

Abstract
Background and purpose
Neurological manifestations in coronavirus disease (COVID)‐2019 may adversely affect clinical outcomes. Severe COVID‐19 and uremia are risk factors for neurological complications. However, the lack of insight into their pathogenesis, particularly with respect to the role of the cytokine release syndrome (CRS), is currently hampering effective therapeutic interventions. The aims of this study were to describe the neurological manifestations of patients with COVID‐19 and to gain pathophysiological insights with respect to CRS.
Methods
In this longitudinal study, we performed extensive clinical, laboratory and imaging phenotyping in five patients admitted to our renal unit.
Results
Neurological presentation included confusion, tremor, cerebellar ataxia, behavioral alterations, aphasia, pyramidal syndrome, coma, cranial nerve palsy, dysautonomia, and central hypothyroidism. Notably, neurological disturbances were accompanied by laboratory evidence of CRS. Severe acute respiratory syndrome coronavirus‐2 (SARS‐CoV‐2) was undetectable in the cerebrospinal fluid (CSF). Hyperalbuminorrachia and increased levels of the astroglial protein S100B were suggestive of blood−brain barrier (BBB) dysfunction. Brain magnetic resonance imaging findings comprised evidence of acute leukoencephalitis (n = 3, one of whom had a hemorrhagic form), cytotoxic edema mimicking ischaemic stroke (n = 1), or normal results (n = 2). Treatment with corticosteroids and/or intravenous immunoglobulins was attempted, resulting in rapid recovery from neurological disturbances in two cases. SARS‐CoV2 was undetectable in 88 of the 90 patients with COVID‐19 who underwent Reverse Transcription‐PCR testing of CSF.
Conclusions
Patients with COVID‐19 can develop neurological manifestations that share clinical, laboratory and imaging similarities with those of chimeric antigen receptor T‐cell‐related encephalopathy. The pathophysiological underpinnings appear to involve CRS, endothelial activation, BBB dysfunction, and immune‐mediated mechanisms.
Keywords: corticosteroids, COVID‐19, cytokine, encephalitis, intravenous immunoglobulins, kidney, neurological disorders
Introduction
Coronavirus disease 2019 (COVID‐19) caused by severe acute respiratory syndrome coronavirus‐2 (SARS‐CoV‐2) can be complicated by neurological manifestations that have an adverse impact on morbidity and mortality [1, 2, 3]. A study from China reported preliminary evidence that neurological manifestations may be present in up to 36% of hospitalized cases [1]. Severe neurological manifestations occurred in 10% of patients during their hospital stay, with severe disease and uremia acting as potential risk factors [1]. Unfortunately, the clinical detection of neurological symptoms in patients with COVID‐19 remains challenging because of the severity of concomitant respiratory and systemic manifestations. Moreover, the lack of insight into the presentation and pathogenesis of neurological complications is currently hampering effective therapeutic interventions [3].
Similar to other coronaviruses, SARS‐CoV‐2 theoretically has the potential to penetrate the central nervous system (CNS) through hematogenous or retrograde neuronal routes [4]. However, the question as to whether SARS‐CoV‐2 may cause neurological manifestations through a direct neuropathic effect or by promoting a hyperinflammatory reaction mounted by the host's immune system in the form cytokine release syndrome (CRS) remains to be established [5]. It is also noteworthy that high serum levels of interleukin (IL)‐6 have been observed to be a strong predictor of mortality in patients with COVID‐19 [6].
In this longitudinal study conducted in a renal unit, we describe five patients with severe COVID‐19 who presented severe neurological manifestations. Extensive clinical, laboratory and imaging phenotyping was performed to gain pathophysiological insights that may guide clinical decision‐making, especially with respect to CRS.
Patients and methods
Patients
Between 9 March and 9 April 2020, a total of 2284 patients were hospitalized with COVID‐19 at the Strasbourg University Hospital (Strasbourg, France). Of these, 328 were directly admitted to the intensive care unit and 58 to our renal unit. The distribution of the underlying disorders in our 58 patients was as follows: previous kidney transplantation (n = 37); end‐stage renal disease (n = 16); acute kidney injury (n = 4); and hyponatremia (n = 1). Seven cases (12.7%) developed severe neurological manifestations. In the context of clinical care, five of the patients underwent extensive laboratory and imaging characterization. The remaining two (presenting with coma and seizures) were excluded because of the lack of extensive laboratory and magnetic resonance imaging (MRI) data. SARS‐CoV‐2 infection was confirmed in all cases by Reverse Transcription (RT)‐PCR assays targeting the RNA‐dependent RNA polymerase (RdRp) viral gene from nasopharyngeal swab specimens. According to current French law, ethical approval for retrospective studies conducted in the context of clinical care can be waived.
Clinical evaluation
All five patients underwent extensive clinical evaluation, aimed at excluding other causes of neurological impairment, such as alcohol or drug intoxication, metabolic disorders, hypoxemia, thiamine or vitamin B12 deficiencies, epilepsy, hypothermia, sepsis, antibiotic overdose, hypercapnic encephalopathy, hepatic encephalopathy, and CNS infections. All patients underwent lumbar puncture to screen for the presence of SARS‐CoV‐2 in the cerebrospinal fluid (CSF). If serum laboratory findings showed signs of CRS, patients received dexamethasone (20 mg/day for 5 days in all cases, followed by individualized dosing).[7] Antibiotic and antithrombotic prophylaxis was offered to all participants.
Serum laboratory markers
As of the beginning of the COVID‐19 pandemic in France, longitudinal assessments of numerous laboratory markers were conducted as an integral part of our clinical practice in an effort to identify the occurrence of CRS, which was defined as a peak in levels of proinflammatory molecules (IL‐6, C‐reactive protein, ferritin) and indices of cytolysis (e.g. lactate dehydrogenase). Markers of inflammation, CRS, cell lysis, coagulation and thrombotic microangiopathy were measured. Levels of serum thyroid hormones were also assessed. Serum levels of S100B protein, an established astroglial marker[8], were quantified on a Roche e411 analyzer (F. Hoffmann‐La Roche AG, Basel, Switzerland), whereas IL‐6 was measured with a chemiluminescent immunoassay (Lumipulse G600 II; Fujirebio, Tokyo, Japan) [9].
Laboratory analysis of cerebrospinal fluid
To investigate whether SARS‐CoV‐2 was detectable in the CSF, Reverse Transcription (RT)‐PCR assays were conducted according to current guidelines (Institut Pasteur, Paris, France; World Health Organization technical guidance). The threshold limit of detection was 10 copies per reaction. Biochemical and immunological analyses of CSF were also performed.
Brain MRI
All patients underwent brain MRI on a 3‐Tesla MRI scanner. The following protocols were applied: three‐dimensional T1‐weighted spin‐echo, three‐dimensional fluid‐attenuated inversion recovery (FLAIR; both with and without gadolinium), diffusion‐weighted imaging, susceptibility‐weighted imaging, and two‐dimensional FLAIR post‐contrast imaging.
Results
General characteristics, clinical course and MRI findings
Five patients with severe COVID‐19 were included in the study: a 71‐year‐old woman (case 1); a 64‐year‐old man (case 2); a 53‐year‐old woman (case 3); a 51‐year‐old man (case 4); and a 67‐year‐old man (case 5). No patient had a known history of neurological disease or previous neurological manifestations. Cases 3 and 4 had severe COVID‐19‐related acute kidney injury. Table 1 summarizes the general characteristics, clinical features, neurological disturbances, MRI findings, EEG results, treatment approaches, and outcomes of the five study patients. Neurological presentation included confusion (n = 5), tremor (n = 5), cerebellar ataxia (n = 5), behavioral alterations (n = 5), aphasia (n = 4), pyramidal syndrome (n = 4), coma (n = 2), cranial nerve palsy (n = 1), and central hypothyroidism (n = 3). Case 1 died from coma and secondary infection.
Table 1.
General characteristics, clinical features, neurological manifestations, MRI findings, EEG results, treatment approaches, and outcomes of the five patients studied
| Characteristic | Patient 1 | Patient 2 | Patient 3 | Patient 4 | Patient 5 |
|---|---|---|---|---|---|
| Age, years/sex | 71/F | 64/M | 53/F | 51/M | 67/M |
| Medical history | |||||
| Hypertension | Yes | Yes | Yes | No | Yes |
| Mellitus diabetes | No | Yes | Yes | No | No |
| Smoking | No | Yes | No | No | Yes (stopped) |
| Dyslipidemia | Yes | Yes | No | No | No |
| Sleep apnea | Yes | Yes | No | No | No |
| BMI (kg/m2) | 31 | 29 | 30 | 31 | 20 |
| Renal status |
ESRD Polycystic kidney disease Peritoneal dialysis |
ESRD Diabetic nephropathy Peritoneal dialysis |
AKI stage 3, hemodialysis with kidney recovery | AKI stage 3 –kidney recovery |
KTR (C3 glomerulopathy) GFR = 33 ml/min/1.73 m2 |
| Days from symptom onset at hospitalization | 7 | 8 | 7 | 7 | 6 |
| COVID‐19 symptoms at hospitalization | Fever, dyspnea, cough, myalgia | Fever, dyspnea, cough, diarrhea, myalgia | Fever, dyspnea | Fever, dyspnea, anorexia, hypotension | Fever, dyspnea, cough, myalgia |
| Neurologic signs at hospitalization | Confusion | Headache, confusion, minor aphasia, tremor | Headache | None | Headache, anosmia, dysgueusia |
| Severity of respiratory involvement | Severe | Severe | Critical | Critical | Severe |
| Neurological features |
Confusion, agitation, tremor, pyramidal syndrome, coma, dysautonomia, decerebration, Death |
Confusion, agitation, tremor, cerebellar ataxia, aphasia, apraxia, pyramidal syndrome, coma, dysautonomia | Confusion, agitation, tremor, cerebellar ataxia, mild aphasia, behavioral alterations, cognitive disturbances | Confusion, agitation, tremor, cerebellar ataxia, pyramidal syndrome, behavioral alterations, cognitive disturbances | Drop in visual acuity, VI cranial nerve palsy, cerebellar ataxia, behavioral alterations, pyramidal syndrome |
| Central hormonal dysfunction |
Central hypothyroidism Low levels of FSH, LH, ACTH |
Central hypothyroidism | No | No | Central hypothyroidism |
| MRI features |
Acute leukoencephalitis. Symmetric FLAIR and DWI white matter hyperintensities predominantly in subcortical white matter |
Acute leukoencephalitis and cytotoxic edema. FLAIR and DWI white matter hyperintensities in middle cerebellar peduncles, an acute mm‐scale cytotoxic edema on the posterior left frontal lobe, that persisted 16 days later excluding ischaemic stroke |
Normal |
Acute hemorrhagic leukoencephalitis. FLAIR hyperintensities and micro‐hemorrhagic lesions in the splenium of the corpus callosum |
Normal |
| EEG features |
EEG1: diffuse slow wave spikes. EEG2: asymmetric slow wave spikes and right occipital focus without seizure |
EEG1: global and diffuse signal slowdown EEG2: slow bilateral delta elements organized in bursts or predominant opposite bifrontal diversions with bilateral 5−6 Hz theta band elements. |
Normal | N/A | N/A |
| Antiviral treatment at hospitalization | No | Lopinavir‐ritonavir | Hydroxychloroquine | Hydroxychloroquine | Hydroxychloroquine |
| Antiepileptic treatment | Levetiracetam | Oxazepam | No | No | No |
| CS, IVIg | CS | CS, IVIg | No | No | CS |
| Neurological outcome | Temporary improvement after CS, relapse, coma and death | Improvement after CS, relapse, rapid improvement with IVIg | Spontaneous and gradual improvement | Spontaneous and gradual improvement | Rapid improvement with CS |
ACTH, Adrenocortical Hormone; AKI, acute kidney injury; BMI, body mass index; CS, corticosteroids; DWI: diffusion‐weighted imaging; EEG, electroencephalogram; ESRD, end‐stage renal disease; F, female; FLAIR, fluid‐attenuated inversion recovery; FSH, follicle stimulating hormone GFR, glomerular filtration rate; IVIg, intravenous immunoglobulins; KTR, kidney transplantation recipient; LH, luteinizing hormone; M, male; N/A, not available. AKI was staged according to the kidney disease improving global outcome (KIDGO) criteria.
A timeline of the neurological and respiratory course according to symptom onset is shown in Fig. 1a–e. Neurological disturbances occurred in the second week after COVID‐19 onset (24−48 h after respiratory degradation) in cases 1, 2 and 5. In cases 2 and 3, the exact date of onset of neurological signs was not assessable because of the patients' critical conditions requiring mechanical ventilation; therefore, their assessment was conducted after extubation. This issue highlights the paramount importance of biomarkers for early identification of neurological disturbances. Neurological disorders may also play a role in the onset of agitation and may result in a prolonged ventilation time. Brain MRI findings were heterogeneous and included evidence of acute leukoencephalitis (cases 1, 2 and 4; the latter with a hemorrhagic form), cytotoxic edema mimicking an ischaemic stroke (case 2), and normal results (cases 3 and 5; Fig. 2).
Figure 1.

Panels a−e. Temporal course of clinical and laboratory variables in the five patients studied. Diagnostic investigations and administered drugs are also reported. All longitudinal data are shown with respect to the date of COVID‐19 symptom onset (D0). Red stars denote the days on which levels of cytokine release syndrome‐related inflammatory biomarkers reached a peak. % pm infiltration, percentage of lung infiltration on chest scan; CSF, cerebrospinal fluid; DM, dexamethasone; EEG, electroencephalogram; GCS, Glasgow coma scale; LBP, low blood pressure; MP, methylprednisolone; oLBP, orthostatic hypotension; sd, syndrome; Tmax, maximum body temperature; N/A, not available, VA, visual acuity. [Colour figure can be viewed at wileyonlinelibrary.com]
Figure 2.
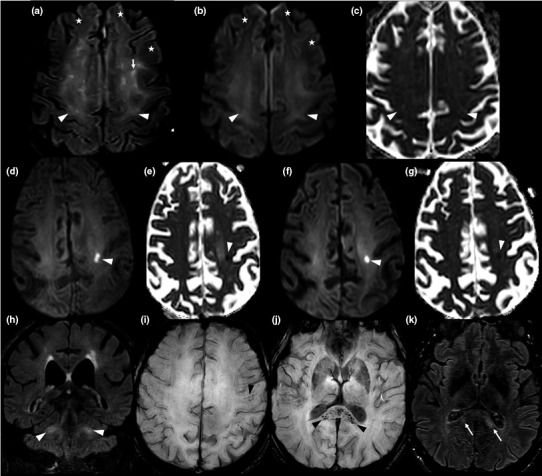
Brain magnetic resonance imaging (MRI) findings in the five patients studied. Case 1: Axial fluid‐attenuated inversion recovery (FLAIR)‐weighted MRI (a), axial diffusion‐weighted MRI (b) and axial apparent diffusion coefficient (ADC) map (c). MRI images obtained on day 21; cross‐sections through motor areas, frontal and parietal lobes. Diffuse bilateral, symmetric white matter FLAIR hyperintensities with mild hyperintense 'ground glass' areas (arrow heads) and frank hyperintense areas (conventional radiological FLAIR hyperintensities; arrows). All abnormal FLAIR areas appeared hyperintense in the diffusion sequence and were characterized by a gradient: frank hyperintense FLAIR lesions had a higher intensity than 'ground glass' areas. On the ADC map, 'ground glass' areas were iso‐ or hypointense, whereas frank hyperintense FLAIR areas were hyperintense. Abnormal FLAIR hyperintensities were preferentially localized to subcortical white matter of motor areas and showed a bilateral symmetric distribution. The anterior frontal white matter appeared normal on FLAIR and diffusion‐weighted imaging (DWI) sequences (a, b: stars). Case 2: Axial DWI (d) and axial ADC map (e): cross‐sections through frontal and parietal lobes on day 17. Axial DWI (f) and axial ADC map (g): cross‐sections through frontal and parietal lobes on D33. Coronal FLAIR weighted MRI on day 33 (h): cross‐section through middle cerebellar peduncles. The first MRI (on D17) revealed an acute hyperintense DWI lesion, with hypointensity on ADC map, suggestive of a cytotoxic edema in the left frontal lobe (d, e) and deemed initially compatible with an acute stroke. However, this lesion maintained a similar aspect following 16 days (f, g), casting doubts on its ischaemic origin. Persistence of middle cerebellar peduncles hyperintensities was also evident (h). Case 4: Axial susceptibility‐weighted imaging MRI at day 31: cross‐section through motor areas, frontal and parietal lobes (i) and the splenium (j). Axial FLAIR weighted MRI (k): cross‐section through the splenium. Multifocal microbleeds were evident in the splenium (j: black arrow heads) and in the white matter/gray matter junction (i: black arrow head), with an apparent perivascular distribution in the Virchow‐Robin spaces. These lesions were associated with hyperintensities on FLAIR weighted sequence (k: arrows). Day 0 = date of COVID‐19 symptom onset.
Serum laboratory markers
The temporal course of CRS biomarkers and viral loads is presented in Fig. 3a–e. Neurological manifestations occurred simultaneously with the peak in CRS serum markers (i.e. C‐reactive protein, IL‐6 and lactate dehydrogenase) in cases 1, 2 and 5. The peak in CRS markers for cases 2 and 3 occurred while the patients were under mechanical ventilation, and serve as potential biomarkers. Serum levels of the astroglial marker, S100B protein, were increased at the time of CRS, reflecting an increased permeability of the blood−brain barrier (BBB), and returned to their reference range when neurological symptoms and signs of hyperinflammation regressed (Fig. 4). Detailed laboratory data are reported in Table S1. Circulating anticoagulant was detected in cases 1, 2 and 4 but not in case 5. Antiphospholipid antibodies were present in case 1 but undetectable in the remaining four cases. Antinuclear antibodies were assessed in cases 2 and 5 and found to be positive. Serum total complement as well as C3 and C4 were within the reference ranges.
Figure 3.
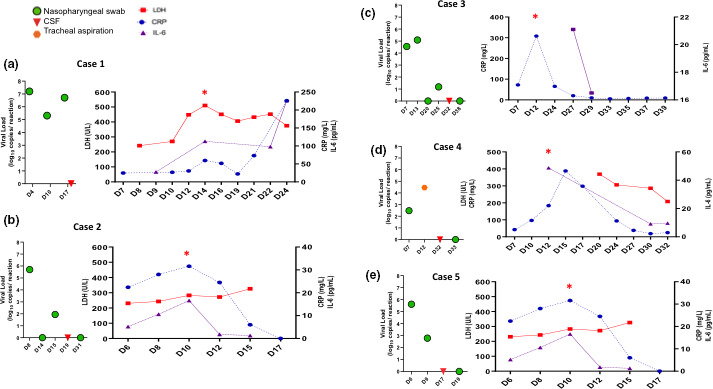
Temporal course of viral loads and biomarkers of cytokine release syndrome (CRS) in the five study patients. Red stars denote the days on which levels of CRS‐related inflammatory biomarkers reached a peak. Reference values: interleukin (IL)‐6, <4 ng/l; lactate dehydrogenase (LDH), 120−246 UI/l; C‐reactive protein (CRP) <4 mg/l. All longitudinal data are shown with respect to the date of COVID‐19 symptom onset (D0). [Colour figure can be viewed at wileyonlinelibrary.com]
Figure 4.
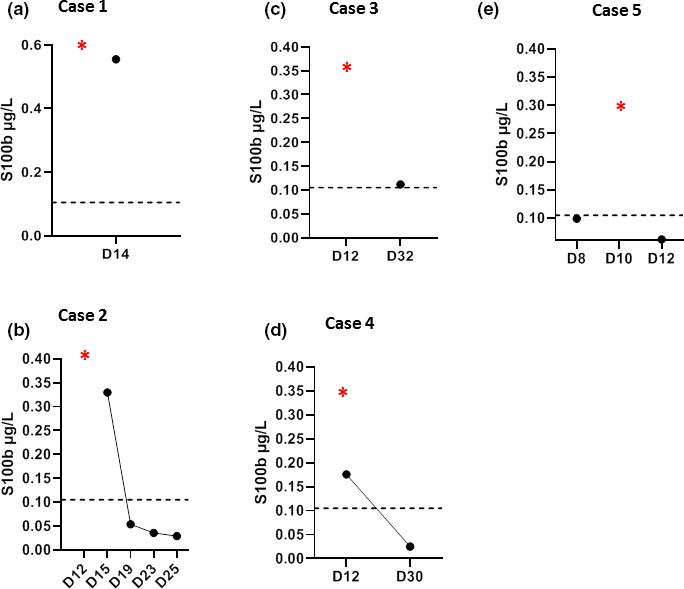
Temporal course of serum S100B (µg/l) levels in the five patients studied. Values above the dotted line indicate high values (>0.105 µg/l). Red stars denote the days on which levels of cytokine release syndrome‐related inflammatory biomarkers reached a peak. All longitudinal data are shown with respect to the date of COVID‐19 symptom onset (D0). [Colour figure can be viewed at wileyonlinelibrary.com]
CSF analysis
SARS‐CoV‐2 was undetectable in the CSF for all patients (Fig. 3). According to data from our Department of Virology, SARS‐Cov‐2 was undetectable in 88 of the 90 patients with COVID‐19 who underwent RT‐PCR testing of CSF (personal communication).
Cerebrospinal fluid/serum albumin index was measured in four patients and found to be increased in three, suggesting the presence of BBB dysfunction. Neither CSF cells nor immunoglobulin G intrathecal synthesis was evident. A CSF increase in IL‐6 levels was observed in two patients at the time of lumbar puncture. With the exception only of case 5, oligoclonal bands were undetectable. Antineuronal antibodies were absent in all patients (Table S2).
Treatment approaches and outcomes
The treatment approaches and outcomes of the five study patients are shown in Fig. 1. Therapeutic attempts with corticosteroids and intravenous immunoglobulins (IVIg) are described below.
Case 1
Corticosteroids were given 14 days after symptom onset to tackle CRS, followed by a temporary improvement, with regression of neurological disturbances (Fig. 1a). Unfortunately, the patient experienced a second worsening of her neurological state on day 19 [Glasgow Coma Scale (GCS) = 8] and she died from coma and secondary infection on day 24.
Case 2
Two days after admission, the patient representing case 2 was switched to hemodialysis in an effort to decrease blood urea nitrogen, but no improvement in neurological condition was observed (ultimately excluding uremic encephalopathy). Further deterioration occurred on day 13, when he presented with a diffuse pyramidal syndrome, mixed fluent and non‐fluent aphasia, anterograde amnesia, cerebellar ataxia, a dysexecutive syndrome, and dysautonomia. After a 5‐day course of dexamethasone, an improvement of both consciousness and respiratory condition was observed (Fig. 1b). Four days after withdrawal of dexamethasone, there was a further neurological relapse with appearance of Cheyne‐Stokes breathing (GCS = 10). Because of the life‐threatening neurological condition, dexamethasone was reintroduced along with IVIg for 5 days. A complete and rapid clinical response was observed. A second cycle of IVIg has been planned.
Case 3
The neurological state of the patient representing case 3 spontaneously and gradually improved until discharge on day 40 after symptom onset.
Case 4
Neurological disturbances gradually and spontaneously regressed with recovery of orientation, memory function and motor function in case 4. Although ataxia and nystagmus improved partially, pyramidal syndrome and affective symptoms persisted on discharge of the patient on day 35.
Case 5
A 67‐year‐old man with a history of kidney transplantion was admitted on day 6 with moderate dyspnea. Mycophenolate mofetil was withdrawn, followed by administration of hydroxychloroquine.
On day 9, worsening of respiratory condition with signs of CRS was evident, prompting dexamethasone administration. On day 11, he developed acute neurological alterations consisting of bilateral reduced visual acuity, horizontal diplopia on the left side, predominant right cerebellar ataxia, tetrapyramidal syndrome, and personality change. Visual assessment revealed 7/10 right visual acuity drop, 1/10 left visual acuity drop in a previously amblyopic eye, a left abduction deficiency, and a left cervical stiff neck caused by sixth nerve palsy. Pupillary light reflexes and fundus examination were normal. Signs of neurological deterioration were paralleled by laboratory evidence of CRS. On treatment with methylprednisolone (500 mg/day for 3 days), both neurological disturbances and respiratory symptoms improved. Dexamethasone was subsequently given for 10 days to tackle CRS (Fig. 1e). On day 60, all neurological signs had regressed except slight palsy of left nerve VI.
Discussion
Severe disease and uremia are potential risk factors for neurological complications in patients with COVID‐19 [1]. Hypertension, obesity and diabetes, which are known risk factors for severe forms of COVID‐19[10], occur frequently in patients with renal disorders.
The present longitudinal study provides an in‐depth clinical, laboratory and imaging characterization of five patients with COVID‐19 who developed severe neurological disturbances. The main findings can be summarized as follows. First, the clinical presentation of CNS involvement included confusion, agitation, tremor, impaired consciousness, dysexecutive syndrome, pyramidal syndrome, cerebellar ataxia, cranial nerve palsy, dysautonomia, and central hormonal dysfunction (mainly in the form of hypothyroidism). The observation that the onset of neurological manifestations occurred in the second week after COVID‐19 symptoms onset, i.e. simultaneously with the worsening of respiratory condition, is also noteworthy. The systemic presentation was reflective of clinical symptoms related to CRS (i.e. fever, headache, myalgia, occasional rash, respiratory failure and, in some cases, multiorgan failure) [11].
Second, the laboratory evaluation demonstrated that SARS‐CoV‐2 was undetectable in the CSF in our patients (as it was in 98% of patients who performed this test in our hospital). Signs and symptoms of CNS impairment were accompanied by evidence of CRS in serum samples, with marked elevation of serum IL‐6. Third, we found evidence of increased BBB permeability, as demonstated by the presence of hyperalbuminorrachia. Moreover, elevation of serum levels of the S100B protein, an astroglial marker, was found to occur simultaneously with CRS. Finally, MRI findings were heterogeneous and included evidence of acute leukoencephalitis (n = 3, of whom one had a hemorrhagic form), cytotoxic edema mimicking an ischaemic stroke (n = 1), or normal results (n = 2). Notably, MRI findings are frequently normal both in influenza‐related acute encephalitis and autoimmune encephalitis [12].
Our findings of CRS‐associated neurological disturbances in COVID‐19 expand previous observations showing that hyperinflammation is associated with deterioration of respiratory function and death [5, 6]. Our data may have relevant implications with respect to clinical management. Because reliable detection of neurological symptoms may be unfeasible in patients on ventilation support, laboratory evidence of CRS should alert clinicians to a potential risk of CNS involvement. Notably, the onset of neurological manifestations was also paralleled by an increase in serum levels of S100B protein (reflecting increased BBB permeability) [8]. Elevated serum S100B levels have been previously reported in acute encephalitis and immune effector cell neurotoxicity syndrome (ICANS) [13, 14, 15].
Cytokine release syndrome is a potentially fatal complication of various infectious (e.g. influenza, SARS, Epstein−Barr virus‐associated hemophagocytic lymphohistiocytosis) and non‐infectious diseases (e.g. multiple organ dysfunction syndrome, multiple sclerosis). Moreover, it can have a iatrogenic origin during the course of T‐ and B‐lymphocyte‐engaging therapies such as chimeric antigen receptor (CAR)‐T‐cell immunotherapy, rituximab and immune check‐point inhibitors [11, 16]. CRS is triggered by an initial release of proinflammatory cytokines from activated T and/or B cells; this event, in turn, activates bystander immune cells and endothelial cells to produce proinflammatory molecules. Engagement of other immune cells follows in the context of a self‐perpetuating positive feedback loop. In this scenario, excess production of IL‐6 from activated macrophages is a key event. CRS‐driven neurological disturbances have been described for the first time following CAR‐T cell therapy and are termed ICANS. Remarkably, the clinical, laboratory and imaging features of severe ICANS closely resemble those shown by the five patients with COVID‐19 described in the present series [13, 17, 18]. Brain MRI in ICANS revealed the presence of acute T2/FLAIR hyperintensities suggestive of interstitial edema of varying severity and small (mm‐scale) ischaemic strokes [13]. One of our cases had a lesion in the splenium of the corpus callosum, a finding in accordance with previous data reported in ICANS [19] and encephalitis complicating acute viral infections (e.g. influenza and mumps) [12]. In ICANS, hemorrhagic forms (similar to the findings of patient 4 in our series) have been described [18]. More importantly, proinflammatory cytokines such as IL‐6 have been shown to lead to endothelial damage and BBB dysfunction in this clinical entity [13, 18].
All of these findings, coupled with the existing evidence, provide a pathophysiological model (Fig. 5) according to which COVID‐19‐associated neurological manifestations are driven by CRS and consequent immune response. Certain neurological complications of COVID‐19 may be caused by endothelial activation with subsequent increased BBB permeability, edema, [20] and penetration of proinflammatory cytokines that can activate microglial cells in the CNS. The resulting neuroinflammatory response may lead to reactive gliosis accompanied by infiltration of CD68+ monocytes/macrophage [21] and release of S100B protein. This pathophysiological model is in accordance with an autopsy report in COVID‐19 showing a range of white matter pathology with an anatomical distribution compatible with MRI lesions observed in our patients (i.e. subcortical areas, corpus callosum, middle cerebellar peduncle) and the lack of typical features of viral encephalitis. Rare neocortical microscopic organizing infarcts were also identified, suggesting a vascular origin followed by secondary myelin loss [22].
Figure 5.
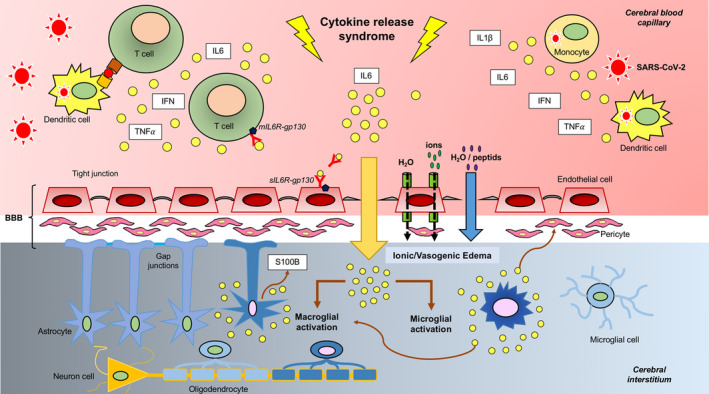
Cytokine release syndrome (CRS)‐associated encephalitis in COVID‐19: pathophysiological model. In severe cases of COVID‐19, SARS‐CoV‐2 induces CRS during the second week of infection. Following viral infection, macrophages, dendritic cells, other immune cells and endothelial cells become activated and produce large amounts of proinflammatory molecules [including interleukin (IL)‐6, IL‐1β, tumor necrosis factor (TNF)‐α, interferon (IFN)‐α/β, and IFN‐γ]. IL‐6 acts as a master mediator of a self‐perpetuating proinflammatory loop that results in lymphocyte activation (via the cis‐pathway) followed by a massive release of cytokines (cytokine storm). The trans‐pathway of activation may alter endothelial permeability, resulting in blood−brain barrier (BBB) dysfunction [20, 27]. In turn, an altered BBB permeability may lead to edema and even red blood cells extravasation (potentially accounting for the hemorrhagic form of acute leukoencephalitis observed in case #4). This sequence of events closely resembles those occurring in immune effector cell neurotoxicity syndrome [18]. Proinflammatory cytokines can leak through a dysfunctional BBB, ultimately activating microglial cells (brain tissue‐resident macrophages). This may, in turn, lead to a secondary inflammatory response in the macroglia accompanied by the release of S100B, an astroglial protein reflecting both glial activation and BBB dysfunction. The resulting neuroinflammatory response can yield to reactive gliosis accompanied by infiltration of CD68+ monocytes/macrophage [21, 22]. ; gp130, glycoprotein 130; sIL6R, soluble interleukin‐6 receptor. [Colour figure can be viewed at wileyonlinelibrary.com]
In keeping with our management strategies to tackle CRS, three of our patients with neurological disturbances received corticosteroids one patient received IVIg, with improvement. This is in accordance with two cases of encephalopathy in COVID‐19 that showed improvement after steroid [23] and IVIg [24]. Blockade of IL‐6 with tocilizumab, an anti‐IL‐6 receptor (IL‐6R) monoclonal antibody, has been also approved to treat CRS complicating CAR‐T cell therapy in the absence of neurological manifestations. Although tocilizumab may be effective against CRS, this molecule is not clinically useful to treat neurotoxicity in the context of CAR‐T cell therapy and can even exacerbate this condition [11, 13].This paradoxical effect may be explained by the transient rise in serum IL‐6 observed following tocilizumab administration and by the poor penetration of this antibody into the CSF [25].
Administration of IVIg during the early course of severe COVID‐19 may improve clinical outcomes through their immunomodulatory effects against the self‐perpetuating positive inflammatory loop typical of CRS [18, 19]. Interestingly, high‐dose IVIg may reduce in vitro and in vivo production of IL‐6 in other infectious or autoimmune diseases [20, 21]. In the present series, rapid recovery was observed after administration of IVIg (0.4 g per kg of weight per day for five consecutive days) in case 2.
Our findings need to be interpreted in the context of some limitations. Notably, this is a case series and some neurological manifestations occurring in patients with COVID‐19 might have been absent in our patients. Because lumbar puncture was performed during the second phase of COVID‐19, CSF analysis may have failed to detect SARS‐CoV‐2 RNA. Similar findings have been previously reported for tick‐borne encephalitis virus, which has been shown to be detectable in the blood by PCR during the preclinical phase only [26].
These caveats notwithstanding, we provide a pilot description of the peculiar clinical, laboratory, and imaging findings of COVID‐19‐associated neurological disturbances in a small but well‐characterized group of patients admitted to a renal unit. The clinical manifestations were chiefly driven by peripheral CRS, absent direct CNS invasion by SARS‐CoV‐2. Pending future confirmation, our data indicate that corticosteroids aimed at tackling CRS [27] and IVIg may be effective to control severe neurological disturbances in patients with COVID‐19.
Disclosure of conflicts of interest
The authors declare no financial or other conflicts of interest.
Supporting information
Table S1. Laboratory findings of the five study patients at hospitalization and during follow‐up
Table S2. Laboratory analysis of cerebrospinal fluid in the five study patient
Acknowledgments
Funding information: This study was conducted in the context of clinical care and did not receive specific funding.
Data availability statement
Data sharing is not applicable to this article as no datasets were generated or analysed during the present study.
References
- 1. Mao L, Jin H, Wang M, et al. Neurologic manifestations of hospitalized patients with coronavirus disease 2019 in Wuhan, China. JAMA Neurol 2019; 77: 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Helms J, Kremer S, Merdji H, et al. Neurologic features in severe SARS‐CoV‐2 infection. N Engl J Med 2020; 382: 2268–2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Romoli M, Jelcic I, Bernard‐Valnet R, et al. The infectious disease panel of the European academy of neurology a systematic review of neurological manifestations of SARS‐CoV‐2 infection: the devil is hidden in the details. Eur J Neurol 2020; 27: 1712–1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wu Y, Xu X, Chen Z, et al. Nervous system involvement after infection with COVID‐19 and other coronaviruses. Brain Behav Immun 2020; 87: 18–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mehta P, McAuley DF, Brown M, Sanchez E, Tattersall RS, Manson JJ. HLH across speciality collaboration, UK COVID‐19: consider cytokine storm syndromes and immunosuppression. Lancet 2020; 395: 1033–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ruan Q, Yang K, Wang W, Jiang L, Song J. Clinical predictors of mortality due to COVID‐19 based on an analysis of data of 150 patients from Wuhan, China. Intensive Care Med 2020; 46: 846–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Alberici F, Delbarba E, Manenti C, et al. Management of patients on dialysis and with kidney transplant during SARS‐COV‐2 (COVID‐19) pandemic in Brescia, Italy. Kidney Int Rep 2020; 5: 580–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Michetti F, D’Ambrosi N, Toesca A, et al. The S100B story: from biomarker to active factor in neural injury. J Neurochem 2019; 148: 168–187. [DOI] [PubMed] [Google Scholar]
- 9. Glady L, Lavaux T, Charchour R, Lacorte J‐M, Lessinger J‐M. Interleukin‐6 chemiluminescent immunoassay on Lumipulse G600 II: analytical evaluation and comparison with three other laboratory analyzers. Clin Chem Lab Med 2020; 58: 229–231. [DOI] [PubMed] [Google Scholar]
- 10. Henry BM, Lippi G. Chronic kidney disease is associated with severe coronavirus disease (COVID‐19) infection. Int Urol Nephrol 2019; 52: 1193–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Garcia Borrega J, Gödel P, Rüger MA, et al. In the eye of the storm: immune‐mediated toxicities associated with CAR‐T cell therapy. Hemasphere 2019; 3: e191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Venkatesan A, Michael BD, Probasco JC, Geocadin RG, Solomon T. Acute encephalitis in immunocompetent adults. Lancet 2019; 393: 702–716. [DOI] [PubMed] [Google Scholar]
- 13. Gust J, Finney OC, Li D, et al. Glial injury in neurotoxicity after pediatric CD19‐directed chimeric antigen receptor T cell therapy. Ann Neurol 2019; 86: 42–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Leis AA, Stokic DS, Petzold A. Glial S100B is elevated in serum across the spectrum of West Nile virus infection. Muscle Nerve 2012; 45: 826–830. [DOI] [PubMed] [Google Scholar]
- 15. Tsukahara H, Fujii Y, Matsubara K, et al. Prognostic value of brain injury biomarkers in acute encephalitis/encephalopathy. Pediatr Int 2013; 55: 461–464. [DOI] [PubMed] [Google Scholar]
- 16. Tisoncik JR, Korth MJ, Simmons CP, Farrar J, Martin TR, Katze MG. Into the eye of the cytokine storm. Microbiol Mol Biol Rev 2012; 76: 16–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lee DW, Santomasso BD, Locke FL, et al. ASTCT consensus grading for cytokine release syndrome and neurologic toxicity associated with immune effector cells. Biol Blood Marrow Transplant 2019; 25: 625–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gust J, Hay KA, Hanafi L‐A, et al. Endothelial activation and blood‐brain barrier disruption in neurotoxicity after adoptive immunotherapy with CD19 CAR‐T Cells. Cancer Discov 2017; 7: 1404–1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Santomasso BD, Park JH, Salloum D, et al. Clinical and biological correlates of neurotoxicity associated with CAR T‐cell therapy in patients with B‐cell acute lymphoblastic leukemia. Cancer Discov 2018; 8: 958–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Stokum JA, Gerzanich V, Simard JM. Molecular pathophysiology of cerebral edema. J Cereb Blood Flow Metab 2016; 36: 513–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Xu J, Zhong S, Liu J, et al. Detection of severe acute respiratory syndrome coronavirus in the brain: potential role of the chemokine mig in pathogenesis. Clin Infect Dis 2005; 41: 1089–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Reichard RR, Kashani KB, Boire NA, Constantopoulos E, Guo Y, Lucchinetti CF. Acta Neuropathol. 2020. [DOI] [PMC free article] [PubMed]
- 23. Pilotto A, Odolini S, Stefano Masciocchi S, et al. Steroid‐responsive encephalitis in Covid‐19 disease. Ann Neurol 2020; 88: 423–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Afshar H, Yassin Z, Kalantari S, et al. Evolution and resolution of brain involvement associated with SARS‐ CoV2 infection: a close clinical ‐ paraclinical follow up study of a case. Mult Scler Relat Disord 2020; 43: 102216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nellan A, McCully CML, Cruz Garcia R, et al. Improved CNS exposure to tocilizumab after cerebrospinal fluid compared to intravenous administration in rhesus macaques. Blood 2018; 132: 662–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Taba P, Schmutzhard E, Forsberg P, et al. EAN consensus review on prevention, diagnosis and management of tick‐borne encephalitis. Eur J Neurol 2017; 24: 1214–e61. [DOI] [PubMed] [Google Scholar]
- 27. Jones SA, Rose‐John S. The role of soluble receptors in cytokine biology: the agonistic properties of the sIL‐6R/IL‐6 complex. Biochim Biophys Acta 2002; 1592: 251–263. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Laboratory findings of the five study patients at hospitalization and during follow‐up
Table S2. Laboratory analysis of cerebrospinal fluid in the five study patient
Data Availability Statement
Data sharing is not applicable to this article as no datasets were generated or analysed during the present study.