

INTRODUCTION
A hallmark of mantle cell lymphoma (MCL) is aberrant cyclin D1 expression in tumor cells due to a t(11;14) (q13;q32) chromosomal translocation (Fig. 1A, Table 1),1 which places the CCND1 genes (encoding cyclin D1) under the control of the immunoglobulin heavy chain enhancer.2–5 Constitutive cyclin D1 expression in MCL cells is aberrant because normal mature human B cells express only cyclin D2 or cyclin D3 but no cyclin D1.6 It accelerates the assembly of an active cyclin D–cyclin-dependent kinase 4 (CDK4) complex that drives cell cycle progression through early G1 by phosphorylating Rb and subsequently releasing E2Fs from phosphorylated Rb (CDK6 is barely expressed in MCL cells). In turn, E2Fs transcriptionally regulate genes that promote cell cycle progression through S (CCNA2, PCNA, and TK1), G2–M (CDK1), and cell division as well as a multitude of other genes that modulate cellular functions, such as EZH27 (see Fig. 1A). To ensure that the cell cycle progresses as programmed, the CDK4/6 activity is subject to negative control by 4 physiologic inhibitors (p16INK4a, p15INK4b, p18INK4c, and p19INK4d) (see Fig. 1A).
Fig. 1.
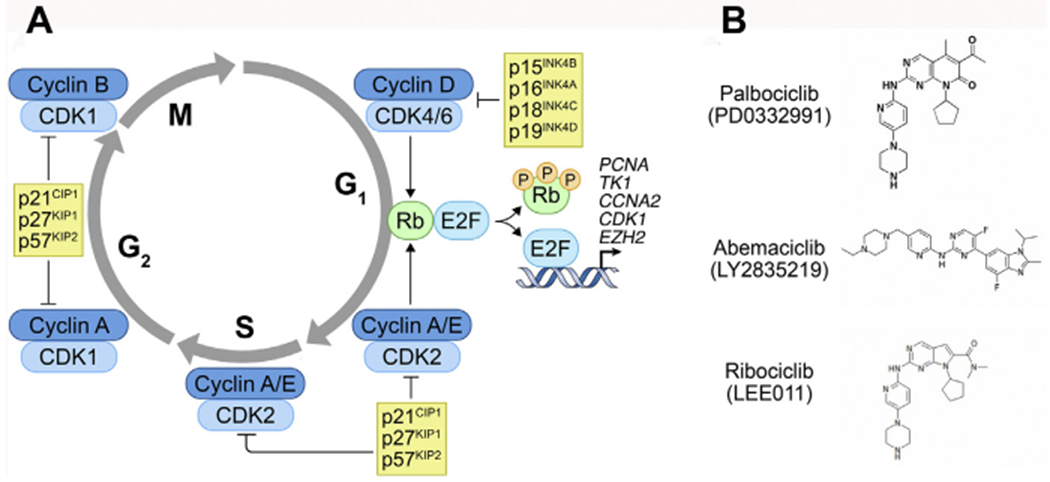
(A) A schema of the mammalian cell cycle. When released from Rb after it is phosphorylated by cyclin D1–CDK4, E2F1 directly activates the transcription of CCNA2 (encoding cyclin A), TK1 (encoding thymidine kinase), PCNA (encoding proliferating cell nuclear antigen), CDK1 (encoding cyclin-dependent kinase I), and EZH2 (encoding EZH2), among other target genes. (B) Three oral small molecule reversible CDK4/6 inhibitors.
Table 1.
Genomic alterations in cell cycle genes in primary mantle cell lymphoma cells
| Gene (Protein) | Locus | Genomic Alterations | Method | References |
|---|---|---|---|---|
| CCND1 | 11q13.3 | t(11;14) (q13;q32) translocation, causing cyclin D1 overexpression | Southern blot, PCR, Sanger sequencing | Williams et al,1 1993 |
| Deletions in 3′-UTR in 7 of15 cases, creating stable truncated mRNAs | qPCR | Wiestner et al,11 2007 | ||
| Point mutations in proximal 3′-UTR in 3 of 15 cases, creating stable truncated mRNAs via premature polyadenylation | Cycle sequencing | Wiestner et al,11 2007 | ||
| Nonsynonymous SNVs in 17 of 90 samples in 5′-UTR or exon 1 | Sanger sequencing | Kridel et al,16 2012 | ||
| Mutations in 10 of 29 cases, most in exon 1, and more common in SOX11− and/or IGHV-mutated MCL | WES | Bea et al,17 2013 | ||
| Mutations in 19 of 102 cases (26 nonsynonymous, all in exon 1) | Targeted sequencing | Meissner et al,15 2013 | ||
| Missense mutations in 9 of 56 cases | WES | Zhang et al,12 2014 | ||
| Mutations in 15 of 176 cases | NGS, Sanger sequencing | Eskelund et al,14 2017 | ||
| Missense mutations in 3 of 16 cases | WES | Yang et al,13 2018 | ||
| Mutations in 2 of 24 cases, all in nonresponders to ibrutinib-venetoclax therapy | WES, WGS, targeted sequencing | Agarwal etal,18 2019 | ||
| CCND2 | 12p13.32 | Rearrangements of Ig light chain enhancer regions in 43 of 56 CCND1− cases, associated with overexpression | FISH | Martin-Garcia et al,19 2019 |
| CCND3 | 6p21.1 | Rearrangements of Ig light chain enhancer regions in 9/56 CCND1− cases, associated with overexpression | WGS, WES, Sanger sequencing , FISH | Martin-Garcia et al,19 2019 |
| CDK4 | 12q14.1 | 12q gains in 9 of 45 cases, associated with CDK4 amplification in 5 of 6 such cases | CGH | Bea et al,21 1999 |
| Copy number gains in 4 of 69 cases, all in highly proliferative blastoid MCL | qPCR | Hernandez et al,20 2005 | ||
| Monoallelic deletions in 2 of 129 cases, gain in 8 cases | qPCR, MLPA | Delfau-Larue et al,25 2015 | ||
| CDKN2A (p16INK4a) | 9p21.3 | Deletions in 15 of 37 cases (9 hemizygous, 6 homozygous) | FISH | Dreyling et al,8 1997 |
| Deletions in 3 of 24 cases (2 homozygous), all associated with aggressive MCL | Southern blot | Pinyol et al,23 1997 | ||
| A148T mutation in 1 of 21 cases | PCR-SSCP | Pinyol et al,23 1997 | ||
| Deletions in 18 of 85 cases, more common in proliferative MCLs | qPCR | Rosenwald et al,9 2003 | ||
| Deletions in 3 of 15 cases | Microarray | Fernandez et al,24 2010 | ||
| Recurrent homozygous deletions, but no mutations | WES | Bea et al,17 2013 | ||
| Deletions in 34 of 134 cases (19 monoallelic, 15 biallelic), associated with poor prognosis | qPCR, MLPA | Delfau-Larue et al,25 2015 | ||
| Deletions in 35 of 176 cases, associated with poor prognosis | ddPCR | Eskelund et al,14 2017 | ||
| Deletions in 15 of 68 cases, associated with shorter OS | Sanger sequencing, WES, SNP microarray | Clot et al,26 2018 | ||
| Missense mutations in 1 of 16 cases | WES | Yang et al,13 2018 | ||
| Deletions in 20 of 42 CCND1− cases (11 homozygous) | Array CGH, CNV assay | Martin-Garcia et al,19 2019 | ||
| RB1 | 13q14.2 | Hemizygous deletions in 15 of 37 cases (9 also with CDKN2A deletion) | FISH | Dreyling et al,8 1997 |
| 13q14 homozygous deletions in 12 of 32 cases | CGH | Pinyol et al,27 2007 | ||
| 13q14 deletion in 1 of 1 case on ibrutinib relapse | WES | Chiron et al,22 2014 | ||
| Mutations in 6 of 56 cases (3 frameshift, 2 missense, 1 nonsense) | WES | Zhang et al,12 2014 | ||
| Deletions in 34 of 131 cases (33 monoallelic, 1 biallelic), gain in 1 case, associated with shorter OS | qPCR, MLPA | Delfau-Larue et al,25 2015 | ||
| CDK2 | 12q13.2 | Monoallelic deletion in 1 of 116 cases, gain in 8 cases | qPCR, MLPA | Delfau-Larue et al,25 2015 |
| CDKN1A (p21Waf1) | 6p21.2 | S31R mutation in 1 of 23 cases | PCR-SSCP | Pinyol et al,23 1997 |
| CDKN1B (p27Kip1) | 12p13.1 | Monoallelic deletions in 13 of 109 cases, gain in 3 cases, associated with shorter OS | qPCR, MLPA | Delfau-Larue et al,25 2015 |
| AURKA | 20q13.2 | Homozygous P31I polymorphisms in 3 of 58 cases | PCR-RFLP | Camacho et al,28 2006 |
| TTK (Mps1) | 6q14.1 | Hemizygous deletions in 6 of 26 cases | qPCR | Camacho et al,28 2006 |
This table lists only reported CNVs, mutations, translocations, and rearrangements of key cell cycle genes in MCL cells; cell cycle genes without reported alterations are not listed.
Abbreviations: Array CGH, array comparative genomic hybridization; copy number variation analysis, quantitative polymerase chain reaction Tagman assay; Cycle sequencing, double-strand direct sequencing; ddPCR, droplet-digital PCR; Ig, immunoglobulin; MLPA, multiplex ligation-dependent probe amplification; NGS, next-generation sequencing; OS, overall survival; PCR-RFLP, restriction fragment length polymorphism polymerase chain reaction; PCR-SSCP, single-strand conformation polymorphism polymerase chain reaction; SNP, single-nucleotide polymorphism; WGS, whole-genome sequencing.
Following the discovery of CCND1 translocation, Dreyling and colleagues8 provided the first evidence that the Rb-p16INK4a axis plays an important role in MCL biology. Fluorescence in situ hybridization (FISH) demonstrated that CDKN2A (encoding p16INK4a) and RB1 (encoding Rb) frequently were deleted (41%) in primary MCL cells and that deletion of CDKN2A, but not RB1, correlated with proliferation as determined by Ki67 expression (see Table 1). Based on gene expression profiling, it was further suggested that deletions of the INK4a/ARF locus determine the tumor proliferation rate and survival in synergy with the abundance of cyclin D1 mRNA.9 Ki67 then was shown to be a prognostic indicator for the patients treated with immunochemotherapy in MCL.10 Collectively, these findings provide a strong rationale to control proliferation of MCL cells by inhibiting CDK4/6, especially in progression disease. Clinical trials targeting CDK4/6 in combination therapy in recurrent MCL have shown promise, but more needs to be done to fully understand the underpinnings and identify resistant biomarkers. To advance precision medicine–based cell cycle therapy, this article reviews genomic aberrations and functions of key cell cycle genes in MCL cells as well as clinical trials of CDK4/6 inhibitors for MCL. Integrative longitudinal functional genomics is discussed as a strategy to discover genomic drivers for resistance in tumor cells and tumor-immune interactions that potentially contribute to the clinical response to palbociclib combination therapy for MCL.
GENOMIC ABERRATIONS IN KEY CELL CYCLE GENES IN MANTLE CELL LYMPHOMA CELLS
G0-G1
The primary cause for aberrant cyclin D1 expression in MCL is the signature t(11;14) (q13;q32) chromosomal translocation1 (Fig. 1, see Table 1). Amplification and mutations of CCND1, however, also contribute to cyclin D1 overexpression associated with poor prognosis. For example, truncating deletions and point mutations in the 3′-UTR region increase the stability of CCND1 mRNAs.11 Numerous other studies have identified mutations of CCND1,11–14 often missense, particularly in exon 1.15–17 In a recent clinical trial of ibrutinib in combination with venetoclax, CCND1 mutations were seen exclusively in nonresponders.18 In cyclin D1–negative MCL cells, rearrangements of immunoglobulin light chain genes with CCND2 or CCND3 led to their overexpression.19
Although cyclin D1 alone has no enzymatic activity, its overexpression promotes early G1 progression by accelerating the assembly of the active cyclin D1–CDK4 complexes in MCL cells This is further fueled by amplifications of CDK4,20,21 which has been associated with tumorigenesis and worsened prognosis in some cases. No deleterious mutations in CDK4 have been reported so far. Nor have there been reports of genomic alterations in CDK6, which is marginally expressed in primary MCL cells.22
Conversely, CDKN2A encoding p16INK4a frequently is deleted in MCL cells, especially in aggressive diseases with poorer outcomes.8,9,13,14,17,19,23–26 Although missense mutations have been detected in rare cases,13,23 hemizygous or homozygous deletions are the dominant genomic alterations in CDKN2A. This results in impaired G1 cell cycle control. Similarly, deletions, but not mutations, are the key alterations in RB1,8,12,22,25,27 resulting in unbridled transcriptional activation of E2Fs and downstream genes to reprogram both the cell cycle and cellular function.
G1-S
Further along the cell cycle, a mutation in CDKN1A encoding p21Waf1, which inhibits various cyclin-CDK complexes in particular CDK2, but not CDK4 or CDK6, has been identified.27 Copy number gains in CDK2 and hemizygous deletions in CDKN1B also were shown to be associated with shorter overall survival.25 CDKN1B encodes p27Kip1, an inhibitor of CDK2–cyclin E(A) that promotes G1 to S transition as well as CDK1–cyclin A(B) for progression through G2 (see Fig. 1A).
G2-M
Homozygous polymorphisms in AURKA (encoding Aurora-A) and hemizygous deletions in TTK (encoding hMPS1) important for the spindle checkpoint and centrosome regulation have been identified,28 although their role in genetic predispositions to lymphoma remains to be clarified.
Overall, the multitude of genomic alterations identified across cell cycle components can individually and collectively contribute to cell cycle dysregulation in MCL cells. Clustering of amplifications (CDK4) and deletions (CDKN2A, RB1) in genes that regulate G1 cell cycle progression and the association of these copy number variations (CNVs) with poorer outcomes further support targeting CDK4 in MCL.
GENOMIC ABERRATIONS IN ATM, TP53, MYC, AND BTK IN MANTLE CELL LYMPHOMA
ATM and TP53 are among the most frequently mutated and deleted genes in MCL cells. ATM, encoding a serine/threonine kinase that activates the DNA damage checkpoint in response to double-strand breaks, is mutated and deleted at a high frequency in MCL cells.15,17,18,29,30 Although its prognostic value has not been consistently significant, inactivating alterations due to truncation or missense mutations involving its PI3K domain have been shown to correlate with increased chromosomal instability.31 TP53 encodes the well-defined tumor suppressor protein p53 that commonly is disrupted in human cancer. Likewise, mutations and deletions of TP53 in MCL are frequent, many of which have been associated with poor prognosis and shorter survival.15,17,18,22,25,29 Moreover, amplifications and deletions in MDM2 encoding a p53-interacting protein and negative regulator have been reported,25,32 and alteration of MDM2 along with CDK4 occurred mainly in highly proliferative MCL with wild-type INK4a/ARF locus.20 A mutation in GTSE1, a p53-binding protein and G2/M checkpoint regulator, also was identified.22
The proto-oncogene MYC is a prominent driver for lymphomagenesis. Burkitt-type 8q24 MYC translocation is frequent in MCL with blastoid features, and amplifications of MYC also contributed to elevated c-Myc expression, increased proliferation, and poor outcomes.33–35
BTK is central to B-cell development and MCL survival. A C481S BTK mutation was identified in MCL cells of patients who progressed after a durable response to the BTK inhibitor ibrutinib but not in those with primary resistance or transient responses to ibrutinib.22 This BTK mutation apparently led to enhanced activation of both BTK and PI3K/AKT in vivo.22 Although infrequent in MCL, BTKC481S was detected in a separate ibrutinib-relapsed patient.36 Finally, PIK3CA was shown to be amplified in MCL cells,37 leading to overactivation of the PI3K/AKT/mTOR pathway that attenuates apoptosis and augments proliferation. These genomic alterations are likely to compound the aberrations in genes that directly control the cell cycle in MCL for cell cycle dysregulation and poorer outcomes.
TARGETING CDK4/6 IN MANTLE CELL LYMPHOMA
Three oral small molecule reversible CDK4/6 inhibitors have been approved by the Food and Drug Administration (FDA) (see Fig. 1B) for treatment of breast cancer, in which cyclin D1 and CDK4 frequently are overexpressed. Palbociclib (PD 0332991), the first selective CDK4/6 inhibitor,38 is highly specific for cyclin D–CDK4 based on a KINOMEScan against 468 serine-threonine kinases, including lipid kinases (Di Liberto M, Huang X, Chen-Kiang S. Targeting CDK4/6, unpublished data, 2020) whereas abemaciclib (LY2835214) appeared significantly less selective.39 The specificity of ribociclib (LEE011)40 is not yet available.
Palbociclib was shown to inhibit CDK4/6 and induce early G1 cell cycle arrest in primary human myeloma cells ex vivo41 and suppress tumor growth in xenografts of various human cancer cell lines in severe combined immunodeficiency mice41–44 and in immunocompetent mouse models of multiple myeloma45 and T-cell acute leukemia.46 Mechanistically, induction of prolonged early G1 arrest (pG1) by sustained CDK4/6 inhibition not only arrested the cell cycle but also restricted the expression of genes programmed for early G1 only.47 This caused an imbalance in gene expression that reprogrammed cancer cells for killing by diverse clinically relevant agents,41,45,47 including inhibitors of PI3K and BTK in primary MCL cells ex vivo22,48 and in animal models. Collectively' these preclinical studies provide compelling evidence for targeting CDK4/6 in human cancer.
PALBOCICLIB
In the first disease-specific single-agent clinical trial, palbociclib not only inhibited CDK4/6 and induced early G1 arrest initially in all patients with previously treated MCL but also elicited clinical responses with tumor regression in some patients, as indicated in preclinical studies49 (Table 2). In this multicenter phase Ib study of 17 patients, an objective response was observed in 3 patients (18%) including 1 complete response (CR) and 2 partial responses (PRs), in addition to 7 patients with stable disease (SD). Although the median progression-free survival (PFS) was 4 months, responding patients experienced a duration of response (DOR) of 18 months or greater. Dual immunohistochemical staining and 18F-fluorothymidine positron emission tomography imaging further confirmed that palbociclib inhibited CDK4/6 and induced G1 arrest in MCL cells.
Table 2.
Summary of CDK 4/6 inhibitor clinical trials in mantle cell lymphoma
| Agent (s) | Specificity | Design | No. of Patients | Results | References |
|---|---|---|---|---|---|
| Palbociclib | CDK4, CDK6 | Phase Ib, multicenter | 17 | ORR 18% (CR 6%) PFS 4 mo | Leonard et al,49 2012 |
| Palbociclib + bortezomib | CDK4, CDK6 | Phase I, single-center | 19 | ORR 24% (CR 6%) SD 30% |
Martin et al,50 2019 |
| Palbociclib + ibrutinib | CDK4, CDK6 | Phase I, multicenter | 27 | ORR 67% (CR 37%) 2-y PFS 59% 2-y OS 61% |
Martin et al,51 2019 |
| Palbociclib + ibrutinib | CDK4, CDK6 | Phase II, multicenter | 61 (estimated) | Pending | NCT03478514 |
| Abemaciclib | CDK4, CDK6 | Phase II, multicenter | 22 | ORR 23% (CR 0%) | Morschhauser et al,55 2014 |
| Ribociclib | CDK4, CDK6 | Phase I, multicenter | 7 MCL (132 total) | ORR 0% SD 0% |
Infante et al,56 2016 |
| AT7519M | CDK1, CDK2 CDK4, CDK5 CDK9 |
Phase II, multicenter | 12 | ORR 27% (PR 18%) mDOR 4.5 mo | Seftel et al,57 2017 |
| Voruciclib | CDK1, CDK4, CDK6, CDK9 | Phase Ib, multicenter | 84 (estimated, including MCL) | Pending | NCT03547115 |
Abbreviations: mDOR, median DOR; OS, overall survival.
Tumor regression in responding patients, particularly in 1 patient with a CR for more than 30 months, was in line with a mechanistic link between palbociclib-induced prolonged early G1 arrest and cell death observed in preclinical studies.47 As the first disease-specific clinical trial of a selective CDK4/6 inhibitor, it laid the groundwork for future studies of CDK4/6 in MCL as well as cancers of solid tissue origin, such as breast cancer.
PALBOCICLIB IN SEQUENTIAL COMBINATION WITH BORTEZOMIB
Leveraging the selectivity of palbociclib, a phase I dose escalation study of palbociclib in sequential combination with bortezomib in patients with previously treated MCL was designed to capitalize on bortezomib killing of MCL cells during palbociclib-induced pG1 as well as synchronous transition of G1-arrested cells into S phase after cessation of palbociclib.50 Bortezomib was administered at a reduced dose (1 mg/m2) during dose escalation of palbociclib with only 4 days of overlap (days 8–11), which potentially minimizes the combined toxicity. Of the 7 patients treated at the optimal dose combination, 4 remained progression-free for greater than 12 months, including 1 patient with a CR that has lasted for more than 7 years while remaining on single-agent palbociclib (see Table 2).
This single-center phase I study demonstrated that selective inhibition of CDK4/6 in sequential combination with reduced-dose bortezomib is biologically active and tolerable in previously treated MCL. Only 1 patient progressed while receiving treatment and this patient subsequently achieved a PR in response to ibrutinib,51 suggesting that the mechanisms mediating clinical responses to palbociclib and ibrutinib are distinct. The maintenance of a durable CR by palbociclib alone after 6 cycles of palbociclib + bortezomib further invokes palbociclib as a potential maintenance treatment after achieving a CR to palbociclib-based combination therapy.
PALBOCICLIB IN COMBINATION WITH IBRUTINIB
Ibrutinib is a standard of care for MCL.52 Approximately half of all patients progressed on treatment during the first year, and this often is associated with a more aggressive disease.53 Induction of pG1 by CDK4/6 inhibition had been shown to overcome ibrutinib resistance in primary human MCL cells expressing the wild-type BTK,22 in part by inactivating NF-κB as well as PI3K through up-regulation of a negative regulator, PIK3IP1, in palbociclib-induced pG1.22,48
On this basis, a multiple institutional clinical trial was undertaken to test if inhibition of CDK4/6 could deepen and prolong the clinical response to ibrutinib while assessing the tolerability. This phase I study of palbociclib + ibrutinib in 27 patients with previously treated MCL demonstrated a CR rate of 37% and median PFS of 25.6 months,51 which appear better than might be expected based on studies of single-agent ibrutinib, despite a comparable objective response rate (ORR) of 67%.54 Among 7 patients with a Ki67 higher than 30%, 5 responded, including 3 patients with a CR. Among 7 patients with a high MIPI, 4 responded, including 1 with a CR.50 The combination had an acceptable safety profile, with a dose-limiting toxicity of grade 3 rash that resulted in discontinuation in 2 patients taking the highest does of palbociclib (125 mg) (see Table 2).
The observed activity was consistent with the hypothesis that induction of pG1 by CDK4/6 inhibition could deepen and prolong the clinical response to ibrutinib, including MCL patients with a high Ki67 and MIPI. A multicenter, phase II study to further characterize the efficacy of this combination regimen, along with longitudinal functional genomics to identify genomic drivers for resistance to palbociclib and ibrutinib is under way (see Table 2).
OTHER CDK4/6 INHIBITORS
Abemaciclib, a broad-spectrum oral CDK4/6 inhibitor, is under study in a single-arm phase II trial for patients with previously treated MCL. Preliminary results in 22 patients treated with abemaciclib showed that 5 patients achieved PR (23%) and 9 patients had SD.55 Despite comparable clinical activity, the adverse effects (diarrhea, nausea, and vomiting) differ considerably from those of palbociclib, potentially due to inhibition of off target serine-threonine kinases besides CDK4 and CDK6. How these features of abemaciclib might contribute to the clinical outcome, tolerability, and durability in combination therapy remain to be determined.
Ribociclib is the third oral FDA-approved CDK4/6 inhibitor for treatment of metastatic breast cancer. A total of 7 patients with MCL were enrolled in a large phase I trial involving patients with a variety of previously treated solid tumors and lymphomas. As expected, myelosuppression was the primary dose-limiting toxicity. There were no responses among the patients with MCL.56
AT7519M is an inhibitor of CDKs 1, 2, 4, 5, and 9. In a phase II single-agent study for patients with previously treated chronic lymphocytic leukemia and MCL, the ORR in 12 MCL patients was 27%, with 2 patients achieving PR (18%) with a DOR of 4.5 months; 6 patients (55%) had SD, including 1 patient who subsequently met PR criteria 9 months after discontinuation of AT7519M with no other therapy. AT7519M had a favorable toxicity profile, with only 2 grade 3 nonhematologic adverse events.57
Voruciclib, a potent inhibitor of CDK9, CDK4, CDK6, and CDK1, is being studied in a phase Ib trial in patients with previously treated B-cell malignancies, including an MCL cohort.58 CDK9 is not a classic cell cycle regulator. It is a transcription factor regulating MCL1 (an antiapoptotic member of the BCL2 family) and MYC, among other targets. Voruciclib potentially may block cell proliferation by inhibiting CDK4, CDK6, and CDK1 and impair cell survival by down-regulating MCL1. Its therapeutic window depends on how inhibiting multiple CDKs can be translated into a clinical response and the tolerability and durability of this broad-spectrum CDK inhibitor.
DISCUSSION
The cell cycle is directional and exquisitely controlled by the balance between positive and negative regulators (see Fig. 1A). Cyclin D1 overexpression in MCL cells alone is insufficient to subvert the cell cycle program, because cyclin D has no enzymatic activity. It predisposes MCL cells, however, to aggressive proliferation and poorer outcome by accelerating the formation of an active cyclin D–CDK4 complex that drives G1 progression. This is exacerbated further by loss of the CDK4/6 inhibitor p16IN4a due to deletions of CDKN2a or increased CDK4 as a consequence of gene amplification, each associated with worsened outcome8,21 (see Table 1). Genomic analyses using various methods, including unbiased whole-exome sequencing (WES), have validated these early studies and established CNV in genes directly regulating the cell cycle (see Table 1) as the driver for cell cycle dysregulation in MCL.
The advent of selective CDK4/6 inhibitors has made it possible to target the cell cycle in mechanism-based clinical trials. Data emerging from 3 completed clinical trials of palbociclib are consistent with the hypothesis that induction of prolonged early G1 arrest by CDK4/6 inhibition not only prevents proliferation of MCL cells but also reprograms them for a deeper and more durable clinical response to the partner drug, including patients with high Ki67 and high MIPI. Durable CR was observed in 1 patient treated with palbociclib for 30 months49; in 1 patient for more than 7 years while on treatment with palbociclib alone after 6 cycles of palbociclib + bortezomib50; and in 10 patients (37%) in the palbociclib + ibrutinib clinical trial,51 for 3 to 5.5 years while on therapy as of 4/2020 (Di Liberto M, Huang X, Chen-Kiang S. Targeting CDK4/6, unpublished data). Rb, the substrate of CDK4/6, is necessary but insufficient for a clinical response to palbociclib therapy. Although preliminary, these findings reinforce the potential of targeting CDK4/6, and the critical importance of defining the mechanisms that discriminate sensitivity from resistance to targeting CDK4/6 in MCL.
Longitudinal integrated analysis of whole-transcriptome sequencing (WTS) and WES of MCL cells isolated from sequential specimens from individual patients before, during, and after treatment represents the best approach to address this question. Such a longitudinal integrated analysis in a single-agent ibrutinib therapy has led to the discovery of a relapse-specific C418S BTK mutation in MCL.22 The ongoing longitudinal functional genomics of palbociclib combination therapies (Di Liberto M, Huang X, Chen-Kiang S. Targeting CDK4/6, unpublished) should shed light on the genomic drivers for resistance to targeting CDK4/6 and BTK. They also could illuminate genes and signaling pathways that are programmed in G1 arrest to maintain a durable clinical response and advance the selection and sequencing of a partner drug(s) with a CDK4/6 inhibitor in combination therapy.
Inhibition of CDK4/6 is not limited to disease or cell lineage, suggesting that the immune landscape and tumor-immune interactions are likely to be dynamically regulated by CDK4/6 inhibition and contribute to the clinical response. Consistent with this possibility, CDK4/6 inhibition has been shown to promote cytotoxic T-cell–mediated clearance of tumor cells in patient-derived mouse model of breast cancer59; increase the infiltration of CD4+ and CD8+ T cells as well as the levels of TH1 cytokines in a mouse model of lung cancer60; and repress a tumor resistance program associated with T-cell exclusion and immune evasion as determined by single-cell RNA sequencing in melanoma tumors.61 Collectively, these studies illustrate the tumor-extrinsic mechanisms by which CDK4/6 inhibitors may enhance antitumor immunity in solid tumors. It will be important to see if these promising preclinical data are recapitulated in lymphoma and translate into better efficacy in patients.
Integrating longitudinal single-cell RNA-sequencing with WTS and WES of purified MCL cells from the phase II palbociclib-ibrutinib clinical trial present an ideal strategy to investigate tumor-immune interactions in the context of a clinical response and shed light on the therapeutic potential of dual CDK4/6 and immune checkpoint inhibition in MCL.
KEY POINTS.
Deletions and amplifications of genes controlling cell cycle progression through early G1 are frequent in mantle cell lymphoma (MCL) cells.
Targeting CDK4/6 with palbociclib appears to deepen and prolong the clinical response to ibrutinib in MCL.
CDK4/6 inhibition may modulate tumor-immune interaction in MCL.
Longitudinal functional genomics of patient specimens represents the best approach to discover resistant biomarkers for CDK4/6 inhibitor therapy.
ACKNOWLEDGMENTS
The authors thank Nicole Zhao for a critical reading of this article and helpful suggestions. This study was supported in part by a Translational Research Grants from V Foundation, United States, (S. Chen-Kiang, M. Di Liberto), NIH/NCI RO1CA18894, United States, (S. Chen-Kiang), MCL-RI Award (MCL7001–18) from The Leukemia & Lymphoma Society, United States, to S. Chen-Kiang. Funding for this project has been provided by the Sarah Cannon Fund at the HCA Foundation (S. Chen-Kiang, X. Huang, M. Di Liberto), and NIH/NCI P01CA21427401, United States, (S. Chen-Kiang, X. Huang, M. Di Liberto).
Footnotes
CONFLICTS OF INTEREST
The authors have no conflicts of interest to declare.
REFERENCES
- 1.Williams ME, Swerdlow SH, Meeker TC. Chromosome t(11;14)(q13;q32) breakpoints in centrocytic lymphoma are highly localized at the bcl-1 major translocation cluster. Leukemia 1993;7:1437–40. [PubMed] [Google Scholar]
- 2.Bosch F, Jares P, Campo E, et al. PRAD-1/cyclin D1 gene overexpression in chronic lymphoproliferative disorders: a highly specific marker of mantle cell lymphoma. Blood 1994;84:2726–32. [PubMed] [Google Scholar]
- 3.de Boer CJ, van Krieken JH, Kluin-Nelemans HC, et al. Cyclin D1 messenger RNA overexpression as a marker for mantle cell lymphoma. Oncogene 1995; 10:1833–40. [PubMed] [Google Scholar]
- 4.Ott MM, Helbing A, Ott G, et al. bcl-1 rearrangement and cyclin D1 protein expression in mantle cell lymphoma. J Pathol 1996;179:238–42. [DOI] [PubMed] [Google Scholar]
- 5.Rosenberg CL, Wong E, Petty EM, et al. PRAD1, a candidate BCL1 oncogene: mapping and expression in centrocytic lymphoma. Proc Natl Acad Sci U S A 1991;88:9638–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bretz J, Garcia J, Huang X, et al. Noxa mediates p18INK4c cell-cycle control of homeostasis in B cells and plasma cell precursors. Blood 2011;117:2179–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kent LN, Leone G. The broken cycle: E2F dysfunction in cancer. Nat Rev Cancer 2019;19:326–38. [DOI] [PubMed] [Google Scholar]
- 8.Dreyling MH, Bullinger L, Ott G, et al. Alterations of the cyclin D1/p16-pRB pathway in mantle cell lymphoma. Cancer Res 1997;57:4608–14. [PubMed] [Google Scholar]
- 9.Rosenwald A, Wright G, Wiestner A, et al. The proliferation gene expression signature is a quantitative integrator of oncogenic events that predicts survival in mantle cell lymphoma. Cancer Cell 2003;3:185–97. [DOI] [PubMed] [Google Scholar]
- 10.Determann O, Hoster E, Ott G, et al. Ki-67 predicts outcome in advanced-stage mantle cell lymphoma patients treated with anti-CD20 immunochemotherapy: results from randomized trials of the European MCL Network and the German Low Grade Lymphoma Study Group. Blood 2008;111:2385–7. [DOI] [PubMed] [Google Scholar]
- 11.Wiestner A, Tehrani M, Chiorazzi M, et al. Point mutations and genomic deletions in CCND1 create stable truncated cyclin D1 mRNAs that are associated with increased proliferation rate and shorter survival. Blood 2007;109:4599–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang J, Jima D, Moffitt AB, et al. The genomic landscape of mantle cell lymphoma is related to the epigenetically determined chromatin state of normal B cells. Blood 2014;123:2988–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang P, Zhang W, Wang J, et al. Genomic landscape and prognostic analysis of mantle cell lymphoma. Cancer Gene Ther 2018;25:129–40. [DOI] [PubMed] [Google Scholar]
- 14.Eskelund CW, Dahl C, Hansen JW, et al. TP53 mutations identify younger mantle cell lymphoma patients who do not benefit from intensive chemoimmunotherapy. Blood 2017;130:1903–10. [DOI] [PubMed] [Google Scholar]
- 15.Meissner B, Kridel R, Lim RS, et al. The E3 ubiquitin ligase UBR5 is recurrently mutated in mantle cell lymphoma. Blood 2013;121:3161–4. [DOI] [PubMed] [Google Scholar]
- 16.Kridel R, Meissner B, Rogic S, et al. Whole transcriptome sequencing reveals recurrent NOTCH1 mutations in mantle cell lymphoma. Blood 2012;119:1963–71. [DOI] [PubMed] [Google Scholar]
- 17.Bea S, Valdes-Mas R, Navarro A, et al. Landscape of somatic mutations and clonal evolution in mantle cell lymphoma. Proc Natl Acad Sci U S A 2013;110: 18250–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Agarwal R, Chan YC, Tam CS, et al. Dynamic molecular monitoring reveals that SWI-SNF mutations mediate resistance to ibrutinib plus venetoclax in mantle cell lymphoma. Nat Med 2019;25:119–29. [DOI] [PubMed] [Google Scholar]
- 19.Martin-Garcia D, Navarro A, Valdes-Mas R, et al. CCND2 and CCND3 hijack immunoglobulin light-chain enhancers in cyclin D1(-) mantle cell lymphoma. Blood 2019;133:940–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hernandez L, Bea S, Pinyol M, et al. CDK4 and MDM2 gene alterations mainly occur in highly proliferative and aggressive mantle cell lymphomas with wild-type INK4a/ARF locus. Cancer Res 2005;65:2199–206. [DOI] [PubMed] [Google Scholar]
- 21.Bea S, Ribas M, Hernandez JM, et al. Increased number of chromosomal imbalances and high-level DNA amplifications in mantle cell lymphoma are associated with blastoid variants. Blood 1999;93:4365–74. [PubMed] [Google Scholar]
- 22.Chiron D, Di Liberto M, Martin P, et al. Cell-cycle reprogramming for PI3K inhibition overrides a relapse-specific C481S BTK mutation revealed by longitudinal functional genomics in mantle cell lymphoma. Cancer Discov 2014;4:1022–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pinyol M, Hernandez L, Cazorla M, et al. Deletions and loss of expression of p16INK4a and p21Waf1 genes are associated with aggressive variants of mantle cell lymphomas. Blood 1997;89:272–80. [PubMed] [Google Scholar]
- 24.Fernandez V, Salamero O, Espinet B, et al. Genomic and gene expression profiling defines indolent forms of mantle cell lymphoma. Cancer Res 2010;70:1408–18. [DOI] [PubMed] [Google Scholar]
- 25.Delfau-Larue MH, Klapper W, Berger F, et al. High-dose cytarabine does not overcome the adverse prognostic value of CDKN2A and TP53 deletions in mantle cell lymphoma. Blood 2015;126:604–11. [DOI] [PubMed] [Google Scholar]
- 26.Clot G, Jares P, Gine E, et al. A gene signature that distinguishes conventional and leukemic nonnodal mantle cell lymphoma helps predict outcome. Blood 2018;132:413–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pinyol M, Bea S, Pla L, et al. Inactivation of RB1 in mantle-cell lymphoma detected by nonsense-mediated mRNA decay pathway inhibition and microarray analysis. Blood 2007;109:5422–9. [DOI] [PubMed] [Google Scholar]
- 28.Camacho E, Bea S, Salaverria I, et al. Analysis of Aurora-A and hMPS1 mitotic kinases in mantle cell lymphoma. Int J Cancer 2006;118:357–63. [DOI] [PubMed] [Google Scholar]
- 29.Greiner TC, Dasgupta C, Ho VV, et al. Mutation and genomic deletion status of ataxia telangiectasia mutated (ATM) and p53 confer specific gene expression profiles in mantle cell lymphoma. Proc Natl Acad Sci U S A 2006;103:2352–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schaffner C, Idler I, Stilgenbauer S, et al. Mantle cell lymphoma is characterized by inactivation of the ATM gene. Proc Natl Acad Sci U S A 2000;97:2773–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Camacho E, Hernandez L, Hernandez S, et al. ATM gene inactivation in mantle cell lymphoma mainly occurs by truncating mutations and missense mutations involving the phosphatidylinositol-3 kinase domain and is associated with increasing numbers of chromosomal imbalances. Blood 2002;99:238–44. [DOI] [PubMed] [Google Scholar]
- 32.Hartmann E, Fernandez V, Stoecklein H, et al. Increased MDM2 expression is associated with inferior survival in mantle-cell lymphoma, but not related to the MDM2 SNP309. Haematologica 2007;92:574–5. [DOI] [PubMed] [Google Scholar]
- 33.Setoodeh R, Schwartz S, Papenhausen P, et al. Double-hit mantle cell lymphoma with MYC gene rearrangement or amplification: a report of four cases and review of the literature. Int J Clin Exp Pathol 2013;6:155–67. [PMC free article] [PubMed] [Google Scholar]
- 34.Felten CL, Stephenson CF, Ortiz RO, et al. Burkitt transformation of mantle cell lymphoma. Leuk Lymphoma 2004;45:2143–7. [DOI] [PubMed] [Google Scholar]
- 35.Au WY, Horsman DE, Viswanatha DS, et al. 8q24 translocations in blastic transformation of mantle cell lymphoma. Haematologica 2000;85:1225–7. [PubMed] [Google Scholar]
- 36.Jain P, Kanagal-Shamanna R, Zhang S, et al. Long-term outcomes and mutation profiling of patients with mantle cell lymphoma (MCL) who discontinued ibrutinib. Br J Haematol 2018;183:578–87. [DOI] [PubMed] [Google Scholar]
- 37.Psyrri A, Papageorgiou S, Liakata E, et al. Phosphatidylinositol 3’-kinase catalytic subunit alpha gene amplification contributes to the pathogenesis of mantle cell lymphoma. Clin Cancer Res 2009;15:5724–32. [DOI] [PubMed] [Google Scholar]
- 38.Fry DW, Harvey PJ, Keller PR, et al. Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol Cancer Ther 2004;3:1427–38. [PubMed] [Google Scholar]
- 39.Gelbert LM, Cai S, Lin X, et al. Preclinical characterization of the CDK4/6 inhibitor LY2835219: in-vivo cell cycle-dependent/independent anti-tumor activities alone/in combination with gemcitabine. Invest New Drugs 2014;32:825–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rader J, Russell MR, Hart LS, et al. Dual CDK4/CDK6 inhibition induces cell-cycle arrest and senescence in neuroblastoma. Clin Cancer Res 2013;19:6173–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Baughn LB, Di Liberto M, Wu K, et al. A novel orally active small molecule potently induces G1 arrest in primary myeloma cells and prevents tumor growth by specific inhibition of cyclin-dependent kinase 4/6. Cancer Res 2006;66:7661–7. [DOI] [PubMed] [Google Scholar]
- 42.Marzec M, Kasprzycka M, Lai R, et al. Mantle cell lymphoma cells express predominantly cyclin D1a isoform and are highly sensitive to selective inhibition of CDK4 kinase activity. Blood 2006;108:1744–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang L, Wang J, Blaser BW, et al. Pharmacologic inhibition of CDK4/6: mechanistic evidence for selective activity or acquired resistance in acute myeloid leukemia. Blood 2007;110:2075–83. [DOI] [PubMed] [Google Scholar]
- 44.Finn RS, Dering J, Conklin D, et al. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast Cancer Res 2009;11:R77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Menu E, Garcia J, Huang X, et al. A novel therapeutic combination using PD 0332991 and bortezomib: study in the 5T33MM myeloma model. Cancer Res 2008;68:5519–23. [DOI] [PubMed] [Google Scholar]
- 46.Sawai CM, Freund J, Oh P, et al. Therapeutic targeting of the cyclin D3:CDK4/6 complex in T cell leukemia. Cancer Cell 2012;22:452–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Huang X, Di Liberto M, Jayabalan D, et al. Prolonged early G(1) arrest by selective CDK4/CDK6 inhibition sensitizes myeloma cells to cytotoxic killing through cell cycle-coupled loss of IRF4. Blood 2012;120:1095–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chiron D, Martin P, Di Liberto M, et al. Induction of prolonged early G1 arrest by CDK4/CDK6 inhibition reprograms lymphoma cells for durable PI3Kdelta inhibition through PIK3IP1. Cell Cycle 2013;12:1892–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Leonard JP, LaCasce AS, Smith MR, et al. Selective CDK4/6 inhibition with tumor responses by PD0332991 in patients with mantle cell lymphoma. Blood 2012;119: 4597–607. [DOI] [PubMed] [Google Scholar]
- 50.Martin P, Ruan J, Furman R, et al. A phase I trial of palbociclib plus bortezomib in previously treated mantle cell lymphoma. Leuk Lymphoma 2019. 10.1080/10428194.2019.1612062:1-5. [DOI] [PubMed] [Google Scholar]
- 51.Martin P, Bartlett NL, Blum KA, et al. A phase 1 trial of ibrutinib plus palbociclib in previously treated mantle cell lymphoma. Blood 2019;133:1201–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang ML, Rule S, Martin P, et al. Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma. N Engl J Med 2013;369:507–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Martin P, Maddocks K, Leonard JP, et al. Postibrutinib outcomes in patients with mantle cell lymphoma. Blood 2016;127:1559–63. [DOI] [PubMed] [Google Scholar]
- 54.Rule S, Dreyling M, Goy A, et al. Outcomes in 370 patients with mantle cell lymphoma treated with ibrutinib: a pooled analysis from three open-label studies. Br J Haematol 2017;179:430–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Morschhauser F, Bouabdallah K, Stilgenbauer S, et al. Clinical Activity of Abemaciclib (LY2835219), a Cell Cycle Inhibitor Selective for CDK4 and CDK6, in Patients with Relapsed or Refractory Mantle Cell Lymphoma. Blood 2014;124:3067. [Google Scholar]
- 56.Infante JR, Cassier PA, Gerecitano JF, et al. A phase I study of the cyclin-dependent kinase 4/6 inhibitor ribociclib (LEE011) in patients with advanced solid tumors and lymphomas. Clin Cancer Res 2016;22:5696–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Seftel MD, Kuruvilla J, Kouroukis T, et al. The CDK inhibitor AT7519M in patients with relapsed or refractory chronic lymphocytic leukemia (CLL) and mantle cell lymphoma. A Phase II study of the Canadian Cancer Trials Group. Leuk Lymphoma 2017;58:1358–65. [DOI] [PubMed] [Google Scholar]
- 58.Dey J, Deckwerth TL, Kerwin WS, et al. Voruciclib, a clinical stage oral CDK9 inhibitor, represses MCL-1 and sensitizes high-risk Diffuse Large B-cell Lymphoma to BCL2 inhibition. Sci Rep 2017;7:18007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Goel S, DeCristo MJ, Watt AC, et al. CDK4/6 inhibition triggers anti-tumour immunity. Nature 2017;548:471–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Deng J, Wang ES, Jenkins RW, et al. CDK4/6 inhibition augments antitumor immunity by enhancing T-cell activation. Cancer Discov 2018;8:216–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jerby-Arnon L, Shah P, Cuoco MS, et al. A cancer cell program promotes T cell exclusion and resistance to checkpoint blockade. Cell 2018;175:984–97.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]