

Abstract
Background
This study was intended to investigate the genomic landscape of the immune microenvironments of brain metastases in breast cancer.
Methods
Three gene expression profile datasets (GSE76714, GSE125989 and GSE43837) of breast cancer with brain metastases were downloaded from Gene Expression Omnibus (GEO) database. After differential expression analysis, the tumor immune microenvironment and immune cell infiltration were analyzed. Then immune-related genes were identified, followed by function analysis, transcription factor (TF)-miRNA–mRNA co-regulatory network analysis, and survival analysis of metastatic recurrence.
Results
The present results showed that the tumor immune microenvironment in brain metastases was immunosuppressed compared with primary caner. Compared with primary cancer samples, the infiltration ratio of plasma cells in brain metastases samples was significantly higher, while the infiltration ratio of macrophages M2 cells in brain metastases samples was significantly lower. Total 42 immune-related genes were identified, such as THY1 and NEU2. CD1B, THY1 and DOCK2 were found to be implicated in the metastatic recurrence of breast cancer.
Conclusions
Targeting macrophages or plasma cells may be new strategies for immunotherapy of breast cancer with brain metastases. THY1 and NEU2 may be potential therapeutic targets for breast cancer with brain metastases, and THY1, CD1B and DOCK2 may serve as potential prognostic markers for improvement of brain metastases survival.
Keywords: Breast cancer, Brain metastases, Immune, Gene
Background
The incidence of brain metastases in cancer patients is rising, which may be due to the improvements in systematic therapies to control extracranial disease and prolong the survive of patients. Thus, patients who previously may have died sooner from other manifestations of the disease may develop brain metastases [1]. Breast cancer is the second most common cause of brain metastases following lung cancer [2]. It has been estimated that 20 to 30% of breast cancers develop brain metastases [3]. Brain metastases are serious complications of cancer with median survival of about 15 months and there is no effective long-term treatment [4, 5]. Therefore, brain metastases have become a major limiting factor in life expectancy and quality of life for many patients [2]. Understanding the biological mechanisms of brain metastases is crucial to predict patients at risk of brain metastases and to identify new therapeutic targets.
The interactions between immune and tumor cells have played an important role in malignant progression [6]. The brain was previously considered as an immunologically privileged organ because the intact brain has almost no lymphocytes [7]. Actually, the central nervous system is an immune specialized site under a tight regulatory network linking astrocytes, microglia, and lymphocytes [8]. T cells and B cells have been found around the tumors of brain metastases [9]. Despite the immune microenvironments of brain metastases in breast cancer have been studied [10, 11], the genomic landscape of breast cancer with brain metastases remains to be investigated.
In this study, we downloaded three gene expression profile datasets of breast cancer with brain metastases from Gene Expression Omnibus (GEO) database and analyzed the tumor immune microenvironment at genetic level (Fig. 1). Some immune-related genes were identified, which may contribute to the development of immunotherapy to treat breast cancer patients with brain metastases.
Fig. 1.

The flow diagram of the analysis
Methods
Data sources
Three expression profile datasets (GSE76714 [12], GSE125989 [1] and GSE43837 [13]) were downloaded from GEO database. GSE76714 included 71 triple negative breast cancer samples, including 48 primary triple negative breast cancer samples and 23 triple negative breast cancer with brain metastases samples, which was based on the platform of GPL14951 Illumina HumanHT-12 WG-DASL V4.0 R2 expression beadchip. GSE125989 contained 16 primary breast cancer samples, 16 paired breast cancer with brain metastases samples. The detection platform was GPL571 [HG-U133A_2] Affymetrix Human Genome U133A 2.0 Array. In GSE43837, there were 19 HER2 + primary breast cancer samples and 19 HER2 + breast cancer with brain metastatic samples. The platform was GPL1352 [U133_X3P] Affymetrix Human X3P Array.
Data downloading and preprocessing
The series matrix file(s) of GSE76714 was downloaded and normalized using quantile normalization with the beadarray library in R. The probe ID was converted into gene symbol using illuminaHumanWGDASLv4 package in R. The probes that did not correspond to gene symbol were removed. For the case where different probes mapped to the same gene, the mean value of different probes was taken as the final expression value of the gene. The FactoMineR package [14] was used for principal component analysis and clustering. The processing flows of GSE125989 and GSE43837 datasets were similar to that of GSE76714. GSE43837 data were transformed by log2(x + 1), and GSE125989 data were normalized by MAS5 algorithm of R language package, and transformed by log2. The microarray annotation packages of the GSE125989 and GSE43837 datasets were u133x3p.db and hgu133a2.db, respectively.
DEGs identification
The modified empirical Bayes t test method provided by limma package [15] (version 3.40.6) was used for differential expression analysis of brain metastasis group vs. cancer group. All RNAs (including mRNAs and lncRNAs) were analyzed to obtain the p value and log fold change (FC). The thresholds of DEGs screening were set as follows: p value < 0.05 and |logFC| > 0.585.
The ggscatter function of ggpubr package [16] (version 0.2.2) was used to draw the volcano plot, and the DEGs with the top 10 |logFC| were labeled in the volcano plot. The clustering heatmaps of DEGs were drawn using the pheatmap package [17] of R language.
Tumor immune microenvironment analysis
The stromal score, immune score and ESTIMATE score of all samples were calculated using the ESTIMATE algorithm [18] (version 1.0.13). The difference of these scores between brain metastatic tumor and primary tumor tissues were analyzed through T test, and the boxplot was drawn by using the R package ggpubr. Additionally, the cytolytic activity score of all samples was calculated and the differences of score between brain metastatic tumor and primary tumor tissues were also analyzed using T test. The boxplot was drawn by the R package ggpubr as well. Validation for the above scores was performed in the GSE125989 and GSE43837 datasets.
Immune cell infiltration abundance analysis
The abundance matrix of immune cells in the samples was estimated using the CIBERSORT deconvolution algorithm [19], and the infiltration abundance of immune cells in the samples was analyzed, with parameters of perm = 200 and QN = FALSE. The proportion difference of immune cell subgroups between two groups was calculated, and relevant landscape map (barplot), clustering heatmap (pheatmap), correlation heatmap (corHeatmap), and violin plot (vioplot) were drawn by R language. The immune cell subgroups with significant differences between groups were screened with threshold of p value < 0.05. The datasets of GSE125989 and GSE43837 were used for validation.
Identification of immune-related DEGs
Using the R corrplot package [20], spearman correlation test was conducted on the DEGs and infiltration abundance of differential immune cell subsets, and the DEGs with p value < 0.05 and correlation coefficient |r| > 0.30 were screened, which were considered as the DEGs related to immune cell subgroup. The ggboxplot function of the ggpubr package in R language was used to plot the boxplot of the expression of immune-related genes between two groups, and the differences of these genes between the two groups were further analyzed by T test. The datasets of GSE125989 and GSE43837 were used for validation.
Function and pathway enrichment analyses
These immune-related DEGs were subjected to Gene Ontology (GO) [21] and KEGG [22] pathway using ClusterProfiler [23] (version 3.12.0). The GO analysis results included biological process (BP), cellular component (CC) and molecular function (MF). The significance threshold was p value < 0.05, and the enrichment number (count) was at least 2. The compareCluster function of the clusterProfiler package was applied to visualize the top 10 GO BP and KEGG enrichment results.
Transcription factor (TF)-miRNA-mRNA co-regulatory network analysis
The miRNAs in the 3′UTR region of immune-related DEGs were predicted using relevant databases (miRWalk3.0 [24], TargetScan [25], miRDB [26], mirTarBase [27]), with a threshold score of > 0.95. Combining the results from each database, miRNAs that were validated (MirTarBase) and predicted in at least one other database were selected as the final mRNA-miRNA relationship pairs. HMDD V3.2 database [28] was used to retrieve the keyword “breast neoplasms” (synonyms of breast cancer) to further validate the predicted miRNAs. Then based on the online database TRRUST [29], the TF-mRNA pairs associated with immune-related DEGs and possible action mode (activation, suppression or unknown) were predicted. The mRNA-miRNA relationship pairs and TF-mRNA relationship pairs were integrated to construct the network using Cytoscape [30]. Additionally, GO and KEGG analyses were performed for the TF and immune-related DEGs in the network.
Breast cancer associated pathway screening and gene annotation
According to the breast neoplasms related pathways included in Comparative Toxicogenomics Database (CTD) [31], further screening was conducted for the KEGG pathways enriched by immune-related genes as well as transcription factors and immune-related genes in the network. The R package pathview [32] (version 1.24.0) was applied to draw the pathway map.
Protein-protein interaction (PPI) network analysis
The interaction between gene coding proteins was predicted and analyzed using STRING database [33] (version 11.0). The input gene set was immune-related genes, and the PPI score was set as 0.4 (medium confidence). After the PPI relation pairs were obtained, Cytoscape software was used to construct a network. Network topology properties (betweenness, closeness and degree) were analyzed using CytoNCA [34] plug-in of Cytoscape software. Functional modules in the network were identified using MCODE [34] plug-in of Cytoscape. Parameters were set as default, and the sub-network modules were screened according to score ≥ 4.
Drug-gene interaction prediction
DGIdb 3.0 [35] (version 3.0.2) database was applied to predict the drug-gene interaction of immune-related genes, and the parameters were set as default. The drug-gene interaction network was constructed through Cytoscape. The predictive drug information was retrieved in the DrugBank [36] database.
Metastatic recurrence survival analysis of key genes
The bc-GenExMiner v4.4 [37] online tool was used for breast cancer gene expression data mining, and the immune-related genes that were verified by data were subjected to metastatic recurrence survival analysis. The parameters were as follows: baseline like (PAM50) and/or triple-negative breast cancer (IHC) prognostic analysis; DNA microarrays samples (n = 10,001); metastatic recurrence; segmentation criteria of median.
Literature retrieval of key genes
The genes in PPI network and TF-miRNA–mRNA network were considered as key genes. The Biopython Python package [38] was used to access the NCBI Entrez database, and GenCLiP 2.0 database [39] was used to summarize the breast cancer associated literatures related to these key genes. The title and abstract were retrieved, and the literatures were considered as relevant literatures if both gene and disease keyword appeared.
Results
Data preprocessing and DEGs analysis
After preprocessing, the overall expression pattern of the samples was similar for each dataset. There was no significant batch effect among the samples in each dataset (Fig. 2a).
Fig. 2.

a Clustergrams of principal component analysis for three datasets. b The volcano plots for differentially expressed genes (DEGs) in three datasets. The red square represents upregulated DEGs, the blue circle represents downregulated DEGs, and the black triangle represents non-DEGs. The transverse dashed line is the p value, and the longitudinal dashed line is the fold change. The genes with the top tenfold changes are shown in the figure. c The heatmap of DEGs in three datasets. Red represents high expression and blue represents low expression
A total of 153 DEGs were obtained from the GSE76714 dataset, including 117 upregulated and 36 downregulated genes. Total 1898 DEGs were obtained from the GSE43837, including 563 upregulated and 1335 downregulated genes. Additionally, 954 DEGs were obtained from the GSE125989, including 311 upregulated genes and 643 downregulated genes. The volcano plots for DEGs are shown in Fig. 2b. The bidirectional hierarchical clustering heatmaps of DEGs is shown in Fig. 2c.
Tumor immune microenvironment analysis
In GSE76714, the stromal score of brain metastases group was significantly lower than that of primary cancer group, while the immune score, ESTIMATE score, and cytolytic activity score showed no significant difference between two groups (Fig. 3a). For the datasets of GSE43837 and GSE125989, the stromal score, immune score, and ESTIMATE score in brain metastases group were significantly lower than that in primary cancer group. Additionally, cytolytic activity scores for the two groups were not significantly different (Fig. 3b, c). The result may indicate the difference of immune microenvironment between two groups.
Fig. 3.

Box plots for stromal score, immune score, ESTIMATE score, and cytolytic activity score between brain metastases (BM) and primary cancer (PC) groups in GSE76714 (a), GSE43837 (b), and GSE125989 (c)
Immune cell infiltration abundance analysis
The infiltration abundance matrix of 22 kinds of immune cells in all samples of GSE76714 was estimated using Cibersort algorithm. The result showed that among the 71 samples, 60 were valid, including 40 cases of PC and 20 cases of BM. The immune cell infiltration abundance for GSE76714 is shown in Fig. 4. The barplot (Fig. 4a) and clustering heatmap (Fig. 4b) showed that the infiltration rates of T cells gamma delta (green), T cells CD4 naive (yellow) and mast cells activated cells (pink) in each sample were relatively high. The correlation heatmap indicated that there existed difference in immune cell infiltration pattern between PC and BM groups (Fig. 4c). For instance, the correlation between NK cell activated and T cell helper in BM was very low (r = 0.02), while it was relatively higher in PC (r = 0.6). The violin plot showed that the infiltration ratio of plasma cells in brain metastases samples was significantly higher than that in primary cancer samples (p = 0.026), while the infiltration ratio of macrophages M2 cells in brain metastases samples was lower than that in primary cancer samples (p = 0.003) (Fig. 4d). For GSE43837 and GSE125989, there was not enough CIBERSORT for analysis.
Fig. 4.

The barplot (a), clustering heatmap (b), correlation heatmap (c), and violin plot (d) of immune cell infiltration in GSE76714
Identification of immune-related genes
Correlation analysis was conducted between macrophages M2 cell infiltration and the expression of DEGs, and 42 immune-related genes were screened. T-test revealed that 27 genes, such as THY1, present significant differences in expression levels between two groups. These genes were significantly enriched in BP term associated with positive regulation of GTPase activity (ALDH1A1, APC2, DOCK2 and THY1). Additionally, other glycan degradation pathway was significantly enriched (Fig. 5).
Fig. 5.

The GO function (a) and KEGG pathway (b) enriched by the immune-related genes
TF-miRNA-mRNA network analysis
A total of 9 miRNA-mRNA pairs were predicted, which included 5 mRNAs and 9 miRNAs, such as miR-520a and miR-361-3p. In addition, 12 TF-mRNA pairs were obtained, involving 8 mRNAs and 11 TFs. Based on the miRNA-mRNA and TF-mRNA intraction pairs, a TF-miRNA-mRNA regulatory network was constructed (Fig. 6a). Function analysis showed that the TFs and mRNAs in the network were significantly enriched in lipid homeostasis, and cholesterol homeostasis associated BP terms (Fig. 6b). Moreover, they were involved in 8 pathways, such as transcriptional misregulation in cancer, PPAR signaling pathway, AMPK signaling pathway, and breast cancer (Fig. 6c).
Fig. 6.

a The constructed transcription factor (TF)-miRNA-mRNA regulatory network. The red round nodes represent upregulated differentially expressed genes (DEGs); the blue round nodes represent downregulated DEGs; the green triangle nodes represent miRNAs; green rhombus nodes represent TF. The gray lines represent the interaction relationship; the red lines represent the activation relationship of TF-mRNA; the blue lines represent the inhibition relationship of TF-mRNA. B and C: The GO function (b) and KEGG pathway (c) enriched by the TF-mRNA
PPI network analysis
Based on the immune-related genes, 46 PPI pairs were obtained and the constructed PPI network included 31 nodes (21 up-regulated and 10 down-regulated ones) (Fig. 7). Among the 31 nodes, NEUROD1, THY1, ALDH1A1, GBX2, MIXL1, CDH8 and ASPN had degrees more than 5, and were considered as hub nodes.
Fig. 7.
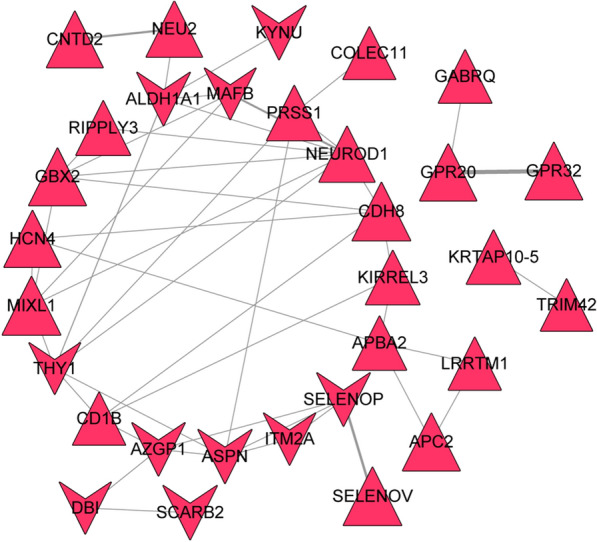
a The constructed protein–protein interaction (PPI) network of differentially expressed genes (DEGs) associated with macrophages M2. The triangle nodes represent upregulated DEGs; the red node represents DEGs associated with macrophages M2. b The thickness of the line is positively correlated to the relationship score obtained by STRING
Drug-gene interaction analysis
A total of 10 drug-gene interaction pairs were identified based on the immune-related genes, which involved 10 drugs (busulfan, retinol, tretinoin, zanamivir, deferoxamine, temazepam, diazepam, oxazepam, bromazepam and nitrazepam) and 5 mRNAs (PRSS1, ALDH1A1, NEU2, NEUROD1 and GABRQ). In detail, GABRQ interacted with temazepam, diazepam, oxazepam, bromazepam and nitrazepam; ALDH1A1 interacted with tretinoin and retinol; NEU2 interacted with zanamivir; PRSS1 interacted with busulfan (Fig. 8).
Fig. 8.

The constructed drug-gene interaction network. The red nodes represent the upregulated differentially expressed genes (DEGs) in macrophages M2. Blue nodes represent downregulated DEGs in macrophages M2. The green nodes represent small molecule drugs
Metastatic recurrence survival analysis of key genes
The Venn diagram of the intersection of DEGs in the three datasets and immune-related genes is shown in Fig. 9a. The genes verified by GSE43817 dataset were DOCK2, HCN4, HASPIN, STK33 and KYNU. The gene verified by GSE125989 dataset was THY1. The genes verified by the two datasets were ASPN and CD1B. These genes were considered as key genes and were performed metastatic recurrence survival analysis. Based on the analysis of the bc-GenExMiner v4.4 database, CD1B, THY1 and DOCK2 were found to affect the metastatic recurrence of triple-negative breast cancer. As shown in Fig. 9b, high expression of THY1 was more likely to cause metastasis of breast cancer, while low expression of CD1B and DOCK2 was likely to cause metastasis of breast cancer.
Fig. 9.

a Venn diagram of differentially expressed genes (DEGs) and DEGs in macrophages M2. b Metastatic recurrence survival curve for CD1B, THY1, DOCK2, HCN4, HASPIN, STK33 and KYNU
Literature retrieval of key genes
Among the key genes, ASPN, DOCK2, THY1 and KYNU were found to be associated with breast cancer based on NCBI Entrez database. Based on the GenCLiP 2.0 database, only THY1 was associated with breast cancer.
Discussion
Brain metastases commonly originate from breast, lung, and melanoma. However, brain metastases are hard to treat because most drugs cannot penetrate the blood brain barrier and often affect multiple areas of the brain [40]. Therefore, identification of new biomarkers may contribute to the development of individualized treatment. The present study is the first time to explore the immune-related genes in breast cancer with brain metastases. The present results showed that the tumor immune microenvironment in brain metastases was immunosuppressed compared with primary caner. Compared with primary cancer samples, the infiltration ratio of plasma cells in brain metastases samples was significantly higher, while the infiltration ratio of macrophages M2 cells in brain metastases samples was significantly lower. Total 42 immune-related genes were identified, such as THY1 (Thy-1 cell surface antigen) and NEU2 (neuraminidase 2). THY1 was a hub protein in the PPI network. NEU2 interacted with zanamivir. CD1B (CD1b molecule), THY1 and DOCK2 (dedicator of cytokinesis 2) were found to be implicated in the metastatic recurrence of breast cancer.
The brain has long been regarded as an immune privileged organ, whereas, this view was revised since a lymphatic vessel network of brain tissues was found in the dura mater in mice [41, 42]. Our study suggested an immune suppressive environment in the brain metastases, partly exemplified by significantly decreased stromal score, immune score, and ESTIMATE score in brain metastases samples compared with that in primary cancer group. Our results were in accordance with a recent study of Kudo et al. [43], who coupled immune gene expression profiling and topological gene–gene network analysis and demonstrated an immune suppressive microenvironment in the brain metastases of non-small-cell lung cancer.
Tumor microenvironment is composed of various nonmalignant stromal cells, among which tumor-associated macrophages are the most prominent type of migratory hematopoietic cells [44]. In breast, the tumor-associated macrophages are primarily pro-tumorigenic M2-like macrophages, which promote the progression and metastasis of breast cancer by releasing various cytokines [45]. Plasma cells can affect antitumor immunity by regulating T-cell responses, or excluding immune-suppressive cell types to provide a permissive tumor microenvironment for CD8+ tumor-infiltrating lymphocytes, the key mediators of antitumor immunity [46]. Our results may devise new strategies for immunotherapy of breast cancer with brain metastases—by targeting macrophages or plasma cells.
In this study, 42 immune-related genes were identified, such as THY1, NEU2, CD1B and DOCK2. THY1 was one of hub proteins in the PPI network. In PPI network, the topological placement of a protein is connected with its biological essentiality. The densely connected hub proteins are more likely to be essential proteins, which is referred to as the “centrality-lethality rule” [47]. THY1 is a glycophosphatidylinositol-anchored protein, which has been proposed to play important roles in cancers [48]. Function analysis showed that THY1 was significantly enriched in function associated with positive regulation of GTPase activity. GTPases can be activated when binding to GTP. Once activated, GTPases carry out many functions in cells, such as the regulation of cell proliferation, apoptosis, and differentiation [49]. It has been reported that in the case of tumor progression, mutations in Ras related small GTPases can increase the proliferation, survival, and adhesion of tumor cells, tending toward a metastatic phenotype [50]. Additionally, high expression of THY1 was more likely to cause metastasis of breast cancer. Taken together, we speculated that THY1 may play a role in brain metastases of breast cancer via positive regulation of GTPase activity. Additionally, it mays sever as a prognostic indicator to predict the metastasis of breast cancer.
NEU2 was involved in the pathway of other glycan degradation. In mammalian tissues, glycans exist in free forms or conjugated forms, which participate in various biological processes, such as host–pathogen interactions, cell migration and metastasis, and initiation of immune response [51]. Study has reported that glycan changes in malignant cells take many forms and mediate key pathophysiological events during various stages of tumor progression [52]. In the tumor environment, glycosylation changes allow tumor cells to usurp many development events, allowing tumor cells to invade and spread throughout the organism. Thus, we speculated that the upregulation of NEU2 may be involved in brain metastases of breast cancer through pathway of other glycan degradation. Drug-gene interaction analysis showed that NEU2 interacted with zanamivir. Zanamivir is an inhibitor of the enzyme neuraminidase, a surface glycoprotein necessary for the replication of type A and B influenza viruses [53]. Its role in cancer is rarely reported. Now we speculated that zanamivir may serve as an antineoplastic drug in breast cancer with brain metastases by targeting NEU2.
CD1B belongs to the group 1 CD1 family of transmembrane glycoproteins, and is associated with major histocompatibility complex class I-like molecules. CD1 molecules regulate the expression of some self- and foreign-lipid antigens to T-cell receptors on T cells [54]. A recent study has indicated that there are different expression patterns of CD1 molecules between tumor cells and normal cells [55]. More recently, low expression of CD1B was reported to be correlated with poorer biochemical recurrence-free survival in prostate cancer. Similar result was found in our study, that was, low expression of CD1B was likely to cause metastatic recurrence of breast cancer.
DOCK2 is a member of the CDM protein family, which can regulate cell motility and cytokine production by activating Rac in mammalian hematopoietic cells. Additionally, DOCK2 plays a critical role in the modulation of the immune system [56]. Hu et al. [57] have reported that low expression of DOCK2 is associated with poorer prognosis of acute myeloid leukemia. Recent study reported that DOCK2 hypermethylation was associated with biochemical recurrence after radical prostatectomy in prostate cancer [58]. To our knowledge, there was no study about the role of DOCK2 in breast cancer. Together with our results, we speculated that DOCK2 may be a prognostic marker of metastatic recurrence in breast cancer.
Despite these findings, there existed a limitation in this study. Due to lack of adequate clinical samples, there was no experimental evidence to support our analysis results. Thus, further experimental studies are needed to confirm our results.
Conclusions
Our study indicated that tumor immune microenvironment in brain metastases of breast cancer was immunosuppressed compared with primary caner. Targeting macrophages or plasma cells may be new strategies for immunotherapy of breast cancer with brain metastases. THY1 and NEU2 may be potential therapeutic targets for breast cancer with brain metastases, and THY1, CD1B and DOCK2 may serve as potential prognostic markers for improvement of brain metastases survival.
Acknowledgements
The authors thank all members in our lab for the excellent technical help.
Abbreviations
- GEO
gene expression omnibus
- FC
fold change
- barplot
relevant landscape map
- pheatmap
clustering heatmap
- corHeatmap
correlation heatmap
- vioplot
violin plot
- GO
gene ontology
- BP
biological process
- MF
molecular function
- TF
transcription factor
- CTD
comparative toxicogenomics database
- PPI
protein–protein interaction
Authors’ contributions
AHW conceived and designed this study. WCL carried out the plan and wrote this paper. HX, CY, JJL and ZYL gave advice and carried out the data analysis. All authors read and approved the final manuscript.
Funding
This work was supported by grants from Guidance Plan of Natural Science Foundation of Liaoning Province (Nos. 201602773; 2019-ZD-0340).
Availability of data and materials
The data that support the findings of this study are available from University of California Santa Cruz Genome Browser and GEO database.
Ethics approval and consent to participate
This work was approved by the Ethical Board of China Medical University.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Iwamoto T, Niikura N, Ogiya R, Yasojima H, Watanabe K-I, Kanbayashi C, Tsuneizumi M, Matsui A, Fujisawa T, Iwasa T. Distinct gene expression profiles between primary breast cancers and brain metastases from pair-matched samples. Sci Rep. 2019;9:1–8. doi: 10.1038/s41598-019-50099-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Witzel I, Oliveira-Ferrer L, Pantel K, Müller V, Wikman H. Breast cancer brain metastases: biology and new clinical perspectives. Breast Cancer Res. 2016;18:8. doi: 10.1186/s13058-015-0665-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Venur VA, Leone JP. Targeted therapies for brain metastases from breast cancer. Int J Mol Sci. 2016;17:1543. doi: 10.3390/ijms17091543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Quigley MR, Fukui O, Chew B, Bhatia S, Karlovits S. The shifting landscape of metastatic breast cancer to the CNS. Neurosurg Rev. 2013;36:377–382. doi: 10.1007/s10143-012-0446-6. [DOI] [PubMed] [Google Scholar]
- 5.Rostami R, Mittal S, Rostami P, Tavassoli F, Jabbari B. Brain metastasis in breast cancer: a comprehensive literature review. J Neurooncol. 2016;127:407–414. doi: 10.1007/s11060-016-2075-3. [DOI] [PubMed] [Google Scholar]
- 6.Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science. 2011;331:1565–1570. doi: 10.1126/science.1203486. [DOI] [PubMed] [Google Scholar]
- 7.Galea I, Bechmann I, Perry VH. What is immune privilege (not)? Trends Immunol. 2007;28:12–18. doi: 10.1016/j.it.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 8.Hudson L, Bragg D, Tompkins M, Meeker R. Astrocytes and microglia differentially regulate trafficking of lymphocyte subsets across brain endothelial cells. Brain Res. 2005;1058:148–160. doi: 10.1016/j.brainres.2005.07.071. [DOI] [PubMed] [Google Scholar]
- 9.Berghoff AS, Lassmann H, Preusser M, Höftberger R. Characterization of the inflammatory response to solid cancer metastases in the human brain. Clin Exp Metas. 2013;30:69–81. doi: 10.1007/s10585-012-9510-4. [DOI] [PubMed] [Google Scholar]
- 10.Duchnowska R, Pęksa R, Radecka B, Mandat T, Trojanowski T, Jarosz B, Czartoryska-Arłukowicz B, Olszewski WP, Och W, Kalinka-Warzocha E. Immune response in breast cancer brain metastases and their microenvironment: the role of the PD-1/PD-L axis. Breast Cancer Res. 2016;18:43. doi: 10.1186/s13058-016-0702-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Harter PN, Bernatz S, Scholz A, Zeiner PS, Zinke J, Kiyose M, Blasel S, Beschorner R, Senft C, Bender B. Distribution and prognostic relevance of tumor-infiltrating lymphocytes (TILs) and PD-1/PD-L1 immune checkpoints in human brain metastases. Oncotarget. 2015;6:40836. doi: 10.18632/oncotarget.5696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Duchnowska R, Jarząb M, Żebracka-Gala J, Matkowski R, Kowalczyk A, Radecka B, Kowalska M, Pfeifer A, Foszczyńska-Kłoda M, Musolino A. Brain metastasis prediction by transcriptomic profiling in triple-negative breast cancer. Clin Breast Cancer. 2017;17:e65–e75. doi: 10.1016/j.clbc.2016.08.008. [DOI] [PubMed] [Google Scholar]
- 13.McMullin RP, Wittner BS, Yang C, Denton-Schneider BR, Hicks D, Singavarapu R, Moulis S, Lee J, Akbari MR, Narod SA. A BRCA1 deficient-like signature is enriched in breast cancer brain metastases and predicts DNA damage-induced poly (ADP-ribose) polymerase inhibitor sensitivity. Breast Cancer Res. 2014;16:R25. doi: 10.1186/bcr3625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lê S, Josse J, Husson F. FactoMineR: an R package for multivariate analysis. J Stat Softw. 2008;25:1–18. [Google Scholar]
- 15.Smyth GK, Ritchie M, Thorne N, Wettenhall J. LIMMA: linear models for microarray data. In: Bioinformatics and Computational Biology Solutions Using R and Bioconductor. Statistics for Biology and Health. 2005.
- 16.Kassambara A. ggpubr:“ggplot2” based publication ready plots. R package version 0.1. 6. 2017.
- 17.Kolde R, Kolde M. Package ‘pheatmap’. 2015.
- 18.Yoshihara K, Shahmoradgoli M, Martínez E, Vegesna R, Kim H, Torres-Garcia W, Treviño V, Shen H, Laird PW, Levine DA. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat commun. 2013;4:1–11. doi: 10.1038/ncomms3612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, Hoang CD, Diehn M, Alizadeh AA. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods. 2015;12:453–457. doi: 10.1038/nmeth.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wei T, Simko V, Levy M, Xie Y, Jin Y, Zemla J. Package ‘corrplot’. Statistician. 2017;56:316–324. [Google Scholar]
- 21.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT. Gene Ontology: tool for the unification of biology. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yu G, Wang L-G, Han Y, He Q-Y. clusterProfiler: an R package for comparing biological themes among gene clusters. Omics. 2012;16:284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dweep H, Sticht C, Pandey P, Gretz N. miRWalk–database: prediction of possible miRNA binding sites by “walking” the genes of three genomes. J Biomed Inform. 2011;44:839–847. doi: 10.1016/j.jbi.2011.05.002. [DOI] [PubMed] [Google Scholar]
- 25.Agarwal V, Bell GW, Nam J-W, Bartel DP. Predicting effective microRNA target sites in mammalian mRNAs. Elife. 2015;4:e05005. doi: 10.7554/eLife.05005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wong N, Wang X. miRDB: an online resource for microRNA target prediction and functional annotations. Nucleic Acids Res. 2015;43:D146–D152. doi: 10.1093/nar/gku1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chou C-H, Shrestha S, Yang C-D, Chang N-W, Lin Y-L, Liao K-W, Huang W-C, Sun T-H, Tu S-J, Lee W-H. miRTarBase update 2018: a resource for experimentally validated microRNA-target interactions. Nucleic Acids Res. 2018;46:D296–D302. doi: 10.1093/nar/gkx1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li Y, Qiu C, Tu J, Geng B, Yang J, Jiang T, Cui Q. HMDD v2 0: a database for experimentally supported human microRNA and disease associations. Nucleic Acids Res. 2014;42:1070–1074. doi: 10.1093/nar/gkt1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Han H, Cho J-W, Lee S, Yun A, Kim H, Bae D, Yang S, Kim CY, Lee M, Kim E. TRRUST v2: an expanded reference database of human and mouse transcriptional regulatory interactions. Nucleic Acids Res. 2018;46:D380–D386. doi: 10.1093/nar/gkx1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Davis AP, Grondin CJ, Johnson RJ, Sciaky D, McMorran R, Wiegers J, Wiegers TC, Mattingly CJ. The comparative toxicogenomics database: update 2019. Nucleic Acids Res. 2019;47:D948–D954. doi: 10.1093/nar/gky868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Luo W, Brouwer C. Pathview: an R/Bioconductor package for pathway-based data integration and visualization. Bioinformatics. 2013;29:1830–1831. doi: 10.1093/bioinformatics/btt285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Szklarczyk D, Morris JH, Cook H, Kuhn M, Wyder S, Simonovic M, Santos A, Doncheva NT, Roth A, Bork P. The STRING database in 2017: quality-controlled protein–protein association networks, made broadly accessible. Nucleic Acids Res. 2016;2:937. doi: 10.1093/nar/gkw937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tang Y, Li M, Wang J, Pan Y, Wu F-X. CytoNCA: a cytoscape plugin for centrality analysis and evaluation of protein interaction networks. Biosystems. 2015;127:67–72. doi: 10.1016/j.biosystems.2014.11.005. [DOI] [PubMed] [Google Scholar]
- 35.Cotto KC, Wagner AH, Feng Y-Y, Kiwala S, Coffman AC, Spies G, Wollam A, Spies NC, Griffith OL, Griffith M. DGIdb 30: a redesign and expansion of the drug–gene interaction database. Nucleic Acids Res. 2018;46:D1068–D1073. doi: 10.1093/nar/gkx1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Law V, Knox C, Djoumbou Y, Jewison T, Guo AC, Liu Y, Maciejewski A, Arndt D, Wilson M, Neveu V. DrugBank 40: shedding new light on drug metabolism. Nucleic Acids Res. 2014;42:D1091–D1097. doi: 10.1093/nar/gkt1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jézéquel P, Frénel J-S, Campion L, Guérin-Charbonnel C, Gouraud W, Ricolleau G, Campone M. bc-GenExMiner 30: new mining module computes breast cancer gene expression correlation analyses. Database. 2013;2013:998. doi: 10.1093/database/bas060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cock PJ, Antao T, Chang JT, Chapman BA, Cox CJ, Dalke A, Friedberg I, Hamelryck T, Kauff F, Wilczynski B. Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics. 2009;25:1422–1423. doi: 10.1093/bioinformatics/btp163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang J-H, Zhao L-F, Lin P, Su X-R, Chen S-J, Huang L-Q, Wang H-F, Zhang H, Hu Z-F, Yao K-T. GenCLiP 2.0: a web server for functional clustering of genes and construction of molecular networks based on free terms. Bioinformatics. 2014;30:2534–2536. doi: 10.1093/bioinformatics/btu241. [DOI] [PubMed] [Google Scholar]
- 40.Joo KM, Park IH, Shin JY, Jin J, Kang BG, Kim MH, Lee SJ. Human neural stem cells can target and deliver therapeutic genes to breast cancer brain metastases. Mol Ther. 2009;17:570–575. doi: 10.1038/mt.2008.290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Louveau A, Smirnov I, Keyes TJ, Eccles JD, Rouhani SJ, Peske JD, Derecki NC, Castle D, Mandell JW, Lee KS. Structural and functional features of central nervous system lymphatic vessels. Nature. 2015;523:337–341. doi: 10.1038/nature14432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Aspelund A, Antila S, Proulx ST, Karlsen TV, Karaman S, Detmar M, Wiig H, Alitalo K. A dural lymphatic vascular system that drains brain interstitial fluid and macromolecules. J Exp Med. 2015;212:991–999. doi: 10.1084/jem.20142290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kudo Y, Haymaker C, Zhang J, Reuben A, Duose D, Fujimoto J, Roy-Chowdhuri S, Solis Soto L, Dejima H, Parra E. Suppressed immune microenvironment and repertoire in brain metastases from patients with resected non-small-cell lung cancer. Ann Oncol. 2019;30:1521–1530. doi: 10.1093/annonc/mdz207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mantovani A, Sica A. Macrophages, innate immunity and cancer: balance, tolerance, and diversity. Curr Opin Immunol. 2010;22:231–237. doi: 10.1016/j.coi.2010.01.009. [DOI] [PubMed] [Google Scholar]
- 45.Gocheva V, Wang H-W, Gadea BB, Shree T, Hunter KE, Garfall AL, Berman T, Joyce JA. IL-4 induces cathepsin protease activity in tumor-associated macrophages to promote cancer growth and invasion. Genes Dev. 2010;24:241–255. doi: 10.1101/gad.1874010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kroeger DR, Milne K, Nelson BH. Tumor-infiltrating plasma cells are associated with tertiary lymphoid structures, cytolytic T-cell responses, and superior prognosis in ovarian cancer. Clin Cancer Res. 2016;22:3005–3015. doi: 10.1158/1078-0432.CCR-15-2762. [DOI] [PubMed] [Google Scholar]
- 47.Ning K, Ng HK, Srihari S, Leong HW, Nesvizhskii AI. Examination of the relationship between essential genes in PPI network and hub proteins in reverse nearest neighbor topology. BMC Bioinformatics. 2010;11:505. doi: 10.1186/1471-2105-11-505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang DH, Yang ZL, Zhou EX, Miao XY, Zou Q, Li JH, Liang LF, Zeng GX, Chen SL. Overexpression of Thy1 and ITGA6 is associated with invasion, metastasis and poor prognosis in human gallbladder carcinoma. Oncol Lett. 2016;12:5136–5144. doi: 10.3892/ol.2016.5341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Heo J. Redox control of GTPases: from molecular mechanisms to functional significance in health and disease. Antioxid Redox Signal. 2011;14:689–724. doi: 10.1089/ars.2009.2984. [DOI] [PubMed] [Google Scholar]
- 50.Spindler V, Schlegel N, Waschke J. Role of GTPases in control of microvascular permeability. Cardiovasc Res. 2010;87:243–253. doi: 10.1093/cvr/cvq086. [DOI] [PubMed] [Google Scholar]
- 51.Liang P-H, Wu C-Y, Greenberg WA, Wong C-H. Glycan arrays: biological and medical applications. Curr Opin Chem Biol. 2008;12:86–92. doi: 10.1016/j.cbpa.2008.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li M, Song L, Qin X. Glycan changes: cancer metastasis and anti-cancer vaccines. J Biosci. 2010;35:665–673. doi: 10.1007/s12038-010-0073-8. [DOI] [PubMed] [Google Scholar]
- 53.Dunn CJ, Goa KL. Zanamivir. Drugs. 1999;58:761–784. doi: 10.2165/00003495-199958040-00016. [DOI] [PubMed] [Google Scholar]
- 54.Beekman E, Porcelli S, Morita C, Behar S, Furlong S, Brenner M. Recognition of a Lipid Antigen by Cd1-Restricted Alpha-Beta (+) T-cells. Nature. 1994;372:691–694. doi: 10.1038/372691a0. [DOI] [PubMed] [Google Scholar]
- 55.Lepore M, de Lalla C, Gundimeda SR, Gsellinger H, Consonni M, Garavaglia C, Sansano S, Piccolo F, Scelfo A, Häussinger D. A novel self-lipid antigen targets human T cells against CD1c + leukemias. J Exp Med. 2014;211:1363–1377. doi: 10.1084/jem.20140410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang L, Nishihara H, Kimura T, Kato Y, Tanino M, Nishio M, Obara M, Endo T, Koike T, Tanaka S. DOCK2 regulates cell proliferation through Rac and ERK activation in B cell lymphoma. Biochem Biophys Res Commun. 2010;395:111–115. doi: 10.1016/j.bbrc.2010.03.148. [DOI] [PubMed] [Google Scholar]
- 57.Hu N, Pang Y, Zhao H, Si C, Ding H, Chen L, Wang C, Qin T, Li Q, Han Y. High expression of DOCK2 indicates good prognosis in acute myeloid leukemia. J Cancer. 2019;10:6088. doi: 10.7150/jca.33244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bjerre MT, Strand SH, Nørgaard M, Kristensen H, Rasmussen AK, Mortensen MM, Fredsøe J, Mouritzen P, Ulhøi B, Ørntoft T. Aberrant DOCK2, GRASP, HIF3A and PKFP hypermethylation has potential as a prognostic biomarker for prostate cancer. Int J Mol Sci. 2019;20:1173. doi: 10.3390/ijms20051173. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from University of California Santa Cruz Genome Browser and GEO database.