

Significance Statement
The EGF receptor (EGFR) ligand amphiregulin (AREG) has emerged as a potent mediator of inflammation. AREG’s tissue-protective and immunosuppressive properties have recently received much attention, but the ligand has another function. In a mouse model of GN, AREG plays an unexpectedly strong proinflammatory rather than protective role. Renal resident cells that secrete AREG enhance the recruitment, proliferation, and activation of tissue-destructive myeloid cells. Importantly, studies in human crescentic GN also revealed strong upregulation of renal AREG expression, indicating clinical relevance of the murine model. These findings contribute to a more balanced understanding of AREG’s biology and help with the selection of patients and timing of AREG/EGFR-directed therapies.
Keywords: macrophages, glomerular disease, immunology, cytokines
Visual Abstract

Abstract
Background
Recent studies have identified the EGF receptor (EGFR) ligand amphiregulin (AREG) as an important mediator of inflammatory diseases. Both pro- and anti-inflammatory functions have been described, but the role of AREG in GN remains unknown.
Methods
The nephrotoxic nephritis model of GN was studied in AREG−/− mice after bone marrow transplantation, and in mice with myeloid cell–specific EGFR deficiency. Therapeutic utility of AREG neutralization was assessed. Furthermore, AREG's effects on renal cells and monocytes/macrophages (M/M) were analyzed. Finally, we evaluated AREG expression in human renal biopsies.
Results
Renal AREG mRNA was strongly upregulated in murine GN. Renal resident cells were the most functionally relevant source of AREG. Importantly, the observation that knockout mice showed significant amelioration of disease indicates that AREG is pathogenic in GN. AREG enhanced myeloid cell responses via inducing chemokine and colony stimulating factor 2 (CSF2) expression in kidney resident cells. Furthermore, AREG directly skewed M/M to a proinflammatory M1 phenotype and protected them from apoptosis. Consequently, anti-AREG antibody treatment dose-dependently ameliorated GN. Notably, selective abrogation of EGFR signaling in myeloid cells was sufficient to protect against nephritis. Finally, strong upregulation of AREG expression was also detected in kidneys of patients with two forms of crescentic GN.
Conclusions
AREG is a proinflammatory mediator of GN via (1) enhancing renal pathogenic myeloid cell infiltration and (2) direct effects on M/M polarization, proliferation, and cytokine secretion. The AREG/EGFR axis is a potential therapeutic target for acute GN.
The EGF receptor (EGFR) pathway is crucially involved in a multitude of cellular processes, such as cell cycle, growth, and differentiation. Given its highly potent effects, agents blocking the EGFR have been established as therapeutics for the treatment of various human malignancies. Recent studies, however, have indicated that targeting the EGFR axis might be used beyond tumor therapy. Treatment with the EGFR-blocking tyrosine kinase inhibitor erlotinib ameliorated immune-mediated experimental arthritis.1 Also, in the renal field, evidence for an important pathophysiologic role of the EGFR signaling cascade is accumulating.2,3 In particular, deficiency of the EGFR ligand HB-EGF was shown to protect from renal tissue injury in acute GN.4 Interestingly, selective abrogation of EGFR signaling in podocytes only resulted in significant reduction of glomerular crescent formation. Most importantly, however, ligand and cell-type nonspecific blockade of the EGFR pathway also strongly ameliorated GN.4 Along the same line, pan-EGFR signaling blockade was shown to ameliorate renal fibrosis,5–8 murine diabetic nephropathy,9 and lupus nephritis in the Fcgr2b−/− model.10 Finally, enhancement of EGFR activity by a naturally occurring mutation resulted in spontaneous development of glomerulopathy.11
Which of the seven known EGFR ligands, apart from HB-EGF, is involved in the individual disease pathogenesis in these models has remained largely unclear. In this respect, amphiregulin (AREG) is of particular interest.12 A number of recent landmark studies have highlighted several unique roles of AREG among the EGFR ligands. Initially, a proinflammatory role was postulated. Overexpression of AREG in keratinocytes resulted in skin inflammation, resembling human psoriasis.13 Later, AREG expression was found to be elevated in various tissues from patients with rheumatoid arthritis and Sjögren's syndrome. Mechanistically, it was demonstrated that AREG stimulated synoviocytes and salivary gland cells to express proinflammatory mediators.14,15 Moreover, a strong profibrotic role of AREG was identified in a model of liver fibrosis.16 Along the same line, AREG was shown to be secreted by tubule cells from kidneys of patients with varying renal diseases. Importantly, the degree of AREG expression correlated with renal fibrosis, and urinary AREG levels were enhanced in patients with acute or chronic kidney injury.17 A very recent study by the same authors, published during preparation of our manuscript, also provided functional in vivo evidence for a profibrotic role of AREG. Cell-specific abrogation of AREG secretion from proximal tubule cells resulted in amelioration of renal fibrosis in the unilateral ureteral obstruction and ischemia reperfusion injury models.18 In stark contrast to these proinflammatory and disease-aggravating effects, AREG seems to possess additional tissue-protective functions.19
Interestingly, AREG was recently found to be secreted by activated regulatory T cells (Tregs). Treg-derived AREG, however, did not contribute to their immune regulatory functions, but rather mediated tissue repair across pathologies of various organs.20–23 This tissue-protective AREG effect seems to be more general in nature and somewhat independent of the leukocyte source. Similar to Treg-derived AREG, secretion by ILC2s (group 2 innate lymphoid cells) was shown to enhance tissue regeneration in models of colitis24 and acute ischemic renal injury.25 In addition, AREG derived from macrophages mediated protective effects in chronic volume overload triggered experimental heart failure26 and lung and liver injury.27 Furthermore, AREG seems to be a central player in defense against helminth infections. Several studies could demonstrate unique roles of the AREG/EGFR axis in enhancing Th2 responses.28,29 This might also be relevant for immune-mediated diseases, in which Th2 immunity is either aggravating, as in asthma, or protective, as in many forms of acute GN. Finally, it has been shown that during inflammation, Tregs upregulate expression of the EGFR and ligation of AREG potently enhances their immunosuppressive capacity.30–34 Similarly, it has been shown that stabilization of EGFR expression on Tregs by the long noncoding RNA lnc-EGFR improves their suppressive function.35 This might be of particular importance for GN because Tregs are central mediators of renoprotection.36–42 In summary, both pro- and anti-inflammatory, as well as tissue-reparative roles for AREG have been described. However, no data exist so far about the in vivo relevance and functions of AREG in GN. We thus aimed to address this aspect, particularly with regards to AREG/EGFR blockade as a potential therapeutic strategy.
Methods
Animals
AREG−/− mice (B6.129P2-Aregtm1Dle) were originally obtained from M.A.A. (Pamplona, Spain).43 LoxP site-flanked EGFRfl/fl mice [B6(Cg)-Egfrtm1Dwt] were provided by P.-L.T. (Paris, France).44 Monocytes/macrophages (M/M)-specific deletion of the EGFR was achieved by crossbreeding with mice expressing LysM-driven Cre recombinase [B6.129P2-Lyz2tm1(cre)Ifo/J]. Specificity of the EGFR knockout on M/M was tested by PCR analysis of genomic DNA and mRNA, prepared from FACS-sorted peritoneal macrophages and T cells (primer sequences available upon request). All mice were on a C57BL/6J background and bred in our facility under specific pathogen-free conditions.
Animal Experiments and Functional Studies
Nephrotoxic nephritis (NTN) was induced in the indicated strains of mice by a single intraperitoneal injection of nephrotoxic sheep serum. Renal and splenic leukocytes were isolated at the indicated time points, as described below, and analyzed by FACS. Urine samples were collected after housing the mice in metabolic cages. Albuminuria was determined by standard ELISA (Bethyl Laboratories). BUN and urinary creatinine were quantified using standard laboratory methods.
For bone marrow transplantation, 9-week-old mice were irradiated with 9Gy. Donor bone marrow cells were isolated as described previously.45 In brief, femoral and humeral bones were dissected and flushed with PBS. Cells were filtered through a 40-µm mesh and washed by centrifugation at 300×g. In total, 1×107 cells were injected into the tail veins of the recipient mice 12 hours after irradiation. NTN was induced 7 weeks after transplantation. For neutralization studies, the accelerated nephrotoxic nephritis model (aNTN) was used. C57BL/6 mice were preimmunized with 0.5 mg sheep IgG in CFA. Five days later, aNTN was induced and anti-AREG (AF989; R&D Systems, Wiesbaden, Germany) or isotype antibodies (AB-108-C; R&D Systems) were intraperitoneally injected at the indicated dose and time points. Organs were removed at day 5 after aNTN induction. Animal experiments were performed according to national and institutional animal care and ethical guidelines and were approved by local committees (approval codes 96/16, 67/17, and 065/2018).
Murine Morphologic Studies
Crescent formation and glomerular necrosis were determined in a minimum of 50 glomeruli per mouse in 2-µm-thick kidney sections stained with periodic acid–Schiff, in a blinded manner. Semiquantitative analysis of tubulointerstitial damage, using a score of 0–4, was performed using 20 randomly selected cortical areas (×200) as published previously.46 Hematoxylin and eosin staining was performed according to standard methods for detection of eosinophil granulocytes. Paraffin-embedded sections were stained with antibodies directed against CD3 (A0452; Dako, Hamburg, Germany), F4/80 (BM8; BMA Biomedicals, Hiddenhausen, Germany), MAC-2 (M3/38; Cedarlane-Laboratories, Burlington, ON, Canada), Foxp3 (FJK-16s; eBiosciences, San Diego, CA), or GR-1 (NIMP-R14; Hycult Biotech, Uden, The Netherlands), and developed with a polymer-based secondary antibody alkaline phosphatase kit (POLAP; Zytomed, Berlin, Germany). Positive cells in 50 glomerular cross-sections and 20 tubulointerstitial high-power fields (magnification ×400) per kidney section were counted in a blinded fashion.
Analyses of Human Renal Biopsy Specimens
Immunostaining of human tissue was performed in 3-µm formalin–acetic acid–alcohol fixed and paraffin-embedded tissue sections of kidney biopsies. Briefly, after dewaxing and rehydration, the sections were incubated overnight at 4°C with the primary antibody rabbit anti-AREG (HPA008720, 1:1000; Atlas Antibodies). Sections were then incubated with polymer-based peroxidase-conjugated antibody (Histofine; Nichirei Biosciences). Staining was revealed with 3,3′-diaminobenzidin (DAB), and counterstained with hematoxylin and eosin. Human renal tissue specimens were obtained from the Pathology Department of Hôpital Européen Georges Pompidou, Assistance Publique-Hôpitaux de Paris, Paris, France. Human tissue was used after obtaining informed consent from all patients, and kidney biopsy collection was approved by the Institut National de la Santé et de la Recherche Médicale Ethics Committee (Institutional Review Board 00003888, approval 13-087; FWA00005831 National Institutes of Health, Office of Human Research Protection) and the local ethics committee (Comité de Protection des Personnes Ile de France IV, Institutional Review Board: 00003835. approval 2015/73NICB). Kidney biopsy specimens were collected in compliance with all relevant ethical regulations, and those with sufficient tissue for immunohistochemical evaluation after the completion of diagnostic workup were included. In total, four biopsy specimens from patients with Goodpasture syndrome and four biopsy specimens from patients with ANCA vasculitis (three microscopic polyangiitis, one granulomatous polyangiitis) were stained. Three control kidneys were obtained from patients who presented with kidney failure, without proteinuria or sediment abnormalities, with a biopsy showing no evidence for glomerular and vascular lesions nor abnormal inflammatory infiltration, and with <10% of interstitial fibrosis.
Isolation of Leukocytes from Various Tissues
Spleens were harvested in HBSS and passed through 70-µm nylon meshes. After lysis of erythrocytes with ammonium chloride, cells were washed and passed through 40-µm meshes. Kidneys were minced and incubated in digestion medium (RPMI 1640 medium containing 10% FCS [Gibco], 1% HEPES, 1% penicillin/streptomycin [Life Technologies, Karlsruhe, Germany], 8 µg/ml collagenase D, and 0.4 µg/ml DNase) at 37°C for 45 minutes. Tissues were then dissociated, using the gentleMACS dissociator (Miltenyi Biotec) to get a single-cell suspension and were centrifuged at 300×g at 4°C for 8 minutes. To further purify the cells, Percoll gradient (37% Percoll; GE Healthcare, Chalfont St Giles, United Kingdom) centrifugation was performed at 500×g at room temperature for 20 minutes. Peripheral blood was drawn into EDTA-coated tubes and red blood cell lysis was performed. Peritoneal macrophages were isolated as described previously.45 Briefly, the peritoneal cavity was lavaged with PBS, 3 days after 1 ml intraperitoneal thioglycollate (Sigma, Steinheim, Germany) injection. Cells were passed over a 40-µm mesh, washed by centrifugation at 300×g for 5 minutes, and resuspended in RPMI 1640 L-glutamine medium (Life Technologies) containing 1% penicillin/streptomycin, 1% HEPES, 10% FCS, and 50 ng/ml M-CSF (Peprotech, Rocky Hill, NJ). After all isolation procedures, cells were counted and prepared for FACS or culture.
Flow Cytometry
Cells were surface-stained for 30 minutes at 4°C with fluorochrome-labeled antibodies against CD45, CD4, CD11c (all BD Biosciences, Franklin Lakes, NJ), CD3, CD8, CD19, Ly6C, Ly6G, CD11b, CCR2, CD206 (BioLegend, San Diego, CA). For intracellular and nuclear staining, samples were processed, using a commercial nuclear staining Kit (Foxp3 staining kit; eBiosciences). Fluorochrome-labeled antibodies against IL-4, IL-13, IL-17, IFNγ, and Foxp3 (eBiosciences) were used. For intracellular cytokine staining, cells were activated with PMA (50 ng/ml; Sigma-Aldrich), ionomycin (1 µg/ml; Calbiochem-Merck, Darmstadt, Germany), and brefeldin A (10 µg/ml; Sigma-Aldrich) for 3.5 hours. LIVE/DEAD staining (Invitrogen Molecular Probes, Eugene, Oregon) was used to exclude dead cells. FACS sorting was performed from single-cell suspensions of spleens and peritoneum by the Institutional FACS Sorting Core Facility.
Mesangium and Tubule Cell Culture
Mouse kidney proximal tubule cells derive from a cell line, which was initially generated by microdissection from mouse proximal tubules and subsequent immortalization by transformation with SV-40 virus.47 Mouse kidney mesangial cells were initially prepared from mouse glomeruli by differential sieving and digestion techniques, followed by immortalization via transformation with SV-40 virus.48 Cells were cultured in DMEM medium (Life Technologies) containing 10% FCS, 100 U/ml penicillin, and 100 mg/ml streptomycin at 37°C with 5% CO2. The cells were incubated in serum-free DMEM medium 24 hours before stimulation. Cells were stimulated with AREG (10–100 ng/ml, 989-AR; R&D Systems), HB-EGF (10–100 ng/ml, SRP6050; Sigma), EGF (30–100 ng/ml, 2028-EG-200; R&D Systems), IL-1β (10 ng/ml, 211–11B), IL-6 (100 U/ml, 216–16), IFNγ (100 ng/ml), IL-17A (10 ng/ml), IL-17F (10 ng/ml), IL-17C (10 ng/ml), TNFα (10 ng/ml, 315–01A), and IL-22 (100 ng/ml) (all Peprotech), alone or with a combination. For AREG/EGFR neutralization experiments, 1.5 µg/ml anti-AREG (AF989), 1.5 µg/ml isotype control (AB-108-C, R&D Systems), or 1 µM erlotinib (Selleckchem, Houston, TX) was added to the culture. Cells were harvested after 6 hours of stimulation and cDNA was prepared for PCR analyses. For assessment of AREG protein secretion, cells were incubated with 50 ng/ml AREG for 6 hours, then the supernatant was carefully removed and cells were washed twice in PBS. After a further 20 hours of incubation in AREG-free medium, levels of AREG protein in the supernatant were determined by standard ELISA (R&D Systems), according to the manufacturer’s instructions.
Macrophage Apoptosis
Peripheral blood from naïve transgenic and control mice was drawn into EDTA-coated tubes and prepared for FACS staining. Cell surface and LIVE/DEAD staining was performed before the staining of phosphatidylserine with fluorochrome-labeled annexin V (BioLegend), according to the manufacturer’s protocol.
Macrophage Phagocytosis Studies
Thioglycollate-induced peritoneal M/M were isolated as described above and seeded in cell culture dishes (1×106 cells/ml) for 2 hours at 37°C and 5% CO2. Cells were washed with PBS and adherent cells were harvested. For assessment of phagocytosis, macrophages were stimulated with or without 50 ng/ml AREG in the presence of FITC-labeled latex beads (LB30–1m; Sigma) for 48 hours at 37°C and 5% CO2 (cell-to-bead ratio of 1:5). Phagocytosis, as indicated by FITC fluorescence of macrophages, was quantified by flow cytometry.
In Vitro M/M Proliferation and Cytokine Expression
For proliferation experiments, 1 ml of macrophage culture was plated at a concentration of 3.3×104/ml on a six-well plate. Cells were cultivated in a humidified incubator with 5% CO2 at 37°C. Colony formation in the presence of AREG, TNFα, HB-EGF, or EGF was evaluated after 5 or 6 days, as indicated. Analysis was performed by counting CFUs, cells per CFU, and cells per well. CFUs were determined as a group of cells with direct cell–cell contacts or a distance of less than one cell in width and a total number of more than three cells. For macrophage cytokine expression analyses, cells were seeded at a concentration of 5×105/ml on a 24-well plate with or without AREG, HB-EGF, or EGF at the indicated concentrations, and the supernatant was harvested after 24 hours. In the case of exogenous AREG supplementation, cells from AREG−/− mice were repetitively stimulated with 50 ng/ml AREG at 0, 4, and 20 hours and the supernatant was harvested at 24 hours. IL-1β was quantified by ELISA (BioLegend), according to the manufacturer’s instructions. For assessment of AREG protein secretion, cells were incubated with 50 ng/ml AREG for 6 hours, then the supernatant was carefully removed and cells were washed twice in PBS. After further 20 hours of incubation in AREG-free medium, levels of AREG protein in the supernatant were determined by standard ELISA (R&D Systems), according to the manufacturer’s instructions. For assessment of M1 differentiation, M/M were in vitro–stimulated for 48 hours, with or without 50 ng/ml AREG, in the presence or absence of 50 ng/ml LPS and subsequently analyzed for Ly6C expression by FACS.
Real-Time Quantitative PCR
Total RNA of renal cortex, spleens, mesangium, and tubule cells or FACS-sorted leukocyte populations was isolated according to a standard TRIzol protocol and purified by utilizing a NucleoSpin kit (Macherey-Nagel, Düren, Germany). Real-time PCR was performed in duplicate for each sample (all primer sequences available upon request), using the SYBR green method. Results were normalized to expression of 18S rRNA, using the ΔCT method. For comparison with baseline levels, the ΔΔCT method was applied.
Statistical Analyses
Results are expressed as mean±SEM. The t test was used for comparison between two groups. In case of three or more groups, one-way ANOVA was used, followed by a post hoc analysis with the Tukey test for multiple comparisons or Dunnett test for comparisons of all time points versus control. A P value <0.05 was considered statistically significant.
Results
AREG Is Expressed by Renal Resident Cells and Upregulated during NTN
In order to evaluate the potential role of the EGFR axis during acute renal inflammation, we first studied expression of its seven ligands during NTN. Although some showed homeostatic (HB-EGF, TGFα, betacellulin) or reduced expression (EGF), only renal AREG mRNA was strongly upregulated (Figure 1A, Supplemental Figure 1A). In naïve kidneys, we found very low AREG expression. During NTN, however, levels massively increased, peaking at days 7–10. Afterward, expression decreased again and reached steady-state conditions (Figure 1A). An overview of the renal expression of the seven EGFR ligands is shown in Figure 1B. In line with the massive induction of renal mRNA expression, we also found serum levels of soluble AREG protein to be increased at days 7 and 10 of NTN (Figure 1C). We next examined splenic mRNA, but found no relevant expression of AREG or regulation of any of the other EGFR ligands during NTN (Supplemental Figure 1B). Given that during NTN AREG was selectively upregulated in the kidney but not in the spleen, we hypothesized that resident renal cells might be the source, which would be in line with recent observations.17,18 In order to analyze this aspect, we started off by studying regulation of AREG expression in two prototypical renal cell populations. Interestingly, in vitro stimulation of mouse mesangium (mMC) and mouse proximal tubule (mPTC) cells with various cytokine mediators known to be important in NTN did not induce AREG mRNA transcription (Supplemental Figure 2, A–C). However, both cell types expressed high levels of AREG mRNA and protein (Figure 1, D–G) in response to stimulation with AREG itself, indicating a positive feed-forward loop. Expression of the EGFR in whole kidney tissue as well as mMC and mPTC was confirmed by quantitative RT-PCR (Supplemental Figure 2, D and E). Addition of an anti-AREG antibody or the tyrosine kinase inhibitor erlotinib both effectively abrogated AREG self-induced expression of AREG mRNA, confirming signaling via the AREG/EGFR pathway (Supplemental Figure 3, A and B). Extensive testing of multiple further cytokine combinations revealed that addition of TNFα to AREG augmented AREG mRNA transcription even more (Supplemental Figure 3B). Given that the expression profile of AREG was unique among all EGFR ligands, we obtained knockout mice (AREG−/−) to further study AREG’s role in NTN. Complete absence of AREG was confirmed by quantitative RT-PCR analyses of nephritic renal tissue. Among all other EGFR ligands, we found only renal EGF expression to be mildly but significantly upregulated in AREG−/− mice, compared with wild-type controls during NTN (Supplemental Figure 4A). Splenic expression of all EGFR ligands was again low and no differences compared with wild-type mice were noted (Supplemental Figure 4B).
Figure 1.
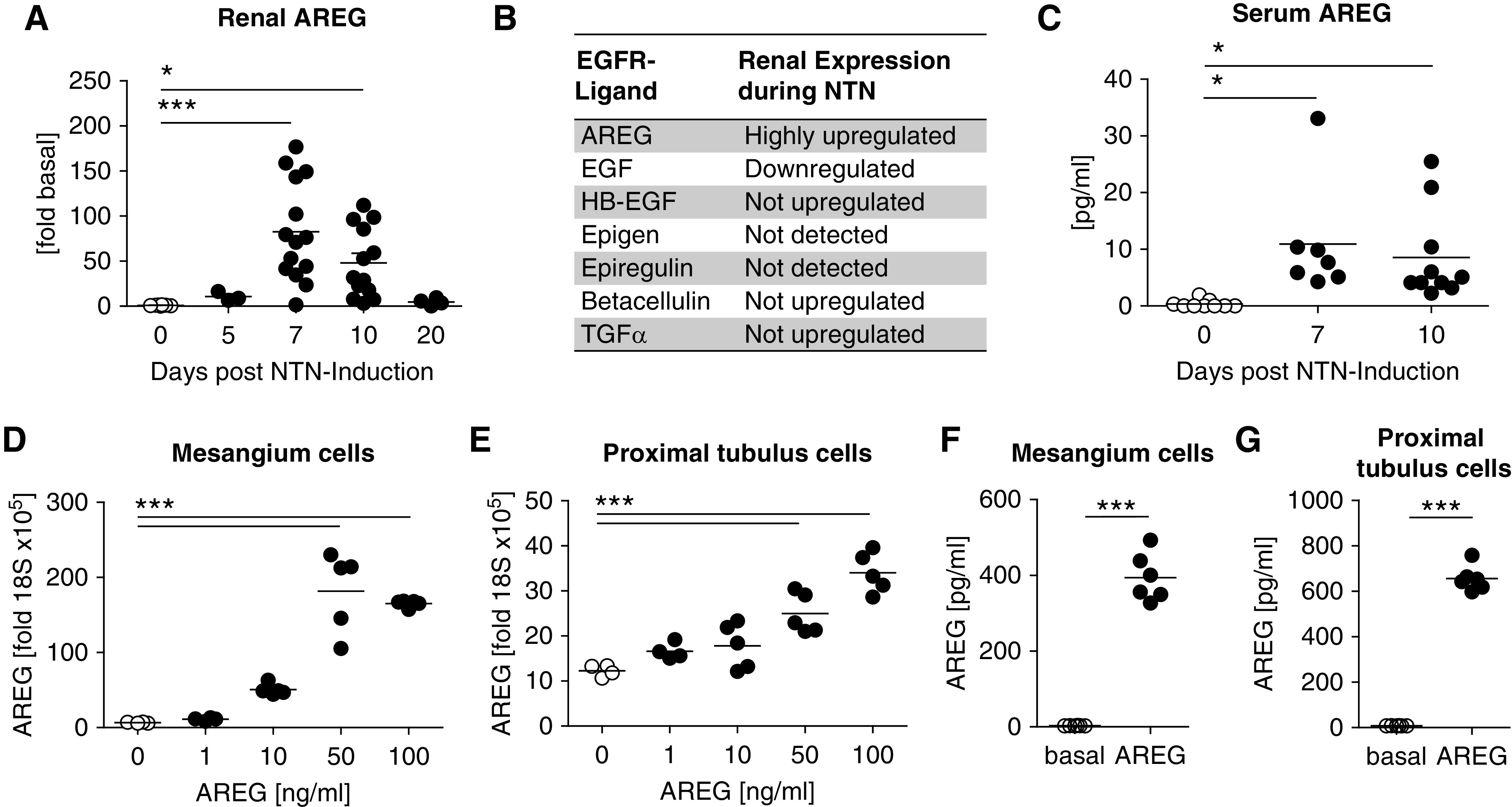
AREG is expressed by renal resident cells and upregulated during NTN. (A) Time course of renal AREG mRNA expression after induction of NTN relative to baseline levels. (B) Overview of renal mRNA expression of the seven known EGFR ligands during NTN. (C) ELISA analyses of soluble AREG protein in the serum at the indicated time points of NTN. (D and E) AREG mRNA expression by cultured (D) mesangial and (E) proximal tubule cells after 6 hours of in vitro stimulation with AREG at the indicated concentrations. (F and G) Levels of soluble AREG protein in the supernatant of cultured (F) mesangial and (G) proximal tubule cells, 20 hours after initial 6-hour in vitro stimulation with AREG. Symbols represent individual animals, horizontal lines show mean values. *P<0.05; ***P<0.001.
AREG Deficient Mice Are Protected from NTN
Because recent studies have suggested broad anti-inflammatory and tissue protective properties, we aimed to examine AREG’s functional role in NTN. Surprisingly however, our analyses at days 7 and 10 showed markedly reduced glomerular crescent formation and interstitial damage (Figure 2, A–C) in AREG−/− mice. In addition, albuminuria (Figure 2D) and BUN (Figure 2E) levels were also significantly lower at both time points, as compared with wild-type controls. Taken together, this demonstrates significant protection from renal histologic and functional injury in AREG−/− mice, indicating a pathogenic rather than protective role for AREG in acute GN.
Figure 2.
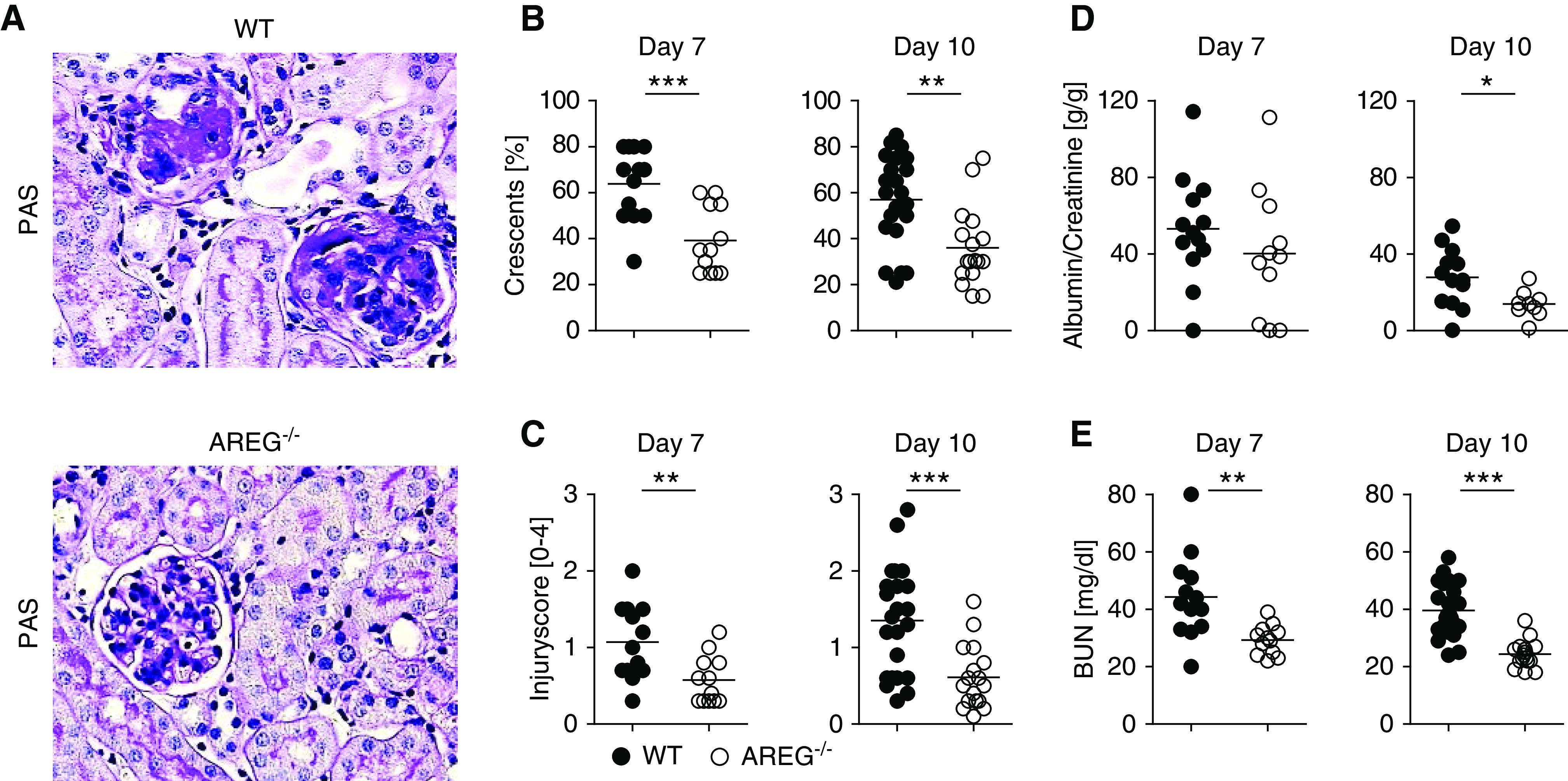
AREG-deficient mice are protected from NTN. (A) PAS-stained sections of nephritic kidneys from the indicated strains of mice at day 10 of NTN (original magnification ×400). (B and C) Quantification of (B) glomerular crescents and (C) interstitial injury at the indicated time points after NTN. (D) Albumin-creatinine ratio in the urine of indicated mouse strains at days 7 and 10 of NTN. (E) BUN levels of indicated mouse strains at 7 and 10 days after NTN induction. Symbols represent individual animals, horizontal lines show mean values. *P<0.05; **P<0.01; ***P<0.001. PAS, periodic acid–Schiff; WT, wild-type.
AREG−/− Mice Show Reduced Renal Myeloid Cell Infiltration and Activation in NTN
We next examined the effect of AREG knockout on renal inflammatory cell infiltration and systemic immunity in NTN. Renal T cell infiltration was not consistently affected and only a mild glomerular reduction at day 7 could be found (Supplemental Figure 5, A–C). However, in the kidneys and also systemically, T cells from AREG−/− mice showed increased production of proinflammatory cytokines (Supplemental Figure 5, D and E). Because this phenomenon could not account for the observed amelioration of renal injury, we looked for further immune alterations, which might overrule the effects of enhanced T cell activation. Renal infiltration of eosinophil granulocytes, another leukocyte population known to be influenced by AREG, was not different between the groups (Supplemental Figure 6, A and B). Interestingly, however, at both 7 and 10 days of NTN, numbers of infiltrating neutrophils were lower in AREG−/− mice (Figure 3, A–C). Similarly, renal glomerular, as well as interstitial M/M infiltration was significantly reduced (Figure 3, D–G). Notably, M/M were not only reduced numerically in AREG−/− mice, but also showed a less inflammatory phenotype with skewing toward M2 polarization. In this regard, we found that renal mRNA expression of the proinflammatory macrophage-secreted cytokine IL-1β was significantly diminished in AREG−/− mice (Figure 3H). Furthermore, our analyses showed reduced proportions of renal Ly6Chi inflammatory M/M (Figure 3, I and J) at both time points. Conversely, M2 characteristic Mannose receptor/CD206 expression on renal M/M was significantly increased in the absence of AREG (Figure 3K, Supplemental Figure 7A). Interestingly, this skewing toward an M2 phenotype was restricted to M/M in the kidney. In contrast, M/M in blood and spleens of AREG−/− mice showed enhanced M1 polarization (Supplemental Figure 7, B and C).
Figure 3.
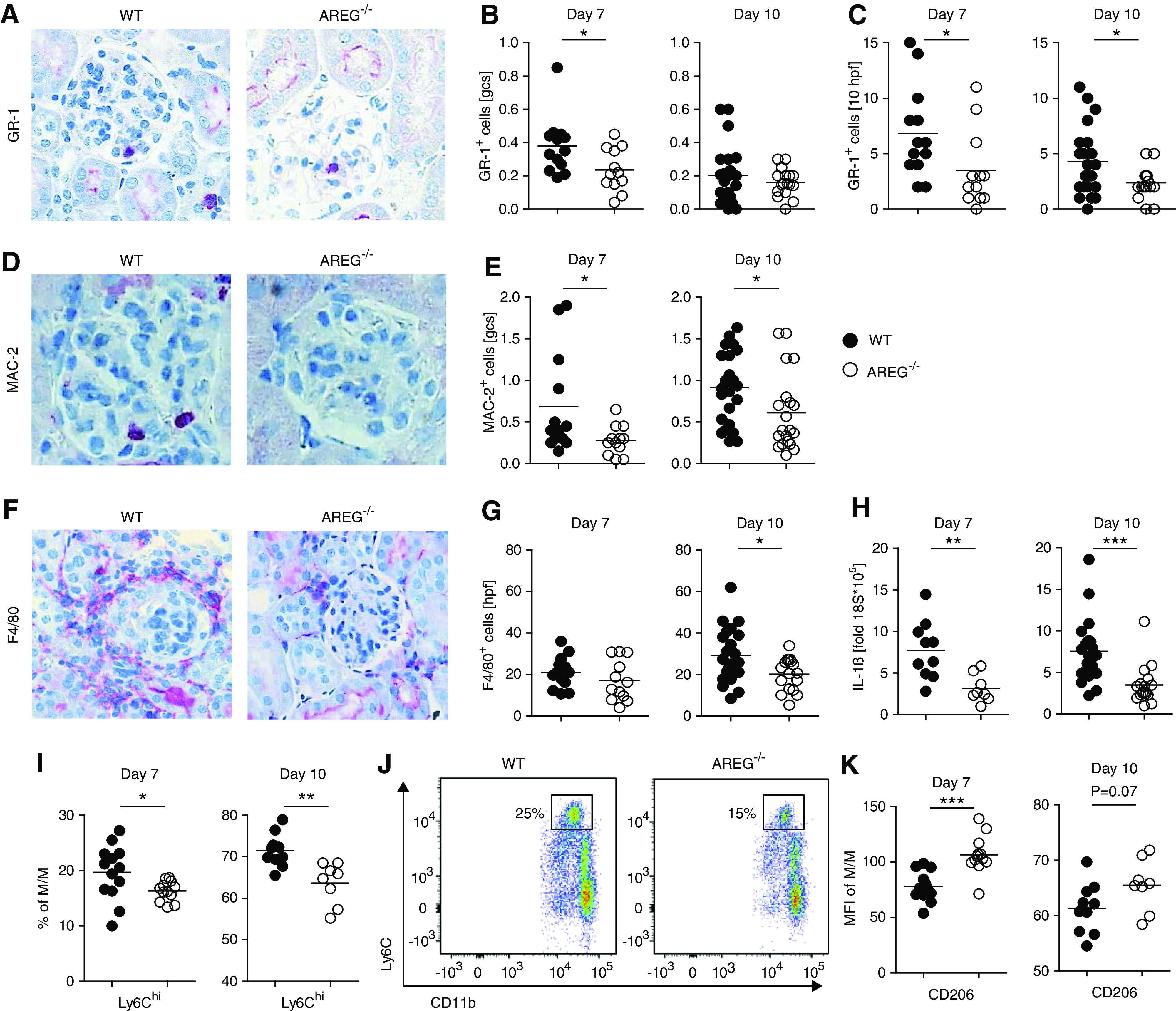
AREG−/− mice show reduced renal myeloid cell infiltration and activation in NTN. (A) Immunohistochemical staining of GR-1+ polymorphonuclear neutrophils (PMN) in kidneys of nephritic mice. (B and C) Quantification of (B) glomerular and (C) interstitial PMN infiltration at the indicated time points after NTN. (D) Immunohistochemical staining of glomerular MAC-2+ M/M in kidneys of nephritic mice. (E) Quantification of glomerular M/M infiltration at the indicated time points after NTN. (F) Immunohistochemical staining of interstitial F4/80+ M/M in kidneys of nephritic mice. (G) Quantification of interstitial M/M infiltration at the indicated time points after NTN. (H) Semiquantitative analysis of renal IL-1β mRNA expression by real-time quantitative PCR at the indicated time points after NTN. (I) FACS quantification of renal Ly6Chigh inflammatory M/M at the indicated time points after NTN. (J) Representative FACS plot of renal Ly6Chigh M/M at day 7 of NTN. (K) Mean fluorescence intensity (MFI) of Mannose receptor/CD206 expression on renal M/M at the indicated time points after NTN (original magnification ×400). Symbols represent individual animals, horizontal lines show mean values. *P<0.05; **P<0.01; ***P<0.001. gcs, glomerular cross section; hpf, high-power field; WT, wild-type.
AREG Induces Myeloid Cell–Attracting Chemokines and Colony Stimulating Factor 2 from Resident Renal Cells
To investigate the mechanisms underlying reduced renal myeloid cell infiltration in AREG−/− mice, we examined renal chemokine production. Indeed, we found significantly diminished renal expression of the neutrophil-attracting CXCL1 and CXCL5, as well as of the macrophage-attracting chemokine MCP-1/CCL2 (Figure 4A). To delineate whether this was a direct effect of AREG, we measured production of these chemokines from in vitro–stimulated resident renal cells. AREG by itself had no effect on expression of chemokines from mMC or mPTC. Because it was reported that TNFα enhances AREG’s effects on human tubule cells,17 we tested this combination. Indeed, simultaneous stimulation potently induced CXCL1 and CCL2 transcription by mMC (Figure 4B) and CXCL1, CXCL5, and CCL2 by mPTC (Figure 4C). The observed enhancement of chemokine production was a specific effect of AREG signaling because upregulation could be blocked by AREG-neutralizing antibodies or inhibition of the EGFR by the tyrosine kinase inhibitor erlotinib (Supplemental Figure 8A). To investigate whether chemokine induction was unique to AREG or shared with other EGFR ligands, we also investigated the effects of HB-EGF and EGF. Indeed, in the presence of TNFα, both HB-EGF and EGF increased expression of CXCL1 and CCL2 from mMC (Supplemental Figure 9, A–D). Furthermore, TNFα plus HB-EGF or EGF also induced chemokine expression from mPTC, albeit significantly less effectively, compared with AREG (Supplemental Figure 10, A–F). Next, we wanted to know whether AREG might not only support myeloid cell recruitment but also differentiation and proliferation of proinflammatory M/M. In this respect, we found the combination of AREG with TNFα to potentiate expression of the myeloid cell colony stimulating factor 2 (CSF2) from mMC and mPTC. This effect was specific for AREG as HB-EGF and EGF stimulation had no effect (Figure 4, D and E).
Figure 4.
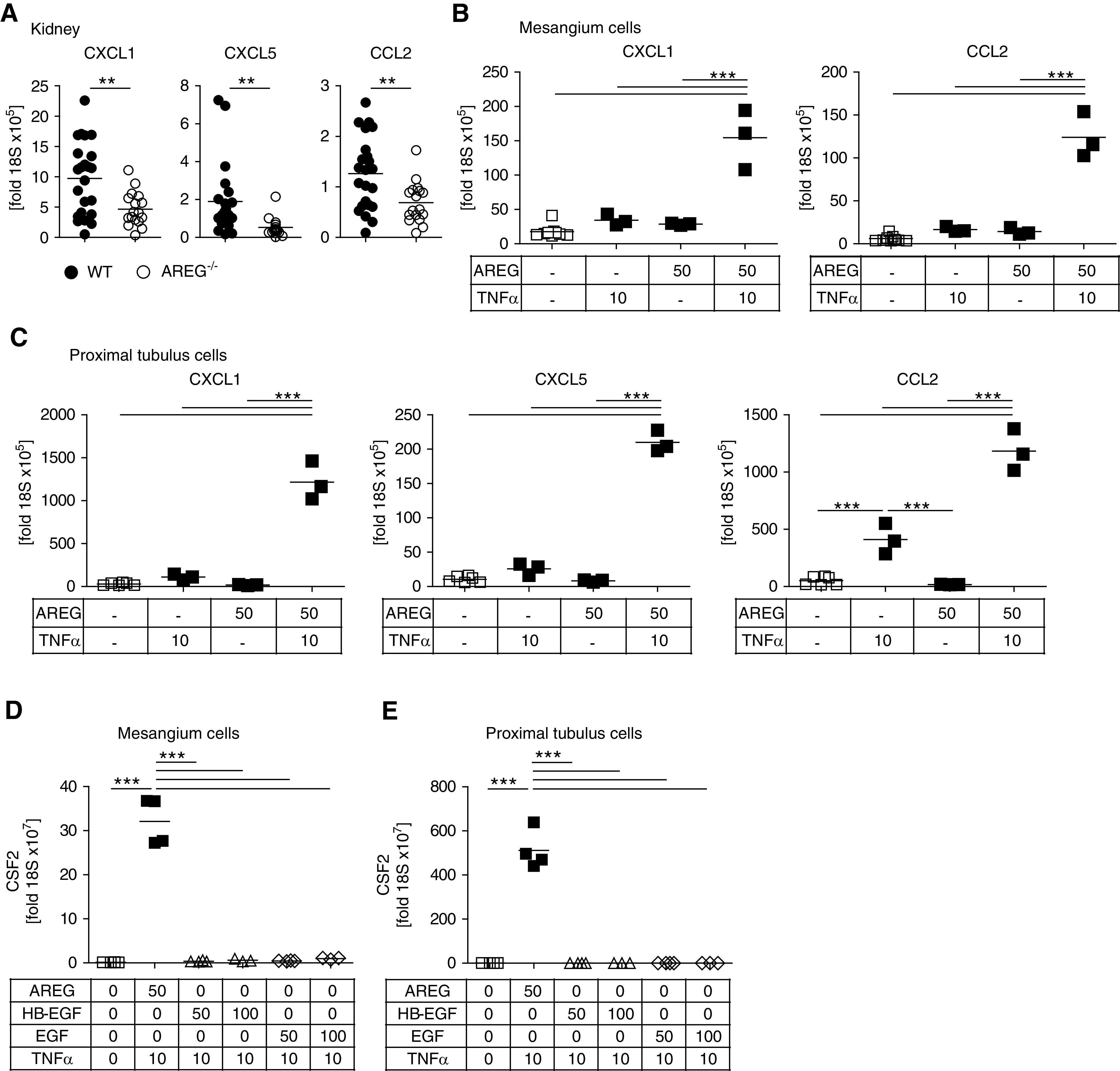
AREG induces myeloid cell–attracting chemokines and CSF2 secretion from resident renal cells. (A) Renal mRNA expression of the indicated chemokines at day 10 after NTN. (B) Expression of the indicated chemokine mRNAs by cultured mMC after stimulation with AREG and/or TNFα at the indicated concentrations in nanograms per milliliter. (C) Expression of the indicated chemokine mRNAs by cultured mPTC after stimulation with AREG and/or TNFα at the indicated concentrations in nanograms per milliliter. (D and E) Expression of CSF2 mRNA by cultured mMC and mPTC after stimulation with AREG, HB-EGF, or EGF in combination with TNFα at the indicated concentrations in nanograms per milliliter. Symbols represent individual animals or cell culture preparations, horizontal lines show mean values. **P<0.01; ***P<0.001. WT, wild-type.
AREG Enhances M/M Proliferation and Induces a Proinflammatory M1 Phenotype
Next, we aimed to study whether AREG might also have direct effects on M/M themselves, because these upregulate the EGFR during inflammation (Supplemental Figure 11A). Indeed, we found AREG to strongly enhance M/M proliferation in vitro (Figure 5A). This effect was AREG-specific and could not be reproduced with HB-EGF or EGF (Figure 5, B and C). Furthermore, we found that AREG also effectively protected M/M from apoptosis in vivo (Figure 5D). We then aimed to analyze whether AREG could also alter the M/M phenotype. Supporting our in vivo observations, we found that in vitro treatment with AREG promoted generation of proinflammatory Ly6Chi M1-type macrophages in both the presence and absence of inflammatory conditions (Figure 5E). AREG stimulation also enhanced M/M production of the highly proinflammatory cytokine IL-1β (Figure 5F). This effect was again specific to AREG as neither HB-EGF nor EGF increased IL-1β levels (Figure 5G). Finally, we found that in addition to promoting M1-type proinflammatory functions, AREG also suppressed the M2 characteristic phagocytic capacity of M/M (Figure 5, H and I). All AREG effects in our in vitro culture systems required release of endogenous AREG as M/M from AREG−/− mice did not respond to treatment with a single AREG dose (Supplemental Figure 11, B–E). Importantly, however, M/M from AREG−/− mice were not generally hyporesponsive because they proliferated equally well in response to TNFα stimulation (Supplemental Figure 11F). Indeed, we found that AREG treatment self-induced AREG expression by M/M similar to the feed-forward loop observed in mMC and mPTC (Supplemental Figure 11G). Interestingly, the requirement for endogenous AREG release could be fully bypassed by supplementation of exogenous AREG (Supplemental Figure 11H).
Figure 5.
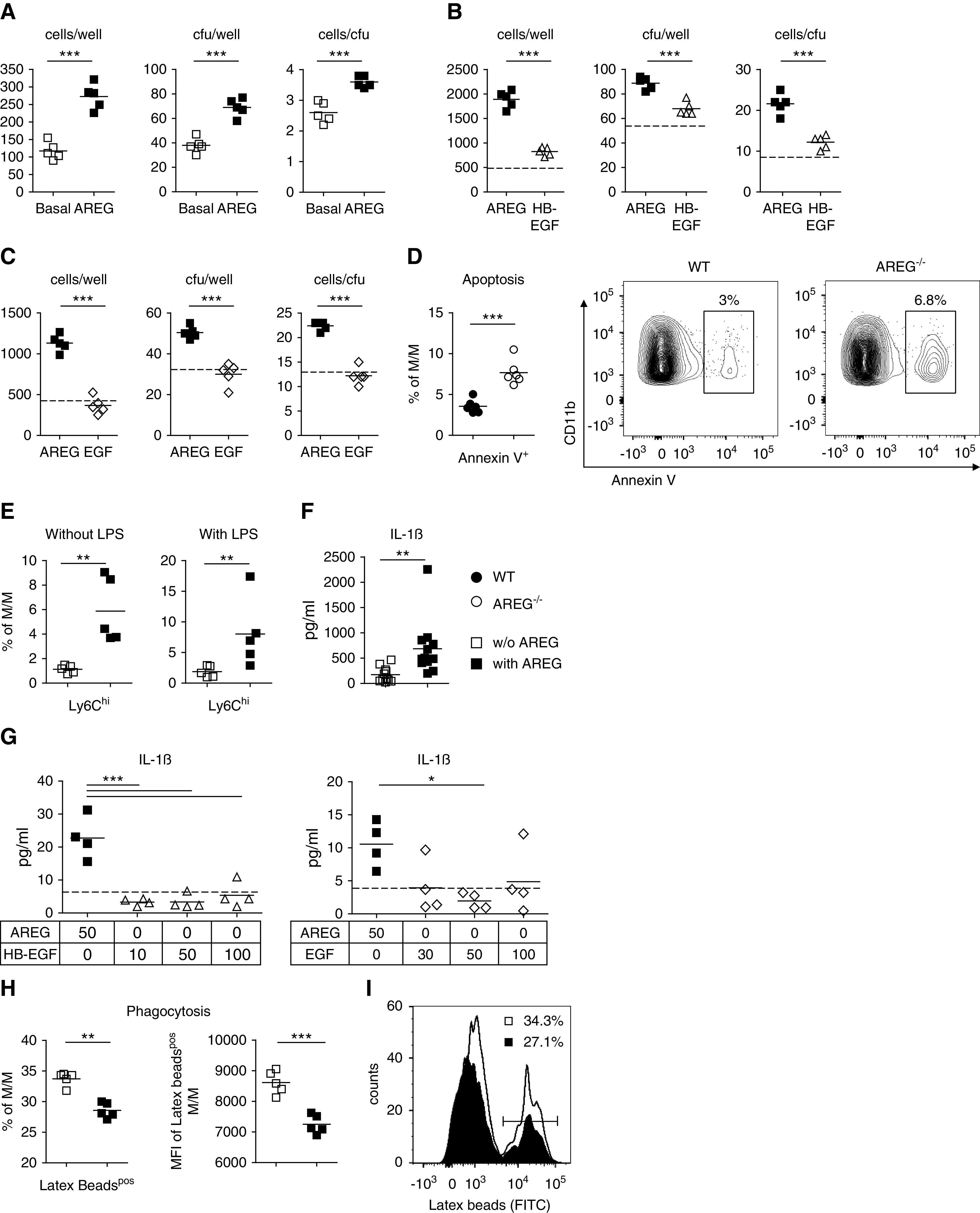
AREG enhances M/M proliferation and induces a proinflammatory M1 phenotype. (A) In vitro M/M proliferation at 6 days after stimulation with 50 ng/ml AREG, as measured by the number of cells per well, CFU per well, and cells per CFU. (B) In vitro M/M proliferation at 6 days after stimulation with 50 ng/ml AREG compared with 50 ng/ml HB-EGF, as measured by the number of cells per well, CFU per well, and cells per CFU. (C) In vitro M/M proliferation at 5 days after stimulation with 50 ng/ml AREG compared with 50 ng/ml EGF, as measured by the number of cells per well, CFU per well, and cells per CFU. Dotted lines in (C and D) indicate basal conditions without addition of EGFR ligands. (D) Quantification and representative FACS plot of apoptosis from blood M/M of naïve mice with the indicated genotypes. (E) FACS quantification of Ly6Chigh M1-type M/M after 48 hours in vitro stimulation with or without LPS, in the presence or absence of 50 ng/ml AREG. (F) IL-1β secretion as measured by ELISA in the supernatant of M/M after 24 hours in vitro stimulation with or without 50 ng/ml AREG. (G) IL-1β secretion as measured by ELISA in the supernatant of M/M after 24 hours in vitro stimulation with 50 ng/ml AREG compared with indicated concentrations of HB-EGF (left) and EGF (right) in nanograms per milliliter. Dotted line indicates IL-1β levels at basal conditions. (H) Percentages of latex beads (FITC)pos M/M (left) and latex bead (FITC) mean fluorescence intensity (MFI) within latex beadpos M/M (right) after 48 hours in vitro culture with or without 50 ng/ml AREG. (I) Representative histogram of latex beads (FITC)-positive CD11b+ M/M. Symbols represent individual animals or cell culture preparations, horizontal lines show mean values. *P<0.05; **P<0.01; ***P<0.001. WT, wild-type.
AREG from Nonhematopoietic Cells Enhances M/M Responses and Aggravates NTN
In order to further investigate the differential roles of hematopoietic versus nonhematopoietic sources of AREG, we next performed bone marrow transplantation experiments. As suggested by our previous findings, loss of AREG from hematopoietic cells (AREG−/− into wild-type) did not result in amelioration of NTN. Loss of AREG from nonhematopoietic cells (wild-type into AREG−/−), however, conferred significant protection from renal injury in comparison with wild-type (wild-type into WT) controls (Figure 6, A and B). Notably, we again found strong effects on M/M polarization. Similar to the situation in AREG pan-knockout mice, loss of AREG selectively from nonhematopoietic cells resulted in skewing of M/M toward an M2 phenotype. Frequencies of M1-type renal Ly6chigh M/M were significantly reduced (Figure 6, C and D), whereas conversely, percentages of CD206+ M2-type M/M (Figure 6, E and F) and M/M expression levels of CD206 (Figure 6G) were enhanced. In contrast to the pronounced effects on M/M, renal CD3+ T cell numbers, as well as Foxp3+ Treg numbers and percentages, were not different between the groups (Supplemental Figure 12, A–D).
Figure 6.
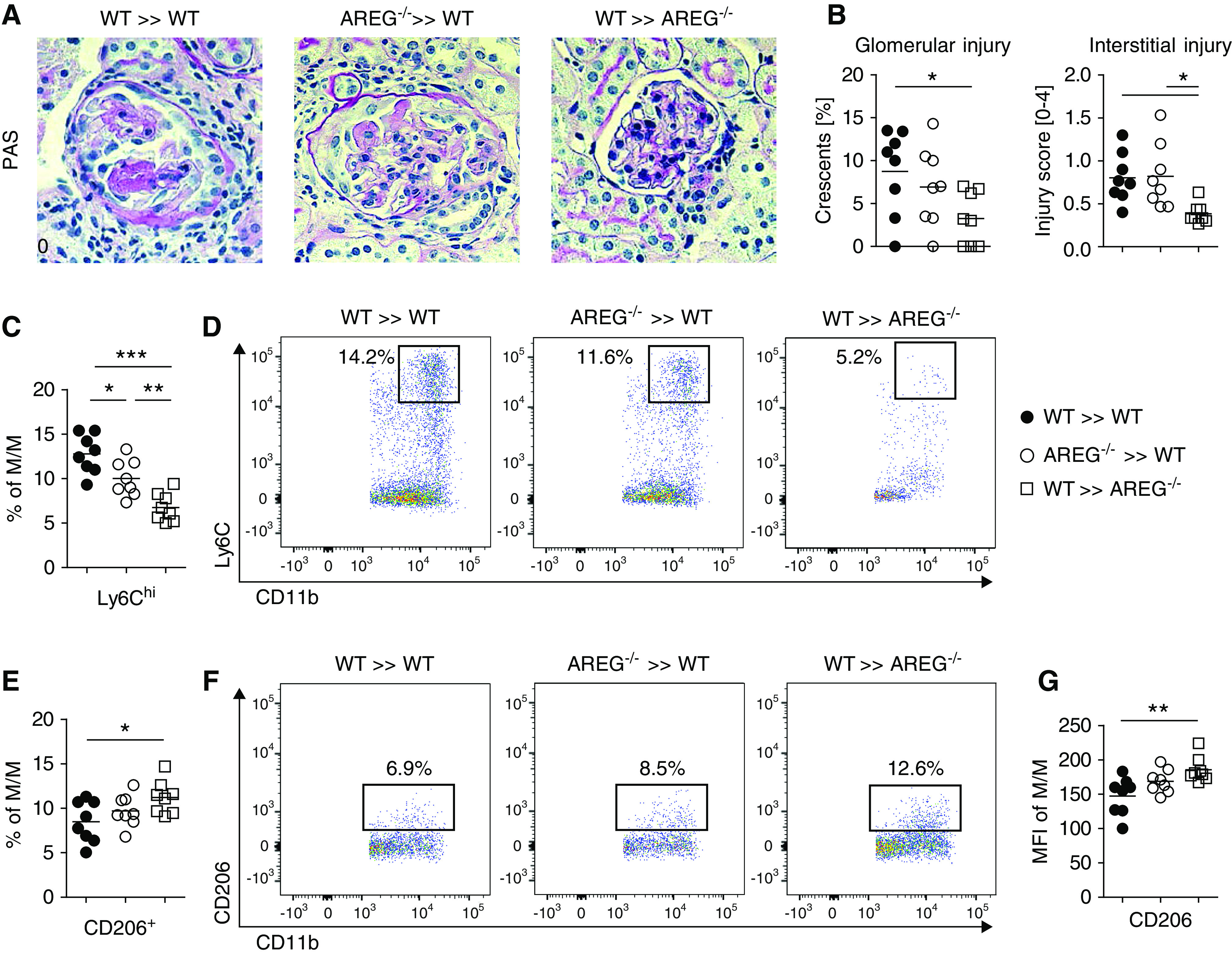
AREG from nonhematopoietic cells enhances M/M responses and aggravates NTN. (A) PAS-stained sections of nephritic kidneys from the indicated bone marrow donor/recipient combinations at day 7 of NTN (original magnification ×400). (B) Quantification of glomerular crescents and interstitial injury. (C) FACS quantification of renal Ly6Chigh inflammatory M/M of the indicated groups of mice. (D) Representative FACS plots of renal Ly6Chigh inflammatory M/M. (E) FACS quantification of renal CD206/Mannose receptor+ M2-type M/M of the indicated groups of mice. (F) Representative FACS plots of renal CD206/Mannose receptor+ M2-type M/M. (G) Mean fluorescence intensity (MFI) of CD206/Mannose receptor within renal M/M. Symbols represent individual animals, horizontal lines show mean values. *P<0.05; **P<0.01; ***P<0.001. PAS, periodic acid–Schiff; WT, wild-type.
AREG Neutralization in the Macrophage-Dependent Effector Phase Ameliorates Nephritis
Because our data pointed toward a role of AREG for proinflammatory M/M activation, we next aimed to provide in vivo evidence. In order to minimize effects on T cell activation in the induction phase of adaptive immunity and more specifically target T cell–activated M/M in the effector phase of nephritis, we switched to the model of aNTN. Wild-type mice were preimmunized with sheep globulin to allow for development of T cell responses against the nephritogenic antigen, and aNTN was subsequently induced. A single dose of 5 µg of anti-AREG antibodies (Figure 7A) 1 day after injection of NTN serum was already sufficient to mildly but significantly ameliorate nephritis (Figure 7B). The degree of protection was dose-dependent because repeated injections of 7.5 µg of anti-AREG antibodies (Figure 7C) in a second set of experiments resulted in more profound reduction of renal tissue injury (Figure 7, D and E). Supporting a role of AREG for M/M responses, we found significantly reduced glomerular and interstitial renal macrophage infiltration in the anti-AREG group (Figure 7, F–I). T cell infiltration, in contrast, remained unaffected (Supplemental Figure 13, A and B). Effects on systemic T cell responses were also minor, in particular when compared with results from constitutively AREG-deficient mice, showing only marginally increased percentages of IFNγ-producing splenic CD4+ T effector cells (Supplemental Figure 13C).
Figure 7.
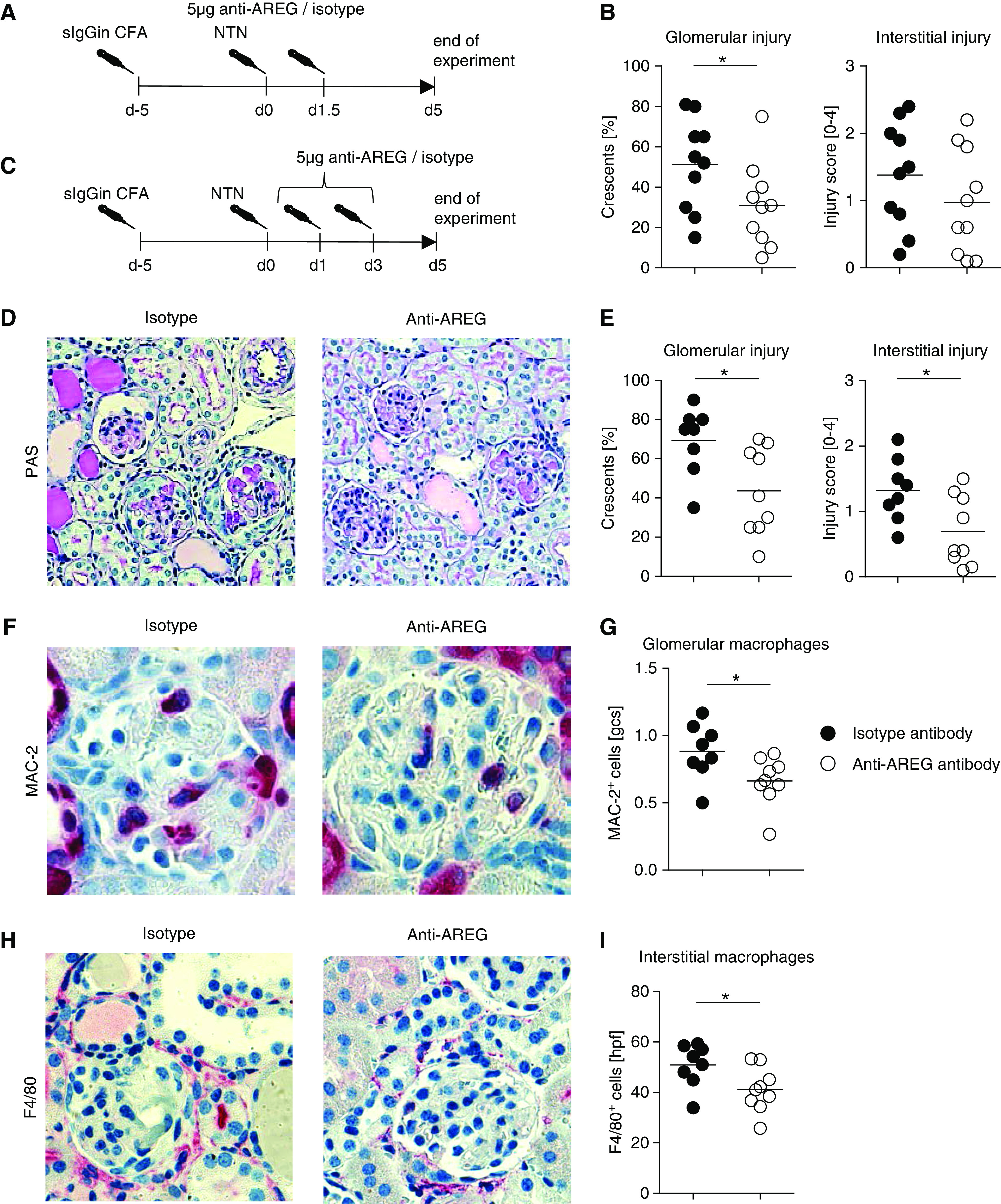
AREG neutralization in the macrophage-dependent effector phase ameliorates nephritis. (A) Overview of the experimental setup. (B) Quantification of glomerular crescents and interstitial injury. (C) Overview of the second experimental setup. (D) PAS-stained sections of nephritic kidneys from anti-AREG or isotype antibody–treated mice at day 5 after induction of aNTN (second experimental setup, original magnification ×200). (E) Quantification of glomerular crescents and interstitial injury from the second experimental setup. (F) Immunohistochemical staining and (G) quantification of glomerular MAC-2+ M/M of the indicated groups from the second experimental setup (original magnification ×400). (H) Immunohistochemical staining and (I) quantification of interstitial F4/80+ M/M of the indicated groups from the second experimental setup (original magnification ×400). Symbols represent individual animals, horizontal lines show mean values. *P<0.05. gcs, glomerular cross section; hpf, high-power field; PAS, periodic acid–Schiff.
Lack of EGFR Signaling in M/M Attenuates Nephritis
Next, we aimed to investigate the functional role of the AREG/EGFR signaling axis directly on M/M in vivo. We thus generated LysMcre×EGFRfl/fl mice, in which lack of EGFR signaling is restricted to M/M. Selective and efficient knockout on M/M was confirmed by PCR of genomic DNA and mRNA from highly purified leukocyte populations (Supplemental Figure 14, A–C). Importantly, after induction of NTN, histologic damage and albuminuria were markedly reduced (Figure 8, A–C) by absence of EGFR signaling on M/M. In line with our previous experiments, we also found renal glomerular and interstitial M/M infiltration to be diminished in LysMcre×EGFRfl/fl mice (Figure 8D). This effect was specific to M/M because renal neutrophil infiltration remained unaltered (Supplemental Figure 14D). Importantly, LysMcre×EGFRfl/fl mice also showed impaired M1-type macrophage responses with significantly reduced proportions of Ly6Chi and CCR2+ M/M (Figure 8, E and F). In addition, we noted significantly lower renal expression of the proinflammatory macrophage-attracting chemokines CCL2 and CCL5, as well as the M1 characteristic inflammatory mediators TNFα and iNOS. Also renal AREG mRNA levels were reduced (Figure 8G). Finally, we studied cell survival and, similar to our findings from AREG−/− mice, we found apoptosis to be increased in M/M from LysMcre×EGFRfl/fl mice (Figure 8H).
Figure 8.
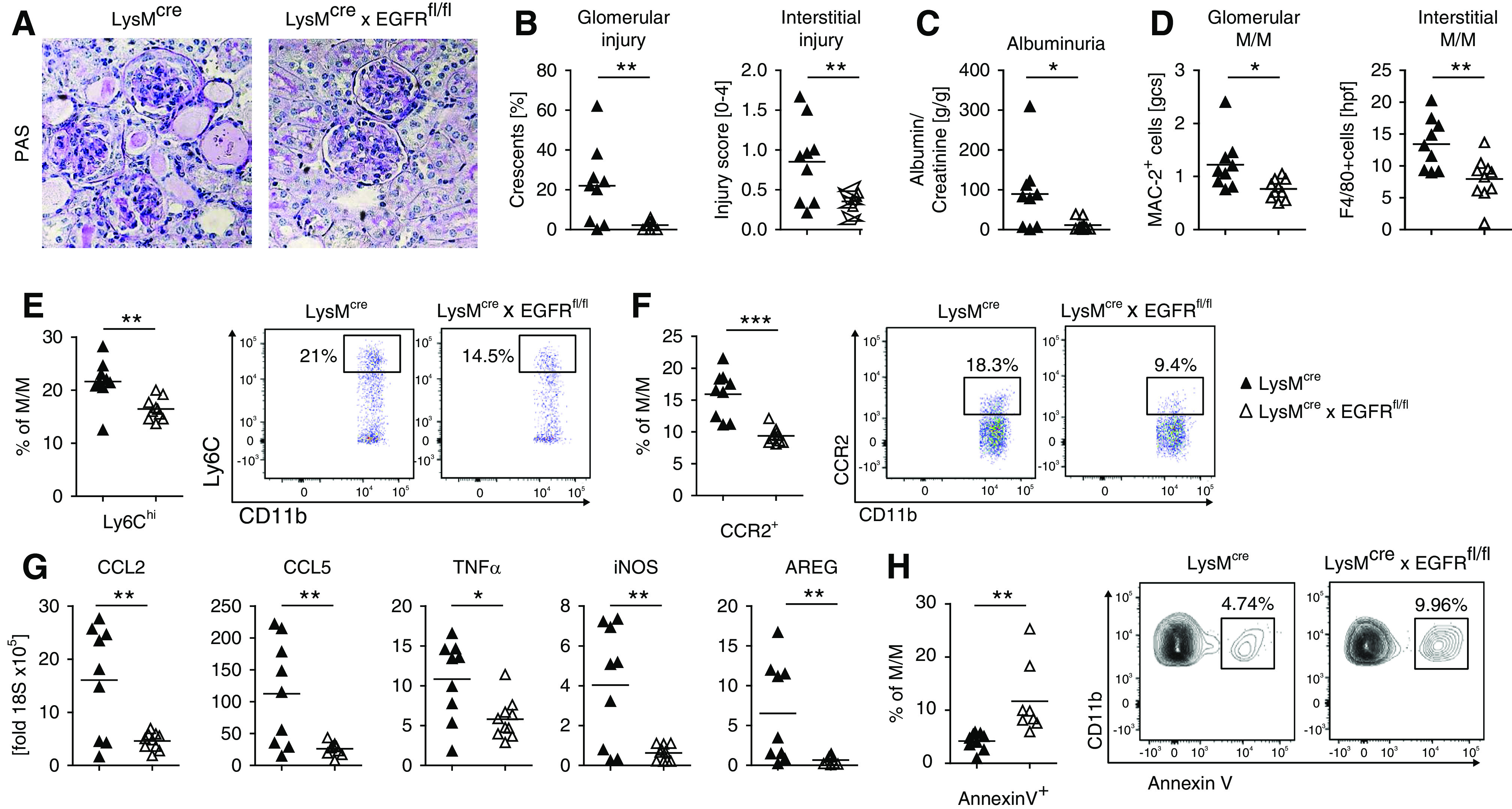
Lack of EGFR signaling in M/M attenuates nephritis. (A) PAS-stained sections of nephritic kidneys from the indicated strains of mice at day 7 (original magnification ×200). (B) Quantification of glomerular crescents and interstitial injury. (C) Albumin-creatinine ratio in the urine of mice from the indicated strains. (D) Quantification of renal glomerular and interstitial M/M infiltration during NTN by immunohistochemistry. (E) Quantification and representative FACS plot of renal Ly6Chigh inflammatory M/M of the indicated strains of mice. (F) Quantification and representative FACS plot of renal CCR2+ inflammatory M/M of the indicated strains of mice. (G) Semiquantitative analyses of renal mRNA expression of the indicated chemokines or cytokines by real-time quantitative PCR. (H) Quantification and representative FACS plot of apoptosis from blood M/M of naïve mice with the indicated genotypes. Symbols represent individual animals, horizontal lines show mean values. *P<0.05; **P<0.01; ***P<0.001. PAS, periodic acid–Schiff.
Renal AREG Expression Is Increased in Human Crescentic GN
Finally, we aimed to assess the potential clinical relevance of our findings for human GN. We thus performed AREG immunostaining on human renal biopsy specimens. In kidneys of control patients, only weak glomerular and tubulointerstitial AREG expression was detected (Figure 9A). Specimens of patients with two different forms of crescentic GN (microscopic polyangiitis and Goodpasture syndrome), however, showed massively increased AREG staining, predominantly in renal resident cells. The strongest positivity was found in areas of glomerular crescents. Furthermore, AREG expression was particularly detected in parietal epithelial cells, podocytes, and tubule cells. Weaker staining was also observed in the periglomerular inflammatory infiltrate (Figure 9, B and C).
Figure 9.
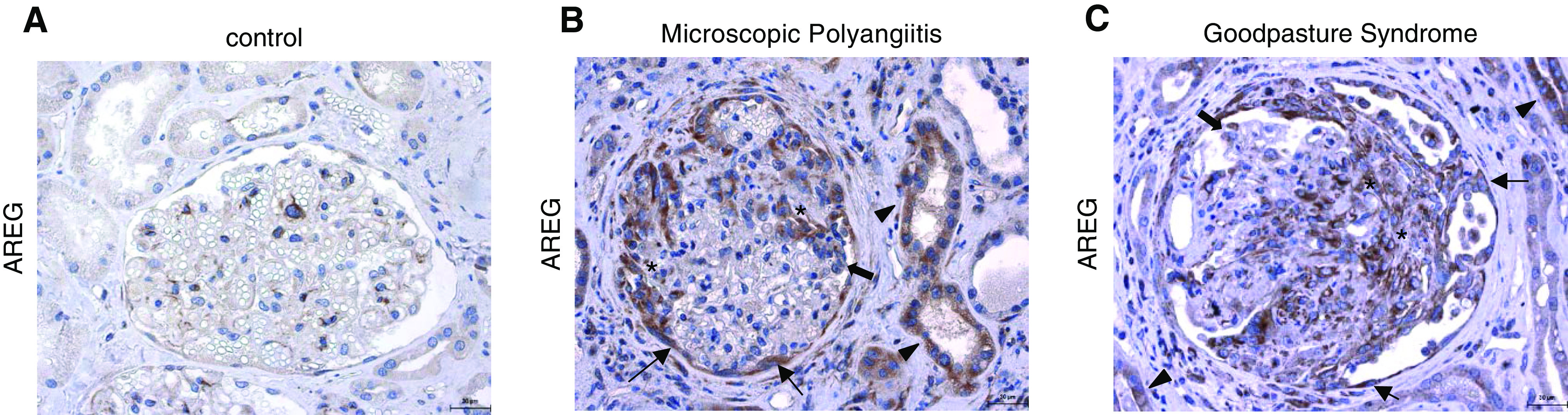
Renal AREG expression is increased in human crescentic GN. AREG immunostaining (brown) of kidney biopsy specimens from (A) control patients, (B) patients with microscopic polyangiitis, and (C) patients with Goodpasture syndrome. Asterisks indicate areas of glomerular crescents, thin arrows indicate parietal epithelial cells, thick arrows indicate podocytes, and arrowheads indicate tubule cells.
Discussion
Our study aimed to evaluate the previously unknown role of AREG in acute GN. Initially, we analyzed expression of the seven known EGFR ligands during the course of NTN. Most of them remained undetected or were constitutively expressed. Interestingly, and in congruence with recent reports from various other renal pathologies,49 renal expression of EGF was significantly reduced. AREG, in contrast, showed a unique and distinctive pattern. Renal mRNA and serum protein levels were highly upregulated during the course of inflammation. This highlights the potential relevance of AREG and indicates that signaling of the different EGFR ligands might not be redundant but could rather result in individual effects, as was previously also suggested by studies of liver inflammation and injury.50,51 As cellular sources for renal AREG, we could in vitro identify mesangium and proximal tubule cells, which resembles the in vivo situation in both mice18 and humans.17 Interestingly, stimulation with numerous well characterized mediators of GN52 could not induce transcription of AREG mRNA. Rather, we found evidence for a feed-forward loop, in which AREG strongly enhances its own secretion. This positive feedback mechanism might initially be sparked by early mediators of renal inflammation, such as TNFα, which we found to enhance AREG’s self-stimulating effects.
Next, we evaluated the functional effects of AREG on the clinical course of acute GN. Studies at day 7 and 10 after nephritis induction, two time points at which renal AREG levels are highest, uniformly showed significant protection from disease in AREG-deficient mice. This finding is remarkable because AREG has recently been described to potently enhance the immunosuppressive function of Tregs.30–34 Furthermore, several studies published in high impact journals could show that AREG derived from Tregs and other immune cells has tissue reparative properties.20–27 We thus aimed to identify the mechanisms by which AREG mediated its surprisingly strong proinflammatory effects in acute GN. In this respect, broad analyses of renal and systemic immune responses revealed the expected enhancement of CD4+ T helper cell activation in AREG−/− mice.30 In contrast, however, we also found a significant reduction of renal myeloid cell infiltration and activation. It thus seems that AREG has strong proinflammatory effects on myeloid cells, which in our acute GN model overrule the suppressive effects on CD4+ T cell responses. Indeed, more detailed studies of M/M showed significant skewing toward an anti-inflammatory M2 macrophage phenotype in the absence of AREG. These changes were restricted to renal, but not systemic M/M, indicating local shaping of the M/M phenotype in the micromilieu of the inflamed kidneys.
In order to shed light on how exactly AREG influences renal M/M responses, we started by studying effects on renal resident cells. Interestingly, renal levels of myeloid cell–attracting chemokines CXCL1, CXCL5, and CCL2 were significantly reduced in AREG-deficient mice. On the other hand, these chemokines were strongly induced after stimulation of both mMC and mPTC with AREG in combination with TNFα. AREG might thus mediate indirect effects on myeloid cell responses via facilitating their renal recruitment. These effects, however, were not unique to AREG, as the prototypic EGFR ligands HB-EGF and EGF could also stimulate chemokine transcription, albeit somewhat less effectively. Next, we sought to evaluate effects of AREG on M/M proliferation and phenotype. Indeed, stimulation of mMC and mPTC resulted in enhanced CSF2/GM-CSF transcription, which is known to support differentiation and proliferation of proinflammatory M1-type M/M.53 Furthermore, direct in vitro stimulation of M/M with AREG much enhanced their survival and proliferation. These previously unknown effects were exclusively induced by AREG but not by HB-EGF or EGF. In line with these data, AREG protected M/M from apoptosis and in vitro stimulation resulted in skewing toward an M1 phenotype with increased proinflammatory IL-1β secretion.
Finally, we also measured phagocytosis, which is a characteristic hallmark of M2-type M/M, and found significant impairment after AREG stimulation. We thus suspected that renal AREG enhances pathogenic myeloid cell responses via two pathways. Firstly, in an indirect fashion, AREG-induced secretion of chemokines from resident renal cells might recruit activated M/M and granulocytes into the kidneys. Future studies analyzing mice with renal cell type–selective EGFR deficiency will help to validate and refine this hypothesis. Secondly, in a direct manner, AREG seems to favor proinflammatory M1-type M/M generation and expansion. Although the indirect effects are likely to be shared among several EGFR ligands, the direct effects on M/M seem to be unique to AREG. It is tempting to speculate that these M/M activating functions might significantly contribute to AREG’s strong profibrotic properties, which are mechanistically still poorly understood.18 In order to further delineate the cellular source of proinflammatory AREG, we next performed bone marrow transplantation experiments. Indeed, mice with AREG deficiency restricted to tissue resident cells showed changes very similar to pan-AREG−/− mice. NTN was significantly ameliorated and M/M showed distinctive skewing toward the M2 phenotype. Wild-type recipients of AREG−/− bone marrow, in contrast, were not protected from nephritis. In conclusion, proinflammatory AREG in our model seems to derive from resident renal cells, rather than from infiltrating leukocytes. This finding is remarkable and supports a novel concept, which might be more broadly applicable. Although leukocyte-secreted AREG was shown to mediate protective effects,20–27,30–34 tissue-derived AREG seems to be profibrotic18 and proinflammatory. The exact cellular sources of AREG, as well as the mechanisms and differential intracellular EGFR signaling pathways underlying this observation clearly warrant further studies.
Next, we wanted to evaluate whether blockade of AREG could be of potential use as a therapy for established GN. In order to predominantly target activated myeloid cells, we made use of the aNTN model.54 Mice were preimmunized with sheep-globulin, allowing unaltered induction of adaptive T cell responses against the subsequently deposited nephritogenic antigen. Nephritis was then induced at day 5 after immunization. This was followed by therapeutic application of anti-AREG antibodies during the effector phase of aNTN, in which T cell–stimulated macrophages are active. Using this protocol, neutralization of AREG indeed predominantly reduced renal macrophage responses, with only minimal effects on T cells. Importantly, AREG blockade resulted in amelioration of renal injury in a dose-dependent manner, indicating potential clinical usefulness. However, our findings also suggest careful timing of AREG-directed interventions. These are more likely to be useful during an acute flare of nephritis but might be less effective, or even deleterious, during chronic T cell–dominated forms of inflammation. Next, we aimed to clarify whether abrogating AREG’s effects specifically on M/M would be sufficient to ameliorate GN. Furthermore, this setup avoids potential bias, which might be caused by altered AREG signaling in T cells, or other yet undefined cellular players. We therefore generated mice with myeloid cell–restricted EGFR deficiency and induced NTN. Supporting our hypothesis and our findings from the AREG-directed interventions, we again found significant amelioration of renal injury in the knockout mice. Importantly, and in line with recent studies on experimental colitis,55 infection,56 and atherosclerosis,57 EGFR silencing in myeloid cells was accompanied by reduced renal M/M influx and skewing toward an anti-inflammatory M2 phenotype. Furthermore, renal M1-type M/M-attracting chemokines and M1 characteristic TNFα and iNOS expression were diminished. Last but not least, similar to our findings from AREG pan-deficient mice, we also found that EGFR signaling protected M/M from apoptosis. Finally, in order to evaluate the potential clinical relevance of our findings for human disease, we analyzed AREG expression in kidneys from patients with two different types of crescentic GN. Regardless of the entity and in line with our findings from the murine studies, strong upregulation of AREG was predominantly found in various types of resident renal cells. Although crescent areas showed strongest staining intensity, single cell types that could be identified included parietal epithelial cells, podocytes and tubule cells. In addition, somewhat weaker staining was also detected in cells of the periglomerular infiltrate. These findings call for future studies to better define and characterize the exact cellular sources of AREG, as well as their individual relevance. Taken together, our data identify AREG as a novel mediator of acute crescentic GN. We show that renal tissue–derived AREG has potent direct and probably also indirect proinflammatory effects on myeloid cell responses by (1) enhancing renal myeloid cell infiltration via induction of chemokine secretion from resident renal cells; and (2) direct effects on M/M by enhancing M1-type polarization, proliferation, and cytokine secretion. Our results thus favor studies evaluating AREG-specific antibody neutralization for the treatment of acute renal inflammation.
Disclosures
All authors have nothing to disclose.
Funding
This work was supported by Deutsche Forschungsgemeinschaft grants STE 1822/6-1 and SFB 1192 TPA03 (to O.M. Steinmetz).
Supplementary Material
Acknowledgments
We thank I. Holtze for her excellent technical help.
Dr. Oliver M. Steinmetz designed the study. Dr. Oliver M. Steinmetz and Dr. Simon Melderis wrote the manuscript. Dr. Julia Hagenstein, Dr. Simon Melderis, Dr. Matthias Tobias Warkotsch, Dr. Julien Dang, Dr. Georg Rudolf Herrnstadt, and Dr. Christoph Benjamin Niehus carried out experiments. Dr. Carmen Berasain, Dr. Matias A. Avila, Dr. Gisa Tiegs, and Dr. Pierre-Louis Tharaux donated mice. Dr. Katrin Neumann, Dr. Matias A. Avila, Dr. Gisa Tiegs, Dr. Pierre-Louis Tharaux, and Dr. Ulf Panzer critically evaluated the manuscript. Dr. Simon Melderis, Dr. Julia Hagenstein, Dr. Julien Dang, Dr. Pierre-Louis Tharaux, and Dr. Oliver M. Steinmetz analyzed the data.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
Supplemental Material
This article contains the following supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2019111215/-/DCSupplemental.
Supplemental Figure 1. Time course of mRNA expression of the indicated EGFR ligands during NTN.
Supplemental Figure 2. Effect of stimulation with various cytokines on AREG mRNA expression.
Supplemental Figure 3. Dependence of AREG mRNA induction on AREG/EGFR signaling.
Supplemental Figure 4. EGFR ligand expression in nephritic AREG−/− mice.
Supplemental Figure 5. Analyses of renal and systemic T cells in NTN.
Supplemental Figure 6. Analyses of renal eosinophils in NTN.
Supplemental Figure 7. CD206 expression on renal M/M and systemic M/M phenotype.
Supplemental Figure 8. Efficacy of neutralizing anti-AREG antibody and erlotinib on chemokine expression.
Supplemental Figure 9. Expression of chemokines from mMC after stimulation with HB-EGF or EGF.
Supplemental Figure 10. Expression of chemokines from mPTC after stimulation with HB-EGF or EGF.
Supplemental Figure 11. EGFR expression on activated M/M and analyses of AREG−/− M/M.
Supplemental Figure 12. Renal T cell and Treg infiltration after bone marrow transplantation.
Supplemental Figure 13. Renal and systemic T cell responses in anti-AREG–treated mice.
Supplemental Figure 14. Specificity of EGFR excision in M/M from LysMCre×EGFRfl/fl mice.
References
- 1.Swanson CD, Akama-Garren EH, Stein EA, Petralia JD, Ruiz PJ, Edalati A, et al.: Inhibition of epidermal growth factor receptor tyrosine kinase ameliorates collagen-induced arthritis. J Immunol 188: 3513–3521, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhuang S, Liu N: EGFR signaling in renal fibrosis. Kidney Int Suppl (2011) 4: 70–74, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Harskamp LR, Gansevoort RT, van Goor H, Meijer E: The epidermal growth factor receptor pathway in chronic kidney diseases. Nat Rev Nephrol 12: 496–506, 2016. [DOI] [PubMed] [Google Scholar]
- 4.Bollée G, Flamant M, Schordan S, Fligny C, Rumpel E, Milon M, et al.: Epidermal growth factor receptor promotes glomerular injury and renal failure in rapidly progressive crescentic glomerulonephritis. Nat Med 17: 1242–1250, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Terzi F, Burtin M, Hekmati M, Federici P, Grimber G, Briand P, et al.: Targeted expression of a dominant-negative EGF-R in the kidney reduces tubulo-interstitial lesions after renal injury. J Clin Invest 106: 225–234, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lautrette A, Li S, Alili R, Sunnarborg SW, Burtin M, Lee DC, et al.: Angiotensin II and EGF receptor cross-talk in chronic kidney diseases: A new therapeutic approach. Nat Med 11: 867–874, 2005. [DOI] [PubMed] [Google Scholar]
- 7.Chen J, Chen JK, Nagai K, Plieth D, Tan M, Lee TC, et al.: EGFR signaling promotes TGFβ-dependent renal fibrosis. J Am Soc Nephrol 23: 215–224, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.François H, Placier S, Flamant M, Tharaux PL, Chansel D, Dussaule JC, et al.: Prevention of renal vascular and glomerular fibrosis by epidermal growth factor receptor inhibition. FASEB J 18: 926–928, 2004. [DOI] [PubMed] [Google Scholar]
- 9.Lee HW, Khan SQ, Khaliqdina S, Altintas MM, Grahammer F, Zhao JL, et al.: Absence of miR-146a in podocytes increases risk of diabetic glomerulopathy via up-regulation of ErbB4 and notch-1. J Biol Chem 292: 732–747, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Qing X, Chinenov Y, Redecha P, Madaio M, Roelofs JJ, Farber G, et al.: iRhom2 promotes lupus nephritis through TNF-α and EGFR signaling. J Clin Invest 128: 1397–1412, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang MZ, Sasaki K, Li Y, Li Z, Pan Y, Jin GN, et al.: The role of the EGF receptor in sex differences in kidney injury. J Am Soc Nephrol 30: 1659–1673, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Berasain C, Avila MA: Amphiregulin. Semin Cell Dev Biol 28: 31–41, 2014. [DOI] [PubMed] [Google Scholar]
- 13.Cook PW, Piepkorn M, Clegg CH, Plowman GD, DeMay JM, Brown JR, et al.: Transgenic expression of the human amphiregulin gene induces a psoriasis-like phenotype. J Clin Invest 100: 2286–2294, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yamane S, Ishida S, Hanamoto Y, Kumagai K, Masuda R, Tanaka K, et al.: Proinflammatory role of amphiregulin, an epidermal growth factor family member whose expression is augmented in rheumatoid arthritis patients. J Inflamm (Lond) 5: 5, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lisi S, Sisto M, Lofrumento DD, Cucci L, Frassanito MA, Mitolo V, et al.: Pro-inflammatory role of Anti-Ro/SSA autoantibodies through the activation of Furin-TACE-amphiregulin axis. J Autoimmun 35: 160–170, 2010. [DOI] [PubMed] [Google Scholar]
- 16.Perugorria MJ, Latasa MU, Nicou A, Cartagena-Lirola H, Castillo J, Goñi S, et al.: The epidermal growth factor receptor ligand amphiregulin participates in the development of mouse liver fibrosis. Hepatology 48: 1251–1261, 2008. [DOI] [PubMed] [Google Scholar]
- 17.Kefaloyianni E, Muthu ML, Kaeppler J, Sun X, Sabbisetti V, Chalaris A, et al.: ADAM17 substrate release in proximal tubule drives kidney fibrosis. JCI Insight 1: e87023, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kefaloyianni E, Keerthi Raja MR, Schumacher J, Muthu ML, Krishnadoss V, Waikar SS, et al.: Proximal tubule-derived amphiregulin amplifies and integrates profibrotic EGF receptor signals in kidney fibrosis. J Am Soc Nephrol 30: 2370–2383, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Santamaría E, Rodríguez-Ortigosa CM, Uriarte I, Latasa MU, Urtasun R, Alvarez-Sola G, et al.: The epidermal growth factor receptor ligand amphiregulin protects from cholestatic liver injury and regulates bile acids synthesis. Hepatology 69: 1632–1647, 2019. [DOI] [PubMed] [Google Scholar]
- 20.Burzyn D, Kuswanto W, Kolodin D, Shadrach JL, Cerletti M, Jang Y, et al.: A special population of regulatory T cells potentiates muscle repair. Cell 155: 1282–1295, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Arpaia N, Green JA, Moltedo B, Arvey A, Hemmers S, Yuan S, et al.: A distinct function of regulatory T cells in tissue protection. Cell 162: 1078–1089, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carney K, Chang YR, Wilson S, Calnan C, Reddy PS, Chan WY, et al.: Regulatory T-cell-intrinsic amphiregulin is dispensable for suppressive function. J Allergy Clin Immunol 137: 1907–1909, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ito M, Komai K, Mise-Omata S, Iizuka-Koga M, Noguchi Y, Kondo T, et al.: Brain regulatory T cells suppress astrogliosis and potentiate neurological recovery. Nature 565: 246–250, 2019. [DOI] [PubMed] [Google Scholar]
- 24.Monticelli LA, Osborne LC, Noti M, Tran SV, Zaiss DM, Artis D: IL-33 promotes an innate immune pathway of intestinal tissue protection dependent on amphiregulin-EGFR interactions. Proc Natl Acad Sci U S A 112: 10762–10767, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cao Q, Wang Y, Niu Z, Wang C, Wang R, Zhang Z, et al.: Potentiating tissue-resident type 2 innate lymphoid cells by IL-33 to prevent renal ischemia-reperfusion injury. J Am Soc Nephrol 29: 961–976, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fujiu K, Shibata M, Nakayama Y, Ogata F, Matsumoto S, Noshita K, et al.: A heart-brain-kidney network controls adaptation to cardiac stress through tissue macrophage activation. Nat Med 23: 611–622, 2017. [DOI] [PubMed] [Google Scholar]
- 27.Minutti CM, Modak RV, Macdonald F, Li F, Smyth DJ, Dorward DA, et al.: A macrophage-pericyte axis directs tissue restoration via amphiregulin-induced transforming growth factor beta activation. Immunity 50: 645–654 e6, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zaiss DM, Yang L, Shah PR, Kobie JJ, Urban JF, Mosmann TR: Amphiregulin, a TH2 cytokine enhancing resistance to nematodes. Science 314: 1746, 2006. [DOI] [PubMed] [Google Scholar]
- 29.Minutti CM, Drube S, Blair N, Schwartz C, McCrae JC, McKenzie AN, et al.: Epidermal growth factor receptor expression licenses type-2 helper T cells to function in a T cell receptor-independent fashion. Immunity 47: 710–722 e6, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zaiss DM, van Loosdregt J, Gorlani A, Bekker CP, Gröne A, Sibilia M, et al.: Amphiregulin enhances regulatory T cell-suppressive function via the epidermal growth factor receptor. Immunity 38: 275–284, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dai K, Huang L, Chen J, Yang L, Gong Z: Amphiregulin promotes the immunosuppressive activity of intrahepatic CD4+ regulatory T cells to impair CD8+ T-cell immunity against hepatitis B virus infection. Immunology 144: 506–517, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yuan CH, Sun XM, Zhu CL, Liu SP, Wu L, Chen H, et al.: Amphiregulin activates regulatory T lymphocytes and suppresses CD8+ T cell-mediated anti-tumor response in hepatocellular carcinoma cells. Oncotarget 6: 32138–32153, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang S, Zhang Y, Wang Y, Ye P, Li J, Li H, et al.: Amphiregulin confers regulatory T cell suppressive function and tumor invasion via the EGFR/GSK-3β/Foxp3 Axis. J Biol Chem 291: 21085–21095, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nosbaum A, Prevel N, Truong HA, Mehta P, Ettinger M, Scharschmidt TC, et al.: Cutting edge: Regulatory T cells facilitate cutaneous wound healing. J Immunol 196: 2010–2014, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jiang R, Tang J, Chen Y, Deng L, Ji J, Xie Y, et al.: The long noncoding RNA lnc-EGFR stimulates T-regulatory cells differentiation thus promoting hepatocellular carcinoma immune evasion. Nat Commun 8: 15129, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wolf D, Hochegger K, Wolf AM, Rumpold HF, Gastl G, Tilg H, et al.: CD4+CD25+ regulatory T cells inhibit experimental anti-glomerular basement membrane glomerulonephritis in mice. J Am Soc Nephrol 16: 1360–1370, 2005. [DOI] [PubMed] [Google Scholar]
- 37.Kluger MA, Luig M, Wegscheid C, Goerke B, Paust HJ, Brix SR, et al.: Stat3 programs Th17-specific regulatory T cells to control GN. J Am Soc Nephrol 25: 1291–1302, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kluger MA, Meyer MC, Nosko A, Goerke B, Luig M, Wegscheid C, et al.: RORγt(+)Foxp3(+) cells are an independent bifunctional regulatory T cell lineage and mediate crescentic GN. J Am Soc Nephrol 27: 454–465, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nosko A, Kluger MA, Diefenhardt P, Melderis S, Wegscheid C, Tiegs G, et al.: T-bet enhances regulatory T cell fitness and directs control of Th1 responses in crescentic GN. J Am Soc Nephrol 28: 185–196, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Diefenhardt P, Nosko A, Kluger MA, Richter JV, Wegscheid C, Kobayashi Y, et al.: IL-10 receptor signaling empowers regulatory T cells to control Th17 responses and protect from GN. J Am Soc Nephrol 29: 1825–1837, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ooi JD, Petersen J, Tan YH, Huynh M, Willett ZJ, Ramarathinam SH, et al.: Dominant protection from HLA-linked autoimmunity by antigen-specific regulatory T cells. Nature 545: 243–247, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Alikhan MA, Huynh M, Kitching AR, Ooi JD: Regulatory T cells in renal disease. Clin Transl Immunology 7: e1004, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Luetteke NC, Qiu TH, Fenton SE, Troyer KL, Riedel RF, Chang A, et al.: Targeted inactivation of the EGF and amphiregulin genes reveals distinct roles for EGF receptor ligands in mouse mammary gland development. Development 126: 2739–2750, 1999. [DOI] [PubMed] [Google Scholar]
- 44.Lee TC, Threadgill DW: Generation and validation of mice carrying a conditional allele of the epidermal growth factor receptor. Genesis 47: 85–92, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kluger MA, Zahner G, Paust HJ, Schaper M, Magnus T, Panzer U, et al.: Leukocyte-derived MMP9 is crucial for the recruitment of proinflammatory macrophages in experimental glomerulonephritis. Kidney Int 83: 865–877, 2013. [DOI] [PubMed] [Google Scholar]
- 46.Steinmetz OM, Summers SA, Gan PY, Semple T, Holdsworth SR, Kitching AR: The Th17-defining transcription factor RORγt promotes glomerulonephritis. J Am Soc Nephrol 22: 472–483, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wolf G, Mueller E, Stahl RA, Ziyadeh FN: Angiotensin II-induced hypertrophy of cultured murine proximal tubular cells is mediated by endogenous transforming growth factor-beta. J Clin Invest 92: 1366–1372, 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wolf G, Haberstroh U, Neilson EG: Angiotensin II stimulates the proliferation and biosynthesis of type I collagen in cultured murine mesangial cells. Am J Pathol 140: 95–107, 1992. [PMC free article] [PubMed] [Google Scholar]
- 49.Isaka Y: Epidermal growth factor as a prognostic biomarker in chronic kidney diseases. Ann Transl Med 4[Suppl 1]: S62, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Berasain C, García-Trevijano ER, Castillo J, Erroba E, Santamaría M, Lee DC, et al.: Novel role for amphiregulin in protection from liver injury. J Biol Chem 280: 19012–19020, 2005. [DOI] [PubMed] [Google Scholar]
- 51.Pardo-Saganta A, Latasa MU, Castillo J, Alvarez-Asiain L, Perugorría MJ, Sarobe P, et al.: The epidermal growth factor receptor ligand amphiregulin is a negative regulator of hepatic acute-phase gene expression. J Hepatol 51: 1010–1020, 2009. [DOI] [PubMed] [Google Scholar]
- 52.Kurts C, Panzer U, Anders HJ, Rees AJ: The immune system and kidney disease: Basic concepts and clinical implications. Nat Rev Immunol 13: 738–753, 2013. [DOI] [PubMed] [Google Scholar]
- 53.Fleetwood AJ, Lawrence T, Hamilton JA, Cook AD: Granulocyte-macrophage colony-stimulating factor (CSF) and macrophage CSF-dependent macrophage phenotypes display differences in cytokine profiles and transcription factor activities: Implications for CSF blockade in inflammation. J Immunol 178: 5245–5252, 2007. [DOI] [PubMed] [Google Scholar]
- 54.Luig M, Kluger MA, Goerke B, Meyer M, Nosko A, Yan I, et al.: Inflammation-induced IL-6 functions as a natural brake on macrophages and limits GN. J Am Soc Nephrol 26: 1597–1607, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lu N, Wang L, Cao H, Liu L, Van Kaer L, Washington MK, et al.: Activation of the epidermal growth factor receptor in macrophages regulates cytokine production and experimental colitis. J Immunol 192: 1013–1023, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hardbower DM, Singh K, Asim M, Verriere TG, Olivares-Villagómez D, Barry DP, et al.: EGFR regulates macrophage activation and function in bacterial infection. J Clin Invest 126: 3296–3312, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zeboudj L, Giraud A, Guyonnet L, Zhang Y, Laurans L, Esposito B, et al.: Selective EGFR (Epidermal Growth Factor Receptor) deletion in myeloid cells limits atherosclerosis-brief report. Arterioscler Thromb Vasc Biol 38: 114–119, 2018. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.