

Significance Statement
Most forms of CKD present with well recognized mineral and bone disorders. It is unknown whether a different biochemical pattern of mineral abnormalities is associated with causes of CKD that manifest with persistent tubular phosphate wasting, such as the lysosomal storage disease nephropathic cystinosis. The authors demonstrate that patients with CKD caused by nephropathic cystinosis have mineral abnormalities that are distinct from those in CKD arising from other causes, including significantly lower levels of fibroblast growth factor-23 (FGF23) and percent tubular reabsorption of phosphate. These findings support the notion that phosphate is a significant driver of increased FGF23 levels in CKD and that mineral abnormalities associated with CKD are likely to vary depending on the underlying renal disease.
Keywords: chronic kidney disease/mineral and bone disorder, Fanconi syndrome, FGF23, nephropathic cystinosis
Visual Abstract

Abstract
Background
The rare lysosomal storage disease nephropathic cystinosis presents with renal Fanconi syndrome that evolves in time to CKD. Although biochemical abnormalities in common causes of CKD–mineral and bone disorder have been defined, it is unknown if persistent phosphate wasting in nephropathic cystinosis is associated with a biochemical mineral pattern distinct from that typically observed in CKD–mineral and bone disorder.
Methods
We assessed and compared determinants of mineral homeostasis in patients with nephropathic cystinosis across the predialysis CKD spectrum to these determinants in age- and CKD stage-matched patients, with causes of CKD other than nephropathic cystinosis.
Results
The study included 50 patients with nephropathic cystinosis-related CDK and 97 with CKD from other causes. All major aspects of mineral homeostasis were differentially effected in patients with CKD stemming from nephropathic cystinosis versus other causes. Patients with nephropathic cystinosis had significantly lower percent tubular reabsorption of phosphate and fibroblast growth factor-23 (FGF23) at all CKD stages, and lower blood phosphate in CKD stages 3–5. Linear regression analyses demonstrated lower FGF23 levels in nephropathic cystinosis participants at all CKD stages when corrected for eGFR and age, but not when adjusted for serum phosphate.
Conclusions
Nephropathic cystinosis CKD patients have mineral abnormalities that are distinct from those in CKD stemming from other causes. Persistently increased urinary phosphate excretion maintains serum phosphate levels within the normal range, thus protecting patients with nephropathic cystinosis from elevations of FGF23 during early CKD stages. These findings support the notion that phosphate is a significant driver of increased FGF23 levels in CKD and that mineral abnormalities associated with CKD are likely to vary depending on the underlying renal disease.
Cystinosis, a rare autosomal recessive lysosomal storage disease, is caused by biallelic loss-of-function mutations in the CTNS gene, which encodes the cysteine transporter protein (cystinosin). The defective function of this transporter leads to the formation and accumulation of cystine crystals in lysosomes,1 causing a multisystemic disease that effects almost all cells and tissues.1,2 Three clinical forms can be distinguished depending on the age of presentation, the severity, and the extent of the disease3,4: Nephropathic infantile form [Online Mendelian Inheritance in Man (OMIM) reference 219800], the most severe and frequent form that effects >90% of patients,5 nephropathic juvenile form (OMIM 219900), and non-nephropathic adult form (OMIM 219750).
Infantile nephropathic cystinosis (NC) is the most common cause of hereditary Fanconi syndrome.5 Deposition of intracellular cystine crystals within the proximal renal tubules causes tubular dysfunction that results in the urinary loss of water, sodium, potassium, bicarbonate, calcium, magnesium, phosphate, amino acids, glucose, and other small molecules.2,3 Patients develop severe dehydration and electrolyte imbalance including hypokalemia, hypocalcemia, metabolic acidosis, hypophosphatemia, and less often hyponatremia.1,5 While glomerular function is generally preserved at diagnosis, without early diagnosis and prompt initiation of appropriate treatment patients develop progressive loss of renal function, and progress to ESKD by approximately 10 years of age.6 Current treatment for NC is cysteamine, an aminothiol that reacts with cysteine to form cysteine-cysteamine complexes that can exit the lysosomes via a lysine transporter. This intervention reduces lysosomal cystine content2,7 and thereby delays various complications caused by abnormal cystine accumulation, including accelerated progression toward ESKD,6,8 and extrarenal manifestations such as distal myopathy, swallowing dysfunction, hypothyroidism, and growth hormone (GH)/GH-releasing hormone deficiency, and thus impaired linear growth.9,10 Supportive measures for Fanconi syndrome include maintenance of adequate fluid, electrolyte and acid-base balance, nutritional support, phosphate supplementation, and hormonal replacement therapy.1,5 However, cysteamine therapy initiated after the first year of life has no significant effect on the renal Fanconi syndrome and thus hypophosphatemia, because the renal tubules are already damaged by that age.1 Consequently, despite therapy, long-term skeletal abnormalities remain highly prevalent in this patient population.11
In non-NC CKD, disturbances of mineral and bone homeostasis (CKD–MBD) are readily detectable during the early disease stages and progress as glomerular function declines. The pathogenesis of CKD–MBD involves a complex cascade of maladaptive events driven by kidney function, which results in bone disease, extraskeletal calcification, and adverse cardiovascular outcomes. Particularly in young children, CKD–MBD can lead to bone abnormalities that including severe bone deformities secondary to increased parathyroid hormone (PTH)-dependent bone turnover, fractures, and delayed growth. It is interesting to note that skeletal abnormalities occur early during the course of CKD, when serum levels of calcium, phosphorus, PTH, and vitamin D are still within the normal range, but circulating fibroblast growth factor-23 (FGF23) levels are already elevated.12
Although abnormalities of mineral homeostasis associated with the most common causes of CKD have been well defined, the effect of persistent phosphate wasting, and the resultant delay in increasing serum phosphate and FGF23 levels, has not been evaluated in patients with NC treated according to current recommendations. Given that FGF23 has been recognized as an important factor in the pathogenesis of CKD–MBD, we predicted that prolonged maintenance of reduced or normal blood phosphate levels, as observed in patients with NC, will have an effect on FGF23 synthesis and secretion, and thus on 1,25-dihydroxyvitamin D (1,25D) and PTH levels. To test this hypothesis, we compared biochemical parameters of mineral ion homeostasis in patients with NC CKD and non-NC CKD with different stages of impaired renal function.
METHODS
Patients
Patients with infantile NC with an eGFR <90 ml/min per 1.73 m2 evaluated at the National Institutes of Health (NIH) Clinical Center between October 2016 and December 2017 under the protocol “Natural History Study of the Use of Cysteamine in the Treatment of Cystinosis” were included. In addition, patients for whom stored blood samples were available for FGF23 determination were also included. The protocol was approved by the National Human Genome Research Institute institutional review board; each adult or parent of a minor gave written informed consent to participate. Patients with nephropathic juvenile and non-nephropathic adult form with a previous renal transplant, and patients treated with dialysis, were excluded.
Patients with NC were matched by age and CKD stage, with patients with common causes of CKD evaluated at the Division of Pediatric Nephrology, University of California, Los Angeles. Every patient/parent(s) gave written consent to participate.
Biochemical Evaluation
Blood phosphate, calcium, albumin, creatinine, and total alkaline phosphatase were measured and interpreted according to the age- and gender-specific normal value ranges. Calcium levels were corrected for serum albumin concentration. Intact PTH concentration was measured by electrochemiluminescence immunoassay (Roche Cobas e601, normal range 16–65 pg/ml; Roche Diagnostics, Basel, Switzerland). 1,25D was measured by chemiluminescencent immunoassay (normal range 19–79 pg/ml; DiaSorin Liaison XL, Saluggia, Italy) in the NC group; in the non-NC group it was determined using liquid chromatography-tandem mass spectrometry (normal range boys/men <16 years: 24–86 pg/ml and ≥16 years: 18–64 pg/ml, girls/women <16 years: 24–86 pg/ml and ≥16 years: 18–78 pg/ml). Carboxy-terminal FGF23, which measures both biologically active/intact FGF23 and carboxy-terminal inactive FGF23 levels, were determined in plasma by immunometric enzyme assay [normal range <100 relative units (RU)/ml as previously reported13; Quidel, San Diego, CA]. Urine creatinine and phosphorous were measured in morning spot samples for percent tubular reabsorption of phosphate (TRP) determination. The eGFR was calculated using the Chronic Kidney Disease Epidemiology Collaboration formula in adults and the Schwartz equation in children.14,15
Statistical Analyses
Descriptive summary statistics were reported in each group of patients (NC versus non-NC) using median [interquartile range (IQR)] and mean (SD) for continuous variables and frequency (percent) for categorical variables after stratifying by CKD stage. Continuous variables were compared between the groups using the Wilcoxon rank sum test or the t test, as appropriate, whereas categorical variables were compared between the groups using the chi-squared/Fisher’s test, as appropriate. The correlations between continuous variables including log FGF23 versus serum phosphate were assessed using the Pearson correlation coefficient separately by group (NC versus non-NC) and the differences in the above correlation coefficients were evaluated using Fisher’s Z-transformations. Linearity was assessed using splines. We evaluated the relationship between FGF23 and diagnosis (NC versus non-NC) using linear regression models before and after adjusting for serum phosphate Z-scores after stratifying for CKD stage. All regression models were additionally adjusted for age and GFR levels as potential confounders. The variable for FGF23 was log transformed for the purpose of the analyses because the above variables on the original scale were skewed, but the log-transformed measures approximated the normal distribution. Statistical significance was defined by P<0.05 for all analyses. Analyses were performed using SAS 9.4 (Copyright © 2016 by SAS Institute Inc., Cary, NC).
RESULTS
Baseline Characteristics
A total of 147 patients were included, 50 with NC and 97 with common causes of CKD (non-NC group). Of the patients with NC, 29 individuals were boys or men (58%). Mean age was 14 years (range, 2–28 years). Disease-causing genetic variants were identified in all patients with NC; 14 (28%) had the most common homozygous 57-kb CTNS deletion, the remaining had either compound heterozygous deletions or other variants. Data about treatment used were collected for 45 out of 50 patients. All patients were receiving cysteamine, 28 (62%) were receiving phosphate supplementation, 31 (69%) calcitriol, and 17 (38%) calcium supplements.
Of the 97 patients in the non-NC group, 56 (58%) were boys or men, with a mean age of 13 years (range, 1–22 years). CKD in this group was due to obstructive or reflux nephropathy (n=44), glomerular disease (n=34), cystic disease (n=5), vascular disease (n=2), tubulointerstitial disease (n=4), or unknown cause (n=8). Thirty-three patients (34%) in this group were receiving calcitriol at the time of evaluation, 10 (10%) phosphate binder, and 13 (13%) calcium supplements.
NC and non-NC groups were matched by age and grouped by CKD stage (P value not significant). CKD stage distribution, age and eGFR of studied patients is shown in Table 1.
Table 1.
Age and glomerular filtration rate in patients with and without NC
| Characteristic | CKD Stage 2 | CKD Stage 3 | CKD Stage 4–5 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| NC | Non-NC | P | NC | Non-NC | P | NC | Non-NC | P | |
| n | 13 | 11 | 19 | 36 | 18 | 50 | |||
| Age (years) | 8.4 (6.6–12.8) | 12.2 (8.6–17.8) | NS | 13.0 (8.0–19.0) | 14.9 (12.5–18.0) | NS | 14.0 (12.2–19.0) | 13.8 (9.4–16.3) | NS |
| eGFR (ml/min per 1.73 m2) | 67.9 (62.7–84.3) | 71.3 (65.6–75.1) | NS | 46.0 (41.6–45.8) | 41.6 (36.1–45.8) | NS | 18.0 (14.6–23.1) | 19.0 (15.0–22.5) | NS |
Values are presented as median with IQR.
Biochemical Characteristics
Overall, in patients with NC, median (IQR) serum phosphorus concentration was 4.1 mg/dl (IQR, 2.8–4.9 mg/dl). Six (12%) patients with NC had age-specific hyperphosphatemia (CKD stages 3a to 5) and thirteen (26%) had hypophosphatemia. Median values of phosphate across CKD stages 2, 3, and 4–5 were 4.2 mg/dl (IQR, 2.7–4.7 mg/dl), 3.7 mg/dl (IQR, 2.3–4.3 mg/dl), and 4.1 (IQR, 3.6–5.5 mg/dl), respectively (Figure 1, Table 2). In comparison, non-NC patients had a median phosphate of 4.9 mg/dl (IQR, 4.2–5.6 mg/dl), significantly higher than the NC group (P<0.001), with median values of phosphate across CKD stages 2, 3, and 4–5 of 4.5 mg/dl (IQR, 3.5–4.9 mg/dl), 4.5 mg/dl (IQR, 3.9–5.3 mg/dl), and 5.2 (IQR, 4.5–6.0 mg/dl), respectively (Figure 1, Table 2). When comparing data among different CKD stages, serum phosphate was significantly lower in the NC group compared with non-NC in stages 3 and 4–5 (Figure 1, Table 2). Age-adjusted phosphate Z-scores were also significantly lower in NC stage 3 compared with non-NC. In addition, both in the NC and the non-NC group, serum phosphate had a significant negative correlation with eGFR (rho=−0.31, P<0.05 and rho=−0.45, P<0.05, respectively). The correlations of age-adjusted serum phosphate with eGFR were −0.42 (P<0.05) and −0.44 (P<0.05) for NC and non-NC groups, respectively.
Figure 1.
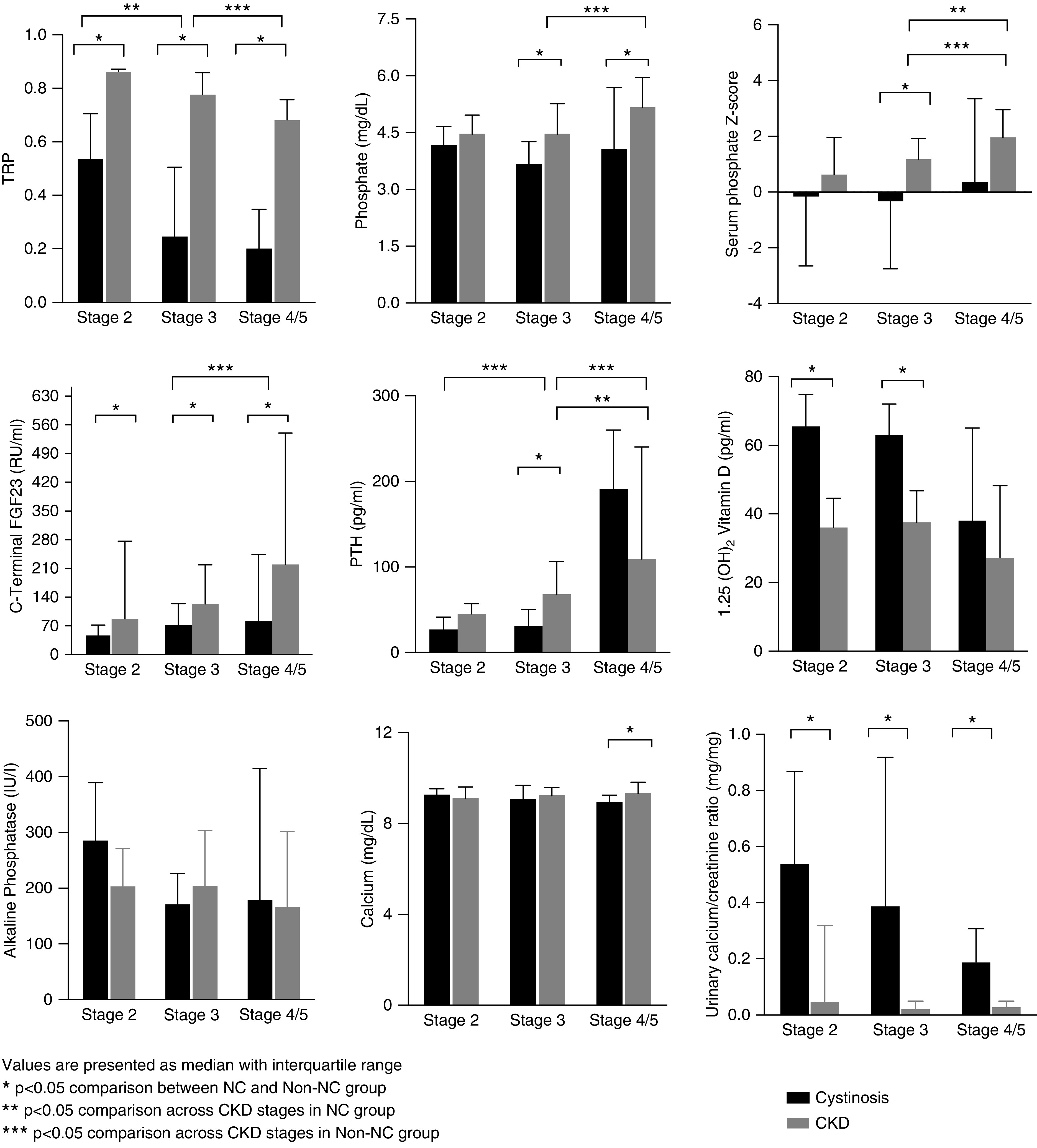
CKD-MBD biochemical parameters in patients with and without NC. Displayed are the biochemical paratmeters of patients with nephropathic cystinosis (black bars, Cystinosis), compared to those with non-cystinontic renal failure chronic kidney disease (gray bars, CKD). The groups are sorted by stage of renal failure, as indicated. Statistically significant differences between groups are indicated.
Table 2.
CKD-MBD biochemical parameters in patients with and without NC
| CKD Stage 2 | CKD Stage 3 | CKD Stage 4/5 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Parameter | Normal Range | NC | Non-NC | P | NC | Non-NC | P | NC | Non-NC | P |
| TRP | 0.85–0.95 | 0.5 (0.4–0.7) | 0.9 (0.8–0.9) | <0.05 | 0.3 (0.0–0.5) | 0.8 (0.8–0.9) | <0.05 | 0.2 (0.1–0.3) | 0.7 (0.6–0.8) | <0.05 |
| Phosphate (mg/dl) | a | 4.2 (2.7–4.7) | 4.5 (3.5–4.9) | NS | 3.7 (2.3–4.3) | 4.5 (3.9–5.3) | <0.05 | 4.1 (3.6–5.5) | 5.2 (4.5–6.0) | <0.05 |
| Phosphate score | −0.2 (−2.4–−0.8) | 0.7 (−0.9–1.8) | NS | −0.4 (−2.5–0.6) | 1.2 (−0.1–1.9) | <0.05 | 0.4 (−0.3–2.9) | 2.0 (0.8–3.0) | NS | |
| FGF23 (RU/ml) | <100 | 48.0 (31.0–103.0) | 87.0 (71.2–247.5) | <0.05 | 72.5 (35.0–123.0) | 123.5 (89.0–209.8) | <0.05 | 81.0 (38.2–187.0) | 210 (131.5–525.5) | <0.05 |
| PTH (pg/ml) | 15–65 | 26.9 (20.5–36.7) | 45 (23.0–54.5) | NS | 30.8 (21.9–48) | 68.0 (48.5–104.0) | <0.05 | 191.0 (111.8–256.1) | 109.5 (87.8–240.0) | NS |
| 1,25 (OH)D | b | 65.5 (56.0–74.2) | 36.0 (23.8–41.0) | <0.05 | 63 (54.5–69.0) | 37.6 (28.1–44.3) | <0.05 | 38.0 (28.0–56.0) | 26.4 (16.6–44.5) | NS |
| Calcium (mg/dl) | 8.6–10.2 | 9.2 (9.0–9.4) | 9.2 (9.1–9.5) | NS | 9.2 (8.8–9.5) | 9.3 (9.0–9.6) | NS | 8.8 (8.5–9.0) | 9.4 (8.9–9.9) | <0.05 |
| ALP (U/L) | c | 288 (135–392) | 206 (66–274) | NS | 1747 (97–229) | 207 (118–306) | NS | 181 (116–417) | 169 (128–304) | NS |
| ALP Z-score | 1.8 (−0.8–0.4) | 1.1 (−0.9–1.2) | NS | 0.2 (−0.2–1.9) | 1.7 (0.2–2.1) | NS | 2.1 (0.5–5.1) | 0.9 (−0.5–2.6) | <0.05 | |
NC and non-NC values are presented as median with IQR. ALP, alkaline phosphata.
Age-specific normal range (mg/dl) 1–3 years: 3.1–6.0; 4–12 years: 3.0–5.7; 13–17 years: 2.5–5.1; and >17 years: 2.5–4.5.
Normal range NC group: 19–79 and non-NC group: boys/men <16 years: 24–86 and ≥16 years: 18–64; girls/women <16 years: 24–86 and ≥16 years: 18–78.
Age-specific normal range (U/L), boys/men 1–3 years: 104–305; 4–6 years: 93–309; 7–9 years: 86–315; 10–12 years: 42–362; 13–15 years: 74–390; 16–17 years: 52–171; and >17 years: 40–130. Age-specific normal range (U/L), girls/women 1–3 years: 108–317; 4–6 years: 96–297; 7–9 years: 69–325; 10–12 years: 51–332; 13–15 years: 50–162; 16–17 years: 47–119; and >17 years: 35–105.
PTH was significantly lower in patients with CKD stage 3 with NC compared with non-NC (Figure 1, Table 2). In the non-NC group, 18%, 57%, and 89% of patients in stages 2, 3, and 4–5 had elevated PTH, respectively. In contrast, most patients with NC with CKD stage 2 or 3 had PTH within the normal range (92% and 87%, respectively), with 88% of patients in stage 4–5 with elevated PTH (Figure 2).
Figure 2.

Prevalence of hyperphosphatemia, hyperparathyroidism, and elevated FGF23.
As shown in Figure 1 and Table 2, FGF23 levels were significantly lower in the NC group compared with patients with non-NC for all CKD stages. In the NC group (n=50), the median FGF23 level was 64.5 RU/ml (IQR, 36.8–133.8 RU/ml); 32% of these patients had elevated FGF23. In contrast, FGF23 levels were elevated in 78% of the non-NC group (n=77) (P<0.05), with a median value of 176.0 RU/ml (IQR, 105.5–327.5 RU/ml). Note that values above normal range were detected in NC groups compared with non-NC groups as follows: 15% versus 38% in stage 2 (P=0.33), 32% versus 73% in stage 3 (P<0.05), and 44% versus 90% in 4–5 stages (P<0.05) (Figure 2).
FGF23 plasma concentration increased in both groups according to the CKD stage (Figure 1). There was also a significant positive correlation between the Z-scores for the serum phosphate levels and the FGF23 concentrations for both groups (r=0.64, P<0.001 and r=0.41, P<0.001, respectively), with no significant difference between both correlations (Figure 3). We then used a linear regression model to evaluate for each CKD stage the FGF23 levels of patients with NC and non-NC, controlled for eGFR and age. This analysis revealed that the NC group had significantly lower FGF23 levels than the non-NC group. Thus, compared with the non-NC group, the patients with NC had FGF23 levels that were reduced by 61% for CKD stage 2 (P=0.04), and by 58% and 52% for CKD stages 3 (P=0.005) and 4–5 (P=0.01), respectively. After the above regression models were adjusted for serum phosphate, the percent differences were diminished and were no longer statistically significant at CKD stages 2 (P=0.25) or 3 (P=0.10).
Figure 3.

Correlation between serum phosphate and carboxy-terminal FGF23 in patients with and without NC.
TRP was significantly lower in the NC group compared with patients with non-NC across all of the CKD stages (Figure 1, Table 2). Median serum calcium levels were significantly different only for both the CKD stage 4 and 5 groups (Figure 1, Table 2). However, the random urinary calcium/creatinine ratio was significantly higher in patients with patients with NC compared with the non-NC group for all CKD stages (P<0.05) (Figure 1). Age- and sex-specific alkaline phosphatase Z-scores did not differ between NC and non-NC groups with CKD stage 2 or 3, but were significantly higher in patients with NC with CKD stages 4–5 [NC Z-score 2.1 (0.5–5.1) versus non-NC Z-score 0.9 (−0.5–2.6), P<0.05] (Table 2).
The median 1,25D level in the NC group (n=32) was 61 pg/ml (IQR, 39–74 pg/ml), with only one patient below the lower normal limit. In the non-NC group (n=48), the median 1,25D value was 34 pg/ml (IQR, 20–47 pg/ml) with low levels in up to 25% of studied patients (P<0.05). While most non-NC patients had 1,25D levels within the normal range in early CKD stages, up to one third of the patients with CKD stages 4–5 had decreased levels. Serum 1,25D concentrations were significantly higher in patients with CKD stages 2 and 3 in the NC compared with the non-NC group (P<0.05) (Figure 1, Table 2).
DISCUSSION
Mineral ion abnormalities encountered in adult and pediatric patients with the most common causes of CKD have been well described.12,16 In this study, we demonstrated significant differences for the indices of mineral ion homeostasis across the CKD spectrum encountered in patients with NC versus non-NC. Specifically, patients with NC CKD had significantly lower TRP, as well as lower blood phosphorus and FGF23 levels at all CKD stages; furthermore, patients with early CKD stages also had lower PTH and higher 1,25D levels. A likely explanation for these differences is a persistent considerable increase in urinary phosphate excretion, delaying the development of hyperphosphatemia and secondary hyperparathyroidism in patients with NC CKD, limiting the rise in FGF23 levels as GFR declined, and helping to prevent a decline in 1-α hydroxylase activity, and thus 1,25D levels.
We characterized the mineral ion abnormalities in patients with NC and found that this group continued to show a significant degree of renal phosphate wasting despite developing CKD. This explained the high percentage of patients with NC CKD with hypophosphatemia, despite phosphate supplementation in most of these patients at the time of evaluation. In addition, all patients with NC had, during the early CKD stages, 1,25D levels that were higher than those in patients with non-NC CKD; furthermore, PTH levels remained within the normal range in most of these patients. Interestingly, only two patients with NC with stage 2 CKD had elevated FGF23 levels, which is in contrast to findings in non-NC patients who had, at comparable CKD stages, elevated FGF23 levels as first evidence of altered mineral ion homeostasis. It is likely that a persistent reduction in tubular phosphate reabsorption allowed FGF23 levels to remain within the normal range, thus providing a plausible explanation for normal 1,25D and PTH levels in most patients with NC with early CKD stages. More frequent supplementation with calcitriol possibly contributed to some patients maintaining normal 1,25D levels in the NC CKD cohort. However, FGF23 levels were considerably lower in the latter group, despite calcitriol therapy that would have been expected to increase FGF23 production.17 Only when CKD advanced further did patients with NC progressively develop hyperphosphatemia despite continuously reduced TRP.
In the non-NC CKD control group, we found progression of biochemical abnormalities similar to those previously described in patients with CKD.12,16 During the early stages, most of these patients presented with phosphate, calcium, and 1,25D levels that were within the normal range; however, more than one third of these patients had elevated FGF23 levels. As CKD progressed, phosphate levels increased and more than one half of these patients had elevated FGF23 levels and one third had low 1,25D levels.
As we recently reported, the changes in mineral ion homeostasis encountered in patients with NC CKD are associated with a significant skeletal phenotype.11 In a group of 30 patients (age 5–44, mean 20) who were investigated at the NIH between 2016 and 2017, and in whom a complete skeletal evaluation was performed, we found that 27% and 32% experienced long bone and/or vertebral fractures, respectively. Consistent with rachitic/osteomalacic bone disease, 50% had significant scoliosis and 64% showed bowing of the lower extremities. Six patients with NC CKD, who had undergone a tetracycline-labeled iliac crest bone biopsy, showed evidence for markedly impaired mineralization. These histomorphometric findings suggest that alkaline phosphatase levels were higher in the NC-CKD cohort, indicating that this biochemical parameter could serve as biomarker for assessing abnormal bone mineralization in this disorder. Because bone consists primarily of calcium and phosphate in the form of hydroxyapatite, it is not surprising that persistent hypophosphatemia resulted in mineralization defects similar to those encountered in patients with hypophosphatemic rickets, yet otherwise normal renal function. As outlined above, we have recently characterized skeletal abnormalities in several patients with NC CKD, demonstrating significant abnormal mineralization.11 Compared with bone biopsies from age-matched patients with CKD without NC, the mineralization defect, characterized by increases in unmineralized bone (osteoid) in conjunction with delayed rates of mineral deposition, was more severe in patients with NC CKD (data not shown). Additional bone biopsy studies are needed to better characterize the long-term consequences of persistent hypophosphatemia on the skeleton in the population of patients with CKD with NC.
Limitations of our study include the fact that 1,25D levels were measured by different techniques in the two groups; however, the reference ranges were only slightly different. In addition, a significant proportion of patients (69% of patients with NC and 34% of patients who were non-NC) in both groups were receiving calcitriol, which may have confounded the results, partially explaining the differences in serum 1,25D levels that were observed for both groups. An additional limitation is the fact that cardiovascular outcomes were not assessed. This was not an end point in this longstanding study, but will be assessed going forward. Future studies will investigate the clinical implications of the different parameters explored in the presented study in other diseases leading to CKD-MBD.
Over the last few years, studies in adult and pediatric patients with CKD have shown that elevated FGF23 and phosphate levels have been associated with several negative clinical outcomes, including left ventricular hypertrophy and increased cardiovascular mortality. Consequently, as these two parameters have been proposed as new modifiable targets, an effort has been made to develop therapeutic strategies that can simultaneously reduce both.18 To date, several studies have assessed the efficacies of different strategies for reducing phosphate and FGF23 levels in patients with CKD, including dietary phosphate restriction,19,20 phosphate binding agents,21–23 or both,24,25, interventions that have shown efficacy in preclinical studies.26 However, the results of several different clinical trials were not particularly encouraging, calling into question the long-term benefits of such interventions. Only one recent prospective study reported that treatment with ferric citrate was associated with decreased serum phosphate concentrations and decreased circulating intact FGF23 levels, possibly because of a combination of a reduction in serum phosphate levels and improved anemia parameters.27 The ferric citrate group also had a lower incidence of a composite end point that included death, initiation of dialysis, or transplantation. Long-term prospective randomized trials in humans with CKD are needed in order to confirm and extend these initial observations. Other pharmacological therapies have also been investigated with promising results.28–30 For example, a recent randomized controlled trial to evaluate the efficacy of tenapanor, a small molecule that inhibits the gastrointestinal sodium hydrogen exchanger and reduces sodium and phosphate absorption, revealed a significant reduction in serum phosphate levels in patients with CKD stage 5.28 Moreover, tenapanor was shown to be associated with significant decreases in phosphate and FGF23 concentrations in patients with CKD.29 In contrast, nicotinamide, a form of vitamin B3 that reduces intestinal sodium-dependent phosphate transporter 2b expression and, therefore, reduces gastrointestinal phosphate absorption, did not significantly alter serum phosphate or FGF23 levels in patients with CKD stage 3b–4.30 Consequently, the benefits of phosphate-lowering therapies remain uncertain and need to be studied further.
Our findings that serum phosphate and, in response, serum FGF23 levels are lower in patients with NC CKD versus non-NC CKD may support the concept that strategies that enhance urinary phosphate excretion may prevent a rise in FGF23 levels and could possibly protect patients from FGF23-dependent morbidity.31
Our study shows that patients with NC, a lifelong disease with persistent urinary phosphate excretion that occurs independently of FGF23 and/or PTH elevation, have lower blood levels of phosphate, FGF23, and PTH compared with other causes of CKD. The biochemical features of patients with NC-CKD are distinct from those encountered in the majority of patients with CKD and support the assertion that the increase in FGF23 levels may be a consequence of the phosphate retention. This would support the notion that phosphate is a significant driver of FGF23 elevation in CKD, and that mineral ion abnormalities associated with CKD are likely to vary depending on the cause of the renal disease.
Disclosures
All authors have nothing to disclose.
Funding
This research was supported in part by the NIH Intramural Research Programs of the National Institute of Dental and Craniofacial Research and the National Human Genome Research Institute, U.S. Public Health Service grants DK-67563 and DK-35423, National Institute of Diabetes and Digestive and Kidney Diseases grants P01 DK011794 (subproject 3) and DK046718 (to H. Jüppner), and NIH/National Center for Advancing Translational Sciences grant UL1TR001881 (to I. Salusky). P. Florenzano received support from Pontificia Universidad Catolica de Chile and The Oscar & Elsa Braun Foundation.
Acknowledgments
The NIH Biomedical Translational Research Information System (BTRIS) was queried to determine and retrieve data sets for this study.
Work in the laboratories of Dr. Michael T. Collins was supported by the Intramural Research Program of the NIH, National Institute of Dental and Craniofacial Research.
Dr. Pablo Florenzano reports grants from Ultragenyx, outside the submitted work. Dr. Rachel I. Gafni reports grants from Ultragenyx, Amgen, Calcilytix, Inozyme, and QED Therapeutics, and other from Ascendis, Bayer, and GACI Global, all outside the submitted work. Dr. Myles Wolf reports personal fees from Akebia, Amag, Amgen, Ardelyx, Diasorin, Keryx, and Lutipold, outside the submitted work. Dr. Michael T. Collins reports grants from Amgen, Calcilytix, and QED Therapeutics, outside the submitted work. Dr. Mary Scott Roberts reports grants from Shire and is an employee of Ultragenyx Pharmaceutical, outside the submitted work.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
References
- 1.Elmonem MA, Veys KR, Soliman NA, van Dyck M, van den Heuvel LP, Levtchenko E: Cystinosis: A review. Orphanet J Rare Dis 11: 47, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gahl WA, Thoene JG, Schneider JA: Cystinosis. N Engl J Med 347: 111–121, 2002. [DOI] [PubMed] [Google Scholar]
- 3.Al-Haggar M: Cystinosis as a lysosomal storage disease with multiple mutant alleles: Phenotypic-genotypic correlations. World J Nephrol 2: 94–102, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Veys KR, Elmonem MA, Arcolino FO, van den Heuvel L, Levtchenko E: Nephropathic cystinosis: An update. Curr Opin Pediatr 29: 168–178, 2017. [DOI] [PubMed] [Google Scholar]
- 5.Emma F, Nesterova G, Langman C, Labbé A, Cherqui S, Goodyer P, et al.: Nephropathic cystinosis: An international consensus document. Nephrol Dial Transplant 29[Suppl 4]: iv87-iv94, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Markello TC, Bernardini IM, Gahl WA: Improved renal function in children with cystinosis treated with cysteamine. N Engl J Med 328: 1157–1162, 1993. [DOI] [PubMed] [Google Scholar]
- 7.Cherqui S, Courtoy PJ: The renal Fanconi syndrome in cystinosis: Pathogenic insights and therapeutic perspectives. Nat Rev Nephrol 13: 115–131, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brodin-Sartorius A, Tête MJ, Niaudet P, Antignac C, Guest G, Ottolenghi C, et al.: Cysteamine therapy delays the progression of nephropathic cystinosis in late adolescents and adults. Kidney Int 81: 179–189, 2012. [DOI] [PubMed] [Google Scholar]
- 9.Kimonis VE, Troendle J, Rose SR, Yang ML, Markello TC, Gahl WA: Effects of early cysteamine therapy on thyroid function and growth in nephropathic cystinosis. J Clin Endocrinol Metab 80: 3257–3261, 1995. [DOI] [PubMed] [Google Scholar]
- 10.Gahl WA, Balog JZ, Kleta R: Nephropathic cystinosis in adults: Natural history and effects of oral cysteamine therapy. Ann Intern Med 147: 242–250, 2007. [DOI] [PubMed] [Google Scholar]
- 11.Florenzano P, Ferreira C, Nesterova G, Roberts MS, Tella SH, de Castro LF, et al.: Skeletal consequences of nephropathic cystinosis. J Bone Miner Res 33: 1870–1880, 2018. [DOI] [PubMed] [Google Scholar]
- 12.Wesseling-Perry K, Pereira RC, Tseng CH, Elashoff R, Zaritsky JJ, Yadin O, et al.: Early skeletal and biochemical alterations in pediatric chronic kidney disease. Clin J Am Soc Nephrol 7: 146–152, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Portale AA, Wolf M, Jüppner H, Messinger S, Kumar J, Wesseling-Perry K, et al.: Disordered FGF23 and mineral metabolism in children with CKD. Clin J Am Soc Nephrol 9: 344–353, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schwartz GJ, Haycock GB, Edelmann CM Jr., Spitzer A: A simple estimate of glomerular filtration rate in children derived from body length and plasma creatinine. Pediatrics 58: 259–263, 1976. [PubMed] [Google Scholar]
- 15.Levey AS, Stevens LA, Schmid CH, Zhang YL, Castro AF III, Feldman HI, et al.: A new equation to estimate glomerular filtration rate [published correction appears in Ann Intern Med 155: 408, 2011]. Ann Intern Med 150: 604–612, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Graciolli FG, Neves KR, Barreto F, Barreto DV, Dos Reis LM, Canziani ME, et al.: The complexity of chronic kidney disease-mineral and bone disorder across stages of chronic kidney disease. Kidney Int 91: 1436–1446, 2017. [DOI] [PubMed] [Google Scholar]
- 17.Georgiadou E, Marketou H, Trovas G, Dontas I, Papaioannou N, Makris K, et al.: Effect of calcitriol on FGF23 level in healthy adults and its dependence on phosphate level. In Vivo 31: 145–150, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Scialla JJ, Wolf M: Roles of phosphate and fibroblast growth factor 23 in cardiovascular disease. Nat Rev Nephrol 10: 268–278, 2014. [DOI] [PubMed] [Google Scholar]
- 19.Moe SM, Zidehsarai MP, Chambers MA, Jackman LA, Radcliffe JS, Trevino LL, et al.: Vegetarian compared with meat dietary protein source and phosphorus homeostasis in chronic kidney disease. Clin J Am Soc Nephrol 6: 257–264, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sigrist M, Tang M, Beaulieu M, Espino-Hernandez G, Er L, Djurdjev O, et al.: Responsiveness of FGF-23 and mineral metabolism to altered dietary phosphate intake in chronic kidney disease (CKD): Results of a randomized trial. Nephrol Dial Transplant 28: 161–169, 2013. [DOI] [PubMed] [Google Scholar]
- 21.Block GA, Wheeler DC, Persky MS, Kestenbaum B, Ketteler M, Spiegel DM, et al.: Effects of phosphate binders in moderate CKD. J Am Soc Nephrol 23: 1407–1415, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gonzalez-Parra E, Gonzalez-Casaus ML, Galán A, Martinez-Calero A, Navas V, Rodriguez M, et al.: Lanthanum carbonate reduces FGF23 in chronic kidney disease Stage 3 patients. Nephrol Dial Transplant 26: 2567–2571, 2011. [DOI] [PubMed] [Google Scholar]
- 23.Covic A, Passlick-Deetjen J, Kroczak M, Büschges-Seraphin B, Ghenu A, Ponce P, et al.: A comparison of calcium acetate/magnesium carbonate and sevelamer-hydrochloride effects on fibroblast growth factor-23 and bone markers: Post hoc evaluation from a controlled, randomized study. Nephrol Dial Transplant 28: 2383–2392, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Isakova T, Gutiérrez OM, Smith K, Epstein M, Keating LK, Jüppner H, et al.: Pilot study of dietary phosphorus restriction and phosphorus binders to target fibroblast growth factor 23 in patients with chronic kidney disease. Nephrol Dial Transplant 26: 584–591, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Isakova T, Barchi-Chung A, Enfield G, Smith K, Vargas G, Houston J, et al.: Effects of dietary phosphate restriction and phosphate binders on FGF23 levels in CKD. Clin J Am Soc Nephrol 8: 1009–1018, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thomas L, Xue J, Murali SK, Fenton RA, Dominguez Rieg JA, Rieg T: Pharmacological Npt2a inhibition causes phosphaturia and reduces plasma phosphate in mice with normal and reduced kidney function. J Am Soc Nephrol 30: 2128–2139, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Block GA, Block MS, Smits G, Mehta R, Isakova T, Wolf M, et al.: A pilot randomized trial of ferric citrate coordination complex for the treatment of advanced CKD. J Am Soc Nephrol 30: 1495–1504, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Block GA, Rosenbaum DP, Yan A, Chertow GM: Efficacy and safety of tenapanor in patients with hyperphosphatemia receiving maintenance hemodialysis: A randomized phase 3 trial. J Am Soc Nephrol 30: 641–652, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Block GA, Rosenbaum DP, Yan A, Greasley PJ, Chertow GM, Wolf M: The effects of tenapanor on serum fibroblast growth factor 23 in patients receiving hemodialysis with hyperphosphatemia. Nephrol Dial Transplant 34: 339–346, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ix JH, Isakova T, Larive B, Raphael KL, Raj DS, Cheung AK, et al.: Effects of nicotinamide and lanthanum carbonate on serum phosphate and fibroblast growth factor-23 in CKD: The COMBINE trial. J Am Soc Nephrol 30: 1096–1108, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Filipski KJ, Sammons MF, Bhattacharya SK, Panteleev J, Brown JA, Loria PM, et al.: Discovery of orally bioavailable selective inhibitors of the sodium-phosphate cotransporter NaPi2a (SLC34A1). ACS Med Chem Lett 9: 440–445, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]