

Here, Ding et al. sought to understand whether and how the ubiquitin–proteasomal system (UPS) contributes to oligodendrocyte dysfunction and repair after white matter injury (WMI). They demonstrate that the E3 ligase VHL interacts with Daam2 and their mutual antagonism regulates oligodendrocyte differentiation during development.
Keywords: CNS development, multiple sclerosis, oligodendrocyte, remyelination
Abstract
Dysregulation of the ubiquitin–proteasomal system (UPS) enables pathogenic accumulation of disease-driving proteins in neurons across a host of neurological disorders. However, whether and how the UPS contributes to oligodendrocyte dysfunction and repair after white matter injury (WMI) remains undefined. Here we show that the E3 ligase VHL interacts with Daam2 and their mutual antagonism regulates oligodendrocyte differentiation during development. Using proteomic analysis of the Daam2–VHL complex coupled with conditional genetic knockout mouse models, we further discovered that the E3 ubiquitin ligase Nedd4 is required for developmental myelination through stabilization of VHL via K63-linked ubiquitination. Furthermore, studies in mouse demyelination models and white matter lesions from patients with multiple sclerosis corroborate the function of this pathway during remyelination after WMI. Overall, these studies provide evidence that a signaling axis involving key UPS components contributes to oligodendrocyte development and repair and reveal a new role for Nedd4 in glial biology.
Oligodendrocytes (OLs) produce and assemble myelin sheaths that insulate axons in the central nervous system (CNS), which are essential for transmitting action potentials and maintaining axonal integrity (Huxley and Stämpfli 1949; Griffiths et al. 1998; Yin et al. 1998; Lappe-Siefke et al. 2003; Edgar et al. 2004). Myelin damage is a hallmark of diseases like multiple sclerosis (MS) in adults as well as periventricular leukomalacia and hypoxic ischemic encephalopathy in infants leading to severe deficits in axonal integrity and function. Loss or dysfunction of OLs leads to a failure in proper remyelination and neuronal impairment, processes that are responsible for the neurological consequences of white matter injury (WMI) (Khwaja and Volpe 2007; Compston and Coles 2008). While several key features are shared between development and regenerative myelination, including developmental milestones (i.e., oligodendrocyte precursor cell proliferation, migration or recruitment, differentiation and myelination) and intrinsic-extrinsic regulatory factors (Fancy et al. 2011a; Bhatt et al. 2014; Gallo and Deneen 2014), the mechanisms by which developmental programs are implemented in WMI repair remain incompletely defined.
Wnt signaling plays a critical role in OL development and myelin repair in the CNS (Shimizu et al. 2005; Fancy et al. 2009; Ye et al. 2009; Langseth et al. 2010; Dai et al. 2014; Lee et al. 2015). Injured white matter is populated with “stalled” oligodendrocyte precursor cells (OPCs), in which enhanced Wnt signaling adversely affects regenerative myelination (Wolswijk 2002; Fancy et al. 2009, 2011b; Reich et al. 2018). A critical step during Wnt signal transduction is the clustering of Wnt receptor complexes into signalosomes (Mao et al. 2001; He et al. 2004; Bilić et al. 2007; Schwarz-Romond et al. 2007; Pan et al. 2008; MacDonald et al. 2009; Qin et al. 2009). Previously, we discovered that the scaffold protein Dishevelled associated activator of morphogenesis 2 (Daam2) clusters Wnt receptor complexes (Lee and Deneen 2012), promotes Wnt signaling activity, and suppresses OL development and repair (Lee et al. 2015). These studies indicate that Daam2 plays a crucial role in Wnt-mediated suppression of OL differentiation.
Our recent studies in gliomas identified an antagonistic relationship between Daam2 and hypoxia-associated von Hippel-Lindau (VHL) during tumorigenesis (Zhu et al. 2017). We found that Daam2 degrades VHL by promoting its polyubiquitination, which leads to a subsequent stabilization of hypoxia-inducible factor (HIF). While the relationship of E3 ubiquitin ligase VHL and its substrate HIF has been well established across a host of developmental and disease conditions, including WMI (Maher and Kaelin 1997; Cockman et al. 2000; Ivan et al. 2001; Hon et al. 2002; Kaelin 2002; Yuen et al. 2014), the mechanisms upstream of VHL are not well characterized.
Here, we used conditional mouse models to discover that Daam2–VHL antagonize one another's function during OL development. To identify mechanisms that control the Daam2–VHL relationship, we performed immunoprecipitation-mass spectrometry analysis for Daam2 and identified Neural precursor cell expressed developmentally down-regulated protein 4 (Nedd4). We show that this E3 ubiquitin ligase regulates Daam2 and VHL expression during OL development by promoting VHL stabilization via K63-linked ubiquitination. Further studies in OL-specific Nedd4 knockout mice revealed that this mechanism is conserved in development and WMI. Indeed, loss of Nedd4 suppresses OPC differentiation and myelination during development and inhibits myelin repair after injury. Our findings show that Nedd4 is a novel regulator of VHL stabilization during OL development and repair in WMI.
Results
Daam2 suppresses VHL expression in oligodendrocytes during development
In order to characterize the relationship between Daam2 and VHL in OL development in vivo, we assessed their expression in the OL lineage during CNS development. Daam2 and VHL are both expressed in the OL lineage (Olig2+), including both OPCs (PDGFRα+) and mature OLs (PLP+) (Supplemental Fig. S1A–E). To examine the association between Daam2 and VHL proteins in OLs, we performed coimmunoprecipitation assays in OPC cell line and primary OPCs and found a strong association via Western blot analysis (Fig. 3B lane 9 vs. 12, below; Supplemental Fig. S1F). Furthermore, Daam2 and VHL transcripts were found to colocalize in Olig2+ cells in vivo by RNA in situ hybridization with immunolabeling, suggesting that Daam2 and VHL are coexpressed in OLs (Supplemental Fig. S1G).
Figure 3.
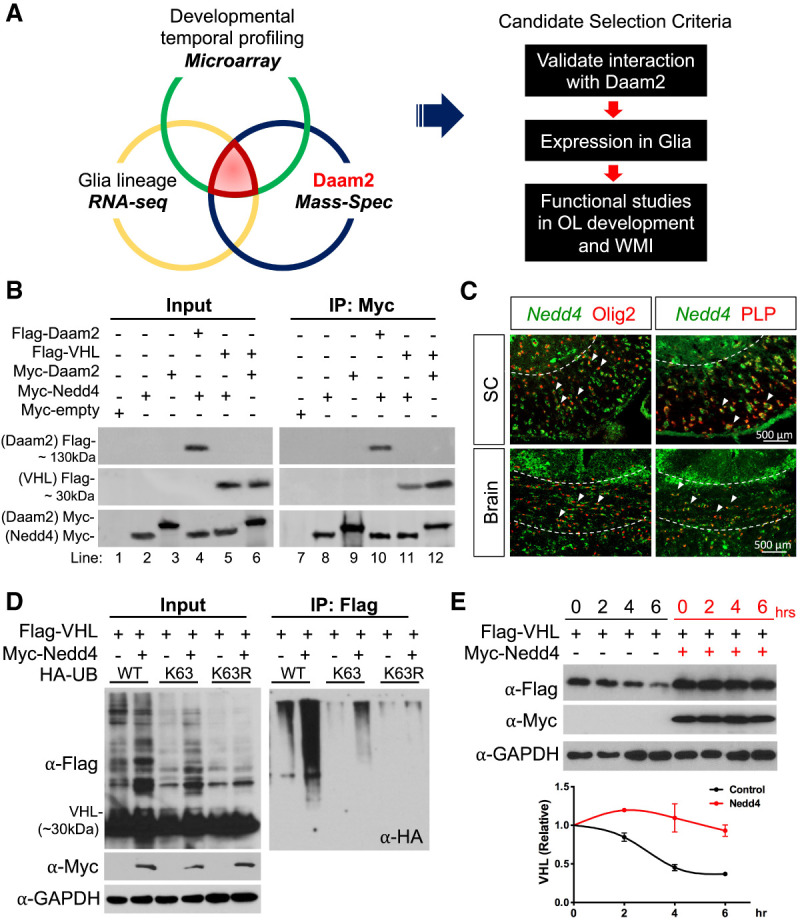
Nedd4 stabilizes the VHL expression during OL development. (A) Screening scheme to identify targeting E3 ligases expressed during glial development (microarray) (Chaboub et al. 2016) and in the glial lineage (RNA-seq data) (Lin et al. 2017) that also interact with endogenous Daam2 in mouse brain (mass spectrometry). Pathway analysis of proteins from the mass spec screen with total protein levels ranked based on intensity-based absolute quantification score (iBAQ). (B) Coimmunoprecipitation of Daam2, VHL, and Nedd4 in the primary OPCs confirms that Daam2 and VHL, VHL and Nedd4, and Daam2–Nedd4 physically associate. (C) Double in situ-Immunofluorescence staining of Nedd4 in the OL lineage in the spinal cord and corpus callosum of the brain in P14 mice. Dashed line indicates white matter. Arrowheads indicate colocalization of Nedd4 with Olig2+ or PLP+ cells. (D) VHL ubiquitination assay. 293T cells were cotransfected with Flag-VHL, Myc-Nedd4, and HA-UbWT (wild type), HA-UbK63 (all six lysine [K] residues mutated to arginine [R] except K63), or HA-UbK63R (K63 residue alone mutated to R, blocking formation of K63-linked polyubiquitin chains). (Left) Representative blots for input samples before IP from three independent repeats. (Right) Representative blot for samples after IP with anti-Flag antibody conjugated beads from three independent repeats. (E) VHL protein stability assay in the presence of Nedd4 upon cycloheximide treatment. Results are mean ± SEM from three independent repeats.
We examined VHL expression in Daam2-depleted OPCs from OL-specific Daam2 conditional knockout mice (see details below) to determine whether the two have an antagonistic relationship during development (Zhu et al. 2017). Indeed, we found that significantly more Olig2+ cells expressed VHL in the absence of Daam2 in both spinal cord and brain (Fig. 1A–F), though the total number of Olig2+ cells remained unchanged (data not shown). Moreover, these Olig2+ cells expressed higher levels of VHL in the absence of Daam2 (see solid white arrows in Fig. 1B,E). To examine whether Daam2 regulates VHL stability in OLs, we performed protein degradation assay in primary OPC cultures from Daam2 knockout mice. We expressed the same amount of VHL protein in Daam2 heterozygous versus knockout primary OPC cultures, then blocked nascent protein synthesis with cycloheximide and measured the relative amount of VHL during OL differentiation. VHL already accumulated to a high level at 0 h and the half-life of VHL was significantly increased in cultured Daam2-deficient OPCs compared with control (Fig. 1G). However, we found no changes of VHL mRNA level by real-time qPCR in Daam2 cKO versus control in vivo (Fig. 1H). Collectively, these in vitro and in vivo observations indicate that Daam2 reduces the protein stability of VHL during OL development.
Figure 1.
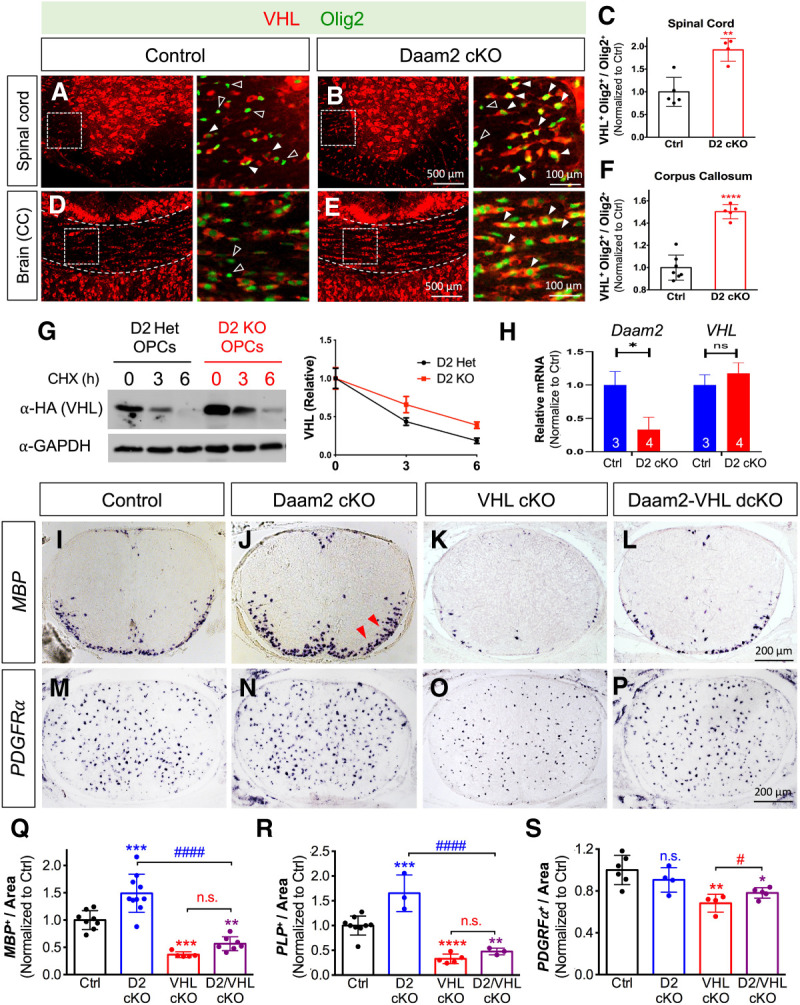
Daam2 suppresses VHL function during OL development. (A–F) Immunofluorescence staining of VHL and Olig2 in the developing spinal cord and corpus callosum (CC) of control (Daam2F/F) versus Daam2 cKO (Sox10-Cre; Daam2F/F) mice at postnatal day 14 (P14). (A,B,D,E) Zoomed-in images are shown in the adjacent panels at the right, which are indicated in the dashed box. Solid and empty arrowheads indicate VHL-expressing versus nonexpressing OLs, respectively. (C,F) VHL-expressing Olig2+ cells were significantly increased in the spinal cord (Student's t-test, [**] P < 0.01) and corpus callosum in Daam2 cKO mice (Student's t-test, [****] P < 0.0001). Data points represent individual animals from three total litters. Plotted values are normalized to control. (G) VHL degradation assay was performed in primary OPCs from Daam2 knockout or heterozygote; OPCs were transfected with plasmids encoding HA-tagged VHL. After 2 d, the amount of VHL was analyzed by immunoblotting after cycloheximide (CHX) treatment for 0, 3, and 6 h. The intensity of VHL/GAPDH from the blots was quantified by ImageJ software and normalized to 0 h. Half-life of VHL in Het OPCs (2.66 h) and KO OPCs (4.77 h). Data were presented as mean ± SEM from three independent repeats. (H) mRNA level of Daam2 and VHL from P0 spinal cords of Daam2 cKO (n = 3) and control mice (n = 4) by real-time qPCR. Plotted values are normalized to control. (I–S) Functional epistatic analysis of Daam2 and VHL loss of function (LOF) during OL development in vivo. (I–P) Representative images of MBP+ mature OLs and PDGFRα+ OPCs via in situ hybridization on P0 spinal cord of Daam2 cKO, VHL cKO, and Daam2–VHL dcKO mice. (Q–S) Quantification of OL and OPC marker expression in single and double LOF. The experiments were performed on at least five animals per genotype, with quantification of at least 6 sections per animal. Each data point represents one animal from at least four litters. Values were normalized to control (one-way ANOVA with multiple comparisons, [**] P < 0.01; [***] P < 0.001; [****] P < 0.0001; [#] P < 0.05; [####] P#### < 0.0001).
VHL rescues Daam2-mediated inhibition of OPC differentiation
Having established that Daam2 negatively regulates VHL expression in OLs, we next assessed the genetic interactions between Daam2 and VHL using mouse models. To determine the consequences of the loss of Daam2 specifically in OLs, we used Sox10-driven Cre to delete Daam2 from a floxed allele (Sox10-Cre+/−; Daam2F/F or Daam2 cKO) where loxP sites flank exon 6 of Daam2. We confirmed 88% Sox10-Cre-derived recombination efficacy in the OL lineage by intercrossing with a ROSA-LoxP-STOP-LoxP-tdTomato reporter, where a frameshift was introduced to result in a null allele (Supplemental Fig. S1H,I). Similarly, we generated conditional OL lineage-specific VHL knockout mice (Sox10-Cre+/−; VHLF/F or VHL cKO) (Yuen et al. 2014) as well as Daam2–VHL double conditional knockout mice (Sox10-Cre+/−; Daam2F/F; VHLF/F or Daam2–VHL dcKO) (Supplemental Fig. S1J). Daam2 cKOs are viable, fertile, and showed no gross changes in appearance compared with controls (littermate Sox10-Cre−; Daam2F/F and Sox10-Cre+; Daam2F/+). However, both VHL cKO and Daam2–VHL dcKO are lethal after birth; therefore, we performed developmental analysis in the spinal cords of Daam2 cKO, VHL cKO, and Daam2–VHL dcKO mice at P0.
We observed an increased number of mature OLs in Daam2 cKO mice, similar to our constitutive knockout mice (Fig. 1I vs. J,Q,R; Supplemental Fig. S2A vs. B; Lee et al. 2015). However, loss of VHL in Daam2 cKO (Daam2–VHL dcKO) reversed this phenotype during OL development (Fig. 1J vs. L,Q,R; Supplemental Fig. S2B vs. D). Analysis of markers for other cell types in the CNS, including GFAP for astrocytes and NeuN for neurons, revealed no significant differences between the single and double cKOs (Supplemental Fig. S2E–N). Of note, loss of VHL alone drastically decreased the number of mature OLs (MBP+ and PLP+) (Fig. 1I vs. K,Q,R; Supplemental Fig. S2A vs. C) and mildly but significantly reduced OPCs (PDGFRα+) (Fig. 1M vs. O,S). Further analysis of this phenotype revealed a significant enrichment of markers for microglia (Iba-1+) (Supplemental Fig. S2O–S) and apoptosis (active-caspase-3) in VHL cKO compared with controls (Supplemental Fig. S2T–X). Intriguingly, the majority of Caspase-3-positive apoptotic cells were co-labeled with neurons (NeuN+), but not OPCs (PDGFRα+) or OLs (PLP+) in VHL cKO (Supplemental Fig. S2Y–AA). Moreover, we did not observe a significant effect on Olig2+ cell proliferation in VHL cKO (Supplemental Fig. S2Y,BB). Therefore, the reduction of PDGFRα labeled OPCs in VHL cKO is not due to cell death or lack of proliferation. Instead, the observations indicate VHL function in OLs influences neuronal survival in a non-cell-autonomous manner during early development. Taken together, our data indicate that Daam2 inhibits OPC differentiation in a cell-autonomous manner by suppressing VHL expression, and that VHL functions downstream from Daam2 during OL development.
To examine this antagonistic relationship at the cellular level, we performed complementary gain-of-function (GOF) experiments using virally transduced primary OPC cultures (Fig. 2A,B). Upon Daam2 viral expression, we observed no changes of OPC numbers at 2 d in vitro (div) (Fig. 2C vs. D,O); however, there were fewer differentiated OLs at 4 div (Fig. 2G,K vs. H,L,P,Q; Lee et al. 2015), indicating that Daam2 overexpression suppresses OPC differentiation. As shown in Figure 1K, in vivo histological analysis established that VHL is necessary for OL maturation. To examine whether VHL alone stimulates OPC differentiation, we overexpressed VHL in primary OPC cultures and observed a significantly increased number of MAG+ and MBP+ OLs at 4 div (Fig. 2G,K vs. I,M,P,Q), but no changes of numbers of PDGFRα+ OPC at 2 div (Fig. 2C vs. E,O). Importantly, VHL expression is sufficient to rescue the inhibition of OPC differentiation by Daam2 overexpression (Fig. 2H,L vs. J,N,P,Q), further supporting the hypothesis that Daam2 counteracts VHL function during OPC differentiation. Collectively, our double loss-of-function (LOF) and GOF studies establish a genetic hierarchy between Daam2 and VHL during OL development in vivo and in vitro.
Figure 2.
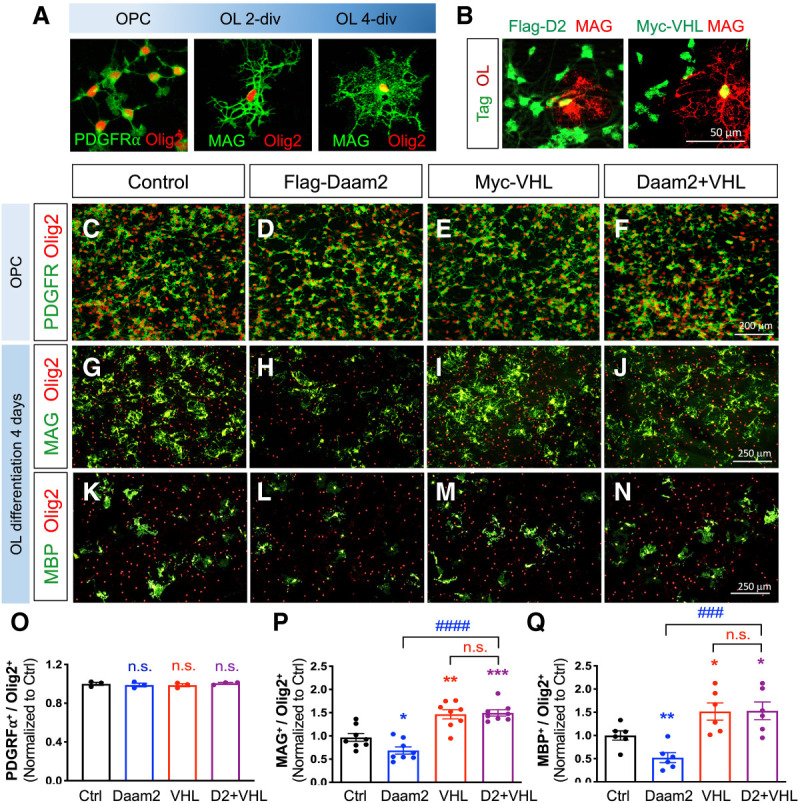
VHL rescues Daam2-induced suppression of OPC differentiation in vitro. (A) Representative images for primary mouse OPC cultures. (B) The expression of Flag and Myc tags are confirmed by immunofluorescence staining, which indicates successful transduction. (C–Q) In vitro overexpression analysis of Daam2 and VHL function during differentiation of OPC cultures. After gene transduction, OPCs were maintained in OPC medium for 2 d (C–F) or in differentiation medium for 4 d (G–N). Cells were then fixed and stained with anti-PDGFRα, anti-MAG and anti-MBP for OPC and mature OL markers, respectively. Anti-Olig2 was also used for labeling the OL lineage cells. (O–Q) Data were acquired from three independent experiments, with values normalized to control (Student's t-test, [*] P < 0.05; [**] P < 0.01; [***] P < 0.001; [###] P < 0.001; [####] P < 0.0001).
Nedd4 is a posttranslational regulator of VHL during OL development
To identify posttranslational modifications that regulate Daam2–VHL function in OLs, we performed Daam2 mass spec using a polyclonal antibody and profiled wild-type and Daam2 constitutive knockout adult mouse brains (2 mo) and cross-compared the candidates with previously reported developmental gene expression data (Fig. 3A; Chaboub et al. 2016; Lin et al. 2017). This comparative analysis revealed a group of candidate genes that are expressed in glial lineages and associate with Daam2 during development and in the adult CNS as well as in vitro (Supplemental Fig. S3A). Since Daam2 promotes VHL ubiquitination-degradation (Zhu et al. 2017) and loss of Daam2 stabilizes VHL in OPCs (Fig. 1G), we focused our analysis on candidate E3 ubiquitin ligases that target the Daam2–VHL complex. Among the candidate E3 ligases (see asterisks indicated in the Supplemental Fig. S3A), Nedd4 is of particular interest as it displays the highest affinity with Daam2 among the candidate factors from our Daam2 mass-spec screen. To validate this relationship, we next examined Nedd4 coexpression and association with Daam2 and VHL in vivo and in vitro. Coimmunoprecipitation assays confirmed that Nedd4 associates with both Daam2 and VHL in primary OPCs and 293T cells in vitro (Fig. 3B; Supplemental Fig. S3B), while double fluorescence in situ-immunostaining results indicate that Nedd4 is coexpressed in OL lineage with Daam2–VHL during CNS development in vivo (Fig. 3C; Supplemental Fig. S3C)
Nedd4 has been shown previously to modify its substrates posttranslationally via ubiquitination in early neuronal development and cancers (Chen and Matesic 2007; Amodio et al. 2010; Drinjakovic et al. 2010; Kawabe et al. 2010; Christie et al. 2012; Ambrozkiewicz and Kawabe 2015; Shao et al. 2018). However, its function in glial development and regeneration is unknown. To examine whether Nedd4 regulates VHL stability through ubiquitination, we performed VHL ubiquitination assays with various lysine mutated Ub constructs in vitro and found that Nedd4 overexpression substantially increased VHL ubiquitination via lysine (K) 63 (UbK63)-linked polyubiquitination. This function was abolished by mutation of the ubiquitin K63 residue to arginine (UbK63R) (Fig. 3D). Ubiquitination at K63 by Nedd4 stabilizes VHL as evidenced by its increased half-life (Fig. 3E). Together these data indicate a novel mechanism where the ubiquitin E3 ligase Nedd4 increases K63 ubiquitination of VHL.
Nedd4 is critical for OL myelination during development
Given that Nedd4 is expressed in OPCs and stabilizes VHL, we examined whether Nedd4 is critical for OL development. We generated conditional null OL-lineage specific Nedd4 mice by crossing Nedd4F/F with the Sox10-Cre line (Sox10-Cre+/−; Nedd4F/F or Nedd4 cKO). Nedd4 cKO mice are smaller in body size and suffer perinatal lethality when compared with littermate controls (wild-type and heterozygous control) (Supplemental Fig. S4A,B). After confirming loss of Nedd4 by immunostaining in the spinal cord of P0 pups (Fig. 4A vs. B), we assessed cellular phenotypes with various OL markers. We found a dramatic decrease in mature OLs expressing PLP and MBP in Nedd4 cKO compared with littermate controls (Fig. 4C,E vs. D,F,K,L), while the number of precursor cells remains relatively unchanged (Fig. 4G vs. H,M; Supplemental Fig. S4S,T). The numbers of active Caspase-3 positive cells increased mainly in the gray matter of Nedd4 cKO (Supplemental Fig. S4C), but there was no change in PDGFRα- and PLP-labeled OL lineage cell death (Supplemental Fig. S4F–I,M). These data indicate that impaired OPC differentiation in Nedd4 cKO is not due to OPC survival during development. Further analysis of markers for other cell types in the CNS revealed no significant differences for astrocytes (GFAP) or neurons (NeuN); however, there was a notable increase for microglia (Iba-1) in Nedd4 cKO compared with control (Supplemental Fig. S4Q–V). In addition, loss of Nedd4 leads to decreased VHL protein levels but no changes in VHL mRNA levels in the white matter of the spinal cord during development (Fig. 4I vs. J,N,O), in agreement with the biochemical data that Nedd4 stabilizes VHL protein but does not impact its transcript.
Figure 4.

OPC-specific Nedd4 KO recapitulates VHL loss-of-function phenotypes. (A–O) Loss-of-function analysis of Nedd4 (Sox10-Cre; Nedd4F/F) in OL development in vivo. (A,B) Confirmation of Nedd4 deletion in the P0 spinal cord by immunofluorescence staining. (C–H) In situ hybridization of mature OL markers for PLP+, MBP+, and OPC for PDGFRα+ cells in Nedd4 cKO versus control (Nedd4F/F) mice. (I,J) Immunostaining of VHL in Nedd4 cKO versus control mice. (K–N) Quantification of the number of PLP+ (Student's t-test, [***] P < 0.0001), MBP+ cells (Student's t-test, [***] P < 0.001), PDGFRα+ cells (Student's t-test, P = 0.11670), and intensity of VHL (Student's t-test, [***] P < 0.0001). Each data point represents individual animal from three litters. Plotted values are normalized to control. (O) mRNA level of Nedd4 from P0 spinal cords of Nedd4 cKO and control by real-time qPCR. Plotted values are normalized to control. (P–S) Electron microscopic analysis of the myelin structure in spinal cord and brain from the Nedd4 loss of function (NG2CreER; Nedd4F/F). Mice were injected with tamoxifen at birth and tissue harvested at P14 for further analysis. (T–W) Statistical analysis of g-ratio and number of myelinated axons in the spinal cord and corpus callosum of NG2CreER; Nedd4F/F (n = 3) versus control animals (n = 4).
To further determine whether myelin integrity is disrupted in the absence of Nedd4 during development, we performed transmission electron microscopy (TEM) on temporally controlled OL-specific conditional null Nedd4 mice by crossing Nedd4F/F with the NG2-CreER line (NG2-CreER+/−; Nedd4F/F); these mice were treated with tamoxifen at P0 and harvested for analysis at P14. Similar to the OL marker analysis that shows an impairment of OPC differentiation in P0 Nedd4 cKO mice (Sox10-Cre-derived), TEM revealed significantly thinner myelin indicated by the larger g-ratio (ratio of the inner axonal diameter to the total outer diameter) and fewer myelinated axons in the spinal cord of NG2-CreER-derived Nedd4 conditional null mice than in control mice (Fig. 4P vs. Q,T,U). Furthermore, we observed thinner myelin and a much lower number of myelinated axons in the corpus callosum of NG2-CreER+/−; Nedd4F/F mice (Fig. 4R vs. S,V,W), suggesting that the corpus callosum is more vulnerable to the loss of Nedd4 during developmental myelination. Together, these data show that Nedd4 is necessary for OL myelination.
Next, we performed complementary overexpression studies in primary OPCs to determine whether Nedd4 stimulates OPC differentiation. We found that overexpression of Nedd4 significantly increases the number of mature OLs expressing MAG and MBP, but no changes in PDGFRα+ OPCs during development (Supplemental Fig. S3D,H vs. F,J,L–N). Interestingly, overexpression of Nedd4 and VHL together shows increased MAG+ mature OLs compared with single overexpression of either Nedd4 or VHL during development (Supplemental Fig. S3G,L), suggesting a cooperative relationship between Nedd4 and VHL to promote OL differentiation. Alternatively, the level of either protein is limiting. Together, our in vivo LOF and in vitro GOF studies indicate that Nedd4 is necessary for OL differentiation and myelination.
Loss of VHL rescues loss of Daam2 following LPC-induced WMI
We next examined whether the developmental mechanism of the Daam2–VHL axis is conserved following white matter injury (WMI). To this end, we used a lysolecithin (LPC)-induced demyelination model to assess the role of the Daam2–VHL axis during myelin repair. To overcome the perinatal lethality of VHL cKO and Daam2–VHL dcKO mice, we generated temporally controlled conditional null alleles in the OL lineage by crossing our existing floxed allele with a NG2-CreER line (NG2-CreER+/−; Daam2F/F, NG2-CreER+/−; VHLF/F, and NG2-CreER+/−; Daam2F/F; VHLF/F). These mice were treated with tamoxifen at 6 wk and Cre recombination and gene deletions were validated at 10 wk (Supplemental Fig. S5). We first examined myelin maintenance for both single and double conditional knockout mice and found no changes in the expression of various cellular markers in 10-wk-old mice (Supplemental Fig. S6), suggesting that short-term deletion of Daam2–VHL axis in OLs in young adult mice does not affect myelin homeostasis. We next injected LPC in the ventral white matter of spinal cords of 10-wk-old mice and analyzed cellular markers for myelin repair at 3 d postlesion (dpl), a well-defined time point for OPC recruitment (Supplemental Fig. S7), and 10 dpl, a time point for OL remyelination initiation when the lesion is repopulated by mature OLs (Fig. 5; Supplemental Fig. S8). We observed no effect on OPC recruitment in the demyelinated lesion site of single or double conditional knockout mice at 3 dpl (Supplemental Fig. S7). However, analysis of OPC differentiation at 10 dpl revealed an increased number of PLP+ and MBP+ mature OLs in the lesion site of NG2-CreER+/−; Daam2F/F mice relative to wild-type controls (Fig. 5F,J vs. G,K,R,S). We also found significantly fewer mature OLs in NG2-CreER+/−; VHLF/F mice with a relatively unchanged number of OPCs, suggesting that VHL is required for OPC differentiation during remyelination after injury (Fig. 5F,J vs. H,L,R,S [for OLs], N vs. P,T [for OPCs]). Importantly, loss of VHL reverses the precocious OPC differentiation that occurs in the absence of Daam2, consistent with VHL functioning downstream from Daam2 (Fig. 5G,K vs. I,M,R,S). In contrast to the altered expression of mature OL markers, single or double conditional knockout mice do not demonstrate any changes in OPC, reactive astrocyte, or microglia populations after WMI (Fig. 5N–Q,T; Supplemental Fig. S8). These findings provide compelling evidence that the genetic hierarchy in which VHL functions downstream from Daam2 during development is conserved in the myelin repair process.
Figure 5.
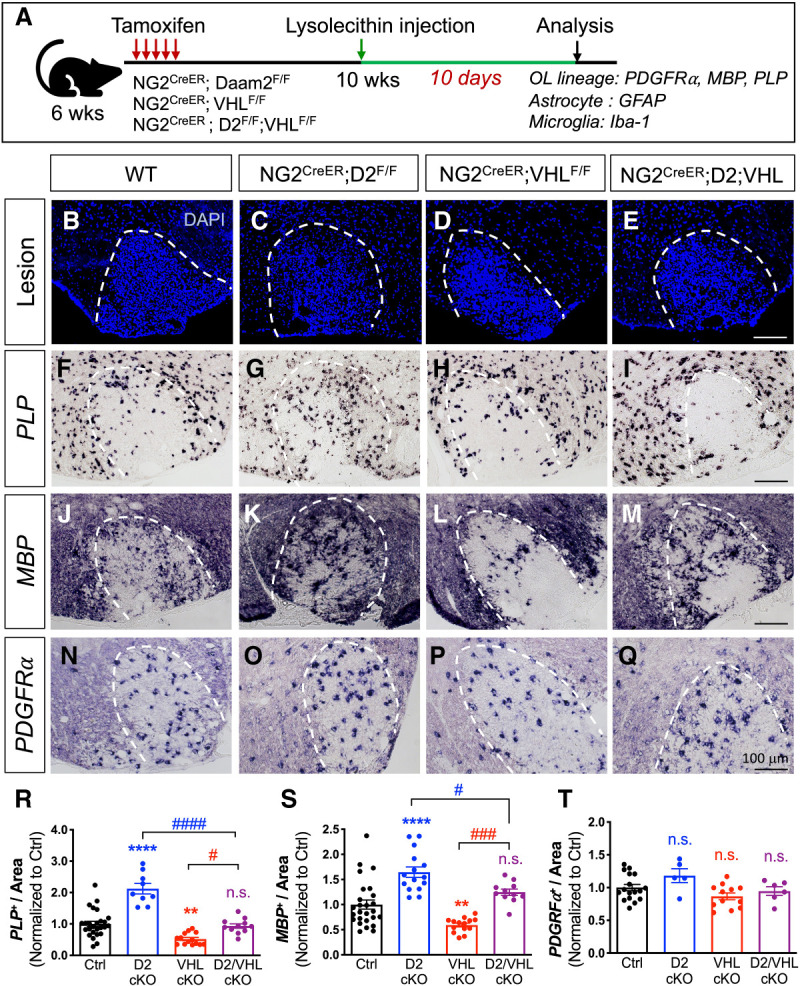
Loss of VHL abolishes the accelerated remyelination by loss of Daam2 in the LPC-induced demyelination model. (A) Experimental scheme of the lysolecithin (LPC)-induced demyelination model. NG2-CreER-derived mutants (NG2-CreER+/−; Daam2F/F, NG2-CreER+/−; VHLF/F, and NG2-CreER+/−; Daam2F/F; VHLF/F) were treated with tamoxifen at 4 wk prior to LPC injection to induce demyelination. Spinal cords were then harvested and analyzed 10 d after LPC injection. (B–E) Lesion area is indicated by the dotted line. (F–Q) In situ hybridization of mature OLs (PLP and MBP) and OPCs markers (PDGFRα) in the lesion. (R–T) Quantification of PLP+, MBP+, and PDGFRα+ in the lesion. Each data point represents individual images from multiple animals. Values were normalized to control (one-way ANOVA with multiple comparisons, α = 0.05; [*] comparison with control; [#] comparison with D2/VHL double LOF; [**] P < 0.01; [***] P < 0.001; [#] P < 0.05; [###] P < 0.001; [####] P < 0.0001).
Nedd4 is required for OPC differentiation after WMI
Given the conserved role of the Daam2–VHL axis in OPC differentiation during remyelination, we next investigated whether Nedd4 is similarly required for this process. We first observed normal myelin maintenance in NG2-CreER+/−; Nedd4F/F mice compared with control littermates (Supplemental Fig. S9). Similar to the studies described in Figure 5A, we next performed LPC-lesioning on NG2-CreER+/−; Nedd4F/F mice and analyzed the extent of OPC differentiation at 3 dpl and 10 dpl. There was no effect on OPC recruitment in the demyelinated lesion site of NG2-CreER+/−; Nedd4F/F mice at 3 dpl (Supplemental Fig. S7). Analysis of OPC differentiation in NG2-CreER+/−; Nedd4F/F mice at 10 dpl revealed a decrease in the number of PLP+ and MBP+ mature cells when compared with wild-type controls (Fig. 6C,E vs. D,F,I,J). However, the number of PDGFRα-expressing precursors was not altered (Fig. 6G vs. H,K). Additionally, reactive astrocytes and inflammatory response markers are unaffected by the loss of Nedd4 (Supplemental Fig. S8). These data indicate that Nedd4 is required for OPC differentiation after WMI.
Figure 6.
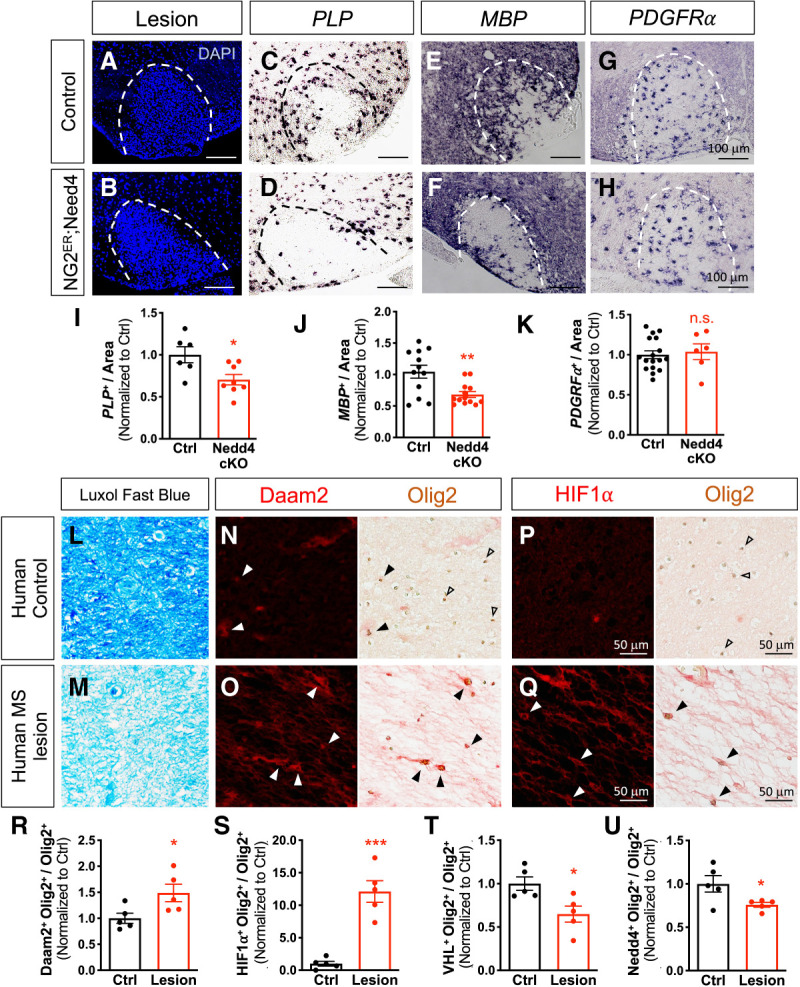
Nedd4 is required for OPC differentiation after WMI and is absent in Olig2+ cells in human MS lesions. (A–H) NG2-CreER; Nedd4F/F and Nedd4F/F mice were treated with tamoxifen at 4 wk prior to LPC injection to induce demyelination. Spinal cords were then harvested and analyzed 10 d after LPC injection. (A,B) Lesion area is indicated by the dotted line. (C–H) In situ hybridization for mature OL (PLP and MBP) and OPC marker (PDGFRα). (I–K) Quantification of PLP+, MBP+, PDGFRα+, and in the lesion. Each data point represents individual images from multiple animals. Values were normalized to control (Student's t-test, [*] comparison with control; [*] P < 0.05; [**] P < 0.01). (L–Q) Histological analysis of healthy human brains and MS lesion tissues. (L,M) Myelin is labeled by luxol fast blue (LFB), showing intact myelin in healthy tissue (L) compared with demyelinated MS lesion tissue (M). Immunohistochemical staining (IHC) of Daam2 (N,O) and HIF1α (P,Q) with Olig2. Solid arrowheads indicate colocalization with Olig2, while empty arrowheads indicate noncolocalization with Olig2. (R–U) The numbers of Daam2, HIF1α, VHL, and Nedd4-positive cells over Olig2-positive cells in the control were counted. Data were acquired from five different tissues per group. Values were normalized to control (Student's t-test, [*] P < 0.01; [***] P < 0.0001).
Daam2 and HIF1α, but not Nedd4 or VHL, are expressed in OPCs in human MS lesions
To determine whether the Daam2–VHL–Nedd4 pathway is operative in human white matter lesions, we performed immunohistochemical analysis of human Daam2, VHL, HIF1α (downstream from VHL), and Nedd4, in combination with Olig2, in postmortem brain samples from five adult MS patients. Healthy white matter versus actively demyelinating lesions were distinguished and confirmed by Luxol Fast Blue staining (confers an intense blue to myelin) (Fig. 6L vs. M). In healthy white matter, Daam2 is expressed in a subset of Olig2+ OL lineage cells (Fig. 6N,R), whereas Nedd4 and VHL are detected in the majority of Olig2+ cells (Fig. 6T,U; Supplemental Fig. S10A, C). In the active plaques, however, Daam2 and HIF1α expression are highly up-regulated in Olig2+ cells (Fig. 6O,Q–S) while Nedd4 and VHL are barely detectable (Fig. 6T,U; Supplemental Fig. S10B,D). These findings support the notion that Daam2 is present in human MS lesions, and that the absence of Nedd4 could contribute to the disease phenotype.
Discussion
The parallels between OL development and postinjury remyelination remain poorly understood. Using lineage-specific LOF approaches, we discovered that Daam2 suppresses OPC differentiation during development and regeneration by down-regulating VHL expression. We found that the Daam2–VHL complex associates with the E3 ubiquitin ligase Nedd4, which in turn functions as a key regulator of VHL stabilization and OPC differentiation during development and repair (Fig. 7). Extending these studies to human WMI, we found increased expression of Daam2 and HIF1α, coupled with reduced expression of VHL and Nedd4 in OPC populations in human MS lesions. Overall, these studies highlight a new and potentially targetable pathway to stimulate the regeneration of OLs after WMI.
Figure 7.
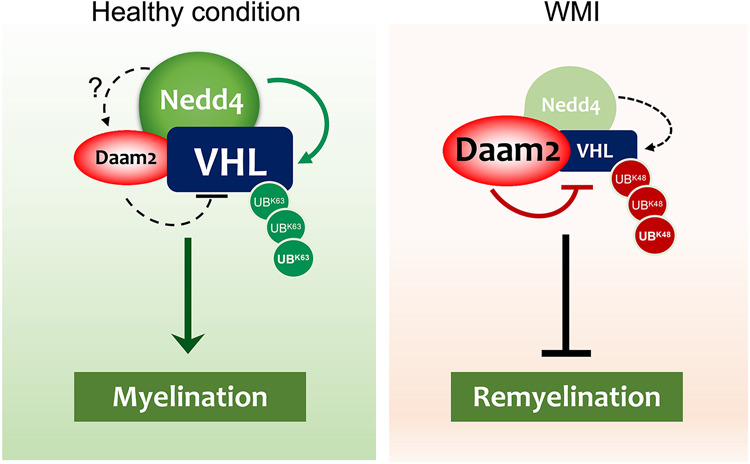
Model for the Daam2–VHL–Nedd4 axis in OL development and WMI. (Left) Under healthy conditions, Nedd4 is expressed in a subset of OPCs where Daam2 is expressed at relatively low levels, and where VHL expression remains high. High VHL levels are protected against proteasomal degradation through Nedd4-mediated K63-linked ubiquitination. (Right) In white matter injury such as MS, Nedd4 expression is lost and Daam2 levels increase. This loss of Nedd4 leads to a decrease in protective K63-linked ubiquitination of VHL, while the coinciding increase in Daam2 promotes VHL degradation through the UPS pathway.
The UPS plays critical roles in the differentiation of multiple cell lineages during development (Sakurai et al. 2006; D'Arca et al. 2010; Opperman et al. 2017; Tsuboi et al. 2018) and is strongly associated with late-onset neurodegenerative diseases (Ciechanover and Brundin 2003; Ross and Pickart 2004; Simpson et al. 2011). Recent reports suggest that a component of the UPS system, Fbxw7, functions through mTOR-dependent mechanisms in OLs in zebrafish (Kearns et al. 2015) and mTOR-independent but c-Jun-dependent pathways in Schwann cells in mice during development (Harty et al. 2019). However, the role of UPS-associated mechanisms, specifically ubiquitin E3 ligases, during OPC differentiation and WMI, remains unknown. Here, we found that the ubiquitin E3 ligase Nedd4 regulates VHL expression during OL development. Furthermore, we showed that Nedd4 promotes OPC differentiation and is required for proper remyelination during WMI. Hence, posttranslational modifications mediated by ubiquitin E3 ligases are critical mechanisms for remyelination upon injury.
Our studies indicate that Nedd4 is required for OL maturation by stabilizing VHL during development; however, whether Daam2 is required for such regulation is unknown. On the one hand, Nedd4 could directly regulate Daam2, which further contributes to the stabilization of VHL. Previous studies have reported that Nedd4 binds to another proximal Wnt signaling component, Dishevelled 1, and subsequently ubiquitinates it, resulting in its degradation (Nethe et al. 2012). Nedd4 may function in a similar manner with respect to Daam2 in OLs. Alternatively, as both Daam2 and VHL are associated with Nedd4, it is also likely that potential competition between Daam2, VHL, and Nedd4 exist. On the other hand, it is possible that Nedd4 stabilizes VHL in a lineage- and stage-specific manner during OL development. VHL is enriched in OPCs while Daam2 is expressed in the majority of mature OLs (Supplemental Fig. S1E). This expression pattern supports the hypothesis that Nedd4 functions differently in OPCs versus OLs through unique patterns of ubiquitination onto different target proteins.
Our studies on Nedd4 represent the first characterization of its role in OL lineage development and myelin repair. Previous studies have shown that Nedd4 regulates neurite growth and brain tumor cell proliferation (Amodio et al. 2010; Drinjakovic et al. 2010; Kawabe et al. 2010; Christie et al. 2012; Eide et al. 2013). Nedd4 germline knockout (Nedd4−/−) is embryonic lethal in mice and heterozygous Nedd4+/− mice exhibit strong deficits in learning and memory, indicating a pivotal role in brain function (Camera et al. 2016). Consistent with the role of Nedd4 as a posttranslational regulator of VHL, we found that loss of Nedd4 phenocopies loss of VHL, including perinatal lethality at P1, smaller body size, impaired OL differentiation, and exaggerated apoptosis, coupled with microglia enrichment. Given the pro-survival effects of VHLand Nedd4 in developmental and injury studies, loss of either VHL or Nedd4 may contribute to a nonpermissive environment for OL myelination and myelin repair.
Finally, the Daam2–VHL relationship was initially discovered in glioma, and it is well established that VHL-HIF signaling plays an essential role in tumorigenesis across a vast spectrum of malignancies. Therefore, our finding that Nedd4 is a key upstream regulator of VHL could have broad implications that extend beyond WMI to include CNS and non-CNS malignancies. In that regard, there is evidence that Nedd4 promotes glioma cell migration and invasion in vitro, thereby contributing to glioma tumorigenesis. It will be important to further delineate whether and how Nedd4 regulates malignancy, perhaps via a mechanism linked to VHL–HIF signaling.
Materials and methods
Mice
To generate the Daam2 conditional knockout mice, we first bred Daam2 floxed (Daam2F/F) mice (loxP flanking exon 6) by crossing the Daam2tm1a(KOMP)Wtsi (KOMP) mouse line with the FLP mouse line. Then Daam2F/F was crossed with Sox10-Cre (JAX 025807) to generate the Daam2 conditional knockout (Daam2 cKO). To confirm the Sox10-Cre-derived recombination efficacy, Daam2 cKO mice were intercrossed with a ROSA-LoxP-STOP-LoxP-tdTomato reporter. We confirmed that 88% of tdTomato-positive cells were colocalized in Olig2+ (n = 7, SD 4%). Similarly, we crossed the VHL floxed (JAX 012933) and Nedd4 floxed (loxP flanking exon 6; provided by Dr. Hiroshi Kawabe) with Sox10-Cre for OL-specific cKO.
To generate OL lineage-specific inducible knockout mice, we crossed the existing floxed alleles (Daam2, VHL, Daam2–VHL, and Nedd4) with NG2-CreER (JAX 008538) and injected tamoxifen (100 mg/kg of body weight) at various time points to induce deletion. To confirm Cre recombination efficacy by tamoxifen injection, we generated NG2-CreER+/−; Daam2F/F mice carrying a ROSA-LoxP-STOP-LoxP-tdTomato reporter, which allowed us to label/isolate the Daam2-depleted OLs after tamoxifen injection. We observed 63% of Tdtomato+ OLs in NG2-CreER+/−; Daam2F/F mice (n = 4, SD 8%). We were not able to generate NG2-CreER+/−; VHLF/F or NG2-CreER+/−; Daam2F/F; VHLF/F mice with a ROSA-LoxP-STOP-LoxP-tdTomato reporter since they shared the same chromosome locus. However, we confirmed adequate deletion of VHL in OLs from NG2-CreER+/−; VHLF/F and NG2-CreER+/−; Daam2F/F; VHLF/F mice by immunostaining. All procedures were approved by the Institutional Animal Care and Use Committee (IACUC) at Baylor College of Medicine and conform to the US Public Health Service Policy on Humane Care and Use of Laboratory Animals.
In situ hybridization and immunofluorescence staining
For both in situ hybridization and immunofluorescence staining, spinal cords and brains of mice were fixed in 4% paraformaldehyde (PFA) overnight, dehydrated in 20% sucrose for cryoprotection, embedded in OCT blocks, and stored at −80°C until ready for sectioning. All slides were sectioned at the thickness of 15 µm and stored at −80°C.
For in situ hybridization, RNA probes of MBP, PLP, PDGFRα, Daam2, VHL, and Nedd4 with DIG and/or FITC labels were generated in-house for detection of the corresponding RNAs by (fluorescence) in situ hybridization. All probes were tested for specificity and sense probes were included in the experiments as a control. Detailed procedures and reagents were described before (Lee et al. 2015). For immunofluorescence staining, tissues were washed in PBS three times for 5 min each, permeabilized with PBST (0.3% Triton in PBS) for 5 min, washed with PBS three times for 5 min each, blocked with 10% normal goat serum in PBST for 1 h at room temperature, and then incubated with primary antibodies overnight at 4°C. The slides were washed in PBS three times for 5 min each, incubated in secondary antibodies for 1 h at room temperature, washed three times for 5 min each with PBS, stained for DAPI, and mounted with VectaShield Antifade mounting medium. The following commercial primary antibodies were used: mouse anti-VHL (1:1000; BD Bioscience), rabbit anti-Olig2 (1:500; Millipore), rabbit anti-GFAP (1:1000; Agilent Dako), mouse anti-NeuN (1:500; Millipore), rabbit anti-Iba-1 (1:1000; Wako) rabbit anti-Caspase 3 (1:1000; R&D), mouse anti-Ki67 (1:1000; BD Bioscience), and rabbit anti-Nedd4 (1:500; Millipore). All secondary antibodies were from invitrogen Alexa Fluor (300, 488, and 568), including goat / donkey anti-rabbit IgG, goat / donkey anti-mouse IgG (H + L), IgG1, IgG2a.
Electron microscopy
As described previously (Hooshmand et al. 2014), mice were perfused with 37°C saline (0.9% NaCl in MilliQ water at pH 7.4) at the speed of 10 mL/min for 5 min and then perfused with 37°C fixative 1 (1% glutaraldehyde + 4% paraformaldehyde in 0.1 M cacodylate) at the speed of 10 mL/min for 5 min. White matter was then dissected out in scintillation vials on ice and postfixed for 1 h in fixative 1 while rotating at 4°C. Tissue was then fixed in lipid fixative for 1 h while rotating at 4°C, washed three times for 15 min each with 0.1 M cacodylate, at 4°C, and fixed in fixative 2 (2% glutaraldehyde in 0.1 M cacodylate) while rotating overnight at 4°C. Tissue was then washed, dehydrated, infiltrated, embedded, and cured for at least 5 d before sectioning and analyzing.
LPC-induced demyelination model of white matter injury
LPC-induced demyelinated lesions were generated in the ventral white matter of the spinal cord of 10-wk-old NG2-CreER-derived inducible conditional mutants. Tamoxifen (100 mg/kg of body weight) was administered five times daily by intraperitoneal injection at 4 wk prior to LPC-induced injury. We then performed the injection of 0.5 µL of 1% lysolecithin in the ventral white matter of the spinal cord via a pulled glass needle attached to the Hamilton syringe. Mice were harvested at multiple time points (between 3 d postlesion [dpl] and 10 dpl) and analyzed. Animals were perfused with 4% PFA. Spinal cords were dissected out and drop-fixed in 4% PFA overnight before being transferred into 20% sucrose. Tissue was embedded in OCT, sectioned, and stored at −80°C before in situ hybridization or immunofluorescence staining was performed as described above.
Primary oligodendrocyte precursor culture
Primary OPC culture was performed as described previously (Lee et al. 2015). Briefly, neural stem cells (NSCs) were harvested and cultured from mouse brains at embryonic day 14.5, maintained as neurospheres in the neural stem cell medium (DMEM/F12 [Gibco], + N2 [Thermo Fisher], B27 [Thermo Fisher], 20 ng/mL EGF [Sigma], 20 ng/mL bFGF [R&D]), and passaged for up to three times before induction of OPC differentiation. For immunocytochemistry, the neurospheres were gently broken down into single-cell suspension and seeded onto poly-D-lysine coated glass coverslips in OPC medium (DMEM/F12, B27, 10 ng/mL bFGF, 10 ng/mL PDGF-aa [PeproTech]). Cells were transduced with lentivirus overexpressing Daam2, VHL, Daam2 + VHL, Nedd4, etc., prior to medium change into OPC medium or OL differentiation medium: (basal chemically defined medium [BDM], 15 nM triiodothyronine [Sigma], 10 ng/mL CNTF [PeproTech], 5 mg/mL N-acetyl-l-cysteine [Sigma]). Upon harvest, OL medium was carefully removed, and cells were washed with ice-cold PBS and then fixed in 4% PFA for 15 min. Immunocytochemistry (ICC) staining was performed with rabbit anti-Olig2 (1:500; Millipore), rat anti-PDGFRα (1:500; Invitrogen) mouse anti-MBP (1:500; BioLegend), mouse anti-MAG (1:500; Millipore), rabbit anti-Flag (1:1000; Sigma), and mouse anti-c-Myc (1:1000; Santa Cruz Biotechnology). For Western blot analysis, neurospheres were gently broken down into single-cell suspensions and seeded into PDL-coated six-well plates in OPC medium. Cells were transfected with constructs expressing genes of interest and maintained in OPC medium or OL differentiation medium for 2 d as described above.
Cell line cultures and biochemical assays
Oli-Neu cells were cultured in DMEM (GenDEPOT) + N1 + 3% heat-inactivated horse serum (Gibco) + 1% PS (penicillin–streptomycin; GenDEPOT). 293T cells were maintained in DMEM + 10% FBS (fetal bovine serum) + 1% PS and passaged with 0.25% 1× Trypsin-EDTA (GenDEPOT) treatment upon confluency. The biochemical studies including coimmunoprecipitation (co-IP), degradation, and ubiquitination assay were performed. Briefly, for co-IP assay, cell lysates were incubated with protein A and/or protein G agarose beads (Thermo Fisher) conjugated with specific antibodies for target protein and incubated overnight at 4°C. Proteins beads were washed three times and boiled with 2× SDS sample buffer for 10 min at 95°C, then Western blot analysis was performed. For the ubiquitination assay, 293T cells were treated with 40 µg/mL MG132 (Selleckchem S2619) for 6 h before harvesting; for the degradation assay, OPCs and 293T cells were treated with 100 µg/mL cycloheximide (Sigma 01810) and collected after 0, 2, 4, or 6 or 0, 3, or 6 h for whole-cell lysates. Antibodies and reagents used for biochemical assays were as follows: rabbit anti-Flag (1:3000; Sigma), mouse anti-c-Myc (1:3000; Santa Cruz Biotechnology), mouse anti-HA (1:3000; Sigma), rat anti-HA-HRP (1:3000; Sigma), mouse anti-Flag-HRP (1:3000; Sigma), rabbit anti-GAPDH (1:3000; GenTex), peroxidase Affinipure goat anti-mouse IgG H + L (1:10,000; Jackson Immuno Research), peroxidase Affinipure goat antirat IgG, light chain (1:10,000; Jackson Immuno Research), and anti-Flag M2 affinity gel (Sigma).
Real-time quantitative PCR (qPCR) analysis
Total RNA from P0 spinal cords was extracted using RNAzol reagent (GenDEPOT). Quantitative real-time PCR was performed in Bio-Rad real-time PCR systems (Bio-Rad) using the amfiSure qGreen Q-PCR Master Mix (GenDEPOT). Primer sequences were as follows: mouse Daam2 (forward: AAAGCCGCATCCACACATCTC, reverse: AGCACTGCAACTTTGGTCTTG), mouse VHL (forward: ACATTGAGGGATGGCACAAAC, reverse: CTCAGCCCTACCCGATCTTAC), and mouse GAPDH (forward: ATGACATCAAGAAGGTGGTG, reverse: CATACCAGGAAATGAGCTTG). Reactions were performed in a 10-µL mixture containing specific primers of each gene, cDNA and amfiSure qGreen Q-PCR Master Mix. Amplification conditions were as follows: 2 min at 95°C, followed by 40 cycles of 15 sec at 95°C and 1 min at 60°C. Relative mRNA expression level was calculated by the threshold cycle (Ct) value of each PCR product and normalized to that of GAPDH by using the comparative 2−ΔΔCt method (Livak and Schmittgen 2001). Results were presented as percent of values in control mice.
Human MS tissue sample and immunohistochemistry
Postmortem MS adult patient brain slices in formaldehyde (demyelinating lesions and healthy controls) were acquired from the Rocky Mountain MS tissue bank. Lesion sites were identified and dissected out by a neuropathologist from Texas Children's Hospital, processed and embedded in paraffin by the Pathology Core of Jan and Dan Duncan Neurological Research Institute. Healthy tissues were acquired from the white matter region adjacent to MS lesions in the same patients. Tissues were sectioned at a thickness of 6 µm. Paraffin-embedded tissue sections were deparaffinized by xylene and rehydrated from 100% to 50% ethanol and PBS. After antigen-retrieval step by citric acid solution (pH 6), the sections were treated with 0.3% H2O2 for 30 min to decrease endogenous peroxidase activity. Tissue sections were blocked with 2.5% horse serum and incubated with goat antihuman Olig2 plus rabbit anti-Daam2 (1:100; LSBio), rabbit anti-HIFα (1:200; Abcam), mouse anti-VHL (1:200; BD Bioscience), or rabbit anti-Nedd4 (1:200; Millipore) in PBS with 0.3% Triton X-100 overnight at 4°C. HRP- and AP-conjugated secondary antibodies (MP-5401, MP-5402, and MP-7405; Vector Laboratories) were then applied to tissue sections for 1 h at RT. The staining was visualized using ImmPACT DAB substrate and VectorRed AP substrate kit (SK4105 and SK-5100; Vector Laboratories). The color images were taken using a light microscope equipped with camera, and the red fluorescence by AP red reaction product was also photographed with a fluorescence microscope (Zeiss).
Quantification and statistics of images
The images were taken with Zeiss Imager.M2m equipped with ApoTome.2, Axiocam 506 mono, and AxioCam MRc. Images were captured using Zen2 software, then exported and analyzed in ImageJ using particle analysis and the cell-counter plug-in. All statistical analysis and quantitative graphs were plotted using Prism 8. Student's t-test was used for comparison between two groups. One-way ANOVA with multiple comparison was used for analyses among four groups. χ2 was conducted to check for lethality of mice. Protein degradation curve was fitted using nonlinear regression (one-phase decay). For all quantitative graphs, individual data point represents individual biological sample with an averaged value measured from at least three technical repeats.
Supplementary Material
Acknowledgments
We thank Diego Cortes for technical assistance. This work was supported by grants from the National Multiple Sclerosis Society (RG-1907-34551 to H.K.L.), the Hilton Foundation (17328 to H.K.L.), National Institutes of Health (NIH)/National Institute of Neurological Disorders and Stroke (R01NS110859-01 to H.K.L.), the Welch Foundation (AU-1971-20180324 to S.-H.Y.), and NIH/National Institute of General Medical Sciences (R01GM114424 to S.-H.Y.). Electron microscopy was supported in part by the Baylor College of Medicine Intellectual and Developmental Disabilities Research Center Neurovisualization Core (U54HD083092) from the Eunice Kennedy Shriver National Institute of Child Health and Human Development. This work was also supported by the Baylor College of Medicine Mass Spectrometry Proteomics Core, Dan L. Duncan Cancer Center, and Jan and Dan Duncan Neurological Research Institute Pathology Core.
Author contributions: X.D., J.J., C.-Y.W., and H.K.L. conceived the project and designed the experiments. X.D., J.J., C.-Y.W., C.D.C., Z.Z., J.M.C., Q.Y., M.W., and A.L.M. performed the experiments. H.K., S.Y.J., and H.J.B. provided reagents. C.A.M. and S.-H.Y. provided expertise. X.D. and H.K.L. wrote the manuscript.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.338046.120.
References
- Ambrozkiewicz MC, Kawabe H. 2015. HECT-type E3 ubiquitin ligases in nerve cell development and synapse physiology. FEBS Lett 589: 1635–1643. 10.1016/j.febslet.2015.05.009 [DOI] [PubMed] [Google Scholar]
- Amodio N, Scrima M, Palaia L, Salman AN, Quintiero A, Franco R, Botti G, Pirozzi P, Rocco G, De Rosa N, et al. 2010. Oncogenic role of the E3 ubiquitin ligase NEDD4-1, a PTEN negative regulator, in non-small-cell lung carcinomas. Am J Pathol 177: 2622–2634. 10.2353/ajpath.2010.091075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatt A, Fan L-W, Pang Y. 2014. Strategies for myelin regeneration: lessons learned from development. Neural Regen Res 9: 1347–1350. 10.4103/1673-5374.137586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilić J, Huang YL, Davidson G, Zimmermann T, Cruciat CM, Bienz M, Niehrs C. 2007. Wnt induces LRP6 signalosomes and promotes dishevelled-dependent LRP6 phosphorylation. Science 316: 1619–1622. 10.1126/science.1137065 [DOI] [PubMed] [Google Scholar]
- Camera D, Coleman HA, Parkington HC, Jenkins TA, Pow DV, Boase N, Kumar S, Poronnik P. 2016. Learning, memory and long-term potentiation are altered in Nedd4 heterozygous mice. Behav Brain Res 303: 176–181. 10.1016/j.bbr.2016.01.054 [DOI] [PubMed] [Google Scholar]
- Chaboub LS, Manalo JM, Lee HK, Glasgow SM, Chen F, Kawasaki Y, Akiyama T, Kuo CT, Creighton CJ, Mohila CA, et al. 2016. Temporal profiling of astrocyte precursors reveals parallel roles for Asef during development and after injury. J Neurosci 36: 11904–11917. 10.1523/JNEUROSCI.1658-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Matesic LE. 2007. The Nedd4-like family of E3 ubiquitin ligases and cancer. Cancer Metastasis Rev 26: 587–604. 10.1007/s10555-007-9091-x [DOI] [PubMed] [Google Scholar]
- Christie KJ, Martinez JA, Zochodne DW. 2012. Disruption of E3 ligase NEDD4 in peripheral neurons interrupts axon outgrowth: linkage to PTEN. Mol Cell Neurosci 50: 179–192. 10.1016/j.mcn.2012.04.006 [DOI] [PubMed] [Google Scholar]
- Ciechanover A, Brundin P. 2003. The ubiquitin proteasome system in neurodegenerative diseases: sometimes the chicken, sometimes the egg. Neuron 40: 427–446. 10.1016/S0896-6273(03)00606-8 [DOI] [PubMed] [Google Scholar]
- Cockman ME, Masson N, Mole DR, Jaakkola P, Chang G-W, Clifford SC, Maher ER, Pugh CW, Ratcliffe PJ, Maxwell PH. 2000. Hypoxia inducible factor-α binding and ubiquitylation by the von Hippel-Lindau tumor suppressor protein. J Biol Chem 275: 25733–25741. 10.1074/jbc.M002740200 [DOI] [PubMed] [Google Scholar]
- Compston A, Coles A. 2008. Multiple sclerosis. Lancet 372: 1502–1517. 10.1016/S0140-6736(08)61620-7 [DOI] [PubMed] [Google Scholar]
- Dai Z-M, Sun S, Wang C, Huang H, Hu X, Zhang Z, Lu QR, Qiu M. 2014. Stage-specific regulation of oligodendrocyte development by Wnt/β-catenin signaling. J Neurosci 34: 8467–8473. 10.1523/JNEUROSCI.0311-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Arca D, Zhao X, Xu W, Ramirez-Martinez NC, Iavarone A, Lasorella A. 2010. Huwe1 ubiquitin ligase is essential to synchronize neuronal and glial differentiation in the developing cerebellum. Proc Natl Acad Sci 107: 5875–5880. 10.1073/pnas.0912874107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drinjakovic J, Jung H, Campbell DS, Strochlic L, Dwivedy A, Holt CE. 2010. E3 ligase Nedd4 promotes axon branching by downregulating PTEN. Neuron 65: 341–357. 10.1016/j.neuron.2010.01.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar JM, McLaughlin M, Yool D, Zhang S-C, Fowler JH, Montague P, Barrie JA, McCulloch MC, Duncan ID, Garbern J, et al. 2004. Oligodendroglial modulation of fast axonal transport in a mouse model of hereditary spastic paraplegia. J Cell Biol 166: 121–131. 10.1083/jcb.200312012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eide PW, Cekaite L, Danielsen SA, Eilertsen IA, Kjenseth A, Fykerud TA, Ågesen TH, Bruun J, Rivedal E, Lothe RA, et al. 2013. NEDD4 is overexpressed in colorectal cancer and promotes colonic cell growth independently of the PI3K/PTEN/AKT pathway. Cell Signal 25: 12–18. 10.1016/j.cellsig.2012.08.012 [DOI] [PubMed] [Google Scholar]
- Fancy SPJ, Baranzini SE, Zhao C, Yuk D-I, Irvine K-A, Kaing S, Sanai N, Franklin RJM, Rowitch DH. 2009. Dysregulation of the Wnt pathway inhibits timely myelination and remyelination in the mammalian CNS. Genes Dev 23: 1571–1585. 10.1101/gad.1806309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fancy SPJ, Chan JR, Baranzini SE, Franklin RJM, Rowitch DH. 2011a. Myelin regeneration: a recapitulation of development? Annu Rev Neurosci 34: 21–43. 10.1146/annurev-neuro-061010-113629 [DOI] [PubMed] [Google Scholar]
- Fancy SPJ, Harrington EP, Yuen TJ, Silbereis JC, Zhao C, Baranzini SE, Bruce CC, Otero JJ, Huang EJ, Nusse R, et al. 2011b. Axin2 as regulatory and therapeutic target in newborn brain injury and remyelination. Nat Neurosci 14: 1009–1016. 10.1038/nn.2855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallo V, Deneen B. 2014. Glial development: the crossroads of regeneration and repair in the CNS. Neuron 83: 283–308. 10.1016/j.neuron.2014.06.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths I, Klugmann M, Anderson T, Yool D, Thomson C, Schwab MH, Schneider A, Zimmermann F, McCulloch M, Nadon N, et al. 1998. Axonal swellings and degeneration in mice lacking the major proteolipid of myelin. Science 280: 1610–1613. 10.1126/science.280.5369.1610 [DOI] [PubMed] [Google Scholar]
- Harty BL, Coelho F, Pease-Raissi SE, Mogha A, Ackerman SD, Herbert AL, Gereau RW, Golden JP, Lyons DA, Chan JR, et al. 2019. Myelinating Schwann cells ensheath multiple axons in the absence of E3 ligase component Fbxw7. Nat Commun 10: 2976 10.1038/s41467-019-10881-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- He X, Semenov M, Tamai K, Zeng X. 2004. LDL receptor-related proteins 5 and 6 in Wnt/β-catenin signaling: arrows point the way. Development 131: 1663–1677. 10.1242/dev.01117 [DOI] [PubMed] [Google Scholar]
- Hon W-C, Wilson MI, Harlos K, Claridge TDW, Schofield CJ, Pugh CW, Maxwell PH, Ratcliffe PJ, Stuart DI, Jones EY. 2002. Structural basis for the recognition of hydroxyproline in HIF-1α by pVHL. Nature 417: 975–978. 10.1038/nature00767 [DOI] [PubMed] [Google Scholar]
- Hooshmand MJ, Anderson AJ, Cummings BJ. 2014. Improved pre-embedded immuno-electron microscopy procedures to preserve myelin integrity in mammalian central nervous tissue. Microscopy: advances in scientific research and education 6: 59–65. [Google Scholar]
- Huxley AF, Stämpeli R. 1949. Evidence for saltatory conduction in peripheral myelinated nerve fibres. J Physiol 108: 315 10.1113/jphysiol.1949.sp004335. [DOI] [PubMed] [Google Scholar]
- Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, Salic A, Asara JM, Lane WS, Kaelin WG. 2001. HIFα targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science 292: 464–468. 10.1126/science.1059817 [DOI] [PubMed] [Google Scholar]
- Kaelin WG. 2002. Molecular basis of the VHL hereditary cancer syndrome. Nat Rev Cancer 2: 673–682. 10.1038/nrc885 [DOI] [PubMed] [Google Scholar]
- Kawabe H, Neeb A, Dimova K, Young SM, Takeda M, Katsurabayashi S, Mitkovski M, Malakhova OA, Zhang D-E, Umikawa M, et al. 2010. Regulation of Rap2A by the ubiquitin ligase Nedd4-1 controls neurite development. Neuron 65: 358–372. 10.1016/j.neuron.2010.01.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearns CA, Ravanelli AM, Cooper K, Appel B. 2015. Fbxw7 limits myelination by inhibiting motor signaling. J Neurosci 35: 14861–14871. 10.1523/JNEUROSCI.4968-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khwaja O, Volpe JJ. 2007. Pathogenesis of cerebral white matter injury of prematurity. Arch Dis Child—Fetal Neonatal Ed 93: F153–F161. 10.1136/adc.2006.108837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langseth AJ, Munji RN, Choe Y, Huynh T, Pozniak CD, Pleasure SJ. 2010. Wnts influence the timing and efficiency of oligodendrocyte precursor cell generation in the telencephalon. J Neurosci 30: 13367–13372. 10.1523/JNEUROSCI.1934-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lappe-Siefke C, Goebbels S, Gravel M, Nicksch E, Lee J, Braun PE, Griffiths IR, Nave K-A. 2003. Disruption of Cnp1 uncouples oligodendroglial functions in axonal support and myelination. Nat Genet 33: 366–374. 10.1038/ng1095 [DOI] [PubMed] [Google Scholar]
- Lee HK, Deneen B. 2012. Daam2 is required for dorsal patterning via modulation of canonical Wnt signaling in the developing spinal cord. Dev Cell 22: 183.–. 10.1016/j.devcel.2011.10.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HK, Chaboub LS, Zhu W, Zollinger D, Rasband MN, Fancy SPJ, Deneen B. 2015. Daam2-PIP5K is a regulatory pathway for Wnt signaling and therapeutic target for remyelination in the CNS. Neuron 85: 1227–1243. 10.1016/j.neuron.2015.02.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C-CJ, Yu K, Hatcher A, Huang T-W, Lee HK, Carlson J, Weston MC, Chen F, Zhang Y, Zhu W, et al. 2017. Identification of diverse astrocyte populations and their malignant analogs. Nat Neurosci 20: 396–405. 10.1038/nn.4493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25: 402–408. 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
- MacDonald BT, Tamai K, He X. 2009. Wnt/β-catenin signaling: components, mechanisms, and diseases. Dev Cell 17: 9–26. 10.1016/j.devcel.2009.06.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maher ER, Kaelin WG Jr. 1997. von Hippel-Lindau disease. Med 76: 381–391. 10.1097/00005792-199711000-00001 [DOI] [PubMed] [Google Scholar]
- Mao J, Wang J, Liu B, Pan W, Farr GH, Flynn C, Yuan H, Takada S, Kimelman D, Li L, et al. 2001. Low-density lipoprotein receptor-related protein-5 binds to Axin and regulates the canonical Wnt signaling pathway. Mol Cell 7: 801–809. 10.1016/S1097-2765(01)00224-6 [DOI] [PubMed] [Google Scholar]
- Nethe M, de Kreuk BJ, Tauriello DVF, Anthony EC, Snoek B, Stumpel T, Salinas PC, Maurice MM, Geerts D, Deelder AM, et al. 2012. Rac1 acts in conjunction with Nedd4 and dishevelled-1 to promote maturation of cell-cell contacts. J Cell Sci 125: 3430–3442. 10.1242/jcs.100925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opperman KJ, Mulcahy B, Giles AC, Risley MG, Birnbaum RL, Tulgren ED, Dawson-Scully K, Zhen M, Grill B. 2017. The HECT family ubiquitin ligase EEL-1 regulates neuronal function and development. Cell Rep 19: 822–835. 10.1016/j.celrep.2017.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan W, Choi SC, Wang H, Qin Y, Volpicelli-Daley L, Swan L, Lucast L, Khoo C, Zhang X, Li L, et al. 2008. Wnt3a-mediated formation of phosphatidylinositol 4,5-bisphosphate regulates LRP6 phosphorylation. Science 321: 1350–1353. 10.1126/science.1160741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin Y, Li L, Pan W, Wu D. 2009. Regulation of phosphatidylinositol kinases and metabolism by Wnt3a and Dvl. J Biol Chem 284: 22544–22548. 10.1074/jbc.M109.014399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reich DS, Lucchinetti CF, Calabresi PA. 2018. Multiple sclerosis. N Engl J Med 378: 169–180. 10.1056/NEJMra1401483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross CA, Pickart CM. 2004. The ubiquitin–proteasome pathway in Parkinson's disease and other neurodegenerative diseases. Trends Cell Biol 14: 703–711. 10.1016/j.tcb.2004.10.006 [DOI] [PubMed] [Google Scholar]
- Sakurai M, Ayukawa K, Setsuie R, Nishikawa K, Hara Y, Ohashi H, Nishimoto M, Abe T, Kudo Y, Sekiguchi M, et al. 2006. Ubiquitin C-terminal hydrolase L1 regulates the morphology of neural progenitor cells and modulates their differentiation. J Cell Sci 119: 162–171. 10.1242/jcs.02716 [DOI] [PubMed] [Google Scholar]
- Schwarz-Romond T, Fiedler M, Shibata N, Butler PJG, Kikuchi A, Higuchi Y, Bienz M. 2007. The DIX domain of dishevelled confers Wnt signaling by dynamic polymerization. Nat Struct Mol Biol 14: 484–492. 10.1038/nsmb1247 [DOI] [PubMed] [Google Scholar]
- Shao G, Wang R, Sun A, Wei J, Peng K, Dai Q, Yang W, Lin Q. 2018. The E3 ubiquitin ligase NEDD4 mediates cell migration signaling of EGFR in lung cancer cells. Mol Cancer 17: 24 10.1186/s12943-018-0784-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu T, Kagawa T, Wada T, Muroyama Y, Takada S, Ikenaka K. 2005. Wnt signaling controls the timing of oligodendrocyte development in the spinal cord. Dev Biol 282: 397–410. 10.1016/j.ydbio.2005.03.020 [DOI] [PubMed] [Google Scholar]
- Simpson JE, Ince PG, Shaw PJ, Heath PR, Raman R, Garwood CJ, Gelsthorpe C, Baxter L, Forster G, Matthews FE, et al. 2011. Microarray analysis of the astrocyte transcriptome in the aging brain: relationship to Alzheimer's pathology and APOE genotype. Neurobiol Aging 32: 1795–1807. 10.1016/j.neurobiolaging.2011.04.013 [DOI] [PubMed] [Google Scholar]
- Tsuboi M, Kishi Y, Yokozeki W, Koseki H, Hirabayashi Y, Gotoh Y. 2018. Ubiquitination-independent repression of PRC1 targets during neuronal fate restriction in the developing mouse neocortex. Dev Cell 47: 758–772.e5. 10.1016/j.devcel.2018.11.018 [DOI] [PubMed] [Google Scholar]
- Wolswijk G. 2002. Oligodendrocyte precursor cells in the demyelinated multiple sclerosis spinal cord. Brain 125: 338–349. 10.1093/brain/awf031 [DOI] [PubMed] [Google Scholar]
- Ye F, Chen Y, Hoang T, Montgomery RL, Zhao X, Bu H, Hu T, Taketo MM, van Es JH, Clevers H, et al. 2009. HDAC1 and HDAC2 regulate oligodendrocyte differentiation by disrupting the β-catenin–TCF interaction. Nat Neurosci 12: 829–838. 10.1038/nn.2333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin X, Crawford TO, Griffin JW, Tu Ph, Lee VM, Li C, Roder J, Trapp BD. 1998. Myelin-associated glycoprotein is a myelin signal that modulates the caliber of myelinated axons. J Neurosci 18: 1953–1962. 10.1523/JNEUROSCI.18-06-01953.1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen TJ, Silbereis JC, Griveau A, Chang SM, Daneman R, Fancy SPJ, Zahed H, Maltepe E, Rowitch DH. 2014. Oligodendrocyte-Encoded HIF function couples postnatal myelination and white matter angiogenesis. Cell 158: 383–396. 10.1016/j.cell.2014.04.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu W, Krishna S, Garcia C, Lin CCJ, Mitchell BD, Scott KL, Mohila CA, Creighton CJ, Yoo SH, Lee HK, et al. 2017. Daam2 driven degradation of VHL promotes gliomagenesis. Elife 6: 1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.