

Abstract
Background -
Dilated cardiomyopathy (DCM) is a genetically heterogeneous cardiac disease characterized by progressive ventricular enlargement and reduced systolic function. Here, we report genetic and functional analyses implicating the RAS signaling protein, SOS1, in DCM pathogenesis.
Methods -
Exome sequencing was performed on 412 probands and family members from our DCM cohort, identifying several SOS1 variants with potential disease involvement. As several lines of evidence have implicated dysregulated RAS signaling in the pathogenesis of DCM, we assessed functional impact of each variant on activation of ERK, AKT, and JNK pathways. Relative expression levels were determined by western blot in HEK293T cells transfected with variant or wild-type human SOS1 expression constructs.
Results -
A rare SOS1 variant [c.571G>A, p.(Glu191Lys)] was found to segregate alongside an A-band Titin (TTN) truncating variant in a pedigree with aggressive, early onset DCM. Reduced disease severity in the absence of the SOS1 variant suggested its potential involvement as a genetic risk factor for DCM in this family. Exome sequencing identified five additional SOS1 variants with potential disease involvement in four other families [c.1820T>C, p.(Ile607Thr); c.2156G>C, p.(Gly719Ala); c.2230A>G, p.(Arg744Gly); c.2728G>C, p.(Asp910His); c.3601C>T, p.(Arg1201Trp)]. Impacted amino acids occupied a number of functional domains relevant to SOS1 activity, including the N-terminal histone fold (HF), as well as the C-terminal RAS-exchange motif (REM), CDC25, and proline-rich (PR) tail domains. Increased pERK expression relative to wild-type levels was seen for all six SOS1 variants, paralleling known disease-relevant SOS1 signaling profiles.
Conclusions -
These data support gain of function variation in SOS1 as a contributing factor to isolated DCM.
Keywords: dilated cardiomyopathy, genetics, genetic heart disease, SOS1 protein, son of sevenless protein
Journal Subject Terms: Cardiomyopathy, Heart Failure, Genetics
Introduction
To date, over 50 genes have been identified as causative or likely causative for dilated cardiomyopathy (DCM)1, 2. While identification of these genes has greatly improved our understanding of the genetic basis for DCM, low diagnostic yields (~37%) of clinical genetic testing panels3 support existence of additional undiscovered genetic causes.
The impetus for the present study originated from identification of a rare Son of Sevenless 1 (SOS1) variant segregating in three severely affected individuals of a large family with early-onset, bilineal DCM (Figure 1A). Exome sequencing of our wider DCM cohort identified a further five SOS1 variants of potential pathogenic significance spanning four additional families (Figure 1B–E). We considered SOS1, a RAS-activating guanine exchange factor (GEF), to be a plausible DCM candidate as animal and human data have previously implicated dysregulated RAS signaling in the pathogenesis of DCM4–8.
Figure 1.
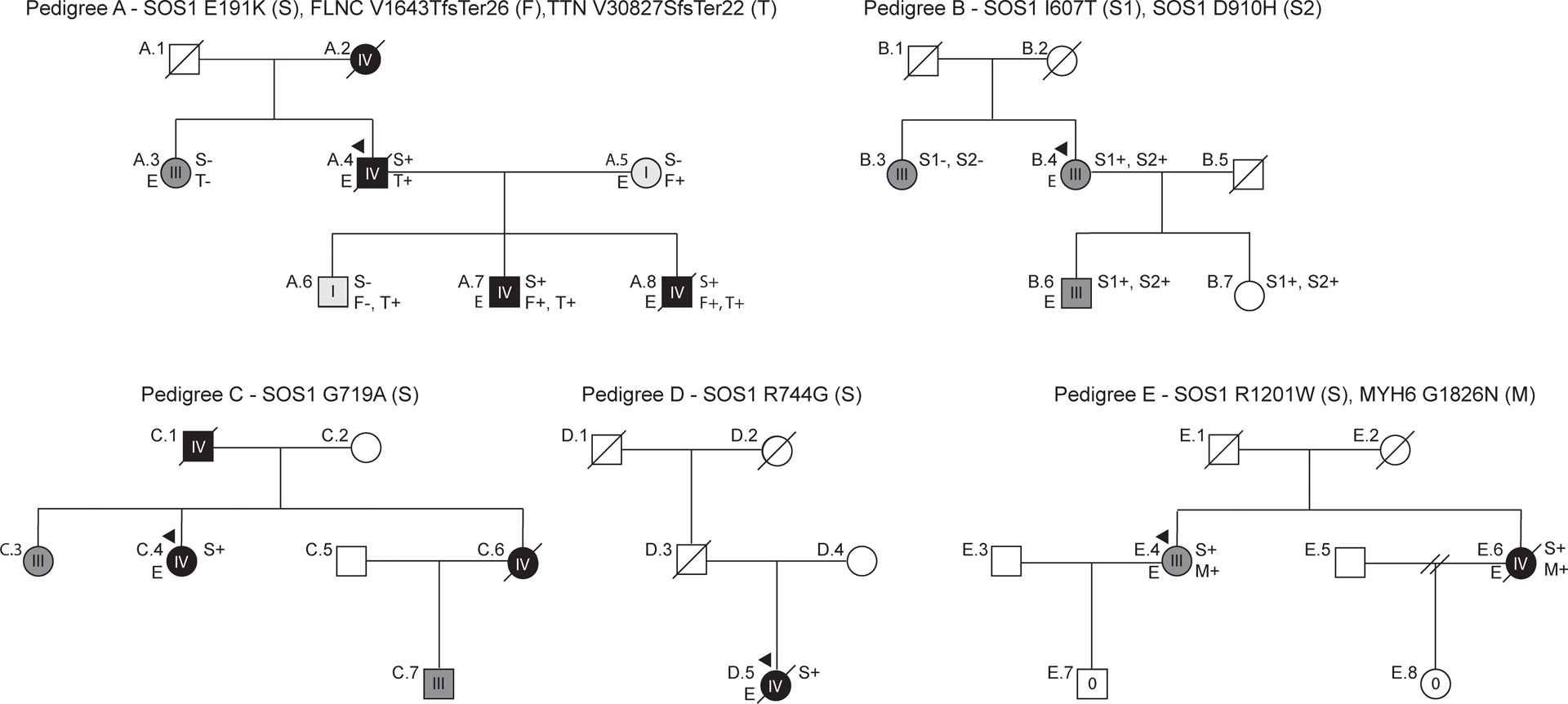
DCM pedigrees with SOS1 variants. Affection status was categorized using an ordinal grading scale. 0 (no cardiovascular disease or evidence of DCM), I (either left ventricular enlargement, LVE, without systolic function or systolic dysfunction without LVE), II (asymptomatic DCM), III (symptomatic DCM with or without medical therapy), IV (DCM requiring advanced interventions or resulting in DCM-related death). In all pedigrees, squares represent males, circles females. Diagonal lines indicate deceased individuals. Presence (+) or absence (−) of identified rare variants are indicated. Individuals for whom exome sequencing was completed are marked with an “E”.
Befitting critical roles for mitogen-activated protein kinase (MAPK) signaling pathways in cardiac development, remodeling, and disease9, SOS1 variants underlie a number of cardiovascular genetic disorders, including Noonan syndrome (NS, Mendelian Inheritance in Man (MIM): 610733), and cardiofaciocutaneous syndrome (CFCS, MIM: 115150). Myocardial involvement is a well-recognized component and while hypertrophic cardiomyopathy (HCM) is more common, DCM has also been reported, including in one patient carrying a p.(Met269Thr) SOS1 variant10. Supporting involvement of SOS1 in non-syndromic disease, targeted sequencing of RAS signaling genes have identified a number of SOS1 variants with possible pathogenic potential in patients with isolated cardiomyopathy11–13.
Clinically-relevant SOS1 variants typically promote increased GEF activity and hyperactivation of downstream RAS effectors14, 15. This proclivity towards gain of function effect is directly related to the structure of the SOS1 protein, which natively exists in an auto-inhibited state governed by complex interplay between six distinct functional domains (Figure 2)16–19. Pathogenic variants disrupt these interactions, promoting sustained RAS activation20. Three N-terminal domains - a histone-like fold (HF) domain, a Dbl homology (DH) domain, and a pleckstrin homology (PH) domain - comprise an auto-inhibitory unit that orients to prevent RAS binding when SOS1 is not associated with the plasma membrane17, 18 (Figure 2A). Membrane association is primarily driven through direct interaction of the proline-rich (PR) tail with Growth factor receptor-bound protein 2 (GRB2)21 while secondary associations are provided by the HF16, 18, and PH22 domains (Figure 2B). Two additional domains: a RAS-exchange motif (REM) and a CDC25 domain, collectively comprise the C-terminal SOS1 catalytic core. The latter domain serves as the site of GDP/GTP exchange and the former as a non-substrate RAS binding site23. When SOS1 is unbound, the DH-PH regulatory unit blocks access of RAS to the REM binding site16, 19 (Figure 2A). Alleviation of auto-inhibition on membrane association frees the REM to bind to RAS, initiating conformational changes that promote binding of a second RAS molecule at the adjacent CDC25 catalytic site (Figure 2B). These structural rearrangements create a positive feedback loop whereby activated RAS from the CDC25 domain can rebind the REM, facilitating further RAS recruitment and activation23. The complexity of these intramolecular interactions provides ample opportunity for genetic disruption and forms the molecular basis for SOS1-related disease.
Figure 2.
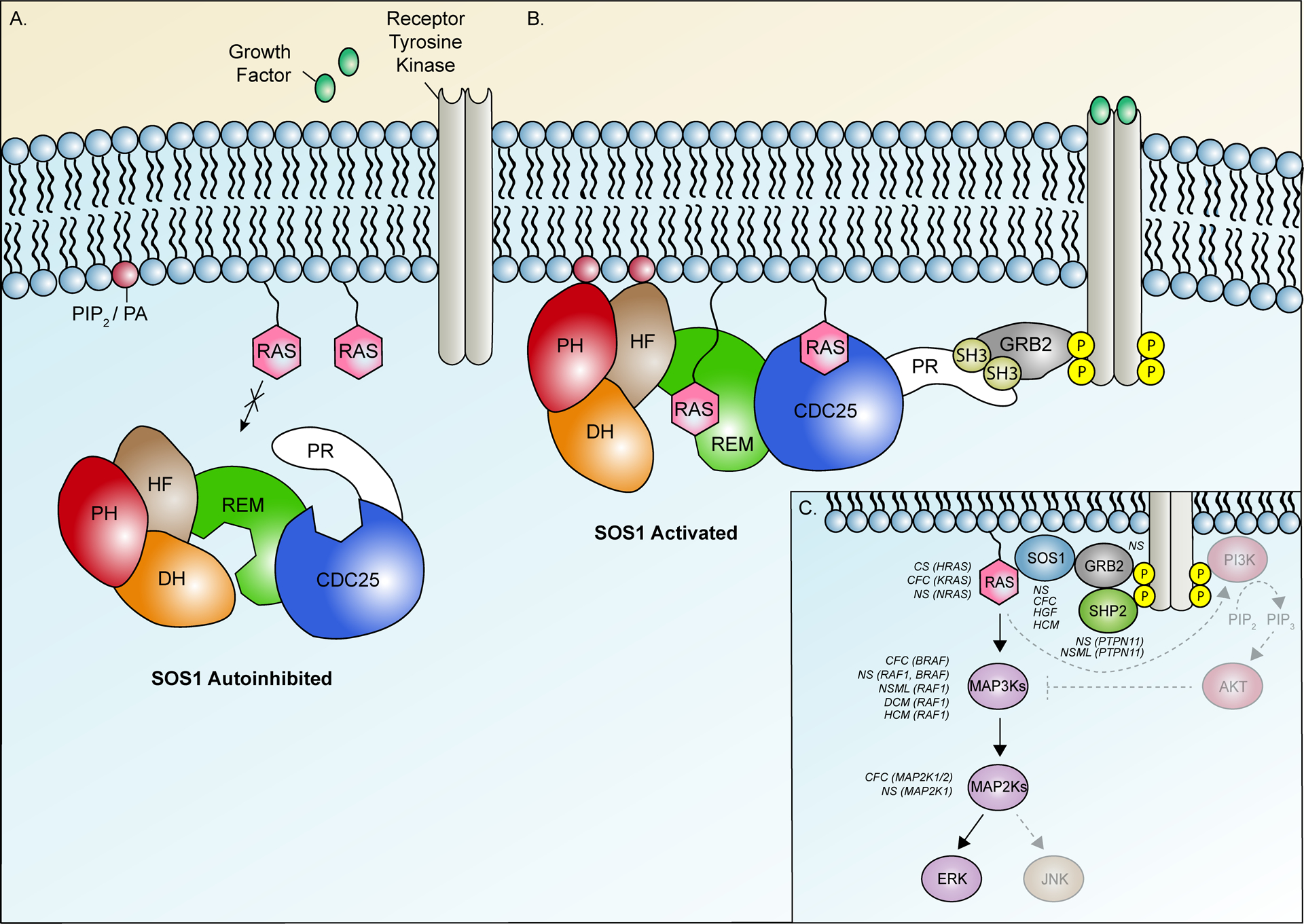
SOS1 activity is governed by complex interactions between six distinct functional domains. (A) SOS1 activity is natively suppressed by a primary auto-inhibitory unit comprised of the N-terminal histone-like fold (HF), Dbl homology (DH) domain, and pleckstrin homology (PH) domains and by secondary mechanisms facilitated by the proline-rich (PR) tail. This closed conformation blocks access of RAS to dual binding sites in the SOS1 catalytic core, comprised of the RAS-exchange motif (REM) and CDC25 domain. (B) Growth factor binding facilitates activation of receptor tyrosine kinases (RTKs) and recruitment of SOS1 to the membrane via direct interaction of the PR tail with GRB2, itself bound to phosphorylated tyrosine residues on the activated RTKs. Secondary membrane associations are further provided by the HF and PH domains via phosphatidylinositol 4,5-bisphosphate (PIP2) and phosphatidic acid (PA). Alleviation of repression by these interactions permit RAS binding to a distal binding site in the REM, initiating additional allosteric changes that recruit a second RAS molecule to the adjacent CDC25 catalytic site. Following GDP/GTP exchange, SOS1 remains strongly associated with the membrane, facilitating recruitment and activation of additional RAS molecules. (C) Simplified overview of RAS signaling pathways and associated disease phenotypes. Solid arrows represent individual steps in the ERK signaling cascade. Hatched arrows represent steps in alternate RAS-mediated AKT and JNK signaling pathways. Disease phenotypes associated with activating variants in individual pathway components are listed in italics. Affected genes are indicated in brackets. Additional abbreviations: CFC, Cardiofaciocutaneous syndrome; CS, Costello syndrome; DCM, Dilated cardiomyopathy; HCM, Hypertrophic cardiomyopathy; HGF, Hereditary gingival fibromatosis; MAP2K, Mitogen-activated protein kinase kinase; MAP3K, Mitogen-activated protein kinase kinase kinase, NS, Noonan syndrome; NSML, Noonan syndrome with multiple lentigines.
A simplified overview of RAS signaling pathway components, including SOS1, is shown in Figure 2C alongside known associated disease phenotypes. In this context, we describe genetic and functional analyses of six SOS1 variants identified in our DCM cohort. Our results provide preliminary evidence for involvement of gain of function SOS1 variants in isolated DCM and support consideration of DCM in the phenotypic spectrum of SOS1-associated genetic disease.
Methods
Complete study methods are provided in the Supplemental Materials. The sequencing and analytical data are available to other researchers upon request for confirmation and/or replication of our analysis. Informed consent was obtained from all study participants following Institutional Review Board approval by the Oregon Health & Science University, the University of Miami, or the Ohio State University.
Results
Exome sequencing of 412 total individuals from our DCM cohort, including 281 probands and 131 of their affected family members, identified six heterozygous missense SOS1 variants of possible pathogenicity segregating in five families of non-Hispanic, European ancestry (Figure 1, Table 1).
Table 1.
Summary of genetic findings.
| Pedigree | Gene | Chr | Transcript | Genomic Position GRCh37 (hg19) | Nucleotide Change | Amino Acid Change | Max ExAC MAF* | Max gnomAD MAF* | dbSNP | Variant Classification† |
|---|---|---|---|---|---|---|---|---|---|---|
| A | SOS1 | 2 | NM_005633.3 | 39281904 | c.571G>A | p.(Glu191Lys) | 0 | 0.0001147 (AFR)(G) | rs886041241 | VUS |
| FLNC | 7 | NM_001458.4 | 128489028 | c.4926_4927 insACGTCACA | p.(Val1643Thrfs*26) | 0 | 0.000009182 (NFE)(E) | Not found | LP | |
| TTN | 2 | NM_001267550.2 | 179413874 | c.92478dupA | p.(Val30827Serfs*22) | 0 | 0 | Not found | LP | |
| B | SOS1 | 2 | NM_005633.3 | 39249749 | c.1820T>C | p.(Ile607Thr) | 0.00003006 (NFE) | 0.00002654 (NFE)(E) | rs758699499 | VUS |
| 2 | NM_005633.3 | 39233616 | c.2728G>C | p.(Asp910His) | 0.00006061 (SAS) | 0.00009806 (SAS)(E) | rs369277679 | VUS | ||
| C | SOS1 | 2 | NM_005633.3 | 39240612 | c.2156G>C | p.(Gly719Ala) | 0.0005556 (NFE) | 0.0003426 (NFE)(E+G) | rs200794965 | VUS |
| D | SOS1 | 2 | NM_005633.3 | 39239427 | c.2230A>G | p.(Arg744Gly) | 0 | 0 | Not found | VUS |
| E | SOS1 | 2 | NM_005633.3 | 39213366 | c.3601C>T | p.(Arg1201Trp) | 0.00006058 (SAS) | 0.00003266 (SAS)(E) | rs752395541 | VUS |
| MYH6 | 14 | NM_002471.3 | 23853739 | c.5476_5477 delGGinsAA | p.(Gly1826Asn) | 0 | 0 | rs878854502 | VUS |
The maximum non-founder minor allele frequency (MAF) and associated population for each variant is reported. A MAF of 0 implies absence of the variant from Exome Aggregation Consortium (ExAC) / Genome Aggregation Database (gnomAD) and sufficient coverage of the genomic position. Reported gnomAD results include both exomes (E) and genomes (G) when available. AFR, African; NFE, non-Finnish European; SAS, South Asian
All six SOS1 variants were considered to be variants of uncertain significance (VUSs) using refined guidelines for DCM variant interpretation24. The five families included 1 simplex and 4 multiplex (i.e., affected members spanning multiple generations) families. DNA was unavailable in some individuals who were reported to be affected. In 2 families (Pedigrees A and E), exome sequencing identified additional rare variants in at least one other known DCM gene. No clinical features consistent with syndromic RASopathies were identified in Families B to E (Table 2). However, presentation of a photograph of Family A (Individuals A.4, A.5, A.6, A.7, A.8) to a research conference of the Department of Molecular and Medical Genetics at the Oregon Health & Science University elicited spontaneous comments from three medical geneticists that individuals A.7 and A.8, both teenagers at the time the photograph was taken, had facial features consistent with NS.
Table 2.
Clinical characteristics of study participants.
| Individual | DCM | Age of Onset (y) | Age at Echo (y) | Height (cm) | LVIDD | Z-score | LVEF | ICD | Hrt. Tx. | Arrhyth. | Vital Status (Age in y, or age at death or heart transplant) | Comments / Additional Features |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pedigree A | ||||||||||||
| A.1 | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | Deceased (d.88, unk.) | No medical records, diabetes (reported), HTN (reported) |
| A.2 | Yes | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | Deceased (d. 58, HF) | No medical records, reported affected |
| A.3 | Yes | 33 | 77 | 160 | 65 | 5.601031 | 52.5 | No | No | Yes | Alive (61) | |
| A.4 | Yes | 44 | 72 | 175 | 62 | 3.036178 | 25 | No | No | Yes | Deceased (d.57, DCM, HF) | Degenerative arthritis, gout, migraines, cholecystectomy |
| A.5 | No | n/a | n/a | 174 | 55 | 2.315058 | 60 | No | No | No | Alive (64) | |
| A.6 | No | n/a | n/a | 181.61 | 54 | 0.709736 | 47.5 | No | No | Yes | Alive (42) | HTN |
| A.7 | Yes | 22 | 22 | 183 | 63.5 | 3.011836 | 20 | Yes | Yes | Yes | Heart transplant (31) | Cardiorenal syndrome |
| A.8 | Yes | 14 | 14 | 165.1 | 75 | 6.30143 | n/a | Yes | Yes | No | Deceased (d. 22), Heart transplant (14) | |
| Pedigree B | ||||||||||||
| B.1 | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | No | No | Deceased (d.74, HF) | HTN (reported) |
| B.2 | n/a | n/a | n/a | n/a | n/a | n/a | n/a | No | No | No | Deceased (d.>89y, aortic dissection) | Alzheimer’s, HTN, cardiomegaly, coronary artery calcifications |
| B.3 | Yes | 56 | 66 | 170.2 | 66 | 5.358739 | 17 | Yes | No | No | Alive (76) | Hx doxorubicin, diabetes, hyperthyroid |
| B.4 | Yes | 61 | 63 | 162.6 | 57 | 3.406839 | 25 | Yes | No | Yes | Alive (75) | Asthma, hx 2mm circumflex coronary aneurysm |
| B.5 | n/a | n/a | n/a | n/a | n/a | n/a | n/a | No | No | No | Deceased (d. 79, unk.) | |
| B.6 | Yes | 45 | 45 | 175.26 | 71 | 5.002545 | 42.5 | Yes | No | No | Alive (54) | WPW, diabetes |
| B.7 | No | n/a | n/a | n/a | n/a | n/a | n/a | No | No | No | Alive (53) | HTN, migraines, hysterectomy |
| Pedigree C | ||||||||||||
| C.1 | Yes | 69 | 69 | n/a | 78 | n/a | n/a | No | No | Yes | Deceased (d.70, HF) | |
| C.2 | No | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | No Medical Records |
| C.3 | Yes | 35 | 45 | 175.26 | 59 | 3.364278 | 40 | No | No | No | Alive (57) | HTN, hypothyroid |
| C.4 | Yes | 25 | 25 | 167.64 | 54 | 2.31695 | 25 | Yes | No | Yes | Alive (54) | MVP repair, breast neurofibromas, esophagitis, anemia |
| C.5 | No | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | Alive (unk.) | No medical records |
| C.6 | Yes | 35 | 35 | n/a | n/a | n/a | n/a | No | No | Yes | Deceased (d.42, SCD) | No medical records, reported affected |
| C.7 | Yes | 22 | 22 | n/a | n/a | n/a | 20 | No | No | Yes | Alive (30) | No medical records, reported affected |
| Pedigree D | ||||||||||||
| D.1 | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | Deceased (unk.) | No medical records |
| D.2 | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | Deceased (unk.) | No medical records |
| D.3 | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | Deceased (unk.) | No medical records |
| D.4 | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | Alive (unk.) | No medical records |
| D.5 | Yes | 35 | 35 | 144 | 70 | 7.591059 | 27.5 | No | No | No | Deceased (d.49, unk.) | |
| Pedigree E | ||||||||||||
| E.1 | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | Deceased (d.80) | No medical records |
| E.2 | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | Deceased (d.80) | No medical records |
| E.3 | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | Alive (unk.) | No medical records |
| E.4 | Yes | 60 | 60 | 158.75 | 55 | 3.031521 | 25 | No | No | Yes | Alive (77) | |
| E.5 | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | Alive (unk.) | No medical records |
| E.6 | Yes | 47 | 59 | 172.72 | 70 | 6.170558 | 20 | Yes | No | Yes | Deceased (d.75, unk.) | Cataracts, cholecystectomy, bladder suspension |
| E.7 | No | n/a | n/a | 178 | 47.7 | −1.86712 | 66 | No | No | No | Alive (53) | |
| E.8 | No | n/a | n/a | 157.48 | 48 | 0.950447 | 50 | No | No | No | Alive (49) | |
Abbreviations: Arrhyth, arrhythmia; DCM, dilated cardiomyopathy; HF, heart failure; Hrt Tx, heart transplant; HTN, hypertension; Hx, history; ICD, implantable cardioverter defibrillator; LVEF, left ventricular ejection fraction; LVIDD, LV internal dimension in diastole; MVP, mitral valve prolapse; n/a, not applicable; SCD, sudden cardiac death; unk, unknown; WPW, Wolff-Parkinson-White Syndrome.
Pedigree A
A paternally inherited p.(Glu191Lys) variant in SOS1 predicted to impact the HF domain was identified in a large family with severe, early-onset DCM and arrhythmia (Pedigree A). The SOS1 variant co-segregated with a truncating A-band TTNtv [p.(Val30827Serfs*22)], present in three of the most severely affected family members: (A.4), who died of heart failure at 57 years, and two of his sons (A.7, A.8), both of whom required transplantation at an early age (30 years and 14 years, respectively). An additional son (A.6) with a borderline low left ventricular ejection fraction (LVEF) of 48% but no other indications of DCM carried only the TTNtv, suggesting that this variant was unlikely to explain the totality of disease in affected family members. An additional maternally inherited truncating FLNC variant [p.(Val1643ThrfsTer26)] was identified in both A.7 and A.8, which also likely contributed to their DCM phenotype even though the remainder of the FLNC carriers in the maternal pedigree had less overt disease. Greatest disease severity - DCM requiring transplantation, implantable cardioverter defibrillator (ICD)-placement, or resulting in DCM-related death - was observed in individuals carrying all three variants.
Pedigree B
Two C-terminal SOS1 variants - p.(Ile607Thr), affecting the REM domain, and p.(Asp910His), impacting the CDC25 domain - were inherited in cis in a family with DCM and conduction system disease (Pedigree B). Both variants were carried by the proband (B.4), her severely affected son (B.6), who also had a history of Wolff-Parkinson-White (WPW) syndrome, and her clinically unaffected and possibly non-penetrant daughter (B.7). One additional family member with advanced DCM, arrhythmia, and a history of Doxorubicin treatment was identified (B.3); however, she did not carry either SOS1 variant. Because of her medical history, we are unable to rule out drug-induced cardiotoxicity as the cause of her DCM.
Pedigree C
A p.(Gly719Ala) variant predicted to affect the REM catalytic domain was identified in a female proband (C.4) with peripartum cardiomyopathy (PPCM), diagnosed at 25 years (Pedigree C). She underwent mitral valve replacement at 37 years following increasing severity of symptoms associated with mitral regurgitation and congestive heart failure. The proband’s father (C.1) died at 70 years from heart failure associated with DCM. DNA and medical records were unavailable for the proband’s sister (C.6), who reportedly died at 42 years from sudden cardiac death, as well as for her sister’s son (C.7), who was reported to have severe (LVEF~20%) early onset DCM diagnosed at 25 years. Echocardiographic records confirmed a diagnosis of DCM in the proband’s other affected sister at 39 years (C.3), but again DNA was unavailable.
Pedigree D
A p.(Arg744Gly) variant predicted to affect the REM domain was identified in a proband (D.5) with severe (Grade IV) DCM (Pedigree D). Family history was significant only for a paternal grandmother with reported heart failure at an early age. No additional family members were available for testing.
Pedigree E
A p.(Arg1201Trp) variant predicted to affect the PR tail domain was identified in two sisters with DCM and arrhythmia (Pedigree E). The proband (E.4) and her sister (E.6) both carried the SOS1 variant as well as a p.(Glu1926Asn) variant in MYH6. Absence of single variant carriers precluded analysis of individual variant impact.
Functional Studies of SOS1 Variants.
To determine the effect of each variant on RAS signaling pathways, wild-type and variant SOS1 constructs were expressed in HEK293T cells. Extracellular signal-regulated kinase (ERK), Protein kinase B (AKT), and c-Jun N-terminal kinase (JNK) activations were then determined by western blotting. Phosphorylated ERK (pERK) signals were undetectable in serum-starved cells, but increased following 15 min stimulation with epidermal growth factor (EGF) (Figure 3). Relative to wild-type SOS1 transfections, estimated fold changes in median pERK expression following 15 min EGF stimulation ranged between 1.5 and 2.1 for all six variants (p < 0.04). As expected, comparable increases were observed for both NS-associated variants used as positive controls for ERK activation [p.(Met269Arg) and p.(Glu846Lys)]15, 26 (p ≤ 0.01), but not for the suspected benign p.(Leu791Ile) variant (p = 0.36). In contrast, 95% confidence intervals supported lack of impact of identified SOS1 variants on pAKT (Figure S1) and pJNK (Figure S2) signaling. A <0.5 fold reduction in pJNK activation observed for the p.(Asp910His) variant was considered to be most likely related to reduced SOS1 expression observed in these samples (Figure S2).
Figure 3.
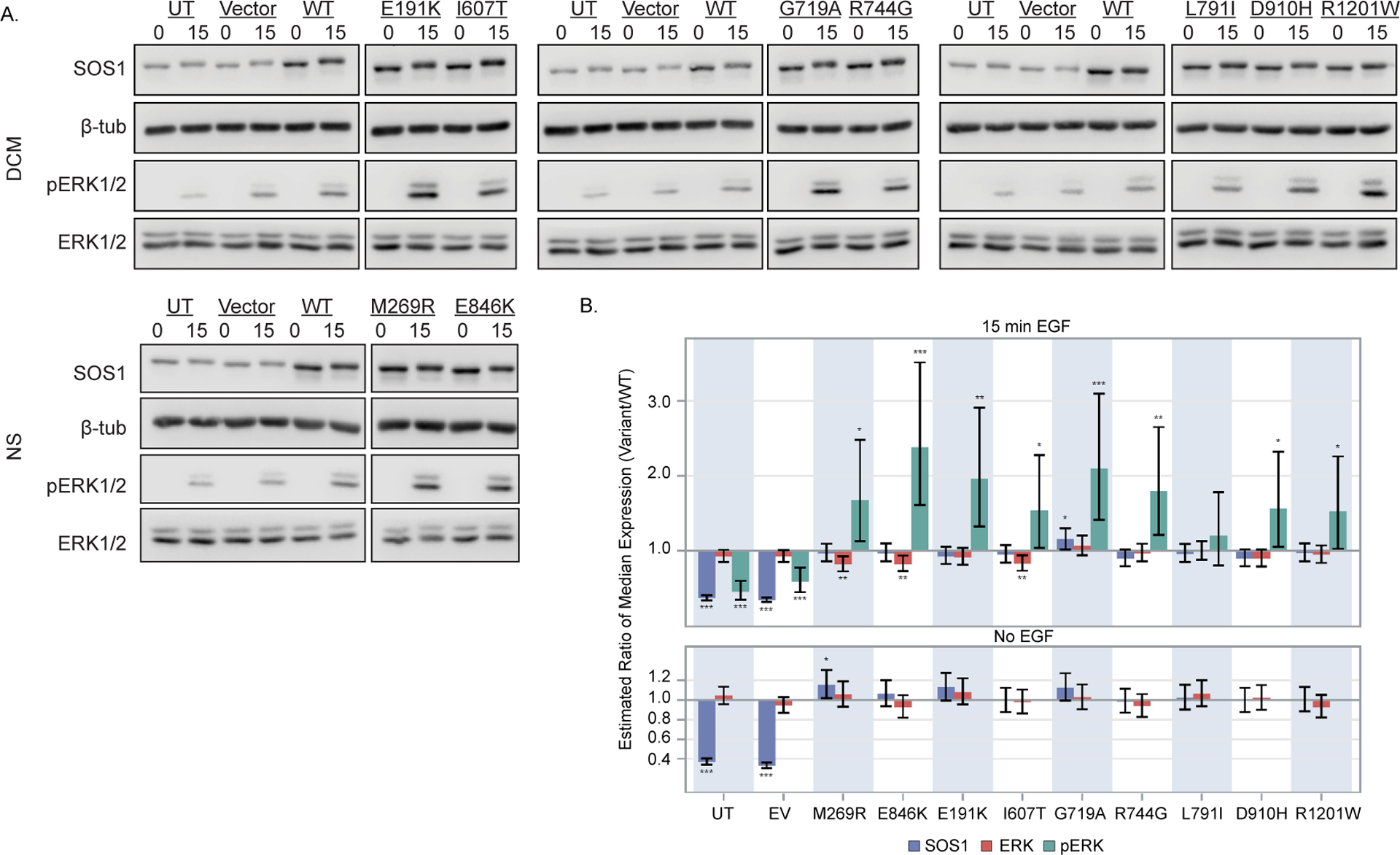
DCM-associated SOS1 variants exhibit gain of function effects on MAPK/ERK signaling. HEK293T cells expressing variant or wild-type (WT) SOS1 were serum starved for 16 hours and alternatively stimulated for 15 minutes with epidermal growth factor (EGF) (15) or left unstimulated (0). (A) Representative immunoblots for each SOS1 variant are shown. Total protein isolates were probed using antibodies specific to phosphorylated ERK (pERK, Cell Signaling #4695, 1:5,000), total ERK (ERK, Cell Signaling #4370, 1:5,000) and SOS1 (Abcam ab140621, 1:10,000). Levels of β-tubulin were used to control for loading (Abcam, ab6046, 1:20,000). Note that pERK and total ERK levels were determined using separate blots generated from equal quantities of the same protein lysate. For figure clarity, SOS1 and β-tubulin are shown for only the pERK blot. Full blots can be found in Figure S3. (B) Differences in ERK activation between each variant and WT SOS1 are shown as estimated ratios of median protein expression (variant/WT) at baseline (no-EGF) and 15 min EGF stimulation. Bars represent point estimates of this ratio from a linear mixed model fit to data from three independent experimental replicates for each variant, and error bars represent pointwise 95% confidence intervals for this ratio. *, **, and *** denote two-sided p ≤ 0.05, 0.01, and 0.001, respectively, for the null hypothesis that this ratio was 1. Relative to WT SOS1, elevated pERK expression was observed for both NS-associated variants used as positive ERK activation controls (M269R, E846K), as well as for all six DCM-associated SOS1 variants. ERK activation for a suspected benign variant detected in our cohort (L791I) did not differ from WT. Differences in pERK expression were not attributable to changes in total ERK or SOS1 expression levels.
Structural Analyses of SOS1 Variants
We next mapped each variant to crystal structures of native and RAS-bound SOS1 [Protein Data Bank (PDB) IDs: 3KSY, 1NVV] and modeled impact on higher level protein structure. Five of the SOS1 variants map to C-terminal domains, including three [p.(Ile607Thr), p.(Gly719Ala), p.(Arg744Gly)] in the REM, one in the CDC25 [p.(Asp910His)], and one in the PR tail [p.(Arg1201Trp)] (Figure 4, Figure 5A). One variant [p.(Glu191Lys)] mapped to the N-terminal HF.
Figure 4.
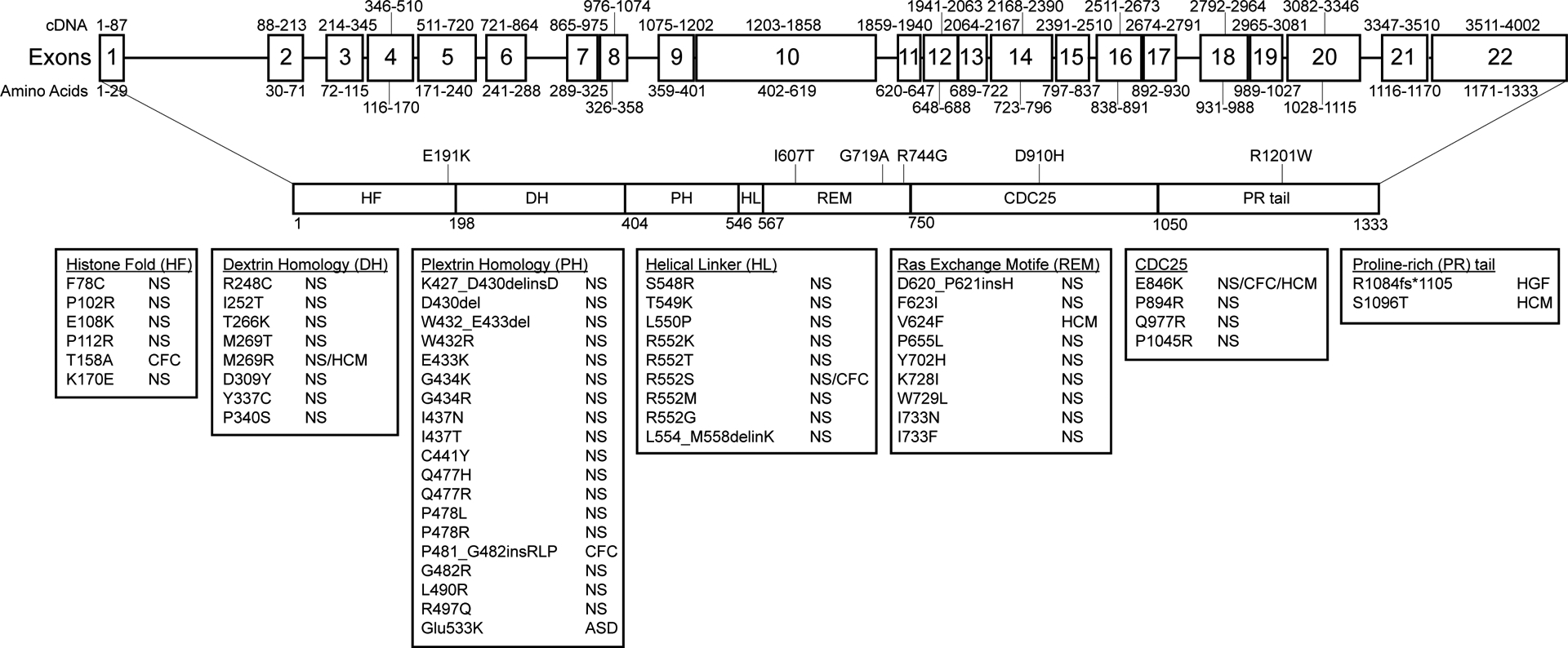
SOS1 gene and protein structure. The SOS1 gene and protein structure is shown. SOS1 is encoded by 22 exons spanning a total of 1333 amino acids and 6 functional domains roughly divided into two functional halves by a short helical linker. Positions and disease associations of reported SOS1 variants are indicated. DCM-associated variants identified in this study are indicated and summarized in Table 1. SOS1 natively exists in an auto-inhibited state that is regulated by a primary N-terminal regulatory unit comprised of a histone-like fold (HF) domain, a Dbl homology (DH) domain, and a pleckstrin homology (PH) domain. Together these domains prevent RAS binding and activation at the C-terminal domains when SOS1 is not actively bound at the plasma membrane. Membrane association is primarily carried out by the proline rich (PR) tail through direct interactions with SH3 domains of GRB2. Two other C-terminal domains, a RAS-exchange motif (REM) and a CDC25 domain, together act as the catalytic core of the SOS1 protein. Alleviation of auto-inhibition following membrane association frees the REM to bind to RAS. Conformational changes promote binding of a second RAS molecule at the adjacent CDC25 catalytic site initiating GDP/GTP exchange and RAS activation. Abbreviations: ASD, Atrial septal defect; CFC, Cardiofaciocutaneous syndrome; HCM, hypertrophic cardiomyopathy; HGF, hereditary gingival fibromatosis; NS, Noonan syndrome.
Figure 5.
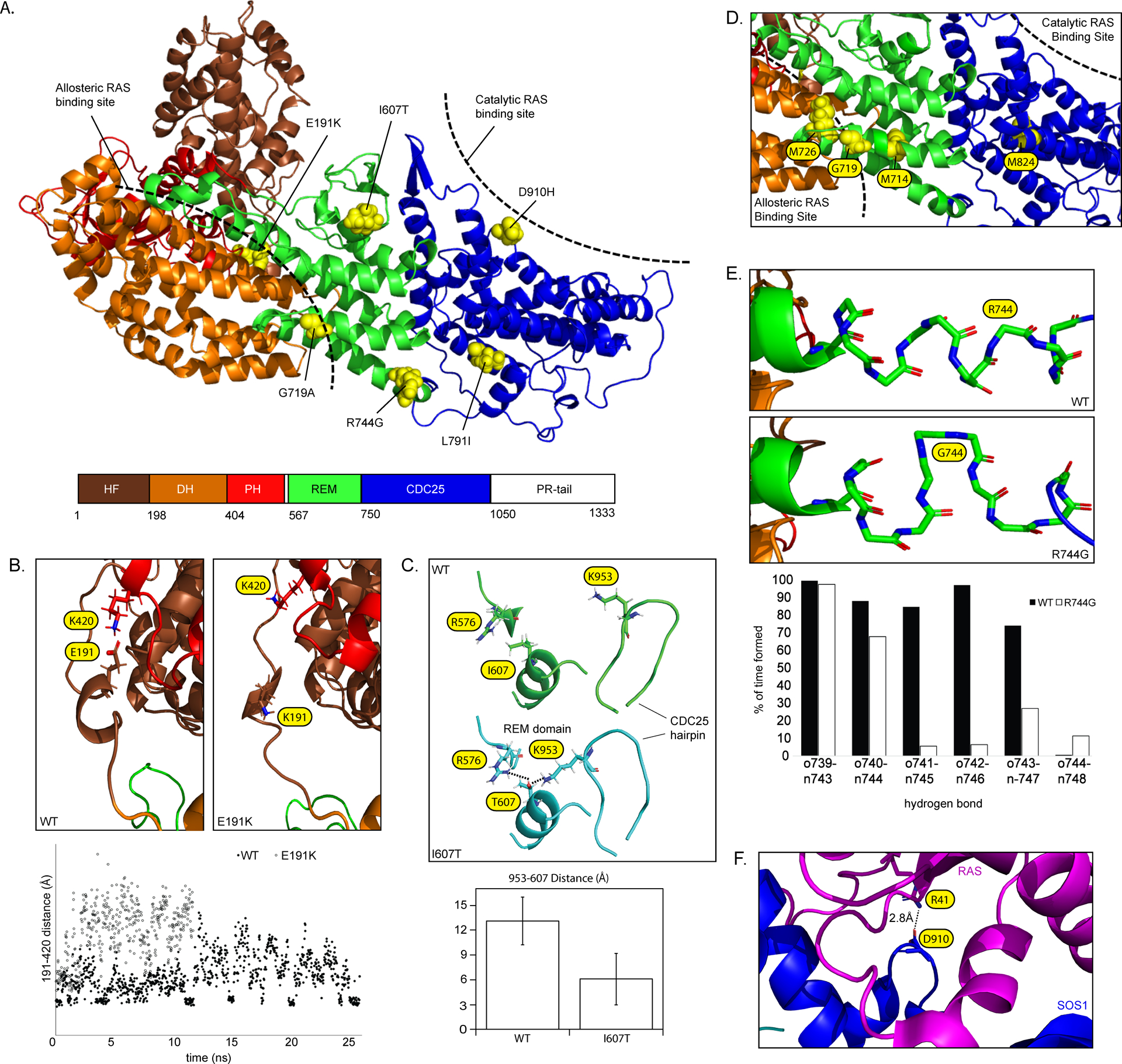
(A) SOS1 domain organization (PDB: 3KSY). Positions of DCM-associated variants identified in this study are indicated. Hatched bars indicate approximate allosteric and catalytic RAS binding sites. (B) Modeling of the wild-type SOS1 suggests the presence of an intermittent electrostatic association between Glu191 and Lys420 (evidenced by multiple short-lived 4 Å distance measurements) that is absent in models of p.(Glu191Lys). (C) Modeling of p.(Ile607Thr) suggests a hydrogen bond rearrangement between the region surrounding Ile607 and the neighboring CDC25 helical hairpin . The addition of an –OH group promotes interactions between both Arg576-Thr607 and Lys953-Thr607 (left) and reorients the CDC25 helical hairpin closer to the REM domain (right). The bar graph displays reduced average distance from the end of the sidechain of Lys953 to Thr607. Average distances with standard deviations represent 1100 independent measurements compiled over 25ns (D) p.(Gly719Ala) is positioned between the allosteric and catalytic RAS binding sites. Other amino acids within this same helix are involved in allosteric changes to the REM domain following RAS binding27. (E) Modeling of p.(Arg744Gly) demonstrates a localized unwinding of the surrounding helix by about 1 turn. (F) Asp910 is located at the SOS1-RAS interface and directly associates with Arg41 of the RAS Switch 1 region28.
The p.(Glu191Lys) variant present in Family A was predicted to impact an unstructured loop bridging the HF and DH domain (Figure 5B). Modeling of the wild-type protein suggests the presence of an intermittent electrostatic association between Glu191 and Lys420 (Figure 5B). This interaction is ablated in p.(Glu191Lys) models. As SOS1 autoinhibition depends on proper orientation of the DH-PH unit relative to the C-terminal catalytic core19, Glu191 may natively act to provide stability to SOS1 through its association with the PH domain. Introduction of the oppositely charged Lys residue may destabilize the wild-type interaction, shifting the loop away from the PH domain and inducing opening of the allosteric RAS binding site.
Molecular models support distinct structural mechanisms for the REM variants identified in Family B. Asp910 directly associates with Arg41 of the RAS Switch 1 region (Figure 5F)28 and borders amino acids important for interactions with Switch 229. While exact mechanisms underlying observed gain of function effects are not known, both positive28, 30 and negative31 impacts on nucleotide exchange rates have been described for RAS Switch 1 variants, highlighting the functional complexity of RAS-SOS1 interactions in this region. Structural modeling of p.(Ile607Thr) supported a hydrogen bond rearrangement between the region surrounding Ile607 and the neighboring CDC25 helical hairpin (Figure 5C). Specifically, the addition of an –OH group was found to promote interactions between both Arg576-Thr607 and Lys953-Thr607. This linking has the effect of repositioning the CDC25 helical hairpin slightly closer to the REM domain. As this area constitutes the primary interface between the REM and CDC2528, and acts as a hinge point for paired rotations of the REM and CDC25 following RAS binding23, 32, this altered hydrogen bond network may promote exposure of the previously occluded catalytic site32.
The REM variants detected in Family C [p.(Gly719Ala)] and D [p.(Arg744Gly)] are predicted to occupy adjacent helices bridging the catalytic and allosteric RAS binding domains. Based on this positioning, neither variant was expected to directly interact with the DH domain or with bound RAS. Nevertheless, nuclear magnetic resonance (NMR) studies have suggested involvement of nearby Met714 in dynamic conformational changes following RAS binding27 (Figure 5D). A functional role for residues of this helix is further supported by elevated ERK signaling and nucleotide exchange rates33 for p.(Tyr702His), a NS-associated variant located at the REM-CDC25 interface. As Arg744 is similarly positioned, it is plausible that p.(Arg744Gly) may also impact SOS1 function by disrupting allosteric communication between the REM and CDC25. While no further structural insights were gained from molecular modeling of p.(Gly719Ala), p.(Arg744Gly) consistently demonstrated a localized unwinding of the helix about 1 turn earlier than in wild-type models (Figure 5E), suggesting local dynamics of this region may play a role in RAS signaling.
The final SOS1 variant, p.(Arg1201Trp), found in Family E, was predicted to impact a highly conserved residue positioned between the second and third of four GRB2 consensus binding motifs within the PR tail. This region adopts a disordered random-coil confirmation in solution34 and cannot be accurately modeled using all-atom simulations. Nevertheless, multiple genetic and functional studies support roles for the PR tail in negatively regulating SOS1 activity35, 36. Thus, the gain of function effect of the Arg1201Trp mutant, while structurally unexplained, is consistent with known functions of the SOS1 C-terminal domain.
Discussion
Exome sequencing completed on 412 individuals with isolated DCM, including 281 probands and 131 of their affected relatives, identified six SOS1 coding variants spanning five families. Detected variants were tested for impact on RAS signaling pathways and were found to result in gain of function effects on ERK signaling with minimal impact on AKT and JNK pathways. Activations were of comparable magnitude to those obtained using two NS-associated variants with previously recognized pathogenicity and functional impact (Table S1). These results extend previous studies4–6 to strengthen the general evidence that dysregulated ERK activation plays a role in pathogenesis of DCM.
This study is one of only a few others11–13 to directly assess SOS1 genetic variation in the context of non-syndromic cardiomyopathy. To date, all reported DCM-associated SOS1 variants, including those reported in this study, have been classified as VUSs (Table S2). Interestingly, several previously reported variants impacted residues located close to functionally significant residues identified in our cohort. In our study, five of six detected SOS1 variants affected C-terminal residues: three in the REM domain [p.(Ile607Thr), p.(Gly719Ala), p.(Arg744Gly), one in the CDC25 domain [p.(Asp910His)], and one in the PR tail [p.(Arg1201Trp)]. While the limited number of identified variants precludes definitive genotype-phenotype determinations, only 18 of the 60 SOS1 missense/nonsense variants currently documented in the Human Gene Mutation Database (HGMD), most of which are NS-associated, localize to C-terminal domains.
A role for activated ERK signaling in development of DCM has previously been established by studies of both failing human hearts and mutant mice models4–6. A number of these animal models impact genes encoding nuclear envelope proteins with known importance to human DCM, including LMNA (lamin A/C) and SYNE1 (nesprin-1). While the exact mechanisms underlying ERK activation are uncertain, it has been hypothesized that RAS signaling may be disrupted through effects on nuclear envelope organization and transport rather than through direct influence on upstream signaling components6. Evidence supporting an alternative, but not necessarily mutually exclusive, mechanism suggests that hyperactivated pERK can act to alter sarcomeric organization through increased phosphorylation and activation of the actin depolymerization factor, cofillin-137. Whether similar disruption of sarcomere actin dynamics are seen with other ERK activating DCM models outside of LMNA remains an untested but important question.
While no consistent changes were seen in AKT and JNK signaling in this study, components of each of these pathways have been reported to be upregulated in dilated human hearts38, 39. This broader signaling complexity is further suggested by results from functional models of RAF18, which implicate AKT activation in development of early onset DCM, and from Tyrosine-protein phosphatase non-receptor type 11 (Ptpn11) mouse models, which develop myocardial thinning evocative of DCM in a setting of reduced ERK and elevated AKT40, 41. Because DCM is only infrequently seen in NS42–44, its occurrence in Ptpn11 mouse models is intriguing. Affected animals develop DCM at 6–8 weeks without any evidence of the cardiac hypertrophy commonly seen in NS. It would be logical to hypothesize that because loss of Ptpn11 function results in DCM, gain of function variants might be associated with HCM in a manner analogous to knock-in models of NS-associated Sos145 and Raf146 variants. However, at least some Ptpn11 knock-in mice models exhibit myocardial thinning evocative of DCM40, 41, 47. These models underscore the fact that we do not yet have a complete picture as to how abnormal ERK signaling acts to promote particular myopathic responses. Recent studies from the Molkentin group48, 49 have supported a role for increased ERK signaling in concentric hypertrophy and decreased ERK signaling in eccentric hypertrophy, while others46, 50 describe pathological eccentric hypertrophy in the context of ERK hyperactivation. One possible explanation for such discrepancies has been raised: ERK activation in other cell types apart from cardiomyocytes is likely to play in a role in shaping the final hypertrophic response. Cardiac fibroblasts, for example, are relevant for cardiomyocyte hypertrophy51 and Raf1L613V knock-in mice, which express the mutant allele in both cell types, develop an eccentric cardiomyopathy characterized by ERK hyperactivation and correctable by postnatal inhibition46, 50. Alongside other emerging studies52, 53, these data suggest that myocyte-focused views of hypertrophy may be too narrow to adequately explain disease mechanisms underlying RASopathies and their associated cardiomyopathies.
Limitations
Because many study participants were recruited from cardiovascular rather than general genetics clinics, subtle findings suggestive of underlying RASopathies may have been missed during evaluation. While it remains a formal possibility that the MYH6 p.(Gly1826Asn) variant detected in Family E may be disease-relevant, a definitive role for MYH6 in DCM has yet to be established and previous reports have considered this variant to be of unlikely or uncertain pathogenic significance54, 55. Current American College of Medical Genetics (ACMG) / Clinical Genome Resource (ClinGen) criteria for variant adjudication focus heavily on single-gene Mendelian paradigms25 and variant classifications will continue to be refined as standards evolve to incorporate a more nuanced understanding of complex disease patterns. It is important to note that ERK activations reported in this study reflect results obtained from a single in vitro system. Additional testing using patient samples or other cell or animal based functional assays will be required before activated ERK signaling can be definitively established as a pathogenic mechanism in these families. Because the identities of precise ERK targets relevant to DCM remain uncertain, these studies will be of great help in determining how aberrant activation of ERK signaling in the heart may differ from pathologic activation in other cell or organ systems.
Conclusions
Our results provide evidence supporting gain of function SOS1 genetic variation as a contributing factor to isolated DCM. Whether the majority of DCM-associated SOS1 variants are sufficient to cause DCM in isolation or more commonly act as genetic modifiers alongside other causative variants remains to be determined. Nevertheless, observed signaling profiles in this study paralleled those previously described for other pathogenic SOS1 variants. While preliminary, these data reflect a well-recognized but poorly understood role for ERK signaling in DCM. By what cellular mechanisms might signaling changes imparted by SOS1 variants promote development of DCM? What are the identities of any relevant downstream signaling targets and how might these differ from those involved in other forms of SOS1-related RASopathy? As clinical and animal-based trials of pathway-specific MAPK inhibitors have already demonstrated therapeutic potential for treatment of DCM, these questions raise intriguing possibilities for precision-medicine approaches to patient care. Large-scale, family-based DCM studies are underway56 and will be invaluable in driving novel gene discovery.
Supplementary Material
Acknowledgments:
We thank all probands and family members for their ongoing participation as well as Dr. Benjamin Neel (New York University Medical Center, NYUMC) for providing pBABE-puro expression vectors. This study was supported in part by computational infrastructure provided by The Ohio State University Division of Human Genetics Data Management Platform and the Ohio Supercomputer Center.
Sources of Funding: NIH RO1 HL58626 and R01 HL128857 (both to Dr Hershberger), and 5 M01 RR000334. Sequencing was provided by the University of Washington Center for Mendelian Genomics (UW-CMG) and was funded by National Human Genome Research Institute (NHGRI) and National Heart, Lung, and Blood Institute (NHLBI) grants UM1 HG006493 and U24 HG008956. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health (NIH).
Nonstandard Abbreviations and Acronyms:
- AKT
Protein kinase B
- ACMG
American College of Medical Genetics
- CFCS
Cardiofaciocutaneous syndrome
- ClinGen
Clinical Genome Resource
- DH
Dbl homology
- DCM
Dilated cardiomyopathy
- LVEF
Left ventricular ejection fraction
- EGF
Epidermal growth factor
- ERK
Extracellular signal-regulated kinase
- ExAC
Exome Aggregation Consortium (ExAC)
- FLNC
Filamin C
- GEF
Guanine exchange factor
- gnomAD
Genome Aggregation Database
- GRB2
Growth factor receptor-bound protein 2
- HCM
Hypertrophic cardiomyopathy
- HGMD
Human Gene Mutation Database
- HF
Histone-like fold
- ICD
Implantable cardioverter defibrillator
- JNK
c-Jun N-terminal kinase
- LMNA
Lamin A/C
- MAPK
Mitogen-activated protein kinase
- MIM
Mendelian Inheritance in Man
- MYH6
Myosin heavy chain 6
- NMR
Nuclear magnetic resonance
- NS
Noonan syndrome
- PDB
Protein Data Bank
- PH
Pleckstrin homology
- PPCM
Peripartum cardiomyopathy
- PR
Proline-rich
- PTPN11
Tyrosine-protein phosphatase non-receptor type 11
- REM
RAS-exchange motif
- SOS1
Son of Sevenless
- SYNE1
Nesprin-1
- TTN
Titin
- TTNtv
Titin truncating variant
- VUS
Variant of uncertain significance
- WPW
Wolff-Parkinson-White
Footnotes
Disclosures: None.
References.
- 1.Hershberger RE, Hedges DJ, Morales A. Dilated cardiomyopathy: the complexity of a diverse genetic architecture. Nat Rev Cardiol. 2013;10:531–47. [DOI] [PubMed] [Google Scholar]
- 2.McNally EM, Mestroni L. Dilated Cardiomyopathy: Genetic Determinants and Mechanisms. Circ Res. 2017;121:731–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pugh TJ, Kelly MA, Gowrisankar S, Hynes E, Seidman MA, Baxter SM, Bowser M, Harrison B, Aaron D, Mahanta LM, et al. The landscape of genetic variation in dilated cardiomyopathy as surveyed by clinical DNA sequencing. Genet Med. 2014;16:601–8. [DOI] [PubMed] [Google Scholar]
- 4.Muchir A, Pavlidis P, Decostre V, Herron AJ, Arimura T, Bonne G, Worman HJ. Activation of MAPK pathways links LMNA mutations to cardiomyopathy in Emery-Dreifuss muscular dystrophy. J Clin Invest. 2007;117:1282–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Muchir A, Reilly SA, Wu W, Iwata S, Homma S, Bonne G, Worman HJ. Treatment with selumetinib preserves cardiac function and improves survival in cardiomyopathy caused by mutation in the lamin A/C gene. Cardiovasc Res. 2012;93:311–9. [DOI] [PubMed] [Google Scholar]
- 6.Zhou C, Li C, Zhou B, Sun H, Koullourou V, Holt I, Puckelwartz MJ, Warren DT, Hayward R, Lin Z, et al. Novel nesprin-1 mutations associated with dilated cardiomyopathy cause nuclear envelope disruption and defects in myogenesis. Hum Mol Genet. 2017;26:2258–2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arimura T, Helbling-Leclerc A, Massart C, Varnous S, Niel F, Lacene E, Fromes Y, Toussaint M, Mura AM, Keller DI, et al. Mouse model carrying H222P-Lmna mutation develops muscular dystrophy and dilated cardiomyopathy similar to human striated muscle laminopathies. Hum Mol Genet. 2005;14:155–69. [DOI] [PubMed] [Google Scholar]
- 8.Dhandapany PS, Razzaque MA, Muthusami U, Kunnoth S, Edwards JJ, Mulero-Navarro S, Riess I, Pardo S, Sheng J, Rani DS, et al. RAF1 mutations in childhood-onset dilated cardiomyopathy. Nat Genet. 2014;46:635–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rose BA, Force T, Wang Y. Mitogen-activated protein kinase signaling in the heart: angels versus demons in a heart-breaking tale. Physiol Rev. 2010;90:1507–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baban A, Olivini N, Lepri FR, Cali F, Mucciolo M, Digilio MC, Calcagni G, di Mambro C, Dallapiccola B, Adorisio R, et al. SOS1 mutations in Noonan syndrome: Cardiomyopathies and not only congenital heart defects! Report of six patients including two novel variants and literature review. Am J Med Genet A. 2019;179:2083–2090. [DOI] [PubMed] [Google Scholar]
- 11.Ceyhan-Birsoy O, Miatkowski MM, Hynes E, Funke BH, Mason-Suares H. NGS testing for cardiomyopathy: Utility of adding RASopathy-associated genes. Hum Mutat. 2018;39:954–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aljeaid D, Sanchez AI, Wakefield E, Chadwell SE, Moore N, Prada CE, Zhang W. Prevalence of pathogenic and likely pathogenic variants in the RASopathy genes in patients who have had panel testing for cardiomyopathy. Am J Med Genet A. 2019;179:608–614. [DOI] [PubMed] [Google Scholar]
- 13.Kaski JP, Syrris P, Shaw A, Alapi KZ, Cordeddu V, Esteban MT, Jenkins S, Ashworth M, Hammond P, Tartaglia M, et al. Prevalence of sequence variants in the RAS-mitogen activated protein kinase signaling pathway in pre-adolescent children with hypertrophic cardiomyopathy. Circ Cardiovasc Genet. 2012;5:317–26. [DOI] [PubMed] [Google Scholar]
- 14.Jang SI, Lee EJ, Hart PS, Ramaswami M, Pallos D, Hart TC. Germ line gain of function with SOS1 mutation in hereditary gingival fibromatosis. J Biol Chem. 2007;282:20245–55. [DOI] [PubMed] [Google Scholar]
- 15.Roberts AE, Araki T, Swanson KD, Montgomery KT, Schiripo TA, Joshi VA, Li L, Yassin Y, Tamburino AM, Neel BG, et al. Germline gain-of-function mutations in SOS1 cause Noonan syndrome. Nat Genet. 2007;39:70–4. [DOI] [PubMed] [Google Scholar]
- 16.Yadav KK, Bar-Sagi D. Allosteric gating of Son of sevenless activity by the histone domain. Proc Natl Acad Sci U S A. 2010;107:3436–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Corbalan-Garcia S, Margarit SM, Galron D, Yang SS, Bar-Sagi D. Regulation of Sos activity by intramolecular interactions. Mol Cell Biol. 1998;18:880–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gureasko J, Kuchment O, Makino DL, Sondermann H, Bar-Sagi D, Kuriyan J. Role of the histone domain in the autoinhibition and activation of the Ras activator Son of Sevenless. Proc Natl Acad Sci U S A. 2010;107:3430–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sondermann H, Soisson SM, Boykevisch S, Yang SS, Bar-Sagi D, Kuriyan J. Structural analysis of autoinhibition in the Ras activator Son of sevenless. Cell. 2004;119:393–405. [DOI] [PubMed] [Google Scholar]
- 20.Lepri F, De Luca A, Stella L, Rossi C, Baldassarre G, Pantaleoni F, Cordeddu V, Williams BJ, Dentici ML, Caputo V, et al. SOS1 mutations in Noonan syndrome: molecular spectrum, structural insights on pathogenic effects, and genotype-phenotype correlations. Hum Mutat. 2011;32:760–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li N, Batzer A, Daly R, Yajnik V, Skolnik E, Chardin P, Bar-Sagi D, Margolis B, Schlessinger J. Guanine-nucleotide-releasing factor hSos1 binds to Grb2 and links receptor tyrosine kinases to Ras signalling. Nature. 1993;363:85–8. [DOI] [PubMed] [Google Scholar]
- 22.Chen RH, Corbalan-Garcia S, Bar-Sagi D. The role of the PH domain in the signal-dependent membrane targeting of Sos. EMBO J. 1997;16:1351–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Margarit SM, Sondermann H, Hall BE, Nagar B, Hoelz A, Pirruccello M, Bar-Sagi D, Kuriyan J. Structural evidence for feedback activation by Ras.GTP of the Ras-specific nucleotide exchange factor SOS. Cell. 2003;112:685–95. [DOI] [PubMed] [Google Scholar]
- 24.Morales A, Kinnamon DD, Jordan E, Platt J, Vatta M, Dorschner MO, Starkey CA, Mead JO, Ai T, Burke W, et al. Variant Interpretation for Dilated Cardiomyopathy: Refinement of the American College of Medical Genetics and Genomics/ClinGen Guidelines for the DCM Precision Medicine Study. Circ Genom Precis Med. 2020;13:e002480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Longoni M, Moncini S, Cisternino M, Morella IM, Ferraiuolo S, Russo S, Mannarino S, Brazzelli V, Coi P, Zippel R, et al. Noonan syndrome associated with both a new Jnk-activating familial SOS1 and a de novo RAF1 mutations. Am J Med Genet A. 2010;152A:2176–84. [DOI] [PubMed] [Google Scholar]
- 27.Vo U, Vajpai N, Flavell L, Bobby R, Breeze AL, Embrey KJ, Golovanov AP. Monitoring Ras Interactions with the Nucleotide Exchange Factor Son of Sevenless (Sos) Using Site-specific NMR Reporter Signals and Intrinsic Fluorescence. J Biol Chem. 2016;291:1703–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hall BE, Yang SS, Boriack-Sjodin PA, Kuriyan J, Bar-Sagi D. Structure-based mutagenesis reveals distinct functions for Ras switch 1 and switch 2 in Sos-catalyzed guanine nucleotide exchange. J Biol Chem. 2001;276:27629–37. [DOI] [PubMed] [Google Scholar]
- 29.Evelyn CR, Duan X, Biesiada J, Seibel WL, Meller J, Zheng Y. Rational design of small molecule inhibitors targeting the Ras GEF, SOS1. Chem Biol. 2014;21:1618–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van den Berghe N, Cool RH, Wittinghofer A. Discriminatory residues in Ras and Rap for guanine nucleotide exchange factor recognition. J Biol Chem. 1999;274:11078–85. [DOI] [PubMed] [Google Scholar]
- 31.Maurer T, Garrenton LS, Oh A, Pitts K, Anderson DJ, Skelton NJ, Fauber BP, Pan B, Malek S, Stokoe D, et al. Small-molecule ligands bind to a distinct pocket in Ras and inhibit SOS-mediated nucleotide exchange activity. Proc Natl Acad Sci U S A. 2012;109:5299–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Freedman TS, Sondermann H, Friedland GD, Kortemme T, Bar-Sagi D, Marqusee S, Kuriyan J. A Ras-induced conformational switch in the Ras activator Son of sevenless. Proc Natl Acad Sci U S A. 2006;103:16692–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Smith MJ, Neel BG, Ikura M. NMR-based functional profiling of RASopathies and oncogenic RAS mutations. Proc Natl Acad Sci U S A. 2013;110:4574–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McDonald CB, Bhat V, Kurouski D, Mikles DC, Deegan BJ, Seldeen KL, Lednev IK, Farooq A. Structural landscape of the proline-rich domain of Sos1 nucleotide exchange factor. Biophys Chem. 2013;175–176:54–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee YK, Low-Nam ST, Chung JK, Hansen SD, Lam HYM, Alvarez S, Groves JT. Mechanism of SOS PR-domain autoinhibition revealed by single-molecule assays on native protein from lysate. Nature Communications. 2017;8:15061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zarich N, Oliva JL, Martinez N, Jorge R, Ballester A, Gutierrez-Eisman S, Garcia-Vargas S, Rojas JM. Grb2 is a negative modulator of the intrinsic Ras-GEF activity of hSos1. Mol Biol Cell. 2006;17:3591–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chatzifrangkeskou M, Yadin D, Marais T, Chardonnet S, Cohen-Tannoudji M, Mougenot N, Schmitt A, Crasto S, Di Pasquale E, Macquart C, et al. Cofilin-1 phosphorylation catalyzed by ERK1/2 alters cardiac actin dynamics in dilated cardiomyopathy caused by lamin A/C gene mutation. Hum Mol Genet. 2018;27:3060–3078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Takeishi Y, Huang Q, Abe J, Che W, Lee JD, Kawakatsu H, Hoit BD, Berk BC, Walsh RA. Activation of mitogen-activated protein kinases and p90 ribosomal S6 kinase in failing human hearts with dilated cardiomyopathy. Cardiovasc Res. 2002;53:131–7. [DOI] [PubMed] [Google Scholar]
- 39.Haq S, Choukroun G, Lim H, Tymitz KM, del Monte F, Gwathmey J, Grazette L, Michael A, Hajjar R, Force T, et al. Differential activation of signal transduction pathways in human hearts with hypertrophy versus advanced heart failure. Circulation. 2001;103:670–7. [DOI] [PubMed] [Google Scholar]
- 40.Tajan M, Batut A, Cadoudal T, Deleruyelle S, Le Gonidec S, Saint Laurent C, Vomscheid M, Wanecq E, Treguer K, De Rocca Serra-Nedelec A, et al. LEOPARD syndrome-associated SHP2 mutation confers leanness and protection from diet-induced obesity. Proc Natl Acad Sci U S A. 2014;111:E4494–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Araki T, Chan G, Newbigging S, Morikawa L, Bronson RT, Neel BG. Noonan syndrome cardiac defects are caused by PTPN11 acting in endocardium to enhance endocardial-mesenchymal transformation. Proc Natl Acad Sci U S A. 2009;106:4736–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kurose A, Oyama K, Murakami Y, Ohyama K, Segawa I, Sawai T. Dilated cardiomyopathy in Noonan’s syndrome: a first autopsy case. Hum Pathol. 2000;31:764–7. [DOI] [PubMed] [Google Scholar]
- 43.Shimizu A, Oku Y, Matsuo K, Hashiba K. Hypertrophic cardiomyopathy progressing to a dilated cardiomyopathy-like feature in Noonan’s syndrome. Am Heart J. 1992;123:814–6. [DOI] [PubMed] [Google Scholar]
- 44.Yu CM, Chow LT, Sanderson JE. Dilated cardiomyopathy in Noonan’s syndrome. Int J Cardiol. 1996;56:83–5. [DOI] [PubMed] [Google Scholar]
- 45.Chen PC, Wakimoto H, Conner D, Araki T, Yuan T, Roberts A, Seidman C, Bronson R, Neel B, Seidman JG, et al. Activation of multiple signaling pathways causes developmental defects in mice with a Noonan syndrome-associated Sos1 mutation. J Clin Invest. 2010;120:4353–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wu X, Simpson J, Hong JH, Kim KH, Thavarajah NK, Backx PH, Neel BG, Araki T. MEK-ERK pathway modulation ameliorates disease phenotypes in a mouse model of Noonan syndrome associated with the Raf1(L613V) mutation. J Clin Invest. 2011;121:1009–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Araki T, Mohi MG, Ismat FA, Bronson RT, Williams IR, Kutok JL, Yang W, Pao LI, Gilliland DG, Epstein JA, et al. Mouse model of Noonan syndrome reveals cell type- and gene dosage-dependent effects of Ptpn11 mutation. Nat Med. 2004;10:849–57. [DOI] [PubMed] [Google Scholar]
- 48.Davis J, Davis LC, Correll RN, Makarewich CA, Schwanekamp JA, Moussavi-Harami F, Wang D, York AJ, Wu H, Houser SR, et al. A Tension-Based Model Distinguishes Hypertrophic versus Dilated Cardiomyopathy. Cell. 2016;165:1147–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kehat I, Davis J, Tiburcy M, Accornero F, Saba-El-Leil MK, Maillet M, York AJ, Lorenz JN, Zimmermann WH, Meloche S, et al. Extracellular signal-regulated kinases 1 and 2 regulate the balance between eccentric and concentric cardiac growth. Circ Res. 2011;108:176–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yin JC, Platt MJ, Tian X, Wu X, Backx PH, Simpson JA, Araki T, Neel BG. Cellular interplay via cytokine hierarchy causes pathological cardiac hypertrophy in RAF1-mutant Noonan syndrome. Nat Commun. 2017;8:15518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Thum T, Gross C, Fiedler J, Fischer T, Kissler S, Bussen M, Galuppo P, Just S, Rottbauer W, Frantz S, et al. MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature. 2008;456:980–4. [DOI] [PubMed] [Google Scholar]
- 52.Lauriol J, Cabrera JR, Roy A, Keith K, Hough SM, Damilano F, Wang B, Segarra GC, Flessa ME, Miller LE, et al. Developmental SHP2 dysfunction underlies cardiac hypertrophy in Noonan syndrome with multiple lentigines. J Clin Invest. 2016;126:2989–3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Josowitz R, Mulero-Navarro S, Rodriguez NA, Falce C, Cohen N, Ullian EM, Weiss LA, Rauen KA, Sobie EA, Gelb BD. Autonomous and Non-autonomous Defects Underlie Hypertrophic Cardiomyopathy in BRAF-Mutant hiPSC-Derived Cardiomyocytes. Stem Cell Reports. 2016;7:355–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Carniel E, Taylor MR, Sinagra G, Di Lenarda A, Ku L, Fain PR, Boucek MM, Cavanaugh J, Miocic S, Slavov D, et al. Alpha-myosin heavy chain: a sarcomeric gene associated with dilated and hypertrophic phenotypes of cardiomyopathy. Circulation. 2005;112:54–9. [DOI] [PubMed] [Google Scholar]
- 55.Hershberger RE, Norton N, Morales A, Li D, Siegfried JD, Gonzalez-Quintana J. Coding sequence rare variants identified in MYBPC3, MYH6, TPM1, TNNC1, and TNNI3 from 312 patients with familial or idiopathic dilated cardiomyopathy. Circ Cardiovasc Genet. 2010;3:155–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kinnamon DD, Morales A, Bowen DJ, Burke W, Hershberger RE, Consortium* DCM. Toward Genetics-Driven Early Intervention in Dilated Cardiomyopathy: Design and Implementation of the DCM Precision Medicine Study. Circ Cardiovasc Genet. 2017;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.