

Abstract
Exosomes are nanosized vesicles secreted by nearly all types of cells and play important roles in intercellular communication. Given their unique and important pharmacological properties, exosomes have been emerging as a new class of cell-free therapeutics. Herein, we describe exosomes developed against epidermal growth factor receptor (EGFR), a key factor in epithelial malignancies, involved in enhanced tumor growth, invasion, and metastasis. The exosomes are genetically modified for displaying two distinct types of monoclonal antibodies on the exosome surface, resulting in novel synthetic multivalent antibodies retargeted exosomes (SMART-Exos) that can simultaneously target tumor-associated human EGFR and T-cell surface CD3 receptor. By redirecting and activating T cells toward attacking EGFR-expressing cancer cells, the designed SMART-Exos exhibit highly potent and specific antitumor activity. In this chapter, the methodologies are outlined for generating and using SMART-Exos for cancer immunotherapy.
Keywords: Exosome, Immunotherapy, Genetic engineering, Monoclonal antibody, EGFR, CD3, Cancer, T cell, Isolation, Flow cytometry, Immunoblot, Confocal microscopy, Cytotoxicity
1. Introduction
Exosomes are nanoscale membranous vehicles secreted by nearly all types of cells and are found in a wide range of tissues and body fluids [1, 2]. By carrying various forms of membrane proteins and soluble cargos including proteins, lipids, and nucleic acids, exosomes function as important mediators for intercellular communication through modulating signaling pathways in target cells and/ or transferring membrane receptors and cargos to recipient cells [3, 4]. Compared with conventional nanoparticles, naturally occurring exosomes are expected to possess excellent biocompatibility, high efficiency for therapeutic delivery, and low immunogenicity [5]. Notably, exosomes are characterized by abundant membrane proteins, critical for their biological functions and therapeutic applications. For instance, tetraspanin CD9 highly expressed on the exosome surface can promote direct membrane fusion with target cells, which facilitate cytosolic delivery of therapeutics [6]. By triggering a “don’t eat me” signal, transmembrane protein CD47 is shown to protect exosomes from clearance by circulating monocytes and macrophages [7]. Given their strong therapeutic potential, exosomes have been emerging as a promising form of nanomedicine. A considerable number of preclinical and clinical studies indicate that exosome-based therapeutics display excellent efficacy for treatment of a variety of human diseases, including cancer, neurodegenerative diseases, liver diseases, ischemic diseases, and immune disorders [8-13].
Currently, different methods are established for loading exosomes with various types of endogenous and exogenous agents for therapeutic delivery [5, 14, 15]. Herein, we develop a new approach for reprogramming exosomes to modulate cellular immunity in a controlled and directed manner. Through genetically displaying two distinct types of monoclonal antibodies on the exosome surface using the transmembrane (TM) domain of human platelet-derived growth factor receptor (PDGFR), a novel class of exosomes is generated, namely synthetic multivalent antibodies retargeted exosomes (SMART-Exos) [16]. By targeting T-cell surface CD3 and cancer cell-associated epidermal growth factor receptor (EGFR), the resulting SMART-Exos not only show tight binding to both EGFR-expressing triple-negative breast cancer (TNBC) cells and T cells but also induce potent and specific killing of EGFR-positive TNBC cells in the presence of nonactivated human peripheral blood mononuclear cells (hPBMCs) (see Note 1). In this chapter, the procedures are elaborated for generation and characterization of SMART-Exo for cancer immunotherapy. It is likely that these methods can be easily adopted for other disease models by utilizing different types of functional antibodies.
2. Materials
2.1. Plasmids
Mammalian expression vector pDisplay.
Anti-EGFR antibody-PDGFR TM domain fusion plasmid: hemagglutinin (HA)-αEGFR-PDGFR TM domain.
Anti-CD3 antibody-PDGFR TM domain fusion plasmid: HA-αCD3-PDGFR TM domain.
Anti-EGFR/CD3 antibody-PDGFR TM domain fusion plasmid: HA-αEGFR-αCD3-PDGFR TM domain.
Anti-CD3/EGFR antibody-PDGFR TM domain fusion plasmid: HA-αCD3-αEGFR-PDGFR TM domain.
ZymoPURE II Plasmid Maxiprep Kit.
2.2. Cell Culturing and Transfection
Expi293F cell line.
Expi293 expression medium.
Opti-Minimal Essential Medium (Opti-MEM) I reduced serum medium.
ExpiFectamine 293 transfection reagent.
125-mL polycarbonate Erlenmeyer flasks.
2.3. Exosome Isolation
Dulbecco’s phosphate-buffered saline (DPBS) (1×).
Benchtop centrifugation instrument: Heraeus Megafuge 40R Refrigerated Centrifuge; rotor: TX-750 Swinging Bucket Rotor; tube: 50-mL centrifuge tubes.
Floor centrifugation instrument: J2–21 Floor Model Centrifuge (Beckman Coulter, Indianapolis, IN, USA); rotor: JA-17 Fixed-Angle Aluminum Rotor; tube: Nalgene Oak Ridge high-speed polypropylene copolymer centrifuge tubes.
Ultracentrifugation instrument: Optima L-80 XP Ultracentrifuge (Beckman Coulter); rotor: Type 70 Ti Fixed-Angle Titanium Rotor, tube: polycarbonate bottle with cap assembly.
Pierce Coomassie (Bradford) Protein Assay Kit.
Synergy H1 Hybrid Multi-Mode Microplate Reader.
2.4. Immunoblots
Phosphate-buffered saline (PBS): 8 g/L NaCl, 0.2 g/L KCl, 1.78 g/L Na2HPO4·2H2O and 0.27 g/L KH2PO4, pH 7.4, filtered with a 0.22-μm disc filter.
4–20% ExpressPlus-PAGE gels.
NuPAGE LDS Sample Buffer (4×).
Trans-Blot SD Semi-Dry Transfer Cell.
ExpressPlus-PAGE gel running buffer: Tris base 6.06 g/L, MOPS 10.46 g/L, SDS 1 g/L, EDTA 0.3 g/L, add deionized water to 1 L.
Immunoblot transfer buffer: 5.82 g/L Tris base, 2.93 g/L glycine, 200 mL/L methanol, add deionized water to 1 L.
Immun-Blot PVDF membrane.
Membrane wash buffer: PBS-T (PBS with 0.1% Tween-20).
Blocking solution: dissolve 5% BSA in PBS-T.
Primary antibodies: anti-HA antibody (clone: 2-2.2.14, Thermo Fisher Scientific), anti-CD63 antibody (clone: H5C6, BioLegend, San Diego, CA, USA), anti-CD81 antibody (clone: 1.3.3.22, Thermo Fisher Scientific), and anti-CD9 antibody (clone: D8O1A, Cell Signaling Technology).
Secondary antibodies: anti-mouse IgG-HRP and anti-rabbit IgG-HRP.
SuperSignal West Pico PLUS chemiluminescent substrate.
ChemiDoc Touch Imaging System.
2.5. Flow Cytometry Analysis
Jurkat cell line.
MDA-MB-468 cell line.
RPMI-1640 medium with 10% fetal bovine serum (FBS).
DPBS, 1× (Mediatech).
0.05% Trypsin-EDTA (1×).
LSR II Flow Cytometer.
Anti-HA antibody (clone: 2-2.2.14, Thermo Fisher Scientific).
Alexa Fluor 488-labeled goat anti-mouse IgG H&L antibody.
Sample wash buffer: DPBS with 2% FBS.
2.6. Confocal Microscopy of Cell–Cell Cross-Linking Mediated by SMART-Exos
Jurkat cell line.
MDA-MB-468 cell line.
RPMI-1640 medium with 10% FBS.
MitoSpy Red.
Carboxyfluorescein succinimidyl ester (CFSE).
Leica SP8 confocal laser scanning microscope.
24-well cell culture plates (sterile, flat bottom).
12-mm circular coverslips (glass).
Microslides (25 × 75 mm, glass).
Fluoromount.
2.7. Cytotoxicity Induced by SMART-Exos
Jurkat cell line.
MDA-MB-468 cell line.
MDA-MB-453 cell line.
RPMI-1640 medium with 10% FBS.
Human peripheral blood mononuclear cells (hPBMCs) from HemaCare.
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) solution: dissolve 5-mg MTT in 1-mL PBS. Sterilize by filtration with a 0.22-μm disc filter.
Lysis buffer (20% SDS in 50% dimethylformamide, pH 4.7).
Synergy H1 Hybrid Multi-Mode Microplate Reader (BioTek).
96-well plates (sterile, flat bottom).
3. Methods
3.1. Plasmid Construction and Preparation
Use the pDisplay vector for constructing SMART-Exos expression plasmids, which contains a human PDGFR TM domain for displaying proteins of interest on the mammalian cell surface. Synthetic genes encoding scFv fragments of αEGFR cetuximab and αCD3 UCHT1 antibodies could be purchased from Integrated DNA Technologies, Inc. (Coralville, IA), in which a (GGGGS)3 linker is inserted between VH and VL regions. To generate single polypeptide encoding dual scFv antibodies, perform overlap extension polymerase chain reactions (PCR) by placing a (GGGGS)3 flexible linker between two individual scFv fragments. Arrange the orientations of variable regions for each scFv or dual scFv antibody as follows: VH-αEGFR-VL-αEGFR (αEGFR scFv), VL-αCD3-VH-αCD3 (αCD3 scFv), VH-αEGFR-VL-αEGFR-VL-αCD3-VH-αCD3 (αEGFR/αCD3 scFv), and VL-αCD3-VH-αCD3-VH-αEGFR-VL-αEGFR (αCD3/αEGFR scFv). Add a hemagglutinin (HA)-tag at the N terminus of the scFv or dual scFv antibody during PCR amplification. Insert the resulting DNA fragments between the N-terminal signal peptide and the TM domain of PDGFR using flanking restriction sites BglII and SalI (Fig. 1). Confirm the generated expression vectors by DNA sequencing.
Prepare transfection-grade plasmids for the sequence-verified expression constructs by using endotoxin-free buffers and ZymoPURE II Plasmid Maxiprep Kits (ZYMO Research).
Fig. 1.

Molecular designs of antibody-PDGFR TM domain fusions for the generation of SMART-Exos. Each fusion protein contains an N-terminal HA epitope tag and flexible (GGGGS)3 linkers
3.2. Transient Transfection
Perform transfection by following the guidelines of Expi293 expression system. Below is a brief outline of the method for transfection of 30 mL of Expi293F cell culture.
On the day prior to transfection (Day -1), determine cell density and viability (see Note 2). To transfect the cells on the following day, split the Expi293F cell culture to a final density of 2 × 106 viable cells/mL with fresh prewarmed Expi293 expression medium and incubate the cells on an orbital shaker (125 rpm/min) for 16–24 h in a 37 °C incubator with a humidified atmosphere of 8% CO2 in air.
On the next day (Day 0), determine cell density and viability (see Note 3) and resuspend cells in fresh prewarmed Expi293 expression medium. For each 30 mL of transfection, add appropriate volume of cell suspension with a total of 7.5 × 107 cells to a sterile 125-mL polycarbonate Erlenmeyer flask and bring up the volume to 25.5 mL by adding fresh prewarmed Expi293 expression medium. Return the cells to the CO2 incubator.
Prepare plasmid DNA/ExpiFectamine 293 complexes. Dilute 30-μg plasmid DNA (see Note 4) with 1.5-mL Opti-MEM I reduced serum medium. Mix gently. Dilute 80-μL ExpiFectamine 293 reagent with 1.5-mL Opti-MEM I reduced serum medium. Mix gently and allow to incubate at room temperature (RT) for 5 min (see Note 5). Add the diluted plasmid DNA to the diluted ExpiFectamine 293 reagent. Mix gently.
Incubate plasmid DNA/ExpiFectamine 293 complexes at RT for 20–30 min, and then slowly transfer the solution to the shaker flask from step 2. Swirl the flask gently during addition. Return the cells to the CO2 incubator.
On the first day after transfection (Day 1, 18–22 h posttransfection), add 150-μL ExpiFectamine 293 transfection enhancer 1 and 1.5-mL ExpiFectamine 293 transfection enhancer 2 to the shaker flask. Gently swirl the flask during addition. Return the cells to the CO2 incubator (see Note 6).
Day 3 and Day 6: Collect exosome-containing cell culture media via centrifugation (see Note 7).
3.3. Exosome Isolation
Purify the expressed SMART-Exos from the collected culture media through differential centrifugation.
Collect cell cultures containing expressed SMART-Exos.
Centrifuge at 100 × g for 10 min at 4 °C to remove residual cells (see Note 8).
Discard pellets and transfer supernatants to clean 50-mL centrifuge tubes and spin at 4000 × g for 30 min at 4 °C to remove dead cells and large debris.
Discard pellets and transfer supernatants to clean centrifuge tubes and spin at 14,000 × g for 50 min at 4 °C to remove small debris and larger vesicles.
Discard pellets and transfer supernatants to clean ultracentrifuge tubes and centrifuge at 371,000 × g (60,000 rpm) in a Type 70 Ti rotor for 2 h at 4 °C (see Note 9).
Discard the supernatants, carefully refill the tubes with PBS to wash the pellets. Repeat this step once.
Resuspend the pellet in PBS (50–200 μL) and transfer to 1.7-mL Eppendorf tubes.
Vortex for 20 min at RT and then centrifuge at 14,000 × g for 15 min at 4 °C to remove precipitation. Repeat the centrifugation step once.
Collect the supernatants and filter with 0.22-μm filters (see Note 10).
Measure protein concentrations using the Coomassie (Bradford) protein assay kits.
Store SMART-Exos at 4 °C or on ice prior to immediate use or at −80 °C for long-term storage (see Note 11).
3.4. Immunoblots
Reduce the isolated SMART-Exos (4 μg of protein in total) with 10-mM dithiothreitol in 1 × LDS sample buffer at 95 °C for 5 min (see Note 12).
Load the sample into each well (up to 40 μL per well for 4–20% ExpressPlus-PAGE gels).
Run the gel at 150 V for 50 min at RT.
Transfer samples to PVDF membranes at 15 V for 35 min at RT (see Note 13).
Wash the PVDF membrane briefly in wash buffer.
Block the PVDF membrane in PBS-T with 5% BSA for 1 h at RT.
Incubate with appropriate primary antibodies in PBS-T at RT.
Wash three times with PBS-T for a total of 30 min.
Incubate with appropriate HRP conjugated secondary antibodies for 1 h at RT.
Wash three times with PBS-T for a total of 30 min.
Visualize the immunoblots using the ChemiDoc Touch Imaging System upon the addition of SuperSignal West Pico PLUS chemiluminescent substrate (Fig. 2).
Fig. 2.
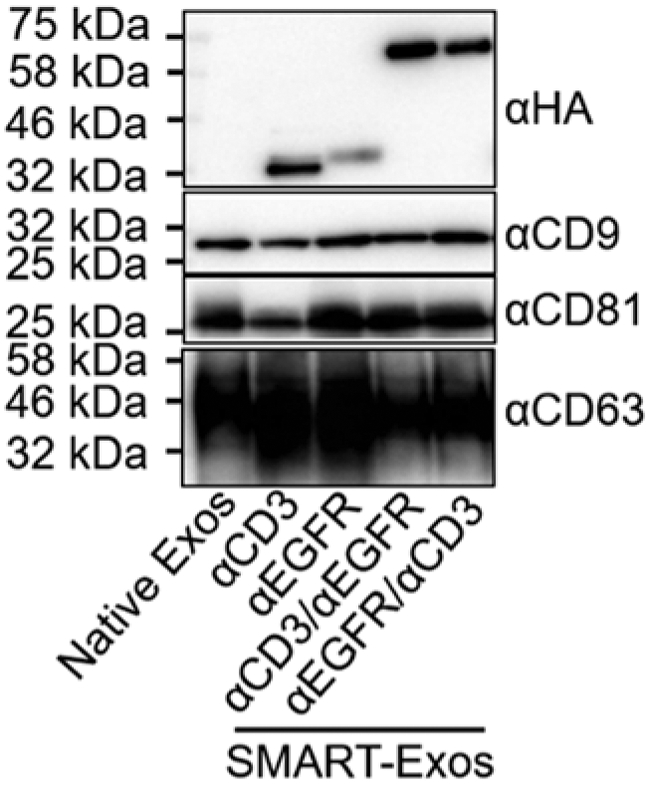
Immunoblot analysis of native exosomes and the generated SMART-Exos using antibodies specific for HA to detect expression of scFv antibodies and for CD9, CD81, and CD63 exosomal marker proteins. A total of 4 μg exosomes (by total protein concentration) for each sample was loaded for immunoblot analysis. (Adapted with permission from J. Am. Chem. Soc. 2018, 140, 48, 16,413–16,417. Copyright (2018) American Chemical Society)
3.5. Flow Cytometry
Harvest cells and wash twice in PBS with 2% FBS at 400 × g for 5 min at 4 °C.
Discard the supernatants and incubate the cells (0.2 million) with exosomes (0.1 mg/mL) in PBS with 2% FBS for 30 min at 4 °C (see Note 14).
Wash twice with PBS containing 2% FBS.
Discard the supernatants and incubate with the anti-HA antibody for 30 min at 4 °C.
Wash twice with PBS containing 2% FBS.
Discard the supernatants and incubate with the Alexa Fluor 488-labeled goat anti-mouse IgG H&L antibody for 30 min at 4 °C.
Wash twice with PBS containing 2% FBS.
Analyze samples using an LSR II Flow Cytometer (Fig. 3).
Fig. 3.

Flow cytometric analysis of the binding of the generated SMART-Exos to MDA-MB-468 cells (EGFR+ CD3−) and Jurkat cells (EGFR− CD3+). (Adapted with permission from J. Am. Chem. Soc. 2018, 140, 48, 16,413–16,417. Copyright (2018) American Chemical Society)
3.6. Confocal Microscopy of Cell–Cell Cross-Linking Mediated by SMART-Exos
Incubate MDA-MB-468 cells and Jurkat cells with 200 nM MitoSpy Red and 5 μM CFSE at 37 °C for 20 min, respectively (see Note 15).
Wash twice with PBS to remove free dyes (see Note 16).
Incubate Jurkat cells (6 × 104) with α-CD3/α-EGFR SMART-Exos (0.1 mg/mL) or a mixture (1:1) of αCD3 SMART-Exos and αEGFR SMART-Exos in 100 μL PBS for 30 min at 4 °C.
Wash Jurkat cells with 1 mL of cold PBS to remove free exosomes.
Mix Jurkat cells (6 × 104) and MDA-MB-468 cells (2 × 104) in 500 μL of RPMI-1640 medium with 10% FBS.
Add cell mixtures into clear bottoms of 24-well plates with 12-mm coverslips at the bottoms and incubate for 5 h at 37 °C (see Note 17).
Gently wash the cells four times with PBS to remove unattached Jurkat cells.
Carefully remove the coverslips from 24-well plates and mount onto slides with fluoromount, then image with a Leica SP8 confocal laser scanning microscope using FITC (for CFSE) and rhodamine (for MitroSpy Red) filters (Fig. 4).
Fig. 4.
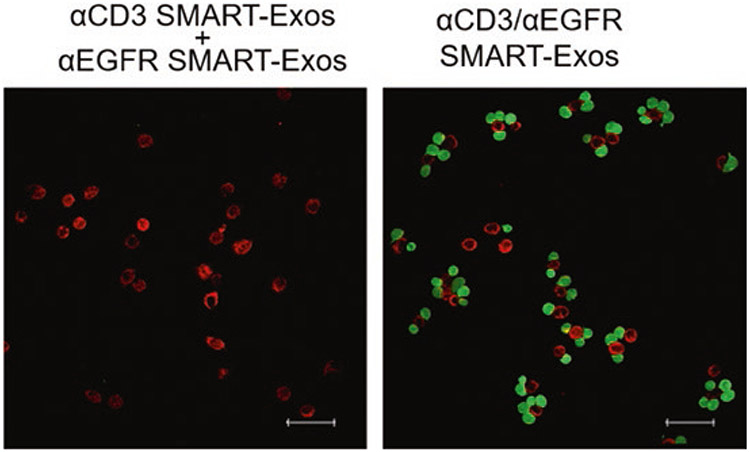
Confocal microscopic analysis of the cross-linking of Jurkat and MDA-MB-468 cells induced by SMART-Exos. Fluorescently labeled target cancer cells (red) and Jurkat cells (green) were mixed at a 1:3 ratio and incubated for 5 h in the presence of αCD3/αEGFR SMART-Exos or a mixture (1:1) of αCD3 SMART-Exos and αEGFR SMART-Exos. Nonbound cells were washed away by PBS. Scale bars: 50 μm. (Adapted with permission from J. Am. Chem. Soc. 2018, 140, 48, 16,413–16,417. Copyright (2018) American Chemical Society)
3.7. Cytotoxicity Induced by SMART-Exos
In this section, we will use a colorimetric MTT assay to measure viability.
Mix cancer cells (target cells) and nonactivated hPBMCs (effector cells) with a ratio of 1:10, then add the cell mixtures into the clear bottoms of 96-well plates (see Note 18).
Prepare SMART-Exos in DPBS with concentrations ranging from 100 μg/mL to 1 pg/mL with a ten-fold serial dilution.
Add 10 μL SMART-Exo dilution into corresponding wells and use 10 μL DPBS as a control.
Incubate the 96-well plates for 40 h at 37 °C.
Wash cells twice with PBS to remove hPBMC suspensions.
Add 10 μL MTT solution with 90 μL culture medium to each well and incubate for 4 h at 37 °C.
Add 100 μL of lysis buffer into each well and incubate for 3 h at 37 °C.
Measure absorbance at 570 nm using a BioTek Synergy H1 Hybrid Multi-Mode Microplate reader.
Calculate cytotoxicity as the percentage of cell viability = [(absorbanceexperimental – absorbancespontaneous average)/(absorbancemaximal viability average – absorbancespontaneous average)] × 100 (Fig. 5).
Fig. 5.

In vitro cytotoxicity of the αCD3/αEGFR SMART-Exos for MDA-MB-468 (EGFR+) or MDA-MB-453 (EGFR−) cells. Nonactivated hPBMCs (effector cells) were incubated with MDA-MB-468 (EGFR+) and MDA-MB-453 (EGFR−) cells (target cells) at an E:T ratio of 10 for 40 h in the presence of αCD3/αEGFR SMART-Exos or a mixture (1:1) of αCD3 SMART-Exos and αEGFR SMART-Exos. After removing hPBMCs suspensions, cell viabilities of target cells were measured through MTT assays. (Adapted with permission from J. Am. Chem. Soc. 2018, 140, 48, 16,413–16,417. Copyright (2018) American Chemical Society)
Acknowledgments
This work was supported, in part, by University of Southern California School of Pharmacy Start-Up Fund for New Faculty, University of Southern California Ming Hsieh Institute for Engineering Medicine for Cancer, American Association of Pharmaceutical Scientists (AAPS) Foundation New Investigator Grant (to Y.Z.), STOP CANCER Research Career Development Award (to Y.Z.), PhRMA Foundation Research Starter Grant in Translational Medicine and Therapeutics (to Y.Z.), P30CA014089 to the USC Norris Comprehensive Cancer Center, and P30DK048522 to the USC Research Center for Liver Diseases.
Footnotes
The αCD3/αEGFR SMART-Exos are expected to induce formation of immunological synapses between T cells and EGFR-expressing tumor cells, leading to the activation of possibly both CD4 and CD8 effector T cells to release cytolytic proteins and inflammatory cytokines for potent anticancer activity, which is independent of antigen presentation by MHC class I molecules or costimulation.
Determine cell number and viability using the trypan blue dye exclusion method. Cell viability should be >95% to proceed with transfection.
To start transfection, the final cell density should be at least 2.5 × 106 cells/mL with >95% viability.
To ensure sterility, DNA should be filtered through a 0.22-μm filter prior to use. A total plasmid DNA of 1.0 μg per mL of culture volume to be transfected is appropriate for most proteins.
Longer incubation time may result in decreased performance.
Enhancer 1 and enhancer 2 can be mixed together just prior to additions to the cell culture.
Cells or media (if recombinant protein is secreted) may be harvested beginning at approximately 48 h posttransfection and assayed for recombinant protein expression. Optimal time for harvesting SMART-Exos depends on the expression levels and stability of the fusion proteins on the exosome surface. For antibody protein fusions, 3–5 days are typical harvest times.
Keep all the centrifugation steps at 4 °C to retain the maximum biological activities of proteins expressed on the exosome surface.
It is recommended that the supernatants be centrifuged at 371,000 × g for 2 h to maximize the yields of exosomes. Ultracentrifugation at lower speeds (e.g., 100,000 × g) for 2 h or longer is also acceptable for this step.
For cell-based assays, the supernatants should be sterilized through filtration with 0.22-μm disc filters. In our experience, there is only a very small percentage of vesicles larger than 200 nm in diameter after the ultracentrifugation step.
Exosomes can be stored on ice or at 4 °C for up to 6 h or at −80 °C for up to 6 months.
Using LDS sample buffer alone allows successful extraction of tetraspanins. However, it could be difficult to extract certain transmembrane proteins from exosomes, which requires optimization of the sample buffer for more efficient extraction, such as using RIPA buffer. Reducing exosomes with DTT can help detect HA-tagged fusion protein and CD9 exosomal marker. But CD81 and CD63 require nonreducing electrophoresis for immunoblotting, possibly due to the disulfide bond-dependent epitopes for their respective antibodies.
The transfer time is dependent on the size of target protein.
Keep all steps at 4 °C to avoid the interference of exosome uptakes by cells. Native exosomes isolated from nontransfected Expi293F cells should also be included as controls.
For CFSE staining, resuspend cells at 10–100 × 106 cells/mL in the 5 μM CFSE working solution in PBS. For MitoSpy Red staining, prepare the working solution with incomplete culture medium and a concentration at 50–250 nM is recommended, if labeling mitochondria for live cell imaging.
Before the wash steps, quench the staining by adding five times of the original staining volume of cell culture medium with 10% FBS.
Incubation at 37 °C for 5 h allows adherent MDA-MB-468 cells to attach to the bottoms of 24-well plates.
Each well should contain 1 × 104 cancer cells and 1 × 105 nonactivated hPBMCs.
References
- 1.Thery C, Zitvogel L, Amigorena S (2002) Exosomes: composition, biogenesis and function. Nat Rev Immunol 2:569–579 [DOI] [PubMed] [Google Scholar]
- 2.Yanez-Mo M, Siljander PR, Andreu Z, Zavec AB, Borras FE, Buzas EI, Buzas K, Casal E, Cappello F, Carvalho J, Colas E, Cordeiro-da Silva A, Fais S, Falcon-Perez JM, Ghobrial IM, Giebel B, Gimona M, Graner M, Gursel I, Gursel M, Heegaard NH, Hendrix A, Kierulf P, Kokubun K, Kosanovic M, Kralj-Iglic V, Kramer-Albers EM, Laitinen S, Lasser C, Lener T, Ligeti E, Line A, Lipps G, Llorente A, Lotvall J, Mancek-Keber M, Marcilla A, Mittelbrunn M, Nazarenko I, Nolte-‘t Hoen EN, Nyman TA, O'Driscoll L, Olivan M, Oliveira C, Pallinger E, Del Portillo HA, Reventos J, Rigau M, Rohde E, Sammar M, Sanchez-Madrid F, Santarem N, Schallmoser K, Ostenfeld MS, Stoorvogel W, Stukelj R, Van der Grein SG, Vasconcelos MH, Wauben MH, de Wever O (2015) Biological properties of extracellular vesicles and their physiological functions. J extracell Ves 4:27066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Colombo M, Raposo G, Thery C (2014) Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu Rev Cell Dev Biol 30:255–289 [DOI] [PubMed] [Google Scholar]
- 4.Jiang XC, Gao JQ (2017) Exosomes as novel bio-carriers for gene and drug delivery. Int J Pharm 521:167–175 [DOI] [PubMed] [Google Scholar]
- 5.Armstrong JP, Holme MN, Stevens MM (2017) Re-engineering extracellular vesicles as smart nanoscale therapeutics. ACS Nano 11:69–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.van Dongen HM, Masoumi N, Witwer KW, Pegtel DM (2016) Extracellular vesicles exploit viral entry routes for cargo delivery. MMBR 80:369–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kamerkar S, LeBleu VS, Sugimoto H, Yang S, Ruivo CF, Melo SA, Lee JJ, Kalluri R (2017) Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature 546:498–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Johnsen KB, Gudbergsson JM, Skov MN, Pilgaard L, Moos T, Duroux M (2014) A comprehensive overview of exosomes as drug delivery vehicles – endogenous nanocarriers for targeted cancer therapy. Biochim Biophys Acta 1846:75–87 [DOI] [PubMed] [Google Scholar]
- 9.Cooper JM, Wiklander PB, Nordin JZ, Al-Shawi R, Wood MJ, Vithlani M, Schapira AH, Simons JP, El-Andaloussi S, Alvarez-Erviti L (2014) Systemic exosomal siRNA delivery reduced alpha-synuclein aggregates in brains of transgenic mice. Mov Disord 29:1476–1485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Oh K, Kim SR, Kim DK, Seo MW, Lee C, Lee HM, Oh JE, Choi EY, Lee DS, Gho YS, Park KS (2015) In vivo differentiation of therapeutic insulin-producing cells from bone marrow cells via extracellular vesicle-mimetic nanovesicles. ACS Nano 9:11718–11727 [DOI] [PubMed] [Google Scholar]
- 11.Fais S, O'Driscoll L, Borras FE, Buzas E, Camussi G, Cappello F, Carvalho J, Cordeiro da Silva A, Del Portillo H, El Andaloussi S, Ficko Trcek T, Furlan R, Hendrix A, Gursel I, Kralj-Iglic V, Kaeffer B, Kosanovic M, Lekka ME, Lipps G, Logozzi M, Marcilla A, Sammar M, Llorente A, Nazarenko I, Oliveira C, Pocsfalvi G, Rajendran L, Raposo G, Rohde E, Siljander P, van Niel G, Vasconcelos MH, Yanez-Mo M, Yliperttula ML, Zarovni N, Zavec AB, Giebel B (2016) Evidence-based clinical use of nanoscale extracellular vesicles in nanomedicine. ACS Nano 10:3886–3899 [DOI] [PubMed] [Google Scholar]
- 12.Gyorgy B, Hung ME, Breakefield XO, Leonard JN (2015) Therapeutic applications of extracellular vesicles: clinical promise and open questions. Annu Rev Pharmacol Toxicol 55:439–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lener T, Gimona M, Aigner L, Borger V, Buzas E, Camussi G, Chaput N, Chatterjee D, Court FA, Del Portillo HA, O'Driscoll L, Fais S, Falcon-Perez JM, Felderhoff-Mueser U, Fraile L, Gho YS, Gorgens A, Gupta RC, Hendrix A, Hermann DM, Hill AF, Hochberg F, Horn PA, de Kleijn D, Kordelas L, Kramer BW, Kramer-Albers EM, Laner-Plamberger S, Laitinen S, Leonardi T, Lorenowicz MJ, Lim SK, Lotvall J, Maguire CA, Marcilla A, Nazarenko I, Ochiya T, Patel T, Pedersen S, Pocsfalvi G, Pluchino S, Quesenberry P, Reischl IG, Rivera FJ, Sanzenbacher R, Schallmoser K, Slaper-Cortenbach I, Strunk D, Tonn T, Vader P, van Balkom BW, Wauben M, Andaloussi SE, Thery C, Rohde E, Giebel B (2015) Applying extracellular vesicles based therapeutics in clinical trials - an ISEV position paper. J Extracell Ves 4:30087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Luan X, Sansanaphongpricha K, Myers I, Chen H, Yuan H, Sun D (2017) Engineering exosomes as refined biological nanoplatforms for drug delivery. Acta Pharmacol Sin 38:754–763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bell BM, Kirk ID, Hiltbrunner S, Gabrielsson S, Bultema JJ (2016) Designer exosomes as next-generation cancer immunotherapy. Nanomedicine 12:163–169 [DOI] [PubMed] [Google Scholar]
- 16.Cheng Q, Shi X, Han M, Smbatyan G, Lenz HJ, Zhang Y (2018) Reprogramming exosomes as nanoscale controllers of cellular immunity. J Am Chem Soc 140:16413–16417 [DOI] [PMC free article] [PubMed] [Google Scholar]