

Abstract
SARS‐CoV‐2 is the causative agent for the ongoing COVID19 pandemic, and this virus belongs to the Coronaviridae family. Like other members of this family, the virus possesses a positive‐sense single‐stranded RNA genome. The genome encodes for the nsp12 protein, which houses the RNA‐dependent‐RNA polymerase (RdRP) activity responsible for the replication of the viral genome. A homology model of nsp12 was prepared using the structure of the SARS nsp12 (6NUR) as a model. The model was used to carry out in silico screening to identify molecules among natural products, or Food and Drug Administration‐approved drugs that can potentially inhibit the activity of nsp12. This exercise showed that vitamin B12 (methylcobalamin) may bind to the active site of the nsp12 protein. A model of the nsp12 in complex with substrate RNA and incoming NTP showed that vitamin B12 binding site overlaps with that of the incoming nucleotide. A comparison of the calculated energies of binding for RNA plus NTP and methylcobalamin suggested that the vitamin may bind to the active site of nsp12 with significant affinity. It is, therefore, possible that methylcobalamin binding may prevent association with RNA and NTP and thus inhibit the RdRP activity of nsp12. Overall, our computational studies suggest that methylcobalamin form of vitamin B12 may serve as an effective inhibitor of the nsp12 protein.
Keywords: inhibitor, nsp12, RNA‐dependent‐RNA polymerase, SARS‐CoV‐2, vitamin B12
Abbreviations
- CDC, Centers for Disease Control and Prevention; Cryo‐EM
cryogenic electron microscopy
- Exon
exonuclease
- FDA
Food and Drug Administration
- GTP, Guanosine triphosphate; kcal/mol
kilocalorie per mole
- LBGFS
limited‐memory Broyden–Fletcher–Goldfarb–Shanno
- MMACHC
methylmalonic aciduria and homocystinuria type C protein
- MMGBSA
molecular mechanics energies combined with generalized born and surface area continuum solvation
- nsp
non‐structural protein
- NTP, Nucleoside triphosphate; OPLS‐AA
optimized potentials for liquid simulations‐all atom
- ORF
open reading frame
- OTC
over‐the‐counter
- PDB
Protein Data Bank
- RMSD
root‐mean‐square deviation of atomic positions
- RdRp
RNA‐dependent RNA polymerase
- SAM
S‐adenosyl methionine
- SDF
spatial data file
- VSGB
variable dielectric surface generalized born
- 3D
three‐dimensional
1. INTRODUCTION
The members of the Coronaviridae family are viruses with positive‐sense single‐stranded RNA genomes. 1 At present, some members of this family, such as SARS, MERS, and SARS‐CoV‐2 represent pathogens of great concern to public health. SARS‐CoV‐2 is responsible for the ongoing pandemic of COVID‐19, which started in the city of Wuhan in China, has now spread to more than 200 countries. At present, there are nearly 9 million confirmed cases of the disease caused by this pathogen, with more than 450,000 fatalities. 2 At present, there are no effective treatments available against COVID‐19, and current medical protocols involve isolating the patient and provide symptomatic treatment for patients with mild disease and oxygen therapy/ventilator support for patients with severe disease.
The genome of COVID‐19 is roughly 30 kB long with a gene at the 5′ end known as orf1ab that encodes for all the polyprotein bearing all the non‐structural proteins. 3 The virus also possesses genes that code for structural proteins, namely spike (S), envelope (E), membrane (M), and nucleocapsid (N). 4 The polyprotein arising from orf1ab may undergo proteolytic processing to give rise to 16 proteins namely nsPs 1–16. 5 Among the cleaved products of the ORF1Ab polyprotein, the proteins of known function include nsp3, which has an adenosine diphosphate‐ribose 1′‐phosphatase activity. 6 The protease activity that is responsible for the cleavage of the polyprotein is present in the nsp5 protein. The nsp12 protein houses the RNA‐dependent RNA polymerases (RdRp) that are responsible for duplication of the genome. The RNA helicase activity that is critical for genome duplication is present in the nsp13 protein. Exoribonuclease (exoN) and N7‐methyltransferase activities are present in the nsp14 protein. 7 The nsp15 protein houses a Nidoviral ribonuclease specific for U, and the nsp16 protein has a S‐adenosyl methionine (SAM)‐dependent O‐methyltransferase activity. 3
There is an urgent need for the identification of new molecules that can reduce viral titers and thus limit the severity of the disease. Toward this end, we have generated a model of the nsp12 molecule from SARS‐CoV‐2(SCV2‐nsp12) and used this model to carry out in silico screening to identify potential inhibitors of the RNA‐dependent‐RNA polymerase activity. Our studies suggest that the methylcobalamin form of vitamin B12 may serve as an effective inhibitor of the RdRp activity of nsp12.
2. METHODS
2.1. Homology modeling of nsp12 and model of the functional ternary complex
The sequence corresponding to nsp12 from a sequence of SARS‐CoV‐2 deposited in Genbank by the CDC (Atlanta, USA) with the accession no. MT044257.1 was used. The nsp12 protein from SARS‐CoV exhibits 97% identity with the corresponding protein from SARS‐CoV‐2. A homology model was generated using the SWISSMOD server, 8 and the structure of nsp12 from the nsp12‐nsp7‐nsp8 complex (6NUR) of SARS‐CoV was used as a template. 9 The validation of the model was carried out by the SWISSMOD server. 8
To generate a computational model of the functional ternary complex (SCV2‐nsp12:RNA:NTP), initially, DALI searches were carried with the model of apo‐ structure to identify structural orthologs of nsp12 bound to RNA and incoming nucleotide. 10 This search showed that, on superimposition, the Q‐beta replicase exhibits significant structural homology in the palm domain. The RNA and incoming nucleotide of the transformed coordinates of the ternary complex of Q‐beta replicase (3AVX) were transferred onto the homology model of SCV2‐nsp12. 11 The local structure in the active site was altered manually to ensure coordination of the cofactor ion by the catalytic residues and the triphosphate moiety of the incoming nucleotide as seen previously for DNA polymerases. 12 The structure prepared in this way was subjected to energy minimization using DESMOND module of the SCHRÖDINGER suite. The structure was minimized in an orthorhombic box containing single point and charge water model and subjected to steepest descent and limited‐memory Broyden–Fletcher–Goldfarb–Shanno (LBGFS) vectors minimization until the difference in energy converged to 0.1 kcal/mol. 13
2.2. In silico screening
The model of SCV2‐nsp12 was imported into the MAESTRO interface of SCHRÖDINGER suite and prepared using the protein preparation wizard program. The prepared structure was used to identify potential binding sites using the SITEMAP program, which uses site score function to rank the possible binding sites according to size, functionality, and extent of solvent exposure on the protein. 14 Sites with a site score value of ≥0.8 were identified and further examined using PyMOL to see if it could inhibit the RNA binding. Residues spanning the most relevant site were used to prepare a receptor grid using the Receptor Grid Generation module in SCHRÖDINGER.
Concurrently various annotated libraries Food and Drug Administration (FDA)‐approved drugs (L1300), natural products (L1400), antiviral compounds (L7000), and drug repurposing compounds (L3800) were downloaded from selleckchem.com in the spatial data file (SDF) format. 15 The library compounds were converted to their accurate energy minimized three‐dimensional (3D) molecular structures using the LIGPREP module in SCHRÖDINGER. LIGPREP was used to expand tautomeric states, ionization states, ring conformations, and stereoisomers of ligand molecules to produce broad chemical and structural diversity from each molecule along with the correction of Lewis structures and ligand order of these molecules.
Using the glide docking module the prepared grid for the binding site in protein and the library of prepared ligands from LIGPREP was docked. GLIDE generates about 5,000 poses for the ligands, which are initially filtered by spatial fit onto the active site of proteins and are checked for the complementarity of interaction using the ChemScore function. The top 400 poses excluding those with an energy difference of more than 100 kcal/mol from the best ligand pose are used for further analysis. The poses that pass this initial filter are minimized with respect to the receptor grid using optimized potentials for liquid simulations‐all atom (OPLS)‐AA non‐bonded ligand‐receptor interaction energy. Once the energy is calculated, the GLIDE Score multi‐ligand scoring function assigned scores to the poses. GLIDE docking was carried out in standard‐precision (SP) mode, and the molecules that bind to the receptors with good docking score and negative binding energy were used for further analysis.
During the revision of the manuscript, the structure of the nsp12‐nsp7‐nsp8 complex (PDB code:6 M71) was released and the coordinates of nsp12 from this structure were used to dock the vitamin B12 using the GLIDE module of SCHRÖDINGER. 16
2.3. Minimization, molecular dynamics, and calculation of binding energies
The SCV2‐nsp12: methylcobalamin complex was then subjected to energy minimization using the DESMOND module of SCHRÖDINGER suite. The system setup program was used to set up an orthorhombic boundary box containing a simple point‐charge water model and 11 Na+ ions to neutralize the system. This system was then minimized using steepest descent and LBGFS vectors with a convergence threshold of 0.1 kcal/mol.
The SCV2‐nsp12:methylcobalamin was subjected to restrained MD for 50 ns to unearth a better pose for the ligand. Different frames, which showed more contacts were subjected to energy minimization. In addition, the binding energy between ligand and nsp12 was calculated using the molecular mechanics energies combined with the generalized born and surface area continuum solvation (MMGBSA) program in the Schrödinger suite. The docked protein and ligand complex were separated manually and loaded as receptor or ligand, respectively, in the MMGBSA module. MMGBSA program minimizes each of them separately as well as in combination using VSGB 2.0 (variable dielectric surface generalized born) solvation model. The energy of binding is then calculated by subtracting the energy of the optimized free receptor and optimized free ligand from the energy of the optimized complex. Further, to check if methylcobalamin could compete with the natural substrates, the binding energy between the modeled nsp12 and RNA plus incoming nucleotide in the model of the ternary complex was also calculated. The binding energy between nsp12 and RNA was also calculated for the newly released structure of the nsp12‐nsp7‐nsp8 in complex with RNA bearing remdesivir monophosphate at the 3′ end of the primer (PDB code:7BV2).
2.4. Analysis
The minimized models of the ternary complex of nsp12 (SCV2‐nsp12:RNA:GTP:2 Ca2+ ions) and the nsp12:methylcobalamin complex were analyzed using CONTACT program in CCP4 suite to identify the residues involved in interactions with the substrates and the vitamin, respectively. 17 The structure of nsp12‐nsp7‐nsp8 in complex with RNA bearing remdesivir monophosphate at the 3′ end of the primer (PDB code:7BV2) was also used to identify the residues of nsp12 that interact with RNA using CONTACT. The models of the complexes were superimposed using the COOT program 18 and all figures were prepared using PyMOL (Schrödinger Inc.)
3. RESULTS
3.1. Model of the SCV2‐nsp12 in its apo‐ and functional state
The SCV2‐nsp12 protein shows a 97% identity with the corresponding protein from SARS (Figure 1). The structure of the SARS‐nsp12 in complex with nsp7 and nsp8 was determined using Cryo‐EM and deposited in the PDB with the accession code 6NUR. The structure of nsp12 from 6NUR was used to generate a computational model of the SCV2‐nsp12 in its apostate using the SWISSMOD server (Figure 2). The server showed that the stereochemistry of the model was good, with 98% residues in the allowed regions and less than 1% residues in the disallowed regions. Swissmod uses a QMEAN (qualitative model energy analysis) to evaluate the models and the score obtained for the model was −0.65, which is good for further analysis. The model encompasses residues 4,509–5,311 of the polyprotein translated from orf1ab of the COVID‐19 genome and also includes two Zn2+ ions. The model shows the presence of the N‐terminal extension (119–397), fingers (398–581 and 628–687), palm (582–627 and 688–815), and thumb (816–919) domains (Figure 2). The SCV2‐nsp12 model showed good structural overlap with nsp12 from the recently released structure of the nsp12‐nsp7‐nsp8 complex (6 M71) 16 and the two sets of coordinates superimposed with an root‐mean‐square deviation of atomic positions (RMSD) value of 0.48 Å.
FIGURE 1.
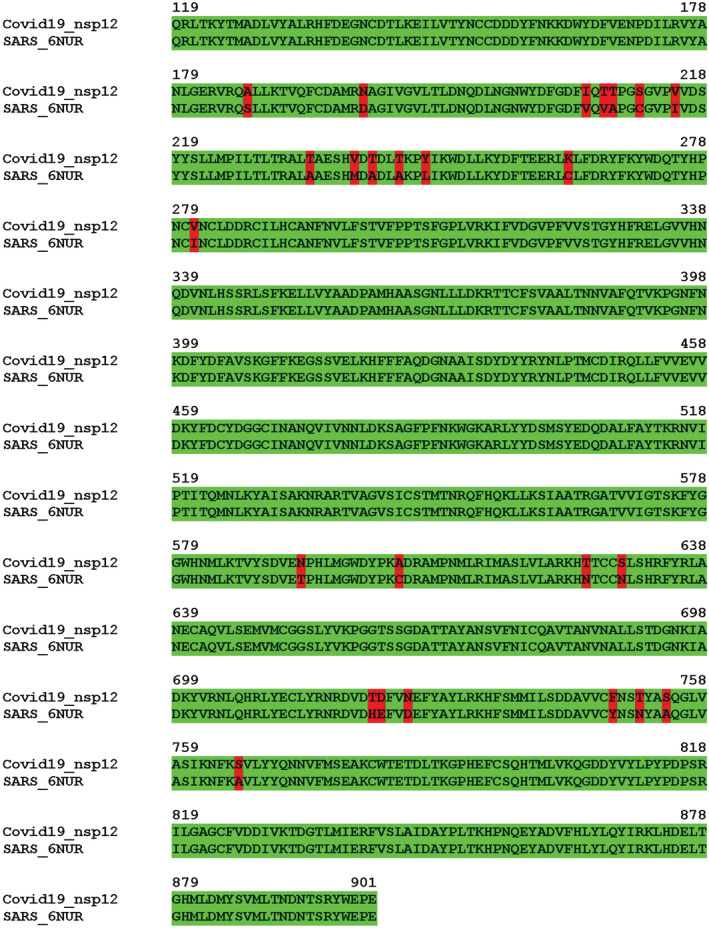
Alignment of the sequence of SCV2‐nsp12 (119–901) with the sequence of the available structure of SARS‐nsp12 (6NUR). The SCV2‐nsp12 sequence exhibits about 97% identity with the corresponding stretch in SARS‐nsp12
FIGURE 2.
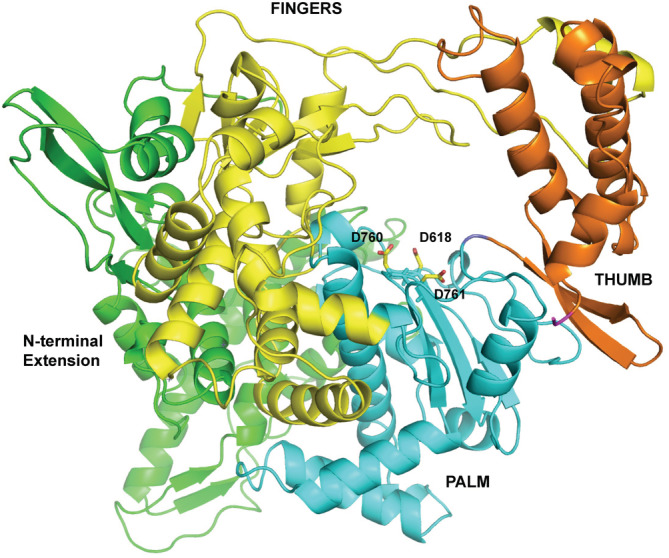
Model of the SCV2‐nsp12 enzyme in its apostate. The N‐terminal extension region and the palm, fingers, and thumb domains are shown in green, cyan, yellow, and orange, respectively. The catalytic residues Asp618, Asp760, and Asp761 are colored according to element and shown in stick representation
To generate the structure of the SCV2‐nsp12 structure in its functional state, initially, a DALI search was carried out to identify structural orthologs of this enzyme. The list of enzymes that showed good superimposition with the nsp12 model was analyzed to identify structures of functional ternary complexes. The structure of Q‐beta replicase in complex with RNA and incoming nucleotide (3AVX) gave a Z‐score of 3.7 and an RMSD of 3.0 in the DALI search. The superimposition was used to generate a model of the SCV2‐nsp12 protein in complex with RNA bearing template C, incoming nucleotide (GTP), and two Ca2+ ions. The model was subjected to energy minimization and converged to minimum energy of −3.97 × 105 kcal/mol. The Ramachandran plot of the minimized model showed that only 1.75% of residues were in the disallowed regions. The model showed the presence of the expected octahedral coordination of one of the cofactor ion between the triphosphate moiety and the catalytic residues D762 and D620 (Figure S1). During the revision of the manuscript, the structure of SCV2‐nsp12 in complex RNA (PDB code:7BV2), solved by Cryo‐EM, was released. 19 The Cryo‐EM structure showed good overlap with the SCV2‐nsp12:RNA:GTP model and the two sets of coordinates superimposed with an RMSD value of 1.01 Å.
3.2. In Silico screening identifies vitamin B12 as a potential binder
The site map program showed four possible binding sites in the SCV2‐nsp12 protein, which were ranked according to their ability to bind to various ligands. Site 2 with a site score of 0.99 was selected over site 1 with a site score of 1.03 as it overlapped with the RNA binding groove and catalytic site of the enzyme. The residues spanning site 2 were used to generate a receptor grid used for molecular docking. Molecular docking was carried out using annotated libraries of molecules that could bind to SCV2‐nsp12 open complex using glide dock program. A total of 8,285 compounds from different libraries were screened to find molecules that can bind to the active site of SCV2‐nsp12. The top hits from each of these libraries were ranked according to their docking score, glide gscore, and glide energy based on the interaction between the protein and ligand (Table 1). Methylcobalamin (vitamin B12) showed the second‐best docking score (−8.193) and is already part of many OTC drug formulations. Since cefoperazone is an antibiotic that functions extracellularly and is associated with adverse side effects, the vitamin molecule was selected for further analysis. 20 , 21 , 22 , 23
TABLE 1.
List of ligands with Docking Score better than −6
| Title | Docking score | Glide gscore | Glide energy |
|---|---|---|---|
| FDA‐approved drugs | |||
| Paclitaxel | −7.579 | −7.58 | −66.241 |
| Fulvestrant | −6.807 | −6.807 | −45.489 |
| Bicalutamide | −6.632 | −6.632 | −40.778 |
| Raltitrexed | −6.085 | −6.149 | −42.758 |
| Antiviral compounds | |||
| Ritonavir | −6.455 | −6.455 | −73.429 |
| Danoprevir | −5.875 | −5.881 | −65.887 |
| Drug repurposing | |||
| Ritonavir | −6.455 | −6.455 | −73.429 |
| Ammonium Glycyrrhizinate | −6.033 | −6.037 | −49.896 |
| Natural products | |||
| Cefoperazone acid | −8.481 | −8.481 | −76.01 |
| Vitamin B12 | −8.193 | −8.263 | −75.794 |
| S3668 Thymopentin | −8.147 | −8.273 | −72.13 |
| S3622 Diammonium Glycyrrhizinate | −7.075 | −7.079 | −67.951 |
| Sennoside A | −7.024 | −7.032 | −77.214 |
| Sennoside B | −7.024 | −7.032 | −77.214 |
| Doripenem hydrate | −6.88 | −7.28 | −44.76 |
| Ampicillin trihydrate | −6.871 | −7.548 | −43.656 |
| Nystatin (Fungicidin) | −6.769 | −6.807 | −74.219 |
| S3815 Mogroside V | −6.754 | −6.754 | −75.913 |
| Cephalomannine | −6.734 | −6.735 | −64.049 |
| Geniposidic acid | −6.73 | −6.742 | −46.101 |
| NATACYN (natamycin) | −6.642 | −6.679 | −60.807 |
| Tigecycline | −6.596 | −6.73 | −56.619 |
It was observed that the docked B12 molecule was a Cobalt free version of the vitamin. Consequently, the correct version of the methylcobalamin ligand, obtained from the complex with human methylmalonic aciduria and homocystinuria type C protein (MMACHC) (3SC0), was docked at the appropriate site in the nsp12 enzyme. 24 , 25 This SCV2‐nsp12:methylcobalamin complex model gave a docking score of −8.749. The model of the complex was subjected to energy minimization and it converged to minimum energy of −4 × 105 kcal/mol.
The docking of methylcobalamin in the active site of nsp12 from the recently determined structure of the nsp12‐nsp7‐nsp8 complex (6 M71) gave a docking score of −8.4 and the structure was subjected to energy minimization and it converged to minimum energy of −4.7 × 105 kcal/mol.
3.3. Molecular dynamics simulation and energy of binding
The SCV2‐nsp12:methylcobalamin was subjected to restrained MD for 50 ns to unearth a better pose for the ligand. Different frames, which showed more contacts were subjected to energy minimization. In addition, the binding energy between ligand and nsp12 was calculated using the MMGBSA method. The best model obtained in the manner showed the presence of one molecule of methylcobalamin bound in the active site of the nsp12 enzyme with a binding energy of −86.3 kcal/mol (Figure 3).
FIGURE 3.
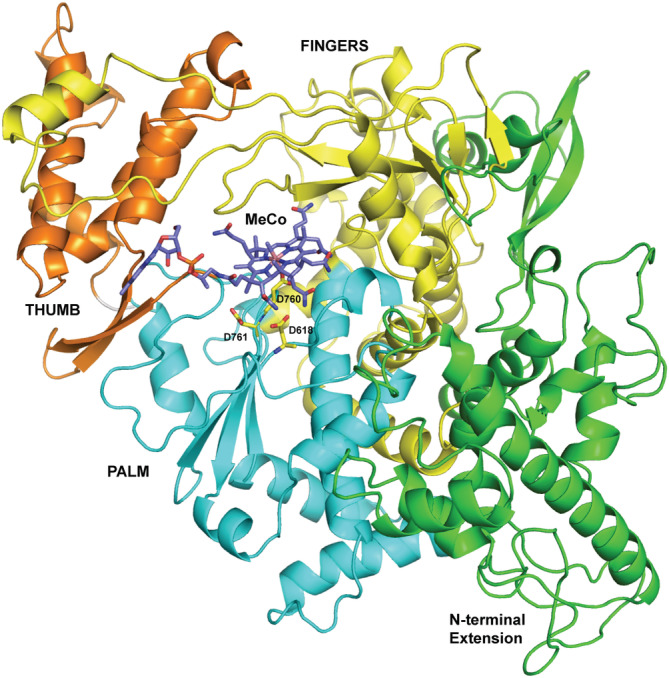
Model of the SCV2‐nsp12 enzyme in complex with methylcobalamin. The vitamin B12 molecule is shown in stick representation and colored according to element. The N‐terminal extension region and the palm, fingers, and thumb domains are shown in green, cyan, yellow, and orange, respectively. The catalytic residues Asp618, Asp760, and Asp761 are colored according to element and shown in stick representation
Using the MMGBSA program, the energy of interaction between SCV2‐nsp12 and RNA + GTP was compared with that of between nsp12 and methylcobalamin. The calculated binding energy for the interaction of RNA and incoming nucleotide with SCV2‐nsp12 is −95 kcal/mol. This value is nearly identical to the binding energy between protein and RNA for the Cryo‐EM structure (7BV2), which was calculated to be −94.2 kcal/mol. In comparison, the calculated binding energy value for the interaction of methylcobalamin with SCV2‐nsp12 is −86.3 kcal/mol. These values suggest that vitamin B12 can bind with a significant affinity at the active site of the SCV2‐nsp12 enzyme.
3.4. Comparison of interactions inSCV2‐nsp12:vitamin B12 complex and the functional ternary complex of SCV2‐nsp12
The model of the ternary complex of SCV2‐nsp12 was analyzed to identify the interacting residues. The residues that form Van der Wall interactions with RNA are Lys411, Asn496, Asn497, Lys500, Ser501, Lys512, Lys545, Tyr5486, Val557, Arg569, Lys577, Ala580, Arg583, Ile589, Gly590, Thr591, Ser592, Lys593, Phe594, Tyr595, Ser682, Gly683, Asp684, Ala685, Ala688, Tyr689, Leu758, Ser759, Asp760, Asp761, Glu811, Cys813, Ser814, Gln815, Pro832, Asp833, Arg836, Ile837, Val848, Leu854, Glu857, Arg858, Val860, Ser861, Leu862, Ile864, Asp865, Asn911, Arg914, and Tyr915. The residues Asn496, Asn497, Lys500, Ser501, Lys512, Arg569, Lys577, Ala580, Arg583, Thr591, Ser592, Lys593, Tyr595, Cys813, Ser814, Asn911, Arg914, and Tyr915 form polar interactions with RNA. In the ternary complex, the residues Lys572, Asp618, Tyr619, Pro620, Lys621, Asp623, Ser682, Asn691, and Asp760 form polar interactions with the incoming nucleotide that is, GTP. Ala622 and Gly683 form Van der Waal's interactions with GTP. The residues Asp618, Tyr619, Asp760, Asp761, and Glu811 also interacts with the incoming nucleotide through the cofactor ions. Asp760, Asp761, and Asp618 are the catalytic residues that are responsible for nucleic acid synthesis reaction.
The structure of SCV2‐nsp12‐nsp7‐nsp8 in complex with RNA (7BV2) superimposed onto the model the ternary complex of SCV2‐nsp12 with an RMSD of 1.01 Å (Figure S2). The Cryo‐EM represents the state after incorporation of remdesivir monophosphate at the 3′ end of the primer but before translocation of the added derivative to the terminal primer nucleotide position. In comparison, the model shows the presence of enzyme, RNA, and incoming nucleotide with the incipient base pair of CTP:GTP present in the active site. The differences observed in the two sets of coordinates can be attributed to the fact that the computational model and Cryo‐EM structure represent the pre‐ and post‐ catalytic states, respectively. In addition, the RNA bound to nsp12 in the Cryo‐EM structure is two base‐pairs longer than the one in the model.
The SCV2‐nsp12:methylcobalamin complex was analyzed to identify the residues that interact with the vitamin B12 (Figure 4). The nsp12 residues that form Van der Waal contacts with the methylcobalamin molecule include Lys438, His439, Arg555, Thr556, Val557, Asp618, Tyr619, Pro620, Lys621, Asp623, Arg624, Ser682, Ser759, Asp760, Asp761, Lys798, Cys813, Ser814, Asp833, Ser835, Arg836, Tyr877. The residues Lys438, Arg555, Asp618, Tyr619, Lys621, Asp623, Arg624, Asp760, Ser814, Ser835, and Arg836 also form polar interactions with the vitamin B12 molecule. Of the interacting residues with vitamin B12 Asp618, Tyr619, Pro620, Lys621, Asp623, Ser682, Asp760, and Asp761 were found to interact with an incoming nucleotide in the model of the ternary complex of SCV2‐nsp12. Similarly, Asp623, Ser682, Ser759, and Asp760 were found to interact with remdesivir in 7BV2 and the interactions with Asp623, Ser682, and Asp760 were conserved in all the three structures. One of the three catalytic residues, Asp760 was found to form polar interactions with the methylcobalamin, remdesivir (7BV2), and GTP(model).
FIGURE 4.
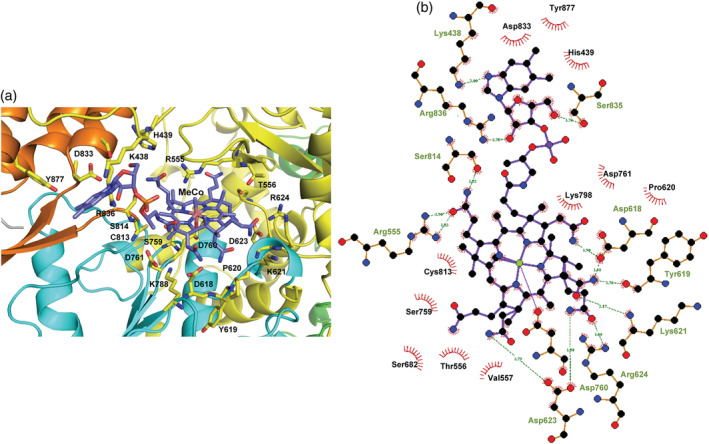
Residues of SCV2‐nsp12 that interact with methylcobalamin. (a) The vitamin B12 (MeCo) molecule is shown in stick representation and colored according to element. The N‐terminal extension region and the palm, fingers, and thumb domains are shown in green, cyan, yellow, and orange, respectively. The interacting residues are colored according to element and shown in stick representation. (b) The LigPlot representation of the interacting residues is displayed here. The polar interactions are shown in the form of dotted lines and the residues involved in hydrophobic interactions are highlighted by arcs with spokes
A superimposition of the model of the ternary complex and that of the nsp12:methylcobalamin complex was carried out to ascertain the level of overlap between the binding sites of the natural substrates of nsp12 and the methylcobalamin molecule. The superimposition showed that the binding site of methylcobalamin overlaps with that of the incoming nucleotide and the terminal primer nucleotide of the RNA substrate (Figure 5). Therefore, the binding of methylcobalamin may inhibit the formation of a functional ternary complex of nsp12 and thus prevent RNA synthesis necessary for replication of the viral genome.
FIGURE 5.
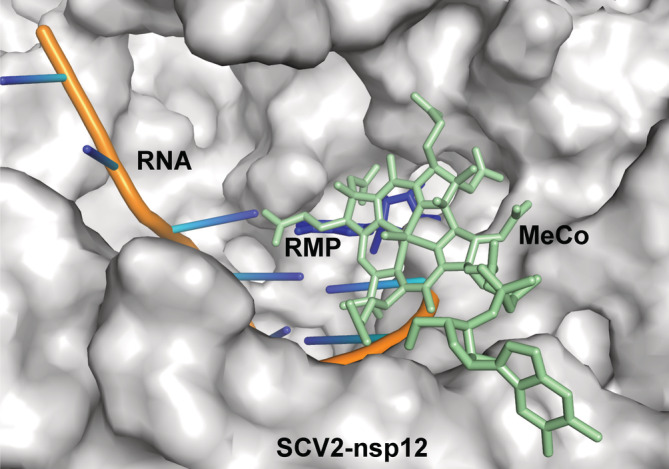
Binding site of methylcobalamin (MeCo) overlaps with that of incoming nucleotide. The Cryo‐EM structure of SCV2‐nsp12 in complex with RNA bearing remdesivir monophosphate (RMP) at the 3′ end of the primer (7BV2) and the model of the SCV2‐nsp12:methylcobalamin complex was superimposed. The surface of the protein molecule is shown and the RNA is shown in cartoon representation. The incoming nucleotide and methylcobalamin are displayed in stick representation and colored blue and light orange, respectively. The vitamin B12 binding site overlaps with that of the incoming nucleotide and the terminal primer nucleotide
4. DISCUSSION
The studies presented here suggest that methylcobalamin may be a possible inhibitor of the RNA‐dependent‐RNA polymerase activity of the SCV2‐nsp12 enzyme. Since this enzyme is critical for the replication of the viral enzyme, the inhibition of this enzyme can result in lower viral titers and reduce the severity of the COVID‐19 disease. The number of patients suffering from COVID‐19 is increasing daily and about 7% of them are in serious condition. Hence, the ability of methylcobalamin to inhibit viral replication should be tested urgently using in vitro and in vivo assays. In addition, given the urgency of the situation and the fact that methylcobalamin is already part of drug formulations, doctors may consider adding or increasing the dosage of methylcobalamin in their current patient care protocols. In addition, methylcobalamin may be prescribed as prophylactic for groups such as medical personnel, the aged or individuals with comorbidities, to reduce the possibility of infection and critical hospital care.
AUTHOR CONTRIBUTIONS
DTN and NN conceived the project. NN carried out the computations, and DTN supervised the work. DTN and NN analyzed the results and wrote the manuscript.
COMPETING INTERESTS STATEMENT
None declared.
Supporting information
Figure S1 Model of the functional ternary complex of the SCV2‐nsp12 enzyme in the complex. The RNA substrate and incoming GTP are shown in stick representation and colored according to element. The N‐terminal extension region and the palm, fingers, and thumb domains are shown in green, cyan, yellow, and orange, respectively. The catalytic residues Asp618, Asp760, and Asp761 are colored according to element and shown in stick representation and the cofactor ions are shown as blue spheres.
{kind=link}
Figure S2 Superimposition of the Cryo‐EM structure of SCV2‐nsp12:RNA and the model of the ternary complex. The backbone of the Cryo‐EM structure is colored green and that of the model is colored cyan. The RNA is shown in the stick representation. The incorporated remdesivir is present in the same location as the incoming nucleotide triphosphate.
{kind=link}
Narayanan N, Nair DT. Vitamin B12 may inhibit RNA‐dependent‐RNA polymerase activity of nsp12 from the SARS‐CoV‐2 virus. IUBMB Life. 2020;72:2112–2120. 10.1002/iub.2359
Funding information Regional Centre for Biotechnology, Grant/Award Number: Intramural Grant
REFERENCES
- 1. Modrow S, Falke D, Truyen U, Schätzl H, Modrow S, et al. Viruses with single‐stranded, positive‐sense RNA genomes. Molecular Virology. Germany: Springer Berlin Heidelberg, 2013; p. 185–349. [Google Scholar]
- 2. Johns Hopkins coronavirus resource center. Baltimore, Maryland: John Hopkins University; 2020. https://coronavirus.jhu.edu/. [Google Scholar]
- 3. Wu F, Zhao S, Yu B, et al. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579:265–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shang W, Yang Y, Rao Y, Rao X. The outbreak of SARS‐CoV‐2 pneumonia calls for viral vaccines. NPJ Vaccines. 2020;5:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Graham RL, Sparks JS, Eckerle LD, Sims AC, Denison MR. SARS coronavirus replicase proteins in pathogenesis. Virus Res. 2008;133:88–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Saikatendu KS, Joseph JS, Subramanian V, et al. Structural basis of severe acute respiratory syndrome coronavirus ADP‐ribose‐1″‐phosphate dephosphorylation by a conserved domain of nsP3. Structure. 2005;13:1665–1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ma Y, Wu L, Shaw N, et al. Structural basis and functional analysis of the SARS coronavirus nsp14‐nsp10 complex. Proc Natl Acad Sci U S A. 2015;112:9436–9441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Biasini M, Bienert S, Waterhouse A, et al. SWISS‐MODEL: Modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014;42:W252–W258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kirchdoerfer RN, Ward AB. Structure of the SARS‐CoV nsp12 polymerase bound to nsp7 and nsp8 co‐factors. Nat Commun. 2019;10:2342–2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Holm L, Laakso LM. Dali server update. Nucleic Acids Res. 2016;44:W351–W355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Takeshita D, Tomita K. Molecular basis for RNA polymerization by Q beta replicase. NatStructMolBiol. 2012;19:229–237. [DOI] [PubMed] [Google Scholar]
- 12. Kottur J, Nair DT. Pyrophosphate hydrolysis is an intrinsic and critical step of the DNA synthesis reaction. Nucleic Acids Research. 2018;46(12):5875–5885. 10.1093/nar/gky402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bowers KJ, Chow E, Xu H, Dror RO, Eastwood MP, et al. Scalable algorithms for molecular dynamics simulations on commodity clusters. Proceedings of the 2006 ACM/IEEE conference on supercomputing, SC'06. New York, NY: ACM Press, 2006; p. 84. [Google Scholar]
- 14. Halgren T. New method for fast and accurate binding‐site identification and analysis. Chem Biolog Drug Design. 2007;69:146–148. [DOI] [PubMed] [Google Scholar]
- 15. Selleckchem.com ‐ Inhibitor expert (inhibitors, compound libraries). Houston, Texas: Selleckchem. https://www.selleckchem.com/. [Google Scholar]
- 16. Gao Y, Yan L, Huang Y, et al. Structure of the RNA‐dependent RNA polymerase from COVID‐19 virus. Science (New York, NY). 2020;368:779–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Winn MD, Ballard CC, Cowtan KD, et al. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr. 2011;67:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Emsley P, Cowtan K. Coot: Model‐building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. [DOI] [PubMed] [Google Scholar]
- 19. Yin W, Mao C, Luan X, et al. Structural basis for inhibition of the RNA‐dependent RNA polymerase from SARS‐CoV‐2 by remdesivir. Science. 2020;368:1499–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Norrby SR. Side effects of Cephalosporins. Drugs. 1987;34:105–120. [DOI] [PubMed] [Google Scholar]
- 21. Carlberg, H. , Alestig, K. , Nord CE, Trollfors B. Intestinal side effects of cefoperazone." J Antimicrob Chemother and 1982; 10: 483‐7. academic.oup.com. [DOI] [PubMed] [Google Scholar]
- 22. Ozen IO, Moralioǧlu S, Karabulut R, Bagbanci B, Turkyilmaz Z, et al. Cefoperazone induced gastro‐intestinal haemorrhage. A case report. Acta Chir Belg. 2008;108:777–778. [DOI] [PubMed] [Google Scholar]
- 23. Strom BL, Schinnar R, Gibson GA, Brennan PJ, Berlin JA. Risk of bleeding and hypoprothrombinaemia associated with NMTT side chain antibiotics: Using cefoperazone as a test case. Pharmacoepidemiol Drug Saf. 1999;8:81–94. [DOI] [PubMed] [Google Scholar]
- 24. Koutmos M, Gherasim C, Smith JL, Banerjee R. Structural basis of multifunctionality in a vitamin B12‐processing enzyme. JBiolChem. 2011;286:29780–29787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Friesner RA, Banks JL, Murphy RB, et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem. 2004;47:1739–1749. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Model of the functional ternary complex of the SCV2‐nsp12 enzyme in the complex. The RNA substrate and incoming GTP are shown in stick representation and colored according to element. The N‐terminal extension region and the palm, fingers, and thumb domains are shown in green, cyan, yellow, and orange, respectively. The catalytic residues Asp618, Asp760, and Asp761 are colored according to element and shown in stick representation and the cofactor ions are shown as blue spheres.
Figure S2 Superimposition of the Cryo‐EM structure of SCV2‐nsp12:RNA and the model of the ternary complex. The backbone of the Cryo‐EM structure is colored green and that of the model is colored cyan. The RNA is shown in the stick representation. The incorporated remdesivir is present in the same location as the incoming nucleotide triphosphate.