

Significance Statement
The combination of exome sequencing with single-cell RNA sequencing can reveal the recipient versus donor origin of each immune cell within human kidney allografts. This approach greatly improves upon previous techniques used to identify and describe leukocyte chimerism within a complex organ, such as Y chromosome identification for sex-mismatched transplants. Exome sequencing and single-cell RNA sequencing of single nucleotide variants indicated that donor-origin macrophages may contribute to the alloimmune response through antigen presentation and signaling, whereas donor-origin T cells remain quiescent. Therefore, teaming these techniques can paint a portrait of the chimerism that may lie behind rejection of a donor kidney.
Keywords: gene expression, transplantation, transcriptional profiling
Abstract
Background
In solid organ transplantation, donor-derived immune cells are assumed to decline with time after surgery. Whether donor leukocytes persist within kidney transplants or play any role in rejection is unknown, however, in part because of limited techniques for distinguishing recipient from donor cells.
Methods
Whole-exome sequencing of donor and recipient DNA and single-cell RNA sequencing (scRNA-seq) of five human kidney transplant biopsy cores distinguished immune cell contributions from both participants. DNA-sequence comparisons used single nucleotide variants (SNVs) identified in the exome sequences across all samples.
Results
Analysis of expressed SNVs in the scRNA-seq data set distinguished recipient versus donor origin for all 81,139 cells examined. The leukocyte donor/recipient ratio varied with rejection status for macrophages and with time post-transplant for lymphocytes. Recipient macrophages displayed inflammatory activation whereas donor macrophages demonstrated antigen presentation and complement signaling. Recipient-origin T cells expressed cytotoxic and proinflammatory genes consistent with an effector cell phenotype, whereas donor-origin T cells appeared quiescent, expressing oxidative phosphorylation genes. Finally, both donor and recipient T cell clones within the rejecting kidney suggested lymphoid aggregation. The results indicate that donor-origin macrophages and T cells have distinct transcriptional profiles compared with their recipient counterparts, and that donor macrophages can persist for years post-transplantation.
Conclusions
Analysis of single nucleotide variants and their expression in single cells provides a powerful novel approach to accurately define leukocyte chimerism in a complex organ such as a transplanted kidney, coupled with the ability to examine transcriptional profiles at single-cell resolution.
Single-cell RNA sequencing (scRNA-seq) technologies offer the ability to measure the transcriptomes of many thousands of cells simultaneously. The resulting data sets have revealed unexpected heterogeneity between cell types and states and new biologic insights.1,2 Natural genetic variations within the coding region of the genome are also captured by scRNA-seq. Using germ line coding sequence data as a reference, this information can be harnessed to identify the origin of each cell from a sample of cells of mixed origin.3
Kidney transplantation offers the greatest survival benefit for patients with ESKD. It was appreciated very early on that acquired tolerance of donor tissues was associated with leukocyte chimerism.4 In the 1960s, long-term, immunosuppression-free graft survival was not uncommon in living related transplants.5 However, with the advent of modern immunosuppression, short-term outcomes improved to >90% at 1 year. Thus, interest in leukocyte chimerism waned and immunosuppression-free transplantation became a rare occurrence. A renewed interest in the relevance of donor leukocyte chimerism has occurred since the 1990s.6 The existence of solid organ transplant–associated graft versus host disease has suggested that donor-derived immune cells can persist and cause pathology years later, but this is extremely rare in kidney, with only six cases reported. Furthermore, donor-derived alveolar macrophages can survive in lung transplants for >3 years.10 Recently, investigators used HLA-disparate lung transplant recipients and FACS analysis to show that donor-derived T cells persist in lung transplantation.11 Also, adoptive transfer of donor-origin T cells has been shown to modulate the alloimmune response.12,13 Other studies report that tissue-resident macrophages are replaced by monocyte-derived cells postinjury.14,15 Whether tissue-resident leukocytes such as macrophages persist and what role they play in kidney is completely unknown. We hypothesized that the immune cell infiltrate in the human kidney transplant is composed of donor- and recipient-origin cells and that the transcriptional patterns of donor-origin immune cells differ from those of recipient cells.
Here we describe scRNA-seq analysis of five kidney transplant biopsies from patients with acute antibody-mediated rejection (ABMR) or nonrejection AKI. We identified all of the major kidney cell types and two main immune cell types (lymphocytes and macrophages). The lymphocyte cluster contained T cells, B cells, and plasma cells. Using whole-exome sequence from recipient and donor for each biopsy sample, we could establish the recipient or donor origin of all of these cells based on expressed single nucleotide polymorphisms. Donor-origin cells made up a substantial proportion of macrophages and lymphocytes. The proportion of donor/recipient cells varies with rejection status for macrophages and with time post-transplant for lymphocytes. Donor-origin macrophages express antigen presentation and complement pathway genes. Donor-origin T cell gene expression correlates with the nonrejecting state. Oxidative phosphorylation genes are differentially expressed in donor-origin T cells compared with recipient T cells, suggesting donor T cells are in a quiescent state.16 This study demonstrates the power of this approach applied to the study of leukocyte chimerism in organ transplantation. Further analysis of donor and host chimerism by scRNA-seq should greatly advance our understanding of the roles donor and recipient immune cells play in the alloimmune response.
Methods
Biopsy Samples
Research core biopsy samples were obtained at the time of indication kidney transplant biopsy at Washington University under an institutional review board–approved protocol. Biopsy tissue was freshly prepared for study.
Single-Cell Isolation and Library Preparation
The renal biopsy was minced into small pieces with a razor blade and incubated at 37°C in freshly prepared dissociation buffer containing 0.25% trypsin and 40 U/ml DNase I. Dissociated cells were harvested every 10 minutes by filtering the cell suspension through a 70-μm cell strainer (pluriSelect) into 10% FBS buffer on ice. The residual biopsy tissue trapped on the cell strainer was dissociated once again with 1 ml dissociation buffer for 10 minutes and passed through the cell strainer into the same FBS buffer from the first collection. We repeated this dissociation procedure three times until most of the tissue had been dissociated into single cells (total dissociation time was approximately 30 minutes). Finally, cells were collected by centrifugation at 400 × g for 5 minutes, resuspended in suspension buffer containing 0.1% BSA, and strained through a 40-μm cell mesh (pluriSelect) to further remove cell clumps and large fragments. Cell viability was approximately 90% for the biopsy specimens used in this study as assessed by Trypan Blue staining.
Bioinformatics and Data Analysis
Cell suspensions were loaded into 10× Chromium lanes for a target cell capture number of 10,000 cells per lane. Two lanes were used per biopsy specimen. The 10× Chromium libraries were prepared according to the manufacturer’s protocol. The 5 Prime kits were used and T cell variable, diversity and joining (VDJ) libraries were also prepared according to the manufacturer’s protocol.
The Illumina HiSeq platform was used to generate paired-end read sequences to a depth of 50,000 reads per cell for gene expression and 5000 reads per cell for T cell VDJ sequence. The Cell Ranger version 3.0 pipeline was used to generate BAM files and gene expression matrices for each biopsy. Expression matrices were used to generate Seurat objects using Seurat version 3 to include cells with genes expressed in at least three cells, 200–2500 genes per cell detected, and <25% mitochondrial genes per cell (Supplemental Figure 1). Each biopsy was initially clustered using an excessive number of principal components and high resolution to create a high number of clusters. Clusters containing two or more different cell type–defining markers were removed as doublet clusters and clusters with a very low average number of genes per cell were removed as debris clusters (see Supplemental Methods, Supplemental References). An integrated data set was created using the standard Seurat version 3 integration analysis pipeline.
The cor() and corrplot() functions in R were used for Spearman correlation of gene expression between donor and recipient T cells and rejecting and nonrejecting T cells, using a significance level of 0.05.
Donor/Recipient Cell Origin: Demuxlet Pipeline
Whole-exome sequencing (WES) was performed on each pair of donor and recipient germline DNA from peripheral blood samples associated with each biopsy specimen. Sequencing of each sample was performed targeting a coverage depth of 50×. Alignment was performed using BWA-MEM. MarkDuplicates with Picard followed by Base Quality Recalibration with GATK was then performed in accordance with the functional equivalence paper published by the Centers for Common Disease Genomics.17 Files were then converted to CRAM format. Variant call format files (.vcf) were created using GATK best-practices filtering for high-quality common variants (allele frequency >0.05). Each combined donor and recipient .vcf and aggregated .BAM file from Cell Ranger output was used as input for the Demuxlet pipeline using Demuxlet default parameters and α of 0, 0.5, and 1.3 Each cell in the final integrated data set was annotated as donor or recipient using the best column from the Demuxlet output data.
Pathway Analysis
Significant differentially expressed genes (adjusted P value <0.05) from the Seurat findmarkers() (minimum percentage of cells with gene=0.25, log fold change threshold of 0.25) were analyzed using the ToppFun online tool. Pathways with a Bonferroni P value >0.05 were removed from the analysis. For T cell pathway analysis, pathways for which only CD3 gene expression was common to the reference pathway gene set were excluded from analysis.
T Cells VDJ Immune Cell Profiling
T cell VDJ sequence was analyzed using VDJ Loupe Browser. Clones were defined as cells with complete VDJ sequence from both α and β chains. Using cell barcodes, T cell clones were identified in the final integrated data set.
Results
Kidney allograft tissue was taken by needle core biopsy from three patients with ABMR and two patients with a nonrejection cause of AKI. All patients diagnosed with ABMR had donor-specific antibodies (DSAs) at the time of biopsy. We included one case of rejection due to donor unique anti-A isoantibody in our ABMR group, assuming this antibody was pathologic (DSA). The other two patients with ABMR had at least one anti-HLA antibody (DSA) with mean fluorescence intensities of at least 5000. Biopsies were performed at 5 days up to 7 years post-transplantation. All patients received anti-thymocyte globulin and methylprednisolone at the time of transplant, followed by maintenance immunosuppression of tacrolimus, mycophenolic acid, and prednisone. One rejecting patient and one nonrejecting patient received additional anti-CD20 antibody infusion at the time of transplantation (Supplemental Table 1).
scRNA-seq of Allograft Biopsies Resolves the Major Kidney and Immune Cell Types
A total of 81,139 cells passed filters with an average of 1124 genes and 2497 transcripts per cell. Unsupervised clustering analysis identified eight individual clusters of kidney cells and two immune cell clusters. One cluster was identified as activated proximal tubular cells because they differentially expressed genes involved in secretion, regulation of apoptosis, and responses to oxidative stress. Interestingly, these cells also have Gene Ontology terms suggesting they are involved in leukocyte activation (Supplemental Figure 2, Supplemental Table 2). We identified 5027 macrophages and 3647 lymphocytes in the same clustering analysis (Figure 1). There was minimal batch effect in this data set (Supplemental Figure 3).
Figure 1.
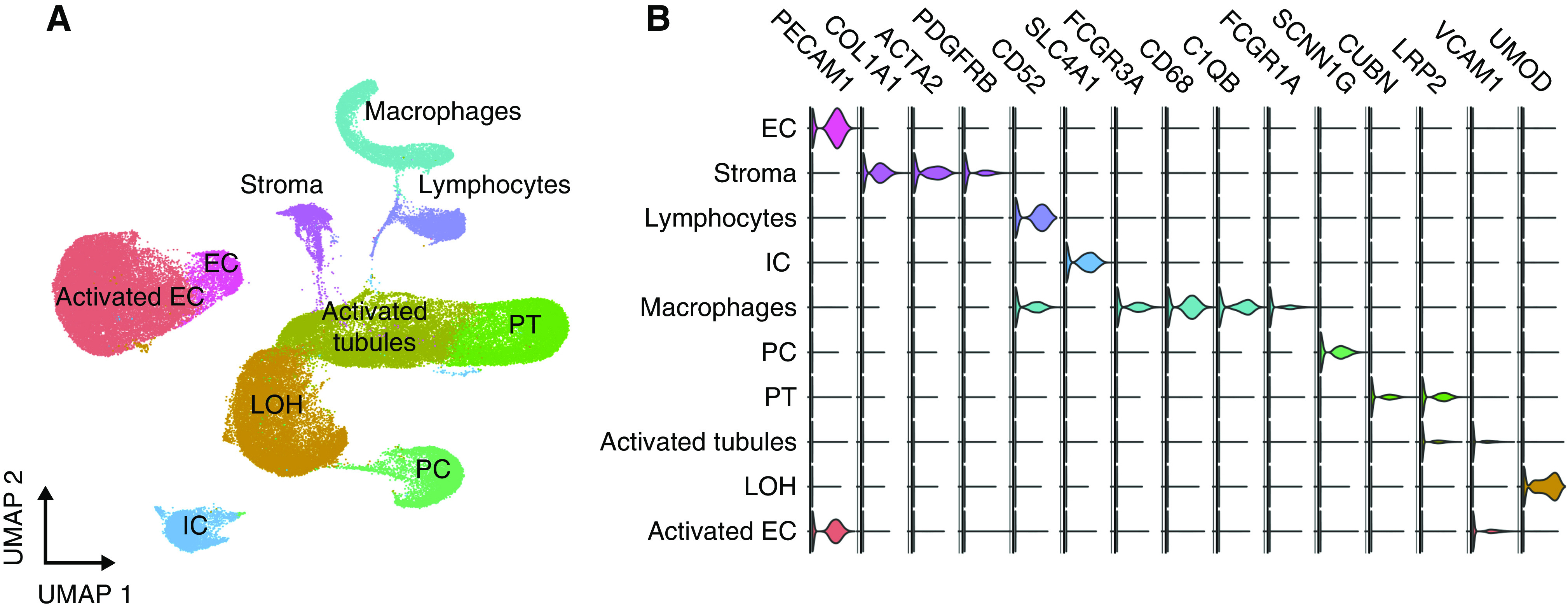
scRNA-seq of 81,139 cells from five kidney allograft biopsies reveals all major kidney and immune cell types. (A) Uniform Manifold Approximation and Projection (UMAP) plot of 81,139 cells from five kidney allograft biopsies. All the main kidney cell types are represented, including activated tubular cells, loop of Henle (LOH), proximal tubule (PT), principal cells (PC), intercalated cells (IC), endothelial cells (EC), stromal cells, macrophages, and lymphocytes. (B) Violin plot demonstrating marker genes for major cell types represented in the whole data set and UMAP plot.
Donor and Recipient Immune Cell Chimerism in the Kidney Allograft
WES was performed on the ten individuals from the five donor-recipient pairs for each biopsy. A single .vcf file containing an average of 120,896 coding single nucleotide variants (SNVs) with high-population minor allele frequency (>5%) was constructed from WES data for each donor and recipient pair. The Demuxlet pipeline3 was applied to each of the five biopsy samples in this data set using the .vcf file as a reference to determine donor or recipient origin of each cell (Figure 2A). The median number of SNVs used per cell to call immune cell origin varied from 71 to 231. The lowest number of SNVs overlapping any read in a cell was 17. However, the lower interquartile ranges were all >50 SNVs. This allowed for 90% accuracy for all cells in the data set and close to 100% accuracy in cells above the lower interquartile range (Supplemental Figure 4).3 Despite manual filtering and removal of doublet cells from the scRNA-seq data (see Supplemental Methods) from each biopsy specimen, the Demuxlet tool identified 3%–10.5% of cells as doublets which allowed for improved quality control of the data set. The macrophage cluster comprised 3798 recipient cells and 1229 donor cells, and the lymphocyte cluster comprised 1528 recipient cells and 2119 donor cells (Figure 2B).
Figure 2.

There is donor and recipient chimerism of macrophages and lymphocytes in the transplanted kidney. (A) The Demuxlet pipeline used to resolve the origin of cells from each biopsy.3 (B) Uniform Manifold Approximation and Projection (UMAP) visualization of 81,139 cells grouped by recipient (red) or donor (green) origin. Inset highlights macrophages (5027 cells) and lymphocytes (3647 cells) grouped by recipient or donor origin.
The ratio of donor/recipient-origin macrophages varied according to rejection status with approximately equal proportions of donor and recipient cells in the two nonrejecting samples and almost complete dominance of recipient cells in rejecting biopsies (Figure 3A). The absolute number of donor- and recipient-origin macrophages in rejecting samples ranged from between four to 39 cells and 514–1258 cells, respectively. In nonrejecting samples, donor- and recipient-origin macrophage numbers ranged from between 217–964 and 389–674, respectively. These differences in donor/recipient ratio suggest the presence of donor macrophages is not simply a function of time post-transplant. As an example, there was a significant proportion (35.8%) of donor macrophages in the nonrejecting allograft at 28 days, whereas recipient macrophages dominate (97%) in the rejecting biopsy at day 11 post-transplant (Figure 3B). In an independent data set, the donor-origin macrophage proportion recovered postrejection treatment, increasing from 3% of total macrophages to 11.7% 1 month post-treatment (Figure 3G). However, in absolute numbers, this was an increase in donor macrophages from 96 to 100 cells.
Figure 3.
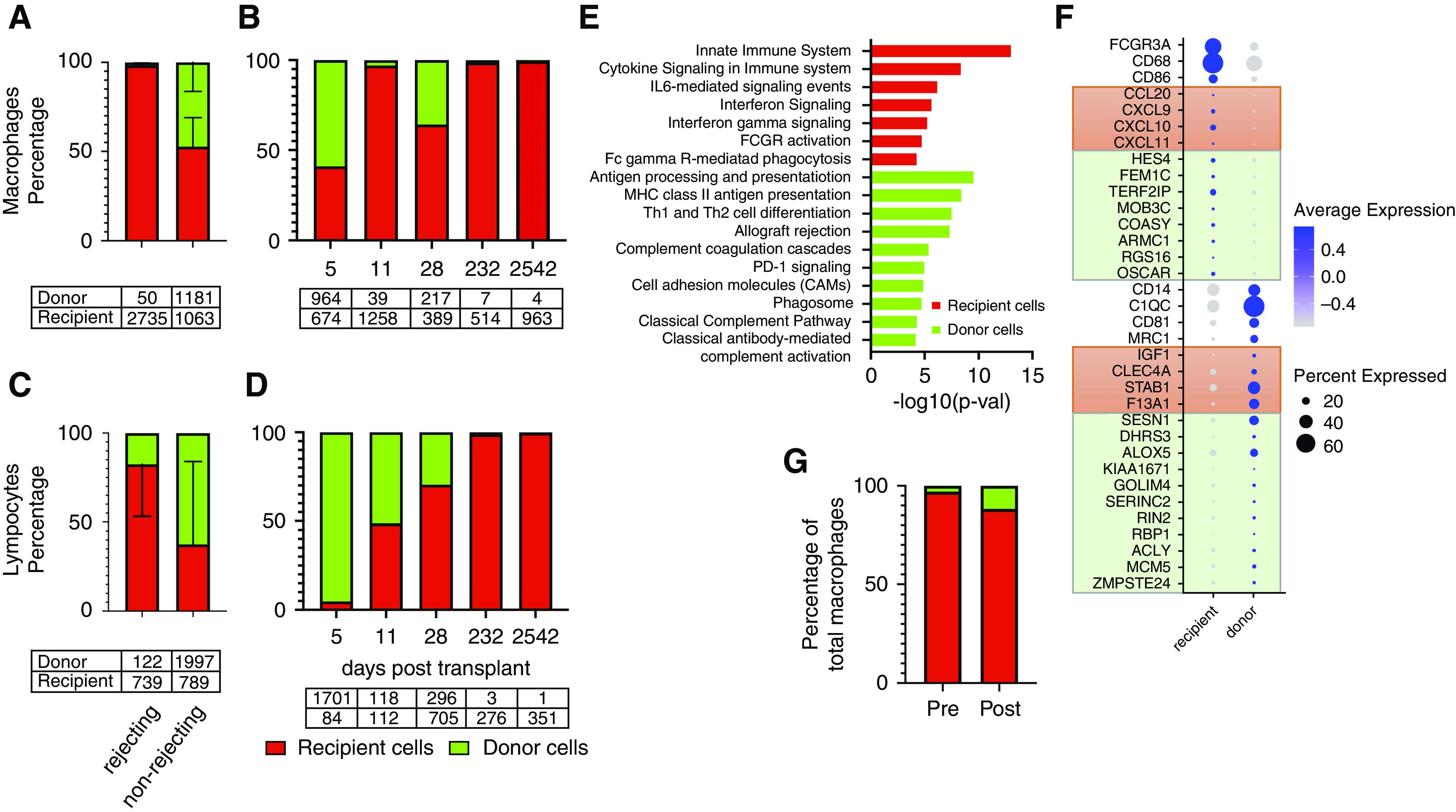
Donor-origin macrophage and lymphocyte populations vary with time and rejection status. (A) Macrophage proportions vary according to rejection status. Recipient-cell error bars in upward direction and donor-cell error bars in downward direction. (B) The proportion of donor/recipient macrophages in each of the five biopsy specimens according to time (days) post-transplant. (C) The proportions of donor and recipient lymphocytes according to rejection status. (D) The proportion of donor/recipient T cells in each of the five biopsies according to time (days) post-transplant. Tables under (A–D) are total cell numbers. (E) Macrophage pathway analysis by cell origin. Differential gene testing between donor and recipient macrophages reveal genes involved in immune-related pathways (ToppGene Suite pathway analysis, Bonferroni P=0.049 or less). (F) Dot plot of genes that define donor and recipient macrophages. Orange boxes include genes differentially expressed in classically activated and wound-healing macrophages, respectively. Light green boxes include highly expressed genes from spectrum of macrophage phenotypes as described by Xue et al.18 (G) Serial biopsy specimens in the same patient at rejection (ABMR) diagnosis (pretreatment) and 1 month post-treatment demonstrates donor macrophage proportion increases postrejection treatment. Pretreatment biopsy was performed 2 years 7 months (945 days) post-transplant and the post-treatment biopsy was performed 1 month after the first biopsy (974 days). MHC, major histocompatibility complex; PD-1, programmed cell death protein 1; p-val, P value; Th1, type 1 CD 4 T helper cell; Th2, type 2 CD 4 T helper cell.
Pathway analysis of differentially expressed genes in recipient versus donor macrophages suggests distinct roles in the alloimmune reaction (Figure 3E, Supplemental Tables 3 and 4). IL-6 signaling, IFN signaling, and FCγR pathway genes are differentially expressed in recipient macrophages. By contrast, complement pathway, antigen presenting, and immunoregulatory genes are differentially expressed in donor macrophages. Recipient and donor macrophages do not fall clearly into either M1 or M2 phenotypes (Supplemental Figure 5). Recipient macrophages differentially express CD16(FCGR3A) and CD68, whereas donor macrophages differentially express CD14 (Figure 3F). Donor macrophages express genes associated with a wound-healing phenotype and recipient macrophages express genes associated with a classically activated macrophage phenotype (Figure 3F, orange boxes).19 A transcriptomics-based network analysis of human macrophages by Xue et al.18 described ten stimulus-specific clusters of activated macrophages that lie along a continuum of M1 to M2 phenotypes. These clusters are represented in both the donor and recipient macrophages (Figure 3F, green boxes). Using this model, top genes expressed by IFNγ– and TNF-stimulated macrophages, FEM1C and TERF2IP, are differentially expressed in recipient macrophages. The top gene expressed by IL-4–stimulated macrophages, KIAA1671, was differentially expressed by donor macrophages.
Lymphocyte Subsets and Donor-Recipient Chimerism in the Kidney Allograft
B cells (n=190), plasma cells (n=231), and T cells (n=3067) could be detected within the lymphocyte cluster (Figure 4A). Cell types were defined by expression of typical markers including CD3E for T cells, MS4A1 (CD20) for B cells, SDC1 (CD138) for plasma cells, and TPSAB1 (tryptase) for mast cells (Figure 4B). Both donor and recipient cells are represented in all lymphocyte subclusters and in mast cells (Figure 4C), as are rejecting and nonrejecting samples (Figure 4D). The T cell cluster represented four T cell subclusters. Subcluster 2 was an activated T cell cluster expressing GZMB, PRF1, and GNLY. Subclusters 0 and 1 expressed IL7R and were likely CD4+ T cells. Neither CD8 nor CD4 were significantly expressed in any T cell subcluster. A fourth subcluster, composed of NKG7-expressing cells, were most likely natural killer T cells. Of the two IL7R+ T cell subclusters, one was predominantly of donor origin and the other was mixed origin (Supplemental Figure 6). A small number of mast cells (n=111) were resolved after subcluster analysis of immune cells.
Figure 4.
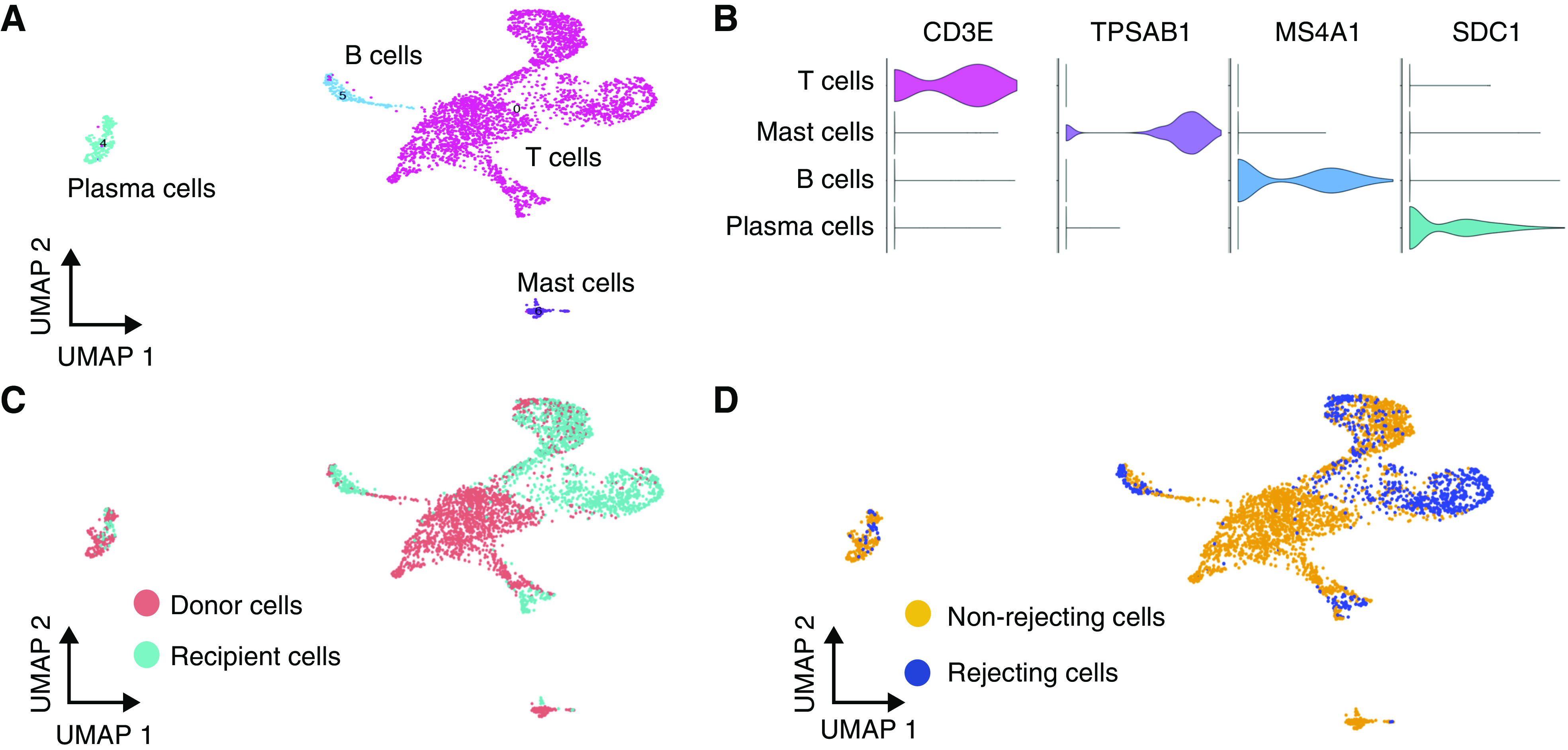
Lymphocytes subcluster into T cells, B cells, plasma cells, and mast cells. (A) Uniform Manifold Approximation and Projection (UMAP) visualization of intragraft lymphocytes subclustered into T cells, B cells, plasma cells, and mast cells. (B) Violin plot of cell type–defining markers: CD3E for T cells, TPSAB1 (tryptase) for mast cells, MS4A1 (CD20) for B cells, and SDC1 (CD138) for plasma cells. (C) UMAP visualization of lymphocytes according to donor or recipient origin, and (D) rejecting or nonrejecting biopsies.
A differential gene expression list was calculated for recipient T cells versus donor T cells, and also for T cells from rejecting biopsies versus T cells from nonrejecting biopsies (Supplemental Table 5). Recipient-origin T cells correlated highly with rejecting T cell transcripts (Spearman correlation coefficient, 0.8). By contrast, donor-origin T cells correlated highly with nonrejecting T cell transcripts (Spearman correlation coefficient, 1.0) (Figure 5A). Conversely, T cells from rejecting samples did not correlate with donor-origin T cells (Spearman correlation coefficient, −0.66) and T cells from nonrejecting samples did not correlate with recipient-origin T cells (Spearman correlation coefficient, −0.66). Many of the top genes defining recipient T cells and rejecting T cells encode for cytotoxic genes such as natural killer cell granule protein 7 (NKG7), granulysin (GNLY), granzyme B (GZMB), and perforin (PRF1) (Figure 5B). Genes differentially expressed in donor-origin T cells and nonrejecting T cells included IL32, CD52, MIF, S100A6, S100A11, and LTB. The products of these genes are both proinflammatory and anti-inflammatory.
Figure 5.
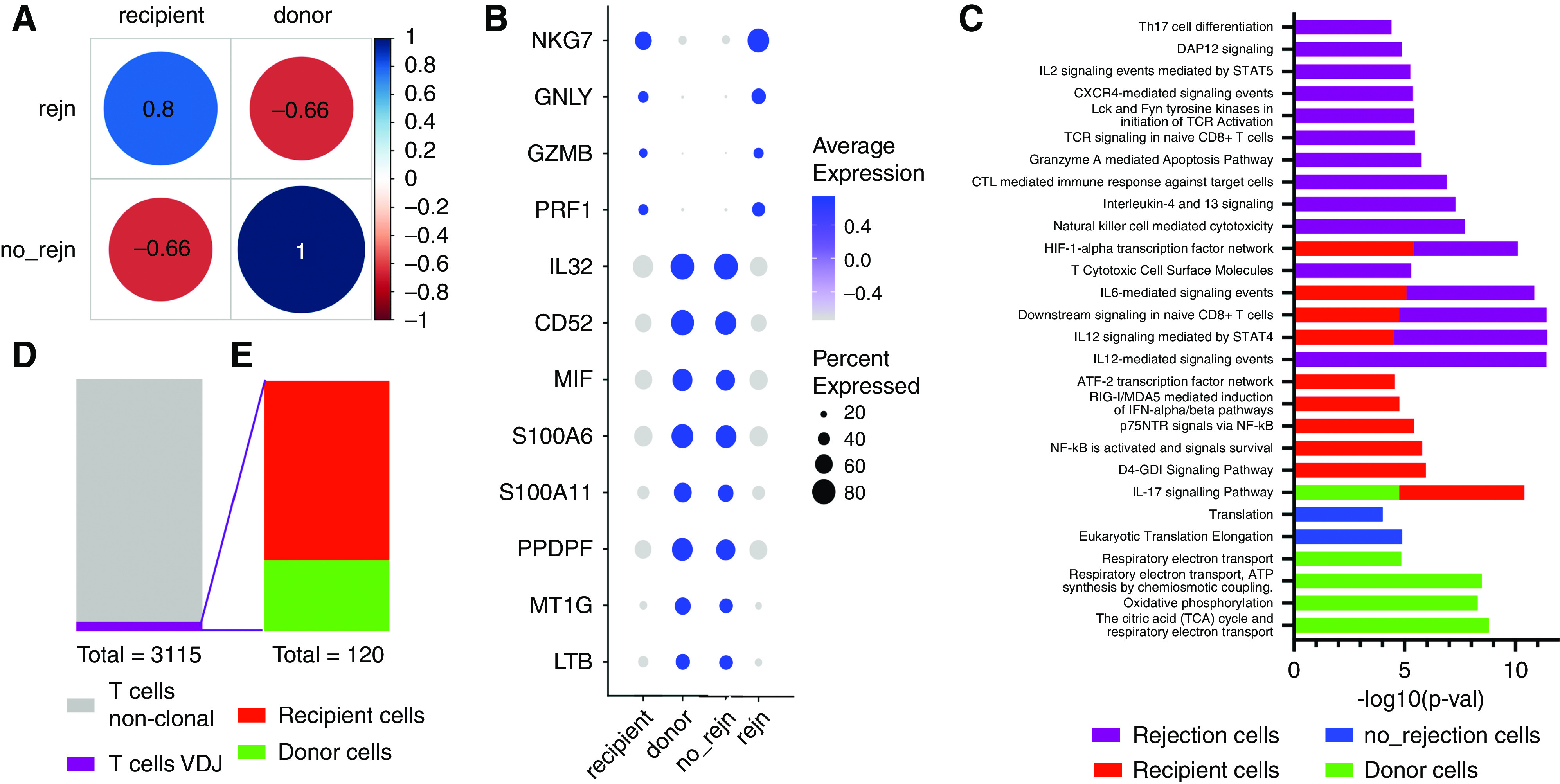
T lymphocyte transcriptional states vary according to donor or recipient origin and rejection status. (A) Spearman correlation analysis of T cells from rejection versus nonrejection samples and recipient versus donor T cells (correlation coefficient are numbers within circles). (B) Select genes from top 20 differentially expressed genes from each group represented in (A): recipient T cells, donor T cells, rejecting T cells (rejn), and nonrejecting T cells (no_rejn). (C) Pathway analysis of significantly differentially expressed genes (Bonferroni P= 0.043 or less). (D) Highlights the proportion (120/3115) of clonal T cells based on VDJ sequencing and (E) proportion of T cell clones from recipient (86) and donor cells (34).
Pathway analysis using differentially expressed genes defining rejecting, nonrejecting, recipient, and donor T cells revealed proinflammatory pathways in rejecting and recipient-origin T cells (Bonferroni P = 0.043 or less) (Figure 5C, Supplemental Table 6). Pathways common to rejecting and recipient cells were IL-12 signaling through STAT4, IL-6 signaling, and downstream signaling in native CD8+ T cells. The HIFα transcription factor pathway was also common to rejecting and recipient T cells. Pathways specific to donor T cells were related to cellular metabolism, specifically oxidative phosphorylation and ATP generation.
T Lymphocyte Clonal Analysis by VDJ Sequencing
We performed single-cell sequencing of VDJ rearrangements in T cells from three of the five biopsy specimens in the current data set (two rejecting and one nonrejecting). Only 120 T cells with VDJ sequence were identified in the final postfilter data set (3.8% of all T cells) (Figure 5D). There were no T cell VDJ sequences from the nonrejecting biopsy identified in the final data set. Of the T cells with VDJ rearrangements sequenced, 34 cells were donor origin and 86 were recipient origin. All the donor-origin VDJ T cells were from one early post-transplantation rejection sample (11 days). The recipient-origin VDJ T cells were from a later rejection sample (6 months and 2 weeks) (Figure 5E).
Discussion
In this proof-of-concept study, we used germline DNA sequencing of donor-recipient pairs and scRNA-seq to determine the degree and nature of leukocyte chimerism in the kidney allograft. We studied biopsy samples from three patients with ABMR and two patients with nonrejection causes of AKI. We demonstrate how this approach can accurately assign donor-recipient origin to each cell from a complex tissue such as the kidney and, coupled with transcriptome analysis, can examine the transcriptional differences between cells of different origin.
Although it is known that donor macrophages can be found in the transplanted organ in the early post-transplant period, it has been assumed that these cells were transient passengers whose presence declines over time.20,21 Furthermore, studies report recipient-derived macrophages accumulate during ischemia reperfusion and that, after resolution of this injury, the macrophage population returned to and remained at low numbers.22 This study showed donor-derived macrophages can remain within the nonrejecting kidney for up to 28 days, despite the infiltration of macrophages associated with the ischemia-reperfusion injury that occurs after organ transplantation (Figure 3, A and B). Our data suggest the donor-derived macrophage population may not simply vary as a function of time post-transplantation but persist depending on rejection events. Donor macrophage proportions varied according to rejection status rather than time (Figure 3B).
Tissue-resident macrophages are derived from functionally distinct subsets of cells derived from embryonic and hematopoietic lineages.23 This distinction suggests there should be phenotypic heterogeneity of tissue-resident macrophages. However, this heterogeneity relies on limited cell markers and animal studies.14 Such tools to determine macrophage origin in human studies have been limited or not possible. The scRNA-seq approach used in this study can accurately determine cell origin and has the potential to better define tissue-resident macrophage phenotypes. We found functional heterogeneity among macrophages in our samples. Functional roles of donor macrophages include wound healing, antigen presentation, and negative regulation of T cells through programmed cell death protein 1. Although classically proinflammatory cytokines are expressed in recipient macrophages, donor-derived macrophages appear to include proinflammatory processes such as complement activation and coagulation (Figure 3, E and F). Complement damage and microthrombosis are known pathologic features of ABMR. Therefore, it was interesting to find F13A1 was differentially expressed in donor macrophages. F13A1 encodes a transglutaminase that crosslinks and stabilizes fibrin. From mouse studies we know resident macrophages are the major source of circulating factor XIII.24 Quantitative proteomic studies of allograft biopsies have shown factor XIII A chain to correlate with degree of fibrosis.25 Further evidence that donor macrophages contribute to pathology in ABMR is their differential expression of C1QA, C1QB, and C1QC. C1q-binding DSAs have been shown to confer a poor prognosis after ABMR.26
In contrast to macrophages, donor lymphocyte numbers decreased as a function of time and by day 232 there are very few donor-origin lymphocytes, suggesting donor/recipient lymphocyte ratios are determined by time post-transplantation (Figure 3, C and D). Recent studies in mice have revealed that memory T cells can reside in nonlymphoid tissue to provide direct antigen recognition and early responses to pathogens.27 These tissue-resident memory T cells have also been described in kidney.28 However, the role of tissue-resident memory T cells in transplantation is unknown. In organs with a larger burden of tissue-resident T cells such as the lung, donor cells were not detectable after 39 days post-transplantation when assessed by serial transbronchial parenchymal biopsies of lung transplant recipients.29 This study demonstrates persistence of a significant number of donor T cells at 28 days after kidney transplantation.
To better understand the functional characteristics of tissue-resident T cells, we performed an in-depth analysis of differentially expressed genes in donor-derived T cells. Interestingly, pathways specific to donor-origin T cells (when compared with recipient T cells) are related to glucose metabolism and energy production, specifically oxidative phosphorylation (Figure 5C). It is known that activated T cells preferentially convert glucose to lactate even when oxygen is not a limiting factor, whereas quiescent T cells generate most of their energy through oxidative phosphorylation.30,31 The byproducts of this increased glycolytic flux in activated T cells are used for the production of nucleotides, fatty acids, and amino acids.32 These data suggest that, compared with recipient T cells, donor T cells are predominantly quiescent which is consistent with our findings of donor T cells correlating with T cells from nonrejecting samples (Figure 5A). Recipient-origin T cell state reflects an effector phenotype typical of acute rejection (Figure 5, B and C). Our analysis, in contrast to donor macrophages, suggest donor T cells do not express proinflammatory genes. Gene Ontology pathway analysis suggested IL-17 signaling in donor T cells but only CD3D, CD3E, and CD2 genes were differentially expressed. Because these genes are expressed in all T cells, more data are therefore required to confirm the relevance of IL-17 signaling in donor T cells.
The lymphocyte cluster (Figure 1) also contained B cells and plasma cells in addition to T cells when subclustering was performed (Figure 4, A and B). There was also a small population of mast cells identified. To better understand this finding, we performed a Spearman correlation that suggested mast cells are as transcriptionally similar to lymphocytes as they are to macrophages (Supplemental Figure 7).
A clonal analysis performed by sequencing the VDJ region of T cells revealed donor-derived T cell clones. The significance of this is not known at this time; however, our ability to identify T cell clones within the kidney, their origin, and analyze their transcriptomes adds another category of data previously not available to this field. These donor-derived clonal T cells are all from an early post-transplant biopsy and may be passenger T cells that regress with time. Whether the VDJ sequences in these T cells code for donor-specific T cell receptors requires further investigation.
In conclusion, we have combined germline DNA sequencing with scRNA-seq of human kidney transplant biopsy specimens to measure the persistence and transcriptional profiles of donor- and recipient-derived leukocytes in ABMR and nonrejecting states. Although further studies will be needed for confirmation, we could easily identify leukocytes from both recipient and donor, establishing the feasibility of this approach in transplantation generally.
Disclosures
All authors have nothing to disclose.
Funding
This work was supported by the Washington University in St. Louis McDonnell Genome Institute 2018 Symposium Pilot Project Fund, NIH/National Center for Advancing Translational Sciences grant UL1TR002345 (to C. Fronick, R. Fulton, B. Humphreys, and A. Malone), and NIH/National Institute of Diabetes and Digestive and Kidney Diseases grants 1K08DK120953 (to A. Malone) and DK103740 and DK107374 (to B. Humphreys).
Supplementary Material
Acknowledgments
Dr. Benjamin D. Humphreys reports personal fees from Celgene, grants from Chinook Therapeutics, personal fees from Chinook Therapeutics, other from Chinook Therapeutics, personal fees from Genentech, personal fees from Indalo Therapeutics, personal fees from Janssen, and grants from Janssen, outside the submitted work. Dr. Joseph P. Gault reports grants from the National Institutes of Health (NIH) and grants from MTS Foundation, outside the submitted work.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
Data Sharing Statement
The National Center for Biotechnology Information Gene Expression Omnibus accession number for the sequencing data reported in this paper is GSE145927.
Supplemental Material
This article contains the following supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2020030326/-/DCSupplemental.
Supplemental Figure 1. Histograms of all droplets with at least one gene per droplet for each sample in the dataset.
Supplemental Figure 2. Gene ontology terms for differentially expressed genes that define stressed tubular cell cluster.
Supplemental Figure 3. UMAP of individual biopsy samples demonstrating degree of batch effect.
Supplemental Figure 4. Total number of SNVs overlapping at least one read in immune cells grouped by five biopsy samples.
Supplemental Figure 5. Gene expression from donor and recipient macrophages correlated with human M1 and M2 macrophage bulk RNA-seq gene expression external datasets.
Supplemental Figure 6. Violin plots and feature plots of common T cell genes across the T cell subclusters.
Supplemental Figure 7. Spearman correlation between mast cell gene expression and macrophage gene expression and with lymphocyte gene expression.
Supplemental Table 1. Clinical Characteristics of the 5 Biopsies.
Supplemental Table 2. Differential gene list defining each cell cluster in the whole dataset.
Supplemental Table 3. Differentially expressed genes for recipient versus donor macrophages.
Supplemental Table 4. Pathway analysis for differential expressed (recipient and donor) macrophages genes using ToppFun tool.
Supplemental Table 5. Differentially expressed genes for recipient versus donor T cells and Differentially expressed genes for T cells from rejecting samples versus T cells from non-rejecting T cells.
Supplemental Table 6. Pathway analysis for differential expressed (recipient vs donor and rejecting vs non-rejecting) T cells genes using ToppFun tool.
References
- 1.Szabo PA, Levitin HM, Miron M, Snyder ME, Senda T, Yuan J, et al.: Single-cell transcriptomics of human T cells reveals tissue and activation signatures in health and disease. Nat Commun 10: 4706, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Plasschaert LW, Žilionis R, Choo-Wing R, Savova V, Knehr J, Roma G, et al.: A single-cell atlas of the airway epithelium reveals the CFTR-rich pulmonary ionocyte. Nature 560: 377–381, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kang HM, Subramaniam M, Targ S, Nguyen M, Maliskova L, McCarthy E, et al.: Multiplexed droplet single-cell RNA-sequencing using natural genetic variation. Nat Biotechnol 36: 89–94, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Billingham RE, Brent L, Medawar PB: Actively acquired tolerance of foreign cells. Nature 172: 603–606, 1953. [DOI] [PubMed] [Google Scholar]
- 5.Starzl TE, Murase N, Demetris AJ, Trucco M, Abu-Elmagd K, Gray EA, et al.: Lessons of organ-induced tolerance learned from historical clinical experience. Transplantation 77: 926–929, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Starzl TE: Chimerism and tolerance in transplantation. Proc Natl Acad Sci U S A 101[Suppl 2]: 14607–14614, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gulbahce HE, Brown CA, Wick M, Segall M, Jessurun J: Graft-vs-host disease after solid organ transplant. Am J Clin Pathol 119: 568–573, 2003. [DOI] [PubMed] [Google Scholar]
- 8.Dunn SP, Krueger LJ, Butani L, Punnett H: Late onset of severe graft-versus-host disease in a pediatric liver transplant recipient. Transplantation 71: 1483–1485, 2001. [DOI] [PubMed] [Google Scholar]
- 9.Guo Y, Ding S, Guo H, Li S, Lu X, Chen Z, et al.: Graft-versus-host-disease after kidney transplantation: a case report and literature review. Medicine (Baltimore) 96: e7333, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nayak DK, Zhou F, Xu M, Huang J, Tsuji M, Hachem R, et al.: Long-term persistence of donor alveolar macrophages in human lung transplant recipients that influences donor-specific immune responses. Am J Transplant 16: 2300–2311, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Snyder ME, Finlayson MO, Connors TJ, Dogra P, Senda T, Bush E, et al.: Generation and persistence of human tissue-resident memory T cells in lung transplantation. Sci Immunol 4: eaav5581, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harper IG, Ali JM, Harper SJ, Wlodek E, Alsughayyir J, Negus MC, et al.: Augmentation of recipient adaptive alloimmunity by donor passenger lymphocytes within the transplant. Cell Rep 15: 1214–1227, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harper IG, Gjorgjimajkoska O, Siu JHY, Parmar J, Mulder A, Claas FHJ, et al.: Prolongation of allograft survival by passenger donor regulatory T cells. Am J Transplant 19: 1371–1379, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Davies LC, Jenkins SJ, Allen JE, Taylor PR: Tissue-resident macrophages. Nat Immunol 14: 986–995, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bajpai G, Bredemeyer A, Li W, Zaitsev K, Koenig AL, Lokshina I, et al.: Tissue resident CCR2- and CCR2+ cardiac macrophages differentially orchestrate monocyte recruitment and fate specification following myocardial injury. Circ Res 124: 263–278, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dumitru C, Kabat AM, Maloy KJ: Metabolic adaptations of CD4+ T cells in inflammatory disease. Front Immunol 9: 540, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Regier AA, Farjoun Y, Larson DE, Krasheninina O, Kang HM, Howrigan DP, et al.: Functional equivalence of genome sequencing analysis pipelines enables harmonized variant calling across human genetics projects. Nat Commun 9: 4038, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xue J, Schmidt SV, Sander J, Draffehn A, Krebs W, Quester I, et al.: Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity 40: 274–288, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mosser DM, Edwards JP: Exploring the full spectrum of macrophage activation. Nat Rev Immunol 8: 958–969, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mannon RB: Macrophages: Contributors to allograft dysfunction, repair, or innocent bystanders? Curr Opin Organ Transplant 17: 20–25, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wyburn KR, Jose MD, Wu H, Atkins RC, Chadban SJ: The role of macrophages in allograft rejection. Transplantation 80: 1641–1647, 2005. [DOI] [PubMed] [Google Scholar]
- 22.Penfield JG, Wang Y, Li S, Kielar MA, Sicher SC, Jeyarajah DR, et al.: Transplant surgery injury recruits recipient MHC class II-positive leukocytes into the kidney. Kidney Int 56: 1759–1769, 1999. [DOI] [PubMed] [Google Scholar]
- 23.Epelman S, Lavine KJ, Randolph GJ: Origin and functions of tissue macrophages. Immunity 41: 21–35, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Beckers CML, Simpson KR, Griffin KJ, Brown JM, Cheah LT, Smith KA, et al.: Cre/lox studies identify resident macrophages as the major source of circulating coagulation factor XIII-A. Arterioscler Thromb Vasc Biol 37: 1494–1502, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mortensen LA, Svane AM, Burton M, Bistrup C, Thiesson HC, Marcussen N, et al.: Proteomic analysis of renal biomarkers of kidney allograft fibrosis-A study in renal transplant patients. Int J Mol Sci 21: 2371, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cozzi E, Biancone L: C1q-binding donor-specific antibody assays help define risk and prognosis in antibody-mediated rejection. Kidney Int 94: 657–659, 2018. [DOI] [PubMed] [Google Scholar]
- 27.Mueller SN, Gebhardt T, Carbone FR, Heath WR: Memory T cell subsets, migration patterns, and tissue residence. Annu Rev Immunol 31: 137–161, 2013. [DOI] [PubMed] [Google Scholar]
- 28.Casey KA, Fraser KA, Schenkel JM, Moran A, Abt MC, Beura LK, et al.: Antigen-independent differentiation and maintenance of effector-like resident memory T cells in tissues. J Immunol 188: 4866–4875, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bittmann I, Dose T, Baretton GB, Müller C, Schwaiblmair M, Kur F, et al.: Cellular chimerism of the lung after transplantation. An interphase cytogenetic study. Am J Clin Pathol 115: 525–533, 2001. [DOI] [PubMed] [Google Scholar]
- 30.Fracchia KM, Walsh CM: Metabolic mysteries of the inflammatory response: T cell polarization and plasticity. Int Rev Immunol 34: 3–18, 2015. [DOI] [PubMed] [Google Scholar]
- 31.Chapman NM, Boothby MR, Chi H: Metabolic coordination of T cell quiescence and activation. Nat Rev Immunol 20: 55–70, 2020. [DOI] [PubMed] [Google Scholar]
- 32.Vander Heiden MG, Cantley LC, Thompson CB: Understanding the warburg effect: the metabolic requirements of cell proliferation. Science 324: 1029–1033, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.