

Abstract
Tobacco smoking is the primary risk factor for lung cancer, driven by the addictive nature of nicotine and the indisputable carcinogenicity of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) as well as other compounds. The integration of lung cancer chemoprevention with smoking cessation is one potential approach to reduce this risk and mitigate lung cancer mortality. Experimental data from our group suggest that kava, commonly consumed in the South Pacific Islands as a beverage to promote relaxation, may reduce lung cancer risk by enhancing NNK detoxification and reducing NNK-derived DNA damage. Building upon these observations, we conducted a pilot clinical trial to evaluate the effects of a 7-day course of kava on NNK metabolism in active smokers. The primary objective was to compare urinary total 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol (NNAL plus its glucuronides, major metabolites of NNK) before and after kava administration as an indicator of NNK detoxification. Secondary objectives included determining kava’s safety, its effects on DNA damage, tobacco use, and cortisol (a biomarker of stress). Kava increased urinary excretion of total NNAL and reduced urinary 3-methyladenine (3-mA) in participants, suggestive of its ability to reduce the carcinogenicity of NNK. Kava also reduced urinary total nicotine equivalents (TNE), indicative of its potential to facilitate tobacco cessation. Plasma cortisol and urinary total cortisol equivalents (TCE) were reduced upon kava use, which may contribute to reductions in tobacco use. These results demonstrate the potential of kava intake to reduce lung cancer risk among smokers.
Keywords: kava, lung carcinogenesis, tobacco exposure, nitrosamine, surrogate biomarker
Introduction
In spite of recent advances in immunotherapy, lung cancer remains the leading cause of cancer-related deaths, with an estimated 140,000 deaths and over 200,000 new cases annually in the U.S. (1). Cigarette smoking is the major risk factor and tobacco cessation is the only strategy to date proven to mitigate lung cancer risk. The success rate of tobacco cessation with currently available therapies, however, remains low (2). Novel approaches, such as the integration of lung cancer prevention in conjunction with tobacco cessation, are urgently needed.
Tobacco smoke contains various carcinogens and co-carcinogens, collectively contributing to the increased lung cancer risk. 4-(Methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) is one of the best characterized tobacco-specific lung carcinogens, demonstrating potent pulmonary carcinogenicity in multiple species (3). Ample evidence support NNK as a key causative factor of human lung adenocarcinoma development (4–7). For instance, by analyzing the prospective samples from the Prostate, Lung, Colorectal, and Ovarian Cancer (PLCO) Screening Trial, Church et al. demonstrated that plasma 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol (NNAL, a major metabolite of NNK) was significantly associated with increased lung cancer risk, even after correcting for age and smoking duration (7). Polymorphisms of genes involved in NNK metabolism (both bioactivation and detoxification) are also associated with differential lung cancer risk (8). Prospective cohort studies found that urinary NNAL was positively associated with lung cancer risk in smokers (9,10), after correcting for cigarettes smoked per day and years of smoking, directly linking NNK exposure to lung cancer development.
The mechanism of NNK carcinogenesis has been well characterized (Fig. 1). Activation of NNK generates two types of reactive species, leading to DNA damage (3). NNK can be reduced to NNAL, the major metabolite in human smokers (11). NNAL is glucuronidated, leading to its detoxification via excretion as NNAL-Gluc (12), which would reduce the carcinogenic risk of NNK (13). At the same time, NNAL can be bioactivated to generate two types of reactive species to form DNA damage. DNA damage is widely accepted as the root cause of NNK-induced carcinogenesis through mutagenesis (3). Some modified DNAs (DNA adducts), however, are not stable and are excreted in urine, which could serve as non-invasive surrogates to estimate the level of DNA damage in tissues. Among various urinary DNA adducts, we and others demonstrated that human urinary 3-methyladenine (3-mA) appeared to be highly tobacco-dependent (14–16) and may be indicative of NNK bioactivation (15).
Figure 1.
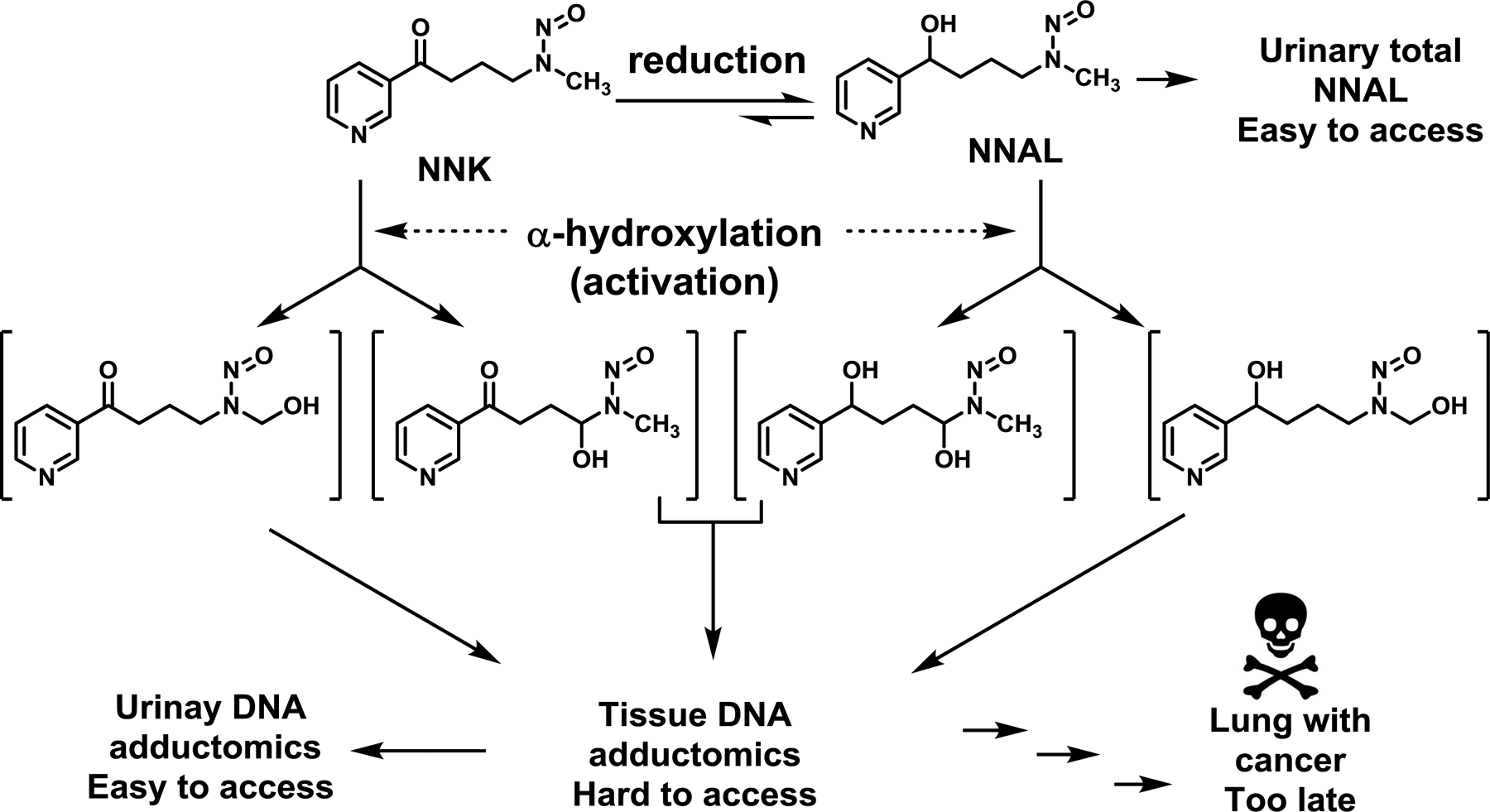
NNK metabolism, resulting in tissue DNA adductomics, urinary DNA adductomics, urinary NNAL excretion and eventually lung cancer formation.
Kava is traditionally consumed by the South Pacific Islanders to promote natural calmness (17,18). It is also marketed as a dietary supplement in the U.S. to help reduce stress. A group of kava-specific lactones (kavalactones, particularly kavain and dihydrokavain, Fig. S1) have been proposed to be responsible for this pharmacodynamic effect (19,20). Epidemiological observations (21,22) and pre-clinical animal studies (23–25) indicate that kava consumption may reduce cancer risk, particularly lung cancer (23,26–28). We recently demonstrated that kava completely blocked NNK-induced lung tumorigenesis in A/J mice (23); dihydromethysticin (DHM, Fig. S1) was identified as the active ingredient with an efficacious dose in A/J mice (28) comparable to doses used in humans as dietary supplements (29). Mechanistically, kava and DHM reduced NNK/NNAL-induced DNA damage in A/J mouse lung tissues (23,28), potentially via enhancing NNAL urinary detoxification (26).
Such mechanistic insight greatly improves the feasibility of kava clinical evaluation because non-invasive NNK metabolites can serve as surrogates of kava’s potential benefits. We herein report a pilot clinical trial in healthy smokers to test the hypothesis that short-term kava administration will result in increased levels of urinary NNAL, reflective of enhanced NNK detoxification. The primary endpoint was a comparison of urinary NNAL before and after a 7-day course of kava. Secondary endpoints included determining kava’s safety, effect on tobacco use and dependence, and effect on stress.
Materials and methods
Chemicals and reagents
Twenty bottles of kava capsules from the same manufacturing lot were obtained directly from Gaia Herbs (Brevard, NC). Each capsule was labeled to contain no less than 75 mg kavalactones. Deuterium-labelled dihydromethysticin (2H-DHM) was synthesized and characterized by both NMR and mass spectrometry (30). [2H4]-nicotine, [2H3]-cotinine, and [2H3]-3-OH-cotinine were purchased from Sigma-Aldrich (St. Louis, MO). [13C6]-NNAL, [2H3]-3-mA, [2H3]-nicotine-N-oxide, [2H4]-cortisol, [2H4]-6β-hydroxycortisol, [2H6]-allo-3α-tetrahydrocortisol, [2H8]-cortisone, and [2H6]-tetrahydrocortisone were purchased from Toronto Research Chemicals (Toronto, ON, Canada). Optima LC/MS grade H2O, HCO2H, CH3OH and CH3CN were purchased from Fisher Scientific (Fair Lawn, NJ). Strata-X cartridges (33 μm, 30 mg/1 mL) were purchased from Phenomenex (Torrance, CA). Oasis MCX cartridges (30 mg) were purchased from Waters (Milford, MA). β-Glucuronidase and other chemicals were purchased from Sigma-Aldrich (St. Louis, MO) unless stated otherwise.
Profiling five major kavalactones
Our method to quantify five major kavalactones (kavain, dihydrokavain, methysticin, DHM, and desmethoxyyangonin) in kava products was published elsewhere (30). Briefly, the dark brown viscous oil in three randomly selected capsules were weighed and dissolved in a solution of DMSO:CH3OH (1:1) to 20 mg/mL. The solution was diluted to a final concentration of 20 pg/μL with 10% CH3OH in H2O. [2H2]-DHM was added as the internal standard at a final concentration of 1.0 pg/μL. The samples (10 μL) were analyzed by ultraperformance liquid chromatography tandem mass spectrometry (UPLC-MS/MS). The amount of these five kavalactones was estimated to be 77 mg/capsule (30). The same method was also used to quantify DHM in the urine samples from the trial participants to assess kava exposure.
Clinical trial
Twenty-one participants completed the clinical trial (Clinicaltrials.gov NCT02500472), conducted at the University of Minnesota. The study design is shown in Figure 2. Eligible participants were adults (≥ 18 years of age) and current smokers (≥ 5 cigarettes per day by self-report). All participants had normal blood counts, electrolytes, and liver function. Those with known hepatobiliary disease or impairment, conditions that might affect kava absorption such as gastric bypass or celiac sprue, major or chronic medical disease, chronic use of medications that could not safely be stopped during the study period, antibiotic use within two months of study enrollment, alcohol dependence, and pregnant/breastfeeding women were excluded. The study was approved by the University of Minnesota Institutional Review Board and was conducted in accordance with recognized ethical guidelines. All participants provided informed, written consent. Demographic information, tobacco use history, medical history, and medication use history were obtained. All participants underwent a screening physical exam by a study physician. Height and weight were measured. Blood for safety or biomarker evaluation were collected at screening, baseline, and on days 4 and 7 of kava intervention. Comprehensive metabolic panel (CMP) and complete blood counts (CBC) were performed at Fairview Diagnostic Laboratories, a CLIA-certified clinical lab. Buffy coat, red blood cell, and plasma were aliquoted and stored at −80°C. Twenty-four hour urine collections were obtained at baseline, and on days 1–2, 4–5 and 6–7 of kava intervention for urinary NNAL and other urinary biomarker analysis, broken down into 0–6 and 6–24 h time periods. Volume was recorded to the nearest millimeter. Urine was aliquoted and stored at ≤ −20°C. Participants were instructed to continue their usual smoking habits, record dietary intake, and keep a smoking diary throughout the kava intervention. Additionally, participants completed the modified Cigarette Evaluation Questionnaire (mCEQ), a validated tool that assesses the degree to which a smoker experiences the reinforcing effects of smoking (31), at baseline and on day 7 of the kava intervention.
Figure 2.
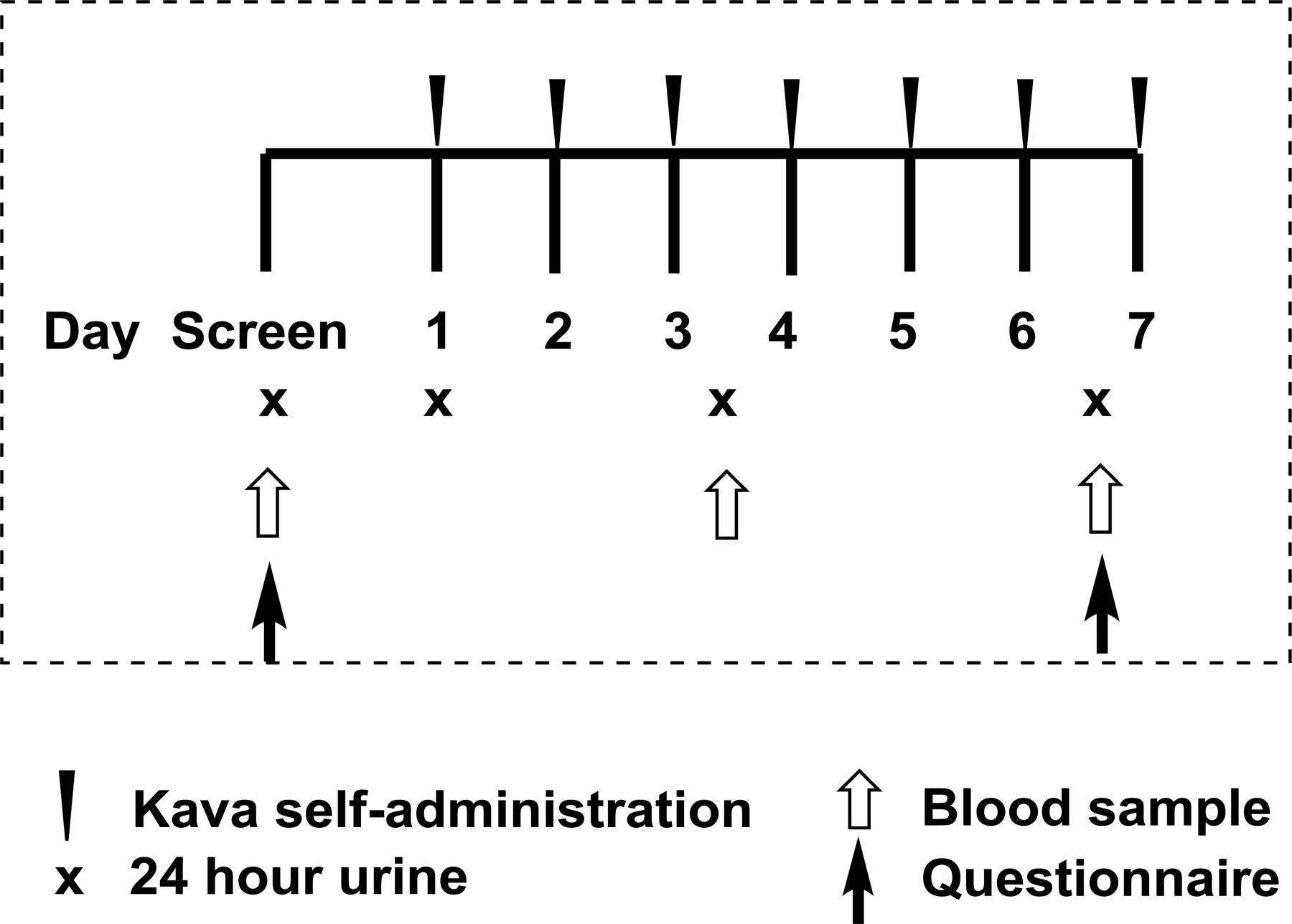
Trial design of pre- and post-one-week kava dietary supplementation among smokers.
The kava manufactured by Gaia Herb was chosen because it had the same preparation as the kava product used in our pre-clinical mouse studies (23,28). Based on the surface area normalization method, the dose of three capsules per day recommended by the manufacturer (standardized to 225 mg kavalactones per 3 capsules) is comparable to the dose utilized in the mouse studies. Kava capsules were stored at room temperature and dispensed by Fairview Drug Service Pharmacy, the institution’s research pharmacy. Participants were asked to take one capsule of kava three times daily at 8:00 ± 2 h, 14:00 ± 2 h and 20:00 ± 2 h for 7 consecutive days. Fasting except water was required at least 1 h before and 1 h after each dose. A dosing diary was maintained by the participant. Compliance was measured by pill counts at study visits, review of the dosing diary, and by measuring urinary DHM. Missed doses were not made up. Participants were asked to abstain from cruciferous vegetables (which contain phytochemicals that may alter NNK metabolism), alcohol, medications (with special attention paid to acetaminophen), supplements and herbs. Topical and inhaled medications were permitted. Adverse events (AE) were assessed at baseline, on day 4 ± 1 day, and on day 7 ± 1 day of kava intervention, and graded according to the National Cancer Institute’s Common Terminology Criteria for Adverse Events (CTCAE) v4.0.
Measurement of urinary total nicotine equivalents (TNE)
Urinary TNE, the sum of nicotine, cotinine, 3’-hydroxycotinine, their glucuronides and nicotine-N-oxide, was determined following a previously established method (32). Briefly, urine sample (2 μL) was diluted with 100 mM ammonium acetate pH 5 (998 μL). Diluted urine (10 μL), 100 mM ammonium acetate pH 5 (200 μL), and internal standards were treated with β-glucuronidase (300 units) at 37 °C for 15 hours. The SPE column was washed with 0.5% HCO2H (2 mL) and CH3OH (1 mL). The compounds were eluted with 2% NH4OH/CH3OH (1 mL), acidified with HCO2H (25 μL) and speed vacuum to dryness. Samples were re-suspended with 100 mM ammonium acetate in 85% CH3OH (50 μL), and 20 μL were injected for UPLC-MS/MS analysis.
Measurement of urinary 3-mA
Urinary 3-mA was quantified as previously described with slight modifications (15). Briefly, urine was diluted 10x with phosphate buffer (100 mM, pH 6.8). The diluted urine sample (20 μL) was mixed with [2H3]-3-mA (100 pg), 0.5% HCO2H (500 μL), and applied to a MCX cartridge, preconditioned with CH3OH (1 mL) and H2O (1 mL). The samples were washed with 0.5% HCO2H (2 mL), CH3OH (0.5 mL) and eluted with 1% ammonium hydroxide in CH3OH (1 mL). The eluents were dried under vacuum and dissolved in 10% CH3OH (30 μL) for UPLC-MS/MS analysis.
Measurement of urinary TCE and plasma cortisol
Urinary TCE was measured as described previously (33). Plasma cortisol was quantified following the same procedures with slight modifications. Briefly, human plasma (10 μL) was mixed with H2O (100 μL), CH2Cl2 (800 μL) and [2H4]-cortisol (2 pg/μL plasma). The CH2Cl2 fraction was dried under vacuum and the residue was dissolved in 10% CH3OH (1 mL), applied to a StrataX cartridge, preconditioned with CH3OH (1 mL) followed by H2O (2 mL). After washing with 10% CH3OH (1 mL) and H2O (4 mL), the analytes were eluted with CH3OH (1 mL), dried under vacuum and re-suspended in 10% CH3OH (50 μL) for UPLC-MS/MS analysis.
Measurement of urinary total NNAL
Urinary total NNAL was quantified following a published procedure (12). Briefly, urine samples (250 μL) with 2 pmols of 13C6-NNAL were incubated with 3000 units of β-glucuronidase enzyme (in 50 μL PBS) overnight at 37°. After incubation, the samples were subjected to primary and secondary extraction using Isolute SLE+ column (Biotage, Part # 820–0140-C) and MCX 1cc (30mg) Extraction Cartridge (Waters, Part # 186000252). The eluted samples were dried under speed-vac and dissolved in 10mM ammonium acetate (30 μL) and analyzed by UPLC-MS/MS.
UPLC-MS/MS analyses
All samples were assayed by targeted UPLC-MS/MS analysis on a Dionex Ultimate 3000 RS and a Q Exactive Hybrid Quadrupole Orbitrap Mass Spectrometer using Parallel Reaction Monitoring (PRM). For TNE analysis, samples were resolved through an Atlantis HILIC column (150 × 2.1 mm, 3 μm) with a 20 min linear gradient from 99% A solvent (CH3CN with 0.05% HCO2H) to 95% B solvents (H2O with 5% CH3CN and 0.05% HCO2H) at a flow rate of 250 μL/min. For 3-mA, the samples were resolved through a Thermo Scientific Acclaim C18 column (150 × 2.1 mm, 3 μm particle size, 100 Å) with a 25-min linear gradient from 99% A (2 mM ammonium acetate in 10% CH3CN) to 80% B (2 mM ammonium acetate in CH3CN with 10% H2O) at a flow rate of 150 μL/min. For cortisol and its metabolites, samples were resolved through an Atlantis dc18 column (150 × 2.1 mm, 3 μm particle size, 100 Å) with a 25-min linear gradient from 99% A (H2O with 1% CH3CN and 0.01% HCO2H) to 99% B (CH3CN with 5% H2O and 0.01% HCO2H) at a flow rate of 150 μL/min. For urinary NNAL, the separation and analysis followed previously published procedures (26).
Measurement of urinary creatinine
Urinary creatinine was measured using a modified Jaffe reaction method (34) by three lab members independently and the mean values were used. Briefly, 1% picric acid (50 μL), 0.75 N potassium hydroxide (50 μL), and diluted urine sample (75 μL) were incubated for 5 min at room temperature and then transferred to a 96-well plate for absorbance measurement. Absorbance was recorded at 520 nm using spectrophotometer.
Method validation
Calibration curves, limit of detection (LOD) and limit of quantification (LOQ) of kavalactones, 3-mA, cortisol and cortisol metabolites have been established as previously described (15,30,33). Performance of these methods were validated by specificity, precision, accuracy, within-day and between-day reproducibility using the pure standards added into the blank matrix at three to four different concentrations on three different days. The methods for urinary TNE and total NNAL quantification were well-established (15,35).
Data analyses
Demographic information was summarized using frequency and percentage. The mCEQ was summarized using mean and standard deviation before and after kava intervention and compared using a two-tailed paired t-test for means. The levels of urinary TNE, NNAL, TCE and 3-mA were normalized by creatinine. The level of total NNAL was also normalized by urinary TNE. The primary endpoint, total urinary NNAL, and other biomarkers of interest were summarized using geometric mean, minimum and maximum due to the skewedness of the data. To investigate the change in these biomarkers before and after kava, a two-sided paired t-test was performed on the log-transformed values. A ratio of geometric means and 95% CI were presented. Measures of liver function were summarized using mean and standard deviation at screening, four days after kava and seven days after kava. To investigate if liver function changed over time, a mixed-effects ANOVA was performed. A significance level of 0.1 was used because of the pilot nature of this clinical trial. Statistical analyses of biomarkers were performed using R (version 3.6.0), GraphPad Prism (version 7.00) and for mCEQ, Excel (version 2016).
Results
Kava compliance, exposure and safety
The characteristics of the 21 eligible participants who completed the study are summarized in Table 1. Compliance was excellent; one subject missed the 8pm dose on Day 2, another subject missed the 8pm dose on Day 6 and one dose on Day 7, and two additional subjects missed one dose each on Day 5, bringing the compliance rate to 92%. DHM, a kava-specific lactone, was not detected in any of the baseline urine samples before kava and was detected in all urine samples after kava initiation (Fig. S2). Overall, kava demonstrated excellent safety. Kava had no impact on liver function (Fig. S3, data for each participant in the Supporting Information, Table S1 – S4). Kava did not affect other parameters in CMPs and CBCs as well. Seven of the 21 participants experienced an AE (Table S5) that was at least possibly attributable to the kava. All were grade 1 and none of these AEs observed occurred in the same participant. These results support 1) the safety and tolerability of short-term kava intervention; 2) the potential use of urinary DHM as a measure of kava exposure; and 3) feasibility of high compliance.
Table 1.
Information of enrolled participants (N = 21).
| Sex | Male | 14 |
| Female | 7 | |
| Age (24–71 years) | 18–30 | 6 |
| 31–40 | 4 | |
| 41–50 | 3 | |
| 51–60 | 4 | |
| >60 | 4 | |
| Race | Caucasian American | 13 |
| African American | 7 | |
| Native American | 1 |
Effect of kava on urinary TNE, mCEQ, plasma cortisol, and urinary TCE
In order to quantitatively and objectively evaluate the impact of kava on the biomarkers of NNK-mediated carcinogenesis risk, the potential effect of kava on tobacco use needs to be characterized. We therefore quantified urinary TNE before and on day 7 of kava ingestion. Nineteen of 21 participants had reductions in 0–6h urinary TNE (Fig. 3A; ratio of geometric means 0.68; 95% CI: 0.57, 0.80; p<0.001; Table S6). The reduction on nicotine, cotinine, 3-hydroxycotine and nicotine-N-oxide (the four components for TNE) by kava was similar (Supporting Information, Fig. S4), suggesting that the reduction of tobacco use by kava is not likely mediated via inhibiting nicotine metabolism. Similar levels of TNE reductions were observed in the 6–24h urine samples (Supporting Information, Fig. S5). There were, however, no changes in self-reported cigarettes per day (CPD) among the participants (Table 2), which is intriguing. The effect of kava on tobacco dependence was assessed using the mCEQ (31). As shown in Table 2, kava significantly reduced mCEQ scores for smoking satisfaction and enjoyment of respiratory tract sensation although the extent of reductions was small. Kava use did not have significant effects on psychological rewards, craving reduction or aversion. It should be noted that the participants enrolled in this pilot trial did not intend to quit smoking while mCEQ was designed for participants active in tobacco cessation.
Figure 3.
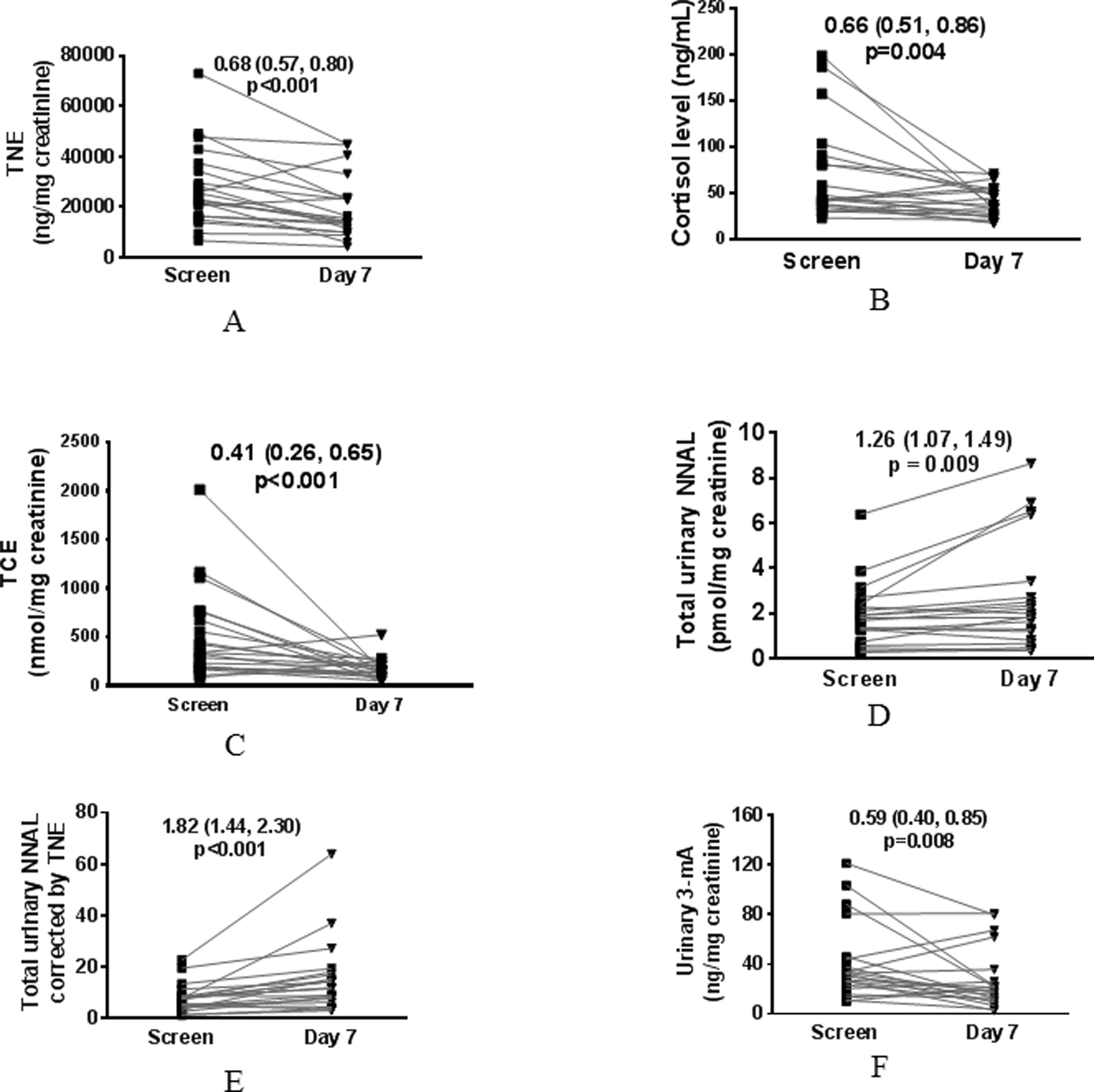
Impact of one-week kava on urinary TNE (A), plasma cortisol (B), urinary TCE (C), urinary total NNAL (D), urinary TNE-corrected total NNAL (E), and urinary 3-mA (F). Two-tailed paired t-test was performed on the log-transformed data for each measure. A ratio of geometric means and 95% CI are presented.
Table 2.
The impact of dietary kava on smoking behaviors based on mCEQ and CPD.
| Factor | Smoking Satisfaction | Psychological Reward | Enjoy Sensation | Craving Reduction | Aversion | CPD |
|---|---|---|---|---|---|---|
| Baseline Mean (SD) | 4.98 (1.42) | 4.10 (1.22) | 4.24 (1.73) | 4.95 (1.66) | 1.52 (0.83) | 16.3 (10.5) |
| 7 Day Mean (SD) | 4.51 (1.35) | 3.77 (1.19) | 3.71 (1.55) | 4.95 (1.36) | 1.40 (0.66) | 16.2 (10.9) |
| Paired t-test p-value (two-tailed) | 0.031 | 0.222 | 0.053 | 1.00 | 0.489 | 0.87 |
To explore the potential mechanisms of kava on tobacco use reduction, we evaluated the impact of kava on plasma cortisol given kava’s relaxing property. Most blood samples were collected between 11 am – 2 pm, reducing circadian influence on plasma cortisol levels. As shown in Fig. 3B, 15 of 21 participants had reduction in plasma cortisol after kava use (ratio of geometric means 0.66; 95% CI: 0.51, 0.86; p=0.004). We also quantified the sum of the major metabolites of cortisol in the urine samples (termed total cortisol equivalent, TCE) following our recently reported methods (33). Kava use resulted in a 59.0% reduction in urinary TCE in the 0–6 h samples (Fig. 3C; ratio of geometric means 0.41, 95% CI: 0.26, 0.65; p<0.001); eighteen of the 21 participants showed reductions). Several studies report that smokers appear to have higher levels of cortisol in comparison to non-smokers (33,36). The elevated cortisol level among smokers, potentially reflecting the levels of mental stress, may contribute to tobacco addiction.
Effect of kava on urinary NNAL and 3-mA
Kava exposure significantly increased the amount of urinary NNAL (Fig. 3D; ratio of geometric means 1.26; 95% CI: 1.07, 1.49; p = 0.009; Table S7); an increase was seen in 15 of the 21 participants. Given the reduction in urinary TNE upon kava use, an indication of potential decrease of tobacco use, urinary NNAL was also analyzed upon urinary TNE correction. The amount of urinary TNE-corrected NNAL nearly doubled (Fig. 3E; ratio of geometric means 1.82; 95% CI: 1.44, 2.30; p<0.001). It should be noted, however, that nicotine and NNK/NNAL have different pharmacokinetics in humans. The differences in their half-lives may also contribute to the changes in TNE-corrected total NNAL. Consistent with the increase in urinary total NNAL, kava use resulted in a significant reduction in urinary 3-mA (Fig. 3F; ratio of geometric means 0.59; 95% CI: 0.40, 0.85; p=0.008); a reduction was seen in 15 of the 21 participants.
Discussion
Our clinical trial is the first to demonstrate an increase in urinary total NNAL among smokers upon kava use, suggesting enhanced NNK/NNAL clearance. This is consistent with kava’s effect in enhancing NNAL detoxification in the NNK-induced lung tumorigenesis A/J mouse model (26). The reduction in urinary 3-mA also supports kava’s potential to reduce NNK-mediated carcinogenesis risk among smokers, although 3-mA may be formed from other potential sources such as unknown methylating agents in tobacco smoke. We unexpectedly observed that kava might be useful to reduce tobacco use. Although only mCEQ was implemented in this trial, it has been validated to assess the degree to which subjects experience the reinforcing effects of smoking (31) and one week kava intervention resulted in significant reductions in mCEQ smoking satisfaction and enjoyment of respiratory track sensation. The reduction in urinary TNE, an objective and quantitative surrogate of tobacco exposure, provides additional evidence that kava might lead to reduced tobacco use. This is particularly promising since the participants were asked not to change their smoking habits during the study intervention. At the same time, there were no changes in self-reported CPD. It is possible that kava use reduced the depth of smoking inhalation among smokers, which may account for this discrepancy. This potential benefit remains to be rigorously evaluated in a placebo-controlled, blinded, randomized trial. Mechanistically, kava use resulted in reduction in nicotine, cotinine, 3-hydroxycotinine and nicotine-N-oxide, suggesting that the effect is unlikely mediated via inhibiting nicotine metabolism. Tobacco use reduction may be mediated by kava’s anti-stress activity, reflected by the significant reductions in plasma cortisol and urinary TCE, respectively. As expected, the cortisol levels (plasma and urine) at baseline were heterogeneous among the smokers, suggesting that they may have different levels of physiologic stress. Interestingly, cortisol reduction was more pronounced in smokers with higher baseline cortisol levels. Furthermore, their cortisol levels were reduced to levels comparable to those in smokers with lower baseline cortisol levels. These dual benefits are consistent with kava’s relaxing properties (17,18), the epidemiological inverse relationship between cancer incidence and kava use (21,22), and the extensive animal data supporting kava’s anxiolytic activity (37) and cancer chemopreventive properties (23–25).
Our trial also demonstrated the feasibility of achieving high compliance of short-term kava use among smokers with an absence of significant safety concerns. These are consistent with the long-standing history of traditional use of kava. Nonetheless, the clinical use of kava as an anxiolytic agent in other international venues was suspended between 2002 – 2014 due to concerns about potential hepatotoxicity (38). Subsequent safety evaluations by the World Health Organization (WHO) and others concluded that the hepatotoxic risk with kava was very low, particularly with proper preparations (17,39–41). Restrictions on kava use in Europe have since been lifted (38). We and others found that flavokavains A and B (Fig. S1), two lipophilic chemicals with higher abundance in low-quality kava cultivars (42), may account for kava’s hepatotoxic risk (43,44). Future rigorous clinical evaluation of kava preparations with well-characterized chemical composition and potentially low (or no) levels of flavokavains A and B, which better mimics its traditional preparation, is necessary.
Our study has several limitations. First, the trial did not have a placebo control group. Second, our sample size was large enough to meet our primary endpoint, but too small to make definitive conclusions. However, this pilot study is a mandatory step to further evaluation of the promising effects we observed in a larger randomized trial. Third, the kava intervention was short. Since tobacco cessation programs typically take weeks to months to achieve complete abstinence, longer trials with longitudinal follow-ups are needed to characterize kava’s potential for sustainable reduction and ideally, complete cessation of tobacco use. Such trials will also provide safety data on long-term kava use. Additionally, regardless of how beneficial an agent may be, thrice daily dosing of any chemopreventive agent or intervention is not realistic. While compliance was high in our study, real-world compliance will be an issue; hence, a simpler dosing schema needs to be developed. Lastly, the mechanisms behinds kava’s potential dual benefits are not fully elucidated. Although kava has been well documented to be anxiolytic, there is no consensus on its active ingredients or its mechanism of action (45–50). Similarly, the mechanism by which kava enhances detoxification of NNK, resulting in higher levels of urinary total NNAL, is not fully elucidated and the upstream target remains to be determined. Such knowledge is critical to support the development of a kava product(s) with an optimal composition. Such knowledge also has the potential to help identify which smokers are more likely to benefit from kava use. It is possible that the anxiolytic compound(s) and the NNK detoxifying compound(s) are different. Kava, comprised of a natural blend of kavalactones, may be better than any of its individual components.
Overall our pilot study supports the hypotheses that kava may lead to increased NNK detoxification, mitigating some of the deleterious effects of tobacco smoking, and reduced satisfaction from smoking. The data generated by this pilot trial support pursuit of further pre-clinical and clinical studies to confirm kava’s benefits, identify the active ingredients, and elucidate its mechanism of action, thus informing the optimal use of kava that will benefit populations at high risk of tobacco-related cancer.
Supplementary Material
Acknowledgement:
We would like to thank Jordan Paladino and Vickie Nguyen for helping sample preparation.
Funding sources
The research reported in this publication was supported by NIH R01CA193278 (CX), University of Minnesota Masonic Cancer pilot program (NF and CX), College of Pharmacy Frank Duckworth Endowment (CX), University of Florida Health Cancer Center Startup fund (CX), P30 CA77598 utilizing the Biostatistics and Bioinformatics Core shared resource of the Masonic Cancer Center, University of Minnesota, the National Center for Advancing Translational Sciences of the National Institutes of Health Award Number UL1TR002494. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Abbreviations
- NNK
4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone
- NNAL
4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol
- TNE
total nicotine equivalents
- TCE
total cortisol equivalents
- PLCO
Prostate, Lung, Colorectal, and Ovarian Cancer Screen Trial
- Gluc
glucuronide
- 3-mA
3-methyladenine
- DHM
dihydromethysticin
- UPLC-MS/MS
ultraperformance liquid chromatography tandem mass spectrometry
- CMP
comprehensive metabolic panel
- CBC
complete blood counts
- ALP
alkaline phosphatase
- ALT
alanine aminotransferase
- AST
asparate aminotransferase
- mCEQ
modified Cigarette Evaluation Questionnaire
- LOD
limit of detection
- LOQ
limit of quantification
- AE
adverse event
- CI
confidence interval
- WHO
World Health Organization
- CPD
cigarettes per day
Footnotes
Disclosure of Potential Conflicts of Interest No potential conflicts of interest were disclosed by the authors.
References:
- 1.Siegel RL, Miller KD, Jemal A Cancer statistics, 2019. CA Cancer J Clin 2019; 69: 7–34. [DOI] [PubMed] [Google Scholar]
- 2.Robles GI, Singh-Franco D, Ghin HL A review of the efficacy of smoking-cessation pharmacotherapies in nonwhite populations. Clin Ther 2008; 30: 800–12. [DOI] [PubMed] [Google Scholar]
- 3.Hecht SS Biochemistry, biology, and carcinogenicity of tobacco-specific N-nitrosamines. Chem Res Toxicol 1998; 11: 559–603. [DOI] [PubMed] [Google Scholar]
- 4.Burns DM, Anderson CM, Gray N Do changes in cigarette design influence the rise in adenocarcinoma of the lung? Cancer Causes Control 2011; 22: 13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hoffmann D, Rivenson A, Hecht SS The biological significance of tobacco-specific N-nitrosamines: smoking and adenocarcinoma of the lung. Crit Rev Toxicol 1996; 26: 199–211. [DOI] [PubMed] [Google Scholar]
- 6.Thun MJ, Lopez AD, Hartge P Smoking-related mortality in the United States. N Engl J Med 2013; 368: 1753–1754. [DOI] [PubMed] [Google Scholar]
- 7.Church TR, Anderson KE, Caporaso NE, Geisser MS, Le CT, Zhang Y, Benoit AR, Carmella SG, Hecht SS A prospectively measured serum biomarker for a tobacco-specific carcinogen and lung cancer in smokers. Cancer Epidemiol Biomarkers Prev 2009; 18: 260–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Murphy SE, Park SL, Balbo S, Haiman CA, Hatsukami DK, Patel Y, Peterson LA, Stepanov I, Stram DO, Tretyakova N, Hecht SS, Le Marchand L Tobacco biomarkers and genetic/epigenetic analysis to investigate ethnic/racial differences in lung cancer risk among smokers. NPJ Precis Oncol 2018; 2: 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yuan JM, Koh WP, Murphy SE, Fan Y, Wang R, Carmella SG, Han S, Wickham K, Gao YT, Yu MC, Hecht SS Urinary levels of tobacco-specific nitrosamine metabolites in relation to lung cancer development in two prospective cohorts of cigarette smokers. Cancer Res 2009; 69: 2990–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yuan JM, Gao YT, Murphy SE, Carmella SG, Wang R, Zhong Y, Moy KA, Davis AB, Tao L, Chen M, Han S, Nelson HH, Yu MC, Hecht SS Urinary levels of cigarette smoke constituent metabolites are prospectively associated with lung cancer development in smokers. Cancer Res 2011; 71: 6749–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carmella SG, Akerkar S, Hecht SS Metabolites of the tobacco-specific nitrosamine 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone in smokers’ urine. Cancer Res 1993; 53: 721–4. [PubMed] [Google Scholar]
- 12.Carmella SG, Le Ka KA, Upadhyaya P, Hecht SS Analysis of N- and O-glucuronides of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol (NNAL) in human urine. Chem Res Toxicol 2002; 15: 545–50. [DOI] [PubMed] [Google Scholar]
- 13.Hecht SS, Chung FL, Richie JP Jr., Akerkar SA, Borukhova A, Skowronski L, Carmella SG Effects of watercress consumption on metabolism of a tobacco-specific lung carcinogen in smokers. Cancer Epidemiol Biomarkers Prev 1995; 4: 877–84. [PubMed] [Google Scholar]
- 14.Hu K, Zhao G, Liu J, Jia L, Xie F, Zhang S, Liu H, Liu M Simultaneous quantification of three alkylatedpurine adducts in human urine using sulfonic acid poly(glycidyl methacrylatedivinylbenzene)-based microspheres as sorbent combined with LC-MS/MS. J Chromatogr B Analyt Technol Biomed Life Sci 2018; 1081-1082: 15–24. [DOI] [PubMed] [Google Scholar]
- 15.Wang Y, Narayanapillai S, Hu Q, Fujioka N, Xing C Contribution of Tobacco Use and 4-(Methylnitrosamino)-1-(3-pyridyl)-1-butanone to Three Methyl DNA Adducts in Urine. Chem Res Toxicol 2018; 31: 836–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tian Y, Hou H, Zhang X, Wang A, Liu Y, Hu Q New validated LC-MS/MS method for the determination of three alkylated adenines in human urine and its application to the monitoring of alkylating agents in cigarette smoke. Anal Bioanal Chem 2014; 406: 5293–302. [DOI] [PubMed] [Google Scholar]
- 17.WHO Kava: a review of the safety of tradtional and recreational beverage consumption. Food and Agriculture Organization of the United Nation 2016; 1: 1–35. [Google Scholar]
- 18.Lasme P, Davrieux F, Montet D, Lebot V Quantification of kavalactones and determination of kava (Piper methysticum) chemotypes using near-infrared reflectance spectroscopy for quality control in vanuatu. J Agric Food Chem 2008; 56: 4976–81. [DOI] [PubMed] [Google Scholar]
- 19.Pearl PL, Drillings IM, Conry JA Herbs in epilepsy: evidence for efficacy, toxicity, and interactions. Semin Pediatr Neurol 2011; 18: 203–8. [DOI] [PubMed] [Google Scholar]
- 20.Weeks BS Formulations of dietary supplements and herbal extracts for relaxation and anxiolytic action: Relarian. Med Sci Monit 2009; 15: RA256–62. [PubMed] [Google Scholar]
- 21.Steiner GG The correlation between cancer incidence and kava consumption. Hawaii Med. J 2000; 59: 420–422. [PubMed] [Google Scholar]
- 22.Henderson BE, Kolonel LN, Dworsky R, Kerford D, Mori E, Singh K, Thevenot H Cancer incidence in the islands of the Pacific. Natl. Cancer Inst. Monogr 1985; 69: 73–81. [PubMed] [Google Scholar]
- 23.Leitzman P, Narayanapillai SC, Balbo S, Zhou B, Upadhyaya P, Shaik AA, O’Sullivan MG, Hecht SS, Lu J, Xing C Kava blocks 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone-induced lung tumorigenesis in association with reducing O6-methylguanine DNA adduct in A/J mice. Cancer Prev Res (Phila) 2014; 7: 86–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Triolet J, Shaik AA, Gallaher DD, O’Sullivan MG, Xing C Reduction in colon cancer risk by consumption of kava or kava fractions in carcinogen-treated rats. Nutr Cancer 2012; 64: 838–46. [DOI] [PubMed] [Google Scholar]
- 25.Tang SN, Zhang J, Jiang P, Datta P, Leitzman P, O’Sullivan MG, Jiang C, Xing C, Lu J Gene expression signatures associated with suppression of TRAMP prostate carcinogenesis by a kavalactone-rich Kava fraction. Mol Carcinog 2016; 55: 2291–2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Narayanapillai SC, von Weymarn LB, Carmella SG, Leitzman P, Paladino J, Upadhyaya P, Hecht SS, Murphy SE, Xing C Dietary Dihydromethysticin Increases Glucuronidation of 4-(Methylnitrosamino)-1-(3-Pyridyl)-1-Butanol in A/J Mice, Potentially Enhancing Its Detoxification. Drug Metab Dispos 2016; 44: 422–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Narayanapillai SC, Lin SH, Leitzman P, Upadhyaya P, Baglole CJ, Xing C Dihydromethysticin (DHM) Blocks Tobacco Carcinogen 4-(Methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK)-Induced O6-Methylguanine in a Manner Independent of the Aryl Hydrocarbon Receptor (AhR) Pathway in C57BL/6 Female Mice. Chem Res Toxicol 2016; 29: 1828–1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Narayanapillai SC, Balbo S, Leitzman P, Grill AE, Upadhyaya P, Shaik AA, Zhou B, O’Sullivan MG, Peterson LA, Lu J, Hecht SS, Xing C Dihydromethysticin from kava blocks tobacco carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone-induced lung tumorigenesis and differentially reduces DNA damage in A/J mice. Carcinogenesis 2014; 35: 2365–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reagan-Shaw S, Nihal M, Ahmad N Dose translation from animal to human studies revisited. FASEB J. 2008; 22: 659–661. [DOI] [PubMed] [Google Scholar]
- 30.Wang Y E. SO; Stacy HM; Narayanapillai SC; Sharma A; Fujioka N; Haddad L; McLaughlin J; Avery BA; Xing C A stable isotope dilution tandem mass spectrometry method of major kavalactones and its applications. PLoS One 2018; 13: e0197940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cappelleri JC, Bushmakin AG, Baker CL, Merikle E, Olufade AO, Gilbert DG Confirmatory factor analyses and reliability of the modified cigarette evaluation questionnaire. Addict Behav 2007; 32: 912–23. [DOI] [PubMed] [Google Scholar]
- 32.Murphy SE, Park SS, Thompson EF, Wilkens LR, Patel Y, Stram DO, Le Marchand L Nicotine N-glucuronidation relative to N-oxidation and C-oxidation and UGT2B10 genotype in five ethnic/racial groups. Carcinogenesis 2014; 35: 2526–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang Y, Fujioka N, Xing C Quantitative profiling of cortisol metabolites in human urine by high-resolution accurate-mass MS. Bioanalysis 2018; 10: 2015–2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Slot C Plasma creatinine determination. A new and specific Jaffe reaction method. Scand J Clin Lab Invest 1965; 17: 381–7. [DOI] [PubMed] [Google Scholar]
- 35.Wang Y, Narayanapillai SC, Hu Q, Fujioka N, Xing C Detection and quantification of 4-hydroxy-1-(3-pyridyl)-1-butanone (HPB) from smoker albumin and its potential as a surrogate biomarker of tobacco-specific nitrosamines exposure and bioactivation. Toxicol Lett 2019; 311: 11–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Badrick E, Kirschbaum C, Kumari M The relationship between smoking status and cortisol secretion. J Clin Endocrinol Metab 2007; 92: 819–24. [DOI] [PubMed] [Google Scholar]
- 37.Garrett KM, Basmadjian G, Khan IA, Schaneberg BT, Seale TW Extracts of kava (Piper methysticum) induce acute anxiolytic-like behavioral changes in mice. Psychopharmacology 2003; 170: 33–41. [DOI] [PubMed] [Google Scholar]
- 38.Kuchta K, Schmidt M, Nahrstedt A German Kava Ban Lifted by Court: The Alleged Hepatotoxicity of Kava (Piper methysticum) as a Case of Ill-Defined Herbal Drug Identity, Lacking Quality Control, and Misguided Regulatory Politics. Planta Med 2015; 81: 1647–53. [DOI] [PubMed] [Google Scholar]
- 39.Teschke R, Qiu SX, Lebot V Herbal hepatotoxicity by kava: update on pipermethystine, flavokavain B, and mould hepatotoxins as primarily assumed culprits. Dig Liver Dis 2011; 43: 676–81. [DOI] [PubMed] [Google Scholar]
- 40.Olsen LR, Grillo MP, Skonberg C Constituents in Kava Extracts Potentially Involved in Hepatotoxicity: A Review. Chemical Research in Toxicology 2011; 24: 992–1002. [DOI] [PubMed] [Google Scholar]
- 41.LiverTox. (2018) Drug Record Kava Kava. https://livertox.nlm.nih.gov/KavaKava.htm. NIH Website.
- 42.Lebot V, Do TK, Legendre L Detection of flavokavins (A, B, C) in cultivars of kava (Piper methysticum) using high performance thin layer chromatography (HPTLC). Food Chem 2014; 151: 554–60. [DOI] [PubMed] [Google Scholar]
- 43.Narayanapillai SC, Leitzman P, O’Sullivan MG, Xing C Flavokawains a and B in kava, not dihydromethysticin, potentiate acetaminophen-induced hepatotoxicity in C57BL/6 mice. Chem Res Toxicol 2014; 27: 1871–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhou P, Gross S, Liu JH, Yu BY, Feng LL, Nolta J, Sharma V, Piwnica-Worms D, Qiu SX Flavokawain B, the hepatotoxic constituent from kava root, induces GSH-sensitive oxidative stress through modulation of IKK/NF-kappaB and MAPK signaling pathways. FASEB J 2010; 24: 4722–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Boonen G, Haberlein H Influence of genuine kavapyrone enantiomers on the GABA-A binding site. Planta Med 1998; 64: 504–6. [DOI] [PubMed] [Google Scholar]
- 46.Dinh LD, Simmen U, Bueter KB, Bueter B, Lundstrom K, Schaffner W Interaction of various Piper methysticum cultivars with CNS receptors in vitro. Planta Medica 2001; 67: 306–311. [DOI] [PubMed] [Google Scholar]
- 47.Yuan CS, Dey L, Wang AB, Mehendale S, Xie JT, Aung HH, Ang-Lee MK Kavalactones and dihydrokavain modulate GABAergic activity in a rat gastric-brainstem preparation. Planta Medica 2002; 68: 1092–1096. [DOI] [PubMed] [Google Scholar]
- 48.Uebelhack R, Franke L, Schewe HL Inhibition of platelet MAO-B by kava pyrone-enriched extract from Piper methysticum Forster (kava-kava). Pharmacopsychiatry 1998; 31: 187–192. [DOI] [PubMed] [Google Scholar]
- 49.Magura EI, Kopanitsa MV, Gleitz J, Peters T, Krishtal OA Kava extract ingredients, (+)-methysticin and (+/−)-kavain inhibit voltage-operated Na+-channels in rat CA1 hippocampal neurons. Neuroscience 1997; 81: 345–351. [DOI] [PubMed] [Google Scholar]
- 50.Jamieson DD, Duffield PH The Antinociceptive Actions of Kava Components in Mice. Clinical and Experimental Pharmacology and Physiology 1990; 17: 495–508. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.