

Assessing protein trafficking in live cells is a core strength of fluorogen activating protein technology. The rapid and specific labeling of a tagged protein of interest, intracellular or surface, with a variety of fluorogenic dye derivatives creates a toolset suitable for scalable, dynamic multi‐color fluorescence experiments and direct quantitative trafficking‐related measurements in single cells and cell populations, using fluorometry, flow cytometry and microscopy.
Keywords: fluorogen activating proteins (FAP), fluorogenic dye, protein trafficking
Abstract
Throughout the past decade the use of fluorogen activating proteins (FAPs) has expanded with several unique reporter dyes that support a variety of methods to specifically quantify protein trafficking events. The platform's capabilities have been demonstrated in several systems and shared for widespread use. This review will highlight the current FAP labeling techniques for protein traffic measurements and focus on the use of the different designed fluorogenic dyes for selective and specific labeling applications.
1. INTRODUCTION
There is a large, growing catalog of protein‐fluorogenic labeling techniques for biologists to use; however, picking which one is best suited for individual research needs, on the small or large scale, is not a simple choice. A single genetically encoded tag, labeled with distinct chemical dyes, can perform different “functional” measurements. Hence, selection of the right dye for the measurement, and use of the right dye and labeling protocol, provides a new experimental avenue to design labeling approaches that generate specific quantitative biological information. There have been several excellent recent reviews on fluorescent and fluorogenic labeling techniques,1, 2, 3, 4, 5, 6, 7, 8, 9 which describe strengths and weaknesses of different methods. Yan and Bruchez, and more recently Xu and Hu have provided an extensive overview of fluorogen activating protein (FAP) technology, broadly. This review discusses the “right dye, right time” experimental design capability of the FAP platform a wide range of different protein trafficking measurements.
Important cell surface proteins, such as receptors and ion channels, are finely controlled in their expression at the cell surface, where they perform significant physiological functions and responses.10, 11, 12 Aberrant cellular trafficking can lead to altered activity and thereby pathological disease states. Understanding the regulated trafficking of these crucial surface proteins is important for elucidating function and correcting disease conditions. Studying plasma membrane (PM) protein dynamics has been revolutionized by using fluorescence, either by labeling with fluorophore conjugated antibodies, pH sensitive green fluorescent protein (GFP) tags, or use of chemical tags such as the SNAP tag, CLIP tag, TMP tag, Halo tag, FAP tag or FLAG (antibody epitope tag), to specifically and quantitatively label surface proteins.13, 14, 15, 16, 17, 18 However, there is a clinical urgency for drugs to treat trafficking‐associated disorders, such as cystic fibrosis, cardiac arrhythmias and some lysosome storage disorders where high‐throughput small‐molecule drug discovery screens have been on‐going to find “hits” that have a desirable effect on a specific disease state. Historically, due to the constraints of direct trafficking detection methods, the most common approach to determine a target effect is through using tools that measure functional changes in protein activity, such as downstream signaling or alterations in ion‐transport. These drug screens do not differentiate between a direct effect on protein activity or regulation of protein trafficking.
Activity of cell‐surface proteins is dependent on both the properties of the protein and the density of protein at the plasma membrane. Being able to independently (and potentially simultaneously) assess activity and trafficking provides a significantly clarified interpretation of the basis of action for genetic and pharmacological perturbations (Figure 1). To include such measurements in high‐throughput screens, detection methods must be readily adaptable to large scale assays, without prohibitive costs or lengthy multi‐step procedures. These requirements are met by the fluorogen activating protein platform, thanks to the exceptional activation of the fluorescence signal upon binding to the tagged target protein, which eliminates the usually required washing steps for fluorescence labeling methods. A number of strategies have been developed for large‐scale measurements of cell surface protein transit using these properties and the recently developed functionally diverse fluorogen dyes.
Figure 1.
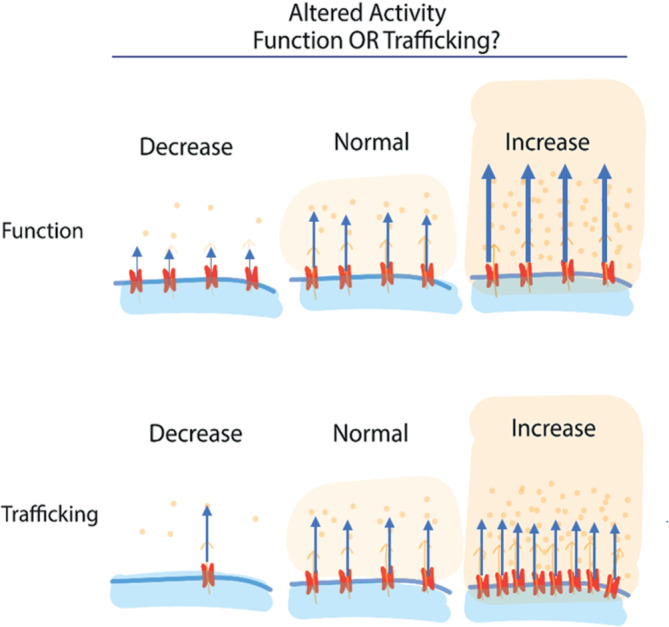
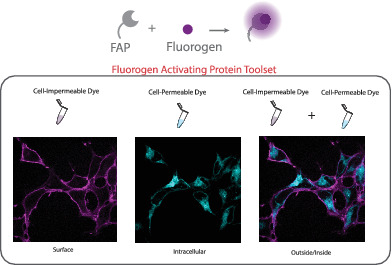
Overall activity can be a result of altered protein function, altered protein expression at a target site (eg, the plasma membrane), or a combination of the two effects
2. FLUOROGEN ACTIVATING PROTEIN PLATFORM
FAP technology was introduced in 2008 as a new class of protein‐dye reporters, combining the selectivity of a genetically encoded single chain antibody ≤25 kDa tag (FAP) with specific fluorogenic dyes, which are nonfluorescent until bound to their cognate FAP.19 The fluorogenic attribute allows for the elimination of wash‐steps for removal of unbound dye. The ability to design and synthesize cell‐permeant and impermeant fluorogenic dyes for cognate receptor tags with distinct colors has made assessing protein trafficking in live cells a core application of the FAP‐fluorogen technology.20, 21 The rapid and specific labeling of a tagged protein of interest (POI), intracellular or surface, with otherwise nonfluorescent green‐emitting “thiazole orange” or far‐red emitting “malachite green” and “dimethylindole red” dye derivatives creates a toolset suitable for scalable, dynamic multi‐color imaging experiments and direct quantitative trafficking‐related measurements, across many distinct fluorescent measurement platforms.22 Over the past several years, a library of distinct fluorogenic malachite green‐based ligands has been demonstrated to bind to a common, FAP tag, the dL5** FAP (eg, Addgene plasmid #73206).5, 7, 19 These dyes include cell‐excluded and cell‐permeable fluorogens (Figures 2, 3 and 4). Recently another lab, Hu and colleagues, has started to develop several new malachite green derivatives.24 Limiting labeling to extracellular FAP tagged protein on the PM is advantageous for selectively observing endocytosis and endosomal trafficking without confounding signal from intracellular POI, previously internalized or throughout the biosynthetic secretory compartments (Figure 2C). Labeling all tagged protein (inside and out) can be a useful tactic for relating the amount of protein at the cell surface with total amount of protein, distinguishing proteostatic or biosynthetic effects from trafficking effects (Figure 2B). Either a matched or a different color cell‐permeable dye can be used sequentially after surface labeling to give a surface/total POI measurement that increases overall mechanistic insight in a variety of multi‐cell or single‐cell assays (Figure 2E).25, 26, 27
Figure 2.
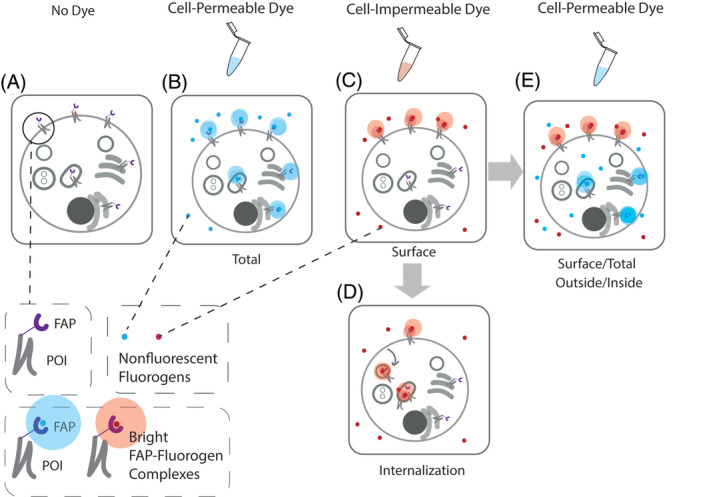
A Snapshot of FAP tagged protein of interest (FAP‐POI) labeled with cell‐excluded or permeable dye without dye wash‐out post labeling. A, No Dye. Absence of fluorescence; B, Use of a cell‐permeant dye labels FAP‐tagged protein throughout the cell, including those in endocytic compartments, as well as those in biosynthetic compartments. This labeling gives a total protein fluorescence measurement. Alternatively (C) use of cell‐excluded dye selectively labels FAP‐tagged proteins at the plasma membrane (surface measurement), which may then be trafficked into the cell by endocytosis, allowing for measurements of internalization (D). E, Sequential labeling of the cells with the cell‐excluded dye followed by a cell permeant dye of the same or different spectral properties allows measurements of the fraction of total protein at the cell surface, at a single cell or population level, depending on the measurement technique
Figure 3.
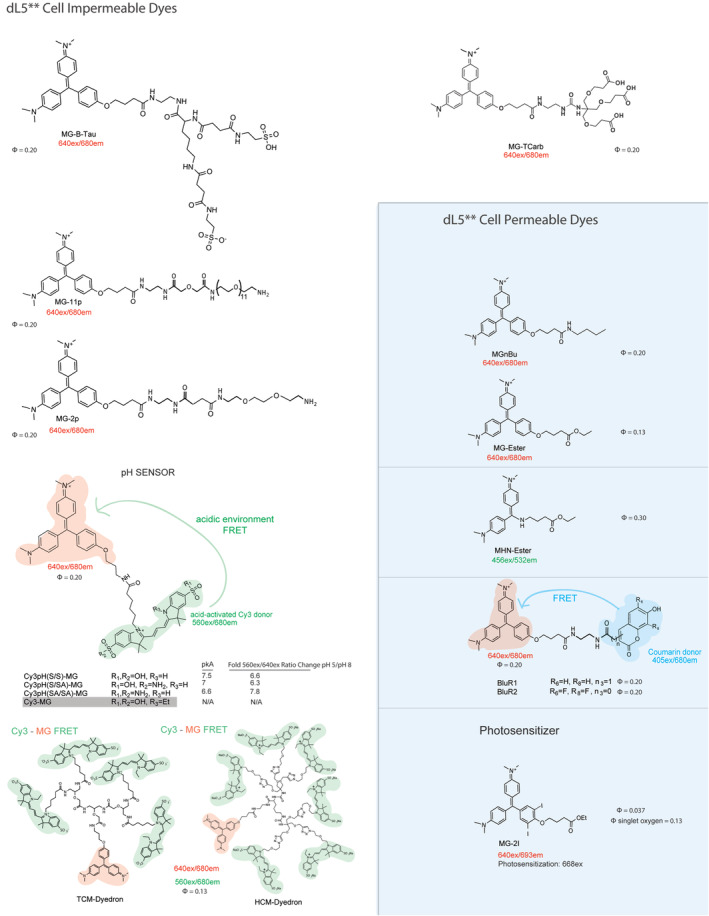
Fluorogenic dyes bound by dL5**. Cell‐excluded and permeable to the mammalian PM. MG2p and MG‐Ester have been used successfully with Mars1Cy and Mars1 FAPs23
Figure 4.
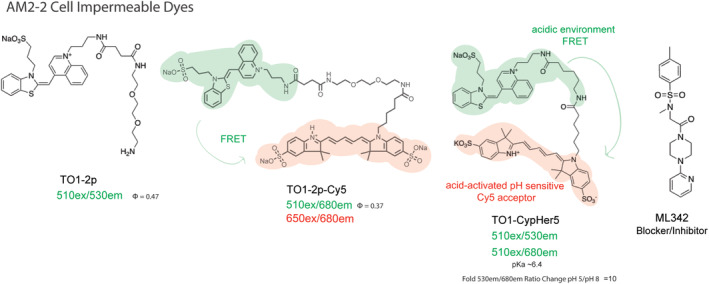
Fluorogenic dyes specific for AM2‐2. Cell‐excluded to mammalian PM. There is no cell‐permeant TO1 analog due to residual DNA binding of the fluorogen within the cell. FRET and pH sensitive variants were developed using Cy5 as an acceptor, with binding selectivity but weak fluorogenic activation upon binding, due to the intrinsic transfer from the donor fluorogen even in the unbound state. Binding to cell surface receptors typically provides adequate concentration and activation in solution to conduct unwashed experiments measuring endocytosis of AM2‐2 labeled receptors using these dyes. The nonfluorescent inhibitor ML342 was identified as a potent inhibitor (approximately 2 nM EC50) of TO1‐2p binding to the AM2‐2 FAP
The FAP platform was developed by screening a yeast display library of single chain variable fragment antibodies (scFv) to find proteins that bind and activate two distinct cell‐excluded fluorogenic dye derivatives: a malachite green with an amido‐diethylene glycol amine linker (MG‐2p) and a sulfonated thiazole orange with a similar linker (TO1‐2p). The dL5** and AM2‐2 FAP tags noncovalently bind MG and TO1 dye derivatives respectively with high affinities (Figures 3 and 4). Later on other scFv tags were identified and characterized for binding and activation of dimethylindole red (DIR) and oxazole thiazole‐blue (OTB) fluorogenic dyes, and near‐infrared dyes, SKC1 and SKCi1 (Figure 5).23, 28, 29, 30 FAP‐dye binding rigidizes the chemical structure, suppressing some bond‐rotations responsible for internal conversion and nonradiative relaxation in the electronic excited state, resulting in increased fluorescence emission. The first synthesized dyes, MG‐2p, MG‐11p and TO1‐2p, possess an hydrophilic diethylene glycol diamine, carrying a positive charge and significant hydration that prevents these dyes from passively crossing the PM and entering the cell.19 A newer cell‐excluded dye, MG‐B‐Tau for dL5**, has been developed that is the current best standard for labeling cell surface proteins. MG‐B‐Tau's two sulfonated groups (net charge of −1) and short hydrophilic linker make it substantially more cell impermeable than the precursor MG dye derivatives.21 For MG binding dL5**, a cell permeant variant has been developed by preparing more hydrophobic linker moieties, such as the MG‐ester or the MG‐nBu (butylamide) dyes. The cell permeability properties imparted by modifications to the linker moieties on the fluorogen can be altered independent of its ability to generate bright fluorescence upon binding the cognate FAP in or on the cell, provided that the fluorogen has inherently low nonspecific activation inside the cell, unfortunately not yet possible for some fluorogen scaffolds like TO1. When available, as in the MG and SC‐fluorogen family, suitable pairs of dyes with nearly identical brightness and spectral properties can be used to quantify cell surface and total protein levels reliably and quickly.
Figure 5.
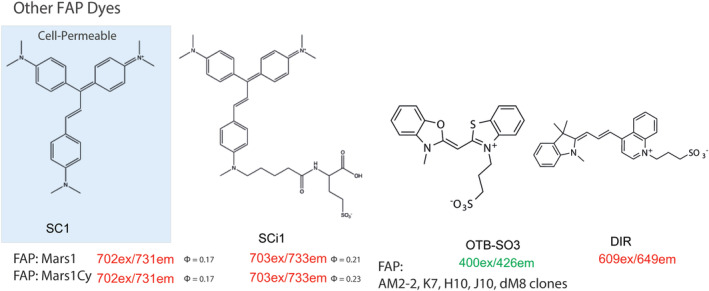
Other fluorogenic dyes that bind to different FAP scFVs
The FAP platform has grown to include ratiometric physiological pH indicator fluoromodules for both AM2‐2 and dL5**. A cell impermeable tandem dye molecule, TO1‐CypHer5, was designed by coupling a pH sensitive, acid activated Cy5 analog with TO1. This reporter uses FRET with two separate emission channels (TO1 510ex/530em and pH dependent FRET 510ex/680em), and functions as a ratiometric pH biosensor where the FRET between donor and acceptor is activated upon internalization into acidic compartments, where the acceptor increases absorption and fluorescence.31 Recently, a new far‐red, genetically Targetable, Ratiometric, Activatable Physiological Indicator Complex (TRApHIC) was developed. In this case, the MG‐acceptor based pH sensor uses FRET with one emission channel (MG 640ex/680em and pH dependent, acid‐activated donor‐excited FRET 560ex/680em using two distinct excitation lasers), and is completely fluorogenic because the dye must be bound for the fluorogen acceptor to act as an emitter, although it is a FRET acceptor in both the bound and unbound states, rendering the donor pH sensor dark until bound to the FAP.
The noncovalent dye‐binding nature of FAP technology is a feature that can allow dye exchanges, as shown in the AM2‐2 TO1‐2p and TO1‐2p‐Cy5 paradigm and could potentially permit bound dye washouts.32 The current developed dL5** MG dyes have subnanomolar Kds, taking hours for dye dissociation. Developing new dyes or FAPs with higher Kds would be an exciting extension for creating dynamic assays and pulse‐chase labeling strategies, as demonstrated recently with the FAST fluorogenic labeling systems, but not yet applied in trafficking studies.33, 34
Different methods have been used to link the FAP protein to cellular target proteins: stable cell lines and transgenic animals have been generated through viral or plasmid‐based over‐expression of a FAP‐tagged protein or via gene editing with CRISPR/Cas9 technology.20, 35 In addition, FAP has been employed as a targetable probe by fusion with a small peptide scaffold affibody (AffiFAP) or covalent linkage to monoclonal antibodies to target endogenous epidermal growth factor receptor (EGFR) expressed on tumor cells.36, 37, 38
3. CELL SURFACE PROTEIN TRAFFICKING MEASUREMENTS: RECEPTORS
G‐protein coupled receptors (GPCRS), epidermal growth factor receptor (EGFR), and the γ‐Aminobutyric acid (GABA) type A receptors (GABAARs) have been the focus of FAP tagged receptor trafficking assay development. This section is organized according to the receptor type that was being investigated utilizing the FAP platform to address their specific trafficking question Table 1. lists the FAP‐receptor based trafficking assay publications. Critical behaviors of surface proteins include biosynthetic processing and secretion to the plasma membrane through the ER and Golgi apparatus, retention at the plasma membrane, endocytosis and recycling or degradation in the steady state or upon stimulation, and, in some cases, retrotranslocation from the endolysosomal pathway into secretory compartments. The FAPs, with suitable fluorogens and addition at times chosen to precede or follow target manipulations within the membrane bounded compartments, have been used to interrogate these processes in a range of trafficking investigations (Figure 6).
Table 1.
FAP‐receptor assay timeline
| Year | Publication (Relevant Method, FAP:Fluorogen) | References |
|---|---|---|
| 2010 | ||
|
22 | |
|
39 | |
| 2012 | ||
|
40 | |
|
41 | |
|
31 | |
| 2013 | ||
|
42 | |
| 2014 | ||
|
32 | |
| 2015 | ||
|
21 | |
|
36 | |
|
23 | |
|
28 | |
| 2017 | ||
|
43 | |
|
44 | |
| 2018 | ||
|
45 | |
| 2019 | ||
|
35 |
Figure 6.
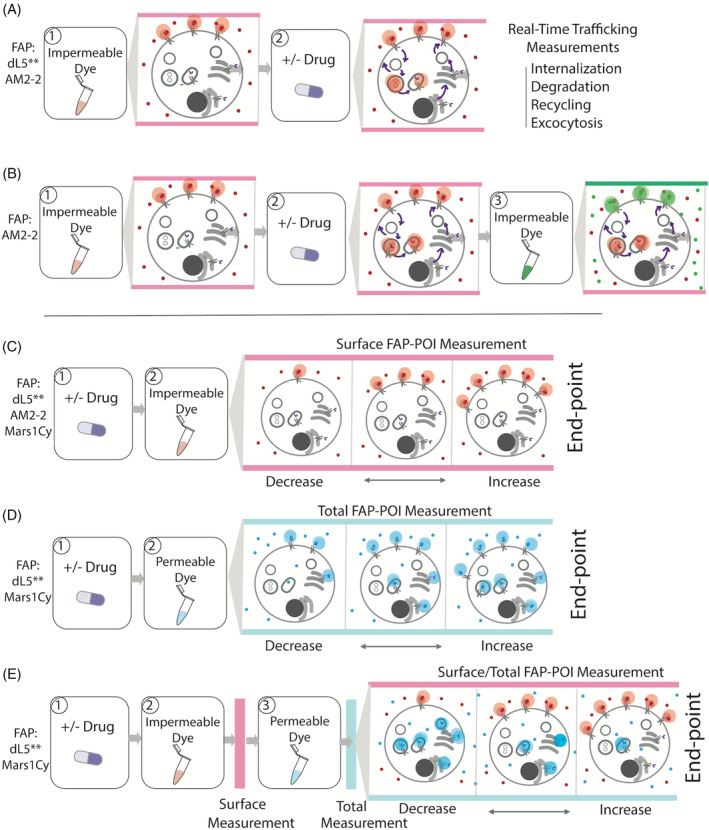
The right dyes at the right time for quantitative assessment of protein trafficking changes without need of wash‐steps. A and B, Real‐time cell surface protein trafficking measurements. C‐E, Detail end‐point trafficking measurements. A, Cell‐impermeable dye is added to the cell followed by drug, allowing for measurement of active trafficking itineraries. Both dL5** and AM2‐2 FAPs and their respective dyes will generate this measurement. B, The self‐checking internalization assay for AM2‐2 FAP consists of first addition of a cell‐excluded fluorogen of one color of moderate affinity, induction of internalization via drug addition, and then competitive displacement of remaining surface FAP‐fluorogen complex with a second cell‐excluded, high‐affinity and different color dye, followed by quantitative assessment of the color ratio. The second dye only labels receptors that are remaining at the plasma membrane, and internalized proteins are protected from displacement. Because all dyes are cell‐excluded, this labeling approach is selective for surface trafficking and does not label proteins contained within biosynthetic compartments.32 C, Drug is added to cells followed by cell‐impermeable dye for a surface FAP‐POI measurement to quantify cell surface POI trafficking changes. dL5**, AM2‐2, Mars1Cy FAPs and their respective dyes will generate this measurement. D, Drug is added to cells followed by cell‐permeable dye for total FAP‐POI measurement. Both dL5** and Mars1Cy FAPs and their respective dyes will generate this measurement. E, Drug is added to cells followed by cell‐impermeable dye for a surface FAP‐POI measurement. Subsequently, a cell‐permeable dye (same color or different color) is added and then total FAP‐POI is measured for the surface/total FAP‐POI. This is a dL5** and Mars1Cy FAP specific assay format, because the TO1 and other fluorogens are not available in cell permeant, highly specific forms
4. G‐PROTEIN COUPLED RECEPTORS
GPCRS are the largest, overarching class of surface signaling protein. Their important physiological roles make them the most heavily investigated drug targets, and the target of the majority of modern pharmaceuticals.46 The surface receptors can be activated by many different ligands, and pair to intracellular G‐proteins, which relay signals and elicit biological effects downstream of receptor activation.47 GPCRs are generally only ligand‐activated on the cell surface, and are desensitized through their removal from the plasma membrane. After GPCR internalization, they are either degraded or sent through the recycling endosomal network to the cell surface. The careful regulation of GPCR traffic to and from the plasma membrane controls their activity and overall biological role.12 Thus, having fluorescent labeling techniques that can report receptor localization and trafficking is crucial for elucidating GPCR specific therapeutic mechanisms.48 GPCRs tagged with fluorescent proteins (FP) at the intracellular c‐terminus have been used as a general observation technique, lacking the ability to quantitatively determine distinct surface signal from collective signal or signal from biosynthetic compartments, except by biochemical or image analysis methods.49 Cell surface receptor labeling with GPCR ligands or antibodies conjugated with radioisotopes or fluorophores has been a long‐standing classical approach for internalization and recycling measurements, although it is unclear whether or not antibodies or ligands remain bound downstream after internalization.50 These techniques can also entail long incubation periods and multiple wash‐steps, constraining the scale of the experiment and adaptation to high‐throughput workflows. The extracellular N‐terminus of GPCRs can be fused with a small peptide tag if it does not interfere with activity, allowing for GPCR fusions with different extracellular tags such as the SNAP tag, Halo tag, FAP tag or FLAG (antibody epitope tag), or ecliptic fluorescent proteins.50, 51, 52, 53, 54, 55, 56, 57 As an extracellular tag, the fluorogenic FAP complex is particularly advantageous due to its simplicity, the quick, selective cell surface protein labeling, and the very high activation ratio upon fluorogen binding to the protein (approximately 20 000‐fold). These properties allow real‐time detection of trafficking events, either to or from the cell surface, in contrast to ecliptic proteins, which revert to the dark state after retrieval from the plasma membrane into acidic vesicles. The development of these measurements using the FAP‐GPCR platform is outlined here.
A specific protein of interest has been the prototypical GPCR, beta‐2‐adrenergic receptor (B2AR), which has widely been used as a foundation for understanding complex GPCRs, and as a classic GPCR test‐bed for new assays.40
The MG‐11p and TO1‐2p dyes were first used to create a framework to detect and quantify GPCR internalization that could be translated to a high‐throughput design.22, 39 There are different FAP‐B2AR labeling strategies to measure changes in cell surface density (Figures 6C and 7). Fluorogen labeling can be performed prior to agonist activation to visualize post‐activation internalization by microscopy or whole‐cell FRET‐measurement with pH sensitive probes, or after receptor activation to measure the loss in signal as an indicator of overall FAP‐B2AR surface downregulation by endocytosis.31 To detect recycling after agonist induced internalization, agonist can be removed and dye added to quantify any increase in cell surface signal, which dynamically increases as internalized FAP‐receptor is returned to the cell surface where it can bind and activate cell‐excluded dye. FAP technology was able to quantifiably and reproducibly detect translocation of cell surface proteins using plate fluorimetry, fluorescence imaging and flow cytometry.39, 40
Figure 7.
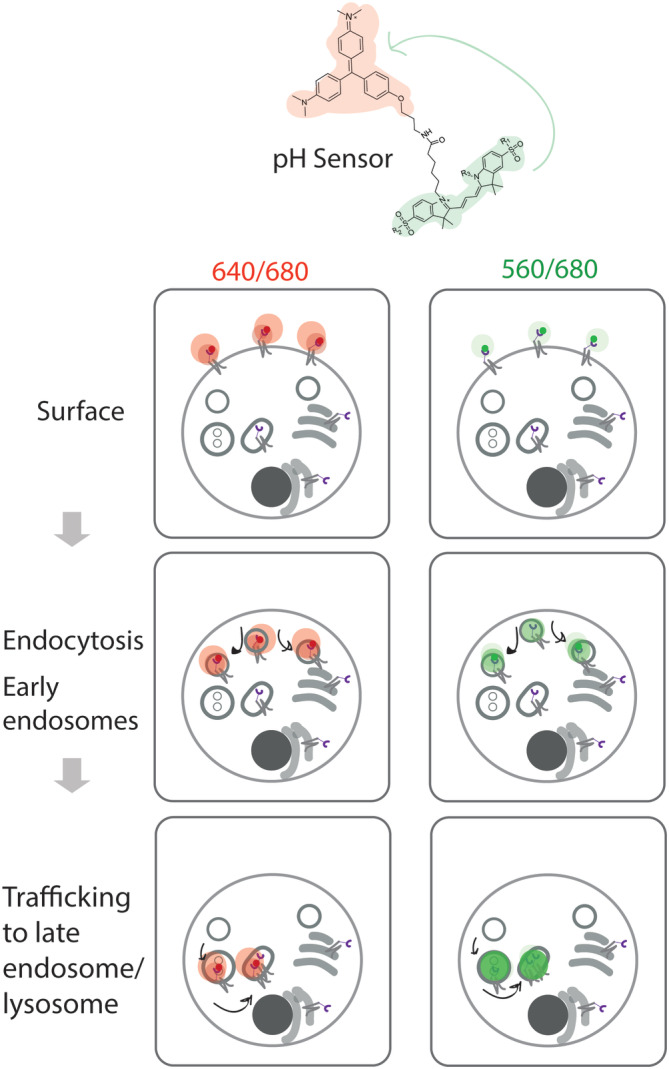
Diagram to show the signal changes related to different steps of Cy3(S/SA)pH‐MG labeled FAP‐B2AR internalization. MG signal (640ex/680em) stays consistent over the trafficking itinerary while pH sensitive FRET signal (560ex/680em) intensity is dependent on the local pH environment
Another assay was designed to measure receptor internalization and recycling with two different cell‐excluded dyes, TO1‐2p‐Cy5 and TO1‐2p (Figure 6B). TO1‐2p has a higher binding affinity than TO1‐2p‐Cy5, allowing bound fluorogen displacement by the higher affinity dye. AM2‐2 FAP‐B2AR was labeled on the surface with TO1‐2p‐Cy5, measured as a red signal, and after agonist incubation, TO1‐2p (green) was added, without or with antagonist. Without antagonist addition, red labeled receptors are present on the cell surface and in endosomal vesicles, and the red surface fluorescence is quickly displaced by TO1‐2p green signal, providing a measurement of overall intracellular accumulation in the red channel. With antagonist addition, red labeled receptors are recycled to the cell surface, where the bound red fluorogen is displaced by the second, green, dye, as a measurement of recycling.32
The newer cell‐excluded dye, MG‐B‐Tau, that is the current best standard for labeling cell surface proteins due to its characteristics that make it highly cell impermeable, demonstrated proficiency to rapidly and selectively label dL5** FAP‐B2AR for quantitative trafficking using flow cytometry and microscopy (Figure 6C).21
The delta opioid receptor was fused to dL5** FAP in order to create a quick labeling scheme with cell‐permeable dye MG‐Ester (Figure 6D).44 The assay design took advantage of the far‐red properties of MG for multicolored imaging with GFP, and worked as a simple, live cell assessment of delta opioid receptor localization in the golgi vs cell surface by colocalization with fluorescent protein markers localized in the Golgi.44
In addition, a user‐friendly, rapid and quantitative high‐throughput screening approach was developed for receptor trafficking utilizing the SCi1 dye and Mars1Cy FAP tag (Figures 5 and 6C).28 This screening platform uses a standard infrared western‐blotting scanner and was validated by assessing the trafficking changes of vasopressin‐2 receptor and dopamine receptor‐2 in response to agonists or antagonists. The method was applied to screen FDA‐approved and drug like small molecules that antagonize leucine‐rich G protein‐coupled receptor 5 (Lgr5) internalization. In this screening study, they reported that glucocorticoids were found to increase Lgr5 expression at the cell surface.28
The FAP platform's ratiometric physiological pH indicator fluoromodules were also tested by GPCR trafficking. The cell impermeable tandem dye molecule, TO1‐CypHer5, functions as a ratiometric pH biosensor where the FRET between donor and acceptor is activated upon internalization into acidic compartments. TO1‐Cypher5 surface labeling was used as an approach to detect the local changes associated with AM2‐2 FAP‐B2AR trafficking after agonist addition (Figure 6A).31 Although suitable for trafficking assays without washing away unbound fluorogen, this tandem dye shows weak, but detectable CypHer5 fluorescence in low‐pH environments regardless of its binding to the FAP‐tagged protein, because the donor is the fluorogen, and FRET efficiency depends only weakly on the quantum yield of the donor. The new far‐red, genetically Targetable, Ratiometric, Activatable Physiological Indicator Complex (TRApHIC) uses the pH sensor dye, Cy3(S/SA)‐MG, with dL5** FAP labeling and is described in Figure 7. Cy3(S/SA)‐MG was shown to quantitatively report dL5** FAP‐B2AR internalization and recycling in response to different agonists, and demonstrated compatibility with Stimulated Emission Depletion (STED) based super resolution microscopy in living cells using a single depletion laser to enhance resolution of the pH probe under both direct (pH independent) and FRET (pH dependent) excitation.45 This pH sensor has subsequently been used for measuring and monitoring different receptors, EGFR and GABAARs, described next.
5. EPIDERMAL GROWTH FACTOR RECEPTOR
The epidermal growth factor receptor (EGFR) function has been heavily investigated in development, physiology and cancer.58 It is a transmembrane glycoprotein that is required for growth, survival, proliferation and differentiation, and plays a large role in metastasis.59 The EGFR pathway relays external signals via a downstream activation cascade of intracellular messengers. EGFR ligand binding induces endocytosis and can be trafficked to the lysosome for degradation or translocation to the nucleus for further signaling. The internalization and degradation of receptor modulates its expression and availability on the cell surface for signaling.10
Wang et al, developed a dL5** fused affibody (AffiFAP) targeting EGFR and showed proof of concept with targeting native receptor on the A431 cell line plasma membrane with a no‐wash dye labeling approach.36 Affibodies are small proteins that can be engineered to selectively bind to targeted molecules, and features rapid tumor uptake and quick blood clearance.60 They have ideal characteristics for molecular imaging and can readily translate the FAP technique to in vivo imaging.
The AffiFAP EGFR probe was labeled with HCM‐dyedron to demonstrate quantitative EGFR surface measurements and receptor endocytosis tracking. Labeling the native receptors with the AffiFAP did not block or stimulate EGFR activation, and reported on changes in receptor surface abundance upon fluorogen labeling prior to and subsequent to stimulation with epidermal growth factor. The AffiFAP system targeting EGFR also showed specific in vivo tumor labeling in mice with MG‐B‐Tau tail vein injection.37
Understanding the EGFR trafficking mechanisms is difficult due to current methods that utilize overexpressed recombinant EGFR and high ligand concentrations that tend to induce a specific mode of receptor internalization over others.35 A recent study by Larsen et al, developed a dL5** FAP‐EGFR assay for high throughput screening of proteins important in the clathrin mediated endocytosis of EGFR.35, 61 This assay design allowed bypass of the difficulties involved with labeled EGFR ligands or EGFR overexpression by using CRISPR/Cas9 technology to maintain endogenous expression levels of dL5** tagged EGFR and response to physiological levels of EGF (sub‐ng/mL).
To report the fraction of dL5** FAP‐EGFR in acidic compartments/lysosomes, the pH sensitive dye, MG‐Bis‐SA was used (Figures 6A and 7).35 They monitored and measured EGFR endocytosis in a 96 well plate high throughput format after siRNA knockdown of human phosphatases. This screen identified new regulatory processes, proteins and demonstrated the capability of the high‐throughput FAP‐TRAPhIC platform for studying endocytosis at native expression and signaling levels.
6. γ‐AMINOBUTYRIC ACID TYPE A RECEPTOR
A FAP tool was designed to track multistage synaptic GABAa Receptor internalization and trafficking, extendable for future use in drug related HT screens (Figures 6B and 7).43 These receptors function as ligand gated chloride channels involved in inhibitory neurotransmission in the brain.62 GABAaR primarily consist of two subunits, with the γ2 subunit being heavily involved in GABAaR function‐and critical for receptor synaptic targeting and cluster maintenance.63 The mechanisms of GABAaR trafficking in response to endogenous and pharmacological molecules has not been fully investigated, which has led Lorzen‐Guertin et al to develop dL5** γ2pHFAP GABAARs as a versatile trafficking sensor. This tool has largely employed the previously described Cy3(S/SA)‐MG dye as a monitor of the trafficking itinerary of γ2pHFAP GABAARs, providing vesicle‐level pH measurements. Using the developed γ2pHFAP GABAAR in conjunction with MG‐B‐Tau pulse labeling they tested the utility of the platform to detect pharmacologically induced changes in trafficking. They found with neuron exposure to a bicuculline‐induced seizure paradigm that GABAAR had enhanced turnover rates and lysosomal targeting that were not detectable by employing pHluorin (pHGFP) fluorescence alone. pHGFP is quenched in the endosomal‐lysosomal acidic compartments, while γ2pHFAP reports pH of the extracellular environment and throughout the endo‐lysosomal trafficking network.43 The platform has established a trafficking paradigm of GABAARs that can be used to assess trafficking changes caused by different agents.
7. CELL SURFACE PROTEIN TRAFFICKING MEASUREMENTS: ION CHANNELS
Ion channels are responsible for the regulated and selective transport of ions across the membrane. The critical functions of ion channels span across a range of pathologies, making them key therapeutic targets, and the second largest class of drug targets after GPCRs.64 Their movements to and from the cell surface regulates their overall function, and changes in trafficking can have drastic impacts on ionic homeostasis. Controlled ion channel activity at the cell surface is important for regulating heartbeat, blood pressure, muscle excitation, neural activity, cell proliferation, bone resorption and salt and water balance.65 Thus, detecting ion channel trafficking gives important mechanistic insights of considerable therapeutic significance in a range of conditions and diseases. The development of fluorescent labeling techniques allowed visualization of ion channel dynamics at the plasma membrane that provides complementary measurements to biochemical assays, with improved mechanistic insight.66 However, designing genetically encoded tagged ion channels is difficult due to potential interference with native channel function, which has greatly hindered the development of ion channel assays with genetic chemical labels.67, 68 Given the limited creation of ion channel FP‐fusions or chemical labeling choices in general, ion channel FAP‐fusion constructs that exhibit native function represent a valuable tool option for labeling cell surface ion channels to assess trafficking and changes in trafficking associated with altered ionic transport.
The ion channels that have been validated and studied with the dL5** FAP platform are the cystic fibrosis transmembrane conductance regulator (CFTR), the large conductance, voltage‐and calcium‐activated potassium (BK) channels, and recently, the K+ inward rectifying channel, Kir2.1, in a yeast model. This section is divided by channel to highlight the use of tailored individualized strategies to address the specific trafficking questions relevant to each of these targets Table 2. Lists the publications of the FAP‐ion channel assay developments.
Table 2.
FAP‐ion channel assay timeline
| Year | Publication (Relevant Method, FAP:Fluorogen) | References |
|---|---|---|
| 2010 | ||
|
39 | |
| 2012 | ||
|
69 | |
|
70 | |
| 2013 | ||
|
71 | |
| 2015 | ||
|
27 | |
| 2016 | ||
|
72 | |
| 2017 | ||
|
73 | |
| 2018 | ||
|
25 | |
|
74 |
8. CYSTIC FIBROSIS TRANSMEMBRANE CONDUCTANCE REGULATOR
The trafficking defective chloride channel, the mutant ΔF508‐CFTR, fails to be exported to the cell surface where its function is to transport chloride ions that are essential regulators of mucus viscosity and layer thickness. The ΔF508 mutation is the most prevalent cause of Cystic Fibrosis (CF), a genetic disease that results in mucus build‐up in the lungs, causing severe breathing difficulties, chronic bacterial lung infections and reduced life expectancy.75 Most CF therapies available only treat the symptoms, and finding therapies to treat the underlying cause of the disease has been extremely challenging. ΔF508‐CFTR is misfolded in the endoplasmic reticulum (ER) and flagged by the quality control machinery and sent for proteosomal degradation.76 However, the mutant protein can form a functional channel if delivered to the cell surface. Therapeutic screens are searching for compounds or targets that can rescue the trafficking defective CFTR by delivering it to the PM. Most corrector HT screens rely on functional chloride transport measurements due the prohibitive costs, time and steps that come with quantifying CFTR at the PM in a large‐scale format, usually by cellular fractionation and western blotting. FAP technology has mitigated other labeling assay limitations by only needing a simple, quick dye addition to label cell surface CFTR and quantify it relative to total expression level. FAP fusion ΔF508‐CFTR has been shown to be useful in high‐throughput assay development utilizing different measurement approaches, including imaging, flow cytometry and multi‐well bulk fluorescent quantitation.
Holleran et al, first developed dL5** FAP‐WT CFTR and dL5** FAP‐ΔF508‐CFTR constructs to establish a novel detection platform, evaluating both N‐terminally tagged and loop‐4 insertion sites for the FAP‐tag. Although both constructs trafficked properly, and responded to correction strategies, the insertion of the protein in the loop‐4 site impaired CFTR glycosylation, so further work was carried out on the terminally labeled construct, although it has an extra transmembrane domain to ensure extracellular exposure of the FAP. The addition of a FAP tagged transmembrane domain to CFTR was demonstrated to not impair its trafficking, anion transport activity or glycosylation.70 Initial work used MG‐11p as a single, fast surface CFTR labeling fluorogen measurable by flow‐cytometry and microscopy (Figure 6C).39 The streamlined system was validated by using corrector compounds and low temperature to rescue FAP‐ΔF508‐CFTR to the cell surface.70 The detection system developed was later used with high‐temporal resolution microscopy to visualize internalization and accumulation of CFTR from the PM, and demonstrated that treatment with C4 or C18 corrector molecules redirects FAP‐ΔF508‐CFTR trafficking from a degradation route to recycling pathway.71 More recently a 96‐well format, HT microscopy based, method was developed as a platform for drug discovery, with Z′ values above 0.7 and read‐time of less than 5 minutes per 96‐well plate. This platform uses MG‐B‐Tau to selectively label FAP‐ΔF508‐CFTR at the PM (Figure 6C).72 To improve experimental sensitivity and mechanistic insight, we have now developed the capability to measure direct CFTR traffic efficiency by measuring both cell surface protein and total protein in the same specimens, in high throughput formats. A surface and total protein HT measurement assay was developed on a plate reader, but could also be implemented using high‐throughput microscopy with on‐board reagent additions without wash steps (Figure 6E). This assay first labels CFTR at the cell surface with MG‐B‐Tau, then after measuring surface signal, a second dye that is cell‐permeable is added to wells. The second dye, MGnBu, is a new MG dye derivative that has the same brightness as MG‐B‐Tau allowing for quantitative collection of the combined signal of MG‐B‐Tau labeling on the surface plus MGnBu intracellular labeling with the same excitation and emission wavelengths, providing a direct measurement of the surface fraction and total protein levels on a sample‐by‐sample or cell‐by‐cell basis. This FAP approach was used to investigate all kinase targets knocked down by siRNA that could enhance CFTR corrector, VX‐809, mediated rescue of ΔF508‐CFTR.25 The versatility of FAP across microscopy, plate reader and flow‐cytometry instruments has allowed for diverse strategies to measure CFTR trafficking and rescue to the plasma membrane at a drug‐discovery and systems‐biology experimental scale, and for detailed mechanistic investigations.
9. LARGE CONDUCTANCE CALCIUM AND VOLTAGE REGULATED POTASSIUM CHANNEL
The BK channel is expressed throughout the central nervous system (CNS), kidney and smooth muscle.77 Reduced function in the CNS can cause epilepsy and seizures.78 Because of its large conductance, small changes to the density of cell surface BK channels can have significant influences on its ability to regulate action potentials and neurotransmitter release.79 To investigate BK channel trafficking, the BKα channel was tagged with dL5** FAP and surface labeled with MG‐2p for comparison to cells where all expressed FAP‐BKα was labeled with cell‐permeable dye, MG‐Ester (Figure 6C, D). Co‐transfecting its beta‐4 subunit down‐regulated its cell surface presence, an effect relieved by cotransfection of the subunit lacking an ER‐retention motif. This study demonstrated that the beta‐4 subunit expression plays a role in reducing BKαs PM density by retaining it in the ER, and established a mechanistic basis for the increased channel currents observed in beta‐4 KO animals.69 Pratt, et al devised a method to quantify surface and internal BK channel pools simultaneously in single cells by using a two color dye FAP labeling system. The fluorogenic Green‐Inside Red‐Outside (GIRO) labeling strategy uses far‐red, cell surface labeling with MG‐B‐Tau in combination with a cell‐permeable green dye, MHN‐Ester. GIRO labeling was able to detect BKα channel surface expression dynamics. This approach recapitulated surface and internal immunofluorescence labeling, with FAP clearly being advantageous over immunofluorescence methods by detecting real‐time changes in live cells, with labeling within minutes.27 Using microscopy and flow‐cytometry, the GIRO method reported BKα channel stability, with a long cell surface half‐life, and that forskolin induced activation of adenylyl cyclase reduced BKα PM density. Recently Pratt et al, demonstrated CRISPR insertion of dL5** FAP into the native BKα genomic locus, analyzing expression patterns in mouse brain tissue, creating a novel mouse model for studying BK channels in live or fixed brain tissue and potentially other tissues.73 In this context, MG‐TCarb became one of the newest dye additions to the cell‐excluded category. MG‐TCarb utilizes a mildly acidic, tricarboxylate moiety configuration to reduce background nuclear DNA dye binding observed in nuclei of some cells in fixed brain tissue labeled with MG‐B‐Tau. The new mouse FAP platform was used to reveal BK channel clustering with beta‐4 subunit expression in the cerebellum, and shows promise for future BK channel trafficking studies.73
10. INWARDLY RECTIFYING POTASSIUM CHANNEL (KIR2.1)
The inwardly rectifying K+ (Kir) channels promote potassium transport into the cell for membrane potential hyperpolarization, and has a significant role in cardiac myocytes, vascular endothelium, smooth muscle cells, skeletal muscle, neurons and in the kidney.80 Dysfunction of the Kir2.1 family members is associated with cardiac arrhythmias and loss‐of‐function mutations in the Kir2.1 gene are linked with Andersen syndrome, a rare form of long QT disorder associated with periodic paralysis with no current standard treatment.80
A study by Hager et al, established a FAP tagged Kir2.1 yeast platform to screen α‐arrestin proteins in regulating Kir2.1 trafficking.74 The screen utilized cell impermeant dye, MG‐B‐Tau, to measure FAP‐Kir2.1 localized at the cell surface, and also cell‐permeable dye, MG‐Ester, to assess the total cellular population (Figure 6C, D). This is the first time that this FAP technique had been used in yeast to differentiate surface and intracellular FAP‐tagged protein pools. The trafficking screen revealed that specific α‐arrestins, Aly1, Aly2 and Ldb19, promote Kir2.1 trafficking to the cell surface. In addition, they determined that two α‐arrestin effectors, Rsp5 ubiquitin ligase and phosphatase calcineurin, also mediate Kir2.1 cell surface localization.74
11. NEW FAP PLATFORM DIRECTIONS: SYNAPSE IMAGING
While most FAP technology developments have focused on select cell surface protein trafficking, recently advances have been made with imaging synapse location and function in complex specimens (mouse brain tissues and fly neuromuscular junction). Synapses are structures comprising a presynaptic and postsynaptic terminal where neurons communicate with each other. A chemical signal is released from the presynaptic terminal via packaged vesicles and quickly diffuses to the close by postsynaptic terminal.81 The packaging and exocytosis of chemical signals to be trafficked is highly regulated and mediated by the synaptic fusion pore or synaptic vesicle fusion with the synaptic plasma membrane.82 Bulgari et al, were able to image dense core vesicle fusion pores without interference from cotransmitting small synaptic vesicles by establishing a FAP tagged proneuropeptide that are released at synapses in the fly neuromuscular junction.83 When these dense core vesicles become open at the pore, the cell impermeable MG dyes are able to label the vesicle‐contained FAP‐proneuropeptide, revealing permeability characteristics of the fusion pore. Uptake of the cell‐excluded fluorogen dye in the absence of electrical stimulation provided the first evidence for spontaneous neuropeptide release, a potential mechanism for regulating neural activity more broadly than at the level of a single synaptic connection.
In addition, a new FAP based method was developed by Kuljis et al, that allows for high‐throughput quantitative imaging of postsynaptic sites across an individual neuron in a complex tissue environment.84 In this work, the FAP was fused to a fragment of neuroligin1 and expressed in rodent brain with a dTomato cell filling protein by viral transduction using site‐specific injection and Cre‐dependent expression. The construct showed clear punctate colocalization with synaptic immunofluorescence, and was a reliable measure of synaptic locations, compatible with input assignment by using distinct color cells to colocalize axonal processes with the identified postsynaptic sites. This allowed identification of patterns of neuronal connectivity that differed between important inhibitory subclasses, with some neuron types preferring to establish somatic and apical dendritic connections while others were biased for connections in more distal dendritic branches. This hierarchical and specified organization has important functional implications for circuit development and function, and raises interesting questions about the development and maintenance of such heterogeneous inter‐ and intra‐cellular architectures.
12. OTHER FLUOROGEN BASED STRATEGIES
There have been many developments in fluorogenic imaging probes alongside with the FAP platform that offer benefits over traditional FP labeling strategies. SNAP, CLIP, Halo, PYP, FAST and TMP tags have illustrated examples of fluorgenic dye probes.13, 33, 85, 86, 87, 88, 89 There have also been several recent advancements in these fluorogen technologies. Silicon‐rhodamine (siR) dyes were created by Johnsson and colleagues as a near‐infrared, cell‐permeable fluorogenic probe compatible with the popular SNAP, CLIP and Halo tags.90 The photostable siR substrates have proved to be invaluable for super‐resolution microscopy, and there continues to be evaluation of silicon rhodamine, rhodamine and carbopyronine dyes for super resolution applications.91 To improve spatial resolution with single molecule microscopy in live‐cells, the Lavis lab at Janelia developed a strategy to enhance the brightness of cell‐permeable fluorophores by minimal structural adjustments. They reported increased quantum yield of tetramethylrhodamine by replacing the N,N‐dialkyl groups with azetidine rings, and expanded it to several other fluorophores: coumarin, naphthalimide, acridine, rhodol, carborhodamine, oxazine and Si‐rhodamine classes.92 These adjusted versatile fluorophores, known as Janelia Fluor (JF) dyes, have also been made photoactivable, compatible with super resolution PALM microscopy and are currently the best “light up” labels for the Halo and SNAP tag.93, 94 These probes are activated upon binding by shifting the equilibrium between the noncolored closed‐form (the lactone form) and the chromophore/fluorophore open form (the zwitterion), the so‐called L‐Z equilibrium, a process distinct from the fluorogen activation discussed above. A red shifted pH sensitive fluorophore has recently been used as a semisynthetic sensor platform with both SNAP tag and specific antibodies for eliminating modification of protein expression in studying endocytosis and exocytosis.95 A pair of dyes with distinguishable spectral properties have been developed for the FAST variants of the photoactive yellow protein.33 These protein‐dye complexes can be rapidly displaced by dye‐exchange approaches, and may be alternate low‐affinity probes for some trafficking applications, if the protein‐ligand interactions can accommodate linker modifications to control cell permeability, and generate environmentally sensitive probes as elaborated for the scFv‐based FAP‐Fluorogen system. In addition, Tebo and Gautier have reported the development of SplitFAST to visualize transient protein‐protein interactions at the plasma membrane or other cellular compartments.96 This designed system uses the “split” fragments of the fluorescence activating and absorption shifting tag, FAST, that specifically and reversibly binds the fluorogen‐like hydroxybenzylidene rhodanine (HBR) analogs. When the two pieces of FAST expressed on POIs interact, they are able to form the correct complex to bind HBR.
An extension of FAP technology was developed using an alternative high‐affinity protein scaffold, designed ankyrin repeat proteins (DARPins). Schütz et al, selected for fluorescence‐activating DARPins using malachite green binding by ribosome display followed by yeast surface display.97 This development showed a new application of DARPins, that these binding proteins can also be selected to bind small organic molecules. The DARPins may be alternate fusion proteins with some binding capacity for established MG fluorogens, in systems where scFv‐FAP fusion impairs normal trafficking or function.
While there have been considerable improvements with FP modification, engineering brighter and photostable FPs is a difficult challenge due to their buried chromophores that are not amenable to chemical modification, and their environment sensitivity, that is, fluorescence quenching at low pH.1 In a breakthrough advancement in de novo design of beta‐barrel protein folds, Dou et al, were able to design beta‐barrel backbone models with cavity shapes that accommodated the fluorogenic compound, DFHBI.98 The generated mini‐fluorescence‐activating proteins (mFAPs), demonstrated a 3‐step approach to creating a de novo design of ligand‐binding proteins. This development method could lead to a new array of backbone structures specifically designed for select fluorogens and increase the spectral range and properties of fluorogenic tools available. These new mFAPs represent another encoded fluorogen activating protein platform amenable to the same rational fluorogen and assay design principles outlined here, with a protein platform completely distinct from and with different advantages and limitations than the scFv based platform.
13. CONCLUDING PERSPECTIVES
The fluoromodule activating system continues to gain new designer dyes, tagged‐proteins and innovative experimental methods to improve detection of protein itineraries, or new trafficking interfaces that can be adapted to a high‐throughput format. The powerful combination of genetic and genomic resources with the robust phenotypic measurements of protein trafficking enabled by the FAP‐fluorogen toolset at the single cell level opens a variety of new research approaches based on library‐driven forward or reverse genetics and clonal selections based on precisely determined trafficking phenotypes and analysis by sequencing. A similar approach may also be applied using new CRISPR libraries or other unbiased genome‐wide disruption and amplification methods. As new fluorogenic hybrid labeling platforms become available, and gain more validation, this labeling and detection approach is having a dramatic impact on the kinds of questions that can be addressed robustly and at large scale in protein trafficking. We hope that new toolkits can continue to provide these kinds of improvements in this and other research areas, and that this review points the way for people who are considering using FAP‐fluorogen technology in their protein trafficking research, and for other probe developers to provide application‐tailored fluorogenic labeling tools that enable new research questions and approaches.
CONFLICT OF INTEREST
M.P.B. is founder and Chief Technology Officer at Sharp Edge Laboratories, Inc., a licensee commercially utilizing the FAP fluorogen technology.
ACKNOWLEDGMENTS
Funding (this work was supported in part by NIH grants R01GM114075 and R01EB017268). The authors would like to thank Dmytro Kolodieznyi for helping with providing dye structures for figures.
Perkins LA, Bruchez MP. Fluorogen activating protein toolset for protein trafficking measurements. Traffic. 2020;21:333–348. 10.1111/tra.12722
Peer Review The peer review history for this article is available at https://publons.com/publon/10.1111/tra.12722/
Funding information National Institute of Biomedical Imaging and Bioengineering, Grant/Award Number: R01EB017268; National Institute of General Medical Sciences, Grant/Award Number: R01GM114075
REFERENCES
- 1. Rodriguez EA, Campbell RE, Lin JY, et al. The growing and glowing toolbox of fluorescent and photoactive proteins. Trends Biochem Sci. 2017;42(2):111‐129. 10.1016/j.tibs.2016.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Li C, Tebo AG, Gautier A. Fluorogenic labeling strategies for biological imaging. Int J Mol Sci. 2017;18(7):e1473 10.3390/ijms18071473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Thorn K. Genetically encoded fluorescent tags. Mol Biol Cell. 2017;28(7):848‐857. 10.1091/mbc.E16-07-0504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jullien L, Gautier A. Fluorogen‐based reporters for fluorescence imaging: a review. Method Appl Fluoresc. 2015;3(4):042007 10.1088/2050-6120/3/4/042007. [DOI] [PubMed] [Google Scholar]
- 5. Yan Q, Bruchez MP. Advances in chemical labeling of proteins in living cells. Cell Tissue Res. 2015;360(1):179‐194. 10.1007/s00441-015-2145-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bruchez MP. Dark dyes‐bright complexes: fluorogenic protein labeling. Curr Opin Chem Biol. 2015;27:18‐23. 10.1016/j.cbpa.2015.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Xu S, Hu H‐Y. Fluorogen‐activating proteins: beyond classical fluorescent proteins. Acta Pharm Sin B. 2018;8(3):339‐348. 10.1016/j.apsb.2018.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kozma E, Kele P. Fluorogenic probes for super‐resolution microscopy. Org Biomol Chem. 2019;17(2):215‐233. 10.1039/c8ob02711k. [DOI] [PubMed] [Google Scholar]
- 9. Péresse T, Gautier A. Next‐generation fluorogen‐based reporters and biosensors for advanced bioimaging. Int J Mol Sci. 2019;20(24):e1642 10.3390/ijms20246142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sorkin A, von Zastrow M. Endocytosis and signalling: intertwining molecular networks. Nat Rev Mol Cell Biol. 2009;10(9):609‐622. 10.1038/nrm2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Barbieri E, Di Fiore PP, Sigismund S. Endocytic control of signaling at the plasma membrane. Curr Opin Cell Biol. 2016;39:21‐27. 10.1016/j.ceb.2016.01.012. [DOI] [PubMed] [Google Scholar]
- 12. Hanyaloglu AC, von Zastrow M. Regulation of GPCRs by endocytic membrane trafficking and its potential implications. Annu Rev Pharmacol Toxicol. 2008;48:537‐568. 10.1146/annurev.pharmtox.48.113006.094830. [DOI] [PubMed] [Google Scholar]
- 13. Sun X, Zhang A, Baker B, et al. Development of SNAP‐tag fluorogenic probes for wash‐free fluorescence imaging. Chembiochem. 2011;12(14):2217‐2226. 10.1002/cbic.201100173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Los GV, Encell LP, McDougall MG, et al. HaloTag: a novel protein labeling technology for cell imaging and protein analysis. ACS Chem Biol. 2008;3(6):373‐382. 10.1021/cb800025k. [DOI] [PubMed] [Google Scholar]
- 15. Miller LW, Cai Y, Sheetz MP, Cornish VW. In vivo protein labeling with trimethoprim conjugates: a flexible chemical tag. Nat Methods. 2005;2(4):255‐257. 10.1038/nmeth749. [DOI] [PubMed] [Google Scholar]
- 16. Keppler A, Gendreizig S, Gronemeyer T, Pick H, Vogel H, Johnsson K. A general method for the covalent labeling of fusion proteins with small molecules in vivo. Nat Biotechnol. 2003;21(1):86‐89. 10.1038/nbt765. [DOI] [PubMed] [Google Scholar]
- 17. Daly CJ, McGrath JC. Fluorescent ligands, antibodies, and proteins for the study of receptors. Pharmacol Ther. 2003;100(2):101‐118. 10.1016/j.pharmthera.2003.08.001. [DOI] [PubMed] [Google Scholar]
- 18. Bizzarri R, Serresi M, Luin S, Beltram F. Green fluorescent protein based pH indicators for in vivo use: a review. Anal Bioanal Chem. 2009;393(4):1107‐1122. 10.1007/s00216-008-2515-9. [DOI] [PubMed] [Google Scholar]
- 19. Szent‐Gyorgyi C, Schmidt BF, Creeger Y, et al. Fluorogen‐activating single‐chain antibodies for imaging cell surface proteins. Nat Biotechnol. 2008;26(2):235‐240. [DOI] [PubMed] [Google Scholar]
- 20. Telmer CA, Verma R, Teng H, Andreko S, Law L, Bruchez MP. Rapid, specific, no‐wash, far‐red fluorogen activation in subcellular compartments by targeted fluorogen activating proteins. ACS Chem Biol. 2015;10(5):1239‐1246. 10.1021/cb500957k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yan Q, Schmidt BF, Perkins LA, et al. Near‐instant surface‐selective fluorogenic protein quantification using sulfonated triarylmethane dyes and fluorogen activating proteins. Org Biomol Chem. 2015;13(7):2078‐2086. 10.1039/c4ob02309a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fisher GW, Adler SA, Fuhrman MH, Waggoner AS, Bruchez MP, Jarvik JW. Detection and quantification of beta2AR internalization in living cells using FAP‐based biosensor technology. J Biomol Screen. 2010;15(6):703‐709. 10.1177/1087057110370892. [DOI] [PubMed] [Google Scholar]
- 23. Zhang M, Chakraborty SK, Sampath P, et al. Fluoromodule‐based reporter/probes designed for in vivo fluorescence imaging. J Clin Invest. 2015;125(10):3915‐3927. 10.1172/JCI81086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhang Q, Wang Q, Sun Y, Zuo L, Fetz V, Hu H‐Y. Superior fluorogen‐activating protein probes based on 3‐indole‐malachite green. Org Lett. 2017;19(17):4496‐4499. 10.1021/acs.orglett.7b02055. [DOI] [PubMed] [Google Scholar]
- 25. Perkins LA, Fisher GW, Naganbabu M, Schmidt BF, Mun F, Bruchez MP. High‐content surface and Total expression siRNA kinase library screen with VX‐809 treatment reveals kinase targets that enhance F508del‐CFTR rescue. Mol Pharm. 2018;15(3):759‐767. 10.1021/acs.molpharmaceut.7b00928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Naganbabu M, Perkins LA, Wang Y, Kurish J, Schmidt BF, Bruchez MP. Multiexcitation fluorogenic labeling of surface, intracellular, and total protein pools in living cells. Bioconjug Chem. 2016;27(6):1525‐1531. 10.1021/acs.bioconjchem.6b00169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pratt CP, He J, Wang Y, Barth AL, Bruchez MP. Fluorogenic green‐inside red‐outside (GIRO) labeling approach reveals adenylyl cyclase‐dependent control of BKα surface expression. Bioconjug Chem. 2015;26(9):1963‐1971. 10.1021/acs.bioconjchem.5b00409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Snyder JC, Pack TF, Rochelle LK, et al. A rapid and affordable screening platform for membrane protein trafficking. BMC Biol. 2015;13:107 10.1186/s12915-015-0216-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gallo E, Jarvik JW. Breaking the color barrier—a multi‐selective antibody reporter offers innovative strategies of fluorescence detection. J Cell Sci. 2017;130(15):2644‐2653. 10.1242/jcs.202952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zanotti KJ, Silva GL, Creeger Y, et al. Blue fluorescent dye‐protein complexes based on fluorogenic cyanine dyes and single chain antibody fragments. Org Biomol Chem. 2011;9(4):1012‐1020. 10.1039/c0ob00444h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Grover A, Schmidt BF, Salter RD, Watkins SC, Waggoner AS, Bruchez MP. Genetically encoded pH sensor for tracking surface proteins through endocytosis. Angew Chem Int Ed Engl. 2012;51(20):4838‐4842. 10.1002/anie.201108107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Fisher GW, Fuhrman MH, Adler SA, Szent‐Gyorgyi C, Waggoner AS, Jarvik JW. Self‐checking cell‐based assays for GPCR desensitization and resensitization. J Biomol Screen. 2014;19(8):1220‐1226. 10.1177/1087057114534299. [DOI] [PubMed] [Google Scholar]
- 33. Plamont M‐A, Billon‐Denis E, Maurin S, et al. Small fluorescence‐activating and absorption‐shifting tag for tunable protein imaging in vivo. Proc Natl Acad Sci U S A. 2016;113(3):497‐502. 10.1073/pnas.1513094113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Li C, Plamont M‐A, Sladitschek HL, et al. Dynamic multicolor protein labeling in living cells. Chem Sci. 2017;8(8):5598‐5605. 10.1039/c7sc01364g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Larsen MB, Perez Verdaguer M, Schmidt BF, Bruchez MP, Watkins SC, Sorkin A. Generation of endogenous pH‐sensitive EGF receptor and its application in high‐throughput screening for proteins involved in clathrin‐mediated endocytosis. Elife. 2019;8:e46135 10.7554/eLife.46135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wang Y, Telmer CA, Schmidt BF, et al. Fluorogen activating protein‐affibody probes: modular, no‐wash measurement of epidermal growth factor receptors. Bioconjug Chem. 2015;26(1):137‐144. 10.1021/bc500525b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wang Y, Ballou B, Schmidt BF, et al. Affibody‐targeted fluorogen activating protein for in vivo tumor imaging. Chem Commun. 2017;53(12):2001‐2004. 10.1039/c6cc09137g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ackerman DS, Altun B, Kolodieznyi D, Bruchez MP, Tsourkas A, Jarvik JW. Antibody‐linked fluorogen‐activating proteins for antigen detection and cell ablation. Bioconjug Chem. 2019;30(1):63‐69. 10.1021/acs.bioconjchem.8b00720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Holleran J, Brown D, Fuhrman MH, Adler SA, Fisher GW, Jarvik JW. Fluorogen‐activating proteins as biosensors of cell‐surface proteins in living cells. Cytometry A. 2010;77(8):776‐782. 10.1002/cyto.a.20925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Saunders MJ, Szent‐Gyorgyi C, Fisher GW, Jarvik JW, Bruchez MP, Waggoner AS. Fluorogen activating proteins in flow cytometry for the study of surface molecules and receptors. Methods. 2012;57(3):308‐317. 10.1016/j.ymeth.2012.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wu Y, Tapia PH, Gouveia K, et al. Inhibitors of FAP‐fluorogen interaction as a multiplex assay tool compound for receptor internalization assays Probe Reports from the NIH Molecular Libraries Program. Bethesda, MD: National Center for Biotechnology Information (US); 2010. [PubMed] [Google Scholar]
- 42. Wu Y, Tapia PH, Fisher GW, Waggoner AS, Jarvik J, Sklar LA. High‐throughput flow cytometry compatible biosensor based on fluorogen activating protein technology. Cytometry A. 2013;83(2):220‐226. 10.1002/cyto.a.22242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lorenz‐Guertin JM, Wilcox MR, Zhang M, et al. A versatile optical tool for studying synaptic GABAA receptor trafficking. J Cell Sci. 2017;130(22):3933‐3945. 10.1242/jcs.205286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Shiwarski DJ, Darr M, Telmer CA, Bruchez MP, Puthenveedu MA. PI3K class II α regulates δ‐opioid receptor export from the trans‐Golgi network. Mol Biol Cell. 2017;28(16):2202‐2219. 10.1091/mbc.E17-01-0030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Perkins LA, Yan Q, Schmidt BF, et al. Genetically targeted ratiometric and activated ph indicator complexes (traphic) for receptor trafficking. Biochemistry. 2018;57(5):861‐871. 10.1021/acs.biochem.7b01135. [DOI] [PubMed] [Google Scholar]
- 46. Hauser AS, Attwood MM, Rask‐Andersen M, Schiöth HB, Gloriam DE. Trends in GPCR drug discovery: new agents, targets and indications. Nat Rev Drug Discov. 2017;16(12):829‐842. 10.1038/nrd.2017.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hill SJ. G‐protein‐coupled receptors: past, present and future. Br J Pharmacol. 2006;147(Suppl 1):S27‐S37. 10.1038/sj.bjp.0706455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hislop JN, von Zastrow M. Analysis of GPCR localization and trafficking. Methods Mol Biol. 2011;746:425‐440. 10.1007/978-1-61779-126-0_25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Barak LS, Ferguson SS, Zhang J, Martenson C, Meyer T, Caron MG. Internal trafficking and surface mobility of a functionally intact beta2‐adrenergic receptor‐green fluorescent protein conjugate. Mol Pharmacol. 1997;51(2):177‐184. 10.1124/mol.51.2.177. [DOI] [PubMed] [Google Scholar]
- 50. Kumagai H, Ikeda Y, Motozawa Y, et al. Quantitative measurement of GPCR endocytosis via pulse‐chase covalent labeling. PLoS One. 2015;10(5):e0129394 10.1371/journal.pone.0129394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Calebiro D, Rieken F, Wagner J, et al. Single‐molecule analysis of fluorescently labeled G‐protein‐coupled receptors reveals complexes with distinct dynamics and organization. Proc Natl Acad Sci U S A. 2013;110(2):743‐748. 10.1073/pnas.1205798110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Levoye A, Zwier JM, Jaracz‐Ros A, et al. A broad G protein‐coupled receptor internalization assay that combines SNAP‐tag labeling, diffusion‐enhanced resonance energy transfer, and a highly emissive terbium cryptate. Front Endocrinol (Lausanne). 2015;6:167 10.3389/fendo.2015.00167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Maurel D, Comps‐Agrar L, Brock C, et al. Cell‐surface protein‐protein interaction analysis with time‐resolved FRET and snap‐tag technologies: application to GPCR oligomerization. Nat Methods. 2008;5(6):561‐567. 10.1038/nmeth.1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Böhme I, Beck‐Sickinger AG. Illuminating the life of GPCRs. Cell Commun Signal. 2009;7:16 10.1186/1478-811X-7-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ward RJ, Xu T‐R, Milligan G. GPCR oligomerization and receptor trafficking. Methods Enzymol. 2013;521:69‐90. 10.1016/B978-0-12-391862-8.00004-1. [DOI] [PubMed] [Google Scholar]
- 56. Kallal L, Benovic JL. Using green fluorescent proteins to study G‐protein‐coupled receptor localization and trafficking. Trends Pharmacol Sci. 2000;21(5):175‐180. 10.1016/S0165-6147(00)01477-2. [DOI] [PubMed] [Google Scholar]
- 57. Tanowitz M, von Zastrow M. A novel endocytic recycling signal that distinguishes the membrane trafficking of naturally occurring opioid receptors. J Biol Chem. 2003;278(46):45978‐45986. 10.1074/jbc.M304504200. [DOI] [PubMed] [Google Scholar]
- 58. Wieduwilt MJ, Moasser MM. The epidermal growth factor receptor family: biology driving targeted therapeutics. Cell Mol Life Sci. 2008;65(10):1566‐1584. 10.1007/s00018-008-7440-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Herbst RS. Review of epidermal growth factor receptor biology. Int J Radiat Oncol Biol Phys. 2004;59(2 Suppl):21‐26. 10.1016/j.ijrobp.2003.11.041. [DOI] [PubMed] [Google Scholar]
- 60. Ståhl S, Gräslund T, Eriksson Karlström A, Frejd FY, Nygren P‐Å, Löfblom J. Affibody molecules in biotechnological and medical applications. Trends Biotechnol. 2017;35(8):691‐712. 10.1016/j.tibtech.2017.04.007. [DOI] [PubMed] [Google Scholar]
- 61. Perez Verdaguer M, Larsen M, Bruchez M, Watkins S, Sorkin A. Analysis of EGF receptor endocytosis using fluorogen activating protein tagged receptor. Bio Protoc. 2019;9(24):e3463 10.21769/BioProtoc.3463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Luscher B, Fuchs T, Kilpatrick CL. GABAA receptor trafficking‐mediated plasticity of inhibitory synapses. Neuron. 2011;70(3):385‐409. 10.1016/j.neuron.2011.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Schweizer C. The γ2 subunit of GABAA receptors is required for maintenance of receptors at mature synapses. Mol Cell Neurosci. 2003;24(2):442‐450. 10.1016/S1044-7431(03)00202-1. [DOI] [PubMed] [Google Scholar]
- 64. Overington JP, Al‐Lazikani B, Hopkins AL. How many drug targets are there? Nat Rev Drug Discov. 2006;5(12):993‐996. 10.1038/nrd2199. [DOI] [PubMed] [Google Scholar]
- 65. Ashcroft FM. From molecule to malady. Nature. 2006;440(7083):440‐447. 10.1038/nature04707. [DOI] [PubMed] [Google Scholar]
- 66. Smyth JW, Shaw RM. Visualizing ion channel dynamics at the plasma membrane. Heart Rhythm. 2008;5(6 Suppl):S7‐S11. 10.1016/j.hrthm.2008.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Bocksteins E, Shepherd AJ, Mohapatra DP, Snyders DJ. Immunostaining of voltage‐gated ion channels in cell lines and neurons—key concepts and potential pitfalls In: Dehghani H, ed. Applications of Immunocytochemistry. IntechOpen; 2012. 10.5772/34817. [DOI] [Google Scholar]
- 68. Zheng J. Handbook of Ion Channels. 1st ed. Boca Raton, FL: Crc Press; 2016:691. [Google Scholar]
- 69. Shruti S, Urban‐Ciecko J, Fitzpatrick JA, Brenner R, Bruchez MP, Barth AL. The brain‐specific Beta4 subunit downregulates BK channel cell surface expression. PLoS One. 2012;7(3):e33429 10.1371/journal.pone.0033429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Holleran JP, Glover ML, Peters KW, et al. Pharmacological rescue of the mutant cystic fibrosis transmembrane conductance regulator (CFTR) detected by use of a novel fluorescence platform. Mol Med. 2012;18:685‐696. 10.2119/molmed.2012.00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Holleran JP, Zeng J, Frizzell RA, Watkins SC. Regulated recycling of mutant CFTR is partially restored by pharmacological treatment. J Cell Sci. 2013;126(Pt 12):2692‐2703. 10.1242/jcs.120196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Larsen MB, Hu J, Frizzell RA, Watkins SC. Simple image‐based no‐wash method for quantitative detection of surface expressed CFTR. Methods. 2016;96:40‐45. 10.1016/j.ymeth.2015.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Pratt CP, Kuljis DA, Homanics GE, et al. Tagging of endogenous BK channels with a fluorogen‐activating peptide reveals β4‐mediated control of channel clustering in cerebellum. Front Cell Neurosci. 2017;11:337 10.3389/fncel.2017.00337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Hager NA, Krasowski CJ, Mackie TD, et al. Select α‐arrestins control cell‐surface abundance of the mammalian Kir2.1 potassium channel in a yeast model. J Biol Chem. 2018;293(28):11006‐11021. 10.1074/jbc.RA117.001293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Ratjen F, Döring G. Cystic fibrosis. Lancet. 2003;361(9358):681‐689. 10.1016/S0140-6736(03)12567-6. [DOI] [PubMed] [Google Scholar]
- 76. Kopito RR. Biosynthesis and degradation of CFTR. Physiol Rev. 1999;79(1 Suppl):S167‐S173. 10.1152/physrev.1999.79.1.S167. [DOI] [PubMed] [Google Scholar]
- 77. Wu RS, Marx SO. The BK potassium channel in the vascular smooth muscle and kidney: α‐ and β‐subunits. Kidney Int. 2010;78(10):963‐974. 10.1038/ki.2010.325. [DOI] [PubMed] [Google Scholar]
- 78. N'Gouemo P. Targeting BK (big potassium) channels in epilepsy. Expert Opin Ther Targets. 2011;15(11):1283‐1295. 10.1517/14728222.2011.620607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Faber ESL, Sah P. Calcium‐activated potassium channels: multiple contributions to neuronal function. Neuroscientist. 2003;9(3):181‐194. 10.1177/1073858403009003011. [DOI] [PubMed] [Google Scholar]
- 80. Hibino H, Inanobe A, Furutani K, Murakami S, Findlay I, Kurachi Y. Inwardly rectifying potassium channels: their structure, function, and physiological roles. Physiol Rev. 2010;90(1):291‐366. 10.1152/physrev.00021.2009. [DOI] [PubMed] [Google Scholar]
- 81. Caire MJ, Varacallo M. Physiology, synapse StatPearls. Treasure Island, FL: StatPearls Publishing; 2019. [PubMed] [Google Scholar]
- 82. Chang C‐W, Chiang C‐W, Jackson MB. Fusion pores and their control of neurotransmitter and hormone release. J Gen Physiol. 2017;149(3):301‐322. 10.1085/jgp.201611724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Bulgari D, Deitcher DL, Schmidt BF, et al. Activity‐evoked and spontaneous opening of synaptic fusion pores. Proc Natl Acad Sci U S A. 2019;116(34):17039‐17044. 10.1073/pnas.1905322116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Kuljis DA, Park E, Telmer CA, et al. Fluorescence‐based quantitative synapse analysis for cell type‐specific connectomics. Eneuro. 2019;6(5). 10.1523/ENEURO.0193-19.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Jing C, Cornish VW. A fluorogenic TMP‐tag for high signal‐to‐background intracellular live cell imaging. ACS Chem Biol. 2013;8(8):1704‐1712. 10.1021/cb300657r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Leng S, Qiao Q‐L, Gao Y, Miao L, Deng W‐G, Xu Z‐C. SNAP‐tag fluorogenic probes for wash free protein labeling. Chin Chem Lett. 2017;28:1911‐1915. 10.1016/j.cclet.2017.03.034. [DOI] [Google Scholar]
- 87. Liu Y, Miao K, Dunham NP, et al. The cation‐π interaction enables a halo‐tag fluorogenic probe for fast no‐wash live cell imaging and gel‐free protein quantification. Biochemistry. 2017;56(11):1585‐1595. 10.1021/acs.biochem.7b00056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Prifti E, Reymond L, Umebayashi M, Hovius R, Riezman H, Johnsson K. A fluorogenic probe for SNAP‐tagged plasma membrane proteins based on the solvatochromic molecule Nile Red. ACS Chem Biol. 2014;9(3):606‐612. 10.1021/cb400819c. [DOI] [PubMed] [Google Scholar]
- 89. Hori Y, Ueno H, Mizukami S, Kikuchi K. Photoactive yellow protein‐based protein labeling system with turn‐on fluorescence intensity. J Am Chem Soc. 2009;131(46):16610‐16611. 10.1021/ja904800k. [DOI] [PubMed] [Google Scholar]
- 90. Lukinavičius G, Umezawa K, Olivier N, et al. A near‐infrared fluorophore for live‐cell super‐resolution microscopy of cellular proteins. Nat Chem. 2013;5(2):132‐139. 10.1038/nchem.1546. [DOI] [PubMed] [Google Scholar]
- 91. Butkevich AN, Mitronova GY, Sidenstein SC, et al. Fluorescent rhodamines and fluorogenic carbopyronines for super‐resolution STED microscopy in living cells. Angew Chem Int Ed Engl. 2016;55(10):3290‐3294. 10.1002/anie.201511018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Grimm JB, English BP, Chen J, et al. A general method to improve fluorophores for live‐cell and single‐molecule microscopy. Nat Methods. 2015;12(3):244‐250, 3 p following 250. 10.1038/nmeth.3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Grimm JB, English BP, Choi H, et al. Bright photoactivatable fluorophores for single‐molecule imaging. Nat Methods. 2016;13(12):985‐988. 10.1038/nmeth.4034. [DOI] [PubMed] [Google Scholar]
- 94. Grimm JB, Muthusamy AK, Liang Y, et al. A general method to fine‐tune fluorophores for live‐cell and in vivo imaging. Nat Methods. 2017;14(10):987‐994. 10.1038/nmeth.4403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Martineau M, Somasundaram A, Grimm JB, et al. Semisynthetic fluorescent pH sensors for imaging exocytosis and endocytosis. Nat Commun. 2017;8(1):1412 10.1038/s41467-017-01752-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Tebo AG, Gautier A. A split fluorescent reporter with rapid and reversible complementation. Nat Commun. 2019;10(1):2822 10.1038/s41467-019-10855-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Schütz M, Batyuk A, Klenk C, et al. Generation of fluorogen‐activating designed ankyrin repeat proteins (FADAs) as versatile sensor tools. J Mol Biol. 2016;428(6):1272‐1289. 10.1016/j.jmb.2016.01.017. [DOI] [PubMed] [Google Scholar]
- 98. Dou J, Vorobieva AA, Sheffler W, et al. De novo design of a fluorescence‐activating β‐barrel. Nature. 2018;561(7724):485‐491. 10.1038/s41586-018-0509-0. [DOI] [PMC free article] [PubMed] [Google Scholar]