

In this study, Yang et al. devised a novel strategy to test the phenotypic impact of individual microRNA–target interactions by disrupting each predicted miRNA-binding site by CRISPR–Cas9 genome editing in C. elegans. They developed a multiplexed negative selection screening approach, in which edited loci are deep sequenced, and candidate sites are prioritized based on apparent selection pressure against mutations that disrupt miRNA binding.
Keywords: CRISPR, in vivo screening, miRNA targets, miRNAs, mutational profiling, negative selection screening, C. elegans
Abstract
Identifying miRNA target genes is difficult, and delineating which targets are the most biologically important is even more difficult. We devised a novel strategy to test the phenotypic impact of individual microRNA–target interactions by disrupting each predicted miRNA-binding site by CRISPR–Cas9 genome editing in C. elegans. We developed a multiplexed negative selection screening approach in which edited loci are deep sequenced, and candidate sites are prioritized based on apparent selection pressure against mutations that disrupt miRNA binding. Importantly, our screen was conducted in vivo on mutant animals, allowing us to interrogate organism-level phenotypes. We used this approach to screen for phenotypic targets of the essential mir-35-42 family. By generating 1130 novel 3′UTR alleles across all predicted targets, we identified egl-1 as a phenotypic target whose derepression partially phenocopies the mir-35-42 mutant phenotype by inducing embryonic lethality and low fecundity. These phenotypes can be rescued by compensatory CRISPR mutations that retarget mir-35 to the mutant egl-1 3′UTR. This study demonstrates that the application of in vivo whole organismal CRISPR screening has great potential to accelerate the discovery of phenotypic negative regulatory elements in the noncoding genome.
microRNAs (miRNAs) play fundamental roles in maintaining appropriate gene expression to ensure normal physiology. miRNAs recognize their multiple target genes primarily through complementarity between the “seed” region of the miRNA (nucleotides 2–8 from the 5′ end) and the 3′UTR of the target. Because of the importance of the seed sequence in target recognition, miRNAs that share an identical seed sequence act redundantly on a common set of target genes, and are together termed a miRNA “family” (Bartel 2009).
Despite great advances in our understanding of the molecular basis of miRNA target recognition and repression, identifying the targets that are regulated in vivo is still a major challenge. Various methods have been developed to identify putative miRNA-binding sites, including prediction algorithms, transcriptional or ribosomal profiling upon miRNA induction, or empirical methods such as cross-linking immunoprecipitation (CLIP) and cross-linking, ligation, and sequencing of hybrids (CLASH) (Quévillon Huberdeau and Simard 2019). However, these methods all generate a long list of candidate genes with their own caveats, including false positives and false negatives from all methods. In vivo validation is necessary to confirm that any given miRNA–target pair is a functional regulatory interaction (Fridrich et al. 2019).
Beyond just identifying the target genes biochemically regulated by a miRNA, understanding a miRNA's most biologically relevant target genes is even more difficult. The biological function of a miRNA or miRNA family can be inferred by its loss-of-function phenotype. Although all target genes of a miRNA lose miRNA-mediated repression in a miRNA loss-of-function context, derepression of only a subset of these genes will have biological consequences (e.g., those with high amplitude derepression or high dosage sensitivity) (Fridrich et al. 2019). Thus, a clear understanding of a miRNA's biological functions requires identifying the subset of the miRNA's biochemical targets whose derepression contributes to the phenotypic outcomes (termed “phenotypic” targets hereafter). Forward genetic screens have historically been a powerful method to identify key phenotypic target genes by using suppression of the miRNA loss-of-function phenotype as a readout. However, in some cases, forward suppressor screens are not effective, such as in the case of the essential mir-35-42 family in C. elegans (Alvarez-Saavedra and Horvitz 2010). Most likely, when this type of screen fails to identify suppressors, this is because either (1) simultaneous mutation of multiple target genes is necessary to achieve suppression or (2) loss of function of the key target gene is lethal (or synthetic lethal with the genetic background), preventing recovery of a suppressed line.
CRISPR has emerged in the last decade as a powerful genome engineering tool to interrogate the function of both protein-coding genes and noncoding regions of the genome. Screens most frequently employ guide RNA (gRNA) libraries that target protein-coding genes to interrogate the consequences of loss-of-function lesions. In some cases, custom-designed gRNA libraries have been used to target noncoding RNAs or regulatory elements in the genome like transcription factor-binding sites (Canver et al. 2015; Fulco et al. 2016; Korkmaz et al. 2016; Rajagopal et al. 2016; Diao et al. 2017; Han et al. 2018). This technology is thus aptly suited to screening individual miRNA-binding sites; by mutating each site to relieve the target from miRNA-mediated repression, those sites whose mutation phenocopies the miRNA loss-of-function mutant could theoretically be identified.
Few cell-autonomous miRNA loss-of-function phenotypes have been identified; more frequently, miRNAs are embedded in genetic pathways that regulate developmental programs or cell type specification (Ambros 2004; Bartel 2018). Thus, in most cases, screening for critical miRNA targets in vivo in a whole organism would be ideal, but CRISPR screens have so far been limited to cell culture or models in which a single tissue is modified. Furthermore, cells and tissues are mosaic for genome edits in previous CRISPR screens, preventing examination of non-cell-autonomous phenotypes. However, screening at the level of mutant animals would be cost-prohibitive in most vertebrate models. Here we developed multiplexed CRISPR screening in the C. elegans system, allowing for whole animal mutant phenotype to be used as a means of selection. This innovation will greatly expand the types of biological processes that can be dissected using CRISPR technology, expanding the scope from only cell-autonomous phenotypes to tissue- and organism-level physiology.
Here we interrogate the essential miRNA–target interactions of the mir-35-42 family of redundant miRNAs. Complete knockout of the mir-35 family (the mir-35-41 cluster and mir-42) causes fully penetrant embryonic or L1 larval lethality (Alvarez-Saavedra and Horvitz 2010). In this context, low-penetrance premature cell death occurs due to derepression of egg-laying defective-1 (egl-1), a proapoptotic BH3-only domain protein (Sherrard et al. 2017). Many other phenotypes have been characterized in the context of mir-35-41 deletion (leaving mir-42 intact), which allows for the bypass of lethality at low temperature. These additional roles for mir-35-41 include promoting germ cell proliferation and fecundity, resistance to radiation and hypoxia, proper sex determination, and dampening of RNA interference (Liu et al. 2011; Massirer et al. 2012; McJunkin and Ambros 2014, 2017; Kagias and Pocock 2015; Benner et al. 2019; Doll et al. 2019; Tran et al. 2019). The lethal phenotype of mir-35 family mutants is still poorly understood: Deep forward mutagenesis screens failed to recover suppressors, suggesting that either multiple target genes contribute to lethality when derepressed or loss-of-function mutations of the critical target gene(s) is lethal (Alvarez-Saavedra and Horvitz 2010).
Because mir-35-42 mutant lethality could not be dissected by traditional genetics, we set out to use a reverse genetic screen to determine the phenotype of derepressing each putative target individually. To this end, we used a multiplexed negative-selection CRISPR screen. Importantly, we used mutational profile at edited sites as a readout of the phenotypic impact of a given site. We identified egl-1 as a target that contributes to embryonic lethality. While egl-1 was implicated previously in precocious cell death in the embryo and radiation-induced germ cell death (Sherrard et al. 2017; Tran et al. 2019), we show that derepression of egl-1 alone is sufficient to induce embryonic lethality and low fertility. Finally, we used CRISPR to restore complementarity of mir-35 and mutant egl-1 3′UTRs and show that this retargeting of an endogenous miRNA restores repression and rescues the deleterious phenotypes. Overall, this work highlights the importance of proper egl-1 repression in embryogenesis and germline function, establishes a platform for multiplexed CRISPR screening in C. elegans, and serves as a roadmap for future efforts to identify and validate functional miRNA targets.
Results
gRNAs targeting multiple loci can be multiplexed in C. elegans germline injection
Our goal is to determine the phenotypic targets of an essential miRNA family. To this end, we sought to use CRISPR to mutate each predicted binding site of the mir-35 family and examine the resulting phenotype. Mutations that prevent repression of phenotypic targets should lead to a partial or complete recapitulation of the miRNA knockout phenotype, whereas mutating the miRNA-binding site in other less important targets or nontargets should have no phenotypic consequences (Supplemental Fig. S1A–D).
Even though efficient CRISPR protocols for introducing insertions or deletions (indels) or homology-directed repair at single locus have been well established in C. elegans (Arribere et al. 2014; Paix et al. 2014; Dokshin et al. 2018), characterizing each binding site mutant individually would be very laborious since miRNAs generally have a large number of predicted targets. We thus set out to test the feasibility of multiplexing gRNAs in C. elegans before embarking upon a large-scale screen. Manual micro-injection into the germline syncytium is so far the only efficient way to deliver gRNAs to C. elegans, yielding heritable mutations in subsequent generations. Theoretically, a pool of multiple gRNAs could be injected into the germline of young adult animals to simultaneously generate indels at multiple loci (Supplemental Fig. S1E).
Since both plasmid-expressed and synthetic gRNAs have been widely used in the C. elegans community, we compared the efficiencies of these methods in a multiplexed context. Plasmid or synthetic guides were injected into a stable Cas9-expressing transgenic strain (EG9615) (see Supplemental Tables S4, S5 for details). To determine how many gRNAs could be multiplexed without losing editing efficiency, we created four simulated pool sizes (mimicking a pool of 4, 10, 20, or 50 gRNAs). To generate these pools, total gRNA concentration was kept constant, and three control gRNAs (dpy-10, nhl-2, and sup-26) were diluted to each represent 25%, 10%, 5%, or 2% of the injected gRNA population (Fig. 1A). Typically, C. elegans genome editing at a locus of interest is carried out by selecting for coediting at a visible marker gene, such as dpy-10 (Arribere et al. 2014). Thus, dpy-10 gRNA was included in all of our simulation pools as a coselection marker. In addition to dpy-10, we included gRNAs targeting the miRNA-binding sites of two previously studied mir-35 target genes, nhl-2 and sup-26 (Kagias and Pocock 2015; McJunkin and Ambros 2017). These three guides were diluted to various extents with a nontargeting gfp gRNA to reach a final concentration of 250 ng/µL (when plasmids were used) or 10 µM (when synthetic gRNAs were used) in all simulated pool sizes (Fig. 1A).
Figure 1.

Mutational profiling coupled with negative selection CRISPR screening for phenotypic mir-35 target sites. (A) Composition of four different simulation pools. Three guide RNAs (targeting sup-26, nhl-2, and selectable marker dpy-10) were diluted 1:4, 1:10, 1:20, and 1:50 by supplementing with gfp guide RNA to maintain a constant total guide RNA concentration. (B) Relationship between dilution factor and indel efficiency (calculated as percent of genotyped alleles in which editing was detected by restriction digest with NciI) (see Supplemental Table S1 for details). (C) Relationship between dilution factor and rate of mutant allele detection per P0 injection. (B,C) Two biological replicates (and mean) are shown. n ≥ 78 per condition. (D) Schematic of mutational profiling of indel positions after CRISPR targeting microRNA-binding sites. (E) Overview of screen workflow. (F) Correlation of indel frequency induced by a given gRNA in two different biological replicates. Spearman's correlation test r and P-value are reported. (G) Distribution of number of alleles detected at each gRNA target site.
Next, we examined the efficiency of coediting with the dpy-10 selectable marker at different simulated pool sizes. Since mutations in the mir-35-binding site in the sup-26 3′UTR are known to have no impact on viability, we examined the rate of editing at the sup-26 locus in Dpy-selected strains to assess the efficiency of coediting. The sup-26 gRNA had a high rate of coediting (∼20%) when it was diluted 1:4 or 1:10 in other gRNAs (Fig. 1B). However, with dilution factors beyond 1:10, the editing efficiency at the sup-26 locus dropped significantly (Fig. 1B). Dilutions higher than 1:10 also decreased the efficiency of editing at our selection marker dpy-10. This reduced the number of edited sup-26 alleles that could be recovered per injected P0 animal since fewer Dpy-selected lines were generated that could be screened for coedits; therefore, screening at higher dilutions would be more laborious (Fig. 1C). Thus, we decided to multiplex gRNAs in pools of 10 since the 1:10 dilution maintained high rates of selectable marker editing and coediting efficiency.
We then tested whether a true complex pool of 10 different gRNAs would behave similarly to the simulated pool with respect to gRNA efficiency. We chose an additional eight genes that are called as putative mir-35 targets by both TargetScan and iCLIP (Jan et al. 2011; Broughton et al. 2016). gRNAs targeting the mir-35-binding sites of these genes were designed and mixed with dpy-10 and sup-26 gRNAs to generate a pool of 10 gRNAs. In addition to the plasmid and synthetic gRNA paradigms tested above, we also tested injection of preassembled RNPs containing synthetic gRNAs and recombinant Cas9. Like in the simulated pools, sup-26 coediting was similar between different delivery modalities, though the overall efficiency was slightly lower (9.5%–13%) than in the simulated pools (Supplemental Fig. S2). Unlike sup-26, other CRISPR targeted loci showed variable rates of editing across methods. Synthetic gRNAs injected into transgenic Cas9-expressing animals performed best, achieving an average of 6.3% editing across all genotyped loci, whereas plasmid-expressed gRNAs or preassembled RNPs both induced an average of 3.7% editing (Supplemental Fig. S2). Therefore, we used synthetic gRNAs and transgenic Cas9 for the multiplexed screen.
Large-scale screening for phenotypic mir-35-binding sites
Having optimized multiplexing conditions, we set out to perform a large-scale screen of all predicted mir-35 family-binding sites to assess their phenotypic impact. We reasoned that deep sequencing to determine the mutational profile at each CRISPR targeted locus would be the ideal readout of the screen because of the somewhat stochastic nature of repair of Cas9-mediated double-strand breaks (DSBs) (Chen et al. 2019; Yang and McJunkin 2020a). When a miRNA-binding site is targeted by CRISPR, some of the resulting indels will disrupt seed binding (hereafter referred to as “seed match-disrupting alleles”) and thus disrupt miRNA-mediated repression of the target (Fig. 1D). Other indels may leave the seed-binding region intact (“seed match-intact alleles”); these would not necessarily impair miRNA binding. If derepression of a target gene causes a deleterious phenotype, then selection for only wild-type animals would deplete seed match-disrupting alleles while maintaining seed match-intact alleles in the population (Fig. 1D). This screen is designed with the assumption that derepression of a target will have a dominant gain of function, and only hemizygous seed match disruption will be necessary for the allele to be counterselected. (Although the screen will be less sensitive for targets whose phenotypic effects are recessive, they may still be detected if the recessive phenotype is very penetrant since an ensemble of F2s is examined for fitness.) For a target gene whose derepression has no phenotypic impact (Fig. 1D, bottom), both seed match-disrupting and seed match-intact alleles would pass through selection for wild-type phenotype. Thus, by generating a mutational profile at each locus, target sites can be prioritized based on the frequency of seed match-disrupting alleles in a population of edited animals selected for wild-type phenotype (Fig. 1D). Notably, along with multiplexing gRNA injection, this deep sequencing strategy further reduces the labor required to complete the screen compared with a one-by-one approach (even with a modest library size of 89 gRNAs) (see Supplemental Fig. S3 for details.)
We performed the screen using all TargetScan targets of the mir-35 family as candidates. TargetScan predicts 89 target genes for the mir-35 seed family, each containing a single predicted binding site (Jan et al. 2011). For each site, we designed one gRNA targeting within or as close as possible to the seed match. We grouped the gRNAs into pools of 10, including dpy-10 gRNA in each pool (Fig. 1E). Because of the syncytial nature of the germline, germ cells may receive more than one gRNA; therefore, we sought to minimize our detection of synthetic effects of two gRNAs in the same pool. To this end, we included each gRNA in two pools with a completely different set of poolmates. This resulted in 20 pools of 10 to assay the 89 target sites (Supplemental Table S7). Pools of synthetic gRNAs were injected into Cas9 transgenic animals, and F1 animals exhibiting dpy-10-associated phenotypes were isolated. Some of the F1 animals in each pool were sterile, had very low fecundity, or laid mostly dead embryos (Supplemental Fig. S4). These deleterious phenotypes might be due to deleterious seed match-disrupting mutations but, since they were observed in all pools, are more likely due to general toxicity of pooled gRNAs resulting from chromosomal rearrangements (Choi and Meyerson 2014; Chen et al. 2015; Iwata et al. 2016). This high rate of general toxicity (average of 35% across pools) highlights the necessity of performing a negative selection screen, since positive selection for dead embryos would include numerous false positives. All F1 plates that were not superficially wild type were discarded, and 30 superficially wild-type strains were collected for each pool (Fig. 1E). Genomic DNA was then isolated from the harvested animals and subjected to site-specific PCRs targeting the edited regions and prepared for deep sequencing (Fig. 1E). In agreement with previous studies that examined CRISPR-mediated indel frequencies in mammalian cell culture (Chari et al. 2015; van Overbeek et al. 2016), we observed that gRNA efficiencies were highly reproducible across biological replicates (Spearman correlation r: 0.829, P < 0.0001) (Fig. 1F). Since the two replicates for a given gRNA were performed with different poolmates, this result also indicates that synthetic effects between guides in the same pool were minimal (Fig. 1F). Most of the gRNAs successfully induced indels at the targeted sequence, whereas 14 gRNAs failed to yield any indels (Fig. 1G). Editing at one locus (bath-40) was not interpretable by deep sequencing due to flanking repetitive sequence.
Complete lack of editing in primary screen is due to low gRNA efficiency, not phenotypic outcomes
Because 14 gRNAs failed to generate indels at their targeted regions, we wondered whether this was due to lack of activity of the gRNAs themselves or to deleterious effects of the mutations generated by these gRNAs. To differentiate between these possibilities, we designed alternative gRNAs targeting these 14 sites and conducted a secondary multiplexed screen in Cas9 transgenic animals, followed by genotyping to assess editing efficiencies (Supplemental Fig. S5A). Eight of the 14 sites (and the previously uninterpretable bath-40) were edited with high efficiency by the second gRNA, indicating that the lack of mutant alleles in the primary screen was due to low activity gRNAs, not phenotypic effects (Supplemental Fig. S5A). Five of the remaining sites were further tested individually by injecting three gRNAs along with a microhomology repair template to mutate the seed match sequence (Paix et al. 2014); this resulted in efficient editing of three of the loci when transgenically expressed Cas9 was used. For three sites (ZK899.1, Y55F3AM.10, and dsh-1), high-efficiency editing was only observed when multiple guides were preloaded in recombinant Cas9 and injected as an RNP with the microhomology donor (Supplemental Fig. S5A). To confirm that seed match-disrupting mutations were difficult to obtain due to low efficiency CRISPR and not phenotypic effects, we quantified the brood sizes and embryonic viability of the seed match-disrupting mutants in Y55F3AM.10 and dsh-1 and observed very small or no difference compared with wild-type animals (Supplemental Fig. S5B). Overall, these experiments indicate that the absence of any mutant alleles (seed match-disrupting or seed match-intact) in the primary screen can be interpreted as low gRNA potency. This emphasizes that the number of mutant alleles at a site is correlated to gRNA efficiency, not phenotypic impact, highlighting the importance of analyzing mutational profile rather than allele number.
Multiple top candidate genes exhibit deleterious phenotypes upon seed match disruption
For the remaining gRNAs, which did successfully generate mutant alleles, we analyzed the mutational profile to determine the percent of alleles that were seed match-disrupting (Fig. 1D). The edited loci were spread across a wide range of values for seed match-disrupting allele frequency (Fig. 2A). Our controls were sup-26, which has no phenotype upon seed match disruption, and nhl-2, which we expected to have a lethal phenotype upon seed match disruption. As expected, sup-26 had a high seed match-disrupting frequency (100%). Surprisingly, nhl-2 also showed a high seed match-disrupting frequency (100%). During the course of this study, we determined in parallel that nhl-2 seed match disruption is actually not deleterious despite our previous results that suggested it would be (McJunkin and Ambros 2017; Yang and McJunkin 2020b). Thus, our screen does not contain a positive control for deleterious phenotype resulting in low frequency of seed match-disrupting alleles.
Figure 2.
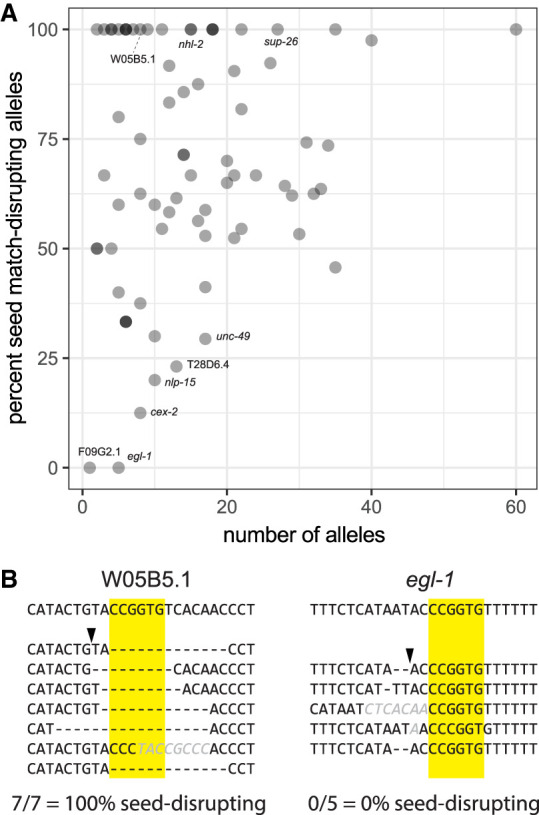
Mutational profiling identifies candidates with low frequency of seed match-disrupting alleles. (A) Distribution of allele number and percent of seed match-disrupting alleles for each targeted site. (B) Indels detected at the W05B5.1 and egl-1 sites. The top line is the reference sequence of each site. The seed sequence matches are highlighted in yellow, and the expected Cas9 cleavage sites are indicated with arrowheads. Dashes represent deleted bases, whereas gray italic text is insertions.
To test whether the negative selection screen we carried out is an effective method for identifying phenotypic binding sites of the mir-35 family, we attempted to validate candidates with the lowest frequencies of seed match-disrupting alleles. We started by attempting to validate the six top-ranked targets by this metric (egl-1, F09G2.9, cex-2, nlp-15, T28D6.4, and unc-49). We did notice that for three of the six sites, the cleavage site predicted by the PAM sequence is located 2 bp outside of the seed sequence (Fig. 2B; Supplemental Fig. S6A,B). We wondered whether all sites that have a predicted cleavage site outside of the seed match also have a low seed match-disrupting allele frequency. Sites with cleavage outside the seed match did show a lower mean seed match-disrupting allele frequency than those with cleavage inside the site (Fig. 2B; Supplemental Fig. S6C). Nonetheless, some of these sites (e.g., W05B5.1 and tsp-20) still showed high seed match-disrupting allele frequencies (100% and 66.7%, respectively) (Fig. 2B; Supplemental Fig. S6C). Thus, while the site of gRNA cleavage is an important consideration in interpretation of seed match-disrupting allele frequency, all high priority sites in our screen merited follow-up studies (see also Discussion).
To determine whether any of the candidates displayed deleterious phenotypes upon seed match disruption, we generated seed match-disrupting mutations at each site using individual site gRNA injections. We examined these six mutant alleles for effects on fecundity and embryonic lethality that partially or fully recapitulate the miRNA mutant phenotype. Of the six candidates, seed-disrupting mutations in egl-1 had the greatest impact on phenotype, reducing brood size and increasing embryonic lethality when compared with wild type (Fig. 3A,B), thus partially phenocopying mir-35 family mutants. A second previously published seed match-disrupting allele of egl-1 (egl-1(on24)) also showed similar phenotypes (Fig. 3B; Tran et al. 2019). Thus, loss of the mir-35-binding site in the egl-1 3′UTR is sufficient to induce phenotypes similar to those observed in mir-35 family mutants, albeit at lower penetrance and expressivity. Seed match mutations in unc-49 also affected both aspects of the phenotype though to a lesser degree, and two other genes affected only brood size (cex-2) or embryonic lethality (T28D6.4) (Fig. 3B). Many of the observed phenotypic effects affected the variance of the phenotype as well as the mean (Fig. 3B). Overall, of the top six screen candidates, two genes were false positives, whereas four genes passed the first round of validation: inducing deleterious phenotypes by individual mutation of the mir-35 family-binding site.
Figure 3.

Validation of top candidates confirms egl-1 as a phenotypic target of mir-35. (A) Schematics of 3′UTR and mir-35 mutant genotypes. (B) Brood count and percent embryonic lethality at 25°C of seven seed match-disrupting mutations in the indicated 3′UTRs, compared with wild type. (C) Brood count and percent embryonic lethality at 25°C of seed match-reversing mutations in the indicated 3′UTRs, in the context of wild type or a mutant mir-35 containing compensatory seed mutations. (B,C) Mean and SEM are shown. One-way ANOVA was conducted to determine significance, followed by post hoc pairwise comparisons with correction for multiple testing (Dunnett's test for comparison of each mutant with wild type in B and Sidak's correction for selected pairwise comparisons is shown in C). In C, all single mutants were compared with wild type, and each 3′UTR mutant was compared with the corresponding mutant crossed into the mir-35(rev) background. P-value is indicated only if <0.05. (*) P < 0.05; (**) P < 0.01; (***) P < 0.001; (****) P < 0.0001.
egl-1 3′UTR mutant phenotypes are due to loss of mir-35-mediated repression
Because 3′UTRs are hubs of regulation and bound by multiple miRNAs and RNA-binding proteins, we wanted to rigorously test that the deleterious phenotypes we observed in the 3′UTR mutants were because of loss of mir-35-mediated repression and not disruption of some other type of regulation. To this end, we designed rescue experiments to restore mir-35 targeting to seed match-mutated 3′UTRs. Our strategy was to first mutate the mir-35 seed match in the 3′UTR of interest by reversing its sequence; this would relieve the mRNA from mir-35 family binding and repression. Then, the seed sequence of mir-35 was also reversed at its genomic locus (along with mutations to preserve the primary miRNA stem-loop structure), and this mutant mir-35 should bind the mutant 3′UTR, rescuing the endogenous pattern of repression (Fig. 3A). This approach takes advantage of the redundant nature of the mir-35 family: While mutant mir-35 is retargeted to mutant 3′UTR, wild-type mir-36-42 remain intact to repress all other mir-35 family target genes.
To this end, we made mutants of egl-1, cex-2, T28D6.4, and unc-49 in which the mir-35 seed match in each gene's 3′UTR is reversed (at the endogenous genomic locus). The egl-1 seed match reversal caused decreased brood size and increased embryonic lethality, similar to other egl-1 seed match-disrupting alleles (Fig. 3C). The effect of unc-49 on brood size was again observed, but its effect on embryonic lethality and cex-2’s effect on brood size fell below statistical significance after correction for multiple testing (Fig. 3C). The new 3′UTR allele of T28D6.4 did not exhibit any effect on embryonic viability (Fig. 3C).
Next we tested whether the phenotypes we observed upon seed match mutation of these 3′UTRs could be rescued by mir-35 retargeting. Each seed match-reversed 3′UTR allele was combined with the mutation in mir-35 that reversed its seed. Double mutants with restored complementarity between egl-1 and mir-35 showed partial rescue of both the brood size and lethality phenotypes (Fig. 3C). Thus, endogenous mir-35 can be retargeted to a mutant egl-1 3′UTR to rescue deleterious phenotypes caused by 3′UTR mutations. Restored mir-35 complementarity did not alter the phenotypes of any of the other 3′UTR mutants (Fig. 3C).
Because mild phenotypes were observed with individual seed match mutations of multiple candidates, we wondered whether combining multiple alleles might have a synthetic effect that more potently phenocopies the miRNA mutant phenotype. Therefore, we constructed a quadruple mutant in which the mir-35 seed match was reversed in egl-1, unc-49, cex-2 and T28D6.4 (Fig. 3C). The quadruple mutant did not display synthetic brood size or embryonic lethality phenotypes (Fig. 3C). When combined with the seed-reversed mir-35 allele, the phenotype was rescued to the same extent as the rescue of the individual egl-1 seed match mutant (Fig. 3C). All together, these data suggest that egl-1 is a phenotypic target of the mir-35 family, while the other candidates are false positives from the screen. While egl-1 was characterized previously as a bona fide mir-35 target gene, we show here that its derepression alone is sufficient for deleterious phenotypes in the absence of stress (Flamand et al. 2017; Sherrard et al. 2017; Tran et al. 2019; Wu et al. 2010). The fact that egl-1 was the top-scoring candidate from the screen (with 0% seed match-disrupting alleles) reinforces the effectiveness of our screening method for prioritizing phenotypic candidate sites. Interestingly, some of the false-positives induced phenotypes upon mutation of the predicted mir-35 seed match. Thus, these sites (such as in unc-49) are likely to be sites of other types of regulation that are predicted as miRNA-binding sites due to their evolutionary conservation.
To confirm the regulatory interaction between mir-35 and egl-1 suggested by the genetic assays, we performed qPCR of egl-1 (as well as unc-49, cex-2, and T28D6.4) in wild type or a respective 3′UTR seed match mutant (Supplemental Table S6). We did not observe significant up-regulation of egl-1 by qPCR (Supplemental Fig. S7). However, a previous report using GFP reporter transgenes demonstrated mir-35 mediated regulation via the egl-1 3′UTR (Sherrard et al. 2017). Reporters may be more sensitive than qPCR in this scenario because they allow for precise comparison of stage-matched embryos. Thus, we engineered the seed match-reversing mutation into a reporter transgene bearing the egl-1 3′UTR (Sherrard et al. 2017). In the wild-type mir-35 background, the reporter with a wild-type egl-1 3′UTR is repressed throughout early and mid-embryogenesis (Fig. 4A,B; Sherrard et al. 2017). When the mir-35 seed match in the egl-1 3′UTR is reversed, the reporter is derepressed (Fig. 4A,B). Introducing the seed-reversed allele of mir-35 reverse restores repression of the reporter with the mutant egl-1 3′UTR (Fig. 4A,B). Thus, consistent with our genetic assays, wild-type or re-engineered mir-35 binds and regulates transcripts containing a complementary seed match in the egl-1 3′UTR.
Figure 4.

egl-1 is a bona fide phenotypic target of the mir-35 family. (A) Analysis of a reporter transgene that expresses nuclear localized GFP::H2B fusion protein under the control of the constitutive mai-2 promoter and the egl-1 3′UTR. Four embryonic stages are shown. For each embryo, DIC image is shown at the left, and GFP::H2B fluorescence is shown at the right. The mir-35 and egl-1 3′UTR genotype are listed at the left of the images. Scale bar, 10 µm. (B) Quantification of GFP intensity of the corresponding genotypes at the four stages shown. Wild-type (no reporter) samples were treated as negative controls, and their average intensity was subtracted from all experimental samples. Mean and SEM are shown. One-way ANOVA was conducted to determine significance, followed by post hoc pairwise comparisons with Sidak's correction for multiple testing. (****) P < 0.0001.
Multiplexing Cas9 RNPs is feasible for future screens
During the course of screening and validation, we made three observations that led us to further test the efficiency of Cas9 RNP injection for multiplexing in future screens. First, RNPs were more efficient than transgenically expressed Cas9 in single-gene experiments (despite being less efficient in our first test of multiplexing). Second, the transgenic Cas9 strain occasionally segregated animals with a non-wild-type appearance, raising concerns that sustained germline expression of Cas9 may increase the rate of genetic drift. Third, the fecundity phenotype of the egl-1(cdb62mut) allele generated in the Cas9 background was more expressive than that of the published allele [egl-1(on24)], which was generated using plasmid-expressed gRNAs (Fig. 3B; Tran et al. 2019). [The fecundity of egl-1(cdb62mut) was quantified after outcrossing to remove the Cas9 transgene itself.] This again raised concerns that the transgenic Cas9 strain may accumulate background mutations.
We wondered whether the low efficiency of some gRNAs in our first test of multiplexing RNPs was due to competition among gRNAs for loading into a limiting amount of Cas9. To test this hypothesis, we loaded each gRNA into Cas9 separately and then mixed the preloaded RNPs together prior to injection. Preloading each gRNA into Cas9 prior to pooling resulted in much higher efficiency, exceeding the efficiency in the Cas9 transgenic strain (Supplemental Fig. S2). Therefore, using recombinant Cas9 with synthetic gRNAs would be an ideal strategy for future screens, as long as gRNAs can be individually preloaded into Cas9 before pooling.
gRNA efficiency correlates with GC content and CrisprScan
Our data set is the largest set of measured gRNA efficiencies in a single experimental setting in C. elegans to our knowledge. Since only egl-1 and unc-49 mutations consistently induced significant deleterious phenotypes, we can infer that other variance in the frequency of indels is primarily due to differing gRNA efficiencies. We therefore analyzed our data (excluding egl-1 and unc-49) to find effective predictors for future gRNA design. Previous studies have suggested that gRNA sequences with a “GG” immediately upstream of the PAM site outperform other gRNAs in C. elegans (Farboud and Meyer 2015); thus, we compared the efficiencies of the “GGNGG” gRNAs with the rest of the population. We observed a slight but nonsignificant increase in indel frequency in the “GGNGG” gRNAs (P-value = 0.29) (Fig. 5A). Since studies in other species have shown that a higher GC content of gRNA sequences is associated with higher editing efficiency (Doench et al. 2014; Wang et al. 2014), we next tested whether this also holds true in C. elegans. A correlation analysis confirmed a significant positive correlation between the frequency of indels introduced by a gRNA and its GC content (Spearman r: 0.4701) (Fig. 5B). Thus, the observed potency of GGNGG gRNAs may be due to their overall higher GC content.
Figure 5.

gRNA efficiency correlates with GC content and CrisprScan scores. (A) Comparison of indel efficiencies between GGNGG PAM sites and others. (B) Correlation between the GC content of each guide RNA and its corresponding indel efficiency. (C,D) Correlation of scores predicted by the indicated algorithm for each guide RNA and its observed indel efficiency. Spearman's correlation test r and P-value are reported.
We also tested how published gRNA prediction algorithms correlated with our gRNA efficiencies. We predicted the on-target score of each gRNA using the IDT design tool, the DRSC algorithm, Rule Set 2, CrisprScan, or GuideScan (Fig. 5C,D; Supplemental Fig. S8; Housden et al. 2015; Moreno-Mateos et al. 2015; Doench et al. 2016; Haeussler et al. 2016; Perez et al. 2017). IDT, DRSC, and GuideScan prediction scores failed to positively correlate with the readout from our experiments (Supplemental Fig. S8). However, the other two models fit much better with our data sets, with the highest performance by CrisprScan (Spearman r = 0.5789, P < 0.0001), which slightly outperformed GC content alone (Fig. 5D). Therefore, where applicable, CrisprScan may help optimize gRNA design for genome editing in C. elegans.
Discussion
Here, we conducted a thorough optimization of multiplexed CRISPR in the C. elegans system, followed by a large-scale in vivo negative selection screen for phenotypic targets of the essential mir-35-42 family. This screen effectively identified egl-1 as a phenotypic target of the mir-35 family. While egl-1 is a known mir-35 family target gene, our screen and validation studies demonstrated for the first time that derepression of egl-1 alone is sufficient to induce embryonic lethality and low fecundity, thus partially phenocopying mir-35 family mutants. We used CRISPR to retarget endogenous mir-35 to mutant egl-1, resulting in rescue of deleterious phenotypes. This type of rescue experiment, which was used to demonstrate let-7's phenotypic target gene (Ecsedi et al. 2015), represents a new best practice for testing miRNA–target relationships since all components of the system are expressed from their endogenous loci.
To our knowledge, this is the first CRISPR screen to be conducted in vivo in whole mutant animals. This innovation allowed us to screen based on organismal phenotypes. While current technologies largely restrict screening to cell-autonomous phenotypes, in vivo screening of mutant animals will greatly expand the physiological processes that can be interrogated by CRISPR. Screening diverse mutant animals rather than a mixed population of cells could enable the study of non-cell-autonomous, tissue-level, developmental, and behavioral phenotypes. An inherent limitation of this in vivo screening method is the need to deliver the multiplexed gRNA mixes to the reproductive system of animals yielding a substantial number of offspring; thus, this method may not be applicable to organisms with few germ cells or small litter size, such as mice.
Like most screening technologies, the type of screen presented here does not have strong potential to exhaustively search for synthetic effects among a large library of gRNAs. Instead, the approach relies on the assumption that mutation of a single miRNA-binding site is sufficient to induce detectable phenotypes, and that those phenotypes will have some similarity to the miRNA mutant phenotype. Of note, all predicted mir-35 family target genes only harbor a single predicted mir-35-binding site, unlike most miRNAs’ targets that bear multiple cognate binding sites. The design of the screen assumes that derepression of the target would occur upon mutation of a single binding site (even in a context where multiple sites are present), which may not be true for all targets.
Multiplexed CRISPR in C. elegans, building on this study, will accelerate many types of studies. Here, we conducted a negative selection screen targeting tiny regulatory elements. Thus, the multiplexing techniques used here should be easily adapted to simpler experimental setups; e.g. positive selection and/or targeting coding sequences. Suppressor screens using gRNAs targeting a set of candidate genes can be conducted, which might be especially useful for RNAi resistant tissues such as neurons (Kamath et al. 2001). In a positive selection setting, pooled gRNAs could be used to look for synthetic effects of lesions in multiple candidate loci. Of course, combining this technique with fluorescent reporters will also allow for screening based on more specific or subtle gene expression changes rather than overtly visible phenotypes. Multiplexing CRISPR will be a valuable addition to the already powerful C. elegans genetic toolkit.
Our screen was a negative selection or “dropout” screen because we expected that mutations in miRNA-binding sites that phenocopied the mir-35 family mutant phenotype would be deleterious or lethal. Negative selection screens are technically challenging due to characteristically low signal to noise. When targeting a very small regulatory region, this problem is exacerbated by the somewhat stochastic nature of CRISPR-mediated mutagenesis. We addressed this by using mutational profiling as a readout of the screen. At each targeted miRNA-binding site, we determined the rate of alleles disrupting miRNA binding that were detected in wild-type-selected animals. Importantly, nearby indels that are not predicted to disrupt miRNA binding act as an internal control for gRNA efficiency at each locus. Therefore, selection pressure against target derepression can be inferred from a low frequency of seed match-disrupting alleles among all alleles. This metric is critically important since the raw number of alleles at each locus primarily reflected not phenotypic effects but variation in gRNA efficiency, which is impacted by multiple factors including gRNA nucleotide composition, genomic context surrounding the targeting site, and chromatin conformation (Doench et al. 2014; Chari et al. 2015; Moreno-Mateos et al. 2015; Horlbeck et al. 2016).
The design of a gRNA library targeting miRNA-binding sites is challenging. The target region is very small (only 6–7 bp to target the seed-binding region). The mir-35 seed match contains three NGG PAMs, making gRNA design straightforward. However, depending on the seed match sequence of interest in future screens, the cleavage site of the gRNA may have to lie outside the seed; in this case, we recommend grouping target loci according to the distance between the cleavage site and the seed match for comparison of seed match-disrupting allele frequency, since cleavage sites farther away from the seed match will inherently have lower seed match-disrupting allele frequencies (Yang and McJunkin 2020a). Notably, the repertoire of targetable PAM sequences is rapidly expanding beyond NGG, with even near-PAMless CRISPR technology (Walton et al. 2020); these advances should alleviate some gRNA design restrictions in the near future.
A second difficulty in gRNA design at miRNA-binding sites is the relatively AT-rich environment of 3′UTRs. High AT content is disadvantageous because it correlates with low gRNA efficiency (Doench et al. 2014; Wang et al. 2014). Furthermore, short oligo(dT) tracts can cause premature transcription termination downstream from the U6 promoters used to drive gRNA expression in vivo (Arimbasseri et al. 2013). We suspect that this contributes to the poor performance of plasmid-expressed gRNAs compared with synthetic gRNAs in our hands. In more GC-rich genomic contexts like coding sequences, plasmid-expressed guides may still be a viable option for multiplexing; moreover, the low cost of plasmid-expressed gRNAs could allow for multiple gRNAs per target site.
Here we optimized the multiplexed delivery of gRNAs to C. elegans, significantly reducing the labor required to screen a large number of regulatory elements. A pool size of 10 yielded the largest number of mutant alleles per labor expended. Here we used transgenically expressed Cas9. However, validation experiments in which recombinant Cas9 RNPs were more efficient prompted us to further optimize multiplexed RNP delivery. Individually loading gRNAs into Cas9 before multiplexing rendered RNPs more effective than transgenic Cas9, and the RNPs are therefore recommended for future screens. Use of RNPs would enable screening in pre-existing complex genetic backgrounds and alleviate any concerns about long-term germline Cas9 expression.
The only miRNA-binding site we identified as a phenotypic mir-35 target was the egl-1 site. This site was the top hit from the screen with 0% seed match-disrupting alleles; therefore, truly negatively selected hits might be expected to have zero seed match-disrupting mutations. Future screens and validation studies will help determine whether this is the case. Future multiplexed screens could also have improvements that aid in the interpretation of the screen readout. When more distinctive phenotypes or fluorescent reporters are used for selection, a phenotype-positive and phenotype-negative population can be isolated. Comparison of the complement of alleles present in these two groups will allow for statistical analysis of the screen results rather than simple ranking. Including a large number of negative control gRNAs could also aid statistical analysis. This screen lacked a positive control, but future screens for miRNA targets inducing deleterious phenotypes can use egl-1 as a positive control. Controls for other phenotypes will depend on the phenotype itself. These could include gRNAs targeting protein-coding genes whose loss of function is known to induce the phenotype of interest or—when the phenotype of interest is altered expression of a reporter transgene—gRNAs targeting the reporter itself.
Furthermore, egl-1 derepression only partially phenocopied mir-35 family mutants, inducing low-penetrance embryonic lethality compared with the complete lethality observed in mir-35-42 mutants. One possibility is that the miRNA mutant phenotype is the result of synthetic effects of derepression of multiple target genes. An exciting possibility would be to conduct the multiplexed screen iteratively, using an increasingly complex genetic background in which mildly phenotypic targets (e.g., egl-1) are already derepressed to eventually reconstruct the complement of targets that together contribute to the phenotype. Alternatively, important targets may be missing from the candidate list predicted by TargetScan. Moving forward, conducting CLASH or chimera-generating Argonaute iCLIP in embryonic samples could help to determine whether a broader set of putative targets should be assayed (Broughton et al. 2016). Overall, this approach does rely on the assumption that derepression of a single miRNA target is sufficient to induce detectable phenotypes. In general, work on phenotypic targets of other miRNA families has supported this assumption (Dorsett et al. 2008; Teng et al. 2008; Ecsedi et al. 2015; Lu et al. 2015; Drexel et al. 2016). However, data supporting the alternative scenario—in which synthetic interactions are necessary to produce phenotypic effects—could be far more difficult to generate.
Finally, the strategy used here, of CRISPR screening followed by mutational profiling, could be applied to screen other types of negative regulatory elements. For instance, if a repressive RNA-binding protein or transcription factor has a distinctive loss-of-function phenotype, then the putative binding sites can be screened to identify phenotypic targets. Binding sites that show a low frequency of consensus binding site mutations (among all mutations at a given site) are high priority candidate phenotypic targets. This general strategy will be applicable not only to C. elegans, but also to cell lines, transplant models, or other model organisms.
Materials and methods
Nematode culture and genetics
Standard culture conditions were used (Stiernagle 2006). Worms were maintained at 20°C except where noted otherwise. Alleles and strains used in this study are listed in Supplemental Tables S4 and S5.
gRNA design and generation
gRNAs targeting the candidate sites predicted by TargetScan were designed to cut in or very close to the seed sequence match, mostly using a PAM site within the seed sequence (see Supplemental Fig. S6; Supplemental Tables S1–S3). gRNA sequences were then either cloned into pRB1017 or ordered from IDT as Alt-R crRNAs (Arribere et al. 2014). See the Supplemental Material for injection mix details.
CRISPR screen and library construction
The customized gRNA library was divided in to 20 subpools, with each pool containing nine to 11 gRNAs (including dpy-10 gRNA), and each gRNA was included in two different pools (see Supplemental Table S7 for pooling strategy). Pools of gRNAs were injected into EG9615, and injected animals were placed at 20°C. Three to four days later, each Dpy or Rol F1 was isolated as an L4 or young adult and placed on a 3-cm plate seeded with 100 µL of OP50. These plates were maintained at 20°C, and 30 wild-type strains for each pool were selected based on visible phenotype and time to starvation. Starved plates were harvested, and genomic DNA was extracted. See the Supplemental Material for more information on pooling, library construction, and sequence analysis.
Image acquisition and analysis
Embryos were obtained by treating gravid hermaphrodites with alkaline hypochlorite solution and mounting them on 2% agarose pads for imaging. All images were acquired at fixed exposure times using the 60× objective lens on a Nikon C2 confocal microscope. Image analysis was performed using Fiji software (Schindelin et al. 2012). Briefly, images of both the GFP and DIC channels were imported into Fiji and synchronized; circles were drawn at the periphery of each nucleus in the DIC channel and copied into the GFP channel. Mean GFP intensities of the circled area were measured and plotted. N2 animals were treated as negative controls, and their average intensities were subtracted from all experimental groups.
Supplementary Material
Acknowledgments
Strains were kindly provided by Barbara Conradt (egl-1 GFP reporters), Brent Derry (egl-1(on24)), and the Caenorhabditis Genetics Center, which is funded by National Institutes of Health Office of Research Infrastructure Programs (P40 OD010440). We thank Helge Grosshans and Joana Vidigal for critical reading of the manuscript. Thank you to the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) Genomics Core for deep sequencing, and to Cameron Palmer for consulting on data analysis. Thank you to the NIDDK Biostatistics Program for consultation. B.Y. and K.M. are funded by the NIDDK Intramural Research Program (1ZIADK075147).
Author contributions: M.S. generated reagents. B.Y. and K.M. conceived the experiments, analyzed the data, and wrote the manuscript. B.Y. performed the experiments.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.339333.120.
References
- Alvarez-Saavedra E, Horvitz HR. 2010. Many families of C. elegans microRNAs are not essential for development or viability. Curr Biol 20: 367–373. 10.1016/j.cub.2009.12.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambros V. 2004. The functions of animal microRNAs. Nature 431: 350–355. 10.1038/nature02871 [DOI] [PubMed] [Google Scholar]
- Arimbasseri AG, Rijal K, Maraia RJ. 2013. Transcription termination by the eukaryotic RNA polymerase III. Biochim Biophys ActaGene Regul Mech 1829: 318–330. 10.1016/j.bbagrm.2012.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arribere JA, Bell RT, Fu BX, Artiles KL, Hartman PS, Fire AZ. 2014. Efficient marker-free recovery of custom genetic modifications with CRISPR/Cas9 in Caenorhabditis elegans. Genetics 198: 837–846. 10.1534/genetics.114.169730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel DP. 2009. MicroRNAs: target recognition and regulatory functions. Cell 136: 215–233. 10.1016/j.cell.2009.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel DP. 2018. Metazoan microRNAs. Cell 173: 20–51. 10.1016/j.cell.2018.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benner LK, Prothro KP, McJunkin K. 2019. The mir-35 family links maternal germline sex to embryonic viability in Caenorhabditis elegans. G3 9: 901–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broughton JP, Lovci MT, Huang JL, Yeo GW, Pasquinelli AE. 2016. Pairing beyond the seed supports MicroRNA targeting specificity. Mol Cell 64: 320–333. 10.1016/j.molcel.2016.09.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canver MC, Smith EC, Sher F, Pinello L, Sanjana NE, Shalem O, Chen DD, Schupp PG, Vinjamur DS, Garcia SP, et al. 2015. BCL11A enhancer dissection by Cas9-mediated in situ saturating mutagenesis. Nature 527: 192–197. 10.1038/nature15521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chari R, Mali P, Moosburner M, Church GM. 2015. Unraveling CRISPR–Cas9 genome engineering parameters via a library-on-library approach. Nat Methods 12: 823–826. 10.1038/nmeth.3473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Li M, Feng X, Guang S. 2015. Targeted chromosomal translocations and essential gene knockout using CRISPR/Cas9 technology in Caenorhabditis elegans. Genetics 201: 1295–1306. 10.1534/genetics.115.181883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, McKenna A, Schreiber J, Haeussler M, Yin Y, Agarwal V, Noble WS, Shendure J. 2019. Massively parallel profiling and predictive modeling of the outcomes of CRISPR/Cas9-mediated double-strand break repair. Nucleic Acids Res 47: 7989–8003. 10.1093/nar/gkz487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi PS, Meyerson M. 2014. Targeted genomic rearrangements using CRISPR/Cas technology. Nat Commun 5: 3728 10.1038/ncomms4728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diao Y, Fang R, Li B, Meng Z, Yu J, Qiu Y, Lin KC, Huang H, Liu T, Marina RJ, et al. 2017. A tiling-deletion-based genetic screen for cis-regulatory element identification in mammalian cells. Nat Methods 14: 629–635. 10.1038/nmeth.4264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doench JG, Hartenian E, Graham DB, Tothova Z, Hegde M, Smith I, Sullender M, Ebert BL, Xavier RJ, Root DE. 2014. Rational design of highly active sgRNAs for CRISPR–Cas9-mediated gene inactivation. Nat Biotechnol 32: 1262–1267. 10.1038/nbt.3026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doench JG, Fusi N, Sullender M, Hegde M, Vaimberg EW, Donovan KF, Smith I, Tothova Z, Wilen C, Orchard R, et al. 2016. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR–Cas9. Nat Biotechnol 34: 184–191. 10.1038/nbt.3437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dokshin GA, Ghanta KS, Piscopo KM, Mello CC. 2018. Robust genome editing with short single-stranded and long, partially single-stranded DNA donors in Caenorhabditis elegans. Genetics 210: 781–787. 10.1534/genetics.118.301532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doll MA, Soltanmohammadi N, Schumacher B. 2019. ALG-2/AGO-Dependent mir-35 family regulates DNA damage-induced apoptosis through MPK-1/ERK MAPK signaling downstream of the core apoptotic machinery in Caenorhabditis elegans. Genetics 213: 173–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorsett Y, McBride KM, Jankovic M, Gazumyan A, Thai T-H, Robbiani DF, Di Virgilio M, Reina San-Martin B, Heidkamp G, Schwickert TA, et al. 2008. MicroRNA-155 suppresses activation-induced cytidine deaminase-mediated Myc-Igh translocation. Immunity 28: 630–638. 10.1016/j.immuni.2008.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drexel T, Mahofsky K, Latham R, Zimmer M, Cochella L. 2016. Neuron type-specific miRNA represses two broadly expressed genes to modulate an avoidance behavior in C. elegans. Genes Dev 30: 2042–2047. 10.1101/gad.287904.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ecsedi M, Rausch M, Großhans H. 2015. The let-7 microRNA directs vulval development through a single target. Dev Cell 32: 335–344. 10.1016/j.devcel.2014.12.018 [DOI] [PubMed] [Google Scholar]
- Farboud B, Meyer BJ. 2015. Dramatic enhancement of genome editing by CRISPR/cas9 through improved guide RNA design. Genetics 199: 959–971. 10.1534/genetics.115.175166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flamand MN, Gan HH, Mayya VK, Gunsalus KC, Duchaine TF. 2017. A non-canonical site reveals the cooperative mechanisms of microRNA-mediated silencing. Nucleic Acids Res 45: 7212–7225. 10.1093/nar/gkx340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fridrich A, Hazan Y, Moran Y. 2019. Too many false targets for microRNAs: challenges and pitfalls in prediction of miRNA targets and their gene ontology in model and non-model organisms. Bioessays 41: 1800169 10.1002/bies.201800169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulco CP, Munschauer M, Anyoha R, Munson G, Grossman SR, Perez EM, Kane M, Cleary B, Lander ES, Engreitz JM. 2016. Systematic mapping of functional enhancer–promoter connections with CRISPR interference. Science 354: 769–773. 10.1126/science.aag2445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haeussler M, Schönig K, Eckert H, Eschstruth A, Mianné J, Renaud JB, Schneider-Maunoury S, Shkumatava A, Teboul L, Kent J, et al. 2016. Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biol 17: 148.. 10.1186/s13059-016-1012-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han R, Li L, Ugalde AP, Tal A, Manber Z, Barbera EP, Chiara VD, Elkon R, Agami R. 2018. Functional CRISPR screen identifies AP1-associated enhancer regulating FOXF1 to modulate oncogene-induced senescence. Genome Biol 19: 118 10.1186/s13059-018-1494-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horlbeck MA, Witkowsky LB, Guglielmi B, Replogle JM, Gilbert LA, Villalta JE, Torigoe SE, Tjian R, Weissman JS. 2016. Nucleosomes impede Cas9 access to DNA in vivo and in vitro. Elife 5: e12677 10.7554/eLife.12677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Housden BE, Valvezan AJ, Kelley C, Sopko R, Hu Y, Roesel C, Lin S, Buckner M, Tao R, Yilmazel B, et al. 2015. Identification of potential drug targets for tuberous sclerosis complex by synthetic screens combining CRISPR-based knockouts with RNAi. Sci Signal 8: rs9 10.1126/scisignal.aab3729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwata S, Yoshina S, Suehiro Y, Hori S, Mitani S. 2016. Engineering new balancer chromosomes in C. elegans via CRISPR/Cas9. Sci Rep 6: 33840 10.1038/srep33840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jan CH, Friedman RC, Ruby JG, Bartel DP. 2011. Formation, regulation and evolution of Caenorhabditis elegans 3′UTRs. Nature 469: 97–101. 10.1038/nature09616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagias K, Pocock R. 2015. microRNA regulation of the embryonic hypoxic response in Caenorhabditis elegans. Sci Rep 5: 11284 10.1038/srep11284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamath RS, Martinez-Campos M, Zipperlen P, Fraser AG, Ahringer J. 2001. Effectiveness of specific RNA-mediated interference through ingested double-stranded RNA in Caenorhabditis elegans. Genome Biol 2: 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korkmaz G, Lopes R, Ugalde AP, Nevedomskaya E, Han R, Myacheva K, Zwart W, Elkon R, Agami R. 2016. Functional genetic screens for enhancer elements in the human genome using CRISPR–Cas9. Nat Biotechnol 34: 192–198. 10.1038/nbt.3450 [DOI] [PubMed] [Google Scholar]
- Liu M, Liu P, Zhang L, Cai Q, Gao G, Zhang W, Zhu Z, Liu D, Fan Q. 2011. mir-35 is involved in intestine cell G1/S transition and germ cell proliferation in C. elegans. Cell Res 21: 1605–1618. 10.1038/cr.2011.102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu L-F, Gasteiger G, Yu I-S, Chaudhry A, Hsin J-P, Lu Y, Bos PD, Lin L-L, Zawislak CL, Cho S, et al. 2015. A single miRNA-mRNA interaction affects the immune response in a context- and cell-type-specific manner. Immunity 43: 52–64. 10.1016/j.immuni.2015.04.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massirer KB, Perez SG, Mondol V, Pasquinelli AE. 2012. The miR-35-41 family of microRNAs regulates RNAi sensitivity in Caenorhabditis elegans. PLoS Genet 8: e1002536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McJunkin K, Ambros V. 2014. The embryonic mir-35 family of microRNAs promotes multiple aspects of fecundity in Caenorhabditis elegans. G3 Genes, Genomes, Genet 4: 1747–1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McJunkin K, Ambros V. 2017. A microRNA family exerts maternal control on sex determination in C. elegans. Genes Dev 31: 422–437. 10.1101/gad.290155.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno-Mateos MA, Vejnar CE, Beaudoin J-D, Fernandez JP, Mis EK, Khokha MK, Giraldez AJ. 2015. CRISPRscan: designing highly efficient sgRNAs for CRISPR–Cas9 targeting in vivo. Nat Methods 12: 982–988. 10.1038/nmeth.3543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paix A, Wang Y, Smith HE, Lee CYS, Calidas D, Lu T, Smith J, Schmidt H, Krause MW, Seydoux G. 2014. Scalable and versatile genome editing using linear DNAs with microhomology to Cas9 sites in Caenorhabditis elegans. Genetics 198: 1347–1356. 10.1534/genetics.114.170423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez AR, Pritykin Y, Vidigal JA, Chhangawala S, Zamparo L, Leslie CS, Ventura A. 2017. GuideScan software for improved single and paired CRISPR guide RNA design. Nat Biotechnol 35: 347–349. 10.1038/nbt.3804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quévillon Huberdeau M, Simard MJ. 2019. A guide to microRNA-mediated gene silencing. FEBS J 286: 642–652. 10.1111/febs.14666 [DOI] [PubMed] [Google Scholar]
- Rajagopal N, Srinivasan S, Kooshesh K, Guo Y, Edwards MD, Banerjee B, Syed T, Emons BJM, Gifford DK, Sherwood RI. 2016. High-throughput mapping of regulatory DNA. Nat Biotechnol 34: 167–174. 10.1038/nbt.3468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. 2012. Fiji: an open-source platform for biological-image analysis. Nat Methods 9: 676–682. 10.1038/nmeth.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherrard R, Luehr S, Holzkamp H, McJunkin K, Memar N, Conradt B. 2017. miRNAs cooperate in apoptosis regulation during C. elegans development. Genes Dev 31: 209–222. 10.1101/gad.288555.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiernagle T. 2006. Maintenance of C. elegans. WormBook 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng G, Hakimpour P, Landgraf P, Rice A, Tuschl T, Casellas R, Papavasiliou FN. 2008. MicroRNA-155 is a negative regulator of activation-induced cytidine deaminase. Immunity 28: 621–629. 10.1016/j.immuni.2008.03.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran AT, Chapman EM, Flamand MN, Yu B, Krempel SJ, Duchaine TF, Eroglu M, Derry WB. 2019. MiR-35 buffers apoptosis thresholds in the C. elegans germline by antagonizing both MAPK and core apoptosis pathways. Cell Death Differ 26: 2637–2651. 10.1038/s41418-019-0325-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Overbeek M, Capurso D, Carter MM, Thompson MS, Frias E, Russ C, Reece-Hoyes JS, Nye C, Gradia S, Vidal B, et al. 2016. DNA repair profiling reveals nonrandom outcomes at Cas9-mediated breaks. Mol Cell 63: 633–646. 10.1016/j.molcel.2016.06.037 [DOI] [PubMed] [Google Scholar]
- Walton RT, Christie KA, Whittaker MN, Kleinstiver BP. 2020. Unconstrained genome targeting with near-PAMless engineered CRISPR–Cas9 variants. Science 368: 290–296. 10.1126/science.aba8853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Wei JJ, Sabatini DM, Lander ES. 2014. Genetic screens in human cells using the CRISPR–Cas9 system. Science 343: 80–84. 10.1126/science.1246981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu E, Thivierge C, Flamand M, Mathonnet G, Vashisht AA, Wohlschlegel J, Fabian MR, Sonenberg N, Duchaine TF. 2010. Pervasive and cooperative deadenylation of 3′UTRs by embryonic microRNA families. Mol Cell 40: 558–570. 10.1016/j.molcel.2010.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang B, McJunkin K. 2020a. CRISPR screening strategies for microRNA target identification. FEBS J 287: 2914–2922. 10.1111/febs.15218 [DOI] [PubMed] [Google Scholar]
- Yang B, McJunkin K. 2020b. The mir-35-42 binding site in the nhl-2 3′UTR is dispensable for development and fecundity. MicroPubl Biol 10.17912/micropub.biology.000241 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.