

Abstract
The oral delivery of bioactive peptides and proteins is prevented by the intestinal epithelial barrier, in which intercellular tight junction complexes block the uptake of macromolecules. Here, we show that anionic nanoparticles induce tight junction relaxation, increasing intestinal permeability and enabling the oral delivery of proteins. This permeation-enhancing effect is a function of nanoparticle size and charge, with smaller (≤ 200 nm) and more negative particles (such as silica) conferring enhanced permeability. In healthy mice, silica nanoparticles enabled the oral delivery of insulin and exenatide, with a 10 U/kg of orally delivered insulin sustaining hypoglycaemia for a few hours longer than a 1 U/kg dose of subcutaneously injected insulin. In healthy, hyperglycaemic and diabetic mice, the oral delivery of 10 U/kg of insulin led to a dose-adjusted bioactivity of, respectively, 35%, 29%, and 23% compared to 1 U/kg subcutaneously injected insulin. The permeation-enhancing effect of the nanoparticles was reversible, non-toxic, and attributable to the binding to integrins on the surface of epithelial cells.
Oral drug delivery, which is painless and convenient, offers superior patient compliance and improved disease outcomes compared to injections. However, delivery challenges have thwarted its successful implementation for decades. Every day, millions of patients across the globe are subjected to injections of macromolecular drugs, such as insulin or heparin. Unfortunately, a fear of injections is pervasive across populations, sometimes surpassing 80% prevalence in both children and adults1,2. An estimated 20%−30% of patients further classify as suffering from severe needle phobia2,3. As a result, up to 33% of diabetic patients report feelings of dread associated with their insulin injections, and 45%−60% report intentionally skipping one or more doses4,5. By enabling painless self-administration, oral dosage forms of insulin and other protein drugs would markedly improve patient experience, compliance, and disease outcomes.
Unfortunately, the physiology of the gastrointestinal tract prevents the clinical translation of most oral protein formulations6. The stomach contains harsh acid and enzymes from which the drug and delivery vehicle must be protected. Most often, this is accomplished by coating with a pH sensitive enteric polymer, which dissolves to expose its cargo only once it reaches the more neutral pH of the intestines7. Should a protein enter the small intestine, it can only be absorbed into the bloodstream by crossing the formidable intestinal epithelial barrier. This barrier consists of a monolayer of epithelial cells that are tightly bound to one another by intercellular protein complexes called tight junctions. Because tight junctions permit the passage only of molecules less than 1 nm in hydrodynamic radius8, proteins and peptides are unable to enter the bloodstream9, and their systemic bioavailability is negligible. Thus, successful oral protein delivery will require permeabilization of the tight junction barrier.
To improve macromolecular drug absorption in the small intestine, many chemical permeation enhancers of the epithelium have been used in preclinical trials10–13. These enhancers manipulate cell membrane permeability or tight junction conformation to increase trans-epithelial protein absorption and bioavailability. For delivery of insulin and other anti-diabetic peptides specifically, several chemical permeation enhancers have entered early stage clinical trials14–16. Unfortunately, no small molecule permeation enhancers have yet advanced through late-stage trials, in part because they are prone to cytotoxicity and have narrow therapeutic windows17–19.
As a potentially safer and more effective alternative to chemical permeation enhancers, some strategies for physically manipulating the epithelial barrier have been examined in preclinical trials. For example, polymer films with surface nanotopography open tight junctions in intestinal cell cultures, improving the transepithelial diffusion of model proteins20. Other studies have shown that ultrasound, through the physical process of cavitation, can temporarily disrupt the intestinal epithelial barrier to systemically deliver protein drugs21. Together, these studies suggest that physical means of intestinal permeation enhancement may offer improved safety profiles when used alone or, potentially, in combination with chemical approaches.
Here, we describe a serendipitous discovery that small, negatively charged particles act as physiochemical permeation enhancers that facilitate the oral delivery of protein. Specifically, the nanoparticles described here act not by moving across the intestinal epithelium as delivery vehicles, but by binding intestinal surface receptors that mediate the opening of tight junctions. This stands in contrast to previous studies for oral insulin delivery using nanoparticles, which primarily fall into two categories: insulin-loaded particles for transcytotic uptake22–26 or insulin-loaded, chemical permeation enhancer-doped particles27,28.
With this surprising revelation that the mere presence of nanoparticles can open tight junctions, we set out to establish the breadth of this effect, its therapeutic potential, and its mechanism. We discovered that small (<100 nm), anionic nanoparticle treatments improve absorption of model drugs across both Caco-2 monolayers mouse intestines in vivo. Further, the oral delivery of two therapeutically-relevant protein drugs, insulin and exenatide, became possible when they were co-administered with silica nanoparticles in mice. Mechanistically, the nanoparticles increase intestinal permeability by binding integrins and activating myosin light chain kinase. Finally, we eliminate several safety concerns typical of permeation enhancers by showing their effect is reversible and does not cause necrosis or inflammation of intestinal tissue.
Nanoparticle library of varied sizes and surface chemistries
A collection of nanoparticles was purchased to probe the effect of size, surface charge, and chemistry on intestinal barrier function. We confirmed the supplier-provided particle properties using dynamic light scattering (DLS) and zeta potential measurements (Table 1). Particle diameters ranged from 20 nm to 1200 nm. Non-functionalised silica particles had the most negative zeta potentials (ranging from −41 to −84 mV), followed by carboxyl-functionalised silica, silver, and gold particles. Polystyrene particles were negligibly charged, and amine-functionalised particles had positive zeta potential (+16 mV).
Table 1 |.
DLS size and zeta potential data for nanoparticles in water at neutral pH. Values for particle number and surface area per mass were obtained from specification sheets provided by the supplier.
| Core | Surface chem. | Nomenclature | Z-average diameter | Zeta pot. | Particles per mass | Surface area | ||
|---|---|---|---|---|---|---|---|---|
| Size (nm) | Material | d.nm | PDI | (mV) | #/mg | cm2/mg | ||
| 1200 | silica | n.f. | 1200 nm SiO− | 1173* | 0.05* | −84.0 | 5.41E+08 | 23 |
| 500 | silica | n.f. | 500 nm SiO− | 536 | 0.08 | −68.9 | 5.88E+09 | 52 |
| 200 | silica | n.f. | 200 nm SiO− | 209 | 0.02 | −57.6 | 1.09E+11 | 137 |
| 100 | silica | n.f. | 100 nm SiO− | 90 | 0.05 | −41.2 | 9.61E+11 | 280 |
| 50 | silica | n.f. | 50 nm SiO− | 49 | 0.04 | −41.4 | 7.81E+12 | 554 |
| 20 | silica | n.f. | 20 nm SiO− | 26 | 0.05 | −57.6 | 7.89E+13 | 1212 |
| 50 | silica | COOH | 50 nm SiO-COO− | 46 | 0.15 | −27.3 | n/a | n/a |
| 50 | silver | PVP | 50 nm Ag− | 54 | 0.14 | −21.4 | 1.47E+12 | 109 |
| 50 | gold | PVP | 50 nm Au− | 61 | 0.09 | −16.3 | 7.64E+11 | 59 |
| 50 | polystyrene | n.f. | 50 nm PS | 58 | 0.06 | 0.2 | n/a | n/a |
| 50 | silica | NH2 | 50 nm SiO-NH3+ | 49 | 0.44 | 15.6 | 7.24E+12 | 547 |
| 50 | FITC-silica | n.f. | 50 nm FITC-SiO− | 56 | 0.20 | −27.2 | n/a | n/a |
| 20 | RhodB-silica | n.f. | 20 nm RhodB-SiO− | 23 | 0.20 | −24.3 | n/a | n/a |
FITC = fluorescein isothiocyanate. RhodB = rhodamine B. n.f. = non-functionalised; PVP = 40,000 MW polyvinylpyrrolidone capping agent.
Values based on T.E.M. measurements and provided by manufacturer due to poor quality of DLS results with large particles.
Anionic particles increased Caco-2 monolayer permeability
To assess the effect of particles on intestinal barrier function in cell culture, we applied silica nanoparticles ranging from 20 to 1200 nm in diameter to the apical side of Caco-2 monolayers, which represent the most common in vitro model of the intestinal epithelium29. For three hours following particle addition, we measured trans-epithelial electrical resistance (TEER), which correlates inversely with permeability18. While all silica nanoparticles reduced TEER values to some degree, 20 nm and 50 nm particles produced the most dramatic effect (Figure 1a). After three hours, monolayers were returned to fresh media, where they recovered their barrier function within 24 hours. 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) and lactate dehydrogenase release (LDH) assays showed no reductions in cell viability (Supplementary Figure 1a and 1b), further confirming that silica nanoparticles induce reversible increases in permeability without permanently destroying the cell monolayers.
Figure 1: Smaller and more negatively charged nanoparticles potently increased intestinal monolayer permeability in vitro.
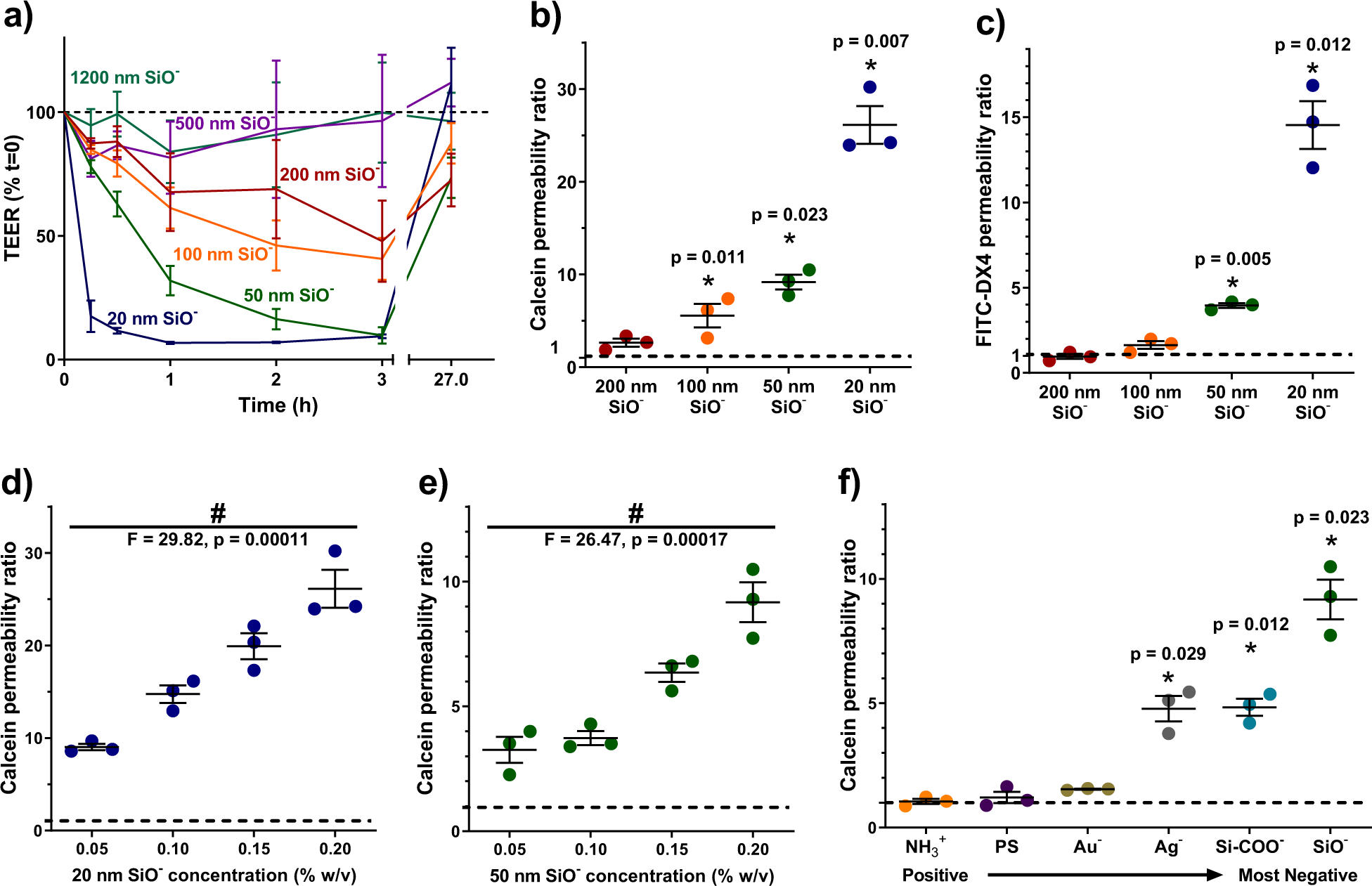
All measurements are expressed as ratios to values for untreated cells (dotted black lines). (a) Smaller silica particles caused greater reductions in the transepithelial electrical resistance (TEER) of Caco-2 monolayers. Particle treatments were removed at three hours, and the monolayers regained their barrier function within 24 hours. (b) Smaller silica particles induced larger permeability increases for the permeation markers calcein and (c) 4 kDa MW FITC-labelled dextran (FITC-DX4). (d) Increases in calcein permeability were dose responsive for both 20 nm particles and (e) 50 nm particles. (f) For a set of 50 nm nanoparticles with varied surface chemistry, those with greater negative surface charge caused greater monolayer permeability to calcein. Neutral and positively charged particles had no significant effect. Statistics bars display arithmetic mean and s.e.m. for biological replicates (n = 3), * p < 0.05 by one-tailed t-test, with respect to untreated cells. # p < 0.05 by one-way ANOVA.
We confirmed that particle size inversely correlated to epithelial permeability for particles ≤ 200 nm by introducing membrane-impermeable diffusion markers to the apical surface of Caco-2 monolayers. Similar to whole proteins, these molecules do not cross the cell membrane, so their accumulation below the epithelial barrier reflects opening of the tight junctions, and has been shown to correlate well with TEER for marker sizes from 180 Da to at least 10 kDa17,18. The transport of the markers calcein (Figure 1b) and 4 kDa fluorescein isothiocyanate labelled dextran (FITC-DX4) (Figure 1c) were most effectively improved by 20 nm silica particles. Efficacy diminished with increasing particle diameter. Silica nanoparticles exhibited dose-dependent effects on intestinal cells over a treatment range of 0.05 – 0.2% w/v, with 20 nm (Figure 1d) and 50 nm (Figure 1e) particles improving calcein permeability up to 26- and 10-fold, respectively.
In addition to particle size, particle surface chemistry was directly linked to permeation enhancing ability. Only particles with negative surface chemistries – including non-functionalised silica, carboxylated silica, gold, and silver – significantly reduced TEER (Supplementary Figure 1b) and increased calcein permeability through the intestinal monolayers (Figure 1f). Particles with neutral or positive charge had little effect on Caco-2 cells.
Particles improved absorption of oral macromolecules in mice
We next assessed particle activity in vivo by orally dosing mice with silica nanoparticles and the macromolecule dextran (4 kDa, FITC-labelled). Because dextran does not contain any acid- or enzyme-labile bonds, it does not require protection in the GI tract. The 4 kDa molecular weight approximates that of common therapeutic peptides, such as insulin, exenatide, and calcitonin16. Particle-induced absorption enhancing effects were monitored by measuring systemic blood serum FITC levels. Unexpectedly, only the 50 and 100 nm particles improved the oral delivery of dextran (Figure 2a). Despite being the most effective treatment in vitro, the 20 nm silica nanoparticles did not substantially affect intestinal absorption in mice.
Figure 2: 50 nm anionic nanoparticles reversibly permeabilized intestinal epithelia in mice.
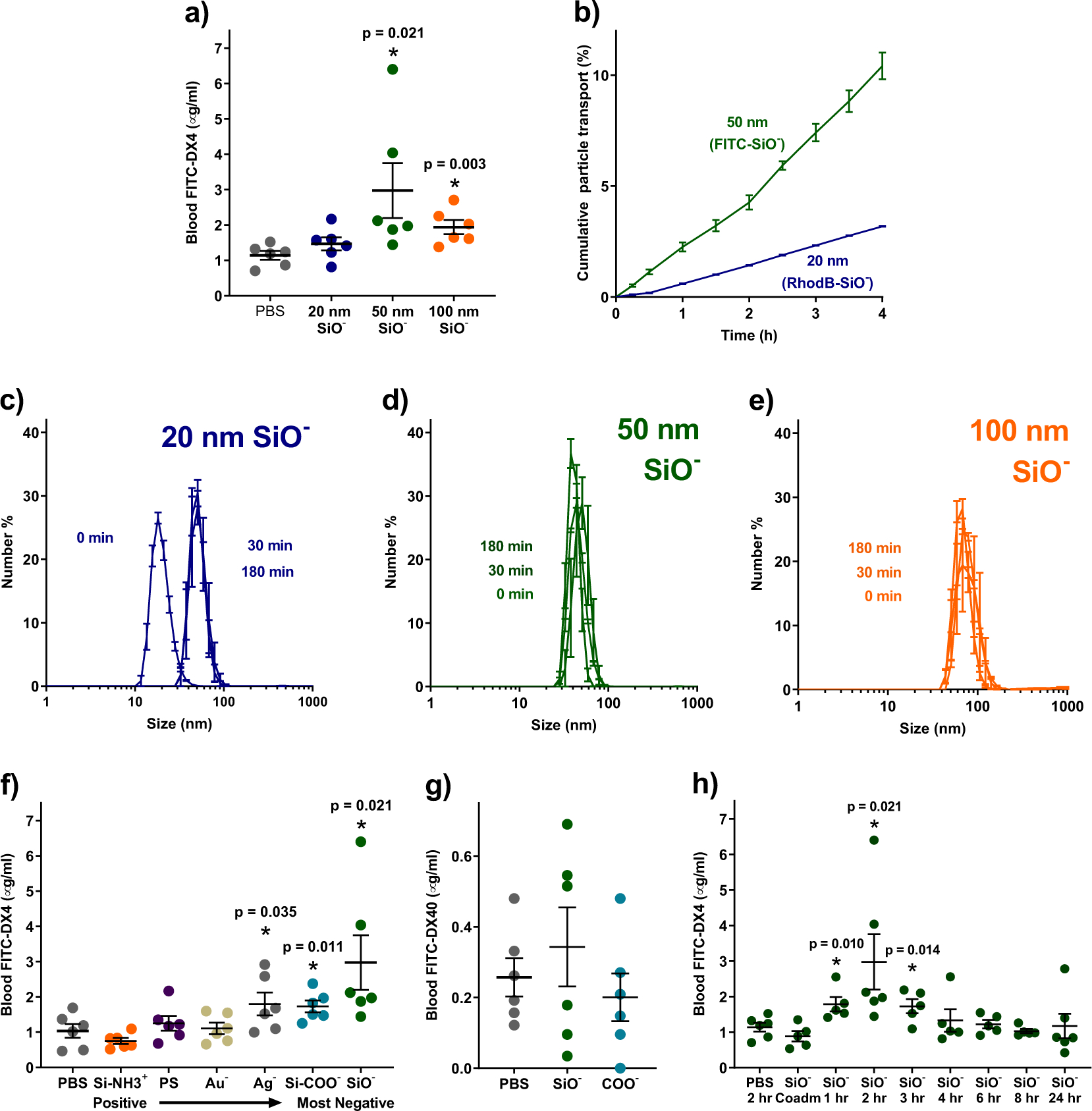
(a) Oral treatment with silica particles improved absorption of 4 kDa, FITC-labelled dextran (FITC-DX4), with 50 nm particles being most effective. Despite their efficacy in vitro, 20 nm silica particles were not effective in mice. (b) This discrepancy was likely caused by the mucus present in vivo. Diffusion of 20 nm particles through model mucus was hindered compared to 50 nm particles. (c) Intestinal mucins bound 20 nm particles within 30 minutes of exposure, increasing their apparent size. (d) By contrast, 50 nm and (e) 100 nm silica particles did not bind to mucin or change size in its presence. (f) Negatively charged particles improved absorption of 4 kDa dextran in mice, while positive and neutral particles did not. (g) Particles did not improve absorption of larger molecules, such as 40 kDa dextran (FITC-DX40). (h) Particle-induced permeability of mouse intestines began to increase within an hour after particle treatment and returned to normal levels within four to six hours of treatment. Statistics bars display arithmetic mean and s.e.m. for biological replicates (n = 3 for mucus studies and n =5 – 6 for mouse dextran studies). * p < 0.05 by one-tailed t-test, with respect to PBS control mice.
We hypothesised that the difference between in vitro and in vivo efficacy of 20 nm particles resulted from the mucus layer that lines the intestines but is not present in the Caco-2 model. To test this, we placed fluorescent silica particles on top of a permeable support that had been coated with a gel of Type II mucin, the predominant component of mucus in the intestines30. The particles used were internally doped with FITC or rhodamine B (RhodB) to track their accumulation beneath the mucus layer without affecting surface chemistry and interactions. Interestingly, the 20 nm particles diffused through the mucus at less than one-third the rate of 50 nm particles (Figure 2b), which is consistent with reports that particles with similar surface characteristics can diffuse disparately through mucus31.
We next demonstrated that 20 nm particles are likely trapped by the mucus due to binding interactions with the mucin proteins. To do this, we mixed 20, 50, and 100 nm silica particles each with a 1% (w/v) solution of type II mucin proteins and tracked their size over time via DLS. The 20 nm particles “grew” to three times their original size within thirty minutes (Figure 2c), indicating that the mucin bound to their surfaces. The narrow size distribution suggested that the particles were not aggregating with one another. In contrast, 50 nm (Figure 2d) and 100 nm silica particles (Figure 2e) did not increase in size at all, suggesting that neither binding nor aggregation occurred in the presence of the mucin. Based on these data, all further in vivo work was conducted with 50 nm particles, as they offered the best compromise between permeation-enhancing efficacy and reduced mucus binding.
We also investigated whether particle surface charge has the same dramatic effect in vivo as it did in cell culture. Mice were orally gavaged with 50 nm particles of varied surface chemistry, followed by 4 kDa, FITC-labelled dextran. As in cell culture experiments, oral dextran delivery correlated with the strength of the negative charge on particles (Figure 2f). Neutral and positively-charged particles caused no significant change in dextran uptake.
Next, we assessed two potential safety concerns for any permeation enhancer. One potential concern is that the enhancers might permit the passage of bacteria or digestive by-products into systemic circulation. The data in Figure 2g suggest that this would be an unlikely issue for silica nanoparticles, as 40 kDa FITC-labelled dextran did not experience increased transport when delivered with silica nanoparticles. Intestinal bacteria are much larger than 40 kDa dextran, so the permeation enhancing effect of silica is unlikely to allow their transepithelial migration32. The second potential concern pertains to the duration of action of the enhancer, as some chemical permeation enhancers have been shown to permanently disrupt the epithelial barrier12,17–19. Fortunately, the silica particles increased intestinal permeability rapidly, but only for a short duration of time. When 4 kDa dextran was delivered at differing times after particle treatment, ranging from zero hours (coadministration) to twenty-four hours, permeability increased by one hour after treatment (Figure 2h), peaking at two hours. However, the measured intestinal permeability returned to baseline levels within four hours after treatment, and remained there through the rest of the trial, indicating that silica nanoparticle-induced permeation enhancement was transient and reversible.
Silica particles enabled oral protein delivery in mice
Ultimately, we are interested in using silica nanoparticles to enhance the delivery of a functional, therapeutically relevant protein. As a proof-of-concept, we asked whether a protein drug could maintain its activity through silica-assisted intestinal translocation. We chose insulin for these studies, as it is a modestly-sized (5.8 kDa) protein that does not readily undergo transepithelial intestinal transport in healthy animals. Furthermore, its bioactivity is easily assessed by monitoring the depression of blood glucose concentration that results from increased insulin circulation. In a first set of experiments, mice received an oral dose of 50 nm silica nanoparticles, followed by an injection of 1 unit per kilogram body weight (U/kg) dose of insulin directly into the small intestine, circumventing digestion in the stomach. Blood glucose levels were monitored each hour and normalised to each mouse’s blood sugar before the procedure. Mice that received insulin and silica nanoparticles experienced a substantial reduction in blood glucose compared to mice that received insulin and polystyrene nanoparticles (Figure 3a). The same data presented with absolute (rather than relative) values can be found in Supplementary Figure 2a. Further, the silica nanoparticle and insulin combination sustained hypoglycaemia several hours longer than the same 1 U/kg dose of subcutaneous insulin.
Figure 3: Silica nanoparticles enabled oral protein delivery in healthy mice.
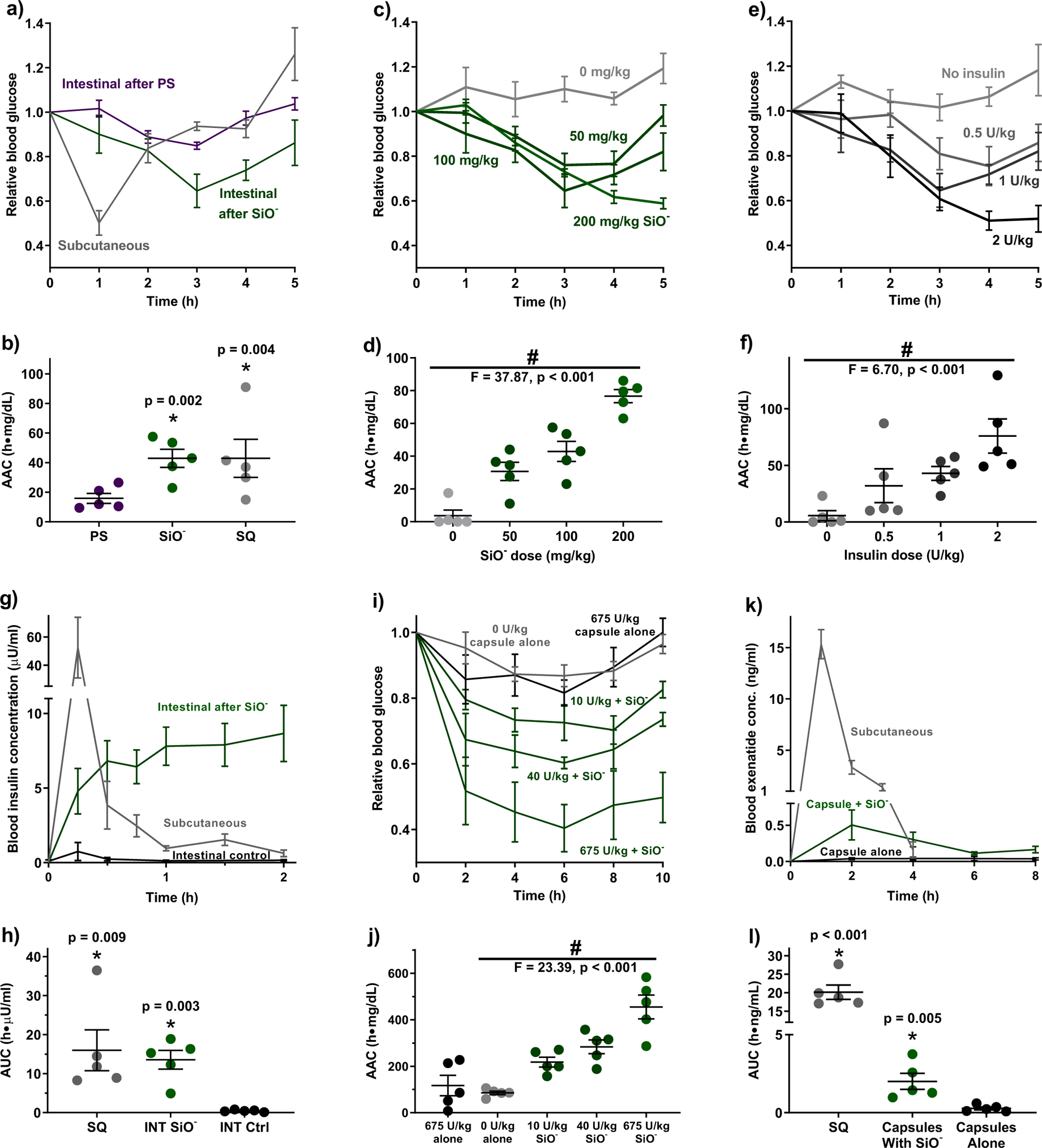
(a) Two hours following oral gavage of silica particles to mice, an intestinal injection of 1 U/kg insulin induced sustained reductions in blood glucose levels. Polystyrene nanoparticles did not enable insulin absorption. A 1 U/kg subcutaneous insulin dose induced a pronounced but brief response. (b) Integrated areas above the curves (AACs) from (a) show that oral delivery with silica nanoparticles and subcutaneous injection resulted in comparable total pharmacodynamic effect. Blood glucose levels and areas above the curves were (c, d) particle dose dependent with a constant insulin dose of 1 U/kg and (e, f) insulin dose dependent with a constant particle dose of 100 mg/kg. (g) Pharmacokinetically, subcutaneous injection caused a spike in blood insulin concentration followed by a rapid decline. By contrast, intestinal administration after silica nanoparticles sustained more modest elevations in serum levels over several hours, leading to (h) nearly equal areas under the blood insulin curves for the two administration methods. (i) Orally delivered insulin induced pronounced and sustained hypoglycaemia at doses as low as 10 U/kg when administered to silica-treated mice. Oral insulin without particles produced no effect compared to the control protein BSA. (j) Particle treatments resulted in multiple-fold increases in the area above the blood glucose curve calculated to 10 hours. (k) Particle treatments enabled systemic uptake of exenatide administered orally in capsules (1 mg/kg), leading to (l) a substantially higher area under the serum concentration curve when compared to capsules administered without silica. Statistics bars display arithmetic mean and s.e.m. for biological replicates (n = 5). * p < 0.05 w.r.t. control by one-tailed t-test. # p < 0.05 by one-way ANOVA.
To compare the total insulin bioactivity between these administration methods, we integrated the areas between each mouse’s glucose curve and its starting blood sugar value. The areas above the curve (AACs) show that pharmacodynamic activity of intestinal insulin in silica-treated mice is comparable to that of subcutaneous insulin (Figure 3b), yielding a relative bioactivity value of 100% (Table 2). Importantly, the more modest but longer-sustained activity of the intestinal insulin indicates that this administration route may be advantageous for drugs that require extended release profiles.
Table 2 |.
Summary of relative bioavailability and relative bioactivity values for proteins delivered with or without silica nanoparticle absorption enhancers in mice. Data is presented as arithmetic average ± standard error.
| Bioactivity | ||||||
|---|---|---|---|---|---|---|
| Drug | Delivery Route | Dose | Silica NPs | rBGmin | AAC | rBA |
| U/kg | % | % | ||||
| Insulin (Healthy) | SQ | 1 | No | 50.1 ± 5.5 | 37.3 ± 13.6 | 100.0 ± 42.3 |
| INT | 1 | No | 100.0 ± 0.0 | 0.0 ± 5.6 | 0.0 ± 0.0 | |
| 1 | Yes | 64.6 ± 7.5 | 37.3 ± 7.5 | 100.0 ± 33.1 | ||
| Oral | 675 | No | 81.6 ± 3.9 | 0.0 ± 0.1 | 0.0 ± 0.0 | |
| 10 | Yes | 70.3 ± 4.3 | 13.2 ± 2.2 | 35.4 ± 11.1 | ||
| 40 | Yes | 60.3 ± 1.8 | 4.9 ± 0.7 | 13.3 ± 4.2 | ||
| 675 | Yes | 40.4 ± 7.2 | 0.5 ± 0.1 | 1.4 ± 0.4 | ||
| Insulin (Hyper-glycaemic) | SQ | 1 | No | 38.4 ± 5.7 | 88.2 ± 17.1 | 100.0 ± 26.6 |
| Oral | 10 | No | 103.1 ± 5.0 | 17.9 ± 11.0 | 0.0 ± 0.0 | |
| 10 | Yes | 75.5 ± 4.7 | 217.3 ± 48.6 | 29.1 ± 8.5 | ||
| Insulin (Diabetic) | SQ | 1 | No | 25.2 ± 8.1 | 98.5 ±44.2 | 100.0 ± 2.4 |
| Oral | 10 | No | 90.1 ± 6.2 | 376.7 ± 34.2 | 0.0 ± 0.0 | |
| 10 | Yes | 63.0 ± 10.7 | 376.7 ± 34.2 | 23.4 ± 4.9 | ||
| Bioavailability | ||||||
| Drug | Delivery Route | Dose | Silica NPs | Cmax | AUC | F |
| U/kg | μU/ml | h * μU/ml | % | |||
| Insulin (Healthy) | SQ | 1 | No | 52.5 ± 21.6 | 16.0 ± 5.2 | 100.0 ± 46.3 |
| INT | 1 | Yes | 8.7 ± 1.9 | 13.6 ± 2.4 | 84.8 ± 31.5 | |
| 1 | No | 0.7 ± 0.6 | 0.5 ± 0.1 | 2.9 ± 1.2 | ||
| U/kg | ng/ml | h * ng/ml | % | |||
| Exenatide (Healthy) | SQ | 1 | No | 15.3 ± 1.4 | 20.2 ± 2.0 | 100.0 ± 13.7 |
| Oral | 1 | Yes | 0.5 ± 0.2 | 2.0 ± 0.5 | 10.0 ± 2.3 | |
| 1 | No | 0.0 ± 0.0 | 0.3 ± 0.1 | 1.4 ± 0.4 | ||
rBGmin = minimum average relative blood sugar achieved. AAC = insulin dose adjusted area above the blood glucose curve. rBA = dose-adjusted relative bioactivity. Cmax = maximum average serum drug concentration achieved. AUC = dose adjusted area beneath the serum concentration curve. F = relative bioavailability.
To ensure that the observed hypoglycaemia was not a procedural artefact, we examined the dose responsiveness of both elements of the delivery system (i.e. the particles and the drug). While maintaining an insulin dose of 1 U/kg, increases in the nanoparticle dose from 50 to 200 mg/kg induced more pronounced and sustained reductions in blood glucose levels (Figure 3c, Supplementary Figure 2b). This trend is also reflected in the AAC calculations (Figure 3d). Because hypoglycaemia did not resolve within 5 hours of treatment, the reported AAC values for the higher dose groups are an underestimate. The delivery system was also dose-dependent on insulin. When particles were administered at a constant dose of 100 mg/kg, increasing insulin dose from 0.5 – 2 U/kg correlated with increased magnitude and duration of hypoglycaemia (Figures 3e–f, Supplementary Figure 2c).
Next, we assessed the pharmacokinetics of intestinally administered insulin by quantifying serum insulin concentrations using enzyme-linked immunosorbent assay (ELISA). Mice that received 1 U/kg subcutaneous insulin injections experienced large spikes in blood insulin concentration within fifteen minutes that returned to normal levels shortly after two hours (Figure 3g). By contrast, mice that received the same intestinal insulin along with 200 mg/kg silica nanoparticles demonstrated more moderate elevations in blood insulin that persisted beyond two hours. Intestinal insulin with no particle treatment gave no substantial increase in serum insulin levels. Integrating the areas under the concentration curve for both administration routes yields approximately the same total serum values (Figure 3h) and nearly 100% relative bioavailability for the intestinal insulin (Table 2). This compares favourably to another promising oral protein delivery technology - CAGE ionic liquid – which registered 51% relative bioavailability upon intestinal injection11.
Interestingly, the integrated bioavailability of the insulin appears to much better predict its relative bioactivity than does the peak serum concentration. Such a discrepancy between total insulin activity and maximal systemic insulin concentration is common when comparing subcutaneous to oral insulin systems33,34, and is likely due to first-pass liver processing of all material absorbed by the intestines. This uptake leads to a rapid decline in hepatic glucose output and more stable regulation of blood sugar than that achieved by injected insulin35. Thus, patients treated with oral insulin should experience superior glycaemic control and significantly reduced risk of dangerous hypoglycaemic episodes when compared to patients treated with subcutaneous insulin15.
Given the successful proof-of-concept that silica nanoparticles facilitate intestinal delivery of insulin, we next demonstrated that particles enable fully oral delivery. To protect insulin in the upper gastrointestinal tract, it was loaded into mouse-specific (Size M) gel capsules along with the protease inhibitor aprotinin, which improves protein survival in digestive fluids36. Furthermore, loaded capsules were coated with Eudragit L100–55, a pH-responsive polymer that remains intact at low pH (e.g. in the stomach) and dissolves to provide a burst release of drug within 15 minutes in the pH 5 to 5.5 conditions of mouse small intestine37 (Supplementary Figure 3). Mice weighing at least 30 g were used for capsule experiments to ensure that their GI tracts would be large enough for the material to transit, and a subcutaneous injection of 1 mg/kg metoclopramide hydrochloride was given at the time of capsule administration to induce gastric emptying into the small intestine.
We tested insulin at three doses: 675 U/kg to observe maximal effect given the packing limitations of the mouse capsules, 40 U/kg to closely compare with many of the oral insulin systems in literature22–24,26,34,38, and 10 U/kg to probe how low of an oral insulin dose could produce an observable therapeutic effect. When delivered orally to mice treated with 200 mg/kg silica nanoparticles, these insulin capsules provoked intense, sustained hypoglycaemia that lasted at least ten hours past administration (Figure 3i, Supplementary Figure 2d). In contrast, maximum dose insulin capsules given to mice without nanoparticle treatment did not affect blood glucose or corresponding AACs when compared to control capsules containing only BSA and aprotinin (Figure 3j).
To confirm that the silica nanoparticles promote the intestinal absorption of protein drugs beyond insulin, we next sought to deliver the anti-diabetic peptide exenatide. Exenatide mimics native hormones to help stabilise blood sugar and is injected subcutaneously for long-term management of type 2 diabetes39. To orally deliver this peptide, exenatide-loaded capsules (1 mg/kg dose) were enterically coated for protection against the stomach environment, and washed down the oesophagus with either silica nanoparticle suspension (200 mg/kg), or saline for a negative control. Blood samples, which were analysed for exenatide concentration via ELISA, showed that the particle treatments greatly improved exenatide uptake compared to the peptide capsules without particle treatment (Figure 3k). When compared to the same dose of subcutaneously administered exenatide, the silica-assisted, orally delivered peptide achieved 10% bioavailability (Figure 3l and Table 2). While this is lower than bioavailability following intestinal injection due to less spatial control and particle-protein co-localization, it does compare closely with oral protein delivery by promising technologies in the literature. For example, nanoparticles that exploit the bile salt pathway for intestinal cell uptake can reach 15.9% oral bioavailability, and Gastrointestinal Permeation Enhancement Technology (GIPET) assisted tablets achieve 8–9% oral delivery of 4–6 kDa proteins10. Given that GIPET has advanced into Stage II clinical trials6, and our successful delivery of both insulin and exenatide, we believe that these silica nanoparticle treatments have the potential to translate into the clinic as a versatile technology for the oral administration of small protein drugs.
We also verified that silica nanoparticles enable oral insulin delivery in diabetic mice, which differ in their pharmacodynamic response to insulin compared to healthy animals. To do this, we employed the streptozotocin-induced model of type 1 diabetes. Our cohort included animals that were fully diabetic (fasting blood glucose levels >250 mg/dL) and hyperglycaemic / pre-diabetic (fasting glucose levels between 150 – 249 mg/dL)40. Insulin was administered in capsules or subcutaneously, as previously described, and blood glucose was monitored over 8 hours. In diabetic mice, a 10 U/kg oral dose of insulin co-administered with nanoparticles induced a more consistent, extended hypoglycaemic effect (Figure 3a–b) with higher areas above the blood glucose curve (Figure 3c) compared to subcutaneous insulin. Similar effects were observed in hyperglycaemic mice (Figure 4d–f).
Figure 4: Silica nanoparticles enabled oral protein delivery in mice with streptozotocin-induced, type 1 diabetes.
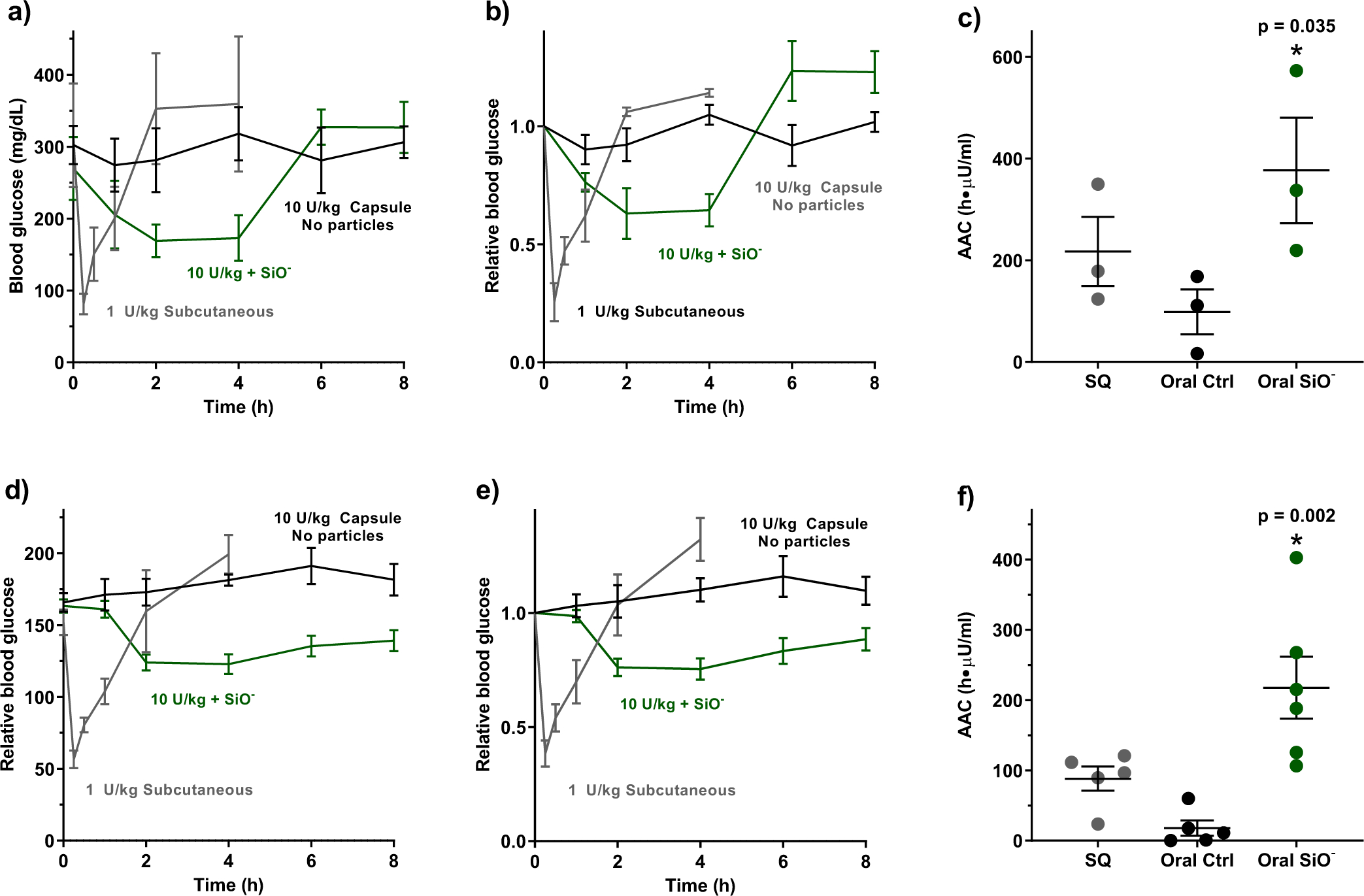
(a, b) In diabetic mice, a 1 U/kg subcutaneous dose of insulin produced a sharp, brief decrease in blood glucose levels. By contrast, a 10 U/kg oral insulin dose induced sustained hypoglycaemia only when co-administered with silica nanoparticles. (c) Particle treatment resulted in a significant increase in the area above the blood glucose curve. (d, e) In hyperglycaemic mice, co-administration of oral insulin and silica nanoparticles facilitated a sustained reduction in blood glucose levels compared to subcutaneous administration, and (f) a substantially larger area above the blood glucose curve. Statistics bars display arithmetic mean and s.e.m. for biological replicates (n = 3 for diabetic mice, n = 5–6 for hyperglycaemic mice). * p < 0.05 w.r.t. control by one-tailed t-test.
Using the blood glucose AACs to compare insulin pharmacodynamics between subcutaneous and oral administrations, we calculated per-dose, relative bioactivities of the insulin capsules (Table 2). The 10 U/kg capsules were approximately 35%, 29%, and 23% bioactive in healthy, hyperglycaemic, and diabetic mice, respectively, compared to the 1 U/kg subcutaneous injection. This compares favourably with many of the most promising technologies for oral protein delivery. For example, starch microgels24 in diabetic rats and permeation enhancer-loaded mucoadhesive intestinal patches34 in healthy rats have achieved up to 7% relative insulin bioactivity. Similarly, nanoparticles particles decorated with penetratin23 or low molecular weight protamine25 resulted in 10–18% relative insulin bioavailability. These comparisons suggest that silica nanoparticles are a particularly promising option for oral insulin delivery with versatility across disease states.
Particles induce integrin-mediated tight junction remodelling
Having demonstrated that silica improves oral absorption and bioactivity of protein drugs, we asked how the nanoparticles increase epithelial permeability. First, we determined that permeabilizing activity was not a colligative property (Supplementary Figure 4a), indicating that activity was not caused by an osmotic pressure gradient. Next, we applied silica particles of different sizes while maintaining the total particle surface area per treatment to determine that the total anionic charge is not a controlling factor (Supplementary Figure 4b). We also examined particles for calcium chelation activity, a mechanism by which some chemical permeation enhancers improve epithelial permeability41,42 (Supplementary Figure 4c). While some particles caused slight decreases in free calcium ions, none approached the full calcium depletion necessary to induce tight junction opening.
Next, we examined the integrin family of epithelial cell surface receptors. Integrins have been implicated in the mechanism of action for mechanical permeation enhancement by nanostructured films43. When these receptors are bound, they can stimulate a variety of signalling pathways that each activate the enzyme myosin light chain kinase (MLCK). Activated MLCK phosphorylates the myosin portion of the cytoskeleton, which then contracts and exerts tension on the tight junctions, causing them to open43,44 (Figure 5a). This concept has previously been exploited for oral insulin delivery by using rationally designed PIP (permeant inhibitor of phosphatase) peptides to inhibit myosin light chain phosphatase, resulting in the greater degree of myosin phosphorylation that induces actin contraction and tight junction opening33. In the intestinal epithelium, because tight junctions represent the major barrier to passive transepithelial diffusion, their opening facilitates protein transport from the intestinal lumen into the body and systemic circulation. The physiologic purpose for this process in the intestinal epithelium is unknown. However, a purpose for integrin-mediated junction-opening has been identified in the endothelium: it aids the migration of immune cells from the blood stream into surrounding tissue. Specifically, the cells undergo integrin binding with the top of the endothelial monolayer, inducing the tight junctions to briefly open before re-forming the barrier45. Based on this behaviour, we hypothesized that the silica nanoparticles were stimulating a similar pathway, binding to the epithelial cells to briefly and reversibly open the junctions, allowing protein absorption.
Figure 5: Silica nanoparticles increased permeability by binding cell surface integrins and inducing tight junction rearrangement.
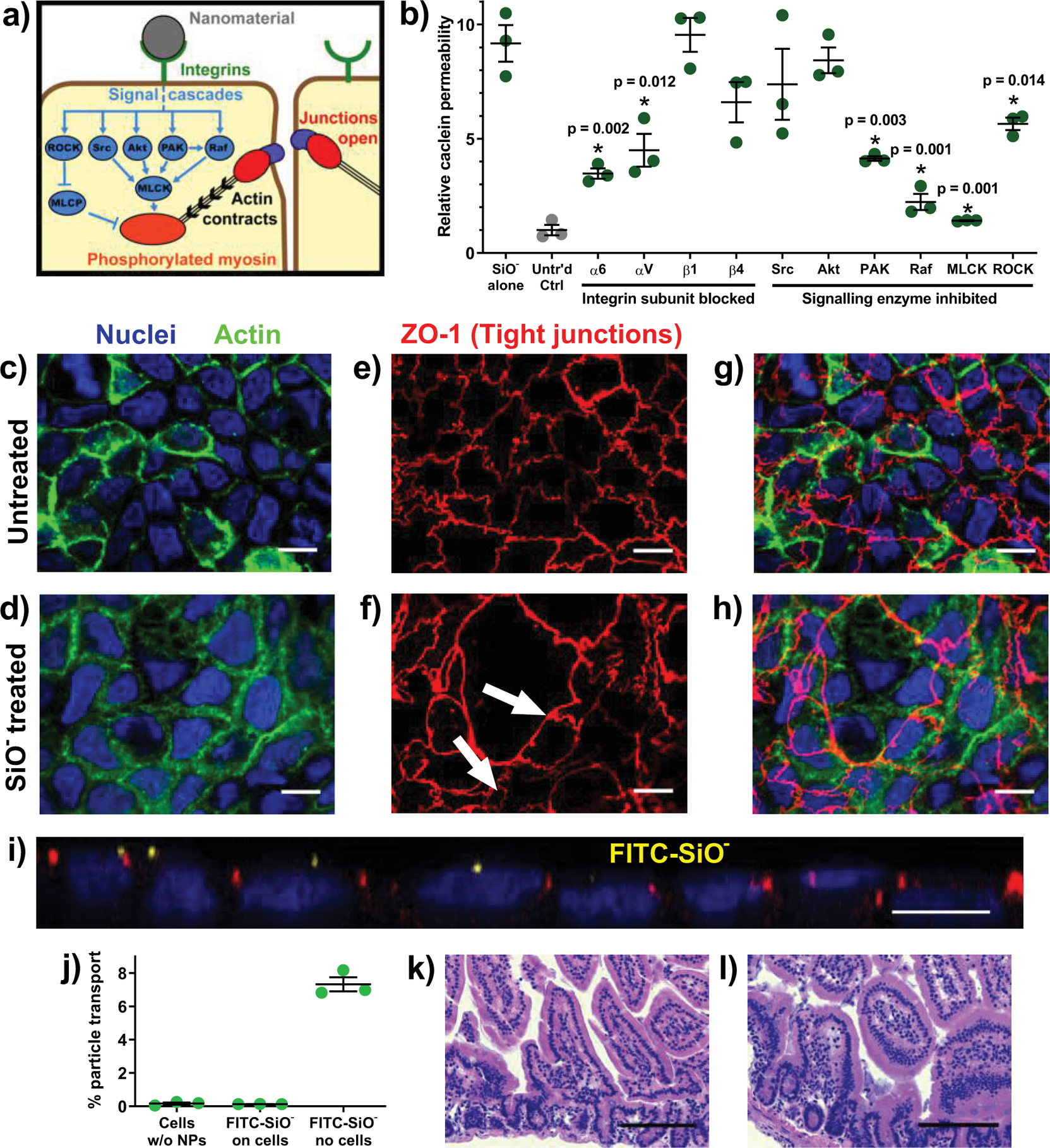
(a) There are several pathways through which integrin activation and myosin light chain phosphorylation are linked to intestinal permeability. (b) The permeation enhancing effect of silica nanoparticles in vitro was reduced by blocking particle binding to integrin α subunits or by inhibiting a subset of intracellular signalling cascade proteins. (c, d) In Caco-2 monolayers imaged at 63x magnification, actin (green) and nuclei (blue) did not rearrange in response to particle treatment. (e) The tight junction protein ZO-1 (red) localised around the perimeter of each untreated cell while (f) silica particle treatment induced ZO-1 rearrangement, creating clusters of cells between which no ZO-1 was present (white arrows). (g) Overlaid images show that the general 1:1 ratio of tight junctions to nuclei in the untreated sample was not maintained in (h) the nanoparticle treated cells, where a single tight junctional boundary encompassed several nuclei. Monolayer images displayed here are a representative sample of over twenty images captured from each treatment. (i) A side view confocal image of a nanoparticle-treated monolayer confirmed that nanoparticles (yellow) localised on top of the cells but did not permeate the tight junctions. (j) FITC-labelled silica nanoparticles did not cross in vitro intestinal barriers. Particles diffused into the basolateral chamber when no epithelial cells were present. (k) Histological analysis of untreated and (l) particle treated mice showed no inflammation and no change in epithelial architecture. Statistics bars display arithmetic mean and s.e.m. for biological replicates (n = 3). * p < 0.05 by one-tailed t-test. White scale bars = 10 μm. Black scale bars = 100 μm.
To test this hypothesis, we first took Caco-2 monolayers and blocked integrins that are highly expressed on intestinal epithelial cells46, including Caco-247, before re-examining the particles’ permeabilizing effects (Figure 5b). Antibodies blocking the integrin alpha subunits ITGα6 or ITGαV were particularly effective at preventing the cells from interacting with the nanoparticles, and the boost to permeability was reduced by approximately half in each case. Neither beta subunit examined appeared to be critical to the nanoparticle binding process, but other integrins may be involved and account for the partial conservation of particle efficacy. Next, we pre-treated monolayers with small molecule inhibitors of known cell signalling enzymes. Inhibitors of rapidly accelerated fibrosarcoma (Raf) or p-21 activated (PAK) kinases were particularly effective at reducing particle efficacy (Figure 5b). Further, treatment with an MLCK inhibitor cut off the signal cascade downstream and completely prevented the particles from increasing monolayer permeability. These data suggest that the particle treatments act via several binding and signalling mechanisms. It is clear, however, that the primary effect derives from MLCK-driven rearrangement of the cytoskeleton, resulting from activation of alpha integrin subunits and subsequent signalling through the Raf and PAK dependent pathways.
To visualise how particle-induced cytoskeletal tension affects tight junction arrangement, we stained Caco-2 monolayers for nucleic acids, actin, and the tight junction protein zonula occludens-1 (ZO-1). There was no apparent rearrangement of nuclei or the actin skeleton (Figure 5 c–d), despite actin rearrangement being a common mechanism of action for intestinal permeation enhancers12,48. However, when compared to ZO-1 patterns in untreated samples, some particle-treated cells formed clusters that did not contain ZO-1 within their junctions (Figure 5 e–h). This rearrangement may create an effective permeation pathway for macromolecule drugs to diffuse between the cells. To the best of our knowledge, this phenomenon has not previously been reported in literature. To quantitatively assess these permeation hotspots, we counted the number of nuclei and ZO-1 loops in both untreated and particle-treated monolayers. Cells exposed to silica nanoparticles had a significantly higher ratio of nuclei to ZO-1 enclosures (1.27 +/− 0.07, mean +/− s.e.m.) than untreated cells (1.05 +/− 0.03), indicating that the particle treatments deplete ZO-1 between epithelial cells.
Silica particles did not cross or damage epithelial tissue
Given that permeation enhancing silica nanoparticles need only to interact with apical cell surface integrins to take effect, we performed a set of studies to determine whether they penetrate or traverse the epithelium. In these experiments, silica nanoparticles containing FITC in their cores were applied to the apical side of Caco-2 monolayers, which were subsequently imaged using confocal microscopy. Composite stacks of monolayer images showed that the particles accumulate at the apical cell surface but do not localise within junctions or inside the cells (Figure 5i). Additionally, the particles did not traverse the monolayers, with only the cell-free membrane supports allowing FITC passage (Figure 5j). Confocal imaging of mouse intestines treated with FITC-doped silica particles further confirmed that particles localise to the apical cell surface, but exhibited no particle penetration through the epithelium and into the centre of the villus within the first two hours after dosing (Supplementary Video 1).This is consistent with previous studies that have shown that negatively charged particles do not readily translocate through epithelial models49,50. These studies add further evidence that silica nanoparticles are acting solely as permeation enhancers and not as delivery vehicles that transport macromolecules across the intestinal barrier.
To assess the integrity of intestinal tissue following treatment, histology was performed on the intestines of untreated and particle-treated mice. There were no significant differences between the control (Figure 5k) and experimental mice (Figure 5l). Further, semi-quantitative analysis by a trained pathologist indicated no difference in immune cell infiltration or inflammatory response between the samples. These results are consistent with several studies that have shown that chronic gastrointestinal exposure to nanosilica does not cause lasting health effects in animal models51. Most notably, a study in rats examined particles ranging from 20 to 100 nm, delivering a particle dose ten times the largest used here (2000 mg/kg vs 200 mg/kg) every day for ninety days. Afterwards, there were no signs of particle uptake into the animals’ bodies and no observable changes in clinical signs, blood biochemistry, or histopathological markers.
Given that our experiments showed efficacy at doses at least as low as 50 mg/kg (Figures 3c and 3d), the particles can be inferred to have a therapeutic window spanning at least 1.5 orders of magnitude. By contrast, many of the most promising chemical permeation enhancers, including piperazines, bile salts, fatty acids, and surfactants, have very narrow therapeutic windows, often less than an order of magnitude concentration in vitro12,13,17,18. Despite their narrow therapeutic windows, several of these species have nonetheless advanced to early stage clinical trials19. We also anticipate that the incorporation of silica nanoparticles into a delivery vehicle, such as a polymer-based intestinal patch34,52,53, will drastically reduce the dose of particles needed to achieve the same protein uptake when scaling up to larger animal models. Whereas the particles and protein drugs are currently spreading throughout the intestines upon administration, we anticipate the same magnitude of effect at lower doses when nanoparticles are co-localised with drug at the surface of the intestinal epithelium.
Outlook
The oral delivery of protein drugs has the potential to improve patient experience and disease outcomes by mitigating the fear and non-compliance associated with injections. Unfortunately, no approved oral protein therapies exist because clinical translation requires high drug bioavailability, and effective permeation enhancement strategies generally induce unacceptable levels of toxicity locally or systemically. Silica nanoparticles offer an alternative permeation enhancement approach that requires both physical elements (nanoparticle size) and chemical elements (negative surface chemistry). It may be this interplay between physical and chemical permeation enhancement that drives the unusual, integrin-binding mechanism of the nanoparticles and promotes reversibility of effect. Silica nanoparticles are one of a number of published strategies to improve insulin bioavailability in rodents. Although insulin bioavailability using silica nanoparticles compares favourably to other strategies, more work will be required in non-human primates to assess interspecies translatability and the potential for clinical studies.
Methods
Materials
Thirteen commercially available nanoparticles were used in this study. Nine were purchased from nanoComposix (San Diego, CA, USA): non-functionalised silica (20, 50, 100, 200, 500 and 1200 nm), 50 nm amine functionalised silica, non-functionalised silver, and gold. Four were purchased from Microspheres-Nanospheres (Cold Spring, NY, USA): 50 nm polystyrene, 50 nm carboxyl functionalised silica, and silica internally doped with FITC (50 nm) or Rhodamine B (20 nm).
For cell culture and in vitro experiments, penicillin/streptomycin, trypsin-ethylenediaminetetraacetic acid (trypsin-EDTA), phosphate buffer saline (PBS), foetal bovine serum (FBS), rat tail Collagen I, calcein, anti-integrin α6 antibodies, anti-integrin β4 antibodies, DAPI, Hoechst 33342, Alexa Fluor® 488 tagged phalloidin, and Alexa Fluor® 594 tagged anti-ZO-1 antibodies were purchased from Life Technologies® (Thermo Fisher subsidiary, Carlsbad, CA, USA). Caco-2 cells and methyl thiazole tetrazolium (MTT) kits were purchased from American Type Culture Collection® (ATCC, Manassas, VA, USA). Dulbecco’s Modified Eagles Medium (DMEM), Falcon® 225 cm2 tissue culture flasks, Corning® HTS 1.0 μm porous support Transwell® plates, Falcon® 24-well plates, Corning® CellBIND® 96-well microplates, sodium butyrate, and MITO+ serum extender were obtained from VWR® (Radnor, PA, USA). FITC-labelled dextrans, Type II mucin from porcine stomach, pepsin, pancreatin, FITC-insulin, FRAX 486, SR 3677, PP2, API-1, AZ628, myosin light chain kinase inhibitor peptide 18 (PIK), Eriochrome® Black T indicator (EBT), EDTA, anti-integrin αV antibodies, and anti-integrin β1 antibodies were purchased from Sigma-Aldrich® (St. Louis, MO, USA).
For mouse experiments, bovine pancreas insulin, bovine serum albumin (BSA), metoclopramide hydrochloride, and streptozotocin were purchased from Sigma-Aldrich®. Exenatide, aprotinin, Aimstrip® Plus blood glucose strips, and blood glucose monitor were obtained from VWR®. Human insulin ELISA kits were purchased from LifeTechnologies®. Exenatide ELISA kits were purchased from Peninsula Laboratories (San Carlos, CA, USA). Mouse sized (M) capsules and dosing kit were supplied by Torpac® (Fairfield, NJ, USA), and Eudragit® L100–55 enteric coating polymers were a gift of Evonik (Essen, Germany).
Particle Characterization
Nanoparticles were diluted in deionised, 0.22 μm filtered water to a solids concentration of 1 mg/mL (w/v) for all DLS and zeta potential characterization. Suspension parameters were measured using a Malvern Zetasizer Nano (Malvern Instruments, UK), using the instrument software’s pre-programmed material properties. Data reported for size and zeta potential are the averages of three technical replicate runs. All other particle characteristics reported were supplied by nanoComposix in particle batch certificates of analysis.
Caco-2 Cell Culture
Caco-2 lines were confirmed mycoplasma free by direct DNA staining with Hoechst 3334254. Cells were cultured in DMEM supplemented with 10% FBS, 100 IU/mL of penicillin, 0.1 mg/mL streptomycin, and 0.25 μg/mL Amphotericin B (“Caco-2 media”). Cultures were incubated at 37°C in a fully humid, 5% CO2 environment. The cells were subcultured with 0.25% trypsin-EDTA and subsequent passaging every 3 to 4 days at ratios between 1:3 and 1:8. Cells at passage numbers 20–50 were utilized for further experiments. The free-to-use PS power calculator (Vanderbilt) was used to determine the minimal sample size for which statistical power was greater than or equal to 0.8, generally n = 3–4 for Caco-2 based experiments.
Toxicity Assays
For the MTT viability assay, Caco-2 cells were seeded in a clear, 96-well plate at a concentration of 105 cells/well. After incubating the plate overnight at 37°C, the media in the wells was aspirated and replaced with treatment solutions (100 μL/well, 0.2% w/v particles). After three hours of exposure, treatments were removed and cells were rinsed with warm PBS. MTT reagent (10 μL/well) and Caco-2 media (100 μL/well) were added to the wells. Three hours later, detergent reagent was added (100 μL/well), and the plate was incubated at room temperature, overnight, in the dark. A BioTek® Synergy2 automated plate reader was used to measure the absorbance of the MTT product in each well. The viability of each treatment is expressed as the ratio of its wells’ absorbance values to the absorbance values of untreated wells.
For the LDH release viability assay, Caco-2 cells were seeded in a 96-well plate at a concentration of 104 cells/well. After incubating the plate overnight at 37°C, the media in the wells was aspirated and replaced with treatment solutions (100 μL/well, 0.2% w/v particles). After three hours of exposure, 50 μL of media from each well was transferred to a new, clear plate and combined with 50 μL/well LDH reaction mixture. Thirty minutes later, stop solution was added (50 μL/well), and the plate was read for absorbance at 680 nm (background signal) and 490 nm (formazan product of LDH activity). Viability percentages are provided by comparing the formazan absorbance of particle-treated cells to that of untreated (negative control) and completely lysed (positive control) cells.
Caco-2 Permeability Experiments
For transepithelial electrical resistance (TEER) and diffusion marker permeability experiments, an established model55–57 of rapid, 3-day Caco-2 intestinal epithelial monolayers was employed. Caco-2 cells were suspended in DMEM supplemented with MITO+ serum extender (basal seeding medium, BSM), seeded at a density of 2×105 cells per well on collagen-coated HTS membrane supports, and incubated for 24–48 hours. The media was then changed to DMEM supplemented with MITO+ and 2 mM sodium butyrate (enterocyte differentiation medium, EDM), and incubated for 48 hours. The TEER was monitored to confirm proper barrier formation, and only monolayers with initial TEER values of at least 150 Ω·cm2 were utilized for TEER or molecular permeability experiments.
HTS inserts containing Caco-2 monolayers were transferred to 24-well plates containing 1 mL DMEM per well and allowed to equilibrate for 30 minutes before recording initial resistance values using a Millicell® voltohmmeter. Nanoparticle treatments were suspended in EDM (0.2 % w/v unless otherwise specified) and applied to the apical chambers, and negative control wells received fresh EDM. TEER readings were taken after 15, 30, 60, 120, and 180 minutes. After 180 minutes, treatments were removed and the monolayers rinsed once with warm PBS before returning to EDM in both the apical and basal chambers for a 24-hour recovery period.
For molecular permeability, fluorescent paracellular diffusion markers were applied at 0.5 mM (calcein) or 0.2 mM (FITC-DX4), into the apical side of the monolayers with the nanoparticle treatments. After one hour, media in the basal chambers was sampled and examined for fluorescence at 495/515 nm (calcein) or 485/515 nm (FITC-DX4) using the plate reader. Application of calibration curves yielded the amount of mass transferred across each monolayer, which was used in the permeability
| equation (1) |
where Papp is the apparent permeability through the monolayer, ΔM is the marker mass in the basal compartment, Ca is the apical marker concentration, A is the monolayer area, and Δt is the time between samples. Permeability measurements are expressed as the ratio of each monolayer’s permeability at 3 hours after treatment addition to its permeability before treatment, normalized to any change in untreated control monolayers during that time.
Integrin Blockade and Signalling Inhibition
For the integrin blockade, Caco-2 monolayers were incubated for an hour before treatment with 25 μg/mL of the specified anti-integrin antibodies. Particle treatments were added without removing the antibodies, and all changes in permeability were normalized to monolayers treated with the antibodies but no particles. For signalling pathway inhibition, the same procedure was employed, adding 10 μM FRAX 486 (PAK inhibitor), 1 μM SR 3677 (ROCK inhibitor), 1 μM PP2 (Src inhibitor), 100 μM API-1 (Akt inhibitor), 2 μM AZ 628 (Raf inhibitor), or 0.33 mM PIK (MLCK inhibitor) to the cell media instead of the antibodies.
Particle Translocation across Mucus Layers
Type II mucin isolated from pig stomach was employed to create the in vitro mucus models, as it is the primary component of intestinal mucus30. Mucus was simulated by dissolving 5% (w/v) mucin in PBS, sonicating, then applying to Transwell® permeable membrane supports (1 μm pore size) to give a 2 mm deep layer. The supports were placed into a basal plate containing 1 mL of PBS in each well, and fluorescent particle suspensions were added to the apical surface of the mucus. Samples were taken from the basal wells over time with PBS replenishment, and read for Rhodamine B (540/625 nm) or FITC (485/515 nm) on the plate reader to determine the fraction of particles transported across the barrier.
Particle-Mucus Interactions
Mucin binding studies were based on previous mucus-binding studies by multiple groups58,59. Briefly, Type II mucin was dissolved in distilled water to a concentration of 10 mg/mL, stirring overnight at room temperature and sonicating to aid dissolution. The solution was then centrifuged for 30 minutes at 850 × g to remove any undissolved solids. Nanoparticles were added to the mucin solution at 1 mg/mL particles, then kept at 37°C with gentle stirring for the remainder of the experiment. At each time point, a sample of nanoparticle and mucin solution was collected and immediately examined for nanoparticle size via dynamic light scattering. Data shown are the averages of three DLS measurements on each sample.
Mouse Studies
All mouse experiments were approved by the institutional animal care and use committee (IACUC) at Carnegie Mellon University (Pittsburgh, PA, USA) under protocol number PROTO201600017, and were performed in accordance with all institutional, local, and federal regulations. C57BL/6 mice were either purchased from Charles River Laboratories (Wilmington, MA, USA) or obtained from an institutionally managed breeding colony. Prior to experiments, mice were housed in cages of no more than six animals, with controlled temperature (25°C), 12 hour light-dark cycles, and free access to food and water. Mice utilized in this study were female and 8–16 weeks (dextran and intestinal insulin, 18–24 g weight range) or 24–30 weeks (protein drug capsules, 30–39 g weight range to ensure capsule passage through the GI tract) old, though only mice within 6 weeks of age are directly compared to one another (i.e. placed on the same graph) for consistency. The free-to-use PS power calculator (Vanderbilt) was used to determine the minimal sample size for which statistical power was greater than or equal to 0.8. (Generally, n=5–6). Mice were fasted 8–12 hours the night before an experiment to limit the variability caused by food matter and faces in the GI tract. Fasting also served to stabilise the animals’ blood sugar for insulin activity experiments, with a starting blood glucose range of approximately 70 to 120 mg/dL. Oral gavages were administered at a volume of 10 ml solution per kg of mouse body weight (10 μl/g). Intestinal and subcutaneous injections were administered at a volume of 1 ml/kg (1 μl/g).
Intestinal Permeability to Dextran
For dextran efficacy studies, fasted mice were orally gavaged with 100 mg/kg nanoparticle solutions (10 mg/kg concentration), then gavaged two hours later with a 60 mg/kg solution of FITC-DX4, at a dose of 600 mg/kg (for a timing study with 50 nm non-functionalised silica, this time step was varied between zero and twenty-four hours). Three hours after the dextran gavage, blood was collected and centrifuged. The serum was removed and examined for FITC concentration by reading for fluorescence on the plate reader (485/515 nm) and comparing to a unique calibration curve for each experiment. For larger macromolecule studies, 40,000 MW dextran (FITC-DX40) was substituted at the same 600 mg/kg concentration.
Intestinal Insulin Delivery
Following ten hours of fasting, mice were orally gavaged with PBS (for control) or nanoparticle suspensions (100 mg/kg unless otherwise specified). Two hours later, their initial blood sugar was measured and the animals were placed under anaesthesia. Their intestines were surgically exposed, and insulin was injected at the predetermined dose (1 unit per kg body weight unless otherwise specified) into the duodenum. The mice were closed and secured with tissue adhesive, then kept under anaesthesia as their blood sugar levels were monitored each hour for five hours. An endpoint at five hours was enforced for all experiments, as the combined effects of the anaesthesia, dehydration, and reduced blood sugar prevented reliable survival beyond that point. For comparison to the current standard of insulin delivery, subcutaneous injections were given at 1 U/kg to additional mice, into the scruff on their necks. To determine areas above the curve for each mouse, trapezoidal integration was used to sum the area between known points on the blood glucose curve and the starting blood glucose value for the individual animal.
To determine specific insulin concentrations, blood samples were collected and separated via centrifugation. The serum was subjected to ELISA analysis per the instructions of the kit manufacturer, and the kit exhibited reliable detection of the bovine insulin used. To calculate areas under the concentration curve for each mouse, trapezoidal integration was used to sum the area between known points on the serum concentration curve and the pre-administration serum concentration for the individual animal.
Capsule Preparation and Characterization
Dry capsule contents for 675 U/kg insulin doses were produced by combining insulin, the protease inhibitor aprotinin, and inactive bovine serum albumin (BSA) filler at a 3:1:1 ratio in aqueous solution, then lyophilizing. Filler for negative control capsules contained just lyophilised aprotinin and BSA (0:1:4). 40 U/kg and 10 U/kg capsule fillers were created by diluting the high-dose insulin powder with the negative control powder. Size M capsules were filled with 2–3 mg filler, and their exact weights recorded. Each capsule was then dip coated 3 times in a 7% (w/v in ethanol) solution of Eudragit® L100–55, drying completely under gentle airflow following each coat. The total dry weight of polymer added to each capsule ranged from 0.4 to 0.8 mg. Capsules to be used for dissolution studies were constructed using FITC-conjugated insulin.
For dissolution trials, capsules were first placed into simulated gastric fluid consisting of 3.2 mg/mL pepsin in 0.3 M NaCl solution, adjusted with HCl to a pH value of 460. Samples were shaken at 120 RPM and 37°C for one hour, and the supernatant fluid sampled for FITC-insulin fluorescence. Capsules were then transferred into simulated intestinal fluid, consisting of 3.2 mg/ml pepsin and 4 mg/ml pancreatin in 0.3 M NaCl and 0.05 M NaHCO3, adjusted to pH 5 or 5.5 with HCl to mimic mouse small intestines37. The samples were then shaken again and sampled for FITC-Insulin in the supernatant every 15 minutes. In each case, the fluorescence was compared to a standard curve prepared in the relevant fluid to obtain an accurate mass of insulin released.
Oral Insulin Delivery with Capsules
Following a ten-hour fasting period, large (> 30 g) mice were orally gavaged with PBS (controls) or 100 mg/kg 50 nm silica nanoparticles, then injected subcutaneously with 5 mg/kg metoclopramide hydrochloride (to stimulate gastric emptying) and orally administered capsules two hours later. Capsules were chosen so small variations in filler weight matched small variations in mouse weight, giving insulin doses within 10% of the designated average dose. The capsules were immediately flushed into the stomach with an additional gavage of PBS or 100 mg/kg silica. Blood glucose was measured every two hours for a total of ten hours, and normalised to each mouse’s reading before capsule administration. From the blood glucose measurements, areas above the curve were calculated as previously described. These areas were used to calculate dose-corrected relative bioactivity as follows:
| (2) |
where β is the insulin dose in U/kg of the capsule treatment being examined, and SQ is subcutaneous injection.
For exenatide delivery using oral capsules, the same scheme was used. Capsule filler was 75:25:1 BSA: aprotinin: exenatide by mass. In the place of blood glucose measurements, blood was collected from each mouse at the predetermined times, and the serum was examined for exenatide concentration via ELISA. Areas under the concentration curve were calculated using trapezoidal integration to sum the area between known points on the serum concentration curve and the pre-administration serum concentration for the individual animal. The ratio of these areas is reported as the bioavailability.
Induction of Type 1 Diabetes in Mice
Mice (female, 23–29 weeks old) were injected with a single, intraperitoneal dose of 180 mg/kg streptozotocin (STZ) to induce type 1 diabetes. Mice were monitored daily, and insulin delivery experiments were performed one week following STZ administration. Animals that exhibited fasting blood sugar of greater than 250 mg/dL were considered to be diabetic, while those with fasting glucose levels between 150 and 249 mg/dL were considered hyperglycaemic, as is common in mild cases of diabetes40.
Confocal Microscopy
Monolayers were rinsed with PBS to remove treatments and fixed in 4% paraformaldehyde. They were permeabilized with 0.2% Triton-X100 and blocked with 0.2% BSA solution to limit non-specific antibody binding, then incubated for one hour with staining solutions. The staining solution contained DAPI (12 μg/mL, 358 nm/461 nm) to mark nucleic acids, AlexaFluor 488® conjugated phalloidin (5 units/mL, 495 nm/518 nm) to bind actin, and AlexaFluor® 594 conjugated Anti-ZO-1 antibodies (50 μg/mL, 590 nm/617 nm) in 0.2% BSA. For monolayers that were treated with FITC-doped particles (490 nm/525 nm) the staining solution was prepared without phalloidin. After staining, the monolayers were mounted on slides using ClearMount™ solution (Invitrogen - Carlsbad, CA, USA) and sealed under coverslips using clear nail polish.
Following treatment with FITC-doped particles (490 nm/525 nm), intestines were gently rinsed with PBS to remove faecal debris and fixed in 4% paraformaldehyde. They were permeabilized with 0.2% Triton-X100 and blocked with 0.2% BSA solution to limit non-specific antibody binding, then incubated for one hour with DAPI (12 μg/mL, 358 nm/461 nm) in 0.2% BSA. After staining, the intestines were gently sliced into small section and mounted onto slides in 0.6 ml PBS, using rubber spacers to prevent the coverslip from crushing the tissue.
Prepared slides were imaged at 10x (intestines) or 63x (monolayers) magnification using a Zeiss LSM 700 confocal microscope with ZEN 2012 SP1 software. Images were captured using a Plan-Apochromat 63x/1.40 Oil DIC objective and an X-Cite Series 120Q laser source exposing at 405, 488, and 555 nm. Since all samples were fixed, images were captured at room temperature and represent a single time point. Images were approximately 101.5 μm × 101.5 μm and were captured with a lateral resolution of approximately 0.3 μm. Z stack images were taken in 0.4 μm slices. No additional processing or averaging was performed to enhance the resolution of the images.
Image J (NIH) image processing software was used to prepare confocal images for publication. Upper and lower thresholds were narrowed slightly to remove background noise and improve visibility of the signals. All images were processed with the same thresholds and display lookup tables (LUTs), which were linear throughout their ranges. Images were converted from their original 16-bit format to RGB colour for saving and arrangement into figures. No other manipulations were performed. Z-stack images were converted to side perspective using the Orthogonal Views tool or assembled into video files using the Stacks tool in ImageJ.
To determine the ratio of nuclei to ZO-1 rings, ten locations were randomly selected from three monolayers each for both untreated and 50 nm, non-functionalised silica-treated cells. All monolayers were seeded from the same population of cells onto the same Transwell® plate. ZO-1 and DAPI were imaged for each location, and the resulting images randomised to blind the counter to their treatment status. Each image was then counted for number of nuclei or number of ZO-1 rings. The samples were then matched with their identities, and the corresponding nuclei to ZO-1 ratios calculated for each sample and each treatment.
Histology
Three particle-treated and three untreated control mice were sacrificed following FITC-DX4 absorption experiments, and their small intestines were immediately excised. The organs were fixed for 24 hours in 4% paraformaldehyde, then transferred to 70% ethanol for shipment to Mass Histology Services (Worcester, MA, USA). There, paraffin sections were prepared and stained with haematoxylin and eosin for histological examination. A semi-quantitative analysis of tissue health, inflammation, and immune cell infiltration for each specimen was also prepared by a certified veterinary pathologist.
Statistics
All data presented are accompanied by an indication of which statistical test was used to determine significance. For experiments in which the direction of effect was known, but the magnitude of the effect was being measured (e.g. permeability increases), one-tailed, Student’s t-tests (*) were applied. For experiments determining if two treatments cause distinct responses from the biological subjects (e.g. difference between same number of two discrete particle sizes), two-tailed, Student’s t-tests (†) were used to calculate relevant p values. In experiments where the same treatment was examined to different degrees (e.g. dose responses), statistics provided are the results of one-way ANOVA (#). All figures display the arithmetic mean of the given “n” number of biological replicates (individual animals or number of in vitro cell culture wells), and error bars display the standard error of the mean.
Reporting summary.
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The authors declare that the data supporting the findings of this study are available within the paper and its Supplementary Information. Tabulated data for research purposes is available from the corresponding author upon request.
Supplementary Material
Acknowledgements
The authors thank Lisa Kasiewicz for her assistance in revising and editing this manuscript. They would also like to acknowledge Dr. Michael Koval (Emory University) for his guidance on examining tight junctions.
N.G.L. would like to acknowledge funding support from the Thomas and Adrienne Klopack Graduate Fellowship and NSF GRFP. This material is based upon work supported by the National Science Foundation Graduate Research Fellowship Program under Grant No. DGE1252522. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the National Science Foundation. The authors also acknowledge support from the National Institutes of Health, grant number 1DP2OD026005-01.
Footnotes
Competing interests
K.A.W. and N.G.L. are registered as inventors on PCT application PCT/US2018/042035, which covers aspects of the technology presented here.
Supplementary information is available for this paper at https://doi.org/10.1038/s41551-019-0465-5
References
- 1.Mclenon J & Rogers MAM The fear of needles : A systematic review and meta ‐ analysis. J. Adv. Nurs 00, 1–13 (2018). [DOI] [PubMed] [Google Scholar]
- 2.Howe CJ, Ratcliffe SJ, Tuttle A, Dougherty S & Lipman TH Needle Anxiety in Children With Type 1 Diabetes and Their Mothers. MCN, Am. J. Matern. Nurs 36, 25–31 (2011). [DOI] [PubMed] [Google Scholar]
- 3.Sokolowski CJ, Giovannitti JA & Boynes SG Needle Phobia: Etiology, Adverse Consequences, and Patient Management. Dent. Clin. North Am 54, 731–734 (2010). [DOI] [PubMed] [Google Scholar]
- 4.Peyrot M, Rubin RR, Kruger DF & Travis LB Correlates of insulin injection omission. Diabetes Care 33, 240–245 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zambanini A, Newson RB, Maisey M & Feher MD Injection related anxiety in insulin-treated diabetes. Diabetes Res. Clin. Pract 46, 239–246 (1999). [DOI] [PubMed] [Google Scholar]
- 6.Moroz E, Matoori S & Leroux JC Oral delivery of macromolecular drugs: Where we are after almost 100years of attempts. Adv. Drug Deliv. Rev 101, 108–121 (2015). [DOI] [PubMed] [Google Scholar]
- 7.Morishita M & Peppas N. a. Is the oral route possible for peptide and protein drug delivery? Drug Discov. Today 11, 905–910 (2006). [DOI] [PubMed] [Google Scholar]
- 8.Shen L, Weber CR, Raleigh DR, Yu D & Turner JR Tight junction pore and leak pathways: a dynamic duo. Annu. Rev. Physiol 73, 283–309 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mrsny RJ Oral drug delivery research in Europe. J. Control. Release 161, 247–253 (2012). [DOI] [PubMed] [Google Scholar]
- 10.Walsh EG et al. Oral delivery of macromolecules: rationale underpinning Gastrointestinal Permeation Enhancement Technology (GIPET). Ther. Deliv 2, 1595–610 (2011). [DOI] [PubMed] [Google Scholar]
- 11.Banerjee A et al. Ionic liquids for oral insulin delivery. Proc. Natl. Acad. Sci 115, 7296–7301 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fein KC, Lamson NG & Whitehead KA Structure-Function Analysis of Phenylpiperazine Derivatives as Intestinal Permeation Enhancers. Pharm. Res 34, 1320–1329 (2017). [DOI] [PubMed] [Google Scholar]
- 13.Bzik VA & Brayden DJ An Assessment of the Permeation Enhancer, 1-phenyl-piperazine (PPZ), on Paracellular Flux Across Rat Intestinal Mucosae in Ussing Chambers. Pharm. Res 33, 2506–2516 (2016). [DOI] [PubMed] [Google Scholar]
- 14.Zijlstra E, Heinemann L & Plum-Mörschel L Oral insulin reloaded: A structured approach. J. Diabetes Sci. Technol 8, 458–465 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Arbit E & Kidron M Oral Insulin Delivery in a Physiologic Context: Review. J. Diabetes Sci. Technol 11, 825–832 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aguirre TAS et al. Current status of selected oral peptide technologies in advanced preclinical development and in clinical trials. Adv. Drug Deliv. Rev 106, 223–241 (2016). [DOI] [PubMed] [Google Scholar]
- 17.Lamson NG, Cusimano G, Suri K, Zhang A & Whitehead KA The pH of Piperazine Derivative Solutions Predicts Their Utility as Transepithelial Permeation Enhancers. Mol. Pharm 13, 578–585 (2016). [DOI] [PubMed] [Google Scholar]
- 18.Whitehead K, Karr N & Mitragotri S Safe and effective permeation enhancers for oral drug delivery. Pharm. Res 25, 1782–1788 (2008). [DOI] [PubMed] [Google Scholar]
- 19.McCartney F, Gleeson JP & Brayden DJ Safety concerns over the use of intestinal permeation enhancers: A mini-review. Tissue Barriers 4, 00–00 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kam KR et al. Nanostructure-mediated transport of biologics across epithelial tissue: Enhancing permeability via nanotopography. Nano Lett 13, 164–171 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schoellhammer CM et al. Ultrasound-mediated gastrointestinal drug delivery. Sci. Transl. Med 7, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fan W et al. Functional nanoparticles exploit the bile acid pathway to overcome multiple barriers of the intestinal epithelium for oral insulin delivery. Biomaterials 151, 13–23 (2018). [DOI] [PubMed] [Google Scholar]
- 23.Zhu X et al. Penetratin derivative-based nanocomplexes for enhanced intestinal insulin delivery. Mol. Pharm 11, 317–328 (2014). [DOI] [PubMed] [Google Scholar]
- 24.Liu L et al. pH- and Amylase-Responsive Carboxymethyl Starch / Poly 2-Isobutyl-acrylic acid) Hybrid Microgels as Effective Enteric Carriers for Oral Insulin Delivery pH- and Amylase-Responsive Carboxymethyl Starch / Poly (2-Isobutyl-acrylic acid) Hybrid Microgels. Biomacromolecules 19, 2123–2136 (2018). [DOI] [PubMed] [Google Scholar]
- 25.Sheng J et al. Enhancing insulin oral absorption by using mucoadhesive nanoparticles loaded with LMWP-linked insulin conjugates. J. Control. Release 233, 181–190 (2016). [DOI] [PubMed] [Google Scholar]
- 26.Shan W et al. Enhanced Oral Delivery of Protein Drugs Using Zwitterion-Functionalized Nanoparticles to Overcome both the Diffusion and Absorption Barriers. ACS Appl. Mater. Interfaces 8, 25444–25453 (2016). [DOI] [PubMed] [Google Scholar]
- 27.Lee JH, Sahu A, Choi W Il, Lee JY & Tae G ZOT-derived peptide and chitosan functionalized nanocarrier for oral delivery of protein drug. Biomaterials 103, 160–169 (2016). [DOI] [PubMed] [Google Scholar]
- 28.Wong CY, Al-Salami H & Dass CR Potential of insulin nanoparticle formulations for oral delivery and diabetes treatment. J. Control. Release 264, 247–275 (2017). [DOI] [PubMed] [Google Scholar]
- 29.Larregieu C. a & Benet LZ Drug discovery and regulatory considerations for improving in silico and in vitro predictions that use Caco-2 as a surrogate for human intestinal permeability measurements. AAPS J 15, 483–97 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Atuma C, Strugala V, Allen, a & Holm, L. The adherent gastrointestinal mucus gel layer: thickness and physical state in vivo. Am. J. Physiol. Gastrointest. Liver Physiol 280, G922–G929 (2001). [DOI] [PubMed] [Google Scholar]
- 31.Grießinger J et al. Methods to determine the interactions of micro- and nanoparticles with mucus. Eur. J. Pharm. Biopharm 96, 464–476 (2015). [DOI] [PubMed] [Google Scholar]
- 32.Bischoff SC et al. Intestinal permeability – a new target for disease prevention and therapy. BMC Gastroenterol. 14, 189 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Taverner A et al. Enhanced paracellular transport of insulin can be achieved via transient induction of myosin light chain phosphorylation. J. Control. Release 210, 189–197 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gupta V et al. Delivery of Exenatide and Insulin Using Mucoadhesive Intestinal Devices. Ann. Biomed. Eng 44, 1993–2007 (2016). [DOI] [PubMed] [Google Scholar]
- 35.Edgerton DS et al. Insulin’ s direct effects on the liver dominate the control of hepatic glucose production. J Clin Invest 116, 521–527 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Morishita I, Morishita M, Takayama K, Machida Y & Nagai T Hypoglycemic effect of novel oral microspheres of insulin with protease inhibitor in normal and diabetic rats. Int. J. Pharm 78, 9–16 (1992). [Google Scholar]
- 37.McConnell EL, Basit AW & Murdan S Measurements of rat and mouse gastrointestinal pH, fluid and lymphoid tissue, and implications for in-vivo experiments. J. Pharm. Pharmacol 60, 63–70 (2008). [DOI] [PubMed] [Google Scholar]
- 38.Lowman AM, Morishita M, Kajita M, Nagai T & Peppas NA Oral delivery of insulin using pH responsive complexation gels. J. Pharm. Sci 88, 933–937 (1999). [DOI] [PubMed] [Google Scholar]
- 39.Genovese S, Mannucci E & Ceriello A A Review of the Long-Term Efficacy, Tolerability, and Safety of Exenatide Once Weekly for Type 2 Diabetes. Adv. Ther 34, 1791–1814 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wu KK & Huan Y Streptozotocin-Induced Diabetic Models in Mice and Rats. Curr. Protoc. Pharmacol 70, 5.47.1–5.47.20 (2015). [DOI] [PubMed] [Google Scholar]
- 41.Sun H, Chow EC, Liu S, Du Y & Pang KS The Caco-2 cell monolayer: usefulness and limitations. Expert Opin. Drug Metab. Toxicol 4, 395–411 (2008). [DOI] [PubMed] [Google Scholar]
- 42.Lopes MA et al. Intestinal absorption of insulin nanoparticles: Contribution of M cells. Nanomedicine Nanotechnology, Biol. Med 10, 1139–1151 (2014). [DOI] [PubMed] [Google Scholar]
- 43.Walsh L et al. Nanotopography Facilitates in Vivo Transdermal Delivery of High Molecular Weight Therapeutics through an Integrin-Dependent Mechanism. Nano Lett 15, 2434–2441 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gilcrease MZ Integrin signaling in epithelial cells. Cancer Lett. 247, 1–25 (2007). [DOI] [PubMed] [Google Scholar]
- 45.Muller WA Mechanisms of Leukocyte Transendothelial Migration. Annu Rev Pathol 6, 323–344 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Beaulieu J-F INTEGRINS AND HUMAN INTESTINAL CELL FUNCTIONS. J. Cell Biol 4, 310–321 (1999). [DOI] [PubMed] [Google Scholar]
- 47.Ölander M, Wiśniewski JR, Matsson P, Lundquist P & Artursson P The Proteome of Filter-Grown Caco-2 Cells with a Focus on Proteins Involved in Drug Disposition. J. Pharm. Sci 105, 817–827 (2016). [DOI] [PubMed] [Google Scholar]
- 48.Salama NN, Eddington ND & Fasano A Tight junction modulation and its relationship to drug delivery. Adv. Drug Deliv. Rev 58, 15–28 (2006). [DOI] [PubMed] [Google Scholar]
- 49.Walczak AP et al. Translocation of differently sized and charged polystyrene nanoparticles in in vitro intestinal cell models of increasing complexity. Nanotoxicology 5390, 1–9 (2014). [DOI] [PubMed] [Google Scholar]
- 50.Yu SH et al. Nanoparticle-induced tight-junction opening for the transport of an anti-angiogenic sulfated polysaccharide across Caco-2 cell monolayers. Acta Biomater. 9, 7449–7459 (2013). [DOI] [PubMed] [Google Scholar]
- 51.Murugadoss S et al. Toxicology of silica nanoparticles: an update. Arch. Toxicol 91, 2967–3010 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Whitehead K, Shen Z & Mitragotri S Oral delivery of macromolecules using intestinal patches: Applications for insulin delivery. J. Control. Release 98, 37–45 (2004). [DOI] [PubMed] [Google Scholar]
- 53.Banerjee A, Lee J & Mitragotri S Intestinal mucoadhesive devices for oral delivery of insulin. Bioeng. Transl. Med 1–9 (2016). doi: 10.1002/btm2.10015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Young L, Sung J, Stacey G & Masters JR Detection of Mycoplasma in cell cultures. Nat. Protoc 5, 929–934 (2010). [DOI] [PubMed] [Google Scholar]
- 55.Chong Saeho; Dando Sandra A.; Morrison RA Evaluation of Biocoat Intestinal Epithelium Differentiation Environment (3-Day Cultured Caco-2 Cells) as an Absorption Screening Model with Improved Screening Productivity. Pharmaceutical Research 14, 1835–1837 (1997). [DOI] [PubMed] [Google Scholar]
- 56.Yamashita S et al. New and Better protocols for a Short-Term Caco-2 Cell Culture System. J. Pharm. Sci 91, 669–679 (2002). [DOI] [PubMed] [Google Scholar]
- 57.Gupta V, Doshi N & Mitragotri S Permeation of Insulin, Calcitonin and Exenatide across Caco-2 Monolayers: Measurement Using a Rapid, 3-Day System. PLoS One 8, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xu Q et al. Impact of Surface Polyethylene Glycol (PEG) Density on Biodegradable Nanoparticle Transport in Mucus ex vivo and Distribution in vivo. ACS Nano 150824093910007 (2015). doi: 10.1021/acsnano.5b03876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pereira De Sousa I et al. Mucus permeating carriers: formulation and characterization of highly densely charged nanoparticles. Eur. J. Pharm. Biopharm 97, 273–279 (2015). [DOI] [PubMed] [Google Scholar]
- 60.Ball RL, Knapp CM & Whitehead K. a. Lipidoid Nanoparticles for siRNA Delivery to the Intestinal Epithelium: In Vitro Investigations in a Caco-2 Model. PLoS One 10, e0133154 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors declare that the data supporting the findings of this study are available within the paper and its Supplementary Information. Tabulated data for research purposes is available from the corresponding author upon request.