

Summary
The Middle East respiratory syndrome coronavirus (MERS‐CoV) emerged in late 2012 in Saudi Arabia. For this study, we conducted a large‐scale comparative genome study of MERS‐CoV from both human and dromedary camels from 2012 to 2019 to map any genetic changes that emerged in the past 8 years. We downloaded 1309 submissions, including 308 full‐length genome sequences of MERS‐CoV available in GenBank from 2012 to 2019. We used bioinformatics tools to describe the genome structure and organization of the virus and to map the most important motifs within various regions/genes throughout the genome over the past 8 years. We also monitored variations/mutations among these sequences since its emergence. Our phylogenetic analyses suggest that the cluster within African camels is derived by S gene. We identified some prominent motifs within the ORF1ab, S gene and ORF‐5, which may be used for barcoding the African camel lineages of MERS‐CoV. Furthermore, we mapped some sequence patterns that support the zoonotic origin of the virus from dromedary camels. Other sequences identified selection pressures, particularly within the N gene and the 5′ UTR. Further studies are required for careful monitoring of the MERS‐CoV genome to identify any potential significant mutations in the future.
Keywords: bioinformatics, coronaviruses, evolution, genome, MERS‐CoV, organization, phylogenetic analysis
1. INTRODUCTION
The Middle East respiratory syndrome (MERS‐CoV) is one of the zoonotic coronaviruses that emerged in the Arabian Peninsula in 2012. 1 MERS‐CoV belongs to the sub‐family Orthocoronavirinae, which includes four genera (Alpha, Beta, Gamma, Delta), family Coronaviridae, and order Nidovirales. 2 The severe acute respiratory syndrome coronavirus (SARS‐CoV), MERS‐CoV, and the newly emerged SARS‐CoV‐2 belong to the Betacoronaviruses. 3 The SARS‐CoV emerged in 2003; the MERS‐CoV was discovered in 2012, and the SARS‐CoV‐2 was reported in late 2019. The time gap between SARS‐CoV and MERS‐CoV is around 9 years, and between the emergence of MERS‐CoV and SARS‐CoV‐2 is 7 years. The continuous emergence of new coronaviruses candidates may be attributed to the features of their genomes. This may be due to several factors, including the low fidelity of their RNA‐dependant RNA polymerases (RdRp), the possibility of recombination, and the high level of expression of their receptors in many mammalian and avian species. 4 Thus, there is an urgent need for the regular monitoring of the genome sequences of coronaviruses from various species of bats, animals, and birds. 5 , 6 , 7 , 8 , 9 , 10 , 11 , 12 The main goal of the current study was to do a comprehensive analysis of the MERS‐CoV genomes available in the public domain from its emergence until late 2019.
The coronavirus genome is composed of a single strand positive‐sense RNA molecule. The MERS‐CoV genome structure and organization is very similar to other members of the family Coronaviridae group II. The genome organization falls into the following order (5′‐UTR‐Gene‐1‐S‐ORF3, ORF4a, ORF4b, ORF5, E, M, N, 3′‐untranslated region (UTR), and the poly (A)‐3′). MERS‐CoV is further classified under the lineage‐C of the Betacoronavirus. MERS‐CoV is further classified into three clades. 13 Clade‐A contains very few numbers of isolates, such as (EMC/2012 and Jordan‐N3/2012). These viruses isolated earlier during the emergence of the virus in the Kingdom of Saudi Arabia (KSA) and Jordon clustered together, while clade B includes the majority of viral isolates from human and some from camel origins. 14 Clade‐C includes viruses of camel origin isolated from various countries in Africa, including Egypt, Morocco, Nigeria, Burkina Faso, and Kenya. 13 , 15 Interestingly, results from the virus neutralization assay revealed that all three clades are closely related, which supports the notion that one vaccine may be able to protect against all three clades. 13 It is believed that MERS‐CoV originated in bats and spilled over to humans via an intermediate host, dromedary camels. 16 , 17 Dromedary camels are the main known reservoir until now. 18 Genome analysis of MERS‐CoV isolates from human and dromedary camel origins revealed a close relationship between each other, suggesting the zoonotic origin of the virus. 14 Like other coronaviruses, MERS‐CoV continues to show some changes at the genome level. Thus, new virus clades and sub‐clades are recently reported of both human and dromedary camel origins. 19 Regular monitoring of the genetic makeup of the virus is very important to track down any potential mutation or recombination.
The main goal of this study was to do a deep bioinformatic analysis of the most available MERs‐CoV genome sequences in Genbank during 2012 to 2019 to understand the evolution of the virus and map any potential changes across the viral genome. Therefore, the development of potential diagnostic assays, vaccines, or therapy should cope with any potential changes over the viral genome. This monitoring may also contribute substantially to the control of the virus by knowing the currently circulating clades in a certain community. An earlier study showed the circulation of three different genotypes of the virus in some patients during 2013 in KSA. 20 One year later, the same group reported the presence of four MERS‐CoV clades during the early emergence of MERS‐CoV. Later, during 2014 three clades were no longer contributing to the reported human cases suggesting their extinction. 19 The main reason behind these changes was the dynamic changes among the S gene of the virus. 19 , 20 Studies on MERS‐CoV isolates from various countries in northern and central Africa revealed that the circulating strains of the virus in dromedary camels from these countries belong to lineage‐C. This lineage is different from the other two lineages reported earlier in the Arabian Peninsula. 13 Although the three lineages have some genetic variations, their antigenic properties remain identical, as shown by the virus neutralization test. 13 Isolates from dromedary camels collected from Nigeria, Burkina Faso, and Morocco clustered together into a new sublineage‐C1 due to shared genetic signatures, including deletions in ORF4b. 13 Thus, there is an urgent need for continued study not only of MERS‐CoV but also other coronaviruses in the context of the human‐animal interface and to understand the biological diversity of coronaviruses.
2. MATERIALS AND METHODS
2.1. Retrieval of the MERS‐CoV sequences
A database of 1309 sequences, including 308 full‐length genome sequences of MERS‐CoV, was downloaded from GenBank, last accessed on August 26, 2019. The human EMC/2012 (accession number: JX869059.2) was used as a reference sequence for downstream analysis.
2.2. Identification of the full‐length genome submissions
MERS‐CoV submissions were considered as a full‐length genome only if they meet three parameters. First, the length of the sequence must be greater than or equal to 30‐kilobases. Second, the submission must have the full 5′ UTR sequence. Third, the submission must have a poly‐A tail even represented by one nucleotide of adenine (A).
2.3. Multiple sequence alignment and single nucleotide polymorphism density analysis
Multiple sequence alignment was conducted using the MAFFT tool (http://ma.cbrc.jp/alignment/soware/). Single nucleotide polymorphism (SNP) density (excluding indels) was counted from the multiple‐sequence alignment of 544 ORF1ab and 744 S gene sequences by use of an in‐house script written in python. For data visualization, Geneious (version 7.1.8) was used.
2.4. Identification of the putative ORFs
To avoid losing open reading frames (ORFs) within different sequences, ORFs were collected by retrieving regions flanked by conserved sequences, as shown in (Table 1). Conserved sequences were obtained from multiple sequence alignment of 308 complete whole MERS‐CoV genomes. Different lengths of ORF4b and ORF8b were calculated at its minimum possible size (300 nt), allowing any start codon (ATG, TTG, or CTG) to initiate the ORF.
TABLE 1.
Flanking sequences used for retrieving ORFs
| ORF | Upstream sequence | Downstream sequence |
|---|---|---|
| ORF1ab | GGGCACATC | NNNNNNNCCAGATTCT |
| S | ATGNTACACTCAGTGT | TTCATGTNCANTAA |
| ORF3 | ACGAACTATNN | CGAACTCT |
| ORF4a | AACGAACTCT | GAAACTGCGC |
| ORF4b | CTACATAAGG | CGAACTATGG |
| ORF5 | ATGGCTTTCT | GCAGCTCTG |
| E | AAACGAACT | CGAACTCCT |
| M | CGAACTCCTNNNNN | GCTCTTTAGT |
| N | TTTCATTGTT | NNNNNNTCAAAGTAAC |
| ORF8 | ATGCCAATTC | GCAGAAACT a |
Note: Bold and underlined sequences are part of the intended ORF region. N is any nucleotide.
Since some sequences showed no wild stop codon at the end of ORF8, 84 downstream nucleotides were included (up to “GGAGCAGTAG”).
2.5. Identification of the cleavage sites for non‐structural proteins
Mapping the cleavage sites of the NSPs among gene‐1 was carried out as previously described. 21
2.6. Phylogenetic analysis
Phylogenetic trees were constructed using MEGA X software 22 using multiple sequence alignment of the whole genome, ORF1ab, S gene, ORF4b, ORF5, E gene, M gene, N gene and ORF8b sequences. The trees were constructed using maximum likelihood methods and the Tamura‐Nei model. Bootstrap analysis (100 pseudo‐replicates) was conducted to evaluate the statistical significance of the inferred trees, and only values greater than 50 were displayed.
Since most isolated sequences did not meet the parameters used in this study for the whole‐genome sequences, we conducted the analysis on individual genes.
For the comparative study of phylogenetic trees, we initially considered 493 sequences greater than 30 kb since most of the isolated sequences (especially isolates from African camels) did not meet the parameters used in this study for the whole‐genome sequences. Then, to make the tree much simpler and easily readable, we narrowed down this selection to be only 57 sequences consisting of one sequence per country and year (2012‐2019), as well as whether the origin was human or dromedary camels.
3. RESULTS
3.1. Genome structure and organization of MERS‐CoV 2012 to 2019
We used 308 full‐length genome sequences after applying filteration parameters. All relevant data of these sequences are summarized in Table S1. The MERS‐CoV genome is around 30 Kb in length. The viral genome is flanked by two UTRs (Figure 1). The 3′ end has a poly (A) tail. About two‐thirds of the viral genome (21 Kb) is occupied with the non‐structural proteins ORF1ab. The 3′ end contains the structural genes interspersed with some other ORFs (Figure 1). The viral genome is organized in the following order (5′‐UTR‐ORF1ab (translated into ORF1a and ORF1b), S, ORF3, ORF4 (a and b), E, ORF5, E, M, N, ORF8b‐UTR‐Poly A‐3′; Figure 1 and Table 2).
FIGURE 1.
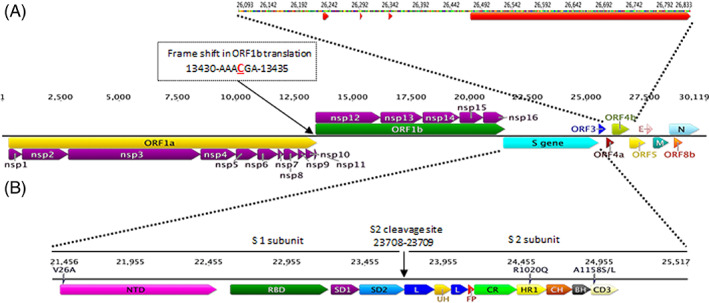
Genome structure and organization of the MERS‐CoV 2012 to 2019. The average full‐length genome sequences of the MERS‐CoV based on the (HCoV‐EMC/2012‐JX869059) is 30 107 bp, excluding the poly A tail at the 3′ end. Central panel: a schematic diagram of MERS‐CoV showing the predicted ORFs and their relative sizes and positions. Mapping the position and sequence of the (−1) ribosomal frameshifting (RFS) in the overlapped region of the ORF1a/ORF1b is shown. Arrows underneath and above ORF1a and ORF1b, respectively, represent positions of the replicase polyproteins (RdRp) pp1a and pp1ab that are predicted to be cleaved by papain‐like proteinases into the 16 non‐structural proteins (NSP‐1‐nap11) or the 3C‐like cysteine proteinase (NSP‐12‐NSP‐16). Top panel: expanded representation of ORF4b. Red arrows are representing the positions of deleted regions (±2 nt) in MERS‐CoV in dromedaries sampled from Burkina Faso, Nigeria, and Morocco. Bottom panel: expanded representation of the S glycoprotein organization showing identified regions in S protein subunits (S1 and S2) and the cleavage site (S1/S2). Amino acid variants in the African camels samples (V26A, R1020Q, and A1158S/L) are shown. NTD, N‐terminal domain; L, linker region; RBD, receptor‐binding domain; S.D., subdomain; U.H., upstream helix; F.P., fusion peptide; C.R., connecting region; H.R., heptad repeat; C.H., central helix; B.H., b‐hairpin
TABLE 2.
MERS‐CoV genes, their locations and protein size
| ORFs | Position (accession number JX869059) | Protein size (aa) | |
|---|---|---|---|
| ORF1ab | Nsp‐1 | 279 to 857 | 193 |
| Nsp‐2 | 858 to 2837 | 660 | |
| Nsp‐3 | 2838 to 8498 | 1887 | |
| Nsp‐4 | 8499 to 10 019 | 507 | |
| Nsp‐5 | 10 020 to 10 937 | 306 | |
| Nsp‐6 | 10 938 to 11 813 | 292 | |
| Nsp‐7 | 11 814 to 12 062 | 83 | |
| Nsp‐8 | 12 063 to 12 659 | 199 | |
| Nsp‐9 | 12 660 to 12 989 | 110 | |
| Nsp‐10 | 12 990 to 13 409 | 140 | |
| Nsp‐11 | 13 410 to 13 451 | 14 | |
| Nsp‐12 | 13 410 to 16 207 | 933 | |
| Nsp‐13 | 16 208 to 18 001 | 598 | |
| Nsp‐14 | 18 002 to 19 573 | 524 | |
| Nsp‐15 | 19 574 to 20 602 | 343 | |
| Nsp‐16 | 20 603 to 21 511 | 303 | |
| S gene | 21 456 to 25 517 | 1353 | |
| ORF3 | 25 532 to 25 843 | 103 | |
| ORF4a | 25 852 to 26 181 | 109 | |
| ORF4b | 26 093 to 26 833 | 246 | |
| ORF5 | 26 840 to 27 514 | 224 | |
| E | 27 590 to 27 838 | 82 | |
| M | 27 853 to 28 512 | 219 | |
| N | 28 566 to 29 807 | 413 | |
| ORF8b | 28 762 to 29 100 | 112 | |
3.1.1. The 5′ UTR
We analyzed 303 sequences of 5′‐UTR region isolated from human and camel (208 camels and 95 humans). The length of 5′ UTR is about 278 nt starting from the first nucleotide of the genome until the starting codon of ORF1ab at the position 279 in the case of the EMC/2012 (accession No.: JX869059). Four strains have deletions of either 9 or 11 nucleotides (Table 3). Two of them were of camel origin while the other two were of human origin, and all were isolated in 2015 in KSA. The multiple sequence alignment of 5′‐UTR sequences revealed three groups (I‐III) based on nucleotides at position 127 and 132 (accession no: JX869059). The 5′‐UTR dromedary camel sequences divided into two groups; group I represents the majority (about 81%) and has nucleotides C and T at 127 and 132 respectively, while group II (represents 19%) has T at both positions at 127 and 132. Strains of human origin divided into three groups. In contrast to samples isolated from camels, group I represents only 3.2% of sequences isolated from humans, while group II represents 46.2%. Group III represents the rest of the sequences, 50.5%, and has nucleotide C in both positions at 127 and 132 (Table 4).
TABLE 3.
Observed unique features of the MERS‐CoV genomes 2012–2019
| ORF | Accession number | Observation |
|---|---|---|
| 5′‐UTR | KT026453.1 and KT026455.1 (Human/Saudi/2015) | Have deletion of 9 nt (at 121‐129 of JX869059) |
| KT368869.1 and KT368879.1 (Camel/Saudi/2015) | Have deletion of 11 nt (121‐131 of JX869059) | |
| ORF1ab | MK462255 (Human/Saudi/2018) | has a deletion of 66 nt (3466‐3531 of JX869059) (within nsp3) |
| MF741827.1 and MF741832.1 (Human/Jordan/2015) | Nsp2 starts with V instead of D (D194V) | |
| S | KJ477102.1 (Camel/Egypt/2013)and MF679171.1 (Camel/UAE/2015) | Have deletion of three consecutive nucleotides (no frame shift) |
| KU710265.1(Human/Saudi/2014) | Has a deletion of 530 nt (2316‐2845 of JX869059.2) | |
| KJ614529, KC776174 (Human/Jordan/2012) and KX108943 (Camel/UAE/2015) | Have African motif R1020Q and A1158S/L | |
| KT806010.1 (Human/Saudi/2015) | Has a mutation found in Korean samples D510G | |
| ORF3 | KY688119 (Human/Saudi/2015) | Has deletion of 41 nt (25 790‐25 830 of JX869059) causing changing in two A.A. and 15AA short ORF3 |
| KT806046 (Human/Saudi/2015) | has deletion of 3AA due to deletion (25693‐25 701 of JX869059) | |
| KX108943 (UAE/camel/2015) | has deletion of 3AA due to deletion (25802‐25 810 of JX869059) | |
| MG923472 (Nigeria/camel/2015) | has deletion of 4AA due to a deletion (25809‐25 820 of JX869059) | |
| MG923473 (Burkina Faso/camel/2015) | Has deletion of 19 A.A. due to deletions of 17 and 25 bp in two positions (25772‐25 788 and 25 806‐25 830 of JX869059) | |
| MF000458.1, MF000459, MF000460.1, MF741837.1, MF741833.1, MF741834.1, MF741835.1, MF741836.1, KU233362.1 (Human/Jordan/2015) | Have deletion of 3AA due to deletion (25 675‐25 683 of JX869059) | |
| ORF4b | MF598715.1, MF598719.1, MF598720.1, MF598721.1, MF598722.1 (UAE/camel/2015) | Have full length gene ORF4b but truncated ORF4b protein (148 AA shorter) due to mutation at 26 388 causing stop codon |
| MF598690.1 (UAE/camel/2015) | Hasefull length gene ORF4b but truncated ORF4b protein (155 AA shorter) due to mutation at 26 366 causing stop codon | |
| KF600612.1, KF600620.1 (Human/Saudi/2012) | Have 144 AA shorter ORF4b due to a deletion of 17 nt (26 544‐26 560 of JX869059) causing a frameshift and stop codon | |
| MK483839.1 (Human/Saudi/2018) | Has full length gene ORF4b with 114 AA shorter ORF4b protein due to SNP at 26 489 creating stop codon mutation | |
| ORF5 | KU851859 (Human/Saudi/2015) | Has 655 nt produced 147 AA due to a deletion of 20 bp (27 227‐27 246 of JX869059) which cause a frame shift |
| KX108941 (Camel/ UAE/2015) | Has 673 nt produces only 7AA ORF5 protein due to deletion of 2 nt (26 859‐26 869 of JX869059) which created stop codon mutation | |
| MG923472, MG923481, MG923480, MG923479, MG923478, MG923477, MG923476, MG923475, MG923474 (Camel/Nigeria/2015‐2016) | Have A19V variants | |
| N | KJ614529 and KC776174 (Human/Jordan/2012) | Have D14Y variant which observed in 10 MERS‐CoV sequences isolated from African camels in 2015 and 2016 |
| KJ650295.1, KJ650296.1, and KJ650297.1 (Camel/Saudi/2013), KT156561 (Human/Oman/2013) | Have L23M variant | |
| MG757604, MG011358, MG011351, MG011345, MG011342, MG011350, MG011349, MG011348, MG011346, MG011344, KX154693, KX154692, KX154691, KX154690, KX154689, KX154688, KX154687, KX154686, KX154685, MH310909, MK129253 | Sequences isolated from human in 2016 to 2018 and have L23M variant | |
| MG923472, MG923481, MG923480, MG923479, MG923478, MG923477, MG923476, MG923475, MG923474 | Isolated from camel from Nigeria in 2015 to 2016 and have G198S variant | |
| MH822886, MK483839, MK462256, MK462255, MK462254, MK462253, MK462252, MK462250, MK462249, MK462248, MK462247, MN120514, MN120513 | Have G198S variant. Thirteen sequences isolated from Human from different regions in Saudi in 2018 and 2019 only (one sample was sequenced in the UK from a traveller from KSA) | |
| ORF8 | JX869059 (Human/Saudi/2012), KJ614529, KC776174 (Human/Jordan/2012), MG011340 (Human/Saudi/2016) | Have a C/T SNP at 28772 of JX869059 which is observed in MERS‐CoV isolated from African Camels only |
TABLE 4.
Grouping of MERS‐CoV‐5′‐UTR sequences based on the nucleotide sequences at positions 127 and 132
| Origin | Position (of JX869059) | % | |
|---|---|---|---|
| 127 | 132 | ||
| Dromedary camels | C | T | 81 |
| T | T | 19 | |
| Humans | C | T | 3.2 |
| T | T | 46.2 | |
| C | C | 50.5 | |
3.1.2. ORF1ab
ORF1ab consists of two overlapping ORFs (ORF‐1a and ORF‐1b). The ORF‐1b is produced by ribosomal frameshifting in which the ribosome steps back one nucleotide and continues reading and producing ORF‐1b (Figure 1). We analyzed 544 sequences of ORF1ab (Table S1). ORF1ab is 21 236 nt in length, and the position of the ribosomal frameshifting is located at 13 433 nt of JX869059. Distribution of polymorphisms (SNPs found in more than one strain in a 50‐bp sliding window) showed that SNP density in ORF1ab varies from 0 to 11 (Figure S1A). From multiple sequence alignment, we identified eight variants seem to be restricted to MERS‐CoV isolated from African camels. All these variants exited in studied African camels only (24 submissions). The eight variants are N581Y in nsp2, I1911T, S2000I, A2333V and I2639L in nsp3, A3361S in nsp5, V3721I in nsp6, and T1169M in nsp13. They are located in regions with SNP density ranging from 4 to 11.
The region of ribosomal frameshifting is 100% conserved across all analyzed sequences (including 15 nt upstream and 67 nt downstream from the frameshifting site; Figure S1B). The phylogenetic tree of 57 MERS‐CoV of both the full‐length genomes and the ORF1ab sequences are shown in Figure 2A,B. Both trees show all African camel samples clustered together in clade C. Furthermore, the distribution of samples in clades A and B is almost identical in both phylogenetic trees of MERS‐CoV whole‐genome and ORF1ab sequences. The total number of different nucleotides between samples in the phylogenetic tree of the full length and ORF1ab sequences is shown in (Table S2).
FIGURE 2.

Phylogenetic analysis of MERS‐CoV full‐length genome and Spike glycoprotein sequences, 2012 to 2019. Phylogenetic analysis of 57 (37 human and 20 camels) MERS‐CoVs full genomes, A, ORF1ab, B, and S gene sequences, C, isolated from 2012 to 2019. The unrooted phylogenetic trees were constructed by the maximum‐likelihood method and bootstrap values calculated from 100 trees. The scale bar represents the tree distance corresponding to 0.001 nucleotide substitution/kb. Numbers at branch nodes indicate bootstrap values greater than 50. Branch lengths represent degrees of diversity between sequences. Clades are denoted, and African camel samples are highlighted in red. Sample information is labeled as the following: Accession number/ Country/Human (Hu) or Camelus dromedary (Cd)/year. Both trees show almost an identical distribution of samples in clades. This phenomenon supports the notion that the cluster of African camels in clade C is derived by ORF1ab and S gene sequences
3.1.3. The non‐structural proteins
ORF1ab is processed into 16 non‐structural proteins (NSPs). The multiple sequence alignment of ORF1a and ORF1b at the amino acid level for the 538 submissions, showed that the start and end codons of all the 16 NSPs are 100% conserved. However, the nsp2 in two submissions of human origin reported from Jordan in 2015, starts with valine (V) instead of aspartic acid (D; Table 3). Details about the NSPs, their locations, as well as sizes, are shown in Table 2.
3.1.4. The spike glycoprotein (S)
We analyzed 744 of the S‐gene sequences (Table S1). The full‐length of the S gene is about 4062 nt. The organization of the MERS‐CoV‐S gene is described in Figure 1B. Only two submissions (MF679171.1 and KJ477102.1) have a deletion of three consecutive nucleotides (no frameshift) at 23 786 to 23 789 and 25 330 to 24 332 of JX869059, respectively. One sequence, KU710265.1, which was extracted from a human from Taif, KSA, has a deletion of 530 nt (at 2316‐2845 of JX869059.2). This deletion causes a frameshift starting from the linker region (L) at residue 772 of JX869059.2. The S1/S2 cleavage site is absolutely conserved across all studied sequences (Figure 1).
The multiple sequence alignment for the full‐length S gene revealed three variants. They seem to be restricted to MERS‐CoV isolated from African dromedary camels from 2013 to 2018. All the analyzed African camel submissions (26 samples) have three substitutions at the same time. The three variants are V26A, R1020Q, and A1158S/L and are located within the N‐terminal domain (NTD), heptad repeat region1 (HR1), and subdomain 3 (SD3), respectively (Table S1). The exception to this observation was one submission isolated from a camel in UAE/2015 (accession No.: KX108943). It is worth noting that the first MERS‐CoV isolate EMC/2012 (accession No.: JX869059) has R1020Q substitution only, and two samples seem to be isolated from the same source (Human/Jordan/2012) ‐ have R1020Q and A1158S substitutions (Table 3). We conducted an SNP density analysis to test whether the three observed variants of the S gene in African camel samples were not in a highly variable region. The distribution of polymorphisms (SNPs found in more than one strain in a 50‐bp sliding window) showed that SNP density in S gene varies from 0 to 12 (Figure S2). The variant V26A is located in a region with a SNP density of five, while both R1020Q and A1158S/L substitutions are located in a region of low SNP density (SNP density = 2). The total number of different nucleotides between samples in the phylogenetic tree of the S gene sequences is shown in Table S2.
The phylogenetic tree of the 57 MERS‐CoV of S gene sequences is presented in Figure 2C. Compared to the phylogenetic tree of MERS‐CoV whole‐genome and ORF1ab sequences (Figure 2A,B), the phylogenetic tree of S gene shows African camel samples in clade C and the same distribution of samples in clades A and B. To verify the notion that the clustering of MERS‐CoV isolated from African camels is influenced by S gene sequences, phylogenetic trees of other structural genes; E, M and N were constructed (Figure S3). None of phylogenetic trees of E, M nor N sequences showed the same distribution of samples in clades as shown in phylogenetic trees of MERS‐CoV whole‐genome, ORF1ab and S gene sequences (Figure 2). It is worth noting that African camel samples in phylogenetic tree of N sequences are clusterd in one clade because they all have a variant N284T. However, only 38% of MERS‐CoV N gene isolated from African camels have variant N284T.
During the MERS outbreak in Korea 2015, mutations in the receptor‐binding domains (RBD), D510G and I529T, was observed. Out of 13 analyzed genomes, 11 had I529T mutation within the RBD, and one had D510G mutation. 23 Both mutations resulted in a reduction in the affinity of these strains to the RBD of human CD26 compared to wild‐type RBD. 23 In our study, I529T mutation was found only in 26 samples, all of them of Korean origin, while the D510G mutation was found in six Korean samples and one Saudi sample sequenced in 2015 (Table S1). There was no sample observed with the two mutations (I529T and D510G) at the same time.
3.1.5. ORF‐3
In the case of ORF‐3, we analyzed 566 sequences (Table S1). The length of ORF3 gene is 312‐nt. However, in some submissions, we found truncated versions of the encoded protein of this ORF due to deletions varying from 9‐nt to 41‐nt (Table 3). Two variants were observed together in ORF3 (L17F and P86L) in about 26% of MERS‐CoV isolated from dromedary camels during 2014 to 2015 only. These two variants were not in the ORF3 sequences isolated from humans before 2015. However, they became more dominant in almost 93% of all the available human‐MERS‐CoV sequences from 2015 until 2019 (Figure 3).
FIGURE 3.

Evolution timeline of MERS‐CoV based on ORF3 sequence analysis. Timelines show the emergence of two variants of the MERS‐CoV‐ORF3 (L17F and P86L of JX869059 ORF3 protein sequence) isolated from humans and camels in 2012 to 2019 and in 2013 to 2019, respectively. Blue arrows represent ORF3 protein sequences with L17 and P86 residues. Red arrows represent the emergence and persistence of the two variants, L17F and P86L in ORF3 protein sequences. The percentages of sequences with the two variants to the total sequences isolated in the same period are shown. The magnification of red arrows does not reflect the actual percentage, but they were magnified for illustration purposes only
3.1.6. ORF‐4a
The ORF‐4 mainly consists of two overlapping ORFs (ORF‐4a and 4b). In the case of ORF‐4a, we analyzed 568 sequences (Table S1). The ORF4a gene is 330‐nt in length, and there is 89‐nt overlapping between ORF4a and ORF4b genes. The ORF4a is highly conserved, but SNP density increases in the last 45 nt (data not shown).
3.1.7. ORF‐4b
We analyzed 575 sequences of the ORF‐4b gene (Table S1). The full‐length of this gene is 741‐nt, which encodes 246 AA. It has been reported previously that MERS‐CoV from dromedaries sampled in Morocco, Burkina Faso, Nigeria, and Ethiopia had deletions in the accessory gene ORF4b. 5 In our study, we found 13 MERS‐CoV strains from dromedary reported from three African countries (Nigeria, Morocco, and Burkina Faso, during 2015 and 2016) have a truncated ORF4b (with a length varies from 14 to 120 AA) due to deletions in several positions of this gene. Interestingly, we noticed some strains have a full‐length ORF4b gene that produces truncated proteins due to mutations that created an early stop codon. For instance, six sequences isolated from camels in UAE, 2015, have full‐length gene ORF4b, but encode 148 AA shorter versions of the ORF4b protein (Table 3). Two human samples reported in KSA in 2012 from Riyadh (KF600612.1) and Bisha (KF600620.1) have the full‐length version of gene ORF4b but encode a 144 shorter version of the ORF4b protein (Table 3). A recent Saudi human sample from Albaha/2018 (MK483839.1) has full‐length gene ORF4b encoding a truncated ORF4b protein (114‐AA shorter) due to a stop codon mutation. All possible ORFs in gene ORF4b of JX869059 and all models of defective genes of ORF4b were considered. Figure 4 revealed that all models of the defective gene ORF4b and the wild type gene ORF4b could produce an identical potential ORF of 90 A.A. in length.
FIGURE 4.

Prediction of all potential ORFs within the MERS‐CoV‐ORF4b sequences, 2012 to 2019. An illustration is showing all putative ORFs of over 300 nucleotides across the wild type ORF4b as well as all the potential models for the defective ORF4b gene. The asterisks are indicating the identical ORFs generated by the wild type ORF4b gene (accession No.: JX869059) and other observed models for the defective ORF4b gene. This phenomenon suggests that the first half of the gene ORF‐4b could be responsible for the functional part of ORF4b protein
3.1.8. ORF‐5
We analyzed 569 MERS‐CoV sequences of ORF5 (Table S1). The full‐ length of gene ORF5 is 675 nt, which encodes a protein of 224 AA in length. We found two strains encoding a truncated ORF5 protein due to deletions causing a frameshift and stop codon (Table 3). All available ORF5 sequences of strains isolated from African camels between 2013 and 2018 have a Q12H variant. The phylogenetic tree of the ORF5 sequences showed the MERS‐CoV isolates from African camels cluster into one clade, but the distributions of other samples differ from their counterparts in the phylogenetic trees of the full‐length genome, ORF1ab and S gene (Figure 2 and Figure S4).
3.1.9. The envelope (E) and membrane (M) proteins
We analyzed 579 sequences of E gene and 575 sequences of M gene (Table S1). The lengths for E protein and M protein are 82‐A.A. and 219‐A.A., respectively. Most of the E and M gene sequences were quite conserved and had no significant observation either at the nucleotide or amino acid level. However, we considered observed variants not significant as they either existed in only one sample or in sequences generated by the same group.
3.1.10. The nucleocapsid (N) protein
The number of retrieved and analyzed sequences of N gene was 565 (Table S1). The full‐length of gene N is 1242 nt, which encodes a protein of 411 AA. We observed four variants within the N gene; one of them seems to be restricted to African camels samples while the others may represent evidence for zoonotic transmission of MERS‐CoV. The first variant, D14Y, was observed in 10 submissions from African camels (45% of analyzed African camel samples), and in two other sequences isolated from humans in Jordan‐2012 (Table 3). The second variant, L23M, was found in three sequences isolated from dromedary camels in 2013 from KSA and one isolate from a human in the Sultanate of Oman‐2013. This variant was found in 21 human sequences; 19 from Saudi‐2016, one from Saudi‐2017, and one from South Korea‐2018 (Table 3). All studied sequences from dromedary camel origins (213 submissions) and human samples up to 2015 (220 samples) have V at the position 178 within N protein except two submissions (JX869059 and NC‐019843), (Human/Saudi/2012) which have L instead of V. However, the third variant, V178A, started to show up in human samples in 2015 up to 2018 (but not before that). Interestingly, the later the date, the more human samples with V178A variant (Figure S5). The fourth variant, G198S, was observed in nine sequences isolated from dromedary camels from Nigeria during 2015 to 2016 only. Then, the same variant was observed in 13 Saudi/human samples from different regions in 2018 and 2019 only (one sample was sequenced in the UK from a patient who traveled from KSA to the UK, accession No.: MH822886; Table 3).
3.1.11. ORF‐8b
We analyzed 598 sequences of gene ORF‐8b (Table S1). The full‐length of the wild type gene ORF8 is 339 nt, which encodes a protein of 112 AA in length. This ORF is located within the N gene (from 28 762 to 29 100 of the JX869059 genome sequencing; Figure 1). Fifty‐eight strains (10% of the analyzed sequences) have a 31 A.A. longer version of the ORF‐8b. This is due to a SNP at the last codon, located at position 29 098 (accession No.: JX869059), which abolishes the wild type stop codon. All these sequences are of human origin reported from KSA during 2015 to 2019 (one submission was sequenced in the UK, accession No.: MH822886). Only one submission was from dromedary camel/Egypt/2013 (KJ477103), has the same length of ORF8 protein due to an SNP at position 29 099 (Table S1). On the other hand, 13 strains have truncated ORF8 protein (34‐AA shorter than the wild type) due to a mutation at position 28 996 of JX869059, changing TCA to a TGA (stop codon). One of these sequences was isolated from a human in Qatar during 2013 while the others were of human origin from KSA in 2014 (one of them was sequenced in Indiana‐KJ813439; Table S1).
All studied African camels (25 samples) have a P4L variant in ORF8b protein. This variant was found in four human samples isolated in 2012 from Saudi and Jordan (Table 3), in addition to one sample isolated later from a human in KSA in 2016 (MG011340).
3.1.12. The 3′ UTR
We analyzed 303 sequences of 3′ UTR after excluding samples with unidentified nucleotides (Ns). The 3′ UTR starts at 29 808, immediately downstream of the N gene on the MERS‐CoV genome. The length of the majority of 3′ UTR sequences is 299 nt, while 3′ UTR in JX869059 and the other 11 sequences (out of 303) is 300 nt in length due to an insertion of one nucleotide at position 30 061 (accession number: JX869059).
4. DISCUSSION
There is continuous emergence of new coronavirus candidates from the clade‐B of the beta coronaviruses. Three major potential zoonotic viruses that belong to this group within the family Coronaviridae have emerged in the past 17 years (SARS‐CoV, MERS‐CoV, and SARS‐CoV‐2). 1 , 24 , 25 The main reason behind this is the dynamic changes in these viruses at the genomic level. Thus, there is a high demand for the continuous monitoring of the genome sequencing of different coronaviruses. The main goal of the current study was to monitor the changes that occurred in MERS‐CoV since its emergence in 2012 until 2019. The viral genome sequencing analysis over a period will give us a quantum leap in our understanding about the virus evolution, origins, the possibility of the emergence of virulent strains, and the selection pressure posed on the virus, which leads to the emergence of novel strains.
4.1. Classification of African camel MERS‐CoV lineage is mainly driven by ORF1ab and S gene sequences
MERS‐CoV isolated from African camels is shown to be phylogenetically distinct from those circulating in the Arabian Peninsula. 13 , 26 , 27 In this study, we showed that the cluster of MERS‐CoV African camels into clade‐C is mainly ORF1ab and S gene dependant. Comparative study of phylogenetic trees of the whole genome, ORF1ab, and S gene kept all tested MERS‐CoV strains in almost the same distribution of within the same clades (A, B, and C), while the phylogenetic tree of other ORFs did not. This observation suggests that the clustering of African camel samples into one clade could be mainly derived by ORF1ab and S gene sequences.
4.2. Potential barcoding of the African MERS‐CoVs isolates through specific motifs and variants within certain genes
We found some important motifs within ORF1ab, S gene and ORF‐5 of the MERS‐CoV isolated from African camels that may be used as evidence for barcoding of African camel strains. We identified a motif within ORF1ab and S gene sequences that seem to be restricted to the MERS‐CoVs isolated from African camel samples. All the analyzed African camel samples have eight and three variants at the same time in ORF1ab and S gene, respectively. Variants within the ORF1ab are located in nsp2, nsp3, nsp5, nsp6, and nsp13. On the other hand, the three variants within the S gene are located in the NTD, heptad repeat region1 (HR1) and subdomain 3 (SD3), respectively. The presence of these three variants in a region of low SNP density supports the notion that they are genuine variants and specific for MERS‐CoV circulating in African camels.
We also found a variant, Q12H, in all ORF5 sequences of MERS‐CoV isolated from African camels only. This variant, in addition to the three in the spike glycoprotein, could be used as potential markers for the African MERS‐CoV isolated from camels. These variants only exist in African camel samples across all analyzed sequences (744 sequences of S gene and 569 sequences of ORF5). Showing the variants that seem to be restricted to African camels in sequences isolated from a human in the early emergence of MERS‐CoV in 2012 suggests that the very start of zoonotic transmission could happen from African camels.
4.3. Potential evidence for zoonotic transmission of MERS‐CoV through the transmission of variants between dromedary camels and humans samples 2012 to 2019
Based on our genome sequencing analysis for the full‐length genome as well as the individual MERS‐CoV genes, we found evidence for zoonotic transmission of MERS‐CoV from camels to humans. The detection of several SNPs in humans after being first observed in dromedary camels is considered strong evidence for the transmission of the virus from camel to human. Furthermore, the prevalence of potential transmitted SNPs in humans may suggest that such variants play important roles in the adaptation of the virus and favors the adaptation of MERS‐CoV to humans. For instance, 26% of analyzed ORF3 sequences isolated from camels in 2015 had L17F and P86L variants. These two variants became dominant in 93% of the ORF3 sequences from humans from 2015 to 2019. However, they did not exist in human samples before 2015. Another example with a longer time interval was observed within N gene sequences isolated from camels in 2013 in KSA, which had L23M variants. Later, the variant existed in 21 human samples in 2016, 2017, and 2018 in KSA and South Korea. In addition, the variant G198S in nine sequences isolated from camels in Nigeria in 2015, and 2016 was observed in 13 human samples reported from KSA in 2018 to 2019, one of them was sequenced independently in the UK The diversity of sample analysis locations supports that the observed variants are genuine.
4.4. Selection pressure within the MERS‐CoV genome is triggered by the frequent emergence of some sequence patterns over the time
We noticed that the V178A variant within the N gene started showing up in human samples from 2015 and became more dominant with the progression of time. The existence of this variant in human samples may highlight the potential immune evasion role of the N protein through inhibition of IFN‐ type I production. 28 Furthermore, the 5′ UTR sequences of MERS‐CoV retrieved from both camels and humans could be divided into three groups based on the nucleotides at positions 127 and 132 (C/T, T/T, and C/C). The motif (T/T) in the minority of camels (19%) represents 46.2% of the 5′ UTR of human samples. In contrast, the motif (C/T) in the majority of camels (81%) represents only 3.2% of human samples. The third group of 5′ UTR is in human samples and represents a selective pressure by converting the (C/T) pattern into (C/C) in 50.5% of all analyzed human samples. This phenomenon suggests that MERS‐CoV circulating in camels with the (T/T) pattern in 5′ UTR, may show more tendency to zoonotic transmission than its counterpart with (C/T) pattern.
4.5. The potential biological consequence of some observed SNPs
A recent study showed that MERS‐CoV from dromedary camels in Nigeria, Burkina Faso, and Morocco have a deletion within the ORF4b gene. 13 In this study, by looking at the ORF4b at both the nucleotide and amino acid sequences levels, we noticed samples (of camel and human origin) with full‐length gene ORF4b but encoding truncated ORF4b protein due to a mutation creating a stop codon. The truncated ORF4b protein expression reduced virus replication compared to the wild type. 13 This may indicate that the full‐length ORF4b protein is not intrinsic for the virus life cycle, at least for the analyzed samples. As a result, all possible ORFs of the wild type ORF4b (JX869059) and all models of the defective gene ORF4b were tested, and we found that the ORF4b (including the wild type) encodes an identical ORF 90 A.A. in length. This suggests that the first half of the ORF4b could translate the essential part of ORF4b protein for the virus. In the case of ORF8b, there were human samples with full‐length genes that encoded either longer or shorter ORF8b protein than the wild type (Table 3). We tested the possibility of a defective version of the ORF8 gene that can produce ORF8 protein with a length close to the wild type by starting from different frames rather than frame‐1, all possible ORFs were tested. There was no defective form of gene ORF8 that produced ORF8 protein with a length close to the wild type (Figure S6). However, the biological consequence behind the variation of ORF8 protein length is still unknown.
5. CONCLUSIONS
Comparative genome sequence analysis of the MERS‐CoV of both dromedary camels and human origins revealed significant evidence for potential barcoding of the African clades based on the S gene sequences. It also provided evidence for the zoonotic origins of the virus from dromedary camels to humans and highlighted the role of selection pressure and compensatory mechanisms in virus genome evolution.
CONFLICT OF INTEREST
The authors have no competing interest.
Abbreviations
- MERS‐CoV
the Middle East respiratory syndrome coronavirus
- SARS‐CoV‐2
the severe acute respiratory syndrome coronavirus‐2
- ORF
open reading frame
- UTR
untranslated region
- RBD
receptor binding domains
- E
envelope protein
- S
spike glycoprotein
- N
nucleocapsid protein
- M
matrix protein
- RNA
ribonucleic acid
- SNP
single nucleotide polymorphism
- KSA
Kingdom of Saudi Arabia
- NT
none‐translated region
- HVR
hypervariable region
- SD
sub‐domains
- UK
United Kingdom
- MSA
multiple sequencing alignment
- AA
amino acids
- NSP
the non‐structural proteins
- UAE
United Arab of Emirates
- MAFFT
multiple alignments using fast Fourier transform
Supporting information
Supplementary Figure S1 Polymorphisms in ORF1ab. (A) Distribution of polymorphisms (SNPs found in more than 1 sample) in gene ORF1ab. SNP counts in a 100‐bp sliding window are shown. (B) An enlarged view of Figure A, from 12 999 to 14 048 nt. The of ORF1b frameshifts positions are shown.
Supplementary Figure S2 Illustrations are showing the distribution of polymorphisms (SNPs found in more than one strain) the spike glycoprotein. SNP counts in a 50‐bp sliding window are shown against their positions in individual components of S glycoprotein. Positions of variants; V26A, R1020Q, and A1158 variants are shown.
Supplementary Figure S3 The phylogenetic trees based on the (E gene, M gene, and N genes) sequences from the same MERS‐CoVs samples (37 human and 20 camels) isolated from 2012 to 2019. The unrooted phylogenetic trees were constructed by the maximum‐likelihood method, and the bootstrap values calculated from 100 trees. The scale bars are representing the tree distance corresponding to 0.001 nucleotide substitution/kb. The numbers at branch nodes indicate bootstrap values greater than 50. Branch lengths represent degrees of diversity between sequences. Clades are denoted (except for E gene), and African camel samples are highlighted in red. Sample information is labeled as follows, Accession number/Country/Human (Hu) or Camelus dromedary (Cd)/year.
Supplementary Figure S4 Phylogenetic analysis of ORF5 and ORF8b of the same MERS‐CoVs sample (37 human and 20 camels) isolated from 2012 to 2019. The unrooted phylogenetic tree was constructed by the maximum‐likelihood method and bootstrap values calculated from 100 trees. The scale bar represents the tree distance corresponding to 0.001 nucleotide substitution/kb. Numbers at branch nodes indicate bootstrap values greater than 50. Branch lengths represent degrees of diversity between sequences. Clades are denoted, and African camel samples are highlighted in red. Sample information is labeled as the following: Accession number/ Country/Human (Hu) or Camelus dromedary (Cd)/year.
Supplementary Figure S5 A bar figure showing the number of N protein sequences having the V178A variant. Graphical representation shows the percentage of N protein sequences isolated from human with the V178A variant in the last 4 years. Each column represents the number of N protein sequences isolated from a human with V178A variant to the total sequences generated in the same year.
Supplementary Figure S6 Diagram showing all ORFs of over 300 nucleotides across the wild type gene ORF8b (accession number JX869059.2) and models of defective gene ORF4b that encode potential truncated ORF8b protein (accession number KF961221.1) or long ORF8b protein (accession number KX154684.1). There is no defective gene ORF8b that produces ORF8 protein with a length close to ORF8 protein generated by the wild type gene ORF8b.
Supplemental Table S1
Supplemental Table S2
ACKNOWLEDGEMENTS
We wish to thank the King Abdul‐Aziz City for Science and Technology (KACST), Saudi Arabia, for their generous funding through the MERS‐CoV research grant program (number 20‐0004), which is a part of the Targeted Research Program (TRP).
Ba Abduallah MM, Hemida MG. Comparative analysis of the genome structure and organization of the Middle East respiratory syndrome coronavirus (MERS‐CoV) 2012 to 2019 revealing evidence for virus strain barcoding, zoonotic transmission, and selection pressure. Rev Med Virol. 2021;31:e2150. 10.1002/rmv.2150
Funding information King Abdul‐Aziz City for Science and Technology (KACST), Grant/Award Number: 20‐0004
REFERENCES
- 1. Zaki AM, van Boheemen S, Bestebroer TM, Osterhaus ADME, Fouchier RAM. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N Engl J Med. 2012;367(19):1814‐1820. [DOI] [PubMed] [Google Scholar]
- 2. de Groot R, Baker SC, Baric R, Enjuanes L, Gorbalenya AE, Holmes KV. Coronaviridae. In: AAQ K, Adams MJ, Carstens EB, Lefkowitz EJ, eds. Ninth Report of the International Committee on Taxonomy of Viruses. San Diego: Elsevier Academic Press; 2012:806‐828. [Google Scholar]
- 3. Cui J, Li F, Shi ZL. Origin and evolution of pathogenic coronaviruses. Nat Rev Microbiol. 2019;17(3):181‐192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hemida MG. Middle East respiratory syndrome coronavirus and the one health concept. PeerJ. 2019;7:e7556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ceraolo C, Giorgi FM. Genomic variance of the 2019‐nCoV coronavirus. J Med Virol. 2020;92(5):522‐528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. De Sabato L et al. Full genome characterization of two novel alpha‐coronavirus species from Italian bats. Virus Res. 2019;260:60‐66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Decaro N, Mari V, Elia G, et al. Full‐length genome analysis of canine coronavirus type I. Virus Res. 2015;210:100‐105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Franzo G, Listorti V, Naylor CJ, et al. Molecular investigation of a full‐length genome of a Q1‐like IBV strain isolated in Italy in 2013. Virus Res. 2015;210:77‐80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Geldenhuys M, Mortlock M, Weyer J, et al. A metagenomic viral discovery approach identifies potential zoonotic and novel mammalian viruses in Neoromicia bats within South Africa. PLoS One. 2018;13(3):e0194527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jonassen CM, Kofstad T, Larsen IL, et al. Molecular identification and characterization of novel coronaviruses infecting graylag geese (Anser anser), feral pigeons (Columbia livia) and mallards (Anas platyrhynchos). J Gen Virol. 2005;86(6):1597‐1607. [DOI] [PubMed] [Google Scholar]
- 11. Nemoto M, Oue Y, Murakami S, et al. Complete genome analysis of equine coronavirus isolated in Japan. Arch Virol. 2015;160(11):2903‐2906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rasmussen TB, Boniotti MB, Papetti A, et al. Full‐length genome sequences of porcine epidemic diarrhoea virus strain CV777; use of NGS to analyse genomic and sub‐genomic RNAs. PLoS One. 2018;13(3):e0193682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chu DKW, Hui KPY, Perera RAPM, et al. MERS coronaviruses from camels in Africa exhibit region‐dependent genetic diversity. Proc Natl Acad Sci U S A. 2018;115(12):3144‐3149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wernery U, Lau SK, Woo PC. Genomics and zoonotic infections: Middle East respiratory syndrome. Rev Sci Tech. 2016;35(1):191‐202. [DOI] [PubMed] [Google Scholar]
- 15. Kiambi S et al. Detection of distinct MERS‐coronavirus strains in dromedary camels from Kenya. Emerg Microbes Infect. 2017;7(1):195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hemida MG, Elmoslemany A, al‐Hizab F, et al. Dromedary camels and the transmission of Middle East respiratory syndrome coronavirus (MERS‐CoV). Transbound Emerg Dis. 2017;64(2):344‐353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Memish ZA et al. Middle East respiratory syndrome coronavirus in bats, Saudi Arabia. Emerg Infect Dis. 2013;19(11):1819‐1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Killerby ME, Biggs HM, Midgley CM, Gerber SI, Watson JT. Middle East respiratory syndrome coronavirus transmission. Emerg Infect Dis. 2020;26(2):191‐198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cotten M, Watson SJ, Zumla AI, et al. Spread, circulation, and evolution of the Middle East respiratory syndrome coronavirus. mBio. 2014;5(1):e01062‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cotten M et al. Transmission and evolution of the Middle East respiratory syndrome coronavirus in Saudi Arabia: a descriptive genomic study. Lancet. 2013;382(9909):1993‐2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Snijder EJ, Bredenbeek PJ, Dobbe JC, et al. Unique and conserved features of genome and proteome of SARS‐coronavirus, an early split‐off from the coronavirus group 2 lineage. J Mol Biol. 2003;331(5):991‐1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33(7):1870‐1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kim DW, Kim YJ, Park SH, et al. Variations in spike glycoprotein gene of MERS‐CoV, South Korea, 2015. Emerg Infect Dis. 2016;22(1):100‐104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Peiris JS et al. The severe acute respiratory syndrome. N Engl J Med. 2003;349(25):2431‐2441. [DOI] [PubMed] [Google Scholar]
- 25. Zhu N et al. A novel coronavirus from patients with pneumonia in China. N Engl J Med. 2019;382(8):727‐733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kandeil A et al. Middle East respiratory syndrome coronavirus (MERS‐CoV) in dromedary camels in Africa and Middle East. Viruses. 2019;(8):717‐733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhang W, Zheng XS, Agwanda B, et al. Serological evidence of MERS‐CoV and HKU8‐related CoV co‐infection in Kenyan camels. Emerg Microbes Infect. 2019;8(1):1528‐1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hu Y, Li W, Gao T, et al. The severe acute respiratory syndrome coronavirus Nucleocapsid inhibits type I interferon production by interfering with TRIM25‐mediated RIG‐I ubiquitination. J Virol. 2017;91(8):e02143‐16. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1 Polymorphisms in ORF1ab. (A) Distribution of polymorphisms (SNPs found in more than 1 sample) in gene ORF1ab. SNP counts in a 100‐bp sliding window are shown. (B) An enlarged view of Figure A, from 12 999 to 14 048 nt. The of ORF1b frameshifts positions are shown.
Supplementary Figure S2 Illustrations are showing the distribution of polymorphisms (SNPs found in more than one strain) the spike glycoprotein. SNP counts in a 50‐bp sliding window are shown against their positions in individual components of S glycoprotein. Positions of variants; V26A, R1020Q, and A1158 variants are shown.
Supplementary Figure S3 The phylogenetic trees based on the (E gene, M gene, and N genes) sequences from the same MERS‐CoVs samples (37 human and 20 camels) isolated from 2012 to 2019. The unrooted phylogenetic trees were constructed by the maximum‐likelihood method, and the bootstrap values calculated from 100 trees. The scale bars are representing the tree distance corresponding to 0.001 nucleotide substitution/kb. The numbers at branch nodes indicate bootstrap values greater than 50. Branch lengths represent degrees of diversity between sequences. Clades are denoted (except for E gene), and African camel samples are highlighted in red. Sample information is labeled as follows, Accession number/Country/Human (Hu) or Camelus dromedary (Cd)/year.
Supplementary Figure S4 Phylogenetic analysis of ORF5 and ORF8b of the same MERS‐CoVs sample (37 human and 20 camels) isolated from 2012 to 2019. The unrooted phylogenetic tree was constructed by the maximum‐likelihood method and bootstrap values calculated from 100 trees. The scale bar represents the tree distance corresponding to 0.001 nucleotide substitution/kb. Numbers at branch nodes indicate bootstrap values greater than 50. Branch lengths represent degrees of diversity between sequences. Clades are denoted, and African camel samples are highlighted in red. Sample information is labeled as the following: Accession number/ Country/Human (Hu) or Camelus dromedary (Cd)/year.
Supplementary Figure S5 A bar figure showing the number of N protein sequences having the V178A variant. Graphical representation shows the percentage of N protein sequences isolated from human with the V178A variant in the last 4 years. Each column represents the number of N protein sequences isolated from a human with V178A variant to the total sequences generated in the same year.
Supplementary Figure S6 Diagram showing all ORFs of over 300 nucleotides across the wild type gene ORF8b (accession number JX869059.2) and models of defective gene ORF4b that encode potential truncated ORF8b protein (accession number KF961221.1) or long ORF8b protein (accession number KX154684.1). There is no defective gene ORF8b that produces ORF8 protein with a length close to ORF8 protein generated by the wild type gene ORF8b.
Supplemental Table S1
Supplemental Table S2