inhibition can exert anti‐inflammatory effects by inhibiting TNF‐α and IL6R cleavage. Moreover, A1AT can regulate neutrophil chemotaxis, degranulation through interactions with neutrophil elastase (NE), IL‐8, and TNF‐α binding to TNFR1 and TNF‐R2 neutrophil receptors, and macrophage activation. Finally, A1AT could inhibit the NE—mediated cleavage of the spike protein of the SARS‐CoV‐2 A2a subtype (not shown)
As mentioned before, TMPRSS2‐mediated SARS‐CoV‐2‐S protein binding is very important for virus entry into host cells. 9 , 29 A1AT efficiently inhibits TMPRSS2 in an in vitro assay, making this a strong candidate as an anti‐Covid‐19 therapy because it could prevent viral entry (Figure 1). 5 More recently, Wettstein et al confirmed the hypothesis that A1AT could inhibit SARS‐CoV‐2 entry. The in vitro assays were performed using Caco2 and Vero cells treated with different concentrations of A1AT, infected with spike‐containing pseudoparticles and wild‐type SARS‐CoV‐2 from France and the Netherlands. A1AT promoted almost complete inhibition of infection in the concentrations of 2 to 4 mg/mL, compatible with “normal variants” physiological levels during infection conditions. In addition, no cytotoxic effect in concentrations up to 8.2 mg/mL, was detected, supporting the possibility of the therapeutic use in patients infected with SARS‐CoV‐2. 6 However, since A1AT inhibits HIV viral infection by binding directly to the virus, its anti‐SARS‐CoV‐2 activity needs further investigation.
A1AT also reduced ADAM17 activity. 4 , 109 ADAM17 is responsible for ACE2 cleavage and an imbalance of the RAS system, leading to inflammation, increased vascular permeability, pulmonary edema, and DIC. 19 , 29 , 57 Reducing its activity might improve the inflammatory condition in Covid‐19 patients. In addition, less ADAM17 shedding could help to control the release of IL‐6R, the release of TNF‐α and consequently, the “cytokine storm,” a phenomenon related to Covid‐19 that increases risk of vascular hyperpermeability, multiorgan failure, and death (Figure 1). 35
ADAM17 down‐regulation can result in less FcyRIIIb release, diminishing neutrophil activation. 4 Importantly, ADAM17 is over‐expressed in diabetic patients 110 ; this could be due to the inhibition of A1AT activity by glycosylation. 93 Therefore, the increased risk among diabetic Covid‐19 patients might be related to the diminished activity of A1AT.
Since pulmonary alveolar macrophages are activated by SARS‐CoV‐2, the possible protective role of A1AT against Covid‐19 might be mediated by the modulation of IL‐8 activity and TNF‐α binding to TNFR1 and 2, preventing neutrophil chemotaxis, exacerbated immune response, and tissue damage (Figure 1). 4 , 86 , 111
Recent studies introduced the idea that air pollution is directly related to Covid‐19 due to the capacity of promoting longevity of SARS‐CoV‐2 particles and inducing respiratory diseases; the most affected regions of Italy and China were also the most polluted. 112 , 113 Nevertheless, this correlation ought to be extensively investigated and bias such as living conditions and cultural behavior of the population of the polluted regions are extremely important and must be taken into consideration.
However, this correlation can be relevant to the role of A1AT in the outcome of Covid‐19, since oxidant agents present in polluted air are able to modify normal variants of A1AT molecules, extinguishing their pulmonary protective function. 64 Interestingly, regions of northern Italy, in addition to being more polluted compared to the south, have higher amounts of defective A1AT alleles. 67
Vianello and Braccioni recently suggested that A1AT deficiency might explain the high Covid‐19 mortality in Italy. 114 The authors observed that the Lombardy region, which suffered 37.8% of the fatalities, also records 47% of all A1AT deficiency cases of Italy. Also, they highlighted inhibition of TMPRSS2 by A1AT as a possible reason for the high mortality rates. 114
A recent clinical analysis with a cohort of 40 individuals showed that A1AT levels are increased in Covid‐19 patients. 7 Usually, the increase in A1AT is directly proportional to the increase in IL‐6, supporting an anti‐inflammatory function. However, Covid‐19 patients requiring intensive care unit (ICU) presented a higher IL‐6:A1AT ratio than Covid‐19 stable patients. Moreover, clinical improvement was observed in ICU patients where the IL‐6:A1AT ratio decreased over the course of the treatment; in contrast, in patients where the ratio remained higher, no improvement was observed. Therefore, the authors indicate that A1AT augmentation therapy might be considered and investigated as a treatment for Covid‐19. 7
Finally, the emergence of A2a SARS‐CoV‐2 subtype with a potential cleavage site for NE, along with the lung‐specific eQTL rs35074065‐delC variant expression that can induce neutrophil infiltration, stresses the possible relevance of A1AT in the pathophysiology of Covid‐19. 14 Indeed, A1AT could inhibit the cleavage of the spike protein of the D614G subtype by NE and diminish neutrophil activation in rs35074065‐delC variant individuals, acting against infection and deleterious effects of neutrophil infiltration (Figure 1).
In this context, it is plausible to consider A1AT as a protective host factor against Covid‐19, not only decreasing SARS‐CoV‐2 entry, but also protecting from the main clinical complications, such as acute inflammation and acute respiratory insufficiency.
6. OPEN QUESTIONS AND CONCLUDING REMARKS
In order to achieve a more comprehensive understanding of A1AT in Covid‐19, is important to address the following concerns:
Is there a correlation between allelic variants, A1AT levels and/or A1AT mutations and worldwide Covid‐19 clinical outcome?
Are there differences in A1AT levels between symptomatic and asymptomatic SARS‐CoV‐2 infection?
Is there any sex or age bias related to A1AT increase in SARS‐CoV‐2 infection?
Can neutrophil elastase improve the SARS‐CoV‐2 A2a subtype infection? If so, is A1AT able to interfere with neutrophil elastase‐mediated cleavage of the spike protein of the SARS‐CoV‐2 A2a subtype?
Is A1AT able to interfere with ADAM17‐mediated ACE2 cleavage?
What A1AT domains are responsible for the inhibition of SARS‐CoV‐2 infection?
Is A1AT capable of inhibiting TMPRSS2‐mediated ACE2 cleavage?
The evidence presented in this review highlights the relevance of the A1AT as a host protective factor, which can inhibit the TMPRSS2‐mediated SARS‐CoV‐2 infection, modulate the deleterious effect of ADAM17 activation and the activity of inflammatory molecules, such as IL‐8, TNF‐α, and neutrophil elastase. In this context, a better understanding of the physiological relevance of A1AT in Covid‐19 may be relevant in the development of new anti‐Covid‐19 approaches.
CONFLICT OF INTEREST
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
AUTHOR CONTRIBUTIONS
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
ACKNOWLEDGEMENTS
The authors would like to thank Donato Zipeto, Bergmann Morais Ribeiro, and Marcos A. S. Silva‐Ferraz for helpful corrections and support work. This work was supported by the Brazilian Foundation for Research Support of the Federal District ‐ Brazil ‐ FAPDF. Grant: 0193001527/2016; 0193001646/2017.
de Loyola MB, dos Reis TTA, de Oliveira GXLM, da Fonseca Palmeira J, Argañaraz GA, Argañaraz ER. Alpha‐1‐antitrypsin: A possible host protective factor against Covid‐19. Rev Med Virol. 2021;31:e2157. 10.1002/rmv.2157
Funding information Fundação de Apoio à Pesquisa do Distrito Federal, Grant/Award Numbers: 0193001527/2016, 0193001646/2017
DATA AVAILABILITY STATEMENT
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
REFERENCES
- 1. Munster VJ, Koopmans M, van Doremalen N, van Riel D, de Wit E. A novel coronavirus emerging in China—key questions for impact assessment. N Engl J Med. 2020;382(8):692‐694. 10.1056/NEJMp2000929. [DOI] [PubMed] [Google Scholar]
- 2. Strnad P, McElvaney NG, Lomas DA. Alpha1‐antitrypsin deficiency. Longo DL, ed. N Engl J Med. 2020;382(15):1443‐1455. 10.1056/NEJMra1910234. [DOI] [PubMed] [Google Scholar]
- 3. Forssmann W‐G, The Y‐H, Stoll M, et al. Short‐term Monotherapy in HIV‐infected patients with a virus entry inhibitor against the gp41 fusion peptide. Sci Transl Med. 2010;2(63):63re3‐63re3. 10.1126/scitranslmed.3001697. [DOI] [PubMed] [Google Scholar]
- 4. Bergin DA, Reeves EP, Meleady P, et al. α‐1 antitrypsin regulates human neutrophil chemotaxis induced by soluble immune complexes and IL‐8. J Clin Invest. 2010;120(12):4236‐4250. 10.1172/JCI41196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Azouz NP, Klingler AM, Rothenberg ME. Alpha 1 antitrypsin is an inhibitor of the SARS‐CoV2–priming protease TMPRSS2. bioRxiv. 2020;077826:1–16. 10.1101/2020.05.04.077826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wettstein L, Conzelmann C, Müller JA, et al. Alpha‐1 antitrypsin inhibits SARS‐CoV‐2 infection. bioRxiv. 2020;183764:1–14. 10.1101/2020.07.02.183764. [DOI] [Google Scholar]
- 7. McElvaney OJ, McEvoy N, McElvaney OF , et al. Characterization of the inflammatory response to severe COVID‐19 illness. Am J Respir Crit Care Med. 2020;1–47. 10.1164/rccm.202005-1583OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Matsuyama S, Ujike M, Morikawa S, Tashiro M, Taguchi F. Protease‐mediated enhancement of severe acute respiratory syndrome coronavirus infection. Proc Natl Acad Sci USA. 2005;102(35):12543‐12547. 10.1073/pnas.0503203102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hoffmann M, Kleine‐Weber H, Schroeder S, et al. SARS‐CoV‐2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181(2):271‐280.e8. 10.1016/j.cell.2020.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Simmons G, Gosalia DN, Rennekamp AJ, Reeves JD, Diamond SL, Bates P. Inhibitors of cathepsin L prevent severe acute respiratory syndrome coronavirus entry. Proc Natl Acad Sci USA. 2005;102(33):11876‐11881. 10.1073/pnas.0505577102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Li H, Wu C, Yang Y, et al. Furin, a potential therapeutic target for COVID‐19. ChinaXiv. 2020. 10.12074/202002.00062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Anand P, Puranik A, Aravamudan M, Venkatakrishnan A, Soundararajan V. SARS‐CoV‐2 selectively mimics a cleavable peptide of human ENaC in a strategic hijack of host proteolytic machinery. bioRxiv. 2020;069476:1–5. 10.1101/2020.04.29.069476. [DOI] [Google Scholar]
- 13. Xia S, Lan Q, Su S, et al. The role of furin cleavage site in SARS‐CoV‐2 spike protein‐mediated membrane fusion in the presence or absence of trypsin. Signal Transduct Target Ther. 2020;5(1):92. 10.1038/s41392-020-0184-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bhattacharyya C, Das C, Ghosh A, et al. Global spread of SARS‐CoV‐2 Subtype with spike protein mutation D614G Is shaped by human genomic variations that regulate expression of TMPRSS2 and MX1 genes. bioRxiv. 2020;075911:1–30. 10.1101/2020.05.04.075911. [DOI] [Google Scholar]
- 15. Hamming I, Timens W, Bulthuis MLC, Lely AT, Navis GJ, Van Goor H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J Pathol. 2004;203(2):631‐637. 10.1002/path.1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tipnis SR, Hooper NM, Hyde R, Karran E, Christie G, Turner AJ. A human homolog of angiotensin‐converting enzyme cloning and functional expression as a captopril‐insensitive carboxypeptidase. J Biol Chem. 2000;275(43):33238‐33243. 10.1074/jbc.M002615200. [DOI] [PubMed] [Google Scholar]
- 17. Mary D, Frank H, Elizabeth B, et al. A novel angiotensin‐converting enzyme–related Carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1‐9. Circ Res. 2000;87(5):e1‐e9. 10.1161/01.RES.87.5.e1. [DOI] [PubMed] [Google Scholar]
- 18. Oudit GY, Zhong J, Basu R, Guo D, Penninger JM, Kassiri Z. Angiotensin converting enzyme 2 suppresses pathological hypertrophy, myocardial fibrosis and diastolic dysfunction. J Card Fail. 2010;16(8):S16. 10.1016/j.cardfail.2010.06.054. [DOI] [PubMed] [Google Scholar]
- 19. Tang N, Li D, Wang X, Sun Z. Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J Thromb Haemost. 2020;18(4):844‐847. 10.1111/jth.14768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jordan RE, Adab P, Cheng KK. Covid‐19: risk factors for severe disease and death. BMJ. 2020;368(m1198):1–2. 10.1136/bmj.m1198. [DOI] [PubMed] [Google Scholar]
- 21. Sama IE, Ravera A, Santema BT, et al. Circulating plasma concentrations of angiotensin‐converting enzyme 2 in men and women with heart failure and effects of renin–angiotensin–aldosterone inhibitors. Eur Heart J. 2020;41(19):1810‐1817. 10.1093/eurheartj/ehaa373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Swärd P, Edsfeldt A, Reepalu A, Jehpsson L, Rosengren BE, Karlsson MK. Age and sex differences in soluble ACE2 may give insights for COVID‐19. Crit Care. 2020;24(1):221. 10.1186/s13054-020-02942-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Circulating Activities of Angiotensin‐Converting Enzyme. Its Homolog, Angiotensin‐Converting Enzyme 2, and Neprilysin in a Family Study | Hypertension. https://www.ahajournals.org/doi/full/10.1161/01.HYP.0000244543.91937.79, Accessed May 30, 2020. [DOI] [PubMed]
- 24. Rice GI, Jones AL, Grant PJ, Carter AM, Turner AJ, Hooper NM. Circulating activities of angiotensin‐converting enzyme, its homolog, angiotensin‐converting enzyme 2, and Neprilysin in a family study. Hypertension. 2006;48(5):914‐920. 10.1161/01.HYP.0000244543.91937.79. [DOI] [PubMed] [Google Scholar]
- 25. Saheb Sharif‐Askari N, Saheb Sharif‐Askari F, Alabed M, et al. Airways expression of SARS‐CoV‐2 receptor, ACE2, and TMPRSS2 is lower in children than adults and increases with smoking and COPD. Mol Ther ‐ Methods Clin Dev. 2020;18:1‐6. 10.1016/j.omtm.2020.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. To KK‐W , Tsang OT‐Y, Leung W‐S, et al. Temporal profiles of viral load in posterior oropharyngeal saliva samples and serum antibody responses during infection by SARS‐CoV‐2: an observational cohort study. Lancet Infect Dis. 2020;20(5):565‐574. 10.1016/S1473-3099(20)30196-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Oudit GY, Kassiri Z, Jiang C, et al. SARS‐coronavirus modulation of myocardial ACE2 expression and inflammation in patients with SARS. Eur J Clin Invest. 2009;39(7):618‐625. 10.1111/j.1365-2362.2009.02153.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Walls AC, Park Y‐J, Tortorici MA, Wall A, McGuire AT, Veesler D. Structure, function, and antigenicity of the SARS‐CoV‐2 spike glycoprotein. Cell. 2020;181(2):281‐292.e6. 10.1016/j.cell.2020.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Heurich A, Hofmann‐Winkler H, Gierer S, Liepold T, Jahn O, Pohlmann S. TMPRSS2 and ADAM17 cleave ACE2 differentially and only proteolysis by TMPRSS2 augments entry driven by the severe acute respiratory syndrome coronavirus spike protein. J Virol. 2014;88(2):1293‐1307. 10.1128/JVI.02202-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ebsen H, Schröder A, Kabelitz D, Janssen O. Differential surface expression of ADAM10 and ADAM17 on human T lymphocytes and tumor cells. Plos One. 2013;8(10):e76853. 10.1371/journal.pone.0076853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sommer A, Kordowski F, Büch J, et al. Phosphatidylserine exposure is required for ADAM17 sheddase function. Nat Commun. 2016;7(1):11523. 10.1038/ncomms11523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hall KC, Blobel CP. Interleukin‐1 stimulates ADAM17 through a mechanism independent of its cytoplasmic domain or phosphorylation at threonine 735. PLoS ONE. 2012;7(2):1–7. 10.1371/journal.pone.0031600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Haga S, Yamamoto N, Nakai‐Murakami C, et al. Modulation of TNF‐ ‐converting enzyme by the spike protein of SARS‐CoV and ACE2 induces TNF‐ production and facilitates viral entry. Proc Natl Acad Sci USA. 2008;105(22):7809‐7814. 10.1073/pnas.0711241105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gooz M. ADAM‐17: the enzyme that does it all. Crit Rev Biochem Mol Biol. 2010;45(2):146‐169. 10.3109/10409231003628015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jose RJ, Manuel A. COVID‐19 cytokine storm: the interplay between inflammation and coagulation. Lancet Respir Med. 2020;8:e46–e47. 10.1016/S2213-2600(20)30216-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ju C, Xia X, Chen R. Expressions of tumor necrosis factor‐converting enzyme and ErbB3 in rats with chronic obstructive pulmonary disease. Chin Med J (Engl). 2007;120(17):1505‐1510. [PubMed] [Google Scholar]
- 37. Lippi G, Henry BM. Chronic obstructive pulmonary disease is associated with severe coronavirus disease 2019 (COVID‐19). Respir Med. 2020;167 –. 10.1016/j.rmed.2020.105941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Worley JR, Hughes DA, Dozio N, Gavrilovic J, Sampson MJ. Low density lipoprotein from patients with type 2 diabetes increases expression of monocyte matrix metalloproteinase and ADAM metalloproteinase genes. Cardiovasc Diabetol. 2007;6(1):21. 10.1186/1475-2840-6-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Standiford TJ, Kunkel SL, Basha MA, et al. Interleukin‐8 gene expression by a pulmonary epithelial cell line. A model for cytokine networks in the lung. doi: 10.1172/JCI114928 [DOI] [PMC free article] [PubMed]
- 40. Chokki M, Mitsuhashi H, Kamimura T. Metalloprotease‐dependent amphiregulin release mediates tumor necrosis factor‐α‐induced IL‐8 secretion in the human airway epithelial cell line NCI‐H292. Life Sci. 2006;78(26):3051‐3057. 10.1016/j.lfs.2005.12.023. [DOI] [PubMed] [Google Scholar]
- 41. Qin C, Zhou L, Hu Z, et al. Dysregulation of immune response in patients with coronavirus 2019 (COVID‐19) in Wuhan, China. Clin Infect Dis. 2020;71(15):762–768. 10.1093/cid/ciaa248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Harada A, Sekido N, Akahoshi T, Wada T, Mukaida N, Matsushima K. Essential involvement of interleukin‐8 (IL‐8) in acute inflammation. J Leukoc Biol. 1994;56(5):559‐564. 10.1002/jlb.56.5.559. [DOI] [PubMed] [Google Scholar]
- 43. Kunkel SL, Standiford T, Kasahara K, Strieter RM. Interleukin‐8 (IL‐8): the major neutrophil chemotactic factor in the lung. Exp Lung Res. 1991;17(1):17‐23. 10.3109/01902149109063278. [DOI] [PubMed] [Google Scholar]
- 44. Trifilieff A, Walker C, Keller T, Kottirsch G, Neumann U. Pharmacological profile of PKF242‐484 and PKF241‐466, novel dual inhibitors of TNF‐α converting enzyme and matrix metalloproteinases, in models of airway inflammation. Br J Pharmacol. 2002;135(7):1655‐1664. 10.1038/sj.bjp.0704616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Schumacher R‐J. ADAM17 activity and IL‐6 trans‐signaling in inflammation and cancer. Cancers. 2019;11(11):1736. 10.3390/cancers11111736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Stolarczyk M, Amatngalim GD, Yu X, Veltman M, Hiemstra PS, Scholte BJ. ADAM17 and EGFR regulate IL‐6 receptor and amphiregulin mRNA expression and release in cigarette smoke‐exposed primary bronchial epithelial cells from patients with chronic obstructive pulmonary disease (COPD). Physiol Rep. 2016;4(16):e12878. 10.14814/phy2.12878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Stolarczyk M, Scholte BJ. The EGFR‐ADAM17 Axis in chronic obstructive pulmonary disease and cystic fibrosis lung pathology. Mediators Inflamm. 2018;2018:1‐22. 10.1155/2018/1067134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Netzel‐Arnett S, Hooper JD, Szabo R, et al. Membrane anchored serine proteases: a rapidly expanding group of cell surface proteolytic enzymes with potential roles in cancer. Cancer Metastasis Rev. 2003;22(2):237‐258. 10.1023/A:1023003616848. [DOI] [PubMed] [Google Scholar]
- 49. Paoloni‐Giacobino A, Chen H, Peitsch MC, Rossier C, Antonarakis SE. Cloning of the TMPRSS2 gene, which encodes a novel serine protease with Transmembrane, LDLRA, and SRCR domains and maps to 21q22.3. Genomics. 1997;44(3):309‐320. 10.1006/geno.1997.4845. [DOI] [PubMed] [Google Scholar]
- 50. Afar DEH, Vivanco I, Hubert RS, et al. Catalytic cleavage of the androgen‐regulated TMPRSS2 protease results in its secretion by prostate and prostate cancer epithelia. Cancer Res. 2001;61(4):1686‐1692. [PubMed] [Google Scholar]
- 51. Bertram S, Heurich A, Lavender H, et al. Influenza and SARS‐coronavirus activating proteases TMPRSS2 and HAT are expressed at multiple sites in human respiratory and gastrointestinal tracts. PLoS ONE. 2012;7(4):1–8. 10.1371/journal.pone.0035876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Asselta R, Paraboschi EM, Mantovani A, Duga S. ACE2 and TMPRSS2 variants and expression as candidates to sex and country differences in COVID‐19 severity in Italy . Social Science Research Network. 2020;1–27. 10.2139/ssrn.3559608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kim TS, Heinlein C, Hackman RC, Nelson PS. Phenotypic analysis of mice lacking the Tmprss2‐encoded protease. Mol Cell Biol. 2006;26(3):965‐975. 10.1128/MCB.26.3.965-975.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bittmann S, Weissenstein A, Moschüring‐Alieva E, Bittmann L, Luchter E, Villalon G. The role of TMPRSS2 and TMPRSS2‐ inhibitors in cell entry mechanism of COVID‐19. J Regen Biol Med. 2020;2(3):1‐3. [Google Scholar]
- 55. Lambert DW, Yarski M, Warner FJ, et al. Tumor necrosis factor‐α Convertase (ADAM17) mediates regulated Ectodomain shedding of the severe‐acute respiratory syndrome‐coronavirus (SARS‐CoV) receptor, angiotensin‐converting Enzyme‐2 (ACE2). J Biol Chem. 2005;280(34):30113‐30119. 10.1074/jbc.M505111200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Gemmati D, Bramanti B, Serino ML, Secchiero P, Zauli G, Tisato V. COVID‐19 and individual genetic susceptibility/receptivity: role of ACE1/ACE2 genes, immunity, inflammation and coagulation. Might the double X‐chromosome in females be protective against SARS‐CoV‐2 compared to the single X‐chromosome in males? Int J Mol Sci. 2020;21(10):3474. 10.3390/ijms21103474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Mahmoud G, Wang K, Anissa V, et al. Angiotensin‐converting enzyme 2: SARS‐CoV‐2 receptor and regulator of the renin‐angiotensin system. Circ Res. 2020;126(10):1456‐1474. 10.1161/CIRCRESAHA.120.317015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Darlington GJ, Astrint KH, Muirhead SP, Desnickt RJ, Smitht M. Assignment of human cvl‐antitrypsin to chromosome 14 by somatic cell hybrid analysis. 4. Proc. Nati, Acad. Sci. 1982;79:870‐873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Köhnlein T, Welte T. Alpha‐1 antitrypsin deficiency: pathogenesis, clinical presentation, diagnosis, and treatment. Am J Med. 2008;121(1):3‐9. 10.1016/j.amjmed.2007.07.025. [DOI] [PubMed] [Google Scholar]
- 60. Greene CM, Miller SDW, Carroll T, et al. Alpha‐1 antitrypsin deficiency: a conformational disease associated with lung and liver manifestations. J Inherit Metab Dis. 2008;31(1):21‐34. 10.1007/s10545-007-0748-y. [DOI] [PubMed] [Google Scholar]
- 61. Stoller JK. a1‐Antitrypsin deficiency 5: intravenous augmentation therapy: current understanding. Thorax. 2004;59(8):708‐712. 10.1136/thx.2003.006544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Soluble aggregates of the human PiZ alpha 1‐antitrypsin variant are degraded within the endoplasmic reticulum by a mechanism sensitive to inhibitors of protein synthesis. The Journal of biological chemistry. 1992;267(15):1072‐1108. https://www.jbc.org/content/267/2/1072.short, Accessed June 9, 2020. [PubMed] [Google Scholar]
- 63. Teckman JH, Blomenkamp KS. Pathophysiology of Alpha‐1 antitrypsin deficiency liver disease. In: Borel F, Mueller C, eds. Alpha‐1 Antitrypsin Deficiency: Methods and Protocols. Humana Press, New York, NY: Springer; 2017:1‐8. 10.1007/978-1-4939-7163-3_1. [DOI] [PubMed] [Google Scholar]
- 64. Serban KA, Petrache I. Alpha‐1 antitrypsin and lung cell. Apoptosis. 2016;13:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. de Serres FJ, Blanco I. Prevalence of α1‐antitrypsin deficiency alleles PI*S and PI*Z worldwide and effective screening for each of the five phenotypic classes PI*MS, PI*MZ, PI*SS, PI*SZ, and PI*ZZ: a comprehensive review. Ther Adv Respir Dis. 2012;6(5):277‐295. 10.1177/1753465812457113. [DOI] [PubMed] [Google Scholar]
- 66. Strnad P, McElvaney NG, Lomas DA. Alpha 1 ‐antitrypsin deficiency. Longo DL, ed. N Engl J Med. 2020;382(15):1443‐1455. 10.1056/NEJMra1910234. [DOI] [PubMed] [Google Scholar]
- 67. Massi G, Cotumaccio R, Auconi P. Alpha‐1‐antitrypsin (α1AT) phenotypes and PiM subtypes in Italy. Evidence of considerable geographic variability. Hum Genet. 1982;61(1):76‐77. 10.1007/BF00291340. [DOI] [PubMed] [Google Scholar]
- 68. Gagliano A, Villani PG, Co' FM, et al. COVID‐19 epidemic in the Middle Province of northern Italy: impact, logistics, and strategy in the first line hospital. Disaster Med Public Health Prep. 2020;24:1–5. 10.1017/dmp.2020.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Deficit di alfa1antitripsina | Centro Nazionale per la diagnosi | Home. http://alfa1antitripsina.it/it, Accessed July 20, 2020.
- 70. Denko CW, Gabriel P. Age and sex related levels of albumin, ceruloplasmin, alpha 1 antitrypsin, alpha 1 acid glycoprotein, and transferrin. Ann Clin Lab Sci. 1981;11(1):63‐68. [PubMed] [Google Scholar]
- 71. Silverman EK, Sandhaus RA. Alpha1‐antitrypsin deficiency. N Engl J Med. 2009;360(26):2749‐2757. 10.1056/NEJMcp0900449. [DOI] [PubMed] [Google Scholar]
- 72. Santangelo S, Scarlata SL, Poeta MJ, Bialas A, Paone G, Antonelli Incalzi R. Alpha‐1 antitrypsin deficiency: current perspective from genetics to diagnosis and therapeutic approaches. Curr Med Chem. 2017;24(1):65‐90. [DOI] [PubMed] [Google Scholar]
- 73. Mast AE, Enghild JJ, Pizzo SV, Salvesen G. Analysis of the plasma elimination kinetics and conformational stabilities of native, proteinase‐complexed and reactive site cleaved serpins: comparison of .alpha.1‐proteinase inhibitor, .alpha.1‐antichymotrypsin, antithrombin III, .alpha.2‐antiplasmin, angiotensinogen, and ovalbumin. Biochemistry. 1991;30(6):1723‐1730. 10.1021/bi00220a039. [DOI] [PubMed] [Google Scholar]
- 74. Guttman O, Baranovski BM, Schuster R, et al. Acute‐phase protein α1‐anti‐trypsin: diverting injurious innate and adaptive immune responses from non‐authentic threats. Clin Exp Immunol. 2015;179(2):161‐172. 10.1111/cei.12476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Ionu R. Fever stress and alpha 1 Antitrypsin. 5. Jurnal de medicinǎ preventivǎ. 2001;9(1):23‐26. [Google Scholar]
- 76. Nielsen CH, Poulsen HK, Teisner B, Thorsen P, Hau J, Westergaard JG. Changes in blood levels of proteinase inhibitors, pregnancy zone protein, steroid carriers and complement factors induced by oral contraceptives. Eur J Obstet Gynecol Reprod Biol. 1993;51(1):63‐71. 10.1016/0028-2243(93)90192-F. [DOI] [PubMed] [Google Scholar]
- 77. Larsson A, Palm M, Hansson L‐O, Basu S, Axelsson O. Reference values for α1‐acid glycoprotein, α1‐antitrypsin, albumin, haptoglobin, C‐reactive protein, IgA, IgG and IgM during pregnancy. Acta Obstet Gynecol Scand. 2008;87(10):1084‐1088. 10.1080/00016340802428146. [DOI] [PubMed] [Google Scholar]
- 78. Madar T, Shahaf G, Sheiner E, et al. Low levels of circulating alpha‐1 antitrypsin are associated with spontaneous abortions. J Matern Fetal Neonatal Med. 2013;26(18):1782‐1787. 10.3109/14767058.2013.801955. [DOI] [PubMed] [Google Scholar]
- 79. Churg A, Wang X, Wang RD, Meixner SC, Pryzdial ELG, Wright JL. Alpha1‐antitrypsin suppresses TNF‐alpha and MMP‐12 production by cigarette smoke‐stimulated macrophages. Am J Respir Cell Mol Biol. 2007;37(2):144‐151. 10.1165/rcmb.2006-0345OC. [DOI] [PubMed] [Google Scholar]
- 80. Pott GB, Chan ED, Dinarello CA, Shapiro L. α‐1‐antitrypsin is an endogenous inhibitor of proinflammatory cytokine production in whole blood. J Leukoc Biol. 2009;85(5):886‐895. 10.1189/jlb.0208145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Shapiro L, Pott GB, Ralston AH. Alpha‐1‐antitrypsin inhibits human immunodeficiency virus type 1. FASEB J. 2001;15(1):115‐122. 10.1096/fj.00-0311com. [DOI] [PubMed] [Google Scholar]
- 82. Lockett AD, Van Demark M, Gu Y, et al. Effect of cigarette smoke exposure and structural modifications on the α‐1 antitrypsin interaction with Caspases. Mol Med. 2012;18(1):445‐454. 10.2119/molmed.2011.00207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Molle WV, Libert C, Fiers W, Brouckaert P. Alpha 1‐acid glycoprotein and alpha 1‐antitrypsin inhibit TNF‐induced but not anti‐Fas‐induced apoptosis of hepatocytes in mice. J Immunol. 1997;159(7):3555‐3564. [PubMed] [Google Scholar]
- 84. Gao W, Zhao J, Kim H, et al. α1‐antitrypsin inhibits ischemia reperfusion‐induced lung injury by reducing inflammatory response and cell death. J Heart Lung Transplant. 2014;33(3):309‐315. 10.1016/j.healun.2013.10.031. [DOI] [PubMed] [Google Scholar]
- 85. Petrache I, Fijalkowska I, Medler TR, et al. α‐1 antitrypsin inhibits Caspase‐3 activity, preventing lung endothelial cell apoptosis. Am J Pathol. 2006;169(4):1155‐1166. 10.2353/ajpath.2006.060058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Bergin DA, Reeves EP, Hurley K, et al. The circulating proteinase inhibitor a‐1 antitrypsin regulates neutrophil degranulation and autoimmunity. 15. [DOI] [PubMed]
- 87. Karnaukhova E. Interactions of α1–proteinase inhibitor with small ligands of therapeutic potential: binding with retinoic acid. Amino Acids. 2010;38(4):1011‐1020. 10.1007/s00726-009-0309-9. [DOI] [PubMed] [Google Scholar]
- 88. Hall JA, Grainger JR, Spencer SP, Belkaid Y. The role of retinoic acid in tolerance and immunity. Immunity. 2011;35(1):13‐22. 10.1016/j.immuni.2011.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Janciauskiene SM, Nita IM, Stevens T. α1‐Antitrypsin, old dog, new tricks: α1‐antitrypsin exerts in vitro anti‐inflammatory activity in human monocytes by elevating cAMP. J Biol Chem. 2007;282(12):8573‐8582. 10.1074/jbc.M607976200. [DOI] [PubMed] [Google Scholar]
- 90. Abecassis A, Schuster R, Shahaf G, et al. α1‐antitrypsin increases interleukin‐1 receptor antagonist production during pancreatic islet graft transplantation. Cell Mol Immunol. 2014;11(4):377‐386. 10.1038/cmi.2014.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Mizrahi M, Cal P, Rosenthal M, et al. Human α1‐antitrypsin modifies B‐lymphocyte responses during allograft transplantation. Immunology. 2013;140(3):362‐373. 10.1111/imm.12149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Azouz NP, Ynga‐Durand MA, Caldwell JM, et al. The antiprotease SPINK7 serves as an inhibitory checkpoint for esophageal epithelial inflammatory responses. Sci Transl Med. 2018;10(444):eaap9736. 10.1126/scitranslmed.aap9736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Hashemi M, Naderi M, Rashidi H, Ghavami S. Impaired activity of serum alpha‐1‐antitrypsin in diabetes mellitus. Diabetes Res Clin Pract. 2007;75(2):246‐248. 10.1016/j.diabres.2006.06.020. [DOI] [PubMed] [Google Scholar]
- 94. Auerbach O, Hammond EC, Garfinkel L, Benante C. Relation of smoking and age to emphysema: whole‐lung section study. N Engl J Med. 1972;286(16):853‐857. 10.1056/NEJM197204202861601. [DOI] [PubMed] [Google Scholar]
- 95. Janoff A, Carp H, Lee D, Drew R. Cigarette smoke inhalation decreases alpha 1‐antitrypsin activity in rat lung. Science. 1979;206(4424):1313‐1314. 10.1126/science.316187. [DOI] [PubMed] [Google Scholar]
- 96. Dziegielewska KM, Guminska M, Matthews N, Saunders NR, Wilkinson G. Reduced levels of Alpha‐1‐antitrypsin in children exposed to high levels of air pollution. Neonatology. 1993;63(5):336‐339. 10.1159/000243950. [DOI] [PubMed] [Google Scholar]
- 97. Sinden NJ, Baker MJ, Smith DJ, Kreft J‐U, Dafforn TR, Stockley RA. α‐1‐antitrypsin variants and the proteinase/antiproteinase imbalance in chronic obstructive pulmonary disease. Am J Physiol‐Lung Cell Mol Physiol. 2015;308(2):L179‐L190. 10.1152/ajplung.00179.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Sohrab S, Petrusca DN, Lockett AD, et al. Mechanism of α‐1 antitrypsin endocytosis by lung endothelium. FASEB J. 2009;23(9):3149‐3158. 10.1096/fj.09-129304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Zhou X, Liu Z, Zhang J, Adelsberger JW, Yang J, Burton GF. Alpha‐1‐antitrypsin interacts with gp41 to block HIV‐1 entry into CD4+ T lymphocytes. BMC Microbiol. 2016;16:1–12. 10.1186/s12866-016-0751-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Zhou X, Shapiro L, Fellingham G, Willardson BM, Burton GF. HIV replication in CD4 + T lymphocytes in the presence and absence of follicular dendritic cells: inhibition of replication mediated by α‐1‐antitrypsin through altered IκBα Ubiquitination. J Immunol. 2011;186(5):3148‐3155. 10.4049/jimmunol.1001358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Ferreira TC d S, Sampaio EP, Argañaraz GA, MVP G, Shapiro L, Argañaraz ER. Increased prevalence of the alpha‐1‐antitrypsin (A1AT) deficiency‐related S gene in patients infected with human immunodeficiency virus type 1. J Med Virol. 2014;86(1):23‐29. 10.1002/jmv.23759. [DOI] [PubMed] [Google Scholar]
- 102. Stephenson SE, Wilson CL, Crothers K, et al. Impact of HIV infection on α1‐antitrypsin in the lung. Am J Physiol‐Lung Cell Mol Physiol. 2017;314(4):L583‐L592. 10.1152/ajplung.00214.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Berman R, Jiang D, Wu Q, Chu HW. alpha1‐Antitrypsin reduces rhinovirus infection in primary human airway epithelial cells exposed to cigarette smoke. Int J Chron Obstruct Pulmon Dis. 2016;11(1):1279. 10.2147/COPD.S105717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Hashemi M, Alavian SM, Ghavami S, et al. High prevalence of alpha 1 antitrypsin phenotypes in viral hepatitis B infected patients in Iran. Hepatol Res. 2005;33(4):292‐297. 10.1016/j.hepres.2005.09.035. [DOI] [PubMed] [Google Scholar]
- 105. Settin A, El‐Bendary M, Abo‐Al‐Kassem R, El Baz R. Molecular analysis of A1AT (S and Z) and HFE (C282Y and H63D) gene mutations in Egyptian cases with HCV liver cirrhosis. J Gastrointest Liver Dis. 2006;15(2):131‐135. [PubMed] [Google Scholar]
- 106. Ferreira TC d S, Queiroz MAF, Argañaraz GA, Ishak R, Vallinoto ACR, Argañaraz ER. A1AT polymorphisms may be associated with clinical characteristics of retrovirus infections in a mixed ethnic population from the Brazilian Amazon region. Int J Infect Dis. 2017;65:67‐71. 10.1016/j.ijid.2017.10.002. [DOI] [PubMed] [Google Scholar]
- 107. Fang L, Karakiulakis G, Roth M. Are patients with hypertension and diabetes mellitus at increased risk for COVID‐19 infection? Lancet Respir Med. 2020;8(4):e21. 10.1016/S2213-2600(20)30116-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Yang X, Yu Y, Xu J, et al. Clinical course and outcomes of critically ill patients with SARS‐CoV‐2 pneumonia in Wuhan, China: a single‐centered, retrospective, observational study. Lancet Respir Med. 2020;8(5):475‐481. 10.1016/S2213-2600(20)30079-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Jedicke N, Struever N, Aggrawal N, et al. Alpha‐1‐antitrypsin inhibits acute liver failure in mice. Hepatology. 2014;59(6):2299‐2308. 10.1002/hep.27024. [DOI] [PubMed] [Google Scholar]
- 110. Salem ESB, Grobe N, Elased KM. Insulin treatment attenuates renal ADAM17 and ACE2 shedding in diabetic Akita mice. Am J Physiol‐Ren Physiol. 2014;306(6):F629‐F639. 10.1152/ajprenal.00516.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Cell pyroptosis, a potential pathogenic mechanism of 2019‐nCoV Infection by Ming Yang:: SSRN. https://papers.ssrn.com/sol3/papers.cfm?abstract_id=3527420, Accessed June 11, 2020.
- 112. Martelletti L, Martelletti P. Air pollution and the novel Covid‐19 disease: a putative disease risk factor. SN Compr Clin Med. 2020;2(4):383‐387. 10.1007/s42399-020-00274-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Conticini E, Frediani B, Caro D. Can atmospheric pollution be considered a co‐factor in extremely high level of SARS‐CoV‐2 lethality in northern Italy? Environ Pollut. 2020;261:114465. 10.1016/j.envpol.2020.114465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Vianello A, Braccioni F. Geographical overlap between alpha‐1 antitrypsin deficiency and COVID‐19 infection in Italy: casual or causal? Arch Bronconeumol. 2020;31:1–2. 10.1016/j.arbres.2020.05.015. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.