

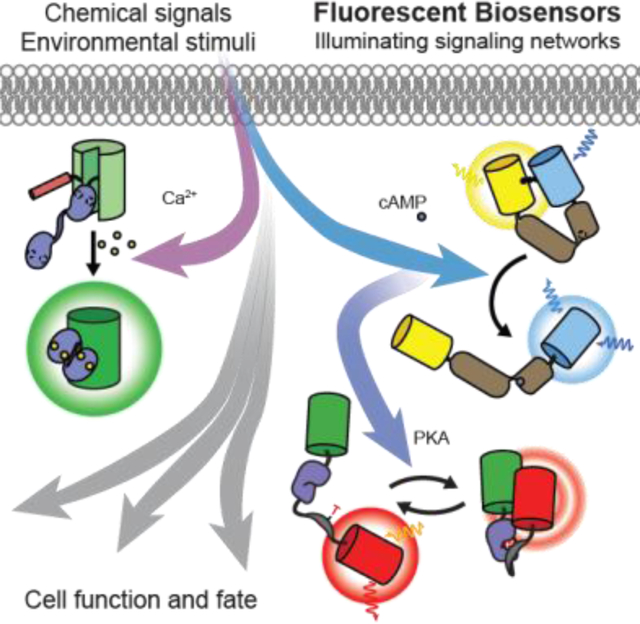
Abstract
Cellular signaling networks are the foundation which determines the fate and function of cells as they respond to various cues and stimuli. The discovery of fluorescent proteins over 25 years ago enabled the development of a diverse array of genetically encodable fluorescent biosensors that are capable of measuring the spatiotemporal dynamics of signal transduction pathways in live cells. In an effort to encapsulate the breadth over which fluorescent biosensors have expanded, we endeavored to assemble a comprehensive list of published engineered biosensors and discuss many of the molecular designs utilized in their development. Then, we review how the high temporal and spatial resolution afforded by fluorescent biosensors has aided our understanding of the spatiotemporal regulation of signaling networks at the cellular and subcellular level. Finally, we highlight some emerging areas of research in both biosensor design and application that are on the forefront of biosensor development.
Graphical Abstract
1. Introduction
One of the most fundamental aspects of life is the ability of cells to respond to external cues originating from other cells or from the environment. Doing so requires cells to exist in a state of dynamic equilibrium, constantly poised to unleash cascades of chemical reactions from which all complex biological phenomena emerge. Through this network of signaling pathways, cells alter their physical state and composition such that they can appropriately determine their fate and perform the functions essential to their role within an organism. In the post-genomic era, wherein the identities of most signaling pathway components are known, elucidating the relationships among these components, and thus the dynamics and regulation of signal transduction, has become one of the foremost avenues towards furthering our understanding of both physiological and pathological cellular function. Because these signaling reactions are often discrete and transitory in nature, the development of fluorescence-based methods capable of monitoring and quantifying signaling dynamics in real time in the endogenous cellular context has proven instrumental to these efforts.
In many ways, fluorescence is an ideal tool for monitoring the behavior of signaling pathways within cells: fluorescence is both rapid, with the absorption and emission of light by a fluorescent molecule (i.e., fluorophore) occurring on the order of nanoseconds, and spatially precise, as the emitted wavelengths are smaller than many cellular structures. Thus, fluorescence-based readouts permit the high-fidelity recording of extremely rapid processes with single-cell and even subcellular spatial resolution. Combined with the development of increasingly sophisticated and sensitive microscopy hardware, fluorescence can be used to nondestructively visualize cellular processes directly in living cells, providing data with a combined temporal and spatial resolution that is not possible through traditional biochemical methods.
Although fluorescence has served as a sensitive in situ label for the better part of a century, beginning with the synthesis of fluorophore-conjugated antibodies and the advent of immunofluorescence1, the first bona fide fluorescent sensors capable of probing intracellular signaling dynamics emerged much later and included covalently labeled versions of endogenous proteins (i.e., “fluorescent analogs”, reviewed in ref. 2) and organic indicator dyes (reviewed in ref. 3). These synthetic probes enabled the first real-time measurements of dynamic changes in cellular parameters such as messenger and ion concentrations. However, this budding field was soon revolutionized by the isolation and engineering of fluorescent proteins (FPs)4–6.
What began with the Aequorea victoria green fluorescent protein (GFP) has expanded through protein engineering and discoveries in other species to encompass a diverse assortment of FPs with unique photophysical properties (recently reviewed in ref 7). Currently, FPs span the colors of the rainbow and beyond, from the near-ultraviolet8 to the near-infrared9, with similarly varying brightness and quantum yield7. In parallel to the development of fluorescent protein technologies, a suite of technologies have been developed for in situ labeling of recombinant proteins (e.g. FlAsH, Halo-Tag, Snap-Tag) (reviewed in ref. [26575238]). These technologies, particularly the development of FPs, opened up new avenues by facilitating the construction of genetically encoded fluorescent biosensors, which can be synthesized directly within cells, expressed in specific subsets of cell populations, and easily fused with different protein tags for targeting to specific subcellular microdomains. As such, a diverse array of genetically encoded fluorescent biosensors have been engineered to monitor the dynamics of various signaling pathway components, including cell-surface receptors, intracellular messengers, and enzymatic effector proteins. Here, we first discuss the different design strategies that have been exploited to develop genetically encoded fluorescent biosensors, including some novel biosensor classes that have been developed recently. To this end, we have endeavored to assemble a comprehensive list of published fluorescent biosensors, which includes over 650 different biosensor variants, as seen in Table 1 and with more detail online at BiosensorDB.ucsd.edu. We then review how fluorescent biosensors have been utilized to examine and dissect the spatiotemporal dynamics of signal transduction and, finally, highlight new avenues of exploration with fluorescent biosensors.
Table 1.
Engineered genetically encoded fluorescent biosensors are organized by their signaling targets. For each signaling target, the different published biosensors are organized into families of related variants (with the family or group name shown in bold). The different read-out mechanisms utilized are shown with the different icons indicating the type of readout for a given biosensor. The different readout mechanisms shown in this table are: Bioluminescence intensity (●), BRET (◓), Intensity-based FRET (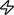 ), FLIM-FRET (♣), FLINC (
), FLIM-FRET (♣), FLINC ( ), Intensity (
), Intensity ( ), Ratiometric (with a reference FP) (◧), Excitation Ratiometric (★), Emission Ratiometric (☆), and Translocation (
), Ratiometric (with a reference FP) (◧), Excitation Ratiometric (★), Emission Ratiometric (☆), and Translocation ( ). Additional details, including biosensor components and FPs involved, etc., can be found in the supplemental materials and at BiosensorDB.ucsd.edu
). Additional details, including biosensor components and FPs involved, etc., can be found in the supplemental materials and at BiosensorDB.ucsd.edu
| Target | Readout Mech | Variants | |
|---|---|---|---|
| Cell Cycle | |||
| Cell Cycle Phases | Inten. () |
Fucci: Fucci(CA)2.1 (2017)207 Fucci4 (2016)208NIR Fucci (2016)209
NIR cell cycle reporter (2016)210
Fucci2.1 (2013)211Fucci2 (2011)212zFucci(2009)213S/G2/M-X(NC) (2008)214Fucci (2008)215
|
|
| G1 Phase | Trans. () |
G1 Phase Biosensor (2009)216
|
|
| S Phase | Trans. () |
S Phase Biosensor (2009)216
|
|
| Cell Environment | |||
| Mechanical Strain | FRET () Ex. Rat.(★) |
HP35: HP35st-TS (2015)217HP35-TS (2015)217
PriSSM (2008)218 ★ stFRET: cpstFRET (2012)219 sstFRET(2011)220stFRET (2008)221TSMod: TSMod F25 (2016)222 TSMod (2010)223
|
|
| pH | Em. Rat. (☆) Rat. (◧) FRET ( ) Inten. ( ) Ex. Rat.(★) |
deGFP: deGFP family (2002)130☆ Dual pH and Cl sensor: ClopHensor(2010)138◧ ExGFP: E1GFP (2007)137 ◧ E2GFP (2006)134 ◧ FluBpH: FluBpH 7.5 (2017)224 FluBpH 6.1 (2017)224
FluBpH 5.7 (2017)224
mtAlpHi (2004)225 Native FP pH Sensitivity: mNect (2009)226 pHVenus(2007)125
CFP-YFP pH FRET (2004)227
EYFP (1998)121 EGFP (1998)120
pHCEC: pHCECSensor01 (2008)228 ◧ pHluorins: pHluorin2 (2011)229 ★Superecliptic-pHluorin (2000)122 ratiometric-pHluorin (1998)117★ Ecliptic-pHluorin (1998)117pHRed (2011)230★ pHTomato (2012)123 pHuji (2014)231 pHusion (2012)232◧ SypHer (2011)233 XFpH: YFpH (2001)234 GFpH (2001)234
|
|
| Membrane Voltage (V+) | Inten. () BRET(◓) Rat. (◧) FRET ( ) |
ArcLight Species Variants: Human Q193 ArcLight (2013)235
Zebrafish Q175 ArcLight (2013)235 Chicken Q175 ArcLight (2013)235 Frog Q174 ArcLight (2013)235
Ci-VSP based: VSD-FR189–188 (2017)236 LOTUS-V (2017)237
◓ Marina (2017)238 tdFlicR1 Δ110AR (2016)239 FlicGR1 (2016)239 ASAP2f (2016)176
tdFlicR-VK-ASAP (2016)239
◧ FlicR1 (2016)177 Bongwoori (2015)240 ArcLightning (2015)241 Nabi2.213 (2015)242 ASAP1 (2014)167 Chimeric VSFP-Butterfly YR (2014)243
Chimeric VSFP-Butterfly CY (2014)243
Mermaid2 (2013)244 ArcLight A242 (2012)245 Chimera Cx (2012)246 VSFP-CR (2012)247
ElectricPk (2012)174 VSFP Butterfly 1.2 (2012)248 VSFP2.3 (2010)249 VSFP2.42 (2010)250 VSFP2.4 (2009)251 VSFP3.1 (2008)172 Mermaid (2008)252 VSFP2.1 (2007)253Danio VSP based: Zahara 2 SE (227D) (2015)254 Zahra 1 SE (227D) series (2015)254
Zahra 1 (2012)255
Zahra 2 (2012)255
Kv based: FlaSH (CFP) + FlaSh (YFP) (2002)256 FlaSh L366A (2002)256
FlaSh IR (2002)256
VSFP1 (2001)257 FlaSh (1997)168
Na Channel based: SPARC (2002)169
Nematostella VSP based: Nema (2012)255 Rhodopsin based: Archon1 (2018)258 Archon2 (2018)258 FRET-opsin Ace1Q-mNeon (2015)259 FRET-opsin Ace2N-mNeon (2015)259 QuasAr2 (2014)260 QuasAr1 (2014)260 Archer1 (2014)261 eFRET GEVI (2014)262
FRET-opsin Mac-mCitrine (2014)263 Arch-EEx variants (2013)264
PROPS (2011)265 Arch(D95N) (2011)266
|
|
| Cellular Analytes | |||
| 2-Oxoglutarate | Inten. () |
mOGsor (2014)267
|
|
| Ammonium | Rat. (◧) Inten. ( ) |
AmTrac: AmTryoshika1;3-LS-F138I -T78H (2017)206◧ AmTryoshika1;3-LS-F138I (2017)206◧ AmTrac (2013)268
|
|
| Arabinose | FRET () |
FLIPara: FLIPara-250n (2008)269
|
|
| Arginine | FRET () |
FRET Arg Reporter (2007)270
|
|
| ATP | BRET(◓) FRET () Inten. ( ) Biolum. (●) Ex. Rat.(★) |
ATeam: BTeam (2016)271◓ ATeam1.03NL (2013)272 GO-Ateam (2011)273 ATeam3.10 (2009)274 ATeam1.03 (2009)274 EAF-ATP (2013)275
Nano-lantern ATP (2012)276● Perceval: PercevalHR (2013)277★ Perceval (2009)278★ QUEEN: QUEEN-2m (2014)279★ QUEEN-7mu (2014)279★ Syn-ATP (2014)280● |
|
| BDNF | FRET () |
Bescell (2008)281
|
|
| cAMP | Inten. () FRET ( ) BRET(◓) Rat. (◧) Biolum. (●) |
cADDis: cADDis-green (2016)182 Camps: Epac2-camps300-cit (2010)282 Epac2-camps300 (2009)283 HCN2-camps (2006)284Epac2-camps (2004)285 Epac1-camps (2004)285 PKA-camps (2004)285CAMYEL (2007)286◓ CUTie (2017)287 Epac-S: Epac-S H188 (2015)288 Epac-S H187 (2015)288 H74 (2011)289 H96 (2011)289 H84: CFP(nd)-EPAC(dDEP/CD)-Venus(d) (2008)290 H81: GFP(nd)-EPAC(ΔDEP)-mRFP (2008)290 H90: CFP(nd)-EPAC(ΔDEP/CD)-cp173Venus(d) (2008)290Epac1 (ΔDEP-CD) based: CEPAC* (2011)291 CFP-Epac(ΔDEP-CD)-YFP (2004)292Flamindo: Pink Flamindo (2017)183 Flamindo2 (2014)180 Flamindo (2013)163
FPX: pPHT-PKA (2015)293◧ ICUE: ICUPID (2011)294 ICUE-YR (2011)294 ICUE3 (2009)295 Rluc-EPAC-YFP (2008)296◓ ICUE2 (2008)297 ICUE1 (2004)298mCRIS based: cit-mCNBD-cer (2013)299
mICNBD-FRET (2016)300 Nano-lantern cAMP: Nano-lantern cAMP1.6 (2012)276● R1α #7 (2016)301 Split Luc cAMP biosensor: 22F (2011)302● Split Luc cAMP biosensor (2008)303● YFP-PKAc + CFP-PKAr: ΔPKA RIIb-CFP + PKAc-YFP (2006)304 R2-Rluc + GFP-C (2006)305◓ R1-Rluc + GFP-C (2006)305◓ YFP-PKAc + CFP-PKAr(R230K) (2004)306 YFP-PKAc + CFP-PKAr (2002)307 PKAc-S65T + PKArII-EBFP (2000)308
|
|
| cGMP | Inten. () FRET ( ) Ex. Rat.(★) |
cGES: Cygnus (2010)309 cGES-DE5 (2006)310
cGKI based: cGi family (2007)311
cGull: Green-cGull (2017)164
CGY: CGY-del1 (2000)312 cygnet: cygnet-2.1 (2005)313 cygnet-2 (2001)314FlincG: H6-FGAM (2013)315 H6-FGB (2013)315 δ-FlincG (2008)165★ |
|
| Citrate | FRET () |
FLIP-Cit: FLIP-Cit-Y (2011)316
|
|
| Carbon monoxide (CO) | Inten. () |
COSer (2012)317
|
|
| Glucose | FRET () |
FLIPglu: FLII12Pglu-Y Series (2008)318 FLIPglu-YΔ13 (2006)319 FLIIXPglu-Y Series (2005)320 FLIPglu-600u-Δ(X) series (2005)320 FLIPglu-Y series (2003)321 GIP: AcGFP1-GBPcys-mCherry (2010)322 GIP C0Yi (2008)323 GIP (2003)324
|
|
| Glutamine | FRET () |
FLIPQ: FLIPQ-TV3.0 (2012)325
|
|
| Heme | FRET () Rat. (◧) Inten. ( ) |
CHY: CH49Y (2017)326 CISDY: CISDY-9 (2015)327 Fluorescence quenching Heme: HS1 (2016)328◧ HS1–M7A (2016)328◧ CG6 (2012)329
|
|
| Histidine | FRET () |
FLIP-HisJ: FLIP-cpHisJ194 (2009)330
|
|
| Insulin | Rat. (◧) | RINS: RINS1 (2017)331◧ | |
| Lactate | FRET () |
Laconic (2013)332
|
|
| Maltose | BRET(◓) FRET ( ) Inten. ( ) |
FLIPmal: GFP2-MBP-RLuc2 (2013)333◓ FLIPmal-YΔ1 (2008)269 FLIPmal-Y Series (2002)334 MBP: PPYF-green (2011)193
|
|
| NADH | Inten. () Rat. (◧) Ex. Rat.(★) |
Frex: RexYFP (2014)335 FrexH (2011)336 Frex (2011)336
NAD+ Biosensor (2016)337
Peredox (2011)201◧ SoNar (2015)197★ |
|
| NADPH | FRET () Ex. Rat.(★) |
Apollo-NADP+ (2016)338 iNap: iNap3 (2017)198★ iNap2 (2017)198★ iNap1 (2017)198★ NADPsor (2016)339
|
|
| O2 | Inten. () FRET ( ) |
dUnOHR hypoxia-reoxygenation sensor (2016)340
FluBO (2012)341
|
|
| Phosphonate | Inten. () |
EcPhnD based: EcPhnD-cpGFP (2011)194
|
|
| Pyruvate | FRET () |
Pyronic: Pyronic (2014)342
|
|
| Ribose | FRET () |
FLIPrib: FLIPrib-Y family (2003)343
|
|
| Sucrose | FRET () |
FLIPsuc: FLIPsuc-Y family (2006)344
|
|
| Trehalose | FRET () |
FLIP: FLIPSuc90μΔ1Venus (2016)345
|
|
| Tryptophan | FRET () |
FLIPW (2007)346
|
|
| Ions | |||
| Ca2+ | BRET(◓) FRET ( ) Inten. ( ) Rat. (D)Ex. Rat.(★) Em. Rat. (☆) Biolum. (®) |
Ca2+
Snapshot: FLARE(2017)347Cal-Light(2017)348 Cameleons: D3 BRET (2015)349◓ D1GO-Cam (2012)350 CaYang1 (2011)351 CaYin1 (2011)351 YC-Nano (2010)352 D3cpv (2006)353 D4cpV (2006)353 D1 (2004)354 YC3.60 (2004)355 YC2.12 (2002)356 YC6.1 (2001)357 YC2.1 (1999)358 Split-Cameleon (1997)359Cameleon 3 (1997)359 Camgaroo: Camgaroo2 (2001)360 Camgaroo1 (1999)144CaMPARI: CaMPARI (2015)361 CASE: Case16 (2007)362 Case12 (2007)362CatchER (2011)363 ddFP Ca2+ sensor (2012)364 ER-GCaMP: ER-GCaMP6-150(2017)365 ER-GCaMP6a-210(2017)365
ER-GCaMP3–373 / GCaMPer-10.19(2015)366 CEPIA1er (2014)367
ER-GECO: R-CEPIA1er (2014)367 GEM-CEPIA1er (2014)367☆ G-CEPIA1er (2014)367FPX Ca2+ sensors: tripartate FPX Ca2+ Sensor (2015)293◧ single polypeptide FPX Ca2+ sensor (2015)293◧ GAP: GAP3 (2016)368 GAP1 (2014)369GCaMP: MatryoshCaMP6s (2017)206◧ sfMatryoshCaMP6s (2017)206◧ sfMatryoshCaMP6s-T78H (2017)206◧ GCaMP-R-3 (2017)202◧ GCaMP-R-6f (2017)202◧ GCaMP-R-6s (2017)202◧ GCaMP3bright (2015)370 GCaMP3fast370 GCaMP2.2Low (2014)371 GCaMP6f (2013)156 GCaMP6s (2013)156 GCaMP6m (2013)156 GCaMP5G (2012)155 G-CaMP 3–8 (Nakai Variants) (2012)372GCaMP-HS(2011)373 GCaMP3 (2009)154 GCaMP2 (2006)153 GCaMP (2001)150GECO: K-GECO1(2018)374 jRGECO1a (2016)162 REX-GECO (2014)196★ LAR-GECO (2014)375 GR-GECO (2013)376 R-GECO1.2 (2013)159 O-GECO (2013)159 CAR-GECO (2013)159 G-GECO1 (2011)158 B-GECO (2011)158 G-GECO1.2 (2011)158 R-GECO1 (2011)158 GEM-GECO (2011)158☆ GEX-GECO (2011)158★ G-GECO1.1 (2011)158Nano-lantern Ca2+: GeNL(Ca2+) (2016)377◓ ONL(Ca2+)(2015)378◓ CNL(Ca2+)(2015)378◓ Nano-lantern Ca2+ (2012)276● Pericam: ratiometric-pericam (2001)148◧ inverse-pericam (2001)148 flash-pericam (2001)148RCaMP: jRCaMP1b (2016)162 jRCaMP1a (2016)162 R-CaMP2 (2015)379 RCaMP1h (2013)160 R-CaMP1.07 (2012)380
TN Ca2+ Sensors: CalfluxVTN (2016)381◓ Twitch series (2014)382 YO-TnC1.0 (2012)383 TN-XXL (2008)384 TN-XL (2006)385 TN-humTnC (2004)386 TN-L15 (2004)386
|
|
| Cd2+ | FRET () |
Cd-FRET: Cd-FRET2 (2011)142
|
|
| Cu+ | FRET () |
Yeast Copper regulator based: Ace1-FRET (2011)387 Mac1-FRET (2011)387 Amt1-FRET (2010)388
|
|
| Halide Ions (Cl−, I−) | FRET () Inten. () |
Clomeleon: SuperClomeleon (2013)389 Cl-sensor (2008)136 Clomeleon (2000)135 Dual pH and Cl sensor: ClopHensor(2010)138 ◧ Halide Sensitive Fluorescent Proteins: Cl-YFP (2014)129 YFP(H148Q, I152L) (2001)128 YFP(H148Q) (2000)126
|
|
| Hg(II) | Inten. () |
eGFP based: eGFP205C (2008)390
IFP based: IFP based Hg sensor (2011)391
|
|
| Phosphate | FRET () |
FLIPPi: FLIPPi-5μ (2006)392 FLIPPi-30m (2006)392
|
|
| Zn2+ | BRET(◓) FRET ( ) |
Atox1 WD4 Based: BLCALWY-1 (2016)393◓ redCALWY (2013)394 eCALWY series (2009)395 CALWY (2007)396 CFP-Atox1 + WD4-YFP (2006)397 Minimal Zinc Finger based: mito-ZifCY1.173(2012)398 His4-Zn2+ sensor (2009)399 Cys2Hys2 - Zn2+ Sensor (2009)399Zap1 based: ZapCY1 (2011)400 ZF1/2-FRET (2006)401ZinCh: BLZinCh-2 (2016)393◓ BLZinCh-3 (2016)393◓ eZinCh-2 (2015)402 eZinCh-1 (2011)142 Cly9–2His (2008)403 ZinCh-x (2007)141
|
|
| Kinases/Phosphatases | |||
| Abl | FRET () |
Abl Indicator: Crk-based Reporter (2001)404
|
|
| Akt | FRET () Trans. ( ) FLIM (♣) |
Akind (2006)405 Akt translocation Reporters: Akt-FoxO3a-KTR-EGFP (2016)110 FoxO1-clover (2015)109AktAR: AktAR2 (2015)406 AktAR (2008)407AktUS (2003)408 BKAR: BKAR v2 (2014)409 BKAR (2005)410Dual Labeled Akt: GFP-PKB-RFP (2007)411 GFP-AKT-YFP (2003)412♣ Eevee-Akt: Eevee-iAkt (2014)413 Eevee-Akt (2011)414ReAktion: ReAktion1 (2007)415
|
|
| AMPK | FRET () |
AMPKAR: bimABKAR (2015)416 ABKAR (2015)417 AMPKAR (2011)418
|
|
| ATM kinase | FRET () |
ATOMIC (2007)419
|
|
| Aurora B kinase | FRET () |
Aurora B sensor: Aurora B sensor (Chu) (2011)420Aurora B Sensor (Fuller) (2008)421
|
|
| Aurora Kinase A | FRET () |
AURKA Biosensor (2016)422
|
|
| B-Raf | FRET () |
Prin-BRaf (2006)423
|
|
| Bcr-Abl | FRET () |
Pickles: Pickles-2.3 (2010)424
|
|
| C-Raf | FRET () |
Prin-CRaf (2005)425
|
|
| CaMKII | FLIM (♣) FRET () |
Camui: Camuiα-mRmC (2016)426♣ ShadowG-Camuiα (2015)427♣ Camuiα-CR (2012)247 green-Camuiα (2009)428♣ mRFP/GFP-Camuiα (2008)429♣ Camuiα (2005)430
|
|
| CaN | FRET () |
CaNAR: CaNAR2 (2014)431 CaNAR1 (2008)432
CaNARi (2014)431 DuoCaN: UniCaN (2015)433 DuoCaN (2015)433
|
|
| CDK1 | FRET () |
CyclinB1-Cdk1 activity sensor: CyclinB1-Cdk1 activity sensor V2 (2014)409 CyclinB1-Cdk1 activity sensor V1 (2010)434
|
|
| CDK2 | Trans. () |
CDK2 Translocation Reporter: DHB-Ven (2013)107
|
|
| DAPK1 | FRET () |
DAPK1(2011)435
|
|
| EGFR | FRET () |
ECaus (2008)281 EGFR Reporter (2001)404^ FLAME: PTB-EYFP, EGFR-ECFP (2004)436 FLAME (2004)436
Picchu-Z (2005)437
|
|
| ERK | FRET () FLIM (♣) BRET (◓) Trans. ( ) FLINC ( ) Rat. (◧) |
EAS: EAS3 (2005)438 EKAR: EKARdual (2017)439♣ bimEKAR (2015)416 EKAR3 (2015)440 EKAREV-TVV (2014)441 EKAR-TVV (2014)441 REV (2013)442◓ EKAR2G (2013)443 EKAREV (2011)414 EKAR (2008)444
ERK-KTR (2014)112 Erkus (2007)445 ERKy (2012)446 FLINC-EKAR1 (2017)447
FPX EKAR (2015)293◧ Miu2 (2006)448
|
|
| FAK | FRET () |
FAK Activation Biosensor: CYFAK413 (2008)449
FAK Autophosphorylation Biosensor (2008)449
FAK sensor (2011)450
|
|
| Fus3 | Trans. () |
Far1-SKARS (2015)113
|
|
| GCK | FRET () |
GCK Activation Biosensors: FRET-GCK (cp173-mCer3/mVen) (2016)451FRET-GCK (mCer/mVen) (2011)452Cerulean-GCK-mCit (2004)173FRET-GCK (2002)453
|
|
| H3–S28p | FRET () |
Histone Phosphorylation reporter (2004)454
|
|
| INSR | BRET(◓) FRET ( ) Inten. ( ) |
Insulin Receptor Activation BRET assay (2001)455◓ Phocus: Phocus-2pp nes (2002)456 Phocus2 (2002)456Sinphos: yellow-sinphos (2004)457 green-sinphos (2004)457cyan-sinphos (2004)457
|
|
| JNK | FRET () Trans. ( ) |
dJUN-FRET (2008)458 JNK-KTR (2014)112 JNKAR: bimJNKAR (2015)416 JNKAR1EV (2011)414 JNKAR1 (2010)459JuCKY (2010)460
|
|
| Kss1 | Trans. () |
Ste7-SKARS: Ste7-SKARS (2015)113
|
|
| LATS | Biolum. (●) | LATS-BS(2018)461 ● | |
| Lck | FRET () |
Lck Activation sensor: CLckY-2 (2009)462
|
|
| MAPK/MK2 | FRET () |
GMB (2001)463
|
|
| MARK | FRET () |
MARK sensor: MARK-AR1 (2011)464
|
|
| Mpk1 | Trans. () |
Mkk2-SKARS (2015)113
|
|
| mTORC1 | FRET () |
TORCAR (2015)406
|
|
| p38 | FRET () Trans. () |
p38 activity reporter (2015)465
p38-KTR (2014)112
|
|
| PAK1 | FRET () |
Pakabi (2009)466
|
|
| PDGFR | FRET () |
PDGFR Biosensor: PDGFR Biosensor(2017)467
|
|
| PDK1 | FRET () |
PARE (2011)468
|
|
| PKA | FLIM (♣) FRET ( ) Biolum. (●) FLINC ( ) Trans. ( ) Inten. ( ) |
AKAR: AKARdual (2017)439♣ AKAR5(2017)469♣ AKAR4.1 (2015)470 FLIM-AKAR (2014)471♣ AKAR-CR (2012)247 bimAKAR (2011)472 LumAKAR (2011)472● AKAR3EV (2011)414 AKAR4 (2011)473 ICUPID (2011)294 AKAR-GR (2011)294 CRY-AKAR (2008)474 AKAR3 (2006)475 AKAR2 (2005)476 AKAR1 (2001)477 ART (2000)478 FLINC-AKAR1 (2017)447 PKA-KTR (2014)112 RLuc-PCA PKA (2007)479● Single Color PKA sensor: GAk (2014)480
|
|
| PKC | FRET () |
CKAR (2003)481 Eevee-PKC (2011)414 KCP: KCAP-1 (2006)482 KCP-2 (2006)482 KCP-1 (2004)483
|
|
| PKD | FRET () |
DKAR (2007)484 G-PKDrep: G-PKDrep live (2012)485 G-PKDrep (2009)486
|
|
| PKM2 | FRET () |
PKAR: PKAR2.3 (2013)487
|
|
| PTEN | BRET(◓) | Rluc-PTEN-YFP (2014)488◓ | |
| ROCK | FRET () |
Eevee-ROCK (2017)489
|
|
| RSK | FRET () |
Eevee-RSK: Eevee-RSK (2011)414
|
|
| RTK | FRET () |
Picchu: PicchuEV (2011)414 Picchu (2001)490
|
|
| S6K | FRET () |
Eevee-S6K: Eevee-S6K(2011)414
|
|
| SAP3K | FRET () |
SAP3K (2009)491
|
|
| Src | FRET () |
Src Indicator: BG-Src1.0 (2013)492 Y0-Src1.0 (2012)383 Src Reporter (ECFP/YPet) (2008)493 Src Reporter (2005)494 Src Indicator (2001)404
Srcus (2007)495
|
|
| ZAP-70 | FRET () |
ROZA (2008)496
|
|
| Neurotransmitters | |||
| 5-HT | FRET () |
5-HT 3A CNiFER: 5-HT 3A LGIC CNiFER (2011)497
|
|
| ACh | FRET () |
ACh CNiFER: α402-nAChR LGIC CNiFER (2011)497 α7 LGIC CNiFER (2011)497 M1-CNiFER (2010)498
|
|
| Dopamine | FRET () |
Dopamine CNiFER: D2 CNiFER (2014)499
|
|
| Glutamate | FRET () Inten. ( ) |
FLIP Glt: FLIP-cpGltI210 (2009)330 FLIPE: FLII81PE-1u (2005)320 FLIPE-Y Surface series (2005)500FLIPE-Y Series (2005)500GluSnFR: superGluSnFR (2008)501 iGluSnFR (2013)166 QBP based: Gln D157N reporter (2010)502
|
|
| NE | FRET () |
NE CNiFER: α1A CNiFER (2014)499
|
|
| Phosphoinositides/Lipids | |||
| 3’ IP | Trans. () FRET ( ) |
GFP AKT domains: GFP-Akt(1999)87GFP-AH(1999)87
Homo FRET mCherry-Akt-PH: mCherry-Akt-PH (2015)503
InPAkt (2005)504
|
|
| DAG | Trans. () FRET ( ) |
Cys1-GFP: C12-GFP(1998)94 Cys1-GFP (1998)92 Daglas: Daglas-mit1(2006)505 Daglas-em1(2006)505
Daglas-pm1(2006)505
DAGR (2003)481 Digda (2008)506
|
|
| IP3 | FRET () BRET(◓) Trans. ( ) |
FIRE: FIRE-3 (2006)507 FIRE-2 (2006)507FIRE-1 (2006)507 fretino: fretino (2015)349◓FRET InsP3 sensor (2015)349 fretino-2 (2005)508
GFP-PH: GFP-PHD (1999)509
IRIS: IRIS-1 (2006)510 LIBRA: LIBRAvI (2009)511 LIBRAvII (2009)511 LIBRAVIIIS (2009)511 LIBRAvIII (2009)511 LIBRA (2004)512
|
|
| PA | FRET () |
Pii: Pii-DK (2010)513
|
|
| PI(4)P | BRET(◓) FRET ( ) |
BRET PI(4)P sensors: SidM-2xP4M (2016)514◓0SH2–2xPH (2016)514◓ Pippi: Pippi-PI(4)P (2008)506
|
|
| PI(3)P | Trans. () |
GFP-FYVE: GFP-FYVE (FENS-1)(2001)90GFP-Pib1p(1998)86GFP-EEA1 (FYVE)(1998)86
GFP-PX: GFP-PX(2001)90
|
|
| PI(3,4)P2 | FRET () |
Pippi-PI(3,4)P2 (2006)405
|
|
| PI(3,5)P2 | Trans. () |
GFP-MLN1 (2013)515
|
|
| PI(4,5)P2 | FRET () Trans. ( ) Rat. (◧) |
CAY (2004)516 CYPHR (2003)481 FP-Tubby (2001)517 FPX PIP2 sensor (2015)293◧ PH(PLCδ): PH(PLCδ)-CFP/YFP (2001)518 PH(PLCδ) - GFP (1998)88 GFP-PH (1998)519Pippi-PI(4,5)P2 (2008)506
|
|
| PIP3 | FRET () Trans. ( ) BRET (◓) |
FLLIP (2003)520 Labeled PH domains: PH(PKB)-GFP (1999)89 PH(GRP1)-GFP (1999)89PIP3 BRET sensor (2012)521◓ |
|
| PS | Trans. () |
2xPH(Evectin2) (2011)522
cPLA2–C2: cPLA2-C2-GFP(2003)95 Lact-C2: mRFP - Lact-C2 (2008)97 GFP - Lact-C2 (2008)97PKC-C2: PKCα-C2-EGFP(2003)95 C2-GFP(1998)94PLCδ1-C2: PLCδ1-C2-EGFP(2002)96
|
|
| Protease | |||
| Atg4a | FRET () |
FRET-LC3B (2012)523
|
|
| Atg4b | FRET () |
FRET-GATE-16 (2012)523
|
|
| Caspase1 | Rat. (◧) | FPX caspase 1 sensor: single polypeptide FPX caspase 1 sensor (2015)293◧ | |
| Caspase3 | Rat. (◧) Inten. ( ) FRET ( ) |
ddFP based: single polypeptide FPX Caspase 3 sensor (2015)293◧Bimolecular FPX Caspase 3 reporter (2015)293◧ddRFP-A1B1-DEVD (2012)364
DEVD: mCitrine-DEVD-mTFP1 (2008)524 mAmetrine-DEVD-tdTomato (2008)524 BFP-DEVD-GFP(2002)525 Sensor C3(2001)526CFP-DEVD-YFP(2000)527DEVK: MiCy-DEVK-mKO (2004)528 EC-RP (2008)529 Far-Red Caspase sensors: mKate2-DEVD-iRFP (2016)530
iProtease: iCasper (2015)531 SCAT3: SCAT3 (2003)532
yDMQDc: yDMQDc (2006)533
|
|
| Caspase8 | FRET () Rat. (◧) |
Bid cleavage sensor (2002)534 FPX caspase 8 sensor: single polypeptide FPX caspase 8 sensor (2015)293◧ IC-RP (2008)529
|
|
| Caspase9 | FRET () |
SCAT9 (2003)532
|
|
| MT1-MMP | FRET () |
MT1-MMP Biosensor: MT1-MMP Biosensor (mCherry/mOrange2) (2010)535MT1-MMP Biosensor (2008)536
|
|
| Receptors/GPCRs | |||
| A1R | FRET () |
A1R SPASM: A1R - FL Gαs SPASM (2017)537A1R - FL Gαi SPASM (2017)537
|
|
| β2-AR | BRET (◓) FRET ( ) Trans. ( ) |
BRET2
β2-AR Activation Probes: β2AR-RLuc Gαs-GFP10 (2005)538◓ β2AR-RLuc GFP-Gβ1 (2005)538◓ β2AR-RLuc GFP-Gγ2 (2005)538◓ β2-AR SPASM: β2-AR FL Gαq SPASM (2017)537 β2-AR FL Gαs SPASM (2017)537 β2-AR-Gα SPASM (2013)539Nb80: Nb80-GFP (2013)105
|
|
| Dictyostelium GPCR |
FRET () |
Labeled G proteins: Gβ-YFP + Gα2-CFP(2001)540
|
|
| DRD2 | Inten () |
iTango: DRD2-iTango(2017)541
|
|
| Gαi | FRET () |
Gαi Sensor: Gαil Sensor v2 (2016)542Gαi3 Sensor v2 (2016)542Gαi2 Sensor v2 (2016)542Gαi3 v1 (2006)543Gαi1 v1 (2006)543Gαi2 v1 (2006)543Bunemann Gαi-Gγ2 (2003)544Bunemann Gαi-Gβ1 (2003)544
|
|
| Gαq | FRET () |
Gαq Sensor: Gαq Sensor (v2) (2011)545Gαq Sensor (vl) (2009)546
|
|
| Gαs | FRET () BRET(◓) Trans. ( ) |
Gαs Sensor (2006)547 Gs Activation BRET Assay: RLucII-117-Gαs + GFPl0-Gγ1(20l 6)548◓ Nb37 based: Nb37-YFP(2017)106
|
|
| α1-AR | FRET () |
α1-AR SPASM: α1-AR FL Gαq SPASM (2017)537
|
|
| α2A-AR | FRET () BRET(◓) |
α2-AR SPASM: α2-AR FL Gαs SPASM (2017)537α2-AR FL Gαi SPASM (2017)537 α2A-AR + labeled G protein: α2A-AR-Venus + Gαi1-122Rluc(2006)549◓α2A-AR-Venus + Gαi1-91Rluc(2006)549◓α2A-AR-Venus + RLuc-Gγ2(2006)549◓ α2A-YFP + CFP-Gγ2(2005)550 α2A-AR activation sensor: α2A-AR-cam (2003)551
|
|
| M1R | FRET () |
M1R Activation Sensor: M1R-EYFP-Cerulean (2009)546
|
|
| Odr-10 | BRET(◓) | OGOR (2011)552◓ | |
| Opsin | FRET () |
Opsin SPASM: Opsin-Gα SPASM (2013)539
|
|
| PTHR | FRET () |
PTHR activation sensor: PTHR-cam (2003)551
|
|
| VEGF | BRET (◓) | BRET VEGF biosensor (2016)553◓ | |
| Redox | |||
| H2O2 | Ex. Rat.(★) Inten. ( ) FRET ( ) |
HyPer: HyPerRed (2014)554★ HyPer-3 (2013)555★HyPer-2 (2011)556★ HyPer (2006)557★ roGFP based: roGFP2-Tsa2ΔCPΔCR (2016)558★ roGFP2-Tsa2 ΔCR (2016)558★ roGFP2-GPx4 (2009)559★ roGFP2-Orp1 (2009)559★ Unnatural Amino Acid based: UFP-Tyr66pBoPhe (2012)560 Yap1 based: PerFRET (2013)561 OxyFRET (2013)561
|
|
| H2S | Inten.() |
pAzF H2S sensors: hsGFP (2014)562 cpGFP-Tyr66pAzF (2012)563
|
|
| NO | FRET () |
FRET-MT (2000)564 sGC based: Piccell (2006)565 NOA-1 (2005)566
|
|
| Organic Hyrdroperoxides |
Inten.() |
OHSer (2010)567
|
|
| ONOO− | Inten.() |
pnGFP (2013)568
|
|
| Redox status | FRET () Ex. Rat.(★) Inten.( ) |
HSP33: HSP-FRET (2006)569 redox-sensitive linker: CY-RL7 (2011)570 RedoxFluor (2010)571 ECFP-RL-EYFP (2008)572roGFP: roGFP1-iX (2008)573★ Grx1-roGFP2 (2008)574★ roGFP1-Rx Family (2006)575★ roGFP2 (2004)139★ roGFP1 (2004)139★ rxRFP: TrxRFP1 (2017)576 rxRFP1.X Sensitivity series (2016)577 rxRFP (2015)578rxYFP: rxYFP-Grx1p (2006)579 rxYFP149202 (2001)580
|
|
| Superoxide | Inten.() |
mt-cpYFP (2008)581
|
|
| Small G-Protein | |||
| Cdc42 | FRET () FLIM (♣) |
(CDC42) GEF Sensor (2003)582 CDC42 Biosensor (2014)583 Cdc42 FRET (2016)426♣ CDC42 Raichu: CDC42 Raichu (2002)584 CRIB Raichu (2002)584Cdc42–2G: Cdc42–2G(2016)585 GDI Cdc42 FLARE (2016)586
|
|
| Rab5 | FRET () |
Rab5 Raichu (2008)587
|
|
| Racl | FRET () |
FLAIR (2000)588 GDI Rac1 FLARE (2016)585 Rac1 Raichu: Rac1 Raichu EV (2011)414 CRIB Raichu (2002)584 Rac1 Raichu (2002)584Rac1–2G (2015)589
|
|
| Ral | FRET () |
Raichu-Ral: Raichu-RalB (2004)590 Raichu-RalA (2004)590
|
|
| Ran | FRET () |
Ran FRET probes: YIC (2002)591 YRC (2002)591
|
|
| Rapl | FRET () |
Rap1 Raichu (2001)592
|
|
| Ras | FRET () FLIM (♣) |
DORA Ras (2015)593 FRas: ShadowY H-Ras Sensor (2017)594♣ ShadowG FRas2-M (2015)427♣ FRas2-M (2013)595♣ FRas2-F (2013)595♣ FRas-F (2006)596♣ FRas (2006)596♣ Ras Raichu: Ras Raichu EV (2011)414 Ras Raichu (2001)592
|
|
| RhoA | FRET () FLIM (♣) |
RhoA FLARE: RhoA DORA (2015)597 RhoA2G (2013)443 RhoA FLARE | RhoA1G (2006)598 RhoA FRET (2016)426♣ RhoA Raichu: RhoA Raichu CR (2012)247 RBD Raichu (2006)599RhoA Raichu (2003)600
|
|
| RhoC | FRET () |
RhoC FLARE (2013)601
|
|
| RhoQ | FRET () |
TC10 Raichu (2006)602
|
|
| RRas | FRET () |
RRas Raichu (2007)603
|
|
| Other Post-Translational Modifications | |||
| βArrestin 2 | BRET(◓) | βArrestin 2 ubiquitination biosensor (2004)604◓ | |
| Histone Acetylation | FRET () |
Histac: Histac-H3K9/14 (2016)605Histac-H4K12 (2011)606Histac-H4K5/8 (2009)607
|
|
| K27H3 methyltransf. | FRET () |
K27 Reporter (2004)608
|
|
| K9H3 methyltransf. | FRET () |
K9 Reporter (2004)608
|
|
| O-GlcNAc transferase | FRET () |
O-GlcNAc Sensor (2006)609
|
|
| Ubiquitination | Inten. () |
REACh-Ubiquitin (2006)610
|
|
| Other Signaling Proteins | |||
| Annexin 4 | FRET () |
NEX4: ORNEX4 (2008)611 CYNEX4 (2006)612
|
|
| Bax | Trans. () |
Bax Translocation reporter (2016)530
|
|
| CaM | FRET () |
BSCaM: BSCaM2 (1999)613 BSCaM1 (1997)614 MLCK-FIP (2002)615
|
|
| CRAC | Trans. () |
PH(crac)-GFP (2002)616
|
|
| CREB | FRET () |
ICAP (2010)617
|
|
| N-WASP | FRET () |
N-WASP BS (2004)618
|
|
| Plasma Membrane ATPase |
FRET () |
PMCA Activity Sensor: BFP-PMCA-GFP (2007)619
|
|
2. Designing Genetically Encoded Biosensors
2.1. The Elements of Genetically Encoded Biosensors
For the purposes of this review, we define genetically encoded fluorescent biosensors as chimeric proteins that can be expressed intracellularly and are engineered to act as sensors for monitoring signal transduction (Table 1). The basic function of any sensor is to translate information on a physical property or state of a system into a measurable readout. For fluorescent biosensors, this means converting the diverse signaling activities, such as the concentration of intracellular messengers, metabolic compounds and other analytes, or localization, conformations and activity of signaling proteins into one of several types of fluorescence signals. Fluorescent biosensors are thus fundamentally defined by two essential components: a sensing unit which detects changes in signal transduction and a reporting unit which conveys these changes in a quantifiable form.
The sensing unit is often derived from an endogenous cellular protein that participates in the signaling pathway of interest and is thus intrinsically sensitive to the target signaling event (e.g., calmodulin to sense changes in Ca2+ concentrations). In other cases, isolated protein domains or peptides sequences can also be combined to confer a biosensor with the desired sensitivity. Meanwhile, the reporting unit typically consists of one or more FP variants (or fragments thereof) coupled to the sensing unit in such a way that signaling-induced changes in the state of the sensing unit alter FP fluorescence behavior. Tinkering with the precise nature of this coupling (i.e., how the sensing unit affects fluorescence) lies at the heart of biosensor design. Indeed, as will be seen below, how FPs are incorporated into biosensors greatly influences the types of signaling activities that can be detected.
2.2. Biosensors for Monitoring Protein Behavior
2.2.1. Protein expression, turnover, and localization
In keeping with the tradition of using fluorescence as a sensitive and specific in situ label, the simplest biosensor designs utilize FPs as passive, genetically encoded reporters for monitoring the dynamics of gene and protein expression, as well as protein turnover and localization, in living cells. Although generally involving little more than fusing an FP-coding sequence directly to a target DNA sequence, this approach is frequently modified to increase the richness of biological information that can be obtained. For example, despite serving as a ready indicator for the presence or absence of gene expression under steady-state conditions, the stability and long-lived fluorescence of FPs (discussed in ref. 10) can nevertheless obscure the detection of endogenous transcriptional dynamics. One solution has been to incorporate destabilizing elements into the FP sequence11–13. A variety of fluorescent “timers”, whose distinct chromophore maturation rates give rise to time-dependent changes in their fluorescence spectra, have also been developed to differentially label newly synthesized genes or proteins14–18, permitting the chronological mapping of numerous dynamic processes, including transcriptional activity15,19; protein trafficking, dynamics, and degradation14,16; and even the intracellular replication and spread of influenza20. Similarly, photoconvertible FPs, where the fluorescence emission spectra can be irreversibly converted to a different wavelength, have been utilized to quantify protein degradation in a manner that is not impacted by production of new FPs [20453865, 17489230, 24817874]. Meanwhile, photoswitchable FPs21 are increasingly used as genetically encoded protein tags in conjunction with live-cell super-resolution imaging techniques22,23, thereby enabling the precise and dynamic visualization of protein localization in living cells.
However, the heterologous overexpression of FP-tagged chimeras can potentially disrupt endogenous cellular processes and induce artifacts related to protein localization and dynamics. As such, researchers are increasingly exploring methods for monitoring the dynamics of proteins expressed at endogenous levels from their native genomic loci. Notable among these approaches has been the recent rise of genome editing techniques, particularly CRISPR/Cas technology (reviewed in detail by refs. 24–26). In general, these techniques are based on the sequencespecific introduction of DNA double-strand breaks that are subsequently repaired via nonhomologous end-joining (NHEJ) or homology-directed repair (HDR). Furthermore, whereas NHEJ is typically error prone, HDR enables the precise, site-specific introduction of exogenous sequences, and numerous recent studies have successfully utilized CRISPR/Cas technology to incorporate FPs at endogenous loci in living cells27–29 and even in vivo30. Nevertheless, the lowefficiency of HDR-mediated genome editing poses a major obstacle to the wider adoption of endogenous protein tagging, as does the lack of HDR in non-dividing cells. Mikuni et al. cleverly skirted the latter issue by targeting embryonic neuronal precursors to achieve endogenous protein tagging via HDR in the brain30, while a number of groups have also utilized homologyindependent repair pathways for efficient FP tagging in both dividing and non-dividing cells31,32. On the other hand, given that HDR efficiency is also influenced by insert size, Kamiyama and colleagues utilized a previously developed split-GFP, in which GFP is divided between the 10th and 11th β-strands into two spontaneously complementing fragments33, as an alternative strategy to improve tagging efficiency34. Here, the target protein is endogenously tagged with the GFP11 “epitope” and becomes fluorescent upon complementation by exogenously expressed GFP1–10. The insertion of tandem epitope arrays can further be used to amplify the fluorescence signal from proteins with low endogenous expression 34.
Recent advances in the development and application of a variety of scaffolds for generating intracellular affinity reagents (e.g., intracellular antibodies or “intrabodies”, reviewed in ref. 35) offer another exciting approach for monitoring endogenous protein dynamics that serves as a potential alternative to genome editing. These include “monobodies” derived from the 10th domain of human type III fibronectin (10FnIII or Fn3), a well-characterized scaffold that is structurally similar to the immunoglobulin VH domain36,37, as well as “nanobodies” derived from the heavy chain-only antibodies present in members of the Camelidae family (e.g., camels, llamas, alpacas) and in Chondrichthyes (e.g., sharks), wherein antigen recognition is mediated by a single variable domain35,38. These small, soluble domains are easily expressed in living cells, and when fused to FPs, represent powerful, genetically encodable tools for the sensitive and specific visualization of endogenous cellular components such as neuronal proteins (PSD-95 and Gephyrin) at excitatory and inhibitory synapses39, endogenous actin dynamics40, Wnt-induced β-catenin translocation41, PARP1 translocation to sites of DNA damage42, and GTP-bound (i.e., active) H-Ras and K-Ras43. [Potential buffering effects need to be taken into consideration]. In addition, the heterologous nature of this approach means that care must be taken to minimize background fluorescence caused by excess, unbound probe, with many studies utilizing stable cell lines to achieve low nanobody expression41,42,44,45, while Gross and colleagues notably employed a transcriptional regulation system to link monobody expression to target protein levels. A recently described “flashbody” design also promises to help increase signal-to-noise ratio (SNR) by coupling antigen binding to the intensity of a circularly permuted FP (cpFP; discussed further in Section 2.3.3.1)46. Thus, the legacy of fluorescence as a cell biological tool appears to have come full circle, with intrabodies essentially marking the emergence of “native” immunofluorescence.
2.2.2. Protein-protein interactions
Proteins rarely operate in isolation; most in fact participate in myriad transient interactions with other proteins, with many functioning as dimers or higher-order oligomers or assembling into multiprotein complexes that behave as dedicated molecular machines. Thus, in addition to marking the presence and location of proteins within a cell, direct FP tagging can also be used to monitor protein-protein interactions (PPIs) by utilizing fluorescence (or Förster) resonance energy transfer (FRET), a photophysical phenomenon wherein an excited “donor” fluorophore (e.g., an FP) non-radiatively transfers its excited state energy to a nearby “acceptor” fluorophore via dipole-dipole interactions (reviewed extensively in refs. 10,47), yielding an increase in “sensitized” fluorescence emission by the acceptor (i.e., acceptor emission upon donor excitation) at the expense of direct emission by the donor.
FRET is critically dependent on the proximity of the donor and acceptor fluorophores. Specifically, FRET efficiency (E) varies with the inverse 6th power of the distance, as defined by the equation
| (1) |
where r is distance between the donor and acceptor and R0, known as the Förster distance, is the characteristic distance at which a given donor/acceptor pair exhibits half-maximal FRET efficiency, which is influenced by photophysical factors such as the degree of overlap between the emission spectrum of the donor and the absorption spectrum of the acceptor, as well as by the spatial alignment of the donor and acceptor dipoles. This proximity dependence allows FRET to serve as an exquisite molecular ruler48,49 that is routinely used to monitor PPIs in living cells, including in recent studies visualizing direct interactions between rate-limiting glucose metabolic enzymes in the formation of multiprotein “glucosomes”50 and between HIV proteins51,52, as well as the formation of heteromeric potassium channels53 and A-kinase anchoring protein (AKAP)mediated signaling complexes54–59.
In addition to FRET, PPIs can also be detected in live cells through the use of proteinfragment complementation assays (PCAs). PCAs are an adaptation of classic studies in which fragments of enzymes such as β-galactosidase were found to spontaneously associate in vitro to regenerate an intact, active enzyme60, with the major distinction being the use of protein fragments that do not spontaneously associate but instead can be induced to reassemble and reconstitute a functional unit when brought into close proximity by a pair of interacting proteins. Concurrent with the development of PCAs based on various enzymes (β-galactosidase, dihydrofolate reductase, β-lactamase)61–64, FPs were also found to withstand dissection into roughly 157- and 81-residue fragments that can reassemble to produce a fluorescent species with the aid of a protein interaction pair 65,66, yielding the widely used biomolecular fluorescence complementation (BiFC) assay (reviewed in refs. 67, 68). A novel, trimolecular fluorescence complementation (TriFC) assay has also been reported featuring a modification of the aforementioned split-FP approach, in which both the 10th and 11th β-strands are dissected out of GFP and individually tagged to proteins of interest69. When brought into close proximity via PPIs, the two strands will reassemble with the separately expressed GFP1–9 sensor domain, thereby generating a fluorescent signal. This approach improves the expression and solubility of the tagged proteins by reducing the bulk of the fused FP fragments and was recently utilized to map PPIs among proteins related to frontotemporal dementia70.
Despite numerous advances in FP engineering, the relatively slow kinetics of FP refolding and chromophore maturation, which exhibit half-times ranging from minutes to hours depending on the specific variant71, can limit the temporal resolution of BiFC. FP fragment complementation is also largely irreversible, such that protein interaction partners essentially become trapped, rendering BiFC ill-suited to the study of dynamic PPIs. Nevertheless, the large dynamic range and irreversible nature of BiFC is in fact a tremendous boon for the detection of weak or transient PPIs. These properties also make BiFC a good fit for high-throughput approaches, and BiFC is widely used for PPI screening, similar to the yeast two-hybrid (Y2H) assay, having recently been used in conjunction with Y2H screening to map PPIs related to 2component phospho-relays in rice72 and to map PPIs in the pathogenic yeast Candida albicans73. When combined with photochromic FPs, BiFC can also be utilized to localize protein interaction pairs in super-resolution, as was done recently to map the interaction between the E. coli proteins MreB and EF-Tu74, to localize dimers of the microtubule plus-end protein EB175, and to visualize HER2-HER3 and STIM1-ORAI1 interactions in living cells76. Although the latter study successfully visualized changes in STIM-ORAI complex formation in response to endoplasmic reticulum (ER) calcium (Ca2+) depletion, FRET remains the preferred method for monitoring dynamic PPIs. For instance, Jean-Alphonse and colleagues used FRET to monitor dynamic changes in the interactions among β-arrestin (βarr) 2, Gβγ, adenylyl cyclase (AC) 2, and the parathyroid hormone receptor (PTHR) in response to agonist stimulation77, while a recent study by Smith et al. used FRET to monitor the interaction between the catalytic and regulatory subunits of 3’,5’-cyclic adenosine monophosphate (cAMP)-dependent protein kinase (PKA) and argue against dissociation of the PKA holoenzyme under physiological conditions78 (discussed further in Section 4.3).
2.3. Biosensors for Monitoring Biochemical Activity Dynamics
As discussed above, the simplest implementation of a genetically encoded fluorescent biosensor is to use a gene or protein of interest as a proxy for itself. Over the years, however, more sophisticated approaches have been devised to leverage the fact that proteins dynamically alter their behavior in response to a multitude of biochemical inputs. Thus, through the considered use of various proteins or protein modules conjugated to one or more FPs, genetically encoded fluorescent biosensors can be designed that variously change their localization, fluorescence intensity, or other spectral properties in response to – and thereby report on – fluctuations in a specific biochemical parameter within the native cellular environment. As such, it has become possible to directly visualize and probe the constant biochemical flux that defines a living cell.
2.3.1. Inducing the translocation of a fluorescent protein
The distribution of proteins within cells is highly dynamic, with proteins often changing their subcellular localization in response to various biochemical signals. As such, the ability of FP tags to report on protein localization can logically be repurposed to utilize the signal-induced change in protein localization as a proxy for the signal itself. Indeed, such translocation-based biosensors were among the earliest genetically encoded probes developed to monitor dynamic biochemical activities in living cells.
This approach has classically been used to monitor the production and distribution of lipid messengers such as phosphoinositides (i.e., inositol phospholipids), which comprise a minute but critical component of cell membranes that are responsible for mediating numerous cellular processes79. Phosphoinositide signaling occurs through the direct recruitment of effector proteins to the plasma membrane, which is mediated by different lipid-binding modules80–82, including pleckstrin homology (PH) domains83, Epsin N-terminal homology domains84, and FYVE domains (named for the proteins Fab1p, YOPB, Vps27p, and EEA1)85. Furthermore, because lipid-binding domains from different effectors have been shown to selectively bind specific phosphoinositides (discussed in depth in ref. 82), such as PI(4,5)P2 by the PH domain from phospholipase C (PLC)δ, PI(3,4,5)P3 by the PH domain of Btk, or PI(4,5)P2/PI(3,4,5)P3 by the PH domain from protein kinase B (PKB)/Akt, a certain degree of selectivity can in theory be achieved based on the chosen binding module, though appropriate controls should nevertheless be employed given that lipid selectivity is typically assessed in vitro.
Lipid sensors are thus constructed by directly fusing the coding sequence of an FP to that of a full-length effector protein86,87 or an isolated lipid-binding module86,88–90, whereby the translocation of the fluorescence signal to and/or from a membrane structure serves as a clear indicator for the visualization of phosphoinositide dynamics (Figure 1A). The application of this type of biosensor has provided numerous insights into the function and regulation of phosphoinositide signaling, particularly with respect to the spatial organization of 3’phosphoinositides during chemotaxis and cell migration (discussed further in Section 4.2.3), while also remaining a straightforward and powerful approach that continues to be utilized to study the molecular details of phosphoinositide signaling. For example, in their study investigating neutrophil migration in live zebrafish embryos, Yoo and colleagues used a translocation-based biosensor in which the PH-domain of Akt is fused to GFP to visualize plasma membrane PI(4,5)P2/PI(3,4,5)P3 accumulation, and thus phosphoinositide 3-kinase (PI3K) activation, at the leading edge in neutrophils migrating towards laser-induced wounds in vivo91. This approach has also been used to detect other lipids in various membrane compartments. For instance, diacylglycerol (DAG), a plasma membrane lipid product formed by the PLC-catalyzed cleavage of PI(4,5)P2, can be detected using the C1 domain of protein kinase C (PKC)92–94, whereas phosphatidylserine can be recognized using C2 domains derived from either PKC94,95, PLCδ96, or the milk glycoprotein lactahedrin97.
Figure 1:
Translocation based fluorescent biosensors.
A) PH domains from different proteins are fused to a FP and translocate to the plasma membrane upon the production of specific phosphoinositides88,89. For example, phosphorylation of PIP2 by PI3K to produce PIP3 at the plasma membrane causes translocation of the biosensor from the cytosol to the plasma membrane. B) Kinase translocation reporters utilize kinase specific substrate sequences within nuclear localization sequences (NLS) and/or nuclear export sequences (NES) to promote the import into the nucleus when dephosphorylated and export out of the nucleus when phosphorylated110,112.
However, the overall generalizability of this biosensor design strategy is somewhat restricted given the limited availability of endogenous binding modules that are capable of being recruited by specific intracellular targets to drive probe translocation. In this regard, nanobodies have emerged as a promising new resource for the construction of translocation-based sensors. In particular, their compactness renders nanobodies amenable to high-throughput library screening98, thereby facilitating the rapid development of novel nanobodies, and the diversity of potential epitopes that can be recognized means that a much wider range of targets can be detected compared with more traditional translocation-based sensors. For example, Rajan et al. recently generated a nanobody to detect endogenous histone H2AX phosphorylated at serine 139 (γ-H2AX), which is frequently used as an intracellular marker of DNA double-strand breaks, and used the resulting GFP-tagged chromobody to directly visualize the endogenous formation of DNA double-strand breaks in living cells99. Specifically, by inducing DNA breaks using laser micro-irradiation, the authors were able to observe the translocation of the γ-H2AX chromobody to break sites. Nanobodies can also be generated that selectively recognize specific protein conformations. Indeed, several nanobodies have been developed that specifically bind to the active forms of G-protein-coupled receptor (GPCR) signaling pathways components, including the β2-adrenergic receptor (β2-AR) (Nb80100 and Nb6B9101), the muscarinic acetylcholine receptor (Nb9–8102), the μ opioid receptor (Nb39103), and the Gαs heterotrimeric G protein subunit104, some of which have already been utilized as translocation-based sensors to probe GPCR signaling in living cells (see, for example, Section 4.1 below)105,106.
In contrast to the above examples, in which the sensing unit intrinsically relocalizes to the site of the target signal, a slightly different implementation of this approach involves triggering the relocalization of a sensor to a predetermined subcellular site. For example, Spencer and colleagues were able to develop a biosensor that exits the nucleus in response to CDK2 activity107 by fusing Venus (YFP) to the C-terminal domain of human DNA helicase B (HDHB), which mediates the cell cycle-dependent nuclear localization of HDHB in response to CDK2dependent phosphorylation108. Gross and Rotwein similarly generated a translocation-based Akt kinase sensor by fusing full-length FoxO1 to the green FP Clover109, whereas Maryu et al. utilized the central region of FoxO3a, which contains the Akt phosphorylation site and phosphoregulated nuclear localization (NLS) and export (NES) sequences, to generate their own Akt kinase sensor110. Regot and colleagues have also demonstrated the generalizability of this approach by engineering a family of optimized kinase translocation reporters (KTRs) based on a minimal translocation domain that contains a kinase-specific substrate peptide fused in tandem to a bipartite NLS111 and an NES, such that phosphorylation inhibits the NLS and activates the NES112 (Figure 1B). This design was successfully applied to develop KTRs for monitoring JNK, p38, extracellular signal-regulated kinase (ERK), and PKA activity112. A similar technology known as synthetic kinase activity relocation sensors (SKARS) was also reported by Durandau et al. based on the phosphorylation-dependent charge disruption of a tandem NLS sandwiched between a MAPK substrate domain and an FP113.
2.3.2. Directly sensitizing the chromophore of a fluorescent protein
GFP represents an ideal fluorescent tag because it requires no exogenous cofactors and instead forms its chromophore intrinsically through the autocatalytic cyclization of three consecutive amino acids (e.g., S65, Y66, and G67 in native Aequorea victoria GFP) encoded in its primary sequence (see ref. 10 for a more detailed overview of GFP chromophore formation). The native chromophore further adopts either of two chemical states, a major “neutral” species in which the phenolic −OH of Y66 is protonated and a minor “anionic” species in which the Y66 phenolic −OH is deprotonated, which are responsible for the dual-excitation, single-emission behavior of wild-type GFP (wtGFP) via conversion of the neutral to the anionic chromophore through excited-state proton transfer (ESPT)114,115. ESPT is driven by an extensive H-bond network that functions as a proton relay to abstract a high-energy proton from the excited chromophore, while alternately serving to stabilize the neutral (i.e., protonated) chromophore in the ground state. As a result, wtGFP fluorescence is largely insulated from external perturbations116,117. Yet many spectrum-shifting GFP mutations (e.g., S65T, Y66H, T203I)5,115,118,119, which disrupt the wild-type H-bond network, were shown to impart marked pH sensitivity to GFP fluorescence, responding rapidly and dynamically to intracellular pH changes and suggesting that GFP may be capable of serving as a genetically encoded pH indicator in live cells120.
This approach represents a conceptual departure from the biosensor designs mentioned in the preceding sections, wherein the FP largely serves as a passive bystander, by instead directly integrating the FP into the detection scheme and making probe fluorescence actively responsive to the detection of the target cellular parameter (e.g., pH), analogous to the behavior of small-molecule fluorescent indicators3. Indeed, Llopis and colleagues were able to utilize different GFP spectral variants (e.g., EGFP, ECFP, and EYFP) with differing pKa values (6.15, 6.4, and 7.1, respectively) to visualize pH changes in HeLa cells, further taking advantage of the subcellular targetability of these genetically encoded probes to specifically monitor pH dynamics within the cytoplasm, Golgi lumen, and mitochondrial matrix121. Miesenböck and colleagues also set out to specifically engineer a GFP-based pH sensor to monitor synaptic vesicle exocytosis by targeting residues known to participate in the H-bond network with Y66 or to otherwise alter the spectral properties of GFP117. The authors scanned these sites with histidines, which had the desired pKa value to detect the pH transition upon vesicle fusion, and also used random mutagenesis to ultimately obtain a “pH-sensitive fluorescent protein” (pHluorin), whose fluorescence intensity was eclipsed at low pH (i.e., “ecliptic” pHluorin). Ecliptic pHluorin is thus dark when localized to the vesicle lumen and rapidly increases in intensity upon exposure to the extracellular space, thereby enabling the visualization of individual fusion events as discrete flashes117. Sankaranarayanan et al. subsequently generated a superecliptic pHluorin variant by incorporating the EGFP mutations F64L and S65T122, while Li and Tsien recently developed a red-fluorescent pH sensor, pHTomato, which they co-imaged alongside GCaMP3 to simultaneously visualize synaptic vesicle and Ca2+ dynamics123.
Of the spectral variants initially derived from GFP, YFP fluorescence intensity displays a particularly high degree of environmental sensitivity, with the numerous amino acid substitutions having introduced gaps around the chromophore and the further substitution of H148 with Gly even producing a solvent channel directly to the chromophore, which substantially increases the already high pKa value (i.e., pH sensitivity) of YFP124. In fact, Tojima and colleagues utilized this H148G substitution to reintroduce pH sensitivity into Venus for use as a pH sensor125. YFP fluorescence is also strongly affected by halide ions, especially Cl− and iodide, with the H148Q mutant being particularly sensitive126,127. Halide ions are able to bind directly to YFP and increase the apparent pKa of the chromophore, leading to decreased YFP intensity at a fixed pH, thus allowing Jayaraman and coworkers to use YFP(H148Q) as an intracellular Cl− sensor126. Galietta and colleagues later set out to improve the halide sensitivity of YFP(H148Q) via random mutagenesis, resulting in variants with enhanced affinity for either Cl− (H148Q/V163S; Kd ~40 mM) or I− (H148Q/I152L; Kd ~2 mM)128. However, the fact that YFP(H148Q) is sensitive to both pH and Cl− can complicate its application as a Cl− sensor. Thus, Zhong et al. also recently performed a series of mutagenic screens to generate monomeric Cl-YFP (EYFP-F46L/Q69K/H148Q/I152L/V163S/S175G/S205V/A206K), which binds Cl− with a Kd of 14 mM and has a pKa of only 5.9129.
As part of their original screen for pH-sensitive GFP variants, Miesenböck et al. also identified a mutant whose dual excitation peaks responded oppositely to pH, with the ratio of intensity at each excitation wavelength ultimately serving as the readout (e.g., “ratiometric” pHluorin)117. Given that fluorescence intensity in cells is often influenced by multiple factors that are unrelated to the parameter being detected (e.g., variable expression, cell shape/thickness, sample illumination, bleaching, etc.), such ratiometric sensors are often advantageous for live-cell imaging because collecting fluorescence at two wavelengths that show opposite changes in response to a cellular parameter largely cancels out these variations, while also providing more quantitative measurements130,131. Thus, Hanson and colleagues developed a family of emission-ratiometric pH sensors called “dual-emission GFPs” (deGFPs), which switch from green to blue emission with decreasing pH130, while Bizzarri and coworkers also found that the GFP F64L/S65T/T203Y/L231H, or E2GFP132,133, can serve as both an excitation- and emission-ratiometric pH sensor134. To generate a ratiometric Cl− sensor, Kuner and Augustine utilized FRET between YFP and a tethered CFP, which is not sensitive to Cl− ions135. In the resulting sensor, Clomeleon, Cl− binding decreases YFP fluorescence, thereby reducing FRET and dequenching CFP emission135. Markova et al. used a similar approach to generate the ratiometric “Cl-sensor”, but instead utilized a YFP mutant with increased Cl− sensitivity136. Interestingly, E2GFP, which contains the T203Y mutation found in YFPs, is also sensitive to halide ions, but does not display an intrinsic ratiometric response137. However, because pH and Cl− differentially affect the E2GFP spectrum, Arosio and colleagues were able to generate a ratiometric sensor capable of monitoring both parameters (ClopHensor) by linking E2GFP to the monomeric RFP DsRedm138.
2.3.3. Engineered modulation of the photophysical behavior of a fluorescent protein
The above examples represent a somewhat unique case in which the FP doubly performs as both the sensing and reporting unit. However, this design scheme is not very generalizable beyond the detection of a few select ions, as most cellular analytes and biochemical reactions do not directly affect the chromophore. Instead, a more universal strategy involves returning to the use of an extrinsic sensing unit that is engineered to couple the detection of a biochemical signal to the modulation of fluorescence behavior. As exemplifiedby the development of certain redox139,140 and metal ion sensors141,142 such as the ER Ca2+ sensor CatchER [21914846; 24836743; 24311573], the sensing unit can be as simple as a few amino acids inserted into the surface of an FP. In broader practice, however, a more sophisticated design is often required in which the sensing unit comprises a conformationally dynamic element capable of adopting either of two states (i.e., conformations) and readily switching between them in response to a specific input signal. Sensing units containing such “molecular switches” can often be generated from intrinsic conformational switches found in many native proteins or artificially engineered by linking isolated proteins or peptides in a two-component sensing unit that contains a “receiver” domain to sense the signal of interest and a “switch” domain to trigger the conformational change. Below, we describe how molecular switches can be utilized to modulate the photophysical behavior of one or more coupled FPs in order to directly visualize a vast array of biochemical and cellular parameters in living cells.
2.3.3.1. Designs based on the modulation of a single fluorescent protein
The stereotypical β-can architecture of an FP not only protects the chromophore from the external environment but is also directly responsible for providing the internal microenvironment that supports fluorescence. Any alteration that distorts this β-can conformation can thus affect chromophore behavior, allowing conformational changes in a coupled molecular switch to directly influence FP fluorescence. One way to achieve conformational coupling is to directly insert an FP into a target protein. Because the native N- and C-termini of an FP are located in close proximity in the folded 3D structure, FP insertion typically leads to minimal perturbation of the native conformation of the target protein, while at the same time rendering the FP sensitive to the conformation of the fused protein such that conformational changes are transmitted into the FP β-can to alter chromophore behavior. Given that the native N- and C-termini of FPs are unstructured, conformational coupling can also be enhanced through the use of circular permutation, which has long been used to study the structure and function of biological macromolecules143 and refers to the rearrangement of the linear sequence of a macromolecule in order to shift the positions of the native termini along the molecule surface without altering the overall 3D structure. Several FPs have been shown to tolerate circular permutation while retaining fluorescence behavior144, with the resulting cpFPs possessing new termini located within the β-can itself; the added rigidity thus improves the transmission of conformational changes into the β-can. As such, inserting an FP into a conformational switch, or vice versa, provides a mechanism to sensitize the photophysical properties of a single FP to a given biochemical parameter (Figure 2).
Figure 2:
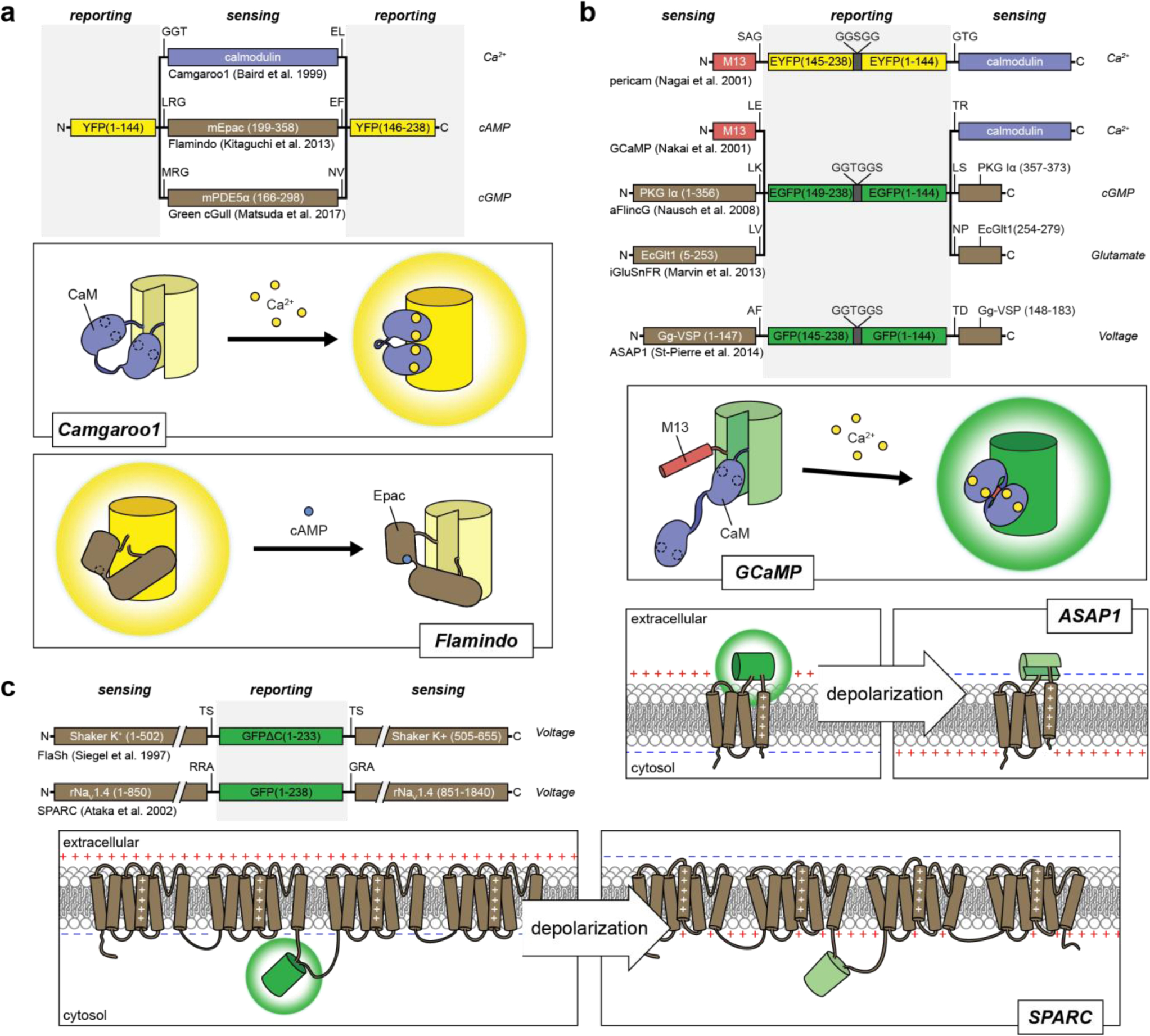
Single FP fluorescent biosensor designs for cellular analytes and membrane potential.
Insertion of a sensing unit into a FP. Calmodulin, mEpac and mPDE5α undergo conformational changes in response to binding Ca2+ 144, cAMP163, and cGMP164, respectively, which perturbs the chromophore and alters the fluorescence. Binding can either lead to an increase in fluorescence, as seen in the Ca2+ biosensor camgaroo1144, or a decrease in fluorescence, as seen in the cAMP biosensor flamindo163. B) Sandwiching a cpFP between sensing units. Biosensors have utilized sensing units that comprise either separate receiver and switch domains148,150 or split proteins that re-constitute during protein folding165–167. For example, GCaMP biosensors utilize separate domains of CaM and M13, where calcium binding to CaM promotes the binding of CaM to M13 and results in a conformational change that leads to an increase in GFP fluorescence. On the other hand, the membrane voltage sensor ASAP1 inserts cpGFP into the voltage-sensing domain of the chicken voltage-sensitive phosphatase Gg-VSP, which reconstitutes after folding and depolarization leads to a conformational change in the 4th transmembrane segment that alters the fluorescence of cpGFP167 C) Insertion of a FP into a voltage-sensitive channel. The conformational changes induced in voltage-sensitive K+ and Na+ channels alter the fluorescence of GFP to act as biosensors of membrane potential168,169.
2.3.3.1.1. Genetically encoded calcium indicators
Advances in live-cell imaging are intimately linked with the study of Ca2+ signaling, which can be attributed to the long history of efforts to visualize Ca2+ dynamics as a proxy for neuronal activity. Indeed, the use of fluorescent indicator dyes was largely popularized by the success of Ca2+ indicators as live-cell probes (also discussed in Section 4.2.1 below), while the discovery of GFP was itself an offshoot of work that yielded another a live-cell Ca2+ probe, the photoprotein aequorin145. Thus, genetically encoded Ca2+ indicators (GECIs) were naturally among the earliest biosensors to be engineered and remain a major focus of biosensor development efforts to this day.
Barring the notable exception of CatchER{21914846} which is based on an engineering Ca2+ binding site, the development of single-FP GECIs has universally centered around calmodulin, a ubiquitous intracellular Ca2+ sensor and major effector of Ca2+ signaling in all eukaryotes (reviewed in refs. 146, 147), which undergoes a switch-like conformational change upon Ca2+ binding and recognition of its target proteins. In their work developing the first cpFPs, for example, Baird and colleagues found that inserting Xenopus laevis calmodulin into EYFP yielded a chimeric protein that displayed an approximately 7-fold increase in fluorescence intensity in the presence of Ca2+ in vitro and functioned as a Ca2+ indicator, nicknamed “camgaroo1”, in living cells144 (Figure 2A). Nagai and coworkers modified this design by utilizing a bipartite molecular switch148. In this scheme, calmodulin was fused to the C-terminus of cpEYFP, while the M13 peptide, which is derived from the calmodulin-binding region of myosin light-chain kinase149, was fused to the N-terminus. Calmodulin forms a compact complex with M13 upon Ca2+ binding, and the M13 peptide thus acts as a switching domain to promote a Ca2+-dependent conformational change in the sensor (Figure 2B). These efforts ultimately yielded a family of GECIs known as “pericams”148 (Table 1).
In a parallel effort, Nakai and coworkers set out to construct a GECI by first testing multiple fusion sites for linking the M13 peptide and calmodulin to the N- and C-termini of cpEGFP in a design scheme similar to that of pericams150. Notably, the two early candidates with the best responses both introduced a 5-residue gap in the β-can structure where the molecular switch is inserted. Subsequent optimization focused on the linker sequences at inter-domain junctions, and the 85th variant tested, which showed the highest basal fluorescence and Ca2+dependent response, was named “GCaMP”150. Tallini et al. were able to improve GCaMP behavior by introducing several mutations previously shown to improve GFP stability (V163A, S175G)151 or promote monomer formation (A206K)152, while also identifying new substitutions (D180Y, V93I) via random mutagenesis153. Adding an N-terminal RSET leader sequence also helped improve thermal stability, resulting in GCaMP2. A mammalian-cell screen for point mutants with increased brightness, dynamic range, and Ca2+ sensitivity then identified two mutations in GFP (M153K, T203V) and a third in calmodulin (N60D), resulting in a 3rd-generation sensor (GCaMP3) with 3-fold higher basal fluorescence and a 2-fold higher dynamic range. GCaMP3 could thus be used to successfully image Ca2+ dynamics in brain slices and in vivo154.
Given the importance of GECIs for imaging neuronal activity, one area of focus for continued engineering efforts has been to enhance the speed and sensitivity of GCaMP responses in neurons to more accurately report the dynamics of action potentials. For instance, Akerboom and colleagues generated a panel of 12 GCaMP5 variants, 3 of which (GCaMP5A, 5G, and 5K) exhibited 2- to 3-fold better SNR and more faithfully recapitulated electrophysiological recordings of in vivo neuronal activity compared with GCaMP3155. Chen et al. extended this work even further by screening for optimized GCaMP5G variants directly in cultured neurons to obtain GCaMP6s, 6m, and 6f with slow, medium, and fast kinetics, respectively156. Of equal importance has been the development of a wider array of GECI color variants, especially red-shifted sensors, which are particularly desirable for in vivo imaging given the reduced phototoxicity, decreased absorption and scattering, and thus deeper tissue penetration, associated with longer-wavelength illumination. Zhao and colleagues, for example, developed a red-shifted GECI by replacing the cpEGFP domain of GCaMP3 with a circularly permuted version of the RFP mApple157, which after several rounds of directed evolution yielded “R-GECO1”158.
However, mApple is known to exhibit substantial photoswitching behavior157, and subsequent studies found that this behavior is preserved in R-GECO1 and its descendants159,160, which significantly increase in intensity upon illumination with blue light, thus potentially limiting their compatibility with optogenetic tools. As part of an independent effort, Akerboom and coworkers160 elected to replace the cpEGFP domain of GCaMP3 with mRuby161. A dimly fluorescent “RCaMP” prototype was subjected to multiple rounds of both random and structureguided mutagenesis to produce a series of improved variants culminating in RCaMP1h. Importantly, despite possessing a lower dynamic range, RCaMP1 did not exhibit any of the complex photophysical effects that characterize mApple-based GECIs. RCaMP1 was successfully employed for dual-color Ca2+ imaging along with GCaMP5G in mixed neuronal/astrocyte cultures, for in vivo imaging, and for combined optogenetics/imaging studies160. Recently, Dana and colleagues were able to further optimize these red GECIs by screening for variants directly in cultured neurons, yielding three improved probes, mRubybased jRCaMP1a and jRCaMP1b and mApple-based jRGECO1a162. Yet despite their promise as powerful tools for interrogating neuronal function in vivo, red GECIs nevertheless continue to lag behind GCaMPs in overall dynamic range, and evidence also suggests that these probes produce multiple species in cells due to incomplete chromophore maturation162. Thus, additional optimization remains a priority for these sensors, likely entailing re-engineering of the RFP itself.
2.3.3.1.2. Genetically encoded voltage indicators
Plasma membrane voltage is a key indicator of electrical activity in neurons. Thus, in addition to GECIs for Ca2+ imaging, the potential to use genetically encodable probes to directly visualize changes in membrane voltage in specific neuronal subsets has also spurred the development of genetically encoded voltage indicators (GEVIs). GEVIs couple changes in membrane potential to changes in fluorescence by taking advantage of the fact that multiple cellular proteins contain a voltage-sensing domain (VSD) that enables them to sense and respond to changes in membrane potential. VSDs consist of a group of four helical segments (S1–S4) that sit in the plasma membrane, with the positively charged S4 undergoing a structural rearrangement in response to voltage changes (otherwise known as “gating”). These conformational changes can be transmitted into the barrel of a coupled FP, thereby allowing a VSD-containing protein to serve as a voltage-dependent molecular switch to control biosensor fluorescence.
Initial efforts relied on voltage-gated ion channels as the source of the VSD. For example, Siegel and Isacoff constructed the very first GEVI by inserting a C-terminally truncated form of GFP into the plasma membrane-proximal region of the cytosolic tail of the Shaker potassium channel, just after the 6th transmembrane segment of the channel168 (Figure 2C). Similar to the reasoning underlying the use of cpFPs in single-FP-base sensors, the unstructured C-terminus of GFP was deleted based on the hypothesis that direct fusion of Shaker to the less flexible β-can would improve conformational coupling and voltage-dependent fluorescence changes. A W434F-mutant form of Shaker, in which the ion-conducting pore is disrupted without affecting channel gating, was also used to prevent overexpression of the sensor from altering cell physiology. However, although the resulting “fluorescent Shaker” (FlaSh) protein displayed a clear decrease in GFP fluorescence upon membrane depolarization in Xenopus oocytes, the kinetics of fluorescence response were ~30-fold slower than the actual gating kinetics of the channel and thus ill-suited to imaging the rapid dynamics of neuronal action potentials168. Ataka and Pieribone similarly constructed a GEVI by using the μI voltage-gated sodium channel as the VSD169. In contrast to potassium channels, sodium channels function as monomers, which offers a wider selection of insertion sites for an FP and also eliminates the risk that the sensor will reconstitute with native channel subunits. After screening 10 different insertion sites, the authors found that inserting full-length EGFP between the 2nd and 3rd transmembrane regions yielded a sodium channel protein-based activity reporting construct (SPARC) (Figure 2C). Importantly, although SPARC exhibited a much smaller fluorescence change compared with FlaSh, the response kinetics were fast enough to report voltage pulses on the order of 2 ms169.
Although successful as a proof of concept, GEVIs based on ion channels can be difficult to express in cells and exhibit poor membrane localization. Thus, subsequent efforts have simplified GEVI architecture by turning toward more compact VSDs, beginning with the discovery of the Cionia intestinalis voltage-sensitive phosphatase (CiVSP)170. Genomic surveys found that this sea squirt (family Ascidiascea) expresses a lipid phosphatase fused to a single VSD, which is homologous to those found in voltage-gated potassium channels yet functions as a monomer. As with the VSDs from voltage-gated channels, the 4th helical segment in the VSD of CiVSP (i.e., Ci-VSD) undergoes a voltage-dependent structural rearrangement within the plasma membrane, which can be directly coupled to an FP, though this requires incorporating point mutations in the VSD to shift voltage sensitivity into the physiological range of membrane potential in neurons171. Notably, electrophysiological studies performed by Lundby and coworkers found that the Ci-VSD produces a very rapid gating current (i.e., current generated by the movement of positively charged segment 4 within the membrane) in response to membrane depolarization172. Thus, by fusing the CFP variant Cerulean173 directly to the C-terminus of Ci-VSD using a short linker, they were able to generate an improved voltage-sensitive fluorescent protein (VSFP) with a robust voltage-dependent fluorescence change and ~1-ms activation kinetics. Taking their inspiration from the development of GECIs, Barnett and colleagues similarly appended cpEGFP to the C-terminus of Ci-VSD174. Screening through 90 constructs that varied either the fusion site between Ci-VSD and cpEGFP or the precise residues surrounding the circular permutation site yielded the sensor “ElectricPk”, which also exhibited a rapid fluorescence response capable of resolving 1-ms voltage pulses174.
More recently, St-Pierre et al. based the design of their single-FP GEVI, “accelerated sensor of action potentials” (ASAP1), on structural studies of the gating mechanism of VSDs167. Specifically, given reports demonstrating a large conformational change within the loop connecting helical segments S3 and S4175, they inserted a circularly permuted superfolder GFP (cpsfGFP)33 into this loop region in the chicken (Gallus gallus) VSD (Gg-VSD) (Figure 2B). The loop connecting S3 and S4 is shorter in Gg-VSD than in Ci-VSD, which the authors reasoned would increase conformational coupling to cpsfGFP. ASAP1 exhibited fast (~2 ms) kinetics, as well as a much higher dynamic range compared with previous GEVIs167. To develop an optimized GEVI that was suitable for use with 2-photon imaging of in vivo neuronal activity in the Drosophila visual system, Yang and colleagues generated variants of ASAP1 by mutating the junction sequence connecting S3 from Gg-VSD to cpsfGFP176. These efforts yielded the improved variant ASAP2f, which produced a larger voltage-dependent fluorescence change in neurons compared with ASAP1 while exhibiting the same fast response kinetics. Abdelfattah et al. also recently reported the development of a red-shifted GEVI with comparable performance characteristics to optimized GFP-based GEVIs177. Similar to the development of R-GECO1, the authors fused cpmApple to the S4 helix at the C-terminus of Ci-VSD and then performed directed evolution. After screening thousands of variants, they obtained “Fluorescent indicator for voltage imaging Red”, or FlicR1, which exhibited sub-millisecond activation and deactivation kinetics and a large dynamic range177. Overall, the development of GEVIs is still an ongoing effort, with even greater improvements in sensitivity and dynamic range hopefully to come in the near future.
2.3.3.1.3. Single fluorescent protein-based indicators for other cellular analytes
The “killer app” of the molecular-switch-as-sensing-unit design is versatility: given the availability of an endogenous effector protein that undergoes a suitable conformational change, this approach can be used to engineer biosensors for monitoring virtually any cellular analyte – or in some cases, ratios of analytes (e.g., ATP/ADP and NADH/NAD+, discussed in {PMID29679217}) – in living cells.
For instance, exchange protein regulated by cAMP (Epac), a Rap1/2 guanine nucleotide exchange factor (GEF) regulated by the ubiquitous intracellular messenger cAMP, has been shown to undergo a major conformational change in response to cAMP binding178,179. Kitaguchi and colleagues were thus able to develop a yellow-fluorescent cAMP sensor by inserting the murine homolog of Epac1 (mEpac1) into Citrine163. A screen of 12 variants featuring different mEpac1 fragments and linkers yielded a successful “fluorescent cAMP indicator” (Flamindo) containing just the mEpac1 cyclic nucleotide binding domain (CNBD) and exhibiting an ~50% cAMP-dependent intensity decrease (Figure 2A). Odaka et al. subsequently improved the response by mutating the N-terminal Citrine-mEpac1 junction, with the resulting Flamindo2 sensor exhibiting an ~75% intensity decrease in response to cAMP180. Tewson and coworkers also constructed “cAMP difference detector in situ” (cADDis), which shows a ~35% cAMP-dependent fluorescence decrease, by inserting a circularly permuted version of the bright green FP mNeonGreen181 into the hinge region of Epac2182. Finally, Harada et al. recently generated the red-shifted “Pink Flamindo” cAMP sensor by inserting the mEpac1 switch from Flamindo into mApple and screening multiple linker variants183. Pink Flamindo, which exhibits a 4-fold cAMP-dependent increase in fluorescence, was successfully co-imaged with a green-fluorescent GECI (G-GECO158), yet it remains to be seen whether future applications of this sensor face the same photoswitching behavior as other mApple-based probes.
Likewise, 3’,5’-cyclic guanosine monophosphate (cGMP) is another key intracellular messenger that regulates numerous cellular processes, especially in relation to vascular and neuronal biology184. As with cAMP, cGMP exerts its biological effects through the regulation of a number of protein targets, such as the cGMP-dependent protein kinase (PKG), which has been shown to undergo isoform-specific conformational changes in response to cGMP binding185,186. In fact, Nausch and coworkers previously found that inserting cpEGFP into the C-terminal tail region of the tandem CNBDs isolated from PKG I was sufficient to yield a cGMP biosensor165. By incorporating the CNBD regions from the α, β, or δ isoforms of PKG I, they were thus able to generate a family of “fluorescent indicators of cGMP”, or FlincGs, that each displayed a range of cGMP-binding affinities while all showing fairly robust cGMP-dependent fluorescence increases in cells. More recently, Matsuda et al. developed a cGMP sensor using another cGMP-binding domain known as a GAF domain164, which is present as an allosteric regulatory domain in many enzymes, including certain phosphodiesterase (PDE) isoforms, and is also known to undergo a conformational change upon cGMP binding187,188. Similar to the design of Flamindo, the authors inserted the GAF-A domain from murine PDE5α (mPDE5α) into Citrine and screened several mPDE5α fragments and linker sequences to obtain the single-FP biosensor “Green cGull”, which exhibited an ~8-fold cGMP-dependent fluorescence intensity increase, as well as 3 orders of magnitude higher affinity for cGMP (~1 μM) compared with cAMP (~1 mM)164.
A number of studies have also explored the potential for bacterial periplasmic binding proteins (PBPs) to serve as scaffolds for biosensor engineering. PBPs are responsible for mediating the uptake of various nutrients and other solutes in Gram-positive bacteria such as Escherichia coli (E. coli) and are therefore capable of binding a wide range of small molecules189,190. Members of this large protein superfamily generally share a similar protein architecture comprising a pair of globular domains that are linked by a hinge region, with ligand binding inducing substantial conformational rearrangement of the globular domains191,192. In one early proof-of-concept study, Marvin and colleagues utilized structural data on the E. coli maltose-binding protein (MBP) to identify candidate sites for the optimal insertion of cpGFP193. Four different insertion sites were tested, and after screening libraries of linker variants, a candidate construct named MBP165-cpGFP.PPYF, or “PPYF”, was obtained that displayed an ~2.5-fold increase in green fluorescence upon maltose binding. Similarly, Alicea et al. were able to generate a genetically encoded phosphonate sensor by inserting cpGFP into the E. coli EcPhnD protein194. Most recently, Marvin and co-workers developed “iGluSnFR”, a single-FP sensor for monitoring the neurotransmitter glutamate that exhibits an up to 4-fold glutamatedependent fluorescence intensity increase in cells166. iGluSnFR was constructed by inserting cpGFP into the E. coli PBP Glt1 (Figure 2B); in keeping with the engineering of the above maltose and phosphonate sensors, a position close to the hinge region of Glt1 was chosen as the insertion point, followed by screening to optimize the inter-domain junction sequences. PBPs thus show considerable promise as biosensor scaffolds and, given the relative ease with which their ligand-binding sites can be computationally redesigned190, will potentially yield a myriad of new single-FP biosensors.
2.3.3.1.4. Engineering single-fluorescent-protein biosensors with a ratiometric readout
The single-wavelength, intensiometric biosensors discussed above are popular for in vivo imaging due to their large dynamic ranges and high SNR, as well as for biosensor multiplexing and all-optical (e.g., optogenetic) studies enabled by their small spectral footprint. Nevertheless, as alluded to previously, dual-wavelength ratiometric sensors are often preferable for cancelling out artifacts related to probe expression and for obtaining quantitative measurements.
In some cases, ratiometric behavior can be obtained as an intrinsic property, in which the molecular switch induces a shift in the excitation or emission spectrum of a single FP. For instance, Nagai et al. were able to generate a ratiometric Ca2+ sensor by introducing an H203F mutation, which increases fluorescence by the protonated YFP chromophore195, into the intensiometric “flash-pericam”148. Screening identified two additional mutations (F46L, H148D) that, along with linker optimization, improved the response to yield a “ratiometric-pericam” that undergoes a Ca2+-dependent shift from ~415 nm to ~495 nm excitation. More recently, a trio of ratiometric GECO-family Ca2+ indictors were also identified through screening that undergo a Ca2+-induced shift from blue (~450 nm) to green (~510 nm) emission upon ~400 nm excitation (“GEM-GECO”), from ~400 nm to ~488 nm excitation with ~510 nm (green) emission (“GEX-GECO”), and from ~575 nm to ~480 nm excitation with ~600 nm (red) emission (“REX-GECO”)158,196. Zhao and colleagues also obtained a ratiometric sensor for monitoring the intracellular NADH/NAD+ concentration ratio after inserting cpYFP into the inter-domain loop of the Thermus aquaticus Rex (T-Rex) protein197. This sensor, named “SoNar”, undergoes an increase in the 420/480 nm excitation ratio upon NADH binding, whereas NAD+ induces the opposite response. By computationally redesigning the T-Rex binding pocket in SoNar, Tao et al. were then able to generate a ratiometric sensor that directly reports NADPH concentrations, yielding a family of “iNap” sensors with a range of affinities198.
Alternatively, a more straightforward approach for generating ratiometric sensors is simply to add a second, spectrally distinct FP as an internal reference. Hung and colleagues, for example, also generated a ratiometric NADH/NAD+ redox sensor, “Peredox”, by inserting the long-Stokes-shift FP cpT-sapphire199 between a tandem dimer of T-Rex proteins and fusing mCherry200 to the C-terminus201. cpT-sapphire exhibits an increase in fluorescence intensity upon NADH binding to T-Rex, whereas mCherry fluorescence remains unaltered, thereby providing a ratiometric readout. Recently, Cho et al. used a similar approach to design ratiometric versions of GCaMP by fusing mCherry to the C-terminus of GCaMPs 3, 6s, and 6f202. However, they included a rigid ER/K α-helical linker203,204 between calmodulin and mCherry, reasoning that this 30-nm spacer would eliminate any potential FRET with cpGFP. The resulting GCaMP-R-3, GCaMP-R-6s, and GCaMP-R-6f sensors all displayed large emission ratio changes and enabled quantitative Ca2+ imaging in cells and tissues. Meanwhile, Ast and coworkers added a twist to this approach by creating a nested FP pair, in which a long-Stokes-shift OFP (LSSmOrange205) is inserted into the cpGFP linker, to develop “GO-Matryoshka” (so named after the famous Russian dolls)206. Importantly, the use of LSSmOrange allows both FPs to be excited at a single wavelength (~440 nm), while the lack of spectral overlap between cpGFP emission and LSSmOrange absorption prevents FRET. This approach successfully yielded the ratiometric Ca2+ sensor MatryoshCaMP6s (derived from GCaMP6s), as well as the ammonium sensor AmTryoshka1;3, which was constructed by inserting GO-Matryoshka into the Arabidopsis thaliana ammonium transporter AtAMT1;3206.
2.3.3.2. Designs based on the modulation of a pair of fluorescent proteins
Far from providing a mere reference signal, a second FP can be directly coupled to the sensing unit such that both FPs function as the reporting unit, with the molecular switch inserted between a pair of FPs to modulate the photophysical interaction between them. Compared with the engineered modulation of a single FP by a molecular switch, this dual-FP sandwich configuration represents a more straightforward and accessible design scheme that is less strictly reliant on extensive protein engineering and, to date, remains the most popular and generalizable approach to biosensor design. The most common implementation of this dual-FP design scheme involves sandwiching a molecular switch between two FPs that constitute a FRET pair. Recall from Section 2.2.2 that FRET efficiency not only falls off with the 6th power of the interfluorophore distance but also requires the donor and acceptor dipoles to be properly oriented relative to one another. In addition to monitoring interactions between proteins (i.e., intermolecular FRET), this proximity and orientation dependence therefore make FRET an excellent reporter of conformational changes within a protein (i.e., intramolecular FRET), allowing the straightforward coupling of a biochemical signal to a change in FRET and forming the basis for a seemingly endless array of genetically encoded FRET-based biosensors.
2.3.3.2.1. FRET sensors for monitoring cellular analytes
Much like engineered single-FP biosensors, FRET-based sensors are often built by incorporating a sensing unit that undergoes an engineered or intrinsic conformational change in response to the binding of an intracellular messenger or other small molecule. As detailed below, this approach has yielded numerous FRET-based sensors engineered to monitor the dynamics of metal ions, cyclic nucleotides, phospholipids, and various other analytes within living cells.
2.3.3.2.1.1. FRET-based indicators of metal ion concentrations
The very first GECIs were also among the first ever genetically encoded FRET-based biosensors. Specifically, Miyawaki and colleagues generated the sensor “cameleon” by sandwiching a Ca2+-sensitive molecular switch, composed of calmodulin linked to the M13 peptide via a diglycine linker, between the FRET donor BFP and the FRET acceptor GFP359. In a design that would later inspire additional classes of FRET-based biosensors (see below), the reversible binding of Ca2+ causes the sensing unit to switch between an extended conformation and a more compact form in which calmodulin essentially engulfs the M13 peptide, thereby altering the relative proximity, and thus the efficiency of FRET, between BFP and GFP. As discussed above, changes in FRET cause inverse changes in the intensity of the acceptor and donor fluorophores, thereby altering the ratio of fluorescence emission and providing a dynamic, quantitative readout of intracellular Ca2+ elevations. However, although cameleon performed well as a Ca2+ indicator in vitro, the low brightness of the BFP donor fluorophore rendered cameleon less effective when expressed in mammalian cells. Substitution of CFP and YFP in place of the BFP/GFP FRET pair was therefore performed to yield “yellow-cameleon”, or “YC” (Figure 3A), which exhibited higher brightness in mammalian cells and was also less affected by cellular autofluorescence359.
Figure 3:
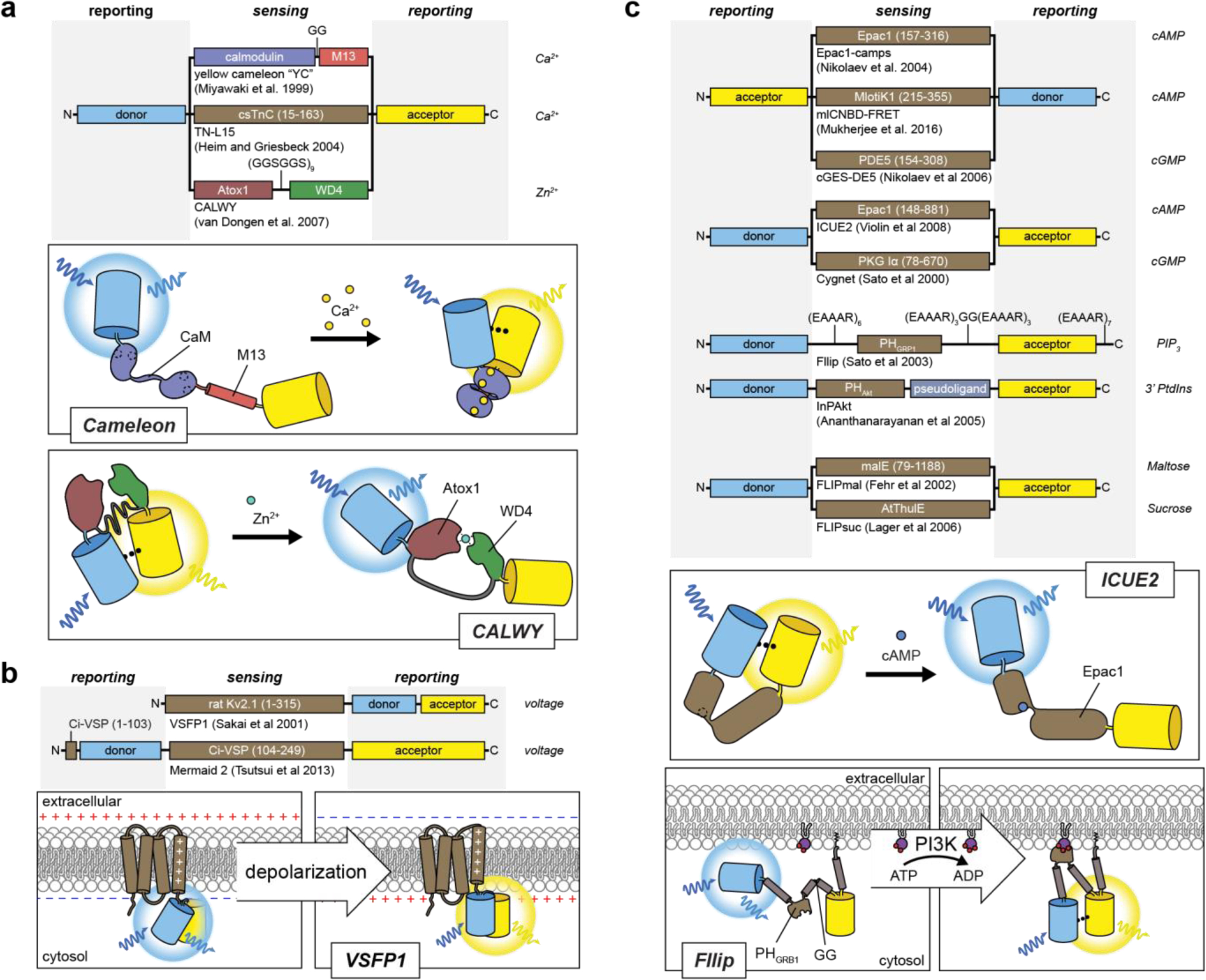
Designs of FRET-based reporters for ions, cellular analytes and membrane potential.
FRET-based metal ion biosensors utilize receiver and switch domains that bind to each other358,396, or endogenous proteins that undergo a conformational change386 in response to binding of metal ions. These conformational changes alter the intramolecular distance and orientation of a pair of FPs and thus changes the amount of energy transferred from the donor FP to the acceptor FP. B) Voltage sensors that utilize a FRET-based readout often rely primarily on changing the orientation of dipoles of FPs244,257. VSFP1 contains a CFP inserted between the 3rd and 4th transmembrane domain and a YFP fused to the C terminal tail of a truncated portion of the rat Kv2.1 channel where cell depolarization induces a conformational shift in the 4th transmembrane domain, thus changing the relative angle of the two dipoles of the fluorescent proteins257. C) Several biosensors for cellular analytes have utilized the design of sandwiching a conformationally switching domain between a FP FRET pair285,297,300,310,312,334,344,504,520. For example, the cAMP biosensor ICUE2 utilizes the conformational change induced by cAMP binding to the CNB domain of Epac1 to increase the intramolecular distance between the donor and acceptor FPs297. Alternatively, having both a PIP3 binding PH domain from GRP1 and a C-terminal membrane localization sequence in combination with engineered rigid α-helical linkers yielded a chimeric protein that exhibits significant conformational changes in response to PIP3 production, leading to changes in FRET between a FRET pair flanking the chimeric protein520.
One of the primary motivations for developing cameleons, as well as all subsequent GECIs, was to enable the direct visualization of highly localized Ca2+ signaling events via subcellular targeting (discussed further in Section 4.2.1). However, early efforts to investigate local Ca2+ elevations in neurons revealed that YC suffers a considerable reduction in Ca2+ sensitivity when targeted to the plasma membrane (discussed in refs. 352, 382). This effect was attributed to intramolecular interactions between the sensor and endogenous calmodulin, which is present at high concentrations in the vicinity of the plasma membrane due to its role as a binding partner for Ca2+ channels620,621. Indeed, a key consideration in biosensor design is the potential for the sensing unit, which is derived from cellular proteins, to interact with endogenous components within cells, which can both perturb biological functions and also disrupt the biosensors response. Thus, to develop a sensor that was less likely to experience these effects, Heim and Griesbeck replaced the calmodulin/M13 molecular switch with one derived from troponin C (TnC), a muscle-specific Ca2+ sensor that plays a far less central role in intracellular Ca2+ signaling, yielding the sensors TN-L15, based on chicken skeletal muscle TnC, and TN-humTnC, based on human cardiac TnC386. Alternatively, Palmer and colleagues opted to retain the original YC design scheme and instead computationally re-engineered the complementary “bumps” and “holes” that make up the calmodulin-M13 binding interface, yielding a panel of designed, or “D”-series, YCs that are largely orthogonal to their endogenous intracellular counterparts, albeit with a notable reduction in affinity353. Importantly, both approaches produced FRET-based GECIs capable of reporting local Ca2+ dynamics at the plasma membrane353,386.
GECIs are highly desirable tools for monitoring neuronal activity, and FRET-based GECIs are no exception. Thus, considerable efforts have been devoted to enhancing the speed and sensitivity of FRET-based GECIs to reliably measure physiological Ca2+ dynamics in living neurons. As alluded to in Section 2.3.3.1.1, the Ca2+-binding affinity of GECIs can be tuned within a fairly broad range by mutating the two Ca2+-binding “EF-hand” motifs located in both of the globular domains of calmodulin147, and this approach has successfully yielded YC variants with Ca2+-binding affinities spanning several orders of magnitude359. In addition, the original calmodulin-M13 switch design used in these probes is based on in vitro studies showing that a hybrid protein in which calmodulin and M13 are fused by a diglycine linker undergoes a Ca2+dependent switch from an extended “dumbbell” shape to a more compact, globular form622. Because the binding of Ca2+-bound calmodulin (Ca2+/CaM) to target peptides is known to slow the dissociation of Ca2+ from calmodulin, thereby increasing apparent Ca2+-binding affinity623, shifting the conformational equilibrium towards the compact form is expected to increase the Ca2+-binding affinity of the hybrid complex, and indeed, the incorporation of a 5-residue linker was found to increase the Kd compared with the original diglycine624. Based on this logic, Horikawa and colleagues were therefore able to generate YC variants with enhanced Ca2+binding affinities by lengthening the calmodulin-M13 spacer from the original 2 to between 3 and 5 residues, yielding “YC-Nano” sensors with dissociation constants as low as 15 nM, which were sensitive enough to visualize spontaneous in vivo neuronal activity in zebrafish embryos352.
Meanwhile, parallel efforts have yielded improved TnC-based GECIs with very fast response kinetics for neuronal imaging. Like calmodulin, TnC is also a “dumbbell”-shaped molecule that contains a pair of EF-hand motifs in each of its globular “head” domains625. Whereas the N-terminal regions specifically bind Ca2+, the C-terminal domains are only partially selective and are capable of binding Mg2+, which also results in more complex Ca2+-binding kinetics626,627. However, by introducing mutations based on the Ca2+-binding sites of calmodulin into EF-hands III and IV within the C-terminal domain of TnC, Mank and colleagues were able to eliminate Mg2+ binding to the TN-L15 sensor and obtain an improved variant, TN-XL, that exhibited very fast Ca2+ binding kinetics, with an off-rate of ~140 ms385. However, these mutations somewhat decreased the affinity of TN-XL for Ca2+ compared with TN-L15 (~2 μM vs ~750 nM), potentially reducing its effectiveness in measuring small neuronal Ca2+ transients. These authors were subsequently able to boost the sensitivity of this probe by replacing N-terminal EF-hands I and II with a duplicate copy of domains III and IV to generate TN-XXL (Kd ~800 nM)384. More recently, Thestrup et al. set out to minimize the structure of TnC-based Ca2+ sensors by reducing the number of Ca2+-binding sites in the molecular switch, reasoning that fewer binding sites would yield a sensor with a simpler, more linear response behavior382. Although initially unsuccessful in obtaining a high-affinity, single-EF-hand derivative of the chicken TnC used in previous TN sensors, the authors were ultimately able to construct a high-affinity Ca2+ sensor by sandwiching a single functional EF-hand from the oyster toadfish Opsanus tau between CFP and YFP. The resulting “Twitch” sensors retained the rapid kinetics of the previous TN-XL/XXL sensors while exhibiting dissociation constants as low as 150 nM382.
In addition to Ca2+, other metal ions also play critical roles in cells, especially transition metal ions, which often function as essential cofactors in enzyme catalysis. Because these ions, like Ca2+, are often toxic, cells have evolved numerous pathways to tightly regulate their intracellular accumulation628, and components of these pathways have proven useful in the development of biosensors to monitor the dynamics of transition metal ion homeostasis in living cells. For example, van Dongen and colleagues set out to generate a copper biosensor based on the Cu(I)-induced dimerization between Atox1, a copper chaperone, and WD4, the 4th copperbinding domain of the “Wilson’s disease” protein (aka ATP7B), a copper-transporting ATPase397. However, although this strategy did indeed yield a Cu(I)-induced change in intermolecular FRET between CFP-Atox1 and W4-YFP, the response was disrupted by the presence of free thiol groups (e.g., DTT), which disrupted the Cu(I)-mediated bridging of Cys residues within these two metallothioneins. Instead, this effort serendipitously yielded the first FRET-based Zn2+ sensor, as these two domains also exhibited a Zn2+-induced FRET change that was insensitive to free thiols397. van Dongen et al. subsequently refined this design by incorporating a flexible linker between the metal-binding domains, yielding the CFP-Atox1Linker-WD4-YFP, or CALWY, sensor (Figure 3A)397. Using a strategy analogous to that described above for Cameleons, Vinkenborg and colleagues then systematically modified the length of this linker to engineer a suite of CALWY sensors with a range of Zn2+-binding affinities{19718032}. Qiao and coworkers were also able to construct a Zn2+ biosensor using the Zn2+-finger (ZF) 1 and 2 domains of the yeast Zap1 transcription factor401. Zn2+ binding causes these domains to adopt a canonical ZF fold structure and form an interdomain complex, and sandwiching this molecular switch between CFP and YFP generated the ZF1/2 sensor. This design was further optimized by Qin and coworkers to yield ZapCY1 and 2 (PMID21502528), while Dittmer et al. similarly utilized a single canonical Cys2His2 ZF domain from the Zif268 transcription factor, which also adopts a folded structure upon Zn2+ binding, to generate a more compact FRET-based Zn2+ biosensor399. Carter and colleagues also recently compared the performance of several Zn2+ sensors, providing a roadmap for obtaining quantitative measurements with minimal perturbation{PMID28758723}, which is crucial given the much smaller labile pools of intracellular Zn2+ and other metal ions versus Ca2+.
2.3.3.2.1.2. FRET-based voltage indicators
The earliest FRET-based GEVIs, like their single-FP counterparts, were developed by utilizing full-length voltage-gated ion channels to provide the molecular switch in the sensing unit. For example, Sakai and colleagues constructed the original, FRET-based VSFP using the Kv2.1 voltage-gated potassium channel257. To convert the channel into a biosensor, the authors joined CFP and YFP via a single-amino-acid linker and fused the resulting “CYFP” reporting unit after the S4 helix of a C-terminally truncated channel mutant. According to this design, the voltage-induced rotation of the S4 helix is expected to alter the relative orientation of CFP with respect to YFP, thereby yielding a change in FRET (Figure 3B), and indeed, the resulting construct exhibited a nearly linear relationship between CFP-sensitized YFP emission (i.e., FRET) and membrane potential257. Dimitrov et al. subsequently developed an improved FRET-based VSFP biosensor family by replacing the Kv2.1 sensing unit with Ci-VSD, which they coupled to a similar tandem CFP-YFP reporting unit fused to the C-terminus of the S4 helix253. Progressively lengthening the spacer between S4 and CFP yielded four variants, VSFP2A-D, with longer linkers producing larger FRET changes. However, the voltage sensitivity of all four variants was well outside the range of physiological membrane potentials observed in mammalian cells, and an R217Q mutation was therefore incorporated into the VSD to produce a sensor (VSFP2.1) with a more physiological response range253. This same design has been carried forward in subsequent FRET-based GEVIs, such as VSFP2.3172; VSFP2.4251, and “Mermaid”252. However, more recent probe designs have reconfigured the coupling between the sensing and reporting units by sandwiching the VSD between a FRET pair (Figure 3B), including VSFP-Butterfly243,248, “Mermaid2”244, and the recent Nabi sensors242.
2.3.3.2.1.3. FRET-based sensors for monitoring cyclic nucleotide dynamics
Most FRET-based cAMP biosensors utilize the intrinsic, cAMP-induced conformational change within either Epac1 or Epac2 as the basis of their sensing unit. For example, Nikolaev and colleagues generated Epac2-camps (“Epac2-based cAMP sensor”) by fusing YFP and CFP to the N- and C-termini of a minimal fragment spanning the second CNBD (CNBD-B) of Epac2285. Epac1-camps was similarly constructed using the solitary cAMP-binding domain of Epac1, which corresponds to CNBD-B from Epac2. Parallel work by DiPilato and coworkers alternatively utilized full-length Epac1 sandwiched between CFP and YFP to generate the “indicator of cAMP using Epac”, or ICUE298, though subsequent versions of ICUE have utilized an N-terminally truncated form of Epac1295,297 (Figure 3C). However, despite the prominence of Epac-based designs, various FRET-based cAMP sensors have also been generated using cAMP-binding domains from other proteins. In fact, the very first genetically encoded FRET-based cAMP sensor, PKA-GFP, was based on the cAMP-induced dissociation of EGFP- and EBFP-tagged PKA catalytic (C) and regulatory (R) subunits308, a design that was based on a previous fluorescent analog2 of PKA, called FlCRhR, featuring fluorescein and rhodamine conjugated to purified PKA C and R629. Surdo and colleagues also recently generated a FRET-based cAMP sensor using the CNBD-B domain of the type II PKA regulatory subunit (RII) by fusing CFP to the C-terminus and inserting YFP into a flexible loop, yielding a “cAMP universal tag for imaging experiments” (CUTie)287. Meanwhile, Mukherjee et al. were able to construct a FRET-based cAMP sensor with nanomolar sensitivity by sandwiching the CNBD from a bacterial cyclic-nucleotide-gated potassium channel630 between CFP and YFP300.
Several different binding domains have also been used as sensing units to construct FRET-based cGMP reporters. Initial designs utilized nearly full-length forms of PKG Iα, which undergoes a cGMP-dependent conformational change to relieve autoinhibition185. For example, both the CGY (“cyan-G kinase-yellow”)312 and cygnet (“cyclic GMP indicator using energy transfer”)314 sensors were constructed by sandwiching N-terminally truncated PKG Iα between CFP and YFP. Because PKG Iα functions as a dimer, these N-terminal truncations were used to remove the dimerization sequences and thus avoid intermolecular FRET between individual sensors. Notably, whereas CGY sensors retain the N-terminal autoinhibitory domain and thus preserve the native regulation of PKG Iα312, cygnet features a larger N-terminal deletion that also removes this domain, necessitating the incorporation of a Thr516Ala substitution to prevent the sensor from being constitutively active314. However, finding that these early designs exhibited slow response kinetics, Nikolaev et al. sought to improve the temporal resolution by engineering simplified molecular switches based on isolated cGMP-binding domains, including the CNBD-B domain of PKG I, the GAF-A domain of PDE2A, and the GAF-B domain of PDE5310. Of these, the “cGMP energy transfer sensor” based on PDE5 (cGES-DE5), showed the best performance, including a large, rapid FRET change capable of reporting cGMP pulses. In contrast, Russwurm and coworkers observed slow in vitro response kinetics when they constructed a FRET sensor incorporating both the GAF-A and B domains of PDE5 and instead engineered a rapidly responding cGMP indicator (cGi) based on CNBD-A and B of PKG311.
2.3.3.2.1.4. FRET-based sensors for lipid messengers
As described in Section 2.3.1, lipid-based messengers were a major focus of early biosensor development, yielding translocation-based fluorescent biosensors for detecting multiple lipid species. However, despite their continued utility, these sensors possess certain limitations, for instance, in their ability to probe lipid production in different subcellular compartments, as their reliance on translocation as a reporting mechanism is incompatible with subcellular targeting. Meanwhile, the development of FRET-based biosensors has provided an alternative reporting strategy that can be combined with targeting to specific subcellular sites.
Like translocation-based fluorescent probes, FRET-based lipid sensors utilize endogenously derived lipid-binding domains. The most commonly used design was originally reported by Sato and colleagues, who generated the FRET-based PI(3,4,5)P3 sensor Fllip (“fluorescent indicator for lipid second messenger”) using three rigid α-helical linkers631 to position the PH-domain of Grp1 (PHGrp1) between CFP and YFP and also anchor the probe to a target membrane520. A key aspect of this design is the presence of a diglycine “hinge” within the linker connecting PHGrp1 and YFP; thus, in the absence of PI(3,4,5)P3, the rigid linkers keep CFP and YFP separated, whereas PHGrp1 binding to PI(3,4,5)P3 causes CFP to “flip” outward and into closer proximity to YFP (Figure 3C). Swapping out the lipid-binding domain has allowed this design to be adapted to monitor other lipid messengers, including PI(4,5)P2 (PHPLCδ)506, PI(4)P (PHFAPP1)506, phosphatidic acid (PHSOS1, Dock2 C-terminal domain)513, and DAG (PKCβ C1 domain)505,506. Alternatively, Ananthanarayanan et al. constructed a FRET-based PI(3,4,5)P3/PI(3,4)P2 sensor by tethering PHAkt to a negatively charged pseudoligand sequence504, based on evidence that PHAkt recognizes head groups via a basic binding pocket632. Sandwiching this switch between CFP and YFP yielded InPAkt (“indicator for phosphoinositides based on Akt”), wherein binding of PHAkt to PI(3,4,5)P3/PI(3,4)P2 displaces the pseudoligand and induces a FRET change, similar to the design of cameleon.
2.3.3.2.1.5. FRET-based sensors for tracking other cellular analytes
Numerous FRET-based biosensors have also been developed to illuminate the spatiotemporal dynamics of several additional analytes that are critical for cellular function. As discussed previously for engineered single-FP sensors (see Section 2.3.3.1.3), the designs of these probes often take advantage of the diverse molecular machinery present in bacterial cells, whose genomes encode a variety of proteins capable of binding intracellular and extracellular analytes with high affinity and specificity. For example, Zhao and colleagues recently developed a FRET-based NADP+ sensor (NADPsor) by sandwiching E. coli ketopantoate reductase, which undergoes a large conformational change due to the binding of NADP+ between its N- and C-terminal domains633,634, between CFP and YFP339. San Martín and coworkers also recently developed FRET-based sensors for lactate (“lactate optical nano indicator from CECs [Centro de Estudios Científicos]”; Laconic) and pyruvate (Pyronic) by similarly sandwiching the E. coli transcription factors LldR and PdhR, respectively, between a FRET pair332,342. Notably, bacterial PBPs have proven to be a particularly rich resource for the development of genetically encoded fluorescent biosensors, given their broad target repertoire and well-characterized conformational changes (see Section 2.3.3.1.3). Both the FLIPE (“fluorescent indicator protein for glutamate”)500 and GluSnFR (“glutamate sensing fluorescent reporter”)501,635 sensors, for instance, are based on the E. coli ybeJ/Glt1 protein, which is predicted to switch between open and closed conformations in response to the binding of glutamate. Meanwhile, PBPs have also yielded several FRET-based sensors that have been used to monitor the concentrations of various sugars, including maltose334, glucose321, ribose343, sucrose344, and more recently, trehalose345. [Add one or two sentences for the hybrid sensors]
2.3.3.2.2. FRET sensors for monitoring enzyme activation/activity
Along with sensing the dynamics of second messengers, metabolites, and other analytes, conformational changes in protein-based molecular switches can also be used to report on the biochemical functions of proteins themselves. Indeed, the sensitivity of FRET to intramolecular conformational changes has enabled the construction of genetically encoded biosensors capable of monitoring not only the activation of proteins in response to specific signaling events but also the endogenous catalytic activity of many key signaling enzymes directly within living cells. Sensors belonging to the former category, much like early FP-based sensor designs (see Section 2.2), typically utilize a full-length protein of interest to serve as a proxy for its own behavior (e.g., activation), whereas sensors in the latter category generally make use of a surrogate substrate that can be catalytically modified by an enzyme of interest as part of an engineered molecular switch. FRET-based biosensors have thus opened a window onto the biochemical behavior of proteins in their native context that extends far beyond the ability to simply track protein expression, localization, and interactions.
2.3.3.2.2.1. FRET-based enzyme activation sensors
Many proteins undergo conformational changes in response to various signaling events (e.g., messenger binding), and while this phenomenon has proven extremely useful as a proxy for monitoring the dynamics of upstream signaling processes (e.g., messenger production), these same conformational changes can also be used to directly probe the activation dynamics of signaling proteins themselves. Often, this approach involves the heterologous overexpression of the protein of interest fused at its N- and C-terminus to a donor and acceptor FP, such that the activation-induced conformational change will alter FRET between the FP pair. This design has been applied to generate activation indicators for a variety of different enzymes, including a number of protein kinases. For example, to study the spatiotemporal regulation of Ca2+/CaM-dependent kinase II (CaMKII), a key signaling enzyme involved in learning and memory, Takao and colleagues appended YFP and CFP to the N- and C-termini of full-length rat CaMKIIα to generate Camuiα430. The binding of Ca2+/CaM switches CaMKII from a closed, autoinhibited state to an open, active conformation, thus inducing a FRET decrease in Camuiα. Similar sensors have also been developed to probe Erk2 activation during MAPK signaling448, the regulation of PKB/Akt activation by phosphoinositides and phosphoinositide-dependent kinase 1 (PDK1)mediated phosphorylation415, and the activation of PDK1 itself468. Recently, the activation of the Ca2+/CaM-dependent protein phosphatase calcineurin (CaN) was similarly studied by sandwiching the full-length CaN catalytic subunit between CFP and YFP431(Figure 4A).
Figure 4:
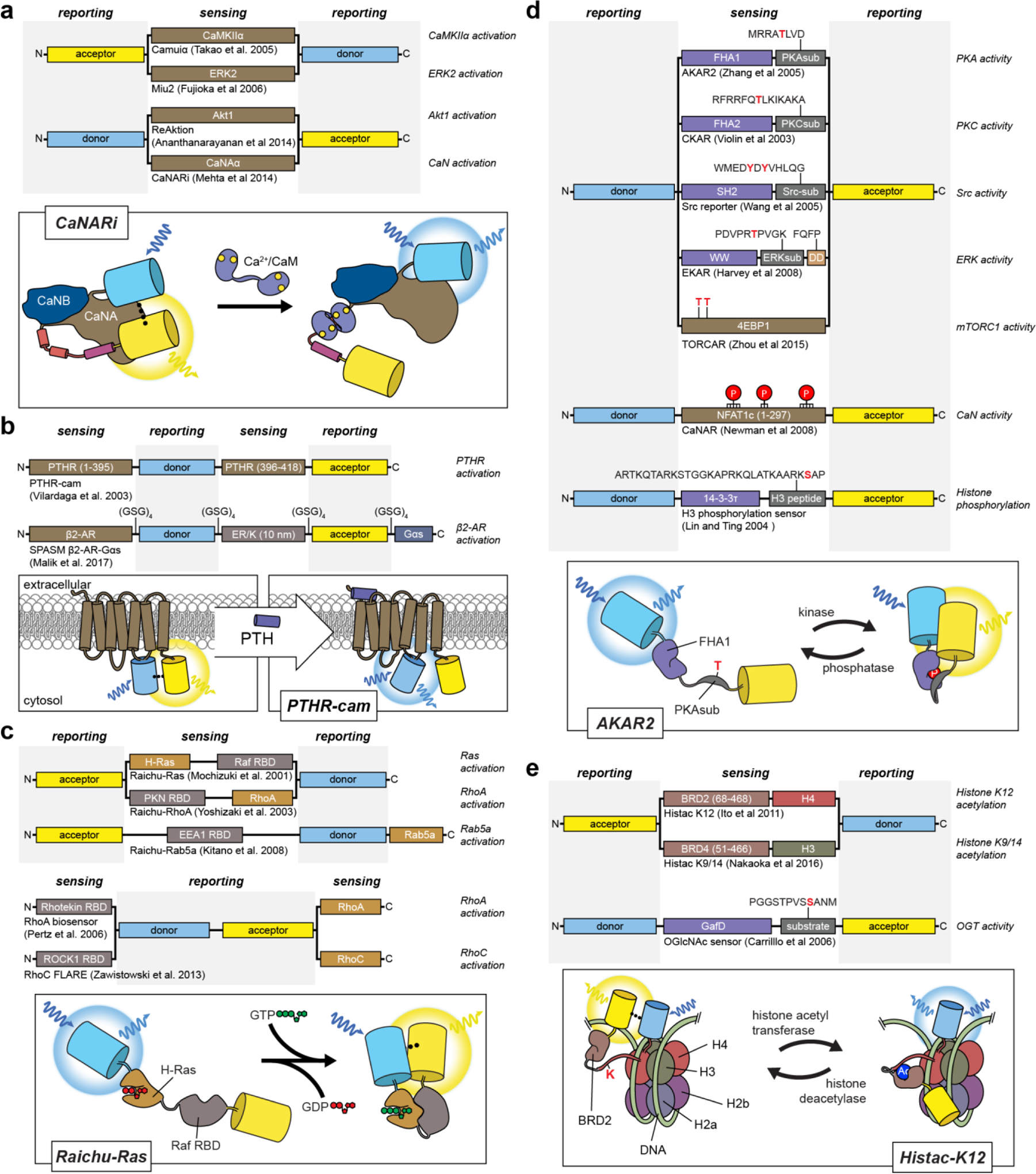
FRET-based biosensor designs for signaling proteins.
Biosensors for signaling enzyme activation consist of whole or truncated portions of the enzyme sandwiched between a FP FRET pair430,431,448,504. For example, the conformational change in the phosphatase CaNAα upon activation by Ca2+ bound CaM increases the distance between the two FPs in CaNARi431. B) The insertion of FPs into a receptor551 or between a GPCR and Gαs537 have been utilized to create FRET-based biosensors of receptor activation. C) Biosensors for small G-protein activation utilize G-protein binding domains (e.g., Raf RBD, PKN RBD, EEA1 RBD), which bind to specific G-proteins upon activation to create a conformational change587,592,598,600,601. D) FRET-based biosensors for kinase activity consist of either an endogenous kinase substrate406 or a kinase substrate sequence paired with a phospho-amino acid binding domain444,454,476,481,494 sandwiched between two FPs, which undergo a conformational change upon phosphorylation. Conversely, the phosphatase biosensor CaNAR uses a fragment of NFAT1c, which is phosphorylated at basal levels and exhibits a conformational shift upon dephosphorylation by CaN432. E) Similarly, biosensors for other PTMs use substrates paired with protein domains that recognize the modified substrates. The Histac biosensors contain a full-length histone, H3 or H4, and a fragment of a bromodomain (BRD)containing protein that binds the acetylated substrate605,606.
Vilardaga and colleagues also previously utilized this biosensor strategy to investigate the activation kinetics of the α2A adrenergic receptor (α2AAR) and PTHR, members of two distinct classes of the GPCR superfamily of seven-transmembrane cell-surface receptors551. To detect the conformational changes that occur during GPCR activation, the authors fused YFP to the C-terminus and inserted CFP within the 3rd intracellular loop of both the α2AAR and the PTHR, as the activation of these GPCRs has been shown to include shifts in the relative position of the 6th transmembrane helix636, which flanks the 3rd intracellular loop (Figure 4B). In fact, several FRET-based sensor designs have been employed to study the activation of GPCRs, which comprise the largest family of signaling proteins and are responsible for mediating the activation of numerous signaling pathways in response to diverse extracellular cues. Activated GPCRs function as GEFs for heterotrimeric G proteins, leading to the dissociation of the GTP-bound α subunit (Gα) from the Gβγ-subunit dimer and downstream signaling. Thus, early studies utilized the decrease in intermolecular FRET between FP-labeled G protein subunits as a proxy for GPCR activation540, an approach that continues to be used637. Meanwhile, others have similarly monitored the association of FP-labeled receptor and the Gαβγ heterotrimer (e.g., ref. 550 and also Section 2.3.3.3). Recently, Malik et al. adapted this approach to devise a unimolecular GPCR activation sensor in which YFP and CFP, separated by a 10-nm ER/K linker (see Section 2.3.3.1.4), are sandwiched between a full-length GPCR and a high-affinity binding peptide derived from a specific Gα subunit539. An updated version of this design incorporates a full-length Gα subunit and permits association with endogenous Gβγ537.
Monomeric G proteins represent another large superfamily of signaling proteins that are crucial regulators of multiple cellular processes, and several FRET-based biosensors have been developed to track the activation dynamics of these small GTPases in living cells. In general, the design of these probes utilizes the intramolecular binding between a full-length GTPase and a tethered binding domain from an effector protein to drive a change in FRET between an FP pair, analogous to the design of cameleon. For example, Mochizuki and colleagues constructed the Raichu (“Ras and interacting protein chimeric unit”) sensor by tethering full-length H-Ras to the Ras-binding domain (RBD) from Raf and sandwiching the resulting molecular switch between YFP and CFP (Figure 4C)592. This modular architecture has proven to be highly generalizable, and substituting H-Ras/Raf-RBD with other cognate binding pairs has yielded a family of Raichu sensors for Rap1592, Rac584, Cdc42584, RhoA600, RalA590, TC10/RhoQ602, and R-Ras603 (Table 1). Notably, while these biosensors play an important role in exploring the regulation of GTPases by GEFs and GTPase-activating proteins (GAPs), they cannot be used to study the specific regulation of Rho-family GTPases by guanosine dissociation inhibitors (GDIs) due to the masking of the GTPase C-terminus638, leading to the development of sensors with reconfigured architectures. For instance, when constructing Raichu-Rab5, Kitano et al. kept the RBD between YFP and CFP but moved full-length Rab5a to the biosensor C-terminus587. Others have opted to completely invert the design by sandwiching a FRET pair between the GTPase and RBD597,598,601. Finally, some designs incorporate only the RBD, eschewing the inclusion of a full-length GTPase component in order to monitor endogenous GTPase activation584,600,618.
2.3.3.2.2.2. FRET-based enzyme activity reporters
Proteins must rapidly and continuously adapt their functions to suit the ever-changing needs of a highly dynamic cellular environment. Covalently altering protein sequences by introducing post-translational modifications allows cells to dynamically modulate virtually all aspects of protein function and greatly expand the functional diversity of the cellular proteome, and FRET-based biosensors have proven particularly apt for visualizing the activities of the enzymes responsible for catalyzing the myriad post-translational modifications taking place within cells. These biosensors are based on the incorporation of a target protein sequence into the sensing unit, which therefore functions as a surrogate substrate for a given enzyme activity. Molecular switching behavior is conferred either by utilizing a protein domain that intrinsically alters the biosensor conformation when modified by the target activity or by tethering a minimal substrate sequence (i.e., receiver domain) to a separate module that recognizes and binds the modified substrate (i.e., switch domain) to drive an activity-induced conformational change. Importantly, biosensors for monitoring enzyme activity are subject to enzymatic amplification, in that a single enzyme is able to react with multiple biosensors, which increases probe sensitivity due to the exponential relationship between the biosensor response and the number of active enzymes. This property, combined with the targetability of these genetically encoded probes to specific subcellular regions, allows FRET-based enzyme activity sensors to sensitively report on the spatiotemporal dynamics of endogenous enzyme activities within the context of their native regulatory environment in living cells.
2.3.3.2.2.2.1. FRET-based sensors for monitoring protease activity
What can be considered the first genetically encoded FRET-based biosensors were generated by sandwiching an oligopeptide containing a protease cleavage site between GFP and BFP, whereby catalytic cleavage of the protease substrate liberates the two FPs and produces a dramatic decrease in intramolecular FRET639,640. Although these initial constructs were built largely as a proof of concept to explore and characterize the occurrence of FRET between different FP color variants, the site-specific cleavage of proteins by various proteases is in fact an essential component of numerous physiological – and pathological – processes, and this simple probe design has thus frequently been replicated to study the dynamics of a number of important cellular proteases. Cysteine-aspartyl proteases (caspases), for example, are key regulators of apoptotic cell death pathways, and several groups have studied the dynamics of both effector and initiator caspases by sandwiching the cleavage sites for caspase-3 (Casp3) (e.g., “DMQD”533 or, more commonly, “DEVD”524–529,532), as well as Casp8 (“IETD”)529 and Casp9 (“LEHD”)532 between a FRET FP pair. Ouyang and colleagues have similarly visualized the activity of membrane type 1 matrix metalloproteinase (MT1-MMP), a membrane-tethered extracellular protease that is upregulated in invading cancer cells535,536. Alternatively, Li et al. utilized full-length versions of the LC3B or GATE-16 proteins sandwiched between CFP and YFP to track the activity of Atg4, a key protease involved in regulating autophagy523.
2.3.3.2.2.2.2. FRET-based sensors for monitoring the dynamics of protein phosphorylation
The reversible phosphorylation of proteins on Ser, Thr, or Tyr residues is arguably the most important post-translational modification involved in regulating protein function in cells and also one of the most abundant, with ~40% of the total human proteome estimated to be phosphorylated by protein kinases at some point in time641. FRET-based genetically encoded kinase activity reporters (KARs) were designed based on the fact that cells contain multiple protein modules, known as phosphoamino acid binding domains (PAABDs), that are capable of specifically recognizing and binding phosphorylated residues within target proteins642–644. Thus, by following the architecture established with cameleon, Zhang and colleagues engineered a bipartite kinase-inducible molecular switch in which a consensus phosphorylation sequence for PKA (LRRASLP, based on the so-called “kemptide” PKA substrate645) was tethered by a flexible linker to the PAABD 14-3-3τ. In the resulting 1st-generation “A-kinase activity reporter” (AKAR1)477, PKA-mediated phosphorylation increased the intramolecular binding between 143–3 and the substrate, thereby inducing a conformational rearrangement and altering the FRET between a flanking CFP and YFP pair477. However, the high affinity of the 14-3-3 domain rendered AKAR1 insensitive to cellular phosphatases, and the largely irreversible FRET response was thus unable to capture the full temporal dynamics of PKA activity. Replacing 14–33τ with the lower-affinity forkhead-associated 1 (FHA1) domain, in conjunction with a modified PKA substrate (LRRATLVD) designed to better fit the preferred recognition sequence for FHA1646, therefore yielded a 2nd-generation AKAR (AKAR2), as well as subsequent variants (AKAR3475 and AKAR4473), with a fully reversible FRET response (Figure 4D)476.
Much like the aforementioned Raichu GTPase sensor (Section 2.3.3.2.2.1), the modular architecture of AKAR proved to be highly generalizable and has yielded a diverse family of FRET-based sensors for monitoring the activity of a multitude of different protein kinases that are too numerous to mention (see Table 1). Biosensor specificity is, of course, determined by the incorporation of a substrate sequence that will be recognized and phosphorylated by the kinase of interest but not by other kinases, while in some cases, additional sequence elements are included to enhance specificity, such docking sequences for MAPKs444,445,459,465. Yet identifying a suitable, kinase-specific phosphorylation sequence can be a difficult task that often requires considerable trial and error, though more systematic approaches may help speed up this process, especially for kinases with poorly defined or unknown substrate preferences. For example, Tsou et al. utilized a positional scanning peptide library screen647 when designing a substrate sequence for their AMP-dependent kinase activity reporter (AMPKAR), although multiple candidate sequences were still tested418. More recently, when designing a FRET-based KAR for Rhoassociated protein kinase (ROCK), Li and colleagues initially trialed several previously characterized substrate sequences before turning to a newly developed kinase-interacting substrate screening (KISS) approach648, in which cell lysates are run though kinase-coated beads to enrich interacting substrates, followed by tandem mass spectrometry to deduce the consensus phosphorylation sequence489. Nevertheless, efforts to engineer a molecular switch can sometimes yield no successful candidates, in which case intrinsic molecular switches can also be used, as in the case of Zhou and coworkers, who utilized the native phosphorylation-induced conformational change in full-length 4EBP1 as the basis for their mTOR complex 1 activity reporter (TORCAR)406.
It is important to remember that phosphorylation is a highly dynamic and reversable PTM where the addition of phosphate groups by kinases is constantly opposed by protein phosphatases which are responsible for catalyzing their removal. While cells typically contain a far smaller variety of protein phosphatases (e.g., mammalian genomes encode roughly 1/5th the number of phosphatases as kinases649), these enzymes are no less important in regulating protein function. Nevertheless, visualizing protein phosphatase activity in living cells has proven to be a far greater challenge compared with monitoring protein kinase activity. For one thing, these enzymes have differing requirements for substrate recognition: kinase specificity is defined by the residues that surround the target phosphorylation site, while phosphatases are far more promiscuous in this regard (a fact related to their smaller numbers), with specificity instead being governed by conserved docking motifs within substrate proteins650 and coordination by regulatory domains651. The chief obstacle, however, is the fact that, unlike KARs, which become phosphorylated, a phosphatase sensor must already be phosphorylated, such that it can respond to being dephosphorylated, and constructing a constitutively “dephosphorylation-competent” molecular switch remains something of a conundrum. Thus far, the only design to successfully overcome this problem is that of the FRET-based CaN activity reporter (CaNAR)431,432, whose intrinsic molecular switch features an endogenous CaN substrate (nuclear factor of activated Tcells, NFAT) that is basally hyperphosphorylated by multiple kinases652. Meanwhile, in the absence of a more generalizable design, the development of genetically encoded phosphatase sensors continues to lag far behind that of kinase sensors.
2.3.3.2.2.2.3. FRET-based sensors for monitoring other post-translational modifications
Cells call upon an extensive repertoire of post-translational modifications to regulate protein function. This is perhaps best embodied by histone proteins, which comprise the core structural scaffold in chromatin and undergo a particularly high degree of post-translational modifications along their N-terminal tails. These modifications provide recognition sites for various chromatin-binding proteins and also control access to DNA by the replication and transcriptional machinery, and understanding the dynamics of histone modification is therefore critical for unraveling the mechanisms of epigenetic regulation. Thus, Lin and Ting constructed a sensor for visualizing the phosphorylation of histone H3 at Ser28, which is catalyzed by numerous kinases653, by engineering a molecular switch composed of the 30 N-terminal amino acids of histone H3 tethered to 14-3-3τ and sandwiched between CFP and YFP454. Histones are also frequently acylated and methylated on lysine residues, and Lin et al. were able to generate a FRET-based biosensor for monitoring the methylation of histone H3 on lysine 9 or 27 by similarly using short peptides spanning these regions608. However, more recent efforts have opted for designs that incorporate full-length histones, rather than just minimal peptides, including sensors developed to monitor the acetylation dynamics of both histone H3605 and histone H4606,607. Again, these sensors feature engineered molecular switches wherein the specific histone is coupled to an appropriate binding domain to drive a conformational change (Figure 4E). As a critical component of these biosensors, chromodomains and bromodomains are compact protein modules that specifically recognize methylated654 and acetylated655 lysines, respectively. Thus, Lin and coworkers utilized chromodomains from HP1 polycomb proteins to recognize methylated histone H3608, while Nakaoka et al. used the bromodomain from the BRD4 protein to construct a histone acetylation (Histac) sensor for histone H3 acetylation605.
Meanwhile, a unique form of glycosylation, involving the specific addition of O-linked N-acetylglucosamine (O-GlcNAc) to Ser or Thr residues in proteins throughout the cell, has emerged over the last several years as a major post-translational modification that is implicated in multiple physiological and pathological processes (recently reviewed in ref. 656). In contrast to many established post-translational modifications, O-GlcNAcylation is exclusively regulated by a single pair of enzymes, namely, O-GlcNAc transferase (OGT), which catalyzes the addition of O-GlcNAc, and O-GlcNAcase, which catalyzes its removal. Furthermore, the reciprocal nature of O-GlcNAcylation and phosphorylation, which often compete for the same sites on many proteins, with O-GlcNAcylation able to block protein phosphorylation, suggests a potentially important role for the dynamic interplay between these modifications in the regulation of intracellular signaling. Thus, to track the spatiotemporal dynamics of O-GlcNAcylation in living cells, Carrillo and coworkers set out to engineer a O-GlcNAc-responsive molecular switch and generate a FRET-based O-GlcNAc sensor609. As the receiver domain, they utilized the OGT substrate sequence from casein kinase II, while the E. coli GafD protein, a monomeric lectin (i.e., carbohydrate-binding protein) that specifically recognizes O-GlcNAc, was selected as the switching domain. Again, the selection of both a specific substrate sequence and an appropriate recognition domain is essential for designing an activity-induced molecular switch. Though currently limited to empirical trial and error, it is hoped that the application of more high-throughput methods, such as molecular evolution, will propel the wider development of FRET-based enzyme activity biosensors.
2.3.3.2.2.3. FRET-based sensors for measuring mechanical forces in cells
Cells are physical entities that engage in a variety of mechanical processes, both extrinsic (e.g., cell-cell/cell-environment interactions) and intrinsic (e.g., chromosome segregation, cytokinesis). These processes generate mechanical forces that provide information on the physical state of the cell and its environment, sending signals to the intracellular biochemical machinery and regulating cell physiology and behavior. Indeed, mechanosensitive signaling is fundamental to virtually all aspects of biology across evolution657. Elucidating the mechanisms by which cells sense and interpret stimuli from mechanical forces (i.e., mechanotransduction) is thus essential to our understanding of physiology and disease. Notably, the past decade has seen the development of several genetically encoded FRET-based biosensors capable of performing in situ measurements of the piconewton (pN)-scale mechanical forces experienced by individual molecules inside living cells. In general, these sensors make use of a tension-sensing module composed of an FP pair tethered to either side of a mechanosensitive element 217,220–223. This linker adopts a compact conformation at rest but becomes elongated under strain, thereby increasing the interfluorophore distance and causing FRET to vary as a function of mechanical tension across the module, which can be inserted within a protein of interest to visualize mechanical forces. For example, Grashoff et al. constructed a tension-sensing module using a molecular nanospring composed of 8 tandem 5-amino-acid repeats from the spider silk protein flagelliform658, which they sandwiched between a FRET pair and inserted between the head and tail domains of the vinculin protein to measure tension across focal adhesions223.
Importantly, given the aforementioned mathematical relationship between distance and FRET efficiency (Eq. 1, Section 2.2.2), these mechanosensing probes can be calibrated in vitro (e.g., using single-molecule force spectroscopy659) to convert quantitative FRET measurements into absolute pN force values. Performing such quantitative force measurements requires the mechanosensitive domain to fulfil certain requirements (reviewed in ref. 660), such as relaxation upon the removal of force (i.e., reversibility) and a lack of hysteresis between elongation and relaxation. Furthermore, the identity of the mechanosensitive domain also determines the range of forces that can be detected. Thus, whereas the 40-amino-acid elastic domain used by Grashoff et al. was sensitive between 1 and 6 pN of force223, Austen and coworkers utilized variants of the 35-amino-acid α-helical vilin headpiece peptide, which undergoes an unfolding transition in response to mechanical tension, to sense forces ranging from 7–10 pN217. More recently, Brenner and colleagues found that a shorter flagelliform peptide comprising only 5 repeats showed a much broader sensitive range, spanning 2–11 pN222. Nevertheless, these probes remain limited to monitoring intracellular forces within a relatively narrow window, while their FRET responses also exhibit somewhat poor dynamic ranges. Meanwhile, rather than relying on modulating the distance between a pair of FPs connected by an elastic linker, Meng and colleagues developed an alternative force sensor design that produces large FRET changes by altering the relative orientation between a pair of linked FPs whose dipoles are (almost) perfectly aligned at rest219. Such continued innovation will be crucial for expanding the utility of FRET-based force biosensors.
2.3.3.2.3. Strategies for optimizing FRET-based biosensor responses
Dynamic range plays a key role in determining the signal strength and detection limit of a genetically encoded fluorescent biosensor. In the case of FRET-based biosensors, this property refers to the degree of difference between the lowest FRET signal in the “off” state and the highest FRET signal in the “on” state; the larger this range, the more likely a small change in the parameter of interest will yield a robustly detectable FRET response. A substantial proportion of biosensor development is thus devoted to optimizing dynamic range, for which two major strategies include minimizing basal FRET in the “open” biosensor conformation and maximizing FRET efficiency in the “closed” conformation. The first strategy essentially entails increasing the separation between the donor and acceptor FPs and is largely accessible only to biosensors that feature an engineered, bipartite molecular switch rather than an intrinsic conformational change. For example, many such FRET-based biosensors feature a short linker between the receiver and switch domains (e.g., cameleon359, AKAR477, and Raichu592). However, in an effort to develop an optimized backbone for constructing FRET-based biosensors, Komatsu and colleagues found that progressively increasing linker length using repeating (SAGG) units yielded proportional decreases in basal FRET signals414. This work yielded the “extension for enhanced visualization by evading extraFRET” (Eevee) system and the corresponding “EV linker”, along with a number of enhanced FRET-based biosensors414.
Conversely, FRET efficiency can be increased by minimizing the interfluorophore distance in the closed sensor conformation. Although this conformation is expected to bring the donor and acceptor FPs into close proximity, conformational dynamics within the sensor may cause this distance to fluctuate. Thus, allowing the FPs to associate may help ensure they achieve the closest possible approach. In fact, Vinkenborg et al. 661 suggested this effect as the mechanism underlying the enhanced FRET behavior of the molecularly evolved CyPet/YPet pair662 and later applied this principle to improve the dynamic range of the CALWY Zn2+ sensor395. Given that CALWY exhibits a FRET decrease upon Zn2+ binding, FP dimerization thus helps stabilize the high-FRET “off” state but, importantly, does not affect the conformational switch or the resulting FRET change. Meanwhile, this approach may conceivably pose a problem in sensor designs where the conformational change brings the donor and acceptor closer together and thus increases FRET, as FP dimerization may yield spurious FRET increases, though previous work has suggested otherwise290,663. Nevertheless, a more straightforward approach is simply to use better FPs. Recall from Section 2.2.2 that R0, the Förster distance, is influenced by the photophysical properties of the donor and acceptor. These include the extinction coefficient (i.e., how efficiently photons are absorbed) and quantum yield (i.e., how efficiently photons are re-emitted), and selecting a donor and acceptor pair that optimizes these parameters can greatly improve FRET, and thus dynamic range. FRET can also be enhanced through the incorporation of tandem acceptor FPs290, as demonstrated recently by Klarenbeek et al. in the development of fourth-generation FRET-based cAMP sensors with dramatically improved dynamic ranges288.
The relative orientation of the donor and acceptor FPs also contributes to the FRET efficiency, as the donor and acceptor dipoles must be properly aligned for maximal energy transfer to occur. Although discussed only in the context of single-FP biosensors thus far, circular permutation can also be used to shift the angle at which an FP is fused to the sensing unit in a FRET-based biosensor. The resulting alterations to the relative orientation of the FP and its chromophore can have a significant impact on FRET efficiency by improving dipole-dipole alignment, which has been frequently used to increase biosensor dynamic range273,295,355,398,475. Using truncated FPs (i.e., deleting the flexible N- or C-terminal regions) can also alter the orientation of the chromophore with respect to the final biosensor structure, as can modifying the linkers connecting the FPs to the sensing unit. However, absent detailed structural information, anticipating how specific changes will affect chromophore orientation is difficult, if not impossible, meaning that various incremental changes must be tested through trial and error. Furthermore, optimization typically involves pursuing multiple strategies and, given the above maze of options, may require testing dozens or even hundreds of candidates. More high-throughput approaches are thus sorely needed. To this end, Belal and colleagues developed a bacterial colony screening system for optimizing FRET-based KARs in which both kinase and sensor candidate are inducibly expressed from a single plasmid409, a potentially important advance given that enzyme activity sensors are left out of current bacterial screening approaches.
2.3.3.2.4. Biosensors based on modulating fluorescent protein dimerization
While controlling FP proximity to modulate FRET remains arguably the most popular reporting strategy for genetically encoded fluorescent biosensors, other designs have also emerged in recent years that utilize alternative fluorescent readouts based on FP proximity (also see Section 2.4.3 below). For example, whereas most FP-based applications favor the use of engineered monomeric variants to avoid artifacts caused by the native propensity of FPs to selfassociate, Alford and colleagues have conversely sought to take advantage of the obligate oligomerization of RFPs, which stabilizes the chromophore and thus enhances brightness152,664,665, in an effort to develop a novel biosensor reporting strategy based on modulating dimerization-dependent increases in RFP intensity364. Starting with a weakly fluorescent, monomeric form of tdTomato200, designated “A”, they performed extensive molecular evolution to identify an “dark” interacting partner, designated “B” and also derived from tdTomato, that would rescue the fluorescence of A upon heterodimer formation. This approach ultimately yielded a dimerization-dependent RFP (ddRFP) that exhibited an ~10-fold intensity increase upon dimer formation, which enabled the straightforward construction of dualFP, single-color Ca2+ and protease activity sensors (Figure 5A)364. Subsequent engineering of this initial ddFP also succeeded in producing ddGFP and ddYFP variants666. Meanwhile, Ding et al. recently observed that the B subunits, which contain no chromophore364, were interchangeable between ddRFP-A (RA) and ddGFP-A (GA)293. This observation spurred the development of yet another novel biosensor readout based on so-called fluorescent protein exchange (FPX). Specifically, these biosensors utilize a molecular switch that is designed to exchange GA-B dimer formation for RA-B dimer formation in response to a biochemical signal, yielding reciprocal (i.e., ratiometric) changes in ddGFP and ddRFP intensity293 (Figure 5A). One potentially significant advantage of FPX is its apparent insensitivity to linker length/composition, suggesting a far more straightforward path for biosensor design than that for FRET-based sensors.
Figure 5:
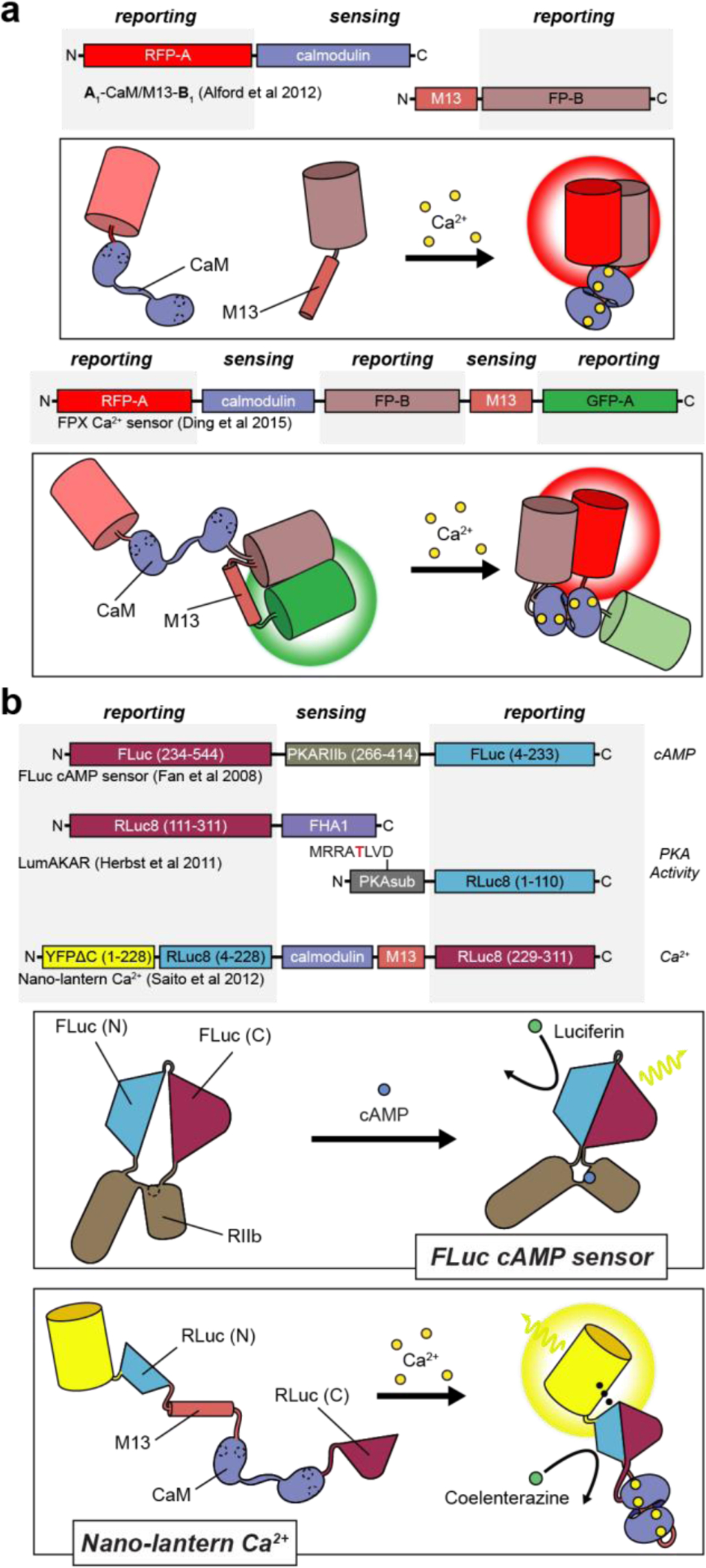
ddFP- and luminescence-based biosensors.
Bimolecular (top) or unimolecular (bottom) Ca2+-biosensor designs based on ddFPs293,364. The dimeriaztion of the dim ddFP partner, FP-B, with either RFP-A or GFP-A increases their fluorescence, and this dimerization is modulated by the Ca2+dependent binding of calmodulin to M13. B) Modulating the structure of split luciferaces by conformational switches has been utilized in cAMP, PKA and Ca2+ biosensors276,303,472. In the FLuc cAMP sensor, the conformational switch induced by binding of cAMP to PKA RIIβ allows the firefly luciferase (FLuc) to properly form, thus allowing the enzyme to catalyze the degradation of luciferin and emit photons303. Similarly, the Nano-lantern Ca2+ biosensor undergoes a Ca2+-dependent reconstitution of renilla luciferace (RLuc), which is then capable of BRET with the adjacent, brighter YFP276.
2.3.3.3. Biosensor designs incorporating luminescent proteins
Bioluminescence, wherein light is generated through an enzyme-catalyzed chemical reaction, represents an alternative biosensing strategy that has in some ways been overshadowed by the popularity of FP technology. Yet bioluminescence offers a number of advantages that support its application in areas where the use of fluorescence-based biosensors continues to pose a challenge (e.g., in vivo). Specifically, the wavelengths used to excite most genetically encoded fluorescent biosensors often cause problems related to autofluorescence, fluorophore bleaching, phototoxicity, and poor tissue penetration due to scattering/absorption within the specimen. In contrast, bioluminescence relies on a broad class of luminescent proteins, known as luciferases, that emit light via the oxidation of a luciferin substrate (e.g., coelenterazine or similar molecules) and therefore require no external illumination. Thus, luminescence-based readouts have the potential to yield reduced background and enhanced contrast and signal-to-noise ratios, and may also lower some of the barriers to in vivo imaging.
Luciferases are present throughout the natural world, most notably in a veritable menagerie of marine organisms667. Among the most commonly used luciferase variants are those derived from the sea pansy Renilla reniformis (Renilla luciferase, RLuc) and the North American firefly Photinus pyralis (firefly luciferase, FLuc), which have been used to construct genetically encoded biosensors that mirror many of the reporting unit configurations found in FP-base probes. For example, based on structural data indicating that FLuc comprises two globular domains that close in a hinge-like motion upon substrate binding668, Fan and colleagues generated a circularly permuted FLuc in which the native N- and C-termini were relocated to opposite sides of this hinge, such that the insertion of a sensing unit would render FLuc activity sensitive to various biochemical signals303. Bridging this gap using either a short “DEVDG” peptide linker or the CNBD-B domain from PKA RIIβ thus yielded luminescent indicators for caspase-3 activity or cAMP accumulation, respectively303 (Figure 5B). Luciferases are also compatible with fragment complementation, offering a sensitive and dynamic readout that contrasts with the irreversible complementation of FP fragments. Indeed, Stefan et al. previously used this approach to monitor PKA activation in living cells by tagging the regulatory and catalytic subunits of PKA with complementary RLuc fragments479. Herbst and colleagues similarly used RLuc fragment complementation to generate luminescent KARS in which the PAABD and substrate peptide are expressed as two separate polypeptides, fused to complementary RLuc fragments, which reassociate upon phosphorylation472. This approach successfully yielded luminescent KARs for monitoring PKA and PKC activity (Figure 5B).
Much like the excited state of an FP chromophore, the high-energy reaction product of luciferin oxidation is also capable of non-radiative energy transfer to a nearby acceptor. Thus, a number of genetically encoded biosensors utilize bioluminescence resonance energy transfer (BRET) as the reporting mechanism, which often requires little more than replacing the donor FP in an existing FRET-based biosensor design with a luciferase. For example, Gulyás and coworkers constructed a BRET-based Ca2+ sensor by incorporating an enhanced version of RLuc669 in place of CFP in the D3 variant of YC349. Over the years, this strategy has similarly yielded BRET-based sensors for monitoring cAMP accumulation286,296, ERK activity442, ATP concentration271, membrane voltage237, and ubiquitination604 (Table 1). BRET-based biosensor assays are also widely used to study the dynamics of GCPR signaling670. Galés et al., for instance, monitored intramolecular BRET between RLuc-tagged GPCRs and GFP-tagged G proteins to probe GPCR activation538, as well as the conformational dynamics of preassembled receptor-G protein complexes549, and Thomsen et al. recently used a similar approach to probe the assembly of GPCR signaling components on the endosomal surface (also discussed in Section 4.1)548. Meanwhile, Dacres and colleagues used intramolecular BRET between a C-terminal RLuc and GFP inserted within the 3rd intracellular loop to directly monitor GPCR activation552, similar to a previous FRET-based design551. Notably, the authors argued that BRET offered a more sensitive readout of GCPR activation compared with FRET552.
Nevertheless, luciferase- and BRET-based sensors have historically underperformed compared with fluorescent biosensors in terms of overall intensity276, despite the fact that BRET can significantly enhance the light output of luciferase (e.g., up to 6-fold higher quantum yield in vitro for RLuc paired with GPF vs. RLuc alone671,672). Poor energy transfer efficiency in most BRET-based probes is one likely explanation for this deficiency. Thus, Saito and colleagues set out to address this problem by developing the novel luminescent protein Nano-lantern276. Nano-lantern comprises an optimized RLuc fused directly to a bright YFP variant (Venus356) and is designed in such a way as to maximize intramolecular BRET. Indeed, at roughly 10 times the brightness of RLuc alone, Nano-lantern was suitable for both live-cell and in vivo imaging. Furthermore, Nano-lantern facilitated the construction of novel luminescence-based biosensors in which a molecular switch is used to modulate Nano-lantern intensity through RLuc fragment complementation, as opposed to direct modulation of BRET efficiency, yielding sensors for Ca2+, cAMP, and ATP (Figure 5B)276. In addition, while some luciferase color variants have been generated through protein engineering673–676, the color spectrum of Nano-lantern can be readily altered by incorporating different FPs as the BRET acceptor377,378. Notably, Suzuki et al. recently developed 5 color variants of a brighter, enhanced Nano-lantern377 that also incorporates the highly optimized NanoLuc variant of Oplophorus gracilirostris luciferase677. Although they have yet to be widely adopted in biosensor design, these and other bright luminescent proteins, such as the recently reported Antares678 and Akaluc679, may be poised to fulfill the promise of bioluminescence-based in vivo imaging of dynamic cellular processes.
2.3.4. Coupled reporter systems
Although genetically encoded biosensors are primarily designed to monitor the dynamics of discrete biochemical events, designs that couple the actions of related biochemical processes can provide spatiotemporal information on complex cellular phenomena, thus greatly expanding the utility of these molecular tools. For example, the reciprocal oscillations of the APCCdh1 and SCFSkp2 E3 ubiquitin ligase complexes mark different cell cycle phases680, and Sakaue-Sawano et al. previously applied this knowledge to generate a “fluorescent ubiquitin-based cell cycle indicator” (Fucci) based on the APCCdh1 and SCFSkp2 substrates Geminin and Cdt1, which accumulate in G1 and S/G2/M, respectively215. Thus, fusing Cdt1 or Geminin fragments to monomeric Kusabira Orange 2 (mKO2) or monomeric Azami Green (mAG) converted the reciprocal nuclear accumulation of orange and green fluorescence into a high-contrast indicator of cell cycle dynamics powerful enough to enable the in vivo monitoring of cell cycle progression213,215,681 (Figure 6A). Fucci has also enabled detailed examinations of the relationships between the cell cycle and processes such as transcriptional activity682 and mitochondrial dynamics683. Meanwhile, several modifications and improvements to Fucci have also been reported to further extend its utility. For instance, Sakaue-Sawano et al. improved the color contrast of Fucci by replacing mKO2 and mAG with mCherry and mVenus, yielding Fucci2212, and more recently developed an alternative indicator system, Fucci(CA), that utilizes changes in the activities of the CUL4Ddp1 and APCCdh1 E3 ligases to indicate cell cycle transitions during interphase (i.e., G1, S, and G2)207. Notably, Bajar and colleagues also recently reported the construction of Fucci4, which utilizes the periodic degradation of 4 spectrally distinct FP-tagged proteins to monitor each individual phase of the cell cycle208 (Figure 6B).
Figure 6:
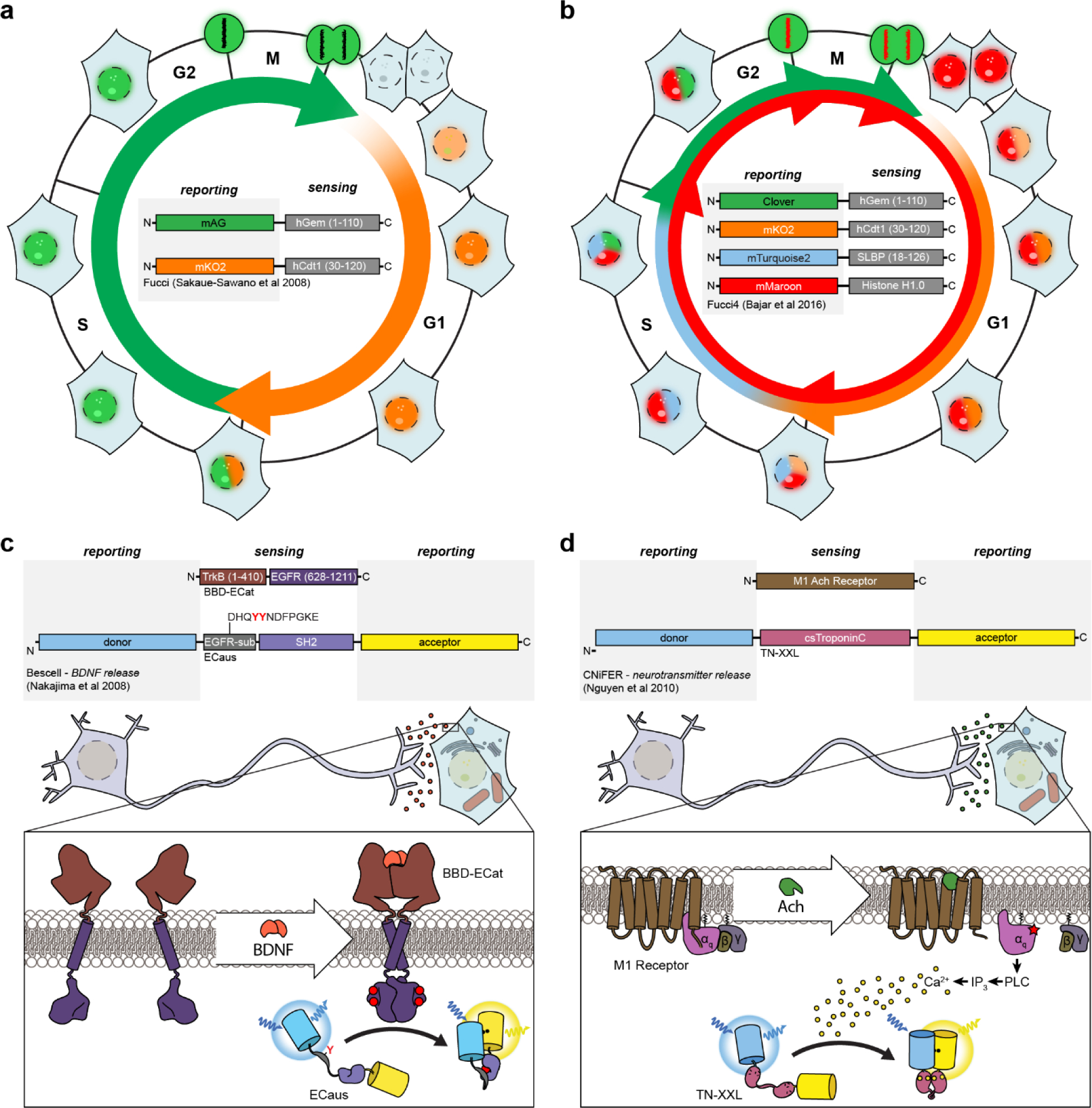
Coupled reporter systems.
(A and B) Biosensors reporting the cell cycle phase of cells utilize fluorescent proteins fused to protein fragments that are selectively degraded during specific phases of the cell cycle208,215. In the Fucci system, Gem is degraded during the G1 and late M phases and, conversely, Cdt1is degraded in S and G2 phases215. The improved Fucci4 adds the condensation of chromatin around Histone H1 to report the M phase, as well as SLBP, which is degraded after S phase, in addition to labeled Cdt1 and Gem from Fucci208. C) The chimeric receptor BBD-ECat, which consists of an extracellular TrkB domain, which dimerizes upon binding BDNF, and an intracellular EGFR domain, is coupled with an EGFR activity reporter ECaus to act as a reporter for BDNF release by neurons281. D) Similarly, the expression of the M1 receptor and the Ca2+ biosensor TN-XXL384 in a co-cultured reporter cell, uses the endogenous coupling of the GPCR M1R to Gαq which, upon receptor stimulation, leads to an increase in intracellular Ca2+ to report the presense of a neurotransmitter, acetylcholine (Ach)498.
Several cell-based indicator systems have also been developed that couple the detection of signaling molecules secreted by one cell to a FRET-based biosensor response in a second cell, thereby providing spatiotemporal information on the dynamics of intercellular rather than intracellular signaling, particularly among neurons. For example, Sato et al.565 developed a reporter system for detecting secreted nitric oxide (NO) based on PK15 cells that stably express the cGMP sensor CGY312. Importantly, PK15 cells endogenously express soluble guanylate cyclase (sGC), which selectively generates cGMP in response to NO binding684. Thus, sGC couples NO detection to the CGY FRET response, with the resulting Piccel (“PK15 indicator cell”) system able to detect picomolar NO release by co-cultured hippocampal neurons565. Using a similar principle, Nakajima and coworkers also developed a “BDNF sensor cell” (Bescell) indicator system that couples BDNF binding by a chimeric receptor tyrosine kinase (BDNFbinding domain fused to EGFR kinase domain) with a FRET-based EGFR activity reporter281 (Figure 6C). Likewise, taking advantage of GPCR coupling to Gαq-containing G proteins, which activate PLC to trigger IP3 production and ER Ca2+ release, Nguyen and colleagues developed a similar approach to visualize endogenous neurotransmitter release using “cell-based neurotransmitter fluorescent engineered reporters” (CNiFERs)498. This system, which utilizes HEK293 cells stably expressing a given metabotropic neurotransmitter receptor along with the high-performance GECI TN-XXL384, has been used to image the in vivo dynamics of cholinergic, dopaminergic, and noradrenergic signaling in mouse brains498,499 (Figure 6D). Thus, the modularity of these designs holds the potential to illuminate the spatiotemporal dynamics of broad range of intercellular signals.
2.4. New Biosensor Classes
Years of tireless innovation have spawned a verdant landscape of genetically encoded fluorescent biosensors capable of visualizing the spatiotemporal dynamics of a diverse and expansive, though by no means exhaustive, array of biochemical, biophysical, and cellular phenomena. Meanwhile, new biosensor designs continue to emerge, often based on novel reporting strategies or biosensor configurations, that not only fill gaps in the existing toolkit but also open up entirely new avenues for exploring previously unanswerable biological questions, thus promising to redefine the reach of these powerful molecular tools.
2.4.1. Sensors based on infrared fluorescent proteins
Decades of engineering have yielded GFP-like FPs in nearly every color of the rainbow7. Notably absent, however, have been variants that excite and emit between 650 nm and 900 nm (i.e., the so-called near-infrared [NIR] window685), wavelengths that are preferable for both cellular and whole-body in vivo fluorescence imaging for the reasons enumerated above (e.g., autofluorescence, phototoxicity, absorbance, and scattering). Yet multiple light-absorbing proteins are present in nature that derive their chromophores from the covalent incorporation of endogenous cellular cofactors686, and several bacterial phytochromes (BphPs), which utilize the heme catabolic intermediate biliverdin (BV) as their chromophore, have been demonstrated to emit photons in the 600 to 700 nm range687–689. BphPs are multidomain photoreceptors that transduce light into signaling activity 690, and Shu and colleagues were able to successfully engineer the first infrared fluorescent protein (IFP) based on a pared-down version of the Deinococcus radiodurans BphP containing just the chromophore-binding GAF and PAS domains691.
Intriguingly, the unique topology and chromophore-binding interactions of IFP are enabling the development of novel biosensors strategies that are not feasible using GFP-like FPs. For example, although numerous genetically encoded fluorescent protease sensors have been developed over the years (see Sections 2.3.3.2.2.2.1 and 2.3.3.2.4), no approach has succeeded in producing a high-contrast, fluorogenic biosensor, wherein the cleaved sensor gains fluorescence. Thus, To and colleagues set out to use IFP as an alternative scaffold for constructing such a reporter531. In BphP, and thus IFP, chromophore formation occurs through the insertion of BV into a binding pocket within the GAF domain, followed by the autocatalytic formation of a thioether bond with a conserved cysteine residue690,692. Noting the proximity of this residue to the BV binding pocket in IFP, To et al. devised a way to control IFP chromophore formation by modulating the distance between the binding pocket and catalytic cysteine within a circularly permuted IFP (cpIFP)531. The original N- and C-termini of IFP were truncated and tethered using a short linker containing a protease cleavage site to physically constrain the catalytic cysteine such that chromophore formation only occurs following cleavage of the linker (Figure 7). Meanwhile, to ensure that cpIFP remained intact following cleavage, splitGFP was added to the new termini introduced between the PAS and GAF domains. This approach yielded a fluorogenic infrared fluorescent caspase reporter (iCasper) for monitoring caspase-3 activity, which allowed the authors to highlight apoptotic cells during morphogenesis and tumor development in Drosophila531.
Figure 7:
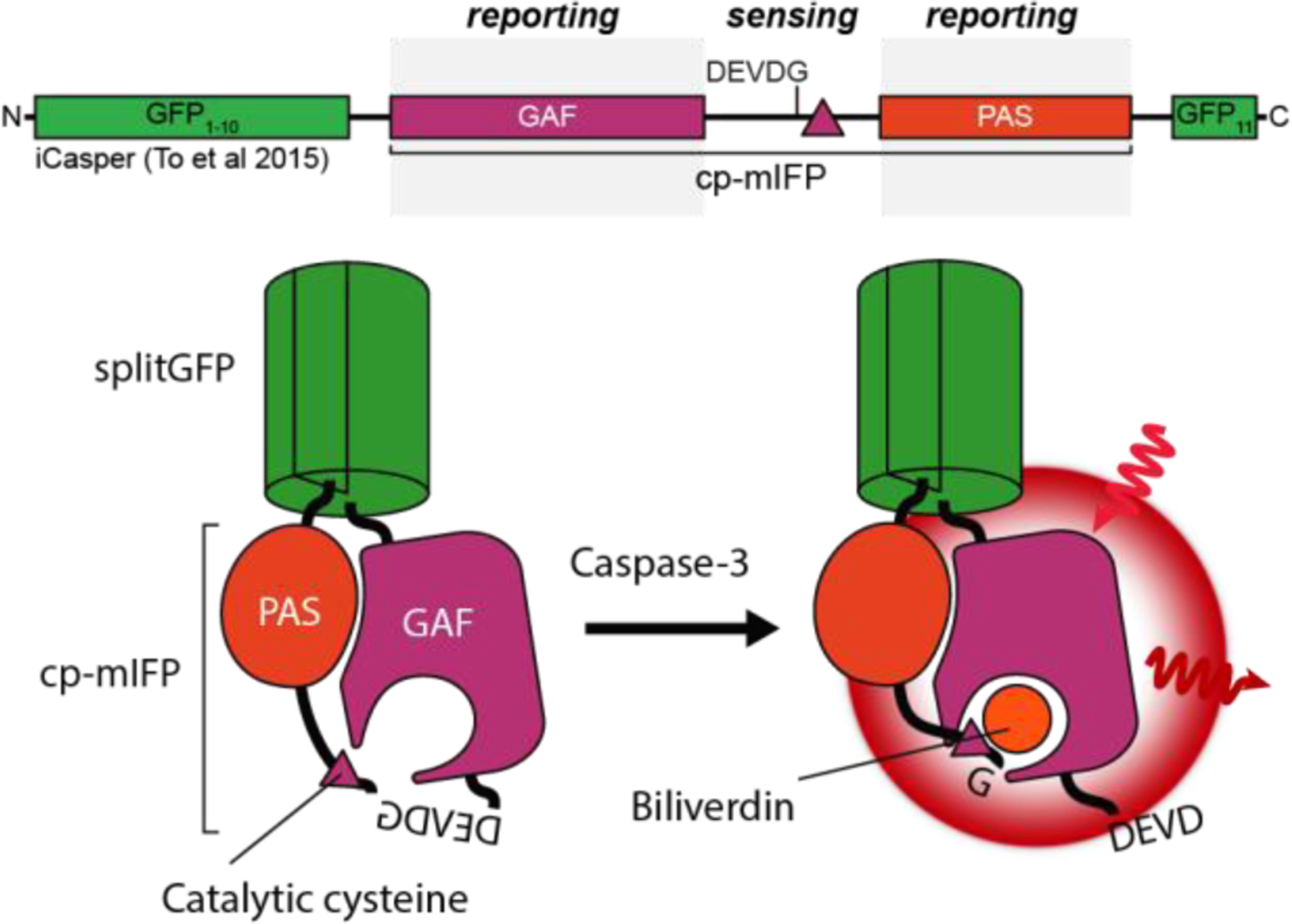
Infrared FP-based Caspase-3 reporter, iCasper.
The introduction of the Caspase-3 cleavage sequence into the circularly permuted mIFP prevents the incorporation of BV into the GAF domain, but cleavage by Caspase-3 liberates the catalytic cysteine to promote the incorporation of BV and the formation of the chromophore531.
One potential concern regarding the wider use of BphP-derived IFPs is their somewhat poor quantum yield and low brightness, which may be further exacerbated by low BV concentrations in certain organisms and cell types693,694. Nevertheless, the development of IFP has ignited a flurry of efforts to develop other alternative FPs, some of which display comparable brightness to that of EGFP209. These advances promise to yield additional novel biosensor designs beyond those currently possible.
2.4.2. Biochemical activity integrators as in vivo snapshot reporters
The idea of a “snapshot” reporter was originally conceived as a way to get around the daunting challenge of performing three-dimensional whole-brain imaging of neuronal activity in order to delineate causal relationships between the firing of specific neuronal subsets and specific behaviors or stimuli (i.e., brain activity mapping)695. Thus, instead of continually reporting the dynamics of neuronal activity (e.g., Ca2+ elevations), the snapshot reporter integrates, or “memorizes”, neuronal activity over an arbitrarily defined period of time by generating a stable signal only in the presence of both activity (e.g., Ca2+) and an external trigger such as light, thereby enabling detailed, retrospective analyses of neuronal firing patterns.
Recently, Fosque and colleagues were able to develop one such snapshot reporter using the single-FP Ca2+ indicator GCaMP as a template361. Specifically, based on a previous report detailing the construction of GR-GECO, a single-FP GECI whose fluorescence emission switches color from green to red upon illumination with 400 nm light376, they set out to similarly engineer a photoconvertible GECI, albeit one whose photoconversion efficiency was modulated by the Ca2+-dependent molecular switch, such that the change in emission wavelength occurs only in response to both Ca2+ and 400 nm light. This was achieved by incorporating a circularly permuted variant of the green-to-red photoconvertible FP mEos2696 into the GCaMP backbone in place of cpEGFP, after which molecular evolution was performed to generate CaMPARI, a calcium-modulated photoactivatable ratiometric integrator361 (Figure 8). During 400 nm illumination, the green fluorescent signal from CaMPARI rapidly changes color to red in proportion to the concentration of Ca2+; thus, the ratio of red-to-green fluorescence intensity provides a permanent snapshot of neuronal activity during the recording period established by the length of illumination361. Zolnik and coworkers also recently demonstrated that the photoconversion rate of CaMPARI can be adjusted by modulating the intensity of 400 nm illumination, thereby tuning the sensitivity of CaMPARI to different thresholds of neuronal activity697. Importantly, CaMPARI also behaves as a traditional single-FP GECI, with the intensities of both the green- and red-fluorescent states decreasing strongly in response to Ca2+361, thereby providing a crucial internal control for any possible effect that the photoconversion light itself may have on the system under investigation.
Figure 8:
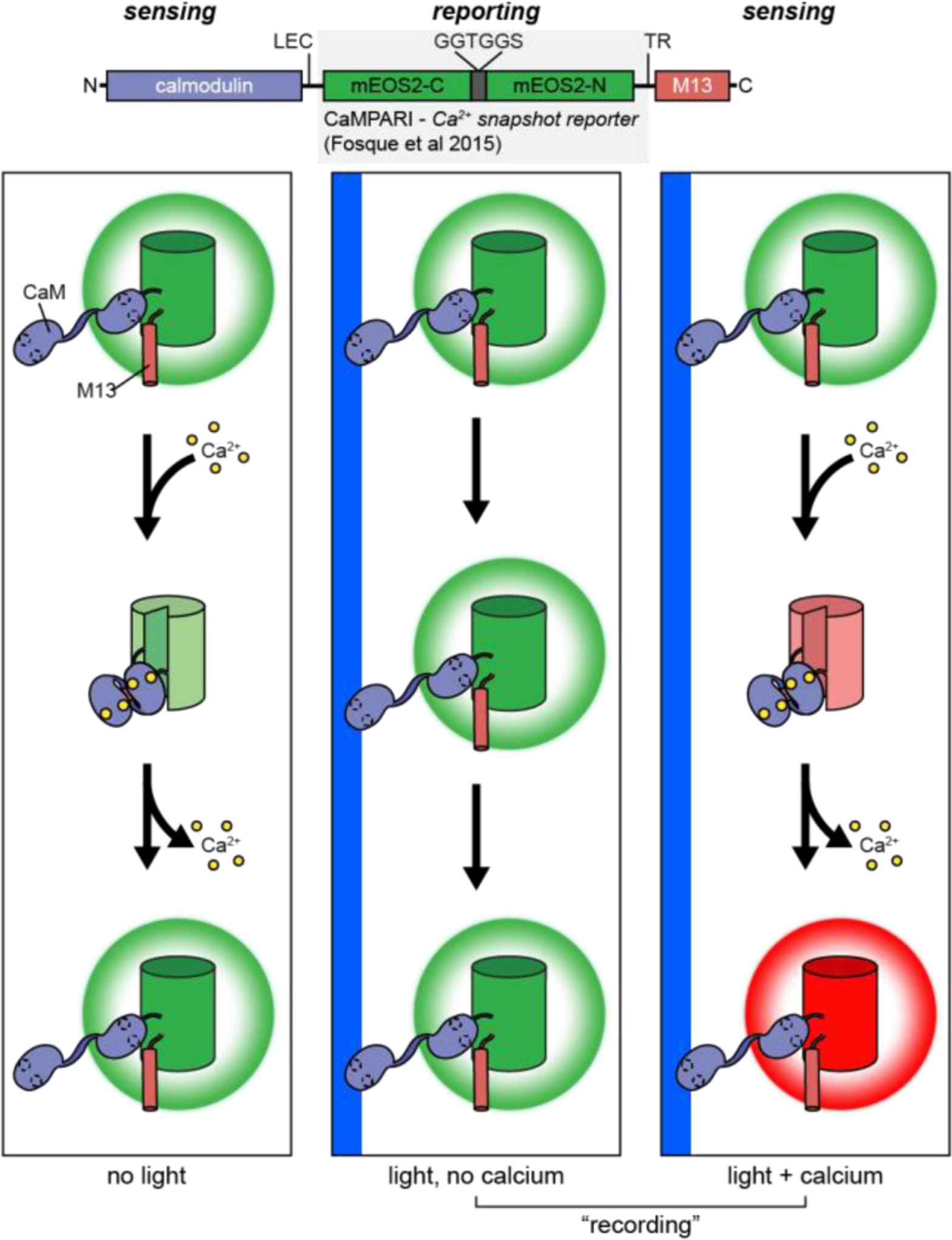
Photoconversion-based snapshot recorder of Ca2+.
CaMPARi acts similarly to other green single color Ca2+ biosensors where the green fluorescence intensity is dependent on the Ca2+ concentration (left), except it can also “record” the presence of high Ca2+ during a snapshot in time. When the CaMPARi biosensor is illuminated with blue/violet light and the intracellular Ca2+ is high, the biosensor will irreversibly convert to a red (right) Ca2+ biosensor361.
Alternatively, a pair of studies347,348 published within the past year have reported the development of snapshot reporters that hew more closely to the original design first articulated by Roger Tsien695. In this scheme, the extrinsic (e.g., light) and intrinsic (e.g., Ca2+) signals are integrated to drive a transcriptional reporter that permanently labels active neurons with high spatiotemporal resolution for post hoc analysis. Thus, although differing somewhat in their particulars, both the Cal-Light system designed by Lee and colleagues348 and the “fast light- and activity-regulated expression” (FLARE) system devised by Wang and coworkers347 feature a three-component, dual-molecular-switch design that functions as an “AND gate” to control the light- and Ca2+-dependent release of a membrane-tethered transcription factor, namely, the tetracycline-inducible transcriptional activator (tTA) (Figure 9). First, these designs utilize a light-dependent molecular switch composed of the light-oxygen-voltage (LOV) domain and C-terminal Jα-helical extension (Jα) from Avena sativa phototropin, wherein blue-light absorption by the flavin-containing LOV domain causes the C-terminal Jα helix to unwind698, which can be used to unmask a specific peptide sequence in response illumination699, in this case a cleavage site for the tobacco etch virus protease (TEVp). Meanwhile, the second, Ca2+-dependent molecular switch utilizes calmodulin binding to the M13 peptide to modulate cleavage of the TEVp substrate, either by controlling the fragment complementation of N- and C-terminal portions of TEVp348 or by simply recruiting TEVp to the cleavage site347. Hence, only the combined presence of both light (to define the recording window) and Ca2+ (in the form of neuronal activity) will permit cleavage of the TEVp substrate and release of the tethered tTA domain to drive the expression of a reporter construct in the nucleus (Figure 9).
Figure 9:
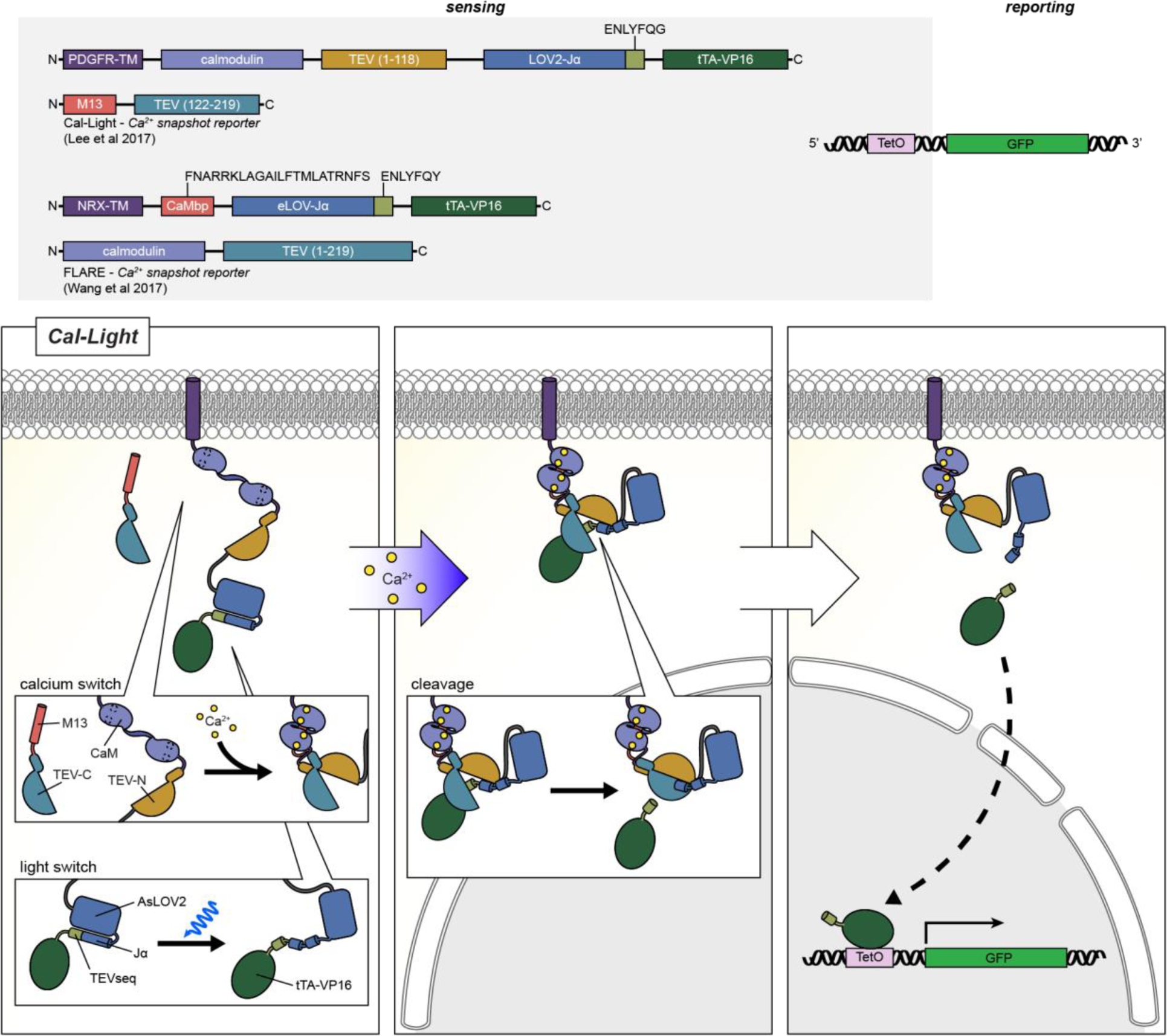
Transcription-based snapshot reporters.
The Cal-Light and FLARE Ca2+ biosensors create an “AND gate” design by combining the optogenetically controlled AsLOV2 domain with a Ca2+ switch controlled split-TEV protease, which leads to the cleavage of the tTa-VP16 transcriptional activator only in the presence of high Ca2+ and blue light347,348. The cleavage of the transcriptional activator will then turn on the expression of a reporter gene such as GFP expression.
Much like CaMPARI, both of these methods enabled active neurons to be specifically and selectively labeled with a fluorescent signal (i.e., FP expression) within the span of the illumination window347,348. Importantly, however, a key advantage of these transcription-based readouts is that any transgene can be placed under the control of the snapshot reporter, thereby facilitating precise functional manipulation of labeled neurons. For instance, the co-expression of a channelrhodopsin was used to permit the targeted inhibition348 or re-activation347 of neurons that had been fluorescently labeled by the snapshot reporter, an ability that is crucial for identifying those neurons whose firing bears a true causal link with a given behavioral response695. Furthermore, Lee and colleagues also developed a similar system for identifying and manipulating neurons that respond to specific neuromodulators by replacing the calmodulin-M13 switch with one based on GPCR-βarr binding541, highlighting the ability of these modular designs to be reconfigured as snapshot reporters for other biochemical events, with the potential for numerous applications beyond neurobiology.
2.4.3. Fluctuation-based biosensors
The biochemical pathways that mediate intracellular signaling are increasingly understood to operate within an exquisitely controlled spatial framework, or activity architecture, that is established through various modes of compartmentalization (discussed in Section 4). Because much of this compartmentalization is ultimately governed on a molecular scale, such as through the assembly of multiprotein signalosomes, genetically encoded fluorescent biosensors capable of visualizing and localizing biochemical activities on a comparable spatial scale are extremely desirable. That being said, a number of super-resolution imaging modalities have been developed that rely on FP photochromism, or the ability of certain FPs to stochastically and reversibly switch between a bright, fluorescent state and a dark, non-fluorescent state under specific illumination conditions (reviewed in ref. 21). FP-based biosensors engineered to modulate this photochromic behavior in response to a biochemical input thus represent one potential strategy for imaging the precise locations of biochemical activities.
Photochromism is thought to involve the disruption of molecular contacts between the FP chromophore and the surrounding β-can, leading to increased conformational flexibility and decreased fluorescence emission21. In addition to exhibiting controlled on/off switching, photochromic FPs also display stochastic intensity fluctuations (i.e., blinking), and Mo et al. recently found that close proximity between the photochromic green FP Dronpa700 and the red FP TagRFP-T157 induced significant fluctuations in TagRFP-T fluorescence intensity447. These intensity fluctuations, which were specifically induced by Dronpa proximity, did not require the Dronpa chromophore but were dependent on surface residues in the Dronpa β-can, suggesting that physical contact with Dronpa deforms the TagRFP-T β-can and directly increases its intrinsic195 blinking behavior. Further analysis revealed that this phenomenon, termed “fluorescence fluctuation increase by contact” (FLINC), was sensitive to the intermolecular distance between Dronpa and TagRFP-T within the range of 5–6 nm, thus lending itself to the construction of a novel class of proximity-based biosensors compatible with super-resolution imaging447. Using FRET-based biosensors as a template, Mo and colleagues substituted the FRET FP pair in AKAR3EV414 with Dronpa and TagRFP-T (Figure 10). When combined with the fluctuation-based super-resolution imaging approach pcSOFI23, the resulting FLINC-AKAR1 sensor was able to map dynamic changes in PKA activity with nanometer precision, facilitating the detailed dissection of PKA compartmentalization447 (see additional discussion in Section 4.3.1). These initial studies hint at the potentially transformative impact of this nascent biosensing strategy.
Figure 10:
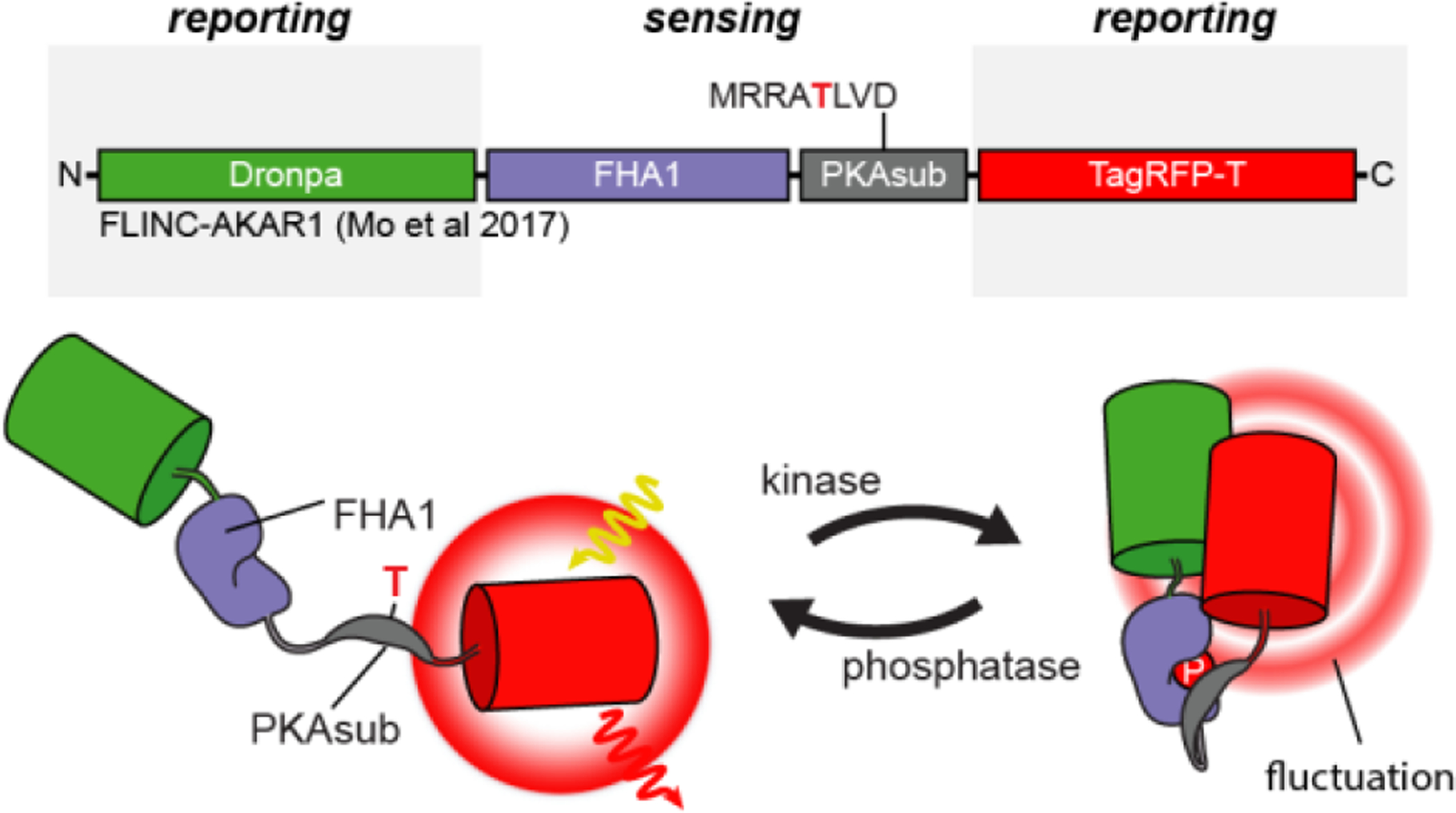
Fluctuation-based PKA biosensor FLINC-AKAR1.
Fluorescence of TagRFP-T is modulated by its interaction with Dronpa through a process termed FLINC, such that when in close proximity to Dronpa, TagRFP-T exhibits increased fluorescence fluctuation. This enables this biosensor to report PKA activity at a sub-diffraction spatial resolution447 through the use of the super-resolution technique pcSOFI23.
Genetically encoded fluorescent biosensors have evolved into a remarkably diverse set of molecular tools capable of probing a broad range of cellular states and biochemical processes. In the following sections, we delve further into the application of genetically encoded fluorescent biosensors by focusing our attention on how this technology has been leveraged to extract detailed information on the temporal and spatial regulation of the intracellular biochemical machinery and thereby elucidate the molecular logic underlying signal transduction networks.
3. Obtaining Temporal Information from Biosensors
The ability of cells to dynamically respond to a vast array of environmental changes and modes of cellular communication has arisen from the evolution of a highly connected network of fairly simple chemical reactions701. Characterizing the dynamics of these biochemical pathways is therefore essential for determining how signaling networks are organized and understanding how cells process external stimuli and make decisions about their fate. Traditionally, quantifying the kinetics of signaling reactions has relied on the use of various in vitro methods such as western blotting or enzymatic assays. Although these methods remain important tools for studying intracellular signaling, genetically encoded fluorescent biosensors offer a number of advantages that make them instrumental for elucidating the temporal dynamics of signaling networks. In particular, because they are genetically encoded, these biosensors can be used to quantitatively track the kinetics of signaling molecules within the native cellular environment. Furthermore, thanks to the rapid, nanosecond timescale of fluorescence, the relatively high brightness of FPs, and the sensitivity of modern digital imaging equipment, the signals from fluorescent biosensors can be monitored with a much a higher temporal resolution than is generally achievable through other in vitro methods. As such, fluorescent biosensors are powerful tools for studying the dynamics of signaling networks across a wide range of timescales. Below, we discuss the types of temporal dynamics that have been observed in cellular signaling and how biosensors have been instrumental in studying these dynamics.
3.1. Kinetics of Individual Reactions
Signaling networks are principally driven by changes in the concentrations of signaling molecules and the reaction rates of signaling enzymes. The most foundational understanding of temporal dynamics can be achieved by identifying the activation and catalysis rates for signal transduction. Historically, these have been studied in purified systems in an in vitro setting. While these assays are able to provide detailed and accurate measurements of the reaction rates of a specific signaling enzyme, they lack the ability to evaluate the dynamics and kinetics of all of the preceding steps that lead to the activation of that enzyme, and any other regulatory mechanisms occurring within the cell. Genetically encoded fluorescent biosensors are able to quantify the kinetics of signal transduction in real time in a living cell. Quantifying these kinetics is the first step to understanding how the connected enzymatic reactions within a network can create the highly dynamic and complex signaling responses seen in nature.
One example of fluorescent biosensors being used to study the activation of signaling proteins has been in the study of GPCR activation kinetics. GPCRs are one of the most drugtargeted protein families702, therefore quantifying the kinetics of their activation is of interest for both understanding their physiological function and evaluating the effects of different drugs. GPCRs consist of a seven-transmembrane receptor that couples to a heterotrimeric G protein comprising α, β and γ subunits703. Ligand binding by the receptor causes a conformational change that promotes the activation of the Gα subunit, which is primarily responsible for downstream signaling, and is canonically considered to dissociate from the Gβγ subunits703. There have been several fluorescent probes developed to quantify the kinetics of different aspects of GPCR activation (Figure 11A). First, the activation of the receptor was quantified by inserting CFP and YFP into the third intracellular loop and intracellular tail, respectively, of the PTHR and α2AAR551. These biosensors showed that the kinetics of the conformational change within the receptor could vary significantly, where PTHR is activated much more slowly than α2AAR, with time constants of 1 second and less than 40 milliseconds, respectively551. After this conformational change occurs, the receptor can then couple with its cognate heterotrimeric G proteins. Quantification of the kinetics up through this step was done by labeling Gγ with CFP and inserting YFP into the intracellular tail of the adenosine receptor A2A (A2AR) and β1adrenergic receptor (β1-AR)547. This association between the receptor and G proteins occurred rapidly where these biosensors both exhibited a time constant near 50 milliseconds547. Finally, this association of the heterotrimeric G protein with the receptor stimulates the exchange of GDP for GTP on the Gα subunit and dissociation of Gα from the Gβγ subunits. Biosensors to study this final step were developed by fusing a Gα subunit and a Gβ or γ subunit with YFP and CFP, respectively544,547. These studies showed that the activation of the Gα subunit was most likely to be rate limiting, where Gαi1 showed a t1/2 of approximately 700 milliseconds in response to an α2A adrenergic agonist544 and Gαs exhibited a time constant near 500 milliseconds in response to A2AR and β1-AR agonists547. The responses from these biosensors were measured at frequencies on the order of 20–75 Hz, which was essential to be able to quantify the kinetics of these rapid inter- and intramolecular changes. Given that Gα, Gβ, and Gγ comprise 23, 5, and 12 different isoforms, respectively, exhaustively testing all of these combinations may be infeasible, but genetically encoded fluorescent biosensors make it possible to test specific combinations543. For example, Gibson and Gilman examined the differences between Gαi isoforms 1, 2 and 3 and Gβ isoforms 1, 2, and 4, showing that Gαi1 exhibited the greatest FRET response to α2-adrenergic receptor stimulation when coupled with Gβ1 or Gβ4 subunits but not Gβ2543. This type of work can provide evidence of subunit isoform preference or specificity and help understand how celltype-specific isoform expression can affect the response of GPCRs704. Similarly, the use of receptor-specific agonists/antagonists with these biosensors can reveal differences in the strength and rate of activation of different G protein isoforms for each receptor542,545,547. Since GPCRs are the first step in many signaling pathways, understanding the kinetics of their activation can help identify rate-limiting steps in a signaling pathway and evaluate potential mechanisms of pharmacological perturbation. These biosensors exemplify the benefits of using genetically encoded fluorescent proteins to study the kinetics of activation because they are able to examine the dynamics of specific proteins within the cellular context at a high temporal resolution.
Figure 11:
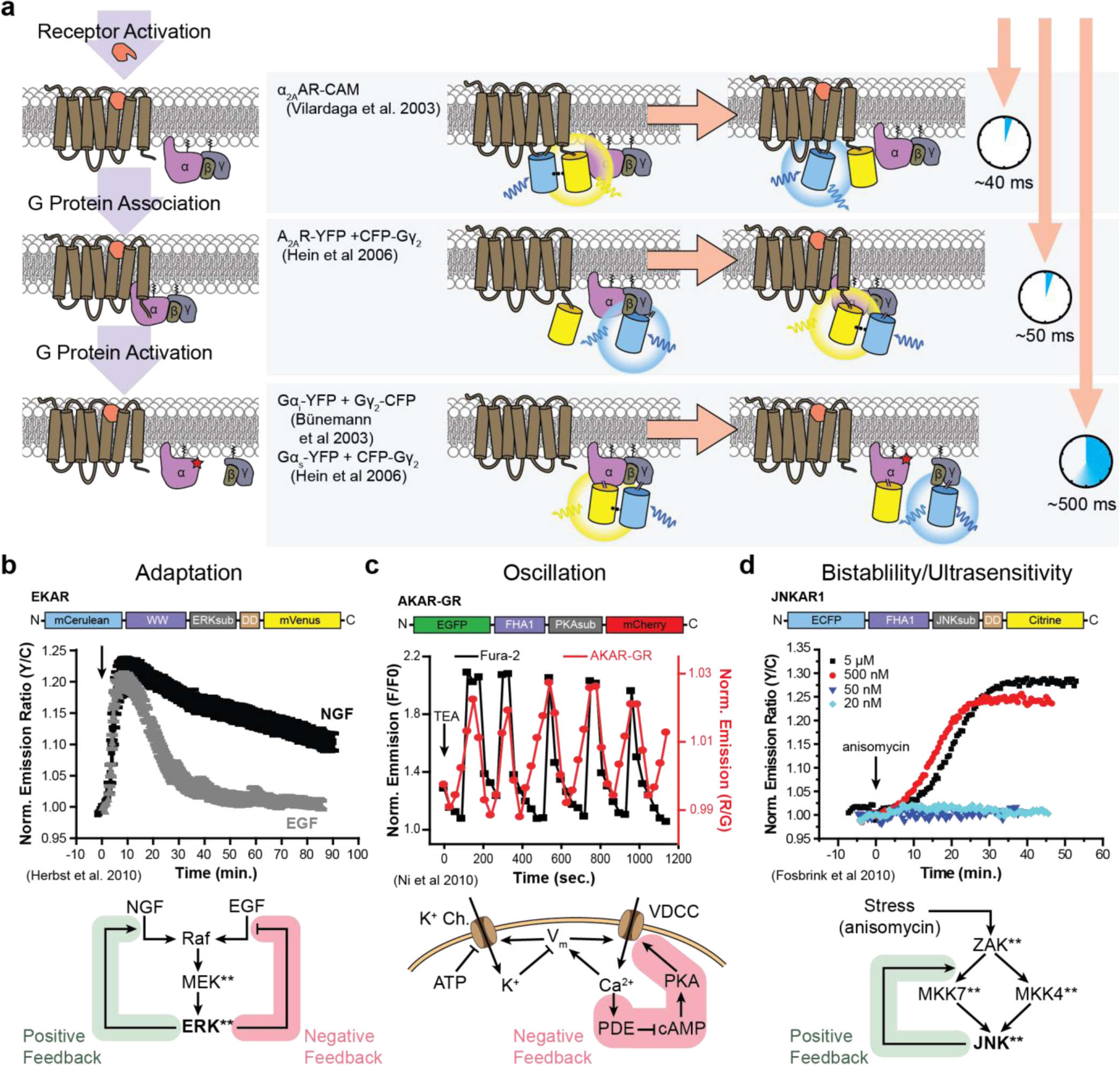
Temporal dynamics quantified by fluorescent biosensors.
The kinetics of GPCR signaling. First, the receptor undergoes a conformational change in response to ligand binding, which was observed to occur with a time constant on the order of 40 ms in the α2-Adrenergic Receptor using the biosensor α2-AR-CAM551. Next, the heterotrimeric G protein associates with the activated receptor. A2A Adenosine Receptor, A2AR, fused with YFP at the C-terminal tail and CFP-labeled Gγ2 exhibited an increase in FRET upon stimulation with adenosine with a time constant of approximately 50 ms547. Finally, the receptor stimulates the exhange of GTP for GDP on the Gα subunit, thus activating Gα and promoting the dissociation of Gα from Gβγ. Biosensors consisting of a CFP-labeled Gγ subunit and either Gαi544 or Gαs547 fused to YFP showed activation time constants on the order of 500 ms. B) Adaptation is the return of a signaling pathway towards its previous state while under continued stimulation. PC12 cells expressing the ERK biosensor EKAR show an adaptive response to EGF stimulation (gray lines) but a much more sustained response to NGF stimulation (black lines), reproduced with permission from Herbst et al. Mol and Cell Bio 2011708. Negative feedback by ERK onto Raf activation is hypothesized to lead to a transient response, whereas positive feedback in the NGF signaling pathway is hypothesized to lead to bistability709. C) Oscillations are the regular or semi-regular cycling between activity/concentration states, as can be seen in the TEA-induced Ca2+ and PKA activity oscillations in MIN6 cells. These oscillations were observed through the simultaneous measurement of Ca2+ concentration with the Ca2+ dye Fura-2 (black line) and a Green/Red variant of AKAR, GR-AKAR, (red line) at the single-cell level in MIN6 cells294. While the inhibition of K+ channels by ATP and the interplay between voltage and Ca2+ are the primary driver of the Ca2+ oscillations, the negative feedback of Ca2+ through cAMP and PKA is hypothesized to strengthen these oscillations and tune their frequencies. Reproduced with permission from Ni et al. Nat Chem Bio 2011294. D) Bistability and ultrasensitivity describe phenomena wherein a signaling pathway is insensitive to stimulation below a certain threshold dose but responds in a switch-like fashion to super-threshold stimuli. HeLa cells expressing the JNK biosensor JNKAR1 did not respond to anisomycin at concentrations of 20 (cyan diamonds), or 50 (blue triangles) nM, but exhibited a strong response to 500 nM (red circle) and 5 μM (black squares), reproduced with permission from Fosbrink et al. PNAS 2010459. In addition to the multistep activation along the JNK signaling pathway, positive feedback by JNK onto the activation of upstream regulator MKK7 is hypothesized to contribute to the ultrasensitive nature of activation by anisomycin710.
In addition to being able to examine the activation of proteins and protein complexes, many biosensors have been developed to evaluate the kinetics of signaling enzyme catalysis. Downstream of the Gαs- and Gαi-mediated regulation of cAMP production is the prototypical kinase PKA. PKA is a tetramer consisting of two regulatory subunits and two catalytic subunits, where binding of cAMP to the regulatory subunits unleashes the catalytic subunit from inhibition by the regulatory subunit. The temporal dynamics of PKA have been the focus of many studies due to its importance in several physiological processes and its utilization of scaffold proteins to coordinate spatiotemporal dynamics705,706. The development of AKARs (section 2.3.3.2.2.2.2) enabled more detailed and specific studies of these signaling dynamics477. First, AKAR1 was able to show the different temporal kinetics of stimulating PKA activation either directly through the activation of ACs by forskolin or indirectly through the activation of the Gαs linked β2adrenergic receptor agonist isoproterenol. The temporal resolution of this early biosensor was a great improvement over what was possible through previous in vitro kinetic assays. Furthermore, biosensors have played an important role in evaluating the effects of signalosomes, coordinated complexes of interconnected signaling proteins, on enzyme kinetics. For example, A-Kinase Anchoring Proteins (AKAPs), a family of scaffold proteins that bind PKA, coordinate the interaction of PKA with downstream substrates and upstream regulators of PKA activity, thus affecting both PKA activity and the kinetics of phosphorylation by PKA707. This effect was directly observed by genetically fusing a PKA regulatory subunit directly to AKAR1, thus mimicking a scaffolded enzyme-substrate complex, which resulted in a nearly 2-fold faster response477. In addition to modulating the temporal dynamics of signal transduction, scaffold proteins control the spatial architecture of signal transduction by anchoring signaling proteins to specific subcellular microdomains. The effects of scaffold proteins on both spatial and temporal dynamics, and the role of biosensors in studying them is discussed further in Section 4.3. The capability of fluorescent biosensors to measure specific enzymatic activities within the cellular environment at a high temporal resolution has made them powerful tools for quantifying the kinetics of signaling protein activation and enzyme catalysis.
3.2. Higher-order Signaling Dynamics
The balance between second messenger production and degradation and signaling enzyme activation and inactivation within signaling networks enable and define the complex dynamics observed within cells. The highly connected nature of signaling networks means that a very diverse array of dynamics can be observed, but these dynamics can be understood in terms of a small set of basic building blocks of signaling dynamics that are defined by how the network is connected711. Furthermore, the organization of the network connections can be grouped into similar topologies, which are called signaling motifs712. Here, we will briefly introduce some of the more common signaling dynamics observed and some network motifs that lead to them, but there are several detailed reviews discussing the details of signaling network topology and their effects on signaling dynamics711–713.
3.2.1. Adaptive responses
One common signaling dynamic observed is what is referred to as adaptation or a transient response. Adaptation can be defined as the resetting of a cellular signaling level to its previous state after stimulation714 (Figure 11B). This type of signaling dynamic can be important for cells to be able to respond to changes in stimulation levels instead of the absolute level of the signal. Previous work has identified that negative feedback loops, where the activation of a downstream signaling enzyme will lead to the inhibition of one or more of its upstream regulators, and incoherent feed forward loops, where an upstream signaling enzyme activates two different pathways that converge on a downstream effector but have opposing effects714, are key motifs involved in signaling adaptation. While these network motifs are needed for a transient signaling dynamics, their existence alone does not guarantee that adaptive signaling will occur; in fact, negative feedback can lead to oscillatory dynamics instead.
Adaptation is by definition a short-lived event which has been found to be susceptible to cell-to-cell variability; therefore, fluorescent biosensors have been critical to observing and understanding these transient dynamics. While these biosensors measure the kinetics of individual reactions or second messenger concentrations, the ability to observe how a given signaling network regulates signaling dynamics over time can provide insights into how the networks architecture can shape the observed response. For example, genetically encoded fluorescent biosensors have been instrumental in identifying how different stimuli can lead to either sustained or adaptive dynamics of extracellular-signal regulated kinase (ERK) signaling. These temporal dynamics are functionally important because the duration of ERK activity has been shown to regulate different cell-fate outcomes in the neuroendocrine PC12 cell line465,709. Earlier in vitro methods measuring ERK activity in response to EGF or NGF had observed that stimulation with EGF resulted in transient ERK activity, whereas NGF stimulation induced a sustained response715,716. These differences in ERK temporal dynamics also correlated with the fact that EGF promoted proliferation while NGF led to PC12 cell differentiation715,716. The desire to obtain a more detailed temporal understanding motivated the development of fluorescent biosensors of both ERK activation, e.g., Mui2 448, and ERK activity, such as EKAR and subsequent improved variants444 (Table 1). When directly comparing the dynamics of ERK activity in response to EGF and NGF stimulation in PC12 cells, Herbst and colleagues were able to use EKAR to accurately calculate a t1/2 for ERK phosphorylation reversal that not only agreed with previous studies (Figure 11B) but also revealed that PKA activity modulated the adaptive signaling dynamics of ERK472. Later work by Ryu et al., using an improved ERK biosensor, EKAR2G443, examined the cell-to-cell variability of ERK dynamics in response to EGF and NGF709. They observed that while the population averages showed a more transient response to EGF than with NGF, there was significant variability between cells. This variability convolutes what uniquely regulates these two signaling cascades, but they found that using a short pulse of growth factor stimulation, instead of sustained stimulation, yielded more uniform responses for both EGF and NGF. Using this experimental design, they observed that the EKAR2G response to low dose NGF stimulation exhibited similar transient responses as low-dose EGF stimulation. But at high doses of NGF, a subset of cells exhibited a sustained response that was not observed with high dose EGF stimulation, suggesting that the NGF signaling pathway may at exhibit bistability, wherein high doses of NGF lead to a switch-like, instead of adaptive, behavior (bistability discussed more in section 3.2.3). This evaluation of cell-to-cell variability is only possible through the use of fluorescent biosensors because these quantitative measurements of signal transduction can be performed at the single-cell level. Furthermore, this detailed analysis was coupled with a computational model of ERK signaling, which demonstrated that repeated pulses of either EGF or NGF could be used to approximate either a transient or sustained response with much less variability across the population. Experimental evaluation of this predicted effect found that pulse frequencies of growth factor stimulation that create a more “sustained-like” ERK response led to more differentiation of PC12 cells, more so for NGF but also observed with EGF. This agreed with earlier work that found that both continuous and pulsatile activation of ERK, using an optogenetic stimulation independent of growth factor, increased PC12 differentiation717. More work will still be needed to identify how these cells are able to integrate the temporal differences between a transient and sustained response into cell fate and gene expression profiles. Additionally, the complexity of these dynamics and the high temporal resolution of fluorescent biosensors has made ERK signaling a prime candidate for interrogation using computational models, which is discussed in Section 5.2.1.
3.2.2. Oscillations
Oscillatory signaling dynamics can be defined as a regular or semi-regular variation in the signaling response (Figure 11C). Oscillations are physiologically critical for several functions such as heart rhythm, insulin secretion and cell cycle timing718. For oscillations to arise, the signaling network must have both negative feedback and delay in the negative feedback719. One of the most common oscillatory dynamics observed has been in intracellular Ca2+ concentrations. These oscillations are driven by the ion channels that form a complex network of feedback mechanisms that can experience a delay due to their dependence on membrane potential or ion concentration720. While these dynamics generally fall under electrophysiological signaling, enzymes and second messengers can be both required for oscillations and regulate the rate of oscillation294,721. But oscillatory dynamics are not exclusive to Ca2+ signaling. For example, nuclear localization of the transcriptional regulator NF-κB has been shown to oscillate due to negative feedback caused by NF-κB promoting the expression of its inhibitor, IκB722. In this system, the time required for both the nuclear translocation of NF-κB and its induction of IκB expression causes a delay between NF-κB activation and feedback inhibition, leading to the oscillatory behavior. Understanding the different mechanisms that can lead to and modulate oscillatory dynamics has been of intense interest for several fields.
Oscillatory signaling dynamics are complex and may be difficult to observe without the use of fluorescent biosensors due to the temporal and single-cell resolution that may be required. For example, cardiac Ca2+ oscillations that regulate the rhythmic contraction of the heart are moderately fast (1–10 Hz depending on species) and homogenous across a population when under pacing723. Other physiological systems that exhibit spontaneous or asynchronous Ca2+ oscillations across a cell population rely more heavily on single-cell measurements of dynamics724,725. While fluorescent dyes such as Fura-2 are commonly used to study Ca2+ dynamics, GECIs can offer selectivity at the cellular and sub-cellular level that is generally not possible using cell-permeable synthetic dyes. For example, GECIs have been expressed in specific neuronal subtypes, e.g., pyramidal neurons154, through the use of cell-specific promoters and have been targeted to specific microdomains through the use of targeting motifs386.
One particularly interesting system that relies on oscillatory signaling is the pulsatile release of insulin by pancreatic β-cells in response to glucose stimulation. Early studies using Ca2+ indicator dyes revealed that the elevation of glucose led to slow Ca2+ oscillations, with insulin release occurring in a similarly oscillatory fashion726. The high temporal resolution of fluorescent biosensors makes it possible to study the kinetics of this oscillatory behavior, such as the frequency and magnitude of the oscillations726. These tools helped determine that the primary mechanism responsible for triggering these Ca2+ oscillations is the increase in the ATP/ADP ratio, due to glycolysis. This increase in ATP in turn inhibits ATP-sensitive potassium channels, leading to oscillatory cellular depolarization and Ca2+ influx727. While Ca2+ oscillations are considered the master regulator of pulsatile insulin secretion, fluorescent biosensors for ATP, glycolysis, cAMP and PKA have shown that these signaling pathways also oscillate in β-cells and can modulate Ca2+ dynamics294,487,728,729. Furthermore, multiplexed imaging using fluorescent biosensors has made it possible to observe how oscillations in different signaling molecules are temporally related and helps elucidate the crosstalk between pathways. Temporal differences between two oscillating signals are described by their phase shift, which is the temporal shift between the waveforms of the two signals. Signals that increase and decrease at the same time are “in phase”, whereas signals that oscillate at the same time but in opposite directions are “out of phase”. In the MIN6 β-cell line, simultaneous measurement of Ca2+ oscillations with PKA activity or cAMP concentrations using fluorescent biosensors showed that PKA and cAMP oscillate out of phase with Ca2+294,729 (Figure 11C). Similarly, simultaneous measurement of ATP and Ca2+ also showed out-of-phase oscillations728. Both of these findings are interesting because both ATP and PKA have been shown to play a role in the formation of Ca2+ oscillations, but these regulatory mechanisms are not static. Furthermore, simultaneous measurement with high temporal resolution allows the identification of which signaling mechanism happens first, providing insights into possible causal relationships that lead to the oscillations. For example, upon glucose stimulation, ATP increases before Ca2+, suggesting that the increase in ATP causes the closure of the KATP channel and subsequently leads to cellular depolarization728. Meanwhile, cross-correlation analysis of the subsequent ATP oscillations showed that Ca2+ began to increase before ATP levels began to drop, indicating that while the increase in ATP may be responsible for initiating Ca2+ oscillations, the subsequent ATP oscillations may in fact be driven by Ca2+ oscillations728. Similar analyses have been performed with faster oscillations (on the order of seconds) in other cell types. For example, the IP3 indicator IRIS1 showed that IP3 increases prior to Ca2+ depolarization caused by glutamate stimulation of metabotropic glutamate receptor 5a (mGluR5a) in HeLa cells expressing exogenous mGluR5a510. These studies highlight the importance of single-cell and high temporal resolution afforded by fluorescent biosensors in decoding the mechanisms and regulation of oscillatory signaling dynamics.
3.2.3. Bistability and ultrasensitivity
The final types of signaling dynamics that we will discuss are the related dynamics of bistability and ultrasensitivity. Bistability generally describes a system that primarily exists in either of two almost discrete states, creating an “all-or-nothing” response to stimulation. Ultrasensitivity, on the other hand, refers to a signaling network that exhibits a “switch-like” response, where low levels of stimulation do not illicit a response, but after a certain threshold, the signaling response is very strong. Usually, this is observed as a dose response curve that has a hill coefficient greater than 1 730. Ultrasensitivity in signaling networks can be important for signaling pathways that may be critical for a response to stimuli that is resistant to noise, such as proliferation or apoptosis. While bistability is a broader term and usually requires ultrasensitivity in the signaling network731, for the purposes of this review, these phenomena will be discussed interchangeably. There are several signaling network factors that can lead to ultrasensitivity, which can make it difficult to identify precisely which elements of a signaling network are responsible732. Two common mechanisms that can lead to the switch-like behavior of an ultrasensitive response are positive feedback, in which a downstream enzyme promotes the activation of its upstream regulators, and multistep activation, wherein an enzyme requires activation via two or more independent steps730–734. One example of an ultrasensitive signaling pathway is MAPK signaling, which contains positive feedback loops and involves a multi-step activation process that requires phosphorylation at two separate sites735,736. The ultrasensitive nature of some MAPK signaling pathways is critical to ensuring that commitment to cell fate decisions or apoptotic programs is not only resistant to errant initiation by noise but also robustly induced when required735,737. Again, the signaling network topology alone is not sufficient to identify ultrasensitivity a priori732, but the ability to study these dynamics at the single cell-level using fluorescent biosensors makes it possible to dissect the key factors that regulate these dynamics.
JNK is a MAPK that is activated by cytokines and environmental stressors and regulates apoptosis738. Early work in Xenopus oocytes had shown, using a very labor-intensive process, that the JNK response to stimulation was a bistable system that exhibited all-or-none responses at the cellular level but appeared to produce a gradated response at the population level due to cellto-cell variability739. The development of a genetically encoded biosensor for monitoring JNK activity dynamics, JNKAR1, greatly improved the ability to study these heterogeneous cell responses in many different cell types459. For example, exposing JNKAR1-expressing HeLa cells to a range of concentrations of the fungal antibiotic anisomycin, which induces ribotoxic stress and leads to the activation of JNK, revealed that cells either maximally responded to super-threshold concentrations or failed to respond at all to sub-threshold concentrations459 (Figure 11D). These data showed that the JNK response to anisomycin has a hill coefficient greater than 9 and thus represents an ultrasensitive response, which conceptually makes sense for a cellular response that regulates apoptosis459. Similar to the regulation of apoptosis by JNK, the regulation of cell division by Aurora B kinase has also been shown to exhibit bistability740. Aurora B kinase activity has been shown to be critical for progression through cytokinesis and exhibits highly dynamic localization throughout cell division741. Intriguingly, multiplexed imaging of an Aurora B kinase biosensor targeted to chromatin along with mCherry-labeled Aurora B kinase showed that during anaphase, Aurora B kinase was highly concentrated at centromeres, whereas the area of high Aurora B kinase phosphorylation extended much farther740. Using in vitro experiments and computational models, Zaytesev et al. determined that this larger area of influence was due to bistability in the activation of Aurora B kinase. This bistability arises due to Aurora B kinase activation occurring mainly through trans-phosphorylation by other Aurora B kinase molecules, while deactivation by dephosphorylation occurs slowly due to a relatively low concentration of phosphatases. Therefore, the re-localization of Aurora B kinase at the centromere allows efficient activation due to the high concentration of Aurora B kinase, which remain active longer even when they diffuse away from the centromere, leading to a larger area of phosphorylation. All of these studies rely on the power of fluorescent biosensors to quantify signaling dynamics at the single-cell level in order to evaluate the bistability or ultrasensitivity of a signaling pathway that may have otherwise hidden in population averages. [add ultrasensitivity of PKA]
3.3. The Importance of Single-cell Readouts
Because fluorescent biosensors are able to quantify signaling dynamics in both time and space, they are able to resolve the dynamic behaviors of individual cells. This property is integral for studying dynamics that are heterogeneous across cell populations and thus difficult to detect using population-based methods such as western blotting. For example, the MIN6 oscillations discussed earlier are largely asynchronous, and the ability to simultaneously measure Ca2+ and the kinetics of other signaling proteins at the single-cell level using fluorescent biosensors was essential for understanding their dynamics294,487,728,729. Indeed, single-cell resolution is often essential for revealing the true dynamics of a signaling pathway that would otherwise be obscured by cell-to-cell variability at the population level, such as ultrasensitive signaling459,740,742. Meanwhile, single-cell studies can also reveal distinct behaviors among cell subsets, such as in recent work by Aoki et al., who used the FRET-based biosensor EKAREV to reveal the presence of stochastic pulses of ERK activity within individual cells, which were found to have a paracrine activating effect on neighboring cells743. Even in cases such as studies of the cell cycle, where it is possible to synchronize cells to the same point in the cell cycle, the rate at which each cell progresses through the cycle will nevertheless exhibit significant cell-tocell variability, and quantification of signaling kinetics at the single-cell level may still be required. Therefore, a genetically encodable biosensor was used to measure Cyclin B1-CDK1 activity through the cell cycle and elucidate cell-to-cell variation during this process409,434. Studying these processes at the single-cell level thus enables a greater understanding of signaling networks than can be achieved through more traditional means.
3.4. How Fast Can We Go?
In most of the examples discussed thus far, a temporal resolution on the order of seconds was adequate to resolve the dynamics of interest. However, numerous signaling processes are known to exhibit very rapid kinetics, such as GPCR activation542,545,547 and neuronal depolarization spikes156,259,379, and a much finer time resolution is thus required in order to faithfully capture the dynamics of these processes. For example, the biosensors developed to study GPCR activation rates, Ca2+ dynamics, and membrane potential are able to achieve temporal resolutions on the order of 100 ms, 1.5 ms and <1 ms, respectively259,379,542. When trying to push fluorescent biosensors to their temporal limit, there are three main factors to consider: the time it takes to collect enough photons, the time it takes for the signal to alter the sensing unit, and the time it takes for the readout mechanism to respond.
The number of photons emitted by a fluorophore is a function of both the molecular brightness, which is a measure of how readily an excitation photon is absorbed and how efficiently that absorbed photon is converted to an emitted photon, and the excitation light intensity744. Although photon gathering is not the limiting factor in most cases when the fluorescent biosensors are overexpressed, in some cases use of the newest and brightest FP variants (e.g., mTurquoise2, mNeonGreen, mScarlet) could increase the signal and enhance temporal resolution542,745,746. A complicating factor is photostability. If the fluorophore is particularly susceptible to light-induced damage, photon gathering could become a limiting factor which in turn limits the temporal resolution when a significant number of fluorophores are lost due to photobleaching. For instance, photostability limited the temporal resolution of the recently developed FLINC super-resolution biosensors where the continuous excitation required for super-resolution imaging causes reversible photobleaching447. Thus, to continue measuring biosensor activity over time, periods of darkness were included between acquisition periods to allow the fluorescent protein to recover.
Another consideration for increasing temporal resolution is how quickly the biosensor responds to signaling changes. The use of GECIs as tools to study Ca2+ dynamics across a diverse array of biological settings has led to several different designs and variants that have been optimized towards specific research goals, such as high Ca2+ sensitivity to observe small changes in Ca2+ or fast biosensor responses to Ca2+ changes to observe individual action potentials (See Section 2.3.3.1.1). While it would be ideal to have a GECI that is both highly sensitive and responds quickly, it has been observed several times that there exists a tradeoff between having fast Ca2+ binding kinetics and having a high biosensor sensitivity156,370,747. For example, among the recently published GCaMP variants, the fast variants GCaMP6f and GCaMP3fast had the higher dissociation constants, 375 nM and 2800 nM, respectively, whereas the slower but brighter GCaMP6s and GCaMP3bright had Kd values of 144 nM and 930 nM, respectively156,370. Through a detailed analysis of the GCaMP kinetics, Helassa et al. observed the binding of the 3rd and 4th Ca2+ to CaM to be much slower than the binding of the first two Ca2+ ions, and in their GCaMP3fast variant, where the 3rd EF hand of CaM was mutated to disable its Ca2+ binding, the kinetics of binding the 3rd (and final) Ca2+ were much faster370. Interestingly, their kinetic analysis showed that the conversion of the calmodulin-peptide bound sensor from dark to fluorescent was one of the slower steps in the activation process. Similar kinetic analysis of the FRET- and troponin-C-based Ca2+ biosensor TN-L15, which only binds one Ca2+ ion, found that an intermediate between the calcium binding and high FRET state is required, suggesting that the conformational change in response to Ca2+binding plays a significant role in the biosensor kinetics748. These kinetics are important considerations for these engineered biosensors as they can affect biosensor design and could be optimized. But for biosensors studying activation using native protein fusions, such as the biosensors of GPCR activation that are comprised of a fluorescently labeled Gαi and Gβ or Gγ, the kinetics of the conformational change upon activation are precisely what is being measured542.
Finally, the time it takes to couple the change in the reporting unit to a change in the readout method can impact the kinetics of fluorescent biosensors. Most unimolecular biosensors are based on an intramolecular conformational change, which has a time-scale ranging from nanoseconds to milliseconds749. Bimolecular biosensors that fully dissociate can be slower than unimolecular biosensors due to diffusion, which can add delays on the order of milliseconds to seconds, but it is still possible to have fast bimolecular biosensors, as the bimolecular GPCR activation biosensors have been used at 100-ms temporal resolution 542,750. Translocation-based biosensors, such as the KTRs112, will be affected by the barriers of diffusion and efficiency of nuclear import and export systems because the readout involves translocation from the nucleus to the cytosol. This is not to say, however, that these types of biosensors are less useful, because they can be used to study signal transduction that occurs on slower time scales (e.g., minutes).
4. Obtaining Spatial Information from Biosensors
Along with providing a detailed view of the kinetics of intracellular signaling, genetically encoded fluorescent biosensors have been essential in dissecting the organization of signaling pathways at the spatial level. Yet in contrast to the obvious role of temporal dynamics in biochemical reaction networks, early frameworks of intracellular signal transduction had no inkling of spatial regulation. The molecular components of signaling pathways were instead thought to be freely diffusible and to move unhindered throughout the cytoplasm (or plasma membrane, in the case of receptors), which logically entails the random, homogeneous distribution and uniform, nonselective activation of signaling molecules within the intracellular space. However, early studies of cAMP/PKA signaling in isolated rat hearts found that only β2AR stimulation, and not prostaglandin E1 (PGE1) stimulation, was able to affect cardiac contractility and alter glycogen metabolism, despite both stimuli eliciting similar elevations in cAMP accumulation and PKA activity751,752. Studies had also revealed that a substantial portion of predominantly type II PKA holoenzyme was associated with the particulate fraction in heart lysates753, and subsequent work found that β2AR stimulation was able to activate this particulate PKA fraction, whereas PGE1 stimulation was not754,755. These results were incompatible with a simple model of freely diffusing messenger and enzyme and suggested that some other process must underlie the ability of hormones to selectively affect specific targets within the same cell.
As a potential solution, the idea of compartmentalized signaling, which postulates that signaling molecules are not randomly distributed within cells but are instead sequestered within different spatial compartments, was proposed to account for these findings (reviewed in ref. 756). However, despite early evidence hinting at compartmentalization, the precise nature of signaling compartments and their underlying molecular mechanisms remained a mystery until the development of advanced optical tools, such as genetically encoded fluorescent biosensors. These tools enabled the non-destructive observation of signaling events within living cells, which preserves the native molecular environment. In addition, fluorescence imaging offers submicron spatial resolution, which in some cases can enable the direct visualization of distinct spatial signaling domains, while further spatial selectivity can be achieved by targeting genetically encoded biosensors to specific subcellular locations, either through the incorporation of various targeting motifs or through direct fusion to a protein of interest. Thus, genetically encoded biosensors are well-suited for directly unraveling the molecular basis of compartmentalized signaling.
Key to achieving compartmentalized signaling is controlling diffusion, which is a major obstacle to signaling specificity. For example, given measured rates of molecular diffusion within the cytosol, signaling molecules are in theory capable of traversing the entire length of a cell on the order of seconds757,758. Thus, any messenger produced or enzyme activated within one subcellular region would not remain there for long, thereby blurring out otherwise local signaling events. Conversely, because diffusion scales with the square-root of time, long-distance signaling processes, such as axonal transmission, would occur extremely slowly if left to diffusion alone. In addition, signaling molecules must efficiently engage in productive interactions and reactions to ensure accurate information flow. Yet cells contain hundreds of different signaling molecules, which are estimated to account for up to 10% of total cellular proteins757,759, and signaling pathways typically involve multiple steps and comprise numerous components. Thus, selectivity becomes increasingly unfeasible if signaling molecules are left free to diffuse and interact randomly. Cells must therefore accomplish three things to lay the foundation for compartmentalized signaling: limit diffusion to preserve local signaling events, augment diffusion to ensure rapid, efficient long-distance signaling, and selectively promote useful interactions while preventing unwanted ones. In the following section, we will discuss how biosensor-based imaging approaches have shed light on the major mechanisms utilized by cells to regulate the diffusion of signaling molecules: 1) physical separation by membranes; 2) confined activation/deactivation by pathway regulators; and 3) association with macromolecular complexes. Through these processes, cells establish the boundaries that define signaling compartments and organize signaling pathways into various spatially regulated domains.
4.1. Membrane-associated Signaling Compartments
4.1.1. Partitioning by membranes
Membranes exist to divide: the plasma membrane divides cells from their external environment, while intracellular membranes further divides cells into distinct, functionally specialized structures (i.e., organelles). Thus, cellular membranes provide ready-made platforms for compartmentalizing the actions of signaling molecules, and genetically encoded biosensors have yielded numerous important insights into the specific local signaling environments associated with membrane-bound compartments.
A number of studies over the past several years, for instance, have called into question the established model of GPCR signaling. In particular, so-called “canonical” GPCR signaling, in which receptors regulate the production of second messengers such as cAMP through coupling to heterotrimeric G proteins, has long been thought to occur exclusively at the plasma membrane and terminate when receptors are bound by βarr and undergo clathrin-mediated endocytosis. Internalized receptors are subsequently thought to engage only in “non-canonical” signaling, such as βarr-dependent MAPK signaling760–762. However, along with recent evidence challenging the role of βarr in non-canonical GPCR signaling763, several biosensors-based imaging studies have revealed that internalized GPCRs remain active and continue to stimulate cAMP production, directly contradicting the classical model of canonical GPCR signaling (Figure 12A).
Figure 12:
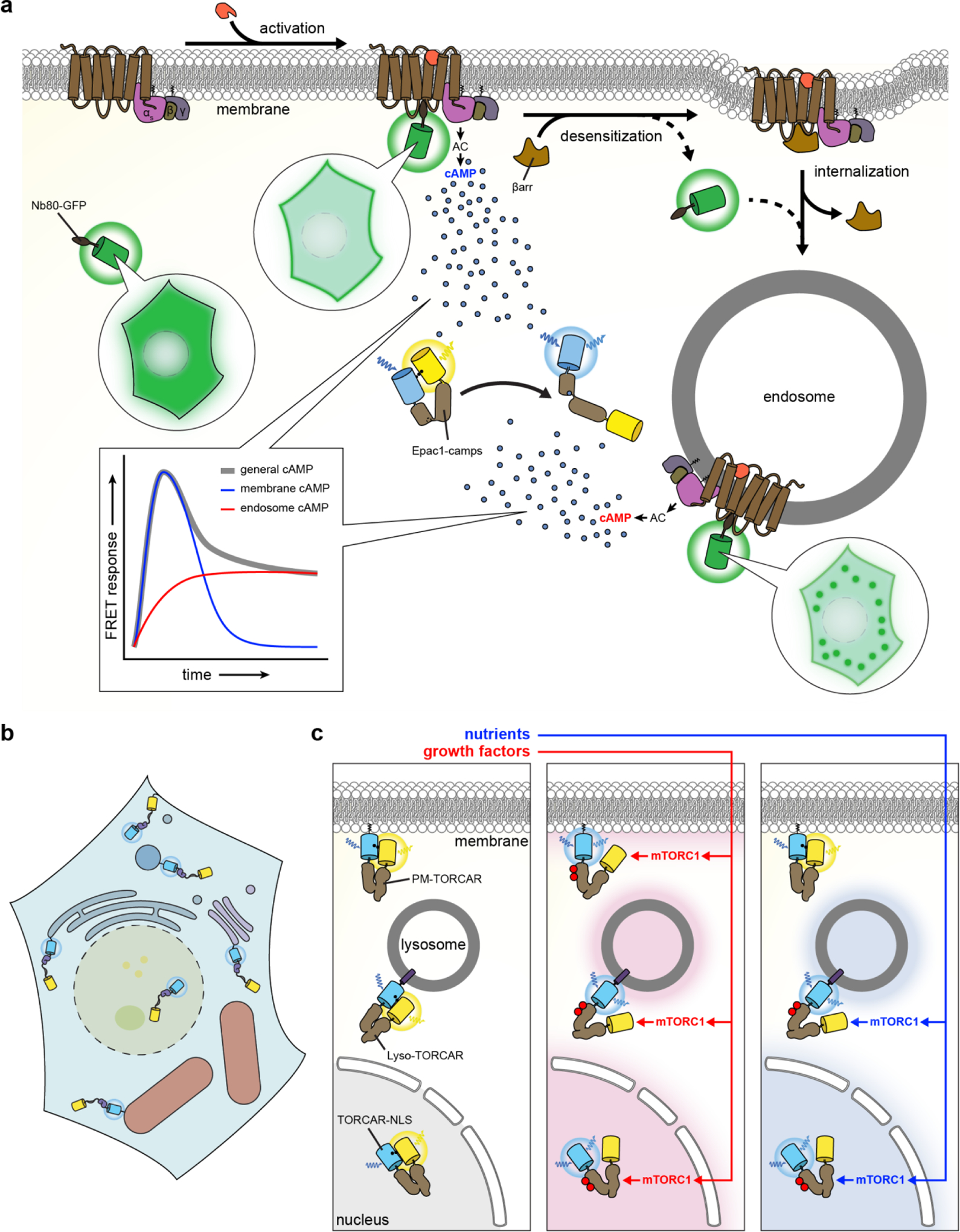
Probing spatial compartmentalization with fluorescent biosensors.
GPCRs are activated at the plasma membrane but can continue to stimulate downstream signaling after internalization. Activation of β2AR at the plasma membrane by an extracellular ligand leads to the production of cAMP. This activation of β2AR can be monitored both directly, by GFP labeled nanobody Nb80 which binds the active conformation of β2AR105, and indirectly, through biosensors of cAMP concentration such as Epac1-camps765. The activation of GPCRs is reversed by the binding of βarrestin (βarr) which attenuates downstream signaling and promotes receptor trafficking to clathrin-coated pits for endocytosis. After internalization, βarr can dissociate from the receptor and some GPCRs have been shown to then continue downstream signaling from the endosome, as shown by Nb80-GFP translocation to the endosomes105 and monitoring of cAMP production after perturbing internalization or blocking the membrane pool of the receptors by using membrane-impermeable antagonists548. B) The fusion of targeting domains to biosensors enables the measurement the signaling dynamics in several different subcellular microdomains by promoting the trafficking and localization of the biosensor to these regions. C) For example, Zhou et al. targeted TORCAR to the plasma membrane (PM-TORCAR), lysosome (Lyso-TORCAR) and nucleus (TORCAR-NLS) to examine the mTORC1 activity in each of these microdomains406. With these targeted biosensors, it was shown that growth factor stimulation leads to mTORC1 activation in all three microdomains, whereas stimulation with nutrients activated mTORC1 at the lysosome and nucleus but not at the plasma membrane406.
In an early study using primary thyroid cells derived from transgenic mice expressing Epac1-camps285, Calebiro et al. set out to investigate cAMP signaling by the thyroid stimulating hormone receptor (TSHR) and found that cAMP production became increasingly sustained in cells treated with longer pulses of TSH764. This transition to sustained cAMP signaling paralleled TSHR internalization, suggesting that these kinetic differences were linked to different spatial compartments. Indeed, co-localization studies confirmed the presence of the cAMP signaling apparatus (e.g., Gαs, ACs) on the same intracellular membrane compartments as internalized TSHR. Furthermore, disrupting receptor internalization led to the loss of sustained cAMP signaling in response to longer TSH pulses, indicating that internalized TSHR continues to promote cAMP signaling from intracellular compartments (e.g., endosomes)764. A parallel study by Ferrandon and colleagues reported similar findings regarding PTHR signaling in HEK293 cells, suggesting the possibility of a more general phenomenon765. Here, the authors used GPCR activation sensors in addition to the Epac1-camps FRET sensor and observed that stimulating cells with PTH1–34 led to prolonged receptor activation and cAMP production. As above, this sustained cAMP production was observed to coincide with the internalization of GFP-tagged PTHR and was blocked by disrupting PTHR internalization765.
Based on these initial reports, Irannejad and colleagues then sought to use a different approach to directly probe the activation state of internalized GPCRs. In this case, the authors transfected HEK293 cells with a GFP-labeled nanobody (Nb80)100 that specifically recognizes the active conformation of the β2AR105. As expected, the initially cytosolic fluorescence signal from Nb80 was observed to translocate to the plasma membrane upon β2AR stimulation using isoproterenol105. However, Nb80 was subsequently observed to re-localize to intracellular puncta containing β2AR, indicating the presence of active receptor on the endosomal surface (Figure 12A). In contrast, a recent study by Thomsen and colleagues using biosensors to monitor both cAMP production and GPCR activation found that the vasopressin type 2 receptor (V2R), but not the β2AR, induced sustained cAMP signaling due to persistent activation of internalized receptors548. V2R, a class B GPCR766,767, strongly associates with βarr, and studies of a hybrid receptor in which the β2AR C-terminal tail was replaced with that of V2R (i.e., β2V2R) have revealed βarr to adopt a so-called “tail” conformation wherein only the receptor tail is bound by βarr, leaving the transmembrane receptor core exposed768. Indeed, using both colocalization and BRET-based PPI assays, Thomsen et al. were able to demonstrate the formation of an internalized GPCR/G protein/βarr “megaplex” by both β2V2R and V2R, suggesting a potential molecular mechanism for persistent signaling by internalized GPCRs548.
These studies establish a new conceptual framework in which internalized GPCRs are a source of sustained cAMP production within the cytosol, raising the question as to the functional significance of such internal signaling. One intriguing possibility is that cAMP generated at endosomes specifically activates PKA within intracellular compartments, such as the nucleus. Indeed, PKA is known to regulate a number of processes in the nucleus, including gene expression769,770 and RNA splicing771–773, and although the cAMP-induced dissociation and diffusion of free PKA C subunit is classically viewed as the sole source of nuclear PKA activity769,770,774,775, a growing body of evidence indicates the existence of a resident pool of PKA holoenzyme within the nucleus776–781. Consistent with this model, Jean-Alphonse et al. used nuclear-targeted Epac1-camps and AKAR3 to reveal that endosomal cAMP production via synergistic β2AR/PTHR activation not only led to higher nuclear cAMP compared with either stimulus alone but also to the rapid induction of nuclear PKA activity by a nuclear-resident pool of PKA holoenzyme77. These results also confirm an earlier study in which nuclear-targeted cAMP and PKA sensors, combined with local manipulation of cAMP production by targeted soluble adenylyl cyclase, demonstrated that cAMP produced within the cytosol or nucleus could specifically and rapidly activate a nuclear pool of PKA, whereas cAMP generated at the plasma membrane could not780. β2AR/PTHR-induced endosomal cAMP production was also required to induce CREB phosphorylation and to promote mineralization in ROS17/2.8 cells, thus underscoring the functional importance of internal cAMP production77. Similarly, Godbole and colleagues, using cAMP and PKA biosensors targeted to either the plasma or Golgi membrane, found that TSHR internalization led to Golgi-localized cAMP/PKA signaling and was important for inducing CREB phosphorylation106.
As alluded to above, this ability to selectively target genetically encoded fluorescent biosensors to different subcellular compartments is instrumental in directly revealing the distinct signaling activities associated with intracellular membranes and in elucidating the mechanisms that shape these unique spatial signaling domains (Figure 12B). In a classic example, Gallegos et al. used targeted versions of CKAR to monitor local PKC activity at the plasma membrane, mitochondrial membrane, Golgi membrane, nucleus, and cytoplasm782. Stimulation with phorbol ester, a DAG mimic that directly activates PKC, revealed that each of these subcellular regions was characterized by distinct PKC activity profiles, which were found to be caused by differences in basal PKC activity or the action of local phosphatases782. Interestingly, the strongest PKC responses were observed on the Golgi membrane, which also displayed sustained PKC activity in response to Gq stimulation, in contrast to transient plasma membrane and cytosolic PKC activity. Further investigation using a translocation-based DAG reporter demonstrated that this sustained activity was directly correlated with sustained DAG accumulation in the Golgi membrane782, and a follow-up study using targeted FRET sensors for both PKC and PKD revealed that prolonged DAG production on the Golgi surface is mediated by Ca2+783.
Similarly, a recent study using CaNAR targeted to different subcellular compartments recently revealed spatial differences in the regulation of CaN activity in response to membrane depolarization-induced cytosolic Ca2+ oscillations in pancreatic β-cells431. In particular, cytosolic CaN activity displayed an integrating response to cytosolic Ca2+ oscillations, wherein each Ca2+ peak produced a step-like increase in CaN activity, while CaN activity located at the ER surface displayed an oscillatory pattern, rising and falling in tandem with cytosolic Ca2+ concentrations. Ultimately, a detailed mechanistic investigation aided by additional targeted sensors found that these spatial differences in CaN activity were the result of differential subcellular access to free Ca2+/CaM 431, which, despite its reputation as a ubiquitous signaling effector, has been shown to be a somewhat limited resource inside cells623,784. This study benefitted from the application of a second-generation CaNAR variant with an improved dynamic range, which is a critical determinant of probe sensitivity. Despite its clear usefulness in probing compartmentalized signaling, subcellular targeting can sometimes negatively impact the dynamic range of a biosensor, and continued improvements to increase dynamic range are therefore essential for detecting local signaling activity and revealing the spatial regulation of signaling pathways by subcellular compartments. For instance, Miyamoto and colleagues recently took advantage of an improved FRET-based AMPK sensor to investigate subcellular AMPK activity. Interestingly, while these authors detected AMPK activity throughout the cell, AMPK activity at different subcellular compartments was found to be associated with distinct isoforms of the AMPK catalytic subunit785. The use of a bimolecular FRET-based AMPK sensor to increase sensitivity at the plasma membrane also allowed Depry et al. to characterize the bidirectional regulation of membrane-localized AMPK activity by PKA416. Meanwhile, the cAMP sensor CUTie was designed for the express purpose of preserving dynamic range upon subcellular targeting in order to study localized signaling events287.
The ongoing development of genetically encoded fluorescent biosensors for detecting new signaling activities also continues to expand our ability to study the spatial regulation of signaling by intracellular compartments. For example, the serine/threonine protein kinase mTORC1 serves as a critical signaling hub overseeing the regulation of cellular metabolism and energy status in response to various extracellular cues786. Although mTORC1 has previously been reported to associate with numerous intracellular compartments787, the precise role of spatial compartmentalization in regulating mTORC1 signaling is unclear. To begin addressing this question, Zhou and colleagues recently developed a first-generation FRET-based reporter for monitoring mTORC1 kinase activity in living cells 406. This probe successfully detected local mTORC1 activity when targeted to different intracellular compartments, including the nucleus, which has been somewhat controversial as a site of mTORC1 activity. The authors also found that while growth factor stimulation elicited mTORC1 activity throughout the cell, amino acid (e.g., nutrient) stimulation appeared to selectively promote lysosomal and nuclear mTORC1 signaling406 (Figure 12C). This more confined activity profile may indicate that the proposed role of nutrient sensing in establishing baseline mTORC1 activity786 is achieved through spatial compartmentalization of the mTORC1 signaling machinery, thus providing important insights into the regulation of mTORC1 signaling by membrane compartments.
4.1.2. Partitioning within membranes
Cells are also thought to achieve an additional layer of spatial compartmentalization within the plasma membrane via the selective partitioning of various signaling proteins into membrane microdomains, nanometer-scale subdivisions of the plasma membrane that are composed of distinct subsets of lipids and other membrane constituents788. In particular, cholesterol- and sphingolipid-enriched “lipid rafts” are strongly implicated in organizing membrane proteins into signaling platforms789. Yet because their small size places them below the diffraction limit of optical microscopy, membrane microdomains are difficult to directly observe and characterize in living cells, rendering their very existence and function the subject of ongoing debate788,790,791. As such, genetically encoded fluorescent biosensors have provided a crucial window into the key biological roles of these subdomains. For example, through the use of a membrane-targeted cAMP biosensor, DiPilato and colleagues observed that disrupting lipid rafts via cholesterol depletion led to enhanced β2AR-induced cAMP production295, while Depry et al. observed a similar effect of raft disruption on PKA activity473, suggesting a negative effect of raft localization on the cAMP/PKA signaling machinery. Even further insights can be gained by virtue of the fact that biosensors can be targeted not just to the plasma membrane generally but also to specific membrane microdomains152. Thus, by comparing the responses of raft- and non-raft-targeted AKAR4, Depry and colleagues found that although the β2AR-stimulated PKA response in lipid rafts was lower, lipid rafts displayed significantly higher resting PKA activity compared with the bulk plasma membrane473.
By similarly comparing the responses of other genetically encoded fluorescent biosensors targeted to different membrane microdomains, researchers have further been able to illuminate key molecular differences that give rise to the distinct signaling behaviors observed in these subcompartments. For example, in a series of studies using genetically encoded FRET-based biosensors to study the phosphoinositide 3-kinase (PI3K)/Akt signaling pathway, Gao and coworkers demonstrated that platelet-derived growth factor (PDGF) stimulation leads to higher Akt activity in lipid rafts due to the increased localization and activation of the upstream kinase PDK1 within rafts compared with the bulk plasma membrane, whereas the negative regulator PTEN was preferentially located outside rafts407,468. The activity of focal adhesion kinase (FAK) was also shown to be higher in lipid rafts compared with non-raft regions in response to either PDGF stimulation or cell adhesion450. Interestingly, Seong et al. found that PDGF-induced raft FAK activity was regulated by Src, which was also reported to be more active in lipid rafts792, whereas adhesion-induced FAK activation in lipid rafts was conversely required to promote Src activity450. This difference in the relationship between raft-localized FAK and Src activity in response to PDGF or cell adhesion signaling may be related to recent observations that PDGFR activity is strongly inhibited by integrin-mediated cell tension sensing within lipid rafts, while bulk plasma membrane PDGFR activity was unaffected467. The studious application of genetically encoded biosensors has thus allowed researchers to cut through the debate and establish the role of membrane microdomains as spatial organizing centers for intracellular signaling pathways.
4.2. Compartmentation by the Activity of Pathway Regulators
Signaling pathways are composed of numerous molecular players that are affected by a number of regulatory processes: negative regulators function to attenuate, antagonize, or otherwise repress signaling within a pathway, while positive regulators serve to prolong or enhance signaling activities. The regulatory connections operating within a signaling pathway can also serve as feedback mechanisms, through which a signaling pathway can dynamically shape its own activity and give rise to various complex features793 (see Section 3.1). When combined with the local activation of signaling enzymes anchored to cellular structures such as membranes, the action of pathway regulators can significantly influence the scope of a signaling event, yielding spatially confined signaling domains of varying sizes.
4.2.1. Calcium signaling
Coupled with advances in optical microscopy and digital image processing, some of the earliest direct studies of spatially regulated Ca2+ signals were propelled by the development of small-molecule fluorescent indicators3, which were much easier to work and with and load into living cells compared with prior approaches and enabled the visualization of Ca2+ signals with high sensitivity and spatiotemporal precision. For example, Sawyer and colleagues used neutrophils loaded with the fluorescent Ca2+ indicator quin2794 to monitor Ca2+ signals in cells undergoing phagocytosis and found that, while Ca2+ levels appeared to be uniform throughout the cytosol in unstimulated cells, neutrophils that were in the process of migrating towards or engulfing a target showed dramatically higher Ca2+ levels in the extending lamellipodia compared with trailing regions of the cell795. Brundage et al. similarly performed time-lapse imaging in chemotaxing eosinophils loaded with another Ca2+ indicator, fura-2131, and found that Ca2+ was highest at the rear of cells that were steadily migrating in a single direction, while a localized burst of Ca2+ at the rear of the cell was observed to precede cell turning796. Meanwhile, in their work with giant squid axons, Smith and colleagues were able to use fura-2 to visualize the formation of sharp Ca2+ gradients in presynaptic terminals in response to trains of presynaptic action potentials797.
To this day, fluorescent Ca2+ indicators remain among the most popular and widely used tools for visualizing and investigating spatially compartmentalized Ca2+ signals. For example, in their study investigating the coordination of growth cone and cell body motility during neuronal migration, Guan and colleagues loaded cultured rat cerebellar granule cells with fluo-4 798 to examine the role of Ca2+ in cells responding to the guidance factor Slit-2 799. Ca2+ is known to be a key regulator of both neuronal migration and growth cone dynamics800–802, and Slit-2 has previously been shown to act via cytosolic Ca2+ elevations to exert a repulsive effect on migrating neurons803,804. In this instance, application of an extracellular gradient of Slit-2 at the front of the advancing growth cone was observed to reverse the direction of granule cell migration in conjunction with the rapid induction of a Ca2+ wave that initiated within the growth cone and travelled quickly along the leading neurite to reach the soma799. Yet the continued popularity enjoyed by fluorescent Ca2+ indicators notwithstanding, genetically encoded Ca2+ indicators (GECIs) have also been widely adopted to visualize intracellular Ca2+ signals. Recent work by Huang et al., for instance, used both Fluo-3 and the high-affinity FRET-based Ca2+ sensor YC-Nano 50 352 to monitor Ca2+ gradients and uncover the role of mechanosensitive Ca2+ channels in regulating cell polarity805. GECIs are also essential for labeling cell populations in vivo, as was done recently by Vargas and colleagues, who expressed a modified form of GCaMP373 in zebrafish somatosensory neurons and observed the passage of two distinct Ca2+ waves through neurons undergoing axonal degeneration806.
These spatially diverse Ca2+ signaling events are governed by various regulatory processes that work in concert to control the persistence and diffusion of Ca2+ fluxes within the cytosol (Figure 13A). Elevations in cytosolic Ca2+ concentrations are initiated via Ca2+ influx across the plasma membrane and release from intracellular stores, and Ca2+ waves and gradients often originate from highly localized Ca2+ elevations. Such “elementary” Ca2+ release events, which assume a variety of aliases in the literature807,808, involve Ca2+ release by individual or clusters of channels and can often be directly visualized using diffusible Ca2+ indicator dyes or GECIs. Early work by Bootman and colleagues, for instance, revealed the induction of Ca2+ waves from individual Ca2+ “puffs” in response to histamine stimulation in fluo-3-loaded HeLa cells809. More recent work by Wei and coworkers also observed discrete Ca2+ “flickers” in migrating human embryonic lung fibroblasts loaded with fluo-4 810. These transient bursts of Ca2+, which were associated with both mechanosensitive TRPM7 channels in the plasma membrane and IP3 receptors (IP3Rs) on the ER surface, were only present in migrating cells and were largely concentrated within the lamellipodium at the leading edge, helping to control cell turning810. Similarly, Sonkusare et al. were able to observe Ca2+ “sparklets” in the vascular endothelial cells of arteries isolated from GCaMP2-transgenic mice811. These discrete Ca2+ release events were associated with the opening of individual plasma membrane TRPV4 channels and were important for the activation of potassium channels to regulate vascular tone811.
Figure 13:
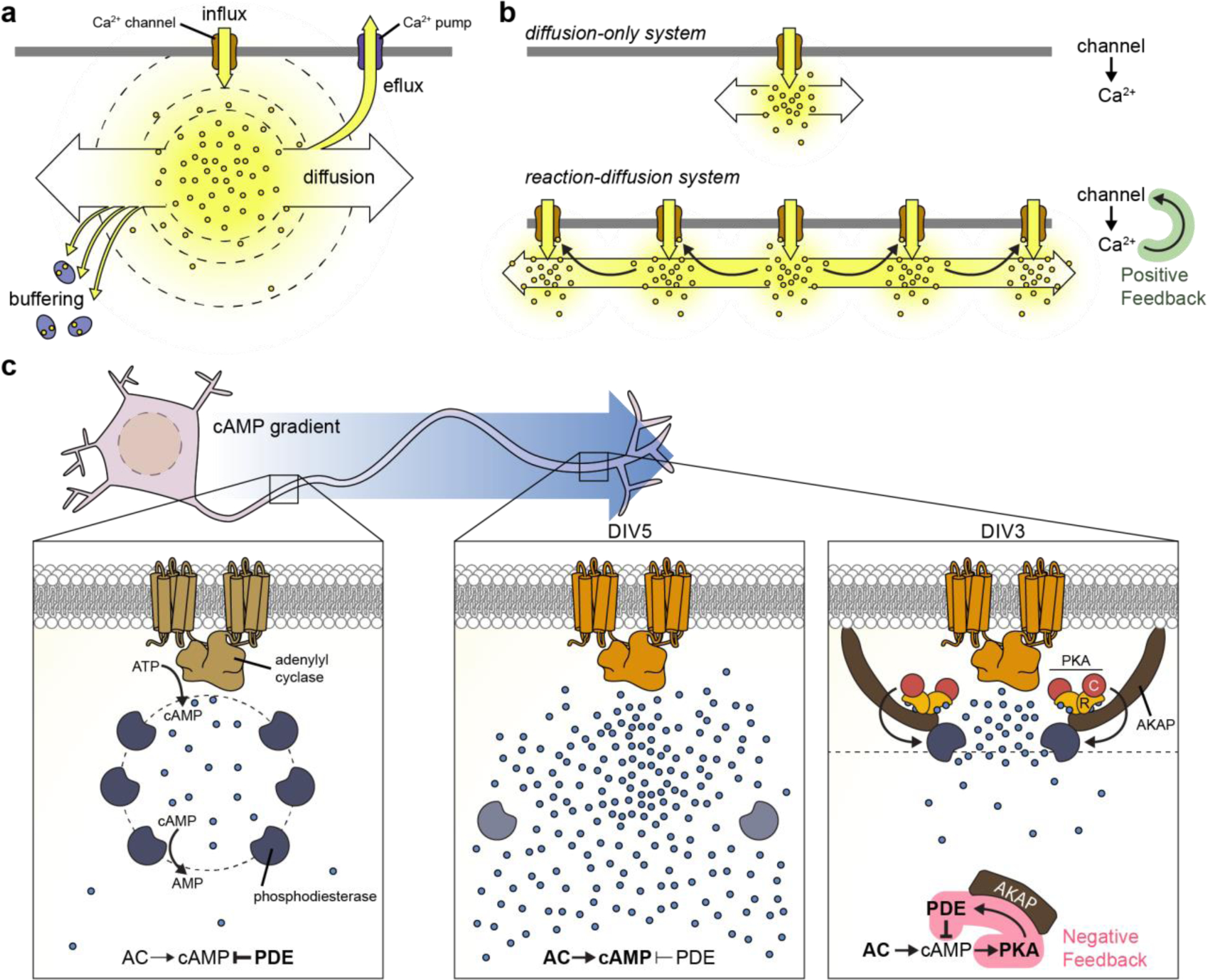
Spatial regulation of diffusible signaling molecules.
Spatial Ca2+ gradients are primarily shaped by 4 effects: the rate of Ca2+ influx into the cytosol from the extracellular environment and intracellular stores, the efflux of Ca2+ out of the cytosol by Ca2+ pumps, Ca2+ binding proteins which buffer the changes in the free Ca2+ concentration and diffusion of Ca2+ through the cytosol. B) While the effect of Ca2+ influx by a single channel may be limited by the rate of Ca2+ flux through the channel and the rate of diffusion (top), Ca2+-induced Ca2+ release (CICR), where an increase in Ca2+ can stimulate the opening of neighboring Ca2+ channels, creates a positive feedback loop that can create a wave of Ca2+ release that spreads faster than possible by diffusion alone (bottom). C) The spatial arrangement of adenylyl cyclases (ACs), which produce cAMP, and phosphodiesterases (PDEs), which degrade cAMP, regulate the formation of gradients of the diffusible messenger cAMP. For example, rat hippocampal neurons at 5 days in vitro (DIV5), when the axons have started to become more thoroughly established, have been observed to exhibit an axon-directed gradient of cAMP accumulation in response to Forskolin stimulation of ACs825. Conversely, at an earlier time point of 3 days in vitro (DIV3), cAMP production was observed to be much more limited in response to Forskolin, which was hypothesized to be due to negative feedback mediated by PKA and PDE, which are localized to distal regions of an axon by an A-Kinase Anchoring Protein (AKAP)825.
More precise monitoring of these Ca2+ channel microdomains can also be achieved by targeting GECIs directly to sites of Ca2+ entry. For example, Tay et al. were able to measure Ca2+ flux immediately in the vicinity of the CaV2.2 voltage-gated Ca2+ channel by using both the FRET-based Ca2+ sensor TN-XL385 fused to the C-terminus of the channel catalytic subunit and total internal reflection fluorescence (TIRF) microscopy to quantify biosensor dynamics exclusively at the plasma membrane812. Because Ca2+ levels near channel openings are anticipated to greatly exceed those generally observed in the cytosol813, it is important to tune the sensitivity of the sensor to match the concentration range being studied and to avoid unwanted probe saturation. Thus, Despa and colleagues developed a modified GCaMP2.2 with a reduced Ca2+-binding affinity (GCaMP2.2Low), which they then used to monitor Ca2+ levels near individual ryanodine receptors (RyRs) in the junctional clefts of adult rat cardiomyocytes371. Using this approach, the authors were able to observe larger and faster Ca2+ elevations in the channel microdomain compared with the bulk cytosol during cardiac contraction371. Targeted sensors can further be used to probe connections between Ca2+ channel microdomains and specific Ca2+ effectors. For instance, Ca2+-sensitive ACs are known to be selectively activated by capacitative Ca2+ entry (CCE, or store-operated Ca2+ entry [SOCE]) versus other forms of Ca2+ influx, suggesting close association with specific Ca2+ channels. Indeed, by targeting GCaMP to AC8, Willoughby et al. observed much stronger CCE-included Ca2+ elevations in the vicinity of AC8 compared with elsewhere in the cell, consistent with the localization of AC8 within a specific Ca2+ channel microdomain814.
Such discrete Ca2+ release events are capable of generating large-scale spatial signaling domains by virtue of the fact that Ca2+ can stimulate its own release via positive feedback on Ca2+ channels such as IP3Rs and RyRs on the ER surface, resulting in Ca2+-induced Ca2+ release (CICR) or “regenerative” fluxes807,815. For example, Bootman and colleagues were able to block the conversion of puffs into waves by disrupting the positive feedback required for CICR809. The rapid Ca2+ wave observed by Guan et al. in response to an applied Slit-2 gradient was similarly blocked by disrupting CIRC via the inhibition of RyR activation799. Local Ca2+ elevations are thereby able to propagate rapidly across long distances because Ca2+ need only diffuse the short distance to a neighboring channel to elicit additional Ca2+ release, and so forth, rather than having to directly diffuse across an entire cell. Such combined reaction-diffusion systems758,816 effectively convert the cytoplasm into an “excitable medium”758,815 capable of spreading Ca2+ signals much more rapidly than diffusion alone (Figure 13B). Together with inter-cell contacts such as gap junctions, this process even allows Ca2+ waves to spread rapidly across groups of cells, as documented by Chifflet and colleagues, who observed Ca2+ waves that traversed across several cells within a monolayer in response to wounding and during regeneration in fluo-4loaded epithelial cells817. Similarly, Sieger and coworkers used GCaMP-expressing transgenic zebrafish to study the molecular mechanisms guiding microglia towards the sites of neuronal injury in the brain and found that targeted neuronal ablation induced a Ca2+ wave capable of traversing the entire brain in a matter of seconds818.
Ultimately, the overall persistence of a Ca2+ signal is determined by the balance between Ca2+ influx and efflux mechanisms, as cytosolic Ca2+ elevations are constantly being antagonized by the steady action of pumps and transporters that work to remove Ca2+ from the cytosol. Yet in addition to control by influx and efflux mechanisms, cytosolic Ca2+ is also heavily buffered by its binding to cytosolic proteins such as parvalbumin, calretinin, and calbindins819. Buffering plays an essential role in controlling the diffusional spread of Ca2+ within the cytosol and has been shown to exert considerable influence on the spatial confines of Ca2+ signals819–822 (Figure 13A). For example, Wang and colleagues visualized the effect of buffering on Ca2+ waves by loading astrocytes with fluo-3 823 and treating them with exogenously applied Ca2+ chelators, finding that both high-affinity Ca2+ chelators and high concentrations of low-affinity chelators significantly attenuated the initiation and propagation of Ca2+ waves820. Dargan and Parker also observed that the binding kinetics of cytosolic Ca2+ buffers have a significant impact on Ca2+ induced by photoreleased (i.e. UV uncaged) IP3 by modulating feedback between and within IP3R clusters, which underlies regenerative Ca2+ release (i.e., CICR), with “slow” buffers promoting more local signals by disrupting feedback between clusters and “fast” buffers promoting global signals by disrupting feedback within clusters while serving as a Ca2+ shuttle to facilitate communication between clusters824.
These studies suggest that the expression of different buffer pools with distinct kinetics may in fact serve to insulate spatially distinct Ca2+ signals in the same cell, such as in the case of Wei and colleagues, whose Ca2+ flickers were observed to co-exist alongside an oppositely oriented global Ca2+ gradient810. Thus, by effectively controlling the persistence and diffusion of cytosolic Ca2+ elevations, cells are able to generate highly specific and precisely tuned spatial signals. It should be noted, however, that fluorescent Ca2+ indicators, which are themselves typically derived from the chemical backbones of Ca2+ chelators3, are also capable of buffering and disrupting Ca2+ waves when loaded at sufficiently high concentrations820, thereby highlighting the delicate balancing act researchers must perform to investigate spatial regulation in signaling.
4.2.2. cAMP signaling
cAMP is another ubiquitous and diffusible intracellular messenger that, like Ca2+, is responsible for controlling a wide array of cellular processes and is capable of forming diverse spatial signaling domains. In one of the earliest examples of the direct visualization of spatially compartmentalized cAMP signaling in living cells, Bacskai and colleagues utilized FlCRhR629 to monitor cAMP levels in sensory neurons of the marine snail Aplysia826. Confocal imaging of cultured Aplysia neurons injected with this probe revealed that bath application of serotonin induced a spatial gradient of elevated cAMP concentrations, with cAMP accumulation rising with increasing distance from the cell body and reaching its highest levels in neuronal processes.
In time, the practical advantages of genetically encoded fluorescent biosensors (e.g., more easily constructed, manipulated, and introduced into cells) would outstrip the utility of fluorescent analogues and also lead to more frequent observations of compartmentalized cAMP signaling. For example, in their study reporting the development of a panel of unimolecular FRET-based cAMP sensors, Nikolaev et al. observed the induction of a cAMP wave that gradually travelled the length of the cell in response to local β2AR stimulation in hippocampal neurons expressing Epac1-camps285. Subsequent work by this same group also revealed the formation of differential cAMP gradients in cells upon local stimulation with either β1AR or β2AR agonists284. Specifically, in adult murine cardiomyocytes derived from transgenic mice expressing another FRET-based cAMP sensor, HCN-camps, local β1AR stimulation produced a large, shallow gradient of cAMP accumulation that spread far into the cell, whereas local β2AR stimulation prompted the formation of a much steeper cAMP gradient that was almost completely confined to the region nearest the stimulation site284. Similarly, Lim and coworkers were able to observe a front-to-back cAMP gradient in migrating CHO cells expressing the cAMP sensor ICUE2, consistent with the key role played by cAMP signaling in cell polarization827. Furthermore, a recent study by Gorshkov and colleagues reported the formation of a developmentally timed spatial cAMP gradient in polarizing rat hippocampal neurons transfected with ICUE3 825. In particular, whereas bath application of forskolin induced a gradient of cAMP accumulation that was oriented towards the developing axon in neurons grown for 5 days in vitro (DIV5), no such gradient was observed in less developed DIV3 neurons825.
Although the compartmentalization of cAMP signaling is less well understood than that of Ca2+, substantial progress has been made over the past 15 years in unravelling the molecular mechanisms governing these spatial domains of cAMP, based on the use of fluorescent biosensors to carefully dissect the contributions of the various processes controlling cAMP persistence (e.g., synthesis and degradation) and diffusion. For instance, cAMP synthesis is typically initiated at the plasma membrane by the GPCR-induced activation of transmembrane ACs, and local cAMP production has previously been implicated as one mechanism capable of generating spatially confined domains of elevated cAMP828. The differential regulation of AC isoforms by various signaling pathways829,830 may in fact yield discrete subcellular cAMP elevations in response to specific inputs, with ACs capable of producing local microdomains of high cAMP concentration that are analogous to the elementary release events and Ca2+ microdomains produced by the opening of small numbers of Ca2+ channels. Indeed, Wachten and colleagues were able to demonstrate this phenomenon directly by expressing variants of the Epac2-camps sensor, which were targeted to the cytosol or plasma membrane or directly fused to AC8, in GH3B6 pituitary cells831. Specifically, treating cells with the hormone vasoactive intestinal peptide, which stimulates AC activity via Gαs-coupled GPCR signaling, strongly increased both global and plasma membrane-adjacent cAMP levels while having minimal effect on cAMP levels in the vicinity of AC8. Conversely, stimulation with thyrotropin-releasing hormone, which promotes Ca2+ release via Gq-coupled signaling, selectively increased cAMP levels near AC8 while actually decreasing cAMP levels globally831.
cAMP signaling terminates with the hydrolysis of cAMP by cyclic nucleotide phosphodiesterases (PDEs), which thereby mediate the persistence of cAMP in cells. Yet the cAMP-degrading activity of PDEs has also come to be regarded as a major mechanism by which cells limit cAMP diffusion832,833. For example, PDE activity was shown to be essential for promoting the aforementioned cAMP gradient in polarizing DIV5 hippocampal neurons, whereas negative feedback signaling through axonal PDE activity actually suppressed the formation of a similar cAMP gradient in DIV3 neurons825 (Figure 13C). PDE activity was also found to be involved in producing the differential β1AR- and β2AR-induced cAMP gradients observed by Nikolaev et al.284. Meanwhile, Maiellaro et al. found that PDE activity was essential for the formation of discrete cAMP gradients within single synaptic boutons in locally stimulated Drosophila sensor neurons834. Similarly, a prior study by Herbst and colleagues demonstrated that PDE3 activity restricted growth factor-induced cAMP accumulation to the plasma membrane in PC12 cells708. Thus, PDEs can be seen to create spatial barriers that constrain the exit of cAMP from within a microdomain, preventing more global accumulation. However, PDEs can inversely restrict the entry of cAMP into subcellular regions, as observed by Monterisi et al., who found that mitochondrially localized PDE2A2 blocked global cAMP signals from activating the FRET-based cAMP sensor H90 835 targeted to the outer mitochondrial membrane836. In fact, in an experiment mirroring studies designed to monitor microdomains of cAMP accumulation, Lohse and coworkers were unable to detect any measureable cAMP accumulation using Epac1-camps fused directly to PDE4A1 837, raising the possibility that PDEs are capable of generating negative cAMP microdomains, or “holes”.
However, our understanding of the molecular pathways regulating cAMP diffusion is complicated by the diffusion of cAMP itself. For example, by using FlCRhR to measure the cytosolic diffusion of microinjected cAMP in Aplysia sensory neurons, Bacskai and colleagues calculated an apparent diffusion coefficient for cAMP of approximately 780 μm2/s 826. Nikolaev et al. employed a similar approach by measuring the cytoplasmic velocity of the cAMP wave generated in response to local β2AR stimulation in Epac1-camps-expressing hippocampal neurons, arriving at an apparent diffusion coefficient of approximately 480 μm2/s 285, while Chen et al. were also able to estimate a cAMP diffusion rate of approximately 270 μm2/s in frog olfactory cilia based on electrophysiological measurements838. These values closely resemble those measured for cAMP diffusing freely in solution839–841, and the idea of rapidly diffusing cAMP has proven difficult to reconcile with the very idea of compartmentalization, let alone the dominant role attributed to PDE activity in forming cAMP compartments. Indeed, mathematical modeling by Lohse et al. based on known catalytic rates indicated that PDEs should be incapable of producing cAMP microdomains with diffusion coefficients of this magnitude, despite their own observations to the contrary837 (for more on computational modeling and fluorescent biosensors, see Section 5). Although some modeling studies have emphasized the importance of PDEs in compartmentalizing cAMP signaling842, still more indicate that PDE activity is insufficient without additional constraints on cAMP diffusion837,843,844. Indeed, recent work has provided evidence that cAMP diffusion is in fact slower than the aforementioned estimates and that some additional processes are responsible for compartmentalizing cAMP signals.
Several additional mechanisms have been proposed to participate in the spatial compartmentalization of cAMP signaling alongside local cAMP production and degradation (reviewed in ref. 845). Bacskai et al., for instance, initially articulated the concept of cell shape as a regulator of cAMP compartmentation based on their studies in Aplysia sensory neurons826, and Neves et al. recently provided a direct demonstration of this process through a combination of live-cell imaging of FRET-based biosensors and mathematical simulations, showing that fine cellular structures (e.g., dendrites) with confined geometries and high surface-to-volume ratios favored cAMP production by transmembrane ACs over cAMP degradation by cytosolic PDEs, thus promoting cAMP accumulation846. The internal geometry of the cell has also previously been suggested to create physical barriers that restrict cAMP diffusion828, and in a recent study, Richards et al. used the FRET-based cAMP sensor H187 288 to reveal that cAMP diffusion rates are slower in adult cardiomyocytes compared with neonatal cardiomyocytes, which contain fewer and less ordered mitochondria compared with adult myocytes and thus present fewer physical barriers to diffusion within the cytoplasm847. Meanwhile, Agarwal and colleagues used correlation spectroscopy analysis to measure the diffusion of fluorescently labeled cAMP in living cells and found that binding to PKA R subunits had a significant influence on cAMP diffusion, highlighting a role for cAMP buffering similar to that of Ca2+ buffering. Notably, both groups reported diffusion coefficients (32 μm2/s 847 and 5 μm2/s 848) that are dramatically lower than previous estimates and are much more compatible with existing models of cAMP compartmentalization.
The work by Agarwal and colleagues is consistent with previous reports in which cAMP binding by PKA was shown to be important for cAMP gradient formation843,849,850, as well as with much earlier indications that a substantial amount of total basal cAMP is bound to PKA in cells753,851. Yet although the idea of cAMP buffering as a heretofore under-appreciated mechanism regulating cAMP diffusion and compartmentalization holds some appeal, particularly given the parallels to Ca2+ buffering, the importance of buffering in cAMP compartmentalization remains unclear. For example, Richards et al. observed little role for buffering in determining cAMP diffusion rates, as saturating intracellular cAMP binding sites did not affect their estimated diffusion coefficients847, though Agarwal et al. conversely observed that intracellular geometry did not significantly affect their measurements of cAMP diffusion848. Furthermore, despite its high abundance and affinity for cAMP, the reported buffering capacity of PKA is quite low compared with Ca2+ buffers845, and cells would likely have to express additional, potentially uncharacterized cAMP-binding proteins to achieve sufficient buffering. Some of these questions may potentially be resolved through the use of molecular tools to perturb cAMP buffering, and to this end, Lefkimmiatis et al. previously developed a genetically encoded cAMP “sponge” that can be introduced into living cells as an exogenous cAMP buffer852. Combining the cAMP sponge with fluorescent cAMP biosensors should enable direct visualization of the effect of different buffering rates on spatial cAMP signaling, similar to previous studies of Ca2+ buffering820. Nevertheless, no single process likely plays a dominant role in cAMP compartmentalization843, and researchers will continue teasing apart the individual contributions of the various molecular mechanisms responsible for directing the formation of compartmentalized signaling domains.
4.2.3. Other signaling molecules
Chemotaxing cells are capable of undergoing rapid polarization even in the presence of very shallow chemoattractant gradients853, implying the amplification of extrinsic spatial differences by the compartmentalization of the intracellular signal transduction machinery, and our understanding of spatial signaling in chemotaxis has greatly benefitted from the use of genetically encoded fluorescent biosensors. For instance, early studies using translocation-based sensors and GFP-tagged fusion proteins revealed the formation of a sharp gradient of 3’ phosphoinositides (3’-PIs, e.g., PI(3,4,5)P3, PI(3,4)P2) towards the leading edge of the plasma membrane in chemotaxing cells854–857. Gradient formation was found to be partially regulated by the inverse localization of PI3K and PTEN, which produce and degrade 3’-PIs, respectively, along the front and back of polarized cells858,859, and to be potentially reinforced by negative feedback between these two enzymes. PI3K signaling also activates a number of small GTPases, including Ras, Rho, Rac, and Cdc42, which in turn stimulate PI3K activity, resulting in a positive feedback loop that further sharpens and amplifies the leading edge 3’-PI gradient860–862.
The use of FRET-based biosensors has similarly revealed gradients of active Cdc42 and Rac at the leading edge in migrating cells584,863, whereas Rho activity is concentrated towards the rear of the cell863,864. These gradients have been shown to be mediated by multiple positive and negative feedback loops, including positive feedback with PI3K 860–862 and mutual antagonism between Rho and Rac activity865, as well as through self-organization via cytoskeletal rearrangements864. Hodgson and colleagues also recently used FRET-based sensors to visualize an inverse spatial relationship between active Cdc42 and Cdc42 that was bound and inhibited by GDIs, revealing another mechanism of spatial control in this pathway586. Interestingly, positive and negative feedback within this network also allow Cdc42 to behave as an excitable medium866, which Yang et al. recently visualized in the form of waves of Cdc42 activation that preceded spontaneous symmetry breaking in unpolarized neutrophils863.
Spatial signaling by the small GTPase Ran has also been investigated using genetically encoded biosensors. The biological functions of Ran GTPase, which regulates multiple processes related to nuclear function867, are largely attributed to the predicted formation of a gradient of GTP-bound (i.e. active) Ran emanating from within the nucleus, due to the anchoring of its GEF, RCC1, to chromatin and the localization of its GAP, RanGAP1, in the cytoplasm. Using FRET-based biosensors that detect endogenous GTP-bound Ran, Kaláb and colleagues were able to confirm the existence of a RanGTP gradient in both Xenopus oocyte extracts and mitotic HeLa cells 591,868, as well as the importance of this gradient in regulating spindle assembly. Lee et al. were further able to combine FRET-based biosensor imaging of RanGTP with a temperaturesensitive RCC1 mutant to observe the loss of the RanGTP gradient surrounding metaphase chromosomes upon acute RCC1 inhibition 869. This study also identified a role for the RanGTP gradient in regulating kinetochore attachment through Aurora B kinase 869, whose spatial signaling during anaphase of the cell cycle had previously been observed by Fuller and colleagues421. Specifically, the use of a chromosomally targeted FRET-based sensor revealed a gradient of Aurora B kinase activity that was shaped by a positive feedback loop between Aurora B activation and spindle microtubules to communicate the location of the spindle midzone421. Employing a similar imaging approach, Liu and coworkers also visualized an Aurora B activity gradient that was essential for sensing chromosome bi-orientation870. Thus, along with Ca2+ and cAMP, studies of numerous signaling pathways have benefited from the use of genetically encoded fluorescent biosensors, which have illuminated the widespread formation of signaling gradients and other dynamic spatial domains of signaling activity within cells.
4.3. Assembly of Macromolecular Signaling Complexes
Given the broad substrate specificities of most signaling enzymes and the sheer number of permutations that can be generated by the intracellular signaling machinery, diffusion alone is clearly insufficient to guarantee that useful interactions will occur among the correct molecular players once a pathway becomes activated. Instead, cells frequently utilize protein-protein interactions to channel signaling activities along specified routes by directly recruiting signaling molecules into productive complexes near their sites of action, thereby ensuring the efficiency and specificity of signal transduction871. One mechanism by which this is achieved is through the use of multivalent scaffolds, which are defined as proteins capable of binding two or more signaling enzymes and tethering them to specific subcellular locations while also coordinating their signaling activities872.
4.3.1. Scaffolding by A-kinase anchoring proteins
AKAPs represent the most well-characterized family of multivalent scaffold proteins in the literature. The various members of this family – comprising more than 50 different proteins with orthologues in numerous species – show no overall structural similarity but nevertheless share common features706,873. In particular, all AKAPs contain a short amphipathic helix that binds to the R subunit dimer of the PKA holoenzyme; while the majority of AKAPs specifically recognize the type II R subunit (RII), some type I (RI)-specific and dual-specificity AKAPs have also been identified. Furthermore, all AKAPs contain targeting sequences that mediate their distinct subcellular localizations, enabling the direct recruitment of PKA to specific compartments throughout the cell706,873. As such, AKAPs play a central role in establishing subcellular domains of PKA activity. For example, Terrin and coworkers used centrosometargeted AKAR3 to observe that AKAP9/AKAP450 is responsible for creating a domain of increased PKA activity in this region874. Wang et al. similarly used cytosol- and membrane-targeted AKAR4 to observe the complete inhibition of kinase activity associated with an AKAP-anchored subpool of PKA at the plasma membrane in cells treated with a cell-permeable peptide that disrupts AKAP-RII binding875. Meanwhile, Schott and colleagues recently performed a study using AN3 CA cancer cells, which do not express AKAP12/gravin, along with subcellularly targeted AKAR3, to reveal that re-expressing wild-type gravin increased membrane-associated PKA activity while decreasing cytosolic PKA activity in response to either β2AR stimulation or direct AC activation using forskolin876. This effect was abolished by the Ca2+-induced intracellular redistribution of gravin and was absent in cells expressing a gravin mutant lacking the RII-binding site, highlighting the importance of anchoring and localization for PKA compartmentalization by AKAPs.
AKAP anchoring has also been demonstrated to yield more complex spatial patterns of PKA signaling. For example, PKA plays an important role in cell migration by phosphorylating the cytoplasmic domain of α4 integrin, which also functions as an atypical, RI-specific AKAP877. Lim et al. therefore used plasma membrane-targeted AKAR3 to investigate the spatial regulation of PKA activity during cell migration and observed the formation of a leading-edge gradient of PKA activity in migrating cells827. This gradient was dependent on anchoring of PKA-RI by α4 integrin, as the mislocalization of type I PKA selectively abolished the PKA activity gradient at the leading edge. Gorshkov and colleagues similarly observed the formation of PKA activity gradient in polarizing DIV5 hippocampal neurons expressing diffusible AKAR4 825. This activity gradient paralleled the axon-directed cAMP gradient also seen in these cells and was abolished by the disruption of AKAP-PKA anchoring. Interestingly, AKAP disruption induced a cAMP gradient in DIV3 cells, suggesting that AKAP anchoring mediates the negative feedback circuit that suppresses the cAMP gradient at this developmental stage825 (Figure 13C). Meanwhile, Mo and colleagues were recently able to obtain an even more detailed view of PKA compartmentalization by imaging the novel super-resolution PKA activity biosensor FLINCAKAR, which revealed the plasma membrane to be decorated with discrete PKA activity microdomains of ~350 nm in diameter following PKA stimulation447. Interestingly, these highactivity puncta were observed to co-localize with clusters of plasma membrane AKAP5 (AKAP79/150) and also disappeared following treatment with a cell-permeable AKAP-disruptor peptide875, suggesting the principal involvement of AKAP anchoring and demonstrating the precision with which AKAPs regulate spatial PKA signaling.
Yet such fine compartmentalization is in apparent conflict with the classical model of a dissociating and diffusing PKA C subunit, raising questions about other mechanisms that may play a role in restricting PKA diffusion, though opinions on this topic diverge considerably. In particular, because the cAMP-triggered dissociation of the PKA C and R subunits is based on early in vitro experiments878–882, some groups have argued that the active C subunit does not in fact dissociate from the R subunits under physiological conditions. Indeed, by imaging fluorescent protein-tagged PKA C and R in living cells, Martin et al. previously reported that AKAP-anchored type I PKA holoenzyme fails to dissociate in response to cAMP alone883. Furthermore, by combining live-cell imaging with biochemical and genetic approaches, Smith and colleagues recently suggested that type II PKA also does not dissociate from AKAP complexes in response to physiological stimuli883, and instead favored a model in which intrinsic disorder within the RII dimer allows the bound, active PKA C subunit to phosphorylate targets within a defined radius884. However, recent chemical cross-linking studies performed by Walker-Gray and colleagues call these results into questions and appear to indicate that the PKA C subunit is capable of dissociating from RII dimers in cells885. Additional findings by these authors support a previously reported model of RII isoform-specific membrane binding via the C subunit myristoyl group886, which restricts C subunit diffusion to within the plane of the membrane and allows for more efficient recapture by RII885. Work by Mo et al. further suggests a mechanism of C subunit recapture and retention, wherein AKAP clustering selectively increases the local concentration of PKA R subunits to further enhance C subunit recapture and spatially confine PKA activity447.
Each AKAP family member is also capable of binding a diverse array of signaling proteins in addition to PKA706,873, allowing individual AKAPs to assemble distinct signaling machines, or macromolecular “signalosomes”, with the potential to produce unique local signaling environments. For example, by using a modified form of AKAR2 containing PKA- and PDE4D3-binding domains to mimic the recruitment of these enzymes by mAKAP, Dodge-Kafka et al. were able to recapitulate the attenuation of local PKA activity by tethered PDE4D3 within the mAKAP complex887. However, this effect was reversed by the activation of ERK5 MAP kinase, which directly phosphorylates and inhibits PDE4D3 and was shown to also be recruited to the mAKAP complex. PDE4D3 itself was further shown to recruit Epac1, which in turn inhibited ERK5 via the activation of Rap1 GTPase887, thus illustrating the ability of AKAPs to assemble tightly integrated signaling machines to provide context-dependent modulation of local PKA activity. Work by Willoughby and colleagues also revealed that AKAP5 directly interacts with and modulates the Ca2+ sensitivity of AC8, thereby generating a subpool of locally regulated AC8 with distinct activation properties55. Hoshi and coworkers similarly investigated the effect of AKAP anchoring on the PKC-mediated inhibition of the KCNQ2 subunit of the Mtype potassium channel888. Fusing the FRET-based PKC activity reporter CKAR directly to AKAP5 revealed that AKAP anchored PKC activity was accelerated compared with PKC activity measured at the general plasma membrane. Moreover, simultaneous FRET and M-current recordings revealed that AKAP-accelerated PKC activity was perfectly timed to match the kinetics of M-current inhibition888, demonstrating the ability of AKAPs to coordinate enzyme activity for more efficient downstream signaling. Subsequent work by Greenwald and colleagues also revealed that this coordination by AKAPs can both accelerate and amplify anchored enzyme activity889.
4.3.2. Molecular scaffolds in other signaling pathways
However, the importance of scaffolding is by no means limited to the PKA signaling pathway871,890. For example, macromolecular complexes have been shown to play a major role in regulating MAP kinase signaling891,892, and Matsunaga-Udagawa et al. previously used a FRET-based biosensor designed to monitor the binding of Ras and Raf-1 to investigate the role of the Shoc2 scaffold protein in regulating Ras/Raf/MEK/ERK pathway activity893. Knocking down Shoc2 led to significantly slower binding between Ras and Raf-1, thereby reducing EGF-induced MEK and ERK phosphorylation by approximately half. Computational modeling further confirmed that Shoc2-binding was required to accelerate Ras/Raf binding and promote downstream pathway activation893. Yoshiki and colleagues similarly used a plasma membrane-targeted FRET-based Raf-1 biosensor, Prin-Raf1, to reveal a role for Shoc2 in the Ca2+dependent regulation of Raf-1 activation894. As above, knocking down Shoc2 decreased plasma membrane recruitment and activation of Raf-1 in response to EGF or following co-activation using an engineered RasGEF and Ca2+ treatment. Based on this line of study, the authors were able to demonstrate that calmodulin mediates Ca2+-dependent Raf-1 activation by controlling Ras binding to Shoc2 894.
More recently, Tobias and coworkers set out to investigate the regulation of the atypical PKC isozyme PKCξ through the use of CKAR-fusion constructs and isoform-selective PKC inhibitors, wherein they were able to determine that binding by the scaffold proteins p62 and Par6 is responsible for both modulating PKCξ activity and also controlling its localization within the cell895. Kroon et al. also recently used a FRET-based sensor to examine local Rac1 activity in epithelial cells exposed to laminar flow and observed persistent Rac activation in the downstream regions of cells exposed to long-term flow stress896. The authors found that the RhoGEF Trio was required to maintain localized Rac activation, but by functioning as a scaffold protein rather than through the direct activation of Rac. Scaffold proteins are thus deeply integrated into the intracellular signaling machinery, and genetically encoded fluorescent biosensors continue to play a unique and crucial role in our efforts to unravel the spatial regulation of signaling activities by molecular scaffolds.
5. Computational Models and Fluorescent Biosensors
As illustrated by the preceding sections, the organization of signaling networks in both space and time can have a level of complexity that makes it difficult to intuitively deduce or infer the composition of a signaling network and the identities of key regulators. In these cases, computational models can be developed to better understand the underlying signaling mechanisms. Computational models are an important tool for quantitatively evaluating hypotheses and making predictions897. Fluorescent biosensors aid the development of computational models by providing a source of temporally and spatially precise measurements of signaling dynamics. Conversely, biosensors are useful tools for the experimental validation of computational predictions. In this section, we will discuss the ways in which quantitative, biosensor-based imaging and mechanistic modeling can be combined to provide new insights into signal transduction.
5.1. Computational Modeling Background
Computational models of cell signaling can be developed to quantitatively evaluate hypotheses, assist in the interpretation of experimental data, or explore conditions that may be infeasible to test experimentally. Here, we will focus on mechanistic models of dynamic cellular signaling, which are based on specific chemical reactions and approximations from first principles701. The development of computational models generally requires the iterative process of constructing a model based on a hypothesized pathway structure, generating model predictions, comparing these predictions with experimental data, and then refining the hypothesized model structure to more accurately represent experimental data898 (Figure 14A). Through this process, biological insights can be obtained from both model failures, which highlight hypothesized network architectures that are unable to capture the signaling dynamics, and successful models, which can be used to dissect the underlying signaling mechanics and generate novel hypotheses. As there are many reviews that focus on the more detailed aspects of computational modeling701,898–901, here we will provide only a brief description of the types of computational models commonly used and how fluorescent biosensor data are integrated into models.
Figure 14:
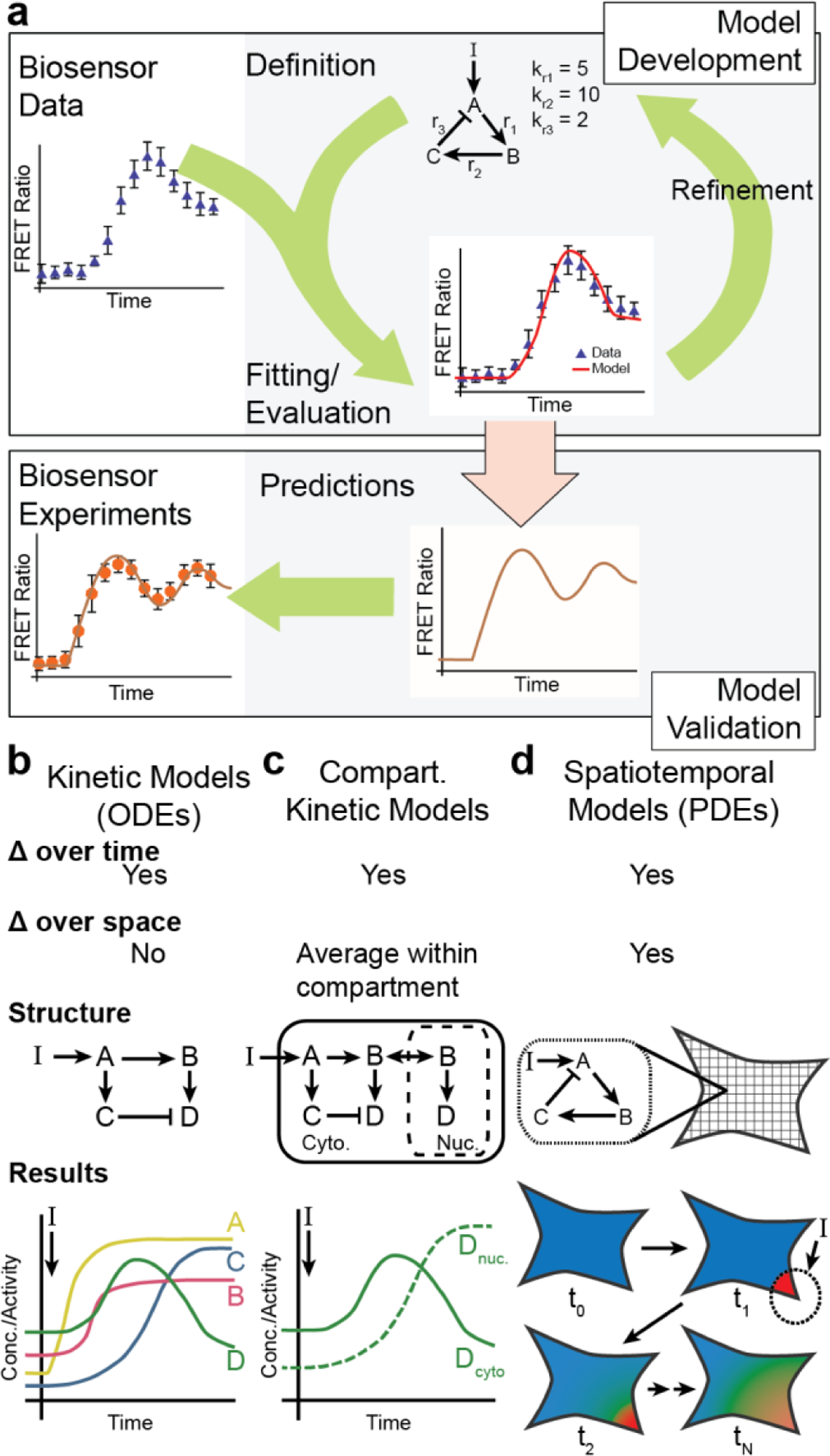
Integrated approach combining biosensor imaging and computational modeling.
A) Computational model development utilizes an iterative approach where the hypothesized structure of the signaling network is implemented into a computational model (top). The results of the computational model are then compared with experimental biosensor data, which serves to approximate unknown model parameters (model fitting) and identify aspects of the experimental data that the model is not capturing. This comparison is then used to refine the hypothesized model structure. This process is iterated until the model adequately reflects the experimental data. This model is then, in turn, capable of generating previously untested conditions (e.g., different stimulation, inhibition of a signaling enzyme) to generate new hypotheses (bottom). Biosensors then serve as a powerful tool to validate these model predictions. B&C) Kinetic computational models simulate changes in the activity and concentration of different signaling reactants as defined by the hypothsized connections within the signaling networks. These models do not directly approximate changes in space (B) but sub-cellular compartments can be defined where specific model species can exchange between compartments (C). D) Spatiotemporal models simulate the changes in signal transduction across both space and time; therefore, the model outputs the model species concentration or activity as it varies across the defined geometry and through time.
There are several types of computational models, and the type utilized is dependent on the system and hypothesis being tested. When evaluating the temporal dynamics of signaling, the most commonly used models are based on ordinary differential equations, or ODEs. Biochemical ODE models are based on differential equations that approximate the change in the concentrations or amounts of signaling molecules (often referred to as model species) over time, where reaction kinetics equations approximate the production and destruction of signaling molecules, as well as changes in state, such as enzyme activation or post-translational modification (Figure 14B). In most cell signaling ODE models, the reaction rate equations are based on mass action kinetics derived from first principles, but in some cases, empirical reaction rate equations are used, especially for components such as gated ion channels. Given an initial state of the signaling molecule concentrations (initial conditions), solutions to this system of equations are numerically approximated over small time increments to simulate changes in signaling dynamics. These ODE models are useful for studying the dynamics of a signaling compartment on average and therefore do not provide spatial information, but subcellular compartments can be defined in these models to evaluate the effect of spatial segregation on the overall signaling dynamics (Figure 14C). Additionally, ODE models are deterministic, in that for a given set of equations and initial conditions, the same solution will be reached every time, though in systems with very small volumes or numbers of reactants, stochastic methods may be necessary902.
When computational models need to consider dynamics in both space and time, partial differential equations (PDEs) are the most common modeling technique and can capture both reaction dynamics and diffusion. PDEs consist of equations that describe the rate of change in the concentrations of molecular species over time, similar to ODEs, while also approximating how these changes occur spatially due to both reactions and diffusion (Figure 14D). In some simplified cases, it is possible to solve these equations analytically837, but the reactions and geometries involved typically require the use of numerical approximation, where the spatial geometry is broken up into small sub-regions and, by taking small steps forward in time from an initial condition, the solution to the PDE is approximated across both space and time. This added complexity requires that in addition to specifying the initial conditions of the molecular species concentration over the whole spatial area, boundary conditions must be applied to define what happens to diffusing species at the edges of the defined geometry. When the geometries studied are cells, the boundaries are most often cell membranes, with the assumption of no diffusion (zero flux) occurring across the membrane, but it is also possible to use other boundary conditions such as a constant concentration or periodic boundary condition. Furthermore, PDE models can examine the temporal dynamics of spatial gradients in 2 or 3 dimensions in biologically relevant geometries such as a given cell shape, which can be integral to study the effects of cell shape and allow for comparison of the spatial changes in biosensor data. Again, in cases of small reaction volumes or low reactant numbers, it may be necessary to utilize a stochastic model architecture to evaluate hypotheses about the spatiotemporal dynamics and the effects that stochasticity may have on these systems903–905.
Finally, data such as signaling dynamics measured by using biosensor-based imaging are integrated into these models as constraints to approximate unknown parameter values and to validate model predictions. One important consideration when comparing experimental data with computational modeling results is that one needs to consider how to reconcile what the experimental measurement means in terms of model outputs. For example, when integrating Ca2+ biosensor dynamics, one must consider that the readout is a change in fluorescence or FRET ratio and that each biosensor has a different sensitivity to Ca2+. Some studies using fluorescent biosensors utilize calibrated biosensor responses based on known standards to relate the biosensor output to quantitative concentrations384,906. While this is possible for biosensors that measure second messenger concentrations, calibration can be time consuming, and for biosensors that measure enzymatic activity, this calibration is not as straightforward given the dependence on enzyme and substrate (biosensor) concentrations and activity levels. Instead, a more common approach is to include the biosensor in the model directly, which requires adding more reactions and species to the model. Furthermore, it is often beneficial for model fitting to experimentally include positive and negative control stimuli to normalize the biosensor response to its dynamic range907.
5.2. Examples of the Integrated Approach
5.2.1. Analysis of temporal dynamics
A combination of computational modeling and fluorescent biosensor-based imaging can be used to study how the kinetics and organization of individual reactions shape the dynamics of signal transduction. Depending on the scope and scale of the study, this approach can be informative for both small and large signaling networks. For example, the PKC biosensor CKAR481 was used in conjunction with computational modeling to evaluate how enzyme-substrate tethering of scaffold proteins alters the kinetics of phosphorylation by kinases889. Biosensor imaging showed that scaffold tethering accelerated and amplified phosphorylation by PKC, and the computational model used these data to quantitatively evaluate a proposed kinetic mechanism to explain the effects of scaffold tethering. This new mechanistic model was able to fit well to the experimental data and was used to predict that the scaffolded enzymatic reaction would be insulated from inhibition from certain types of inhibitors. In turn, the fluorescent biosensors were then used to validate this model prediction by showing that, indeed, AKAP7 tethering insulated phosphorylation by PKC from inhibition by substrate-competitive inhibitors. This example highlights how fluorescent biosensors can provide the data necessary to study the kinetics of individual reactions in computational models and validate the predictions that arise from these models. Similarly, the high temporal resolution afforded by fluorescent biosensors can be indispensable for computational models used to study signal propagation through a network in response to a given stimulus.
While the signaling network motifs described in Section 3.2, such as negative feedback, are known to lead to dynamics such as adaptation and oscillations, many signaling networks contain several different feedback and feedforward loops which can make it difficult to ascertain which aspects of the network are most important. As discussed earlier, ERK can exhibit a transient response to some stimuli, which requires at least one negative feedback mechanism, and fluorescent biosensors have been combined with computational models to understand how these dynamics arise. When the first fluorescent biosensor for ERK activation, Miu2, was developed, the authors built a computational model that aimed to capture the responses of ERK to EGF stimulation448. Within this model, they observed that the magnitude of the ERK response was highly sensitive to the concentration of Raf in silico. This was experimentally validated by knocking down c-Raf and observing that MEK and ERK phosphorylation in response to EGF stimulation decreases with c-Raf knockdown. While computational models of ERK activity had been developed previously, the improved temporal resolution of the fluorescent biosensor compared to western blots improved the accuracy of the model. It should be noted that computational models may not perfectly capture all the observed dynamics, but these discrepancies can provide important clues to identify less understood components of a signaling pathway. For example, Ahmed and colleagues turned to computational models to understand why ERK nuclear localization in response to PDGF stimulation exhibited a much stronger adaptive response than ERK nuclear activity, which is also a slow process and lags the peak in nuclear translocation908. They used EKAR444 and mCherry-labeled ERK2 to simultaneously measure both subcellular ERK activity and ERK shuttling between the cytoplasm and nucleus in response to stimulation. They hypothesized that the competitive interactions between ERK and its different substrates may be responsible for this discrepancy between activation and translocation of ERK and its kinase activity. To test this hypothesis, they developed a computational model that explicitly models the association, phosphorylation and dissociation of ERK substrates, instead of the commonly used Michaelis-Menten enzyme kinetics approximation. The simulations from this new model matched their imaging data, suggesting that the competitive interactions between ERK and its many substrate proteins may limit the amount of ERK available to phosphorylate EKAR in the nucleus, an effect commonly referred to as buffering. Furthermore, this model suggested that most of the nuclear ERK is bound by substrates and also protected from dephosphorylation during the initial phase of the response, which was validated experimentally. These examples highlight the back-and-forth nature of computational model-based examination of fluorescent biosensor dynamics and experimental evaluation of model predictions and how this work can provide insights into the critical mechanisms that underlie the dynamics of cellular signal transduction (Figure 14A).
5.2.2. Evaluation of spatial compartmentalization
The spatial segregation of cellular signaling components into microdomains has been identified as an essential aspect in shaping signaling dynamics, as discussed in Section 4, and the combination of fluorescent biosensors and computational models has been useful in examining how this compartmentalization affects signal transduction. Computational models studying spatial organization of signaling generally come in two forms, compartmentalized kinetic models and models that explicitly consider the spatial gradients of signaling molecules. For the purposes of this review, compartmentalized models are generally the kinetic ODE models discussed before but contain separated compartments that have their own composition and concentration of signaling molecules, and indirectly account for diffusion by incorporating equations to approximate exchange of signaling molecules between compartments. The compartments can be defined to represent organelles, such as the nucleus and mitochondria, subcellular locations, like the plasma membrane or cytoplasm, and signaling microdomains, such as scaffold proteins like AKAPs. Accounting for this spatial organization within models can improve their accuracy and help answer questions about the role of this complex organization, but these models do not explicitly approximate spatial changes in signaling activity; for this, models that account for changes in concentration over both time and space, such as PDE models, are required. Because PDE models can incorporate factors such as cell shape and diffusion coefficients, these models are useful for examining how different factors can lead to and affect the spatial heterogeneity of signaling molecules. Genetically encoded fluorescent biosensors have been critical for providing data on signaling dynamics within microdomains and spatial gradients of signaling activities (see Section 4), and these unique datasets have been instrumental in informing and validating spatiotemporal computational models. As discussed in Section 4.2.2, the spatiotemporal regulation of the cAMP/PKA signaling pathway is intriguing because the tethering of PKA by AKAP scaffolds creates unique signaling microdomains in different locations throughout the cell, whereas its activator cAMP is a diffusible second messenger. This discrepancy had led to two lines of questioning that have benefited from the combination of fluorescent biosensors and computational modeling: 1) how do AKAP-mediated PKA microdomains shape signaling dynamics and 2) what regulates the spatial organization of cAMP concentrations to allow compartmentalized activation by cAMP?
Compartmentalized kinetic models of cAMP/PKA signaling have been utilized to evaluate how the spatial organization of this signaling pathway enables the coordination of several different, sometimes contradictory, phenotypic outcomes. In cardiomyocytes, stimulation of the β-adrenergic receptor as part of the fight-or-flight response leads to the activation of PKA and the phosphorylation of several downstream targets to increase cardiac output909. The diverse array of PKA substrates contributes to several facets of excitation-contraction coupling (ECC), which can sometimes have competing effects on overall cardiac function721. To help comprehend how the different microdomains of cAMP and PKA signaling each contribute to the overall signaling outcome, several computational models have been developed898. For example, recent work by Surdo et al. used both targeted fluorescent biosensors and computational modeling to study how cAMP compartmentalization differs between microdomains and how these differences impact cardiac function287. Their work focused on three PKA microdomains: AKAP79, which coordinates PKA regulation of cAMP production by ACs and Ca2+ influx through the L-type voltage-gated Ca2+ channel (LTCC); AKAP18δ, which localizes PKA to the sarcoplasmic reticulum (SR) to promote the phosphorylation of phospholamban (PLB) to increase the rate of Ca2+ reuptake into the SR; and the myofilament-localized troponin complex, where phosphorylation of the TpnI subunit by PKA reduces its affinity for Ca2+. While PKA activity within the AKAP79 microdomain and phosphorylation of PLB at AKAP18δ increases cardiac Ca2+ amplitude and contraction, phosphorylation of TpnI has the opposite effects of reducing myofilament Ca2+ sensitivity and limiting contraction. By targeting their cAMP biosensor, CUTie, to these different microdomains, Surdo et al. observed that the cAMP response to β-adrenergic stimulation was dampened in the TpnI microdomain compared with the other two microdomains. In order to evaluate how this difference affects cardiac ECC, they expanded a previous computational model to evaluate how this regulation may optimize the tradeoffs of TpnI phosphorylation. They used this model to predict how decreased cAMP may affect the regulation of other myofilament proteins, namely, myosin binding protein C (MyBPC) and titin, by PKA. Interestingly, their model revealed that reducing PKA phosphorylation of TpnI without reducing the phosphorylation of MyBPC and titin resulted in increased myocyte contraction compared with uniform cAMP stimulation across all microdomains. This suggested that this regulation may be specific to TpnI, and indeed, this was experimentally validated by western blots showing that in response to an intermediate dose of isoproterenol, TpnI phosphorylation was reduced but MyBPC phosphorylation was not. This study shows how the targetability of fluorescent biosensors can provide quantitative data within specific compartments, which in turn can yield more detailed and accurate computational models.
On the other hand, the transport and diffusion of signaling molecules throughout the cell can lead to a spatial heterogeneity that is necessary for many critical cellular functions such as migration and cell division910. Fluorescent biosensors are capable of measuring the dynamics of these signaling gradients, and computational models that study the explicit spatial dynamics of signal transduction have helped identify what factors are critical in shaping these complex phenomena. In contrast to the aforementioned compartmentalized models of cAMP/PKA signaling that examined the consequences of cAMP diffusion and PKA activity, several studies have utilized fluorescent biosensors and spatial computational models to understand how cAMP, as a small diffusible second messenger, can be compartmentalized (reviewed in ref. 845). There are 6 (non-exclusive) proposed mechanisms for the compartmentalization of cAMP: localized production by AC, localized degradation by PDEs, physical barriers, cAMP buffering, cell shape and cAMP export845. The ability to control and perturb these mechanisms within computational models has been indispensable in testing these hypotheses, but these models are built on and validated by the experimental evidence afforded by fluorescent biosensors. For example, the role of cell shape in the formation of cAMP gradients in neurons was studied through the development of a spatial computational model along with the cAMP biosensor Epac1-camps846. When neurons expressing Epac1-camps285 were stimulated with a β-adrenergic agonist, cAMP accumulated in dendrites with little change in the cell body846, even though β-adrenergic receptors have been shown to localize to both the dendrites and cell body in vivo911,912. This fluorescent biosensor data was used to constrain and test model parameters within a defined cellular geometry that mimics the experimentally observed cell846. Then, to evaluate the role of cell shape, this parameterized model was tested in an idealized geometry where cellular characteristics that define the shape of a neuron, such as axon diameter, were systematically varied and the effects on cAMP gradients were evaluated. In their model, Neves et al. found that cAMP gradients only formed in axons with smaller diameters, whereas downstream targets such as PKA and MAPK were able to form gradients in larger-diameter axons due to slower diffusion. Later work by Li et al. used a similar approach to examine how the branching of neuronal dendrites affects the transduction of cAMP signaling from the dendrites to the nucleus913. Their model examined how small, localized cAMP production in dendrites, as would occur through confined activation of the dopamine receptor, would be translated to nuclear signaling by the downstream activation of PKA and its substrate DARPP-32. As the location of cAMP release was moved down the dendrite and further away from the nucleus, one would also expect the extent of nuclear PKA activity to decrease monotonically. Surprisingly, the model predicted that the extent of nuclear signaling was higher when cAMP was released in a mid-dendrite location than if cAMP was released in the larger trunks of the dendrite, nearer the cell body. Within their computational model, they were able to vary several dendrite shape parameters, such as dendrite diameter and branching, to test different mechanisms for this observed phenomenon. This analysis led them to postulate that larger-volume dendrites caused the cAMP to become diluted faster, whereas intermediate-size dendrites kept cAMP more concentrated but did not constrain the diffusion of downstream signaling proteins as much as the more distal and smaller dendrite branches. Finally, these computational model predictions were validated using the PKA biosensor AKAR3475, where localized uncaging of cAMP in the medial regions of neuronal dendrites showed a greater nuclear biosensor response compared with cAMP uncaging in the proximal and distal regions of the dendrites. These two examples highlight the effects that cell shape can play in the distribution of cAMP signaling throughout the cell.
While these studies show that cell shape can affect the rate of diffusion, even more fundamentally, the diffusion coefficient of cAMP itself has been questioned both experimentally and computationally837,844,848. For example, using the PKA activity reporter AKAR2476, Saucerman et al. observed that the activation of PKA in the cytosol was slower than expected when cAMP production was stimulated at the plasma membrane compared with cAMP uncaging in the cytosol849. To evaluate the underlying cause of this temporal delay in PKA activation, they developed a computational model that included both cAMP buffering by binding to PKA and compartmentalized PDE activity849. In their model, they found that a much slower cAMP diffusion rate was required when cAMP production was stimulated at the plasma membrane than when cAMP is directly uncaged in the cytosol, suggesting the possible existence of a barrier to cAMP diffusion between the plasma membrane and cytosol849. Similar differences between submembrane and cytosolic cAMP were observed using the cAMP biosensor H30 (aka CFPEpac(δDEP-CD)-YFP 292) in HEK293 cells, but these differences were hypothesized to arise from compartmentalized PDE activity rather than a diffusional barrier914. A stochastic spatial model constructed by Oliveria and colleagues was developed to examine these two hypotheses and revealed that PDE compartmentalization, as opposed to impeded cAMP diffusion, was the key mechanism for cAMP compartmentalization in HEK293 cells 842. Nevertheless, the rates of intracellular cAMP diffusion remain a highly debated area of research, and spatiotemporal dynamic data derived from fluorescent biosensors will be critical in building and testing the spatial signaling models needed to evaluate which mechanisms are most important for establishing signaling gradients.
5.2.3. Analyses of cellular heterogeneity.
By virtue of their ability to reveal single-cell dynamics across a population of cells, fluorescent biosensors can provide information on how communication between cells can affect signal transduction. For example, stochastic pulses of ERK activity were observed in cells expressing the ERK biosensor EKAREV414, and the frequency of these pulses of activity depended on the cell density and would propagate to neighboring cells743. Furthermore, Aoki et al. observed that while basal ERK activity decreased with increasing cell density, the frequency of the stochastic ERK pulses did not monotonically decrease with increasing density. Rather, the frequency exhibited a bell-shaped curve with respect to density, with fewer ERK pulses at both high and low cell densities. Additionally, this biphasic dependence on cell density was correlated with the cell proliferation rate, suggesting that the ERK pulses may act as a rheostat to regulate cell number. In order to evaluate how biological noise, feedback loops and paracrine signaling shape the development of these pulses of high ERK activity, Aoki and colleagues developed a multicellular computational model of ERK activity. In this model, Raf, the upstream activator of ERK, becomes activated through canonical stimulation, noise or paracrine signaling, with both positive and negative feedback loops regulating Raf activity. The model parameters were adjusted to recapitulate the shape and frequency of the experimental biosensor data at different cell densities, after which the simulations suggested that noise in the ERK signaling pathway increased with cell density. This model predicted that only about 20% of the ERK activity pulses can be attributed to paracrine propagation, whereas most of the ERK pules are due to the noise in the system. Indeed, post hoc analysis of the experimental data showed similar fractions of the cell-to-cell propagation-driven ERK activity pulses as predicted. This model suggests that while paracrine signaling has some effects on stochastic ERK pulses, the frequency of these pulses is primarily driven by some yet unidentified cell density-dependent regulation of the ERK signaling pathway.
In another study of the paracrine signaling of ERK, Handly et al. utilized both fluorescent biosensors and a mathematical model to examine how communication between neighboring cells affects the sensing and generation of chemotactic gradients in wound healing915. Cells that are damaged can either actively or passively release ATP from their internal stores, which can activate purinergic receptors and act as a damage-associated molecular pattern (DAMP)916. In the epithelial cells used in this study, ATP activation of the P2Y receptor results in a Ca2+ transient and the release of EGF, which in turn activates EGFR and ERK in a paracrine fashion915. While the Ca2+ response to ATP was variable, the variability of the ERK response was observed to decrease as cells clustered together more densely. Thus, Handly et al. hypothesized that optimal paracrine signaling can lead to optimal localized population averaging, which can provide adequate reduction in variability and increase the SNR of the wound response. To examine this quantitatively, they developed a computational model that incorporated the secretion, diffusion and integration of paracrine signaling to examine the effects of these processes on the variability of the response. When considering a wound assay and the importance of gradient formation for directional guidance, their model predicted that if the effect area of a paracrine signaling ligand were too large, it would reduce the signaling fidelity and thus reduce the SNR by decreasing the signal. The model predicted that an effect distance of approximately 100 μm would maximize the SNR for this system by limiting the noise of cell-to-cell variability while still allowing for enough of a spatial gradient to maintain a strong signal. Indeed, when Handly and colleagues experimentally evaluated this model hypothesis using a novel microfluidic device, the results indicated that on average the extracellular ligand has a proximal effect distance of approximately 100 μm. As we continue to better understand the mechanics underlying cellular signaling dynamics, we will continue to see that the use of fluorescent biosensors may require evaluating how individual cells interact within a population of cells, and computational models will play a key role in helping us understand these interactions.
5.2.4. Information theory
While cellular communication is often thought to act like a volume knob on a speaker, where turning the knob up or down directly results in an increase or decrease in volume, cellular signaling has to compete with a high level of biological variability and noise. Recently, work aiming to understand how biological signaling networks can faithfully process communication in noisy environments via imprecise signaling by chemical reactions has begun to be addressed through fluorescent biosensors and mathematical analyses. One framework that is used to address this question is Shannon information theory, which is a mathematical field of study originally developed to study communication over man-made channels but has been extended to study biological communication917. In information theory, information is conceptually defined as the ability to distinctly identify the state of an input or signal for a given output or response, and vice versa917. In biological terms, this usually entails studying how many distinct levels of stimulus concentrations (e.g., receptor agonists) can be conveyed through a cellular signaling network in order to yield distinct responses (e.g., different gene expression profiles)917. One early example of information theoretic analysis of mammalian signaling was performed by examining the TNF/NF-κB signaling pathway using immunofluorescent labeling of NF-κB and GFP reporter genes918. Interestingly, this work estimated that the TNF/NF-κB signaling pathway is at most capable of distinguishing only two TNF concentrations918. Fluorescent biosensors were used to extend this analysis beyond measurements at a single timepoint to time-course studies. Selimkhanov and colleagues used EKAREV414 targeted to the nucleus to quantify the dynamics of MCF10a cells responding to an array of EGF concentrations to test the hypothesis that temporal dynamics may be able to convey more information 919. Through the use of automated image acquisition and analysis, they were able to quantify the ERK biosensor dynamics in over 825,000 individual cells and showed that when temporal dynamics are considered, instead of a single timepoint, this signaling network is capable of encoding more information from an information theory standpoint 919. A similar analysis of Ca2+ responses to ATP stimulation and the NF-κB nuclear translocation response to LPS stimulation revealed that including the temporal dynamics for these signaling pathways similarly increased the information carrying capacity. These studies suggest that cells may be able to discern more distinct levels of stimulation if the temporal dynamics are integrated into the down-stream signaling outcome. As fluorescent biosensors continue to improve and expand to new signaling domains, these types of analyses will be fundamental in understanding how cells have optimized their ability to function and communicate in a noisy world.
6. Pushing the Field Forward
While genetically encoded biosensors have proven incredibly useful for studying signal transduction, there are several areas in which biosensor-based imaging is highly desirable yet remains a largely unmet need. Below, we highlight several of these areas and discuss both the current obstacles and the ways in which biosensor designs can be and are being adapted to meet these needs and enable more diverse applications.
6.1. Improved Resolution
As discussed in Section 4.3, signaling activities are often organized into discrete microdomains or nanodomains. The submicroscopic size of these functional domains makes them difficult to characterize through biosensor imaging based on current diffraction-limited methods. Nevertheless, a number of super-resolution imaging methods have been developed in recent years (reviewed in refs. 913, 914) that enable virtually diffraction-unlimited imaging of subcellular structures (i.e., “fluorescent nanoscopy”). Thus, one strategy for achieving the direct visualization of signaling activity microdomains is to combine existing fluorescent biosensors with a compatible super-resolution imaging modality.
Existing biosensor designs are perhaps most compatible with super-resolution imaging methods that utilize patterned illumination, such as stimulated emission depletion (STED)922 or structured illumination microscopy (SIM)923. For example, STED involves exciting a diffraction-limited spot while using a second laser to deplete emission within an Airy ring shape to ultimately produce a sub-diffraction-sized spot and allow fluorescence imaging below the diffraction limit920. Because this method primarily requires a highly photostable fluorophore, STED-based biosensor imaging is fairly straightforward and has been used, for example, with the intensity-based H2O2 biosensor HyPer2 924. The cpYFP in HyPer2 was found to be more photostable than TagYFP, EYFP or citrine, thus making it a good candidate for STED imaging. In contrast to the single molecule localization-based super-resolution methods, e.g., stochastic optical reconstruction microscopy (STORM)925 and photo-activation localization microscopy (PALM)926, STED is a super-resolution technique that maintains the fluorescence intensity information in the final image, which can be used as the biosensor readout. With this improved spatial resolution, Mishina and colleagues observed H2O2 microdomains that were as small as 100–200 nm across. Similarly, a recently developed pH biosensor, SRpHi, which is composed of an FP and an organic fluorescent dye that have differing pH sensitivities, was used in conjunction with two-color STED to perform ratiometric super-resolution imaging927. This biosensor was used to study the pH of endosomes, as many structures in the endocytic pathway are <250 nm in size, making it difficult to distinguish closely packed structures. While SRpHi is not a fully genetically encoded fluorescent biosensor, given its requirement for a covalently linked fluorescent dye, the ratiometric nature of the biosensor helps reduce concentration artifacts, and its cellular delivery is facilitated by the endocytosis process itself.
On the other hand, several new biosensors have been developed that are explicitly designed for super-resolution imaging. As alluded to previously (see Sections 2.2.2 and 2.4.3), these biosensor designs largely utilize photoswitchable FPs in conjunction with localization-based super-resolution imaging methods such as PALM926 or stochastic optical fluctuation imaging (SOFI)23,928. In particular, a few BiFC-based strategies have been used to visualize PPIs in super-resolution, including BiFC-PALM based on split versions of either the photoactivatable FP PA-mCherry929 or the green-to-red photoconvertible FP mEos3.2 74, as well as reconstituted fluorescence-based SOFI (refSOFI), which utilizes the photoswitchable FP Dronpa76. However, BiFC offers only a limited ability to track dynamics processes, for instance, due to the relatively slow kinetics of FP reconstitution. RefSOFI may be advantageous in this regard, however, as fluctuations are only observed when overall FP intensity is dim, meaning that super-resolution information can be obtained without having to wait for complete FP maturation76.
Nevertheless, because BiFC is fundamentally irreversible, other designs are needed to fully capture dynamic biochemical processes in super-resolution, such as the recently developed FLINC-based biosensors, which utilizes a molecular switch to control FP proximity and reversibly modulate fluorescence fluctuations447 (discussed in Section 2.4.3). Yet while FLINC is indeed capable of capturing biochemical activity dynamics, such as changes in PKA activity, the temporal resolution of this method is still somewhat constrained. Specifically, each superresolution image in a time-course is itself derived from a series of fluctuation images collected under high-intensity illumination, and the need to collect enough information to construct superresolution images at each time point must be balanced with the need to minimize photobleaching across the experimental time-course. In the current implementation, this balance is achieved by incorporating periods of recovery between sets of imaging frames447. However, improvements in imaging technology and FP engineering will undoubtedly yield increases in temporal resolution, while new biosensor designs will continue to emerge that offer additional strategies for performing dynamic, super-resolution activity mapping.
6.2. Multiplexed Biosensor Imaging
Signaling networks are complex and feature a high degree of connectivity, often integrating several different signaling pathways. The ability to track multiple signaling activities within the same reference frame would therefore be tremendously useful for unraveling this complex web. Multiplexing refers to the ability to send or receive several messages simultaneously, which for fluorescent biosensors entails the ability to measure multiple biosensor readouts with respect to the same frame of reference. In general, this means simultaneously measuring the responses from two or more biosensors within the same cell, though in so-called “computational multiplexing”, measurements obtained from different biosensors expressed in separate cells can be linked through a common fiduciary marker, such as the movement of the plasma membrane930. Nevertheless, the most significant physical constraint on multiplexing remains the limited spectral space available for imaging multiple biosensors in the same cell930–932. Thus, a considerable segment of biosensor development is devoted to reducing the spectral space occupied by fluorescent biosensors.
One strategy to save spectral space for potential multiplexing applications is to expand the engineering of single-color, intensity-based fluorescent biosensors to monitor signaling activities outside the well-developed areas of Ca2+ and voltage biosensors, as exemplified by the recent development of single-color sensors for NADH335–337, cAMP180,183, glutamate166 and certain kinases112 (Table 1). Meanwhile, alternative methods are also being devised to convert dual-wavelength FRET-based biosensors into single-color reporters. First, biosensors have been developed to take advantage of donor fluorescence quenching resonance energy transfer (FqRET), which specifically measures the decrease in donor fluorescence that occurs as a result of energy transfer (Figure 15A). These sensors exploit the fact that the acceptor need not be an FP, as any light-absorbing protein can serve as a FRET acceptor provided it satisfies the conditions for energy transfer. Thus, Ganesan et al. were able to develop a ubiquitination biosensor in which GFP was paired with a YFP-based “dark” acceptor, Resonance Energy-Accepting Chromoprotein (REACh), wherein FqRET allows what would normally be a dual-wavelength FRET probe to provide a single-color readout610. The use of this dark acceptor-based FqRET has since been expanded to other signaling pathways and colors, including a blue cGMP biosensor309, red and green CaMKII biosensors426,428, and red Cdc42 and RhoA biosensors426.
Figure 15:
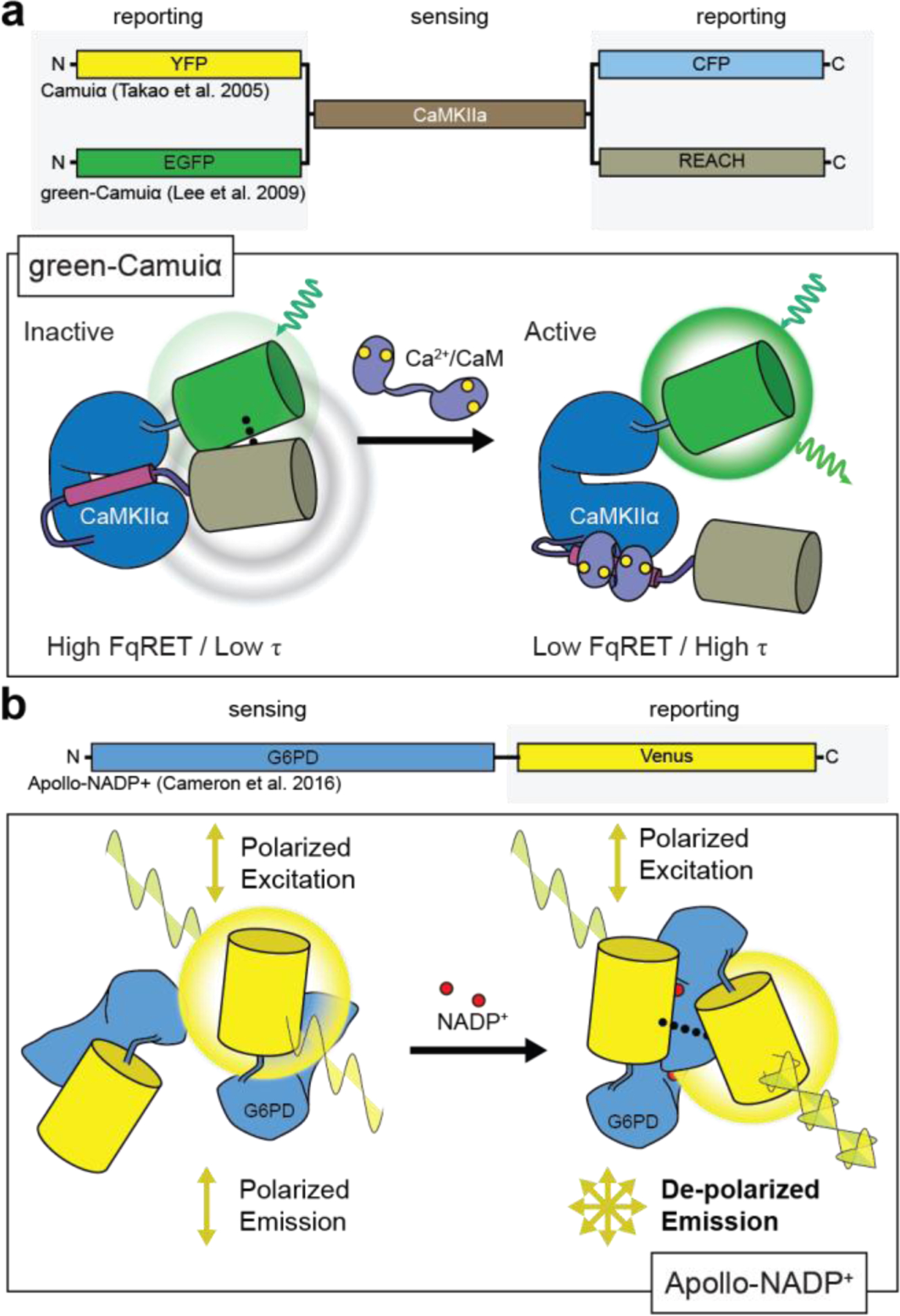
Single-color FRET-based methods.
Fluorescence quenching resonance energy transfer (FqRET)-based CaMKIIα biosensor green-Camuiα428. In the inactive state, the energy from excited EGFP is non-radiatively transferred to the dark acceptor REACh, which then dissipates that energy without emitting a photon. Upon activation, the conformational change moves REACh away from the EGFP, leading to increased EGFP emission. FqRET can also be quantified by fluorescence lifetime imaging (FLIM), where the lifetime, τ, is low in the high-FqRET state and vice versa. B) Homo-FRET-based NADP+ biosensor Apollo-NADP+338. Upon binding NADP+, G6PD dimerizes and thus allow FRET between the two FPs. Fluorescence polarization microscopy can be used where a polarized excitation source will only excite FP in the appropriate dipole orientation, resulting in the emitted photon also being polarized. Conversely, when FRET occurs between these two FPs, the emitted photons exhibit a mix of polarizations and thus decrease the polarized fluorescence signal.
Similarly, homo-FRET, in which excited-state energy is transferred between two fluorophores with similar or identical spectra rather than from a shorter-wavelength fluorophore to a longer-wavelength fluorophore, can also be used to generate single-color biosensors503. Notably, because the donor and acceptor can no longer be distinguished by their emission wavelengths, visualizing the responses from homo-FRET-based biosensors requires measuring changes in fluorescence anisotropy, wherein the loss of polarized fluorescence emission indicates an increase in energy transfer933 (Figure 15B). Two such homo-FRET biosensors, namely, Apollo-NADP+ 338 and mCherry-Akt-PH503, have been successfully multiplexed with biosensors for H2O2 and Ca2+, respectively. Finally, ddFP-based biosensors represent another potential strategy for developing single-color fluorescent biosensors for multiplexed imaging, as evidenced by several proof-of-concept sensors described by Alford et al.364 (see Section 2.3.3.2.4). Thus, as the arsenal of fluorescent probes continues to expand, fluorescent biosensors will increasingly be developed with multiplexing in mind.
6.3. Applications to More Complex Biological Systems
While a majority of the studies discussed in this review have utilized genetically encoded fluorescent biosensors in more in vitro settings, the application of fluorescent biosensors to visualize cellular processes in vivo is becoming increasingly important. In vivo biosensor imaging can provide deeper insights into the behavior of signaling pathways under physiologically relevant conditions934, can be used to study responses to more complex stimuli156, and can be used to correlate signaling dynamics with organ physiology measurements or organismal behaviors935. However, in vivo biosensor imaging faces a number of challenges related to autofluorescence from endogenous cellular components, as well as absorption and scattering of the excitation and emission light936.
As described in Section 2.4.1, mammalian cells and tissues exhibit much less absorption and scattering of light in the NIR region of the spectrum685, and several FPs that are excited and emit in this spectral range have in fact been developed to take advantage of this phenomenon9,937. Fluorescent biosensors that utilize these NIR FPs have thus far been developed for monitoring caspase activity531 and cell cycle progression209,210. For example, the infrared caspase 3 reporter iCasper (Figure 7 and Section 2.4.1) was used to quantify apoptosis in vivo throughout Drosophila development and show that apoptotic cells were spatiotemporally correlated with certain developmental steps531. NIR FPs also enable the development of far-red-shifted FRET sensors, and while an entirely NIR FRET pair has yet to be reported937, FRET between the farred FP mKate2 and the NIR FP iRFP has been successfully utilized in the development of a proof-of-concept caspase-3 biosensor530.
Pending the more widespread development of NIR fluorescent biosensors, a more general approach to achieving higher-quality biosensor imaging in tissues has been to use multi-photon imaging, which is based on the absorption of long-wavelength photons that reach the fluorophore simultaneously (e.g., within less than a femtosecond), thereby triggering excitation and normal fluorescence behavior938,939. Multi-photon imaging reduces background fluorescence due to scattering through the use of longer-wavelength illumination, as well as by reducing the imaging volume of excited fluorophores, thereby facilitating in vivo imaging (for more background on multi-photon imaging see ref. 940). For example, by using two-photon excitation to image GCaMP6 in mouse visual cortex V1 neurons in vivo, Chen and colleagues were able to record Ca2+ transients in the brains of mice presented with different visual stimuli156. While the use of two-photon imaging with this and other single-color, intensity-based fluorescent biosensors, such as the glutamate biosensor iGluSnFr941, has been fairly straight forward, utilizing multi-photon imaging in conjunction with FRET-based biosensors may require some additional biosensor optimization. For example, in order to develop a two-photon-compatible version of AKAR, Tao and colleagues replaced the Cerulean in AKAR4 with mTurquoise to improve the two-photon excitation profile470. Another strategy to facilitate in vivo imaging of genetically encoded biosensors may be to forego the use of excitation light altogether by using luminescent proteins such as the Nano-Lantern chimera developed by Saito et al. (discussed in Section 2.3.3.3), which has been shown to be compatible with whole-body in vivo imaging in mice276. Furthermore, the development of enhanced Nano-Lantern color variants containing red-shifted FPs is expected to yield further improvements through increased tissue penetration by the emitted light377. Recently, Iwano and colleagues used directed evolution of FLuc to generate a luciferase Akaluc that utilizes the infrared substrate AkaLumine-HCl. The akaluc and AkaLumine-HCl pair offers a number of exciting features, including greater catalytic activity and expression levels than the parental pair, excellent bioavailability, and low toxicity679. These improvements, as well as the infrared emission, made it possible to detect single cells in deep tissues of living mice and measure long-term bioluminescence in living marmosets. In the future, integration of Akaluc into a biosensor should enable the in vivo quantification of signaling dynamics in animals while they are free to complete complex tasks.
Finally, genetically encoded fluorescent biosensors may potentially be combined with photoacoustic imaging, in which an excited fluorophore does not emit a photon but instead undergoes thermoelastic dissipation, which can induce an ultrasonic pressure wave942. Although photoacoustic imaging has a lower spatial resolution than fluorescence imaging, it can enable much deeper tissue penetration943. Several FPs have already been utilized in photoacoustic imaging due to their known absorption spectrum, but FPs with low quantum yields tend to work much better because energy that is released as a photon cannot be dissipated thermoelastically943. Interestingly, Li et al. discovered that FRET can be utilized for photoacoustic imaging when an FP such as EGFP is coupled with the photoacoustic-optimized dark fluorescent protein tdUltramarine2, as shown through a proof-of-concept protease biosensor944. While genetically encoded photoacoustic biosensors for signal transduction have not been demonstrated in vivo, these new types of biosensors could enable even deeper in vivo imaging in the near future.
6.4. Translational applications of genetically encodable fluorescent biosensors
As fluorescent biosensors have both expanded in diversity and increased in sensitivity, researchers in both academia and industry have begun to translate genetically encodable biosensors into high-throughput screens and clinical assays. The strengths of fluorescent biosensors, namely, their ability to quantify live-cell kinetics and their subcellular targetability, make them desireable tools for high-throughput screening, but there are often several obstacles that must be overcome to have an efficient and selective high-throughput assay (reviewed in ref. [24357625]). Often one of the most important requirements is a high signal-to-noise ratio (SNR). Fortunately, improvments in both biosensor design and fluorescent protein characteristics over several generations of development can often yield biosensors that exhibit high enough SNR for high-throughput screens. For example, Allen and colleagues demonstrated in a fluorescent platereader based assay that while the Z’ factor of the 2nd generation PKA biosensor AKAR2 [16177793] was less than the industry standard of 0.5, the 3rd generation AKAR3 [16895723] biosensor had a Z’ factor of 0.84 [17163774]. More recently, Zhao et al. developed a NAD+/NADH biosensor, named SoNAR, and used it to screen for metabolism based anti-tumor compounds [25955212]. In a screen of several compound libraries against a lung cancer cell line expressing SoNAR, they found that, surprisingly, a reported AKT inhibitor KP372–1 caused the greatest increase in the NAD+/NADP ratio and was selectively toxic to cancer cell lines. This discovery prompted further probing into the mechanism by which KP372–1 altered metabolism. Using the fluorescent biosensors Hyper [16554833] and roGFP1 [14722062] to measure H2O2 and disulfide redox state, respectively, they showed that KP372–1 potently induced oxidative stress [25955212]. Further work suggests that the NAD(P)H-dependent reactive oxygen species generating enzyme NQO1 modulated both the oxidative stress and the cell toxicity stimulated by KP372–1. This work is one example of how fluorescent biosensors can be powerful tools for drug discovery through both the identification of lead compounds through high-througput screens and the characterization of hits after their identification. [characterization of the effects of the hit compounds on multiple signaling pathways – add the Schultz ref]
Alternatively, the ability to use fluorescent biosensors in higher throughput methods has enabled their use to examine signaling networks from a systems biology perspective. For example, Bakal and colleagues performed an RNAi screen on JNK activity in Drosophila cells expressing the dJUN-FRET biosensor [18927396]. While the initial RNAi screen did provide insight into genes that were essential for basal levels of JNK activity, their approach became even more informative when they extended this work to a combinatorial screen where the RNAi library was screened against cells that had been sensitized by RNAi knockdown of known regulators of the JNK pathway (e.g., ERK, MLK, puckered). This combinatorial approach was therefore capable of providing evidence of the “connectedness” of less known regulators in the signaling network, and when integrated computationaly with bioinformatic and proteomic approaches such as the NetworKIN algorithm [17570479], this data provided a systems level map of the JNK pathway in Drosphila [18927396]. On the other hand, the growing number of fluorescent biosensors means that it is possible to take a systems-level view of signaling by directly examining the responses from several different biosensors to a given stimulus. Taking advantage of this, Kuchenov et al. developed a FRET-based multi-parameter imaging platform where an array of fluorescent biosensors is reverse-transfected into adherent cells in a spatially definied manner [27939899]. In their initial work with this platform, they quantified the simultaneous responses from 40 different fluorescent biosensors to different stimuli of the ERK signaling pathway. When examining the signaling crosstalk between EGF and IGF-1 stimulation using this multiparametric view, they were able to not only idenitify differences in the downstream effects of these two signaling pathways but also query what types of synergies occur during co-stimulation. Their finding that the for some network components, synergies can change from a antagonistic to synergistic in a dose-dependent manner highlights the difficulties in extending studies that examine signaling pathways from a single stimuli towards the complex milieu of stimuli occurring in vivo and the benefit of this type of high-throughput work going forward.
Finally, as both the biosensors and the acquisition of data using fluorescent biosensors becomes more robust, the translation of their use into the clinical setting may become an area of increasing interest in the fluorescent biosensor field. Mizutani and colleagues proposed to use this approach in personalized medicine where they used their improved Bcr-Abl biosensor, pickles2.3, in cancer cells derived from chronic myeloid leukemia (CML) patients [20670950]. Using 293F cells expressing wild type of the Bcr-Abl fusion protein or mutant forms which are resistant to the first-line therapy imatinib, they showed that the biosensor readout decreased in response to imatinib treatment in the wild type Bcr-Abl cells but not the mutant cells. Futhermore, when they mixed these two cell types together at different ratios, the percentage of single cell biosensor readouts that were above an observed threshold was related to the proportion of the mixture that was comprised of the resisitant mutant Bcr-Abl cells. Using this mixture model readout, they showed how different combinations of co-therapy could be tested to find the appropriate treatment to catch both the majority imatinib sensitive population and the smaller resistant population. While this assay would be too work intenstive to be practically applied in the clinic, the advances in biosensor measurement throughput could lead to a time when fluorescent biosensors play a crucial role in both medical diagnostics and personalized medicine.
7. Conclusion
The late Roger Tsien, one of the great pioneers of fluorescent proteins, once said “our work is often described as building and training molecular spies, molecules that will enter a cell or organism and report back to us what the conditions are”945. Genetically encoded fluorescent biosensors have truly become these powerful molecular spies that illuminate the spatiotemporal dynamics of cellular communication and decision making. As the development of fluorescent biosensors has progressed, we have seen both improvements in sensitivity and an expansion of the breadth of signaling pathways that can be studied (Table 1). Here, we have discussed the different categories of biosensor designs utilized to quantify signaling dynamics, including several recently emerging classes of biosensors based on infrared fluorescent proteins, biosensors that act as signal integrators, and biosensors that utilize fluorescence fluctuations for superresolution activity mapping (Section 2). The dynamic nature and fluorescent readout that underlie these designs have enabled the quantification of the kinetics of signal transduction as it occurs throughout the cell. These in situ measurements have transformed our understanding of signaling dynamics ranging from individual reactions to evaluating the origins and regulation of complex signaling phenomena (Section 3). Furthermore, the ability to observe spatial changes in biosensor signals, either through direct spatial quantification or the use of subcellular targeting, has not only confirmed the existence of spatially compartmentalized signaling domains but also provided the means with which to probe the mechanisms that give rise to this intricate spatial organization (Section 4).While the multifaceted nature of signal transduction can make it difficult to directly infer the critical regulators that underlie the observations made using fluorescent biosensors, computational models have been a useful companion to biosensor-based studies, as computational models benefit from the high temporal and spatial resolution of fluorescent biosensors and can in turn can quantitatively evaluate potential hypotheses and probe aspects of biology are not directly measurable experimentally (Section 5). Additionally, the adaptable nature of fluorescent biosensors has made them ideal for validating a number of computational model predictions. Finally, continuing advances are enabling fluorescent biosensor applications beyond the diffraction limit, biosensor multiplexing for simultaneous observation of multiple signaling pathways, and the development of biosensors that are more suitable for in vivo imaging (Section 6). These advances in genetically encoded fluorescent biosensor design will continue to push the boundaries, both spatial and temporal, of the study of signaling dynamics, with the dual goals of expanding our understanding of the native function of signaling networks and translating these tools into the discovery and evaluation of novel therapeutic avenues for a myriad of diseases.
Supplementary Material
8. Acknowledgements
The authors would like to Jason Zhang, Xin Zhou, Jeremiah Keyes, Brian Tenner, Arielle Yoo, Chris Booth and Yanghao Zhong for their help in compiling the database of fluorescent biosensors. Work in this lab is funded by the NIH (F32 GM120798 to E.G. and R35 CA197622, R01 DK073368, R01 GM111665 and R01 MH111516 to J.Z.) and DOD (AFOSR FA9550-18-1-0051 to J. Z.)
11. Biographies
Eric C. Greenwald
Eric Greenwald attended the University of Colorado, where he received a B.S. in Chemical and Biological Engineering, a B.S. in Applied Mathematics, and an M.S. in Chemical Engineering. He then persued a Ph.D. in Biomedical Engineering at the University of Virginia. Under the guidance of Dr. Jeff Saucerman, Eric examined how the coordination of signaling proteins by scaffold proteins changes the underlying kinetic mechanism of signal transduction between a tethered enzyme and substrate. Following graduation, Eric joined Jin Zhang’s lab at the Unviersity of California San Diego, where he is developing tools to increase the throughput of fluorescent biosensor data acquisition.
Sohum Mehta
Sohum Mehta received a B.S. in Biology, with a minor in Fine Arts, from the George Washington University before going on to pursue graduate studies at the Johns Hopkins University, where he received a Ph.D. in Biology. For his thesis work, Sohum studied calcineurin signaling in the yeast Saccharomyces cerevisiae, performing structure-function analyses of the evolutionarily conserved Regulator of Calcineurin (RCaN) protein family. Sohum joined Jin Zhang’s lab as a postdoctoral fellow at the Johns Hopkins University School of Medicine and continues to serve as a senior researcher in the lab at its new home in the University of California San Diego, where his work includes developing novel genetically encoded tools for the sensitive and multiplexed visualization of intracellular signaling.
Jin Zhang
Jin Zhang attended Tsinghua University for her undergraduate studies, and pursued her graduate studies in Chemistry at the U. Chicago. After completing her postdoctoral work at the U. California, San Diego, she joined the faculty of Johns Hopkins University School of Medicine in 2003. She was promoted to Professor of Pharmacology, Neuroscience and Oncology in 2013. In 2015 she moved back to University of California, San Diego and is currently a member of the Moores Cancer Center and a Professor in Departments of Pharmacology, Bioengineering and Chemistry & Biochemistry at UC San Diego. Research in her lab focuses on developing enabling technologies to probe the active molecules in their native environment and characterizing how these active molecules change in diseases including cancer. Professor Zhang has received many awards including the National Institutes of Health (NIH) Director’s Pioneer Award (2009), the John J. Abel Award in Pharmacology from American Society for Pharmacology and Experimental Therapeutics (ASPET) (2012), the Pfizer Award in Enzyme Chemistry from American Chemical Society (2012), and National Institute of Cancer Outstanding Investigator Award (2015). She was elected as a Fellow of the American Association for the Advancement of Science (AAAS) in 2014. She serves on the editorial advisory board of Cell Chemical Biology and is the past-chair of Molecular Pharmacology Division of ASPET and Secretary/TreasurerElect of ASPET.
Footnotes
10 References
- (1).Coons AH The Beginnings of Immunofluorescence. J. Immunol 1961, 87, 499–503. [PubMed] [Google Scholar]
- (2).Taylor DL; Amato PA; Luby-Phelps K; McNeil P Fluorescent Analog Cytochemistry. Trends Biochem. Sci 1984, 9 (3), 88–91. [Google Scholar]
- (3).Tsien RY Fluorescent Probes of Cell Signaling. Annu. Rev. Neurosci 1989, 12 (1), 227–253. [DOI] [PubMed] [Google Scholar]
- (4).Prasher DC; Eckenrode VK; Ward WW; Prendergast FG; Cormier MJ Primary Structure of the Aequorea Victoria Green-Fluorescent Protein. Gene 1992, 111 (2), 229–233. [DOI] [PubMed] [Google Scholar]
- (5).Heim R; Cubitt AB; Tsien RY Improved Green Fluorescence. Nature 1995, 373 (6516), 663–664. [DOI] [PubMed] [Google Scholar]
- (6).Tsien RY The Green Fluorescent Protein. Annu. Rev. Biochem 1998, 67 (1), 509–544. [DOI] [PubMed] [Google Scholar]
- (7).Rodriguez EA; Campbell RE; Lin JY; Lin MZ; Miyawaki A; Palmer AE; Shu X; Zhang J; Tsien RY The Growing and Glowing Toolbox of Fluorescent and Photoactive Proteins. Trends Biochem. Sci 2017, 42 (2), 111–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Tomosugi W; Matsuda T; Tani T; Nemoto T; Kotera I; Saito K; Horikawa K; Nagai T An Ultramarine Fluorescent Protein with Increased Photostability and pH Insensitivity. Nat. Methods 2009, 6 (5), 351–353. [DOI] [PubMed] [Google Scholar]
- (9).Shcherbakova DM; Verkhusha VV Near-Infrared Fluorescent Proteins for Multicolor in Vivo Imaging. Nat. Methods 2013, 10 (8), 751–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Newman RH; Fosbrink MD; Zhang J Genetically Encodable Fluorescent Biosensors for Tracking Signaling Dynamics in Living Cells. Chem. Rev 2011, 111 (5), 3614–3666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Li X; Zhao X; Fang Y; Jiang X; Duong T; Fan C; Huang CC; Kain SR Generation of Destabilized Green Fluorescent Protein as a Transcription Reporter. J. Biol. Chem 1998, 273 (52), 34970–34975. [DOI] [PubMed] [Google Scholar]
- (12).Hackett EA; Esch RK; Maleri S; Errede B A Family of Destabilized Cyan Fluorescent Proteins as Transcriptional Reporters in S. Cerevisiae. Yeast 2006, 23 (5), 333–349. [DOI] [PubMed] [Google Scholar]
- (13).Houser JR; Ford E; Chatterjea SM; Maleri S; Elston TC; Errede B An Improved Short-Lived Fluorescent Protein Transcriptional Reporter for Saccharomyces Cerevisiae. Yeast 2012, 29 (12), 519–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Subach FV; Subach OM; Gundorov IS; Morozova KS; Piatkevich KD; Cuervo AM; Verkhusha VV Monomeric Fluorescent Timers That Change Color from Blue to Red Report on Cellular Trafficking. Nat. Chem. Biol 2009, 5 (2), 118–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Chen M-R; Yang S; Niu W; Li Z-Y; Meng L-F; Wu Z-X A Novel Fluorescent Timer Based on Bicistronic Expression Strategy in Caenorhabditis Elegans. Biochem. Biophys. Res. Commun 2010, 395 (1), 82–86. [DOI] [PubMed] [Google Scholar]
- (16).Khmelinskii A; Keller PJ; Bartosik A; Meurer M; Barry JD; Mardin BR; Kaufmann A; Trautmann S; Wachsmuth M; Pereira G; et al. Tandem Fluorescent Protein Timers for in Vivo Analysis of Protein Dynamics. Nat. Biotechnol 2012, 30 (7), 708–714. [DOI] [PubMed] [Google Scholar]
- (17).Zhu X; Zhang L; Kao Y-T; Xu F; Min W A Tunable Fluorescent Timer Method for Imaging Spatial-Temporal Protein Dynamics Using Light-Driven Photoconvertible Protein. J. Biophotonics 2015, 8 (3), 226–232. [DOI] [PubMed] [Google Scholar]
- (18).Khmelinskii A; Meurer M; Ho C-T; Besenbeck B; Füller J; Lemberg MK; Bukau B; Mogk A; Knop M Incomplete Proteasomal Degradation of Green Fluorescent Proteins in the Context of Tandem Fluorescent Protein Timers. Mol. Biol. Cell 2016, 27 (2), 360–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Miyatsuka T; Matsuoka T. -a.; Sasaki S; Kubo F; Shimomura I; Watada H; German MS; Hara M Chronological Analysis With Fluorescent Timer Reveals Unique Features of Newly Generated -Cells. Diabetes 2014, 63 (10), 3388–3393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Breen M; Nogales A; Baker SF; Perez DR; Martínez-Sobrido L ReplicationCompetent Influenza A and B Viruses Expressing a Fluorescent Dynamic Timer Protein for In Vitro and In Vivo Studies. PLoS One 2016, 11 (1), e0147723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Dedecker P; De Schryver FC; Hofkens J Fluorescent Proteins: Shine On, You Crazy Diamond. J. Am. Chem. Soc 2013, 135 (7), 2387–2402. [DOI] [PubMed] [Google Scholar]
- (22).Nienhaus K; Nienhaus GU Fluorescent Proteins for Live-Cell Imaging with SuperResolution. Chem. Soc. Rev 2014, 43 (4), 1088–1106. [DOI] [PubMed] [Google Scholar]
- (23).Dedecker P; Mo GCH; Dertinger T; Zhang J Widely Accessible Method for Superresolution Fluorescence Imaging of Living Systems. Proc. Natl. Acad. Sci. U. S. A 2012, 109 (27), 10909–10914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Gaj T; Gersbach CA; Barbas CF ZFN, TALEN, and CRISPR/Cas-Based Methods for Genome Engineering. Trends Biotechnol 2013, 31 (7), 397–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Sander JD; Joung JK CRISPR-Cas Systems for Editing, Regulating and Targeting Genomes. Nat. Biotechnol 2014, 32 (4), 347–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Doudna JA; Charpentier E Genome Editing. The New Frontier of Genome Engineering with CRISPR-Cas9. Science 2014, 346 (6213), 1258096. [DOI] [PubMed] [Google Scholar]
- (27).Roberts B; Haupt A; Tucker A; Grancharova T; Arakaki J; Fuqua MA; Nelson A; Hookway C; Ludmann SA; Mueller IA; et al. Systematic Gene Tagging Using CRISPR/Cas9 in Human Stem Cells to Illuminate Cell Organization. Mol. Biol. Cell 2017, 28 (21), 2854–2874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Krentz NAJ; Nian C; Lynn FC TALEN/CRISPR-Mediated eGFP Knock-in Addon at the OCT4 Locus Does Not Impact Differentiation of Human Embryonic Stem Cells towards Endoderm. PLoS One 2014, 9 (12), e114275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Ratz M; Testa I; Hell SW; Jakobs S CRISPR/Cas9-Mediated Endogenous Protein Tagging for RESOLFT Super-Resolution Microscopy of Living Human Cells. Sci. Rep 2015, 5 (1), 9592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Mikuni T; Nishiyama J; Sun Y; Kamasawa N; Yasuda R High-Throughput, High-Resolution Mapping of Protein Localization in Mammalian Brain by In Vivo Genome Editing. Cell 2016, 165 (7), 1803–1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Auer TO; Duroure K; De Cian A; Concordet J-P; Del Bene F Highly Efficient CRISPR/Cas9-Mediated Knock-in in Zebrafish by Homology-Independent DNA Repair. Genome Res 2014, 24 (1), 142–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Suzuki K; Tsunekawa Y; Hernandez-Benitez R; Wu J; Zhu J; Kim EJ; Hatanaka F; Yamamoto M; Araoka T; Li Z; et al. In Vivo Genome Editing via CRISPR/Cas9 Mediated Homology-Independent Targeted Integration. Nature 2016, 540 (7631), 144–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Cabantous S; Terwilliger TC; Waldo GS Protein Tagging and Detection with Engineered Self-Assembling Fragments of Green Fluorescent Protein. Nat. Biotechnol 2005, 23 (1), 102–107. [DOI] [PubMed] [Google Scholar]
- (34).Kamiyama D; Sekine S; Barsi-Rhyne B; Hu J; Chen B; Gilbert LA; Ishikawa H; Leonetti MD; Marshall WF; Weissman JS; et al. Versatile Protein Tagging in Cells with Split Fluorescent Protein. Nat. Commun 2016, 7, 11046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Kaiser PD; Maier J; Traenkle B; Emele F; Rothbauer U Recent Progress in Generating Intracellular Functional Antibody Fragments to Target and Trace Cellular Components in Living Cells. Biochim. Biophys. Acta 2014, 1844 (11), 1933–1942. [DOI] [PubMed] [Google Scholar]
- (36).Koide A; Bailey CW; Huang X; Koide S The Fibronectin Type III Domain as a Scaffold for Novel Binding Proteins. J. Mol. Biol 1998, 284 (4), 1141–1151. [DOI] [PubMed] [Google Scholar]
- (37).Koide A; Koide S Monobodies: Antibody Mimics Based on the Scaffold of the Fibronectin Type III Domain. Methods Mol. Biol 2007, 352, 95–109. [DOI] [PubMed] [Google Scholar]
- (38).Beghein E; Gettemans J Nanobody Technology: A Versatile Toolkit for Microscopic Imaging, Protein-Protein Interaction Analysis, and Protein Function Exploration. Front. Immunol 2017, 8, 771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Gross GG; Junge JA; Mora RJ; Kwon H-B; Olson CA; Takahashi TT; Liman ER; Ellis-Davies GCR; McGee AW; Sabatini BL; et al. Recombinant Probes for Visualizing Endogenous Synaptic Proteins in Living Neurons. Neuron 2013, 78 (6), 971–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Rocchetti A; Hawes C; Kriechbaumer V Fluorescent Labelling of the Actin Cytoskeleton in Plants Using a Cameloid Antibody. Plant Methods 2014, 10 (1), 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Traenkle B; Emele F; Anton R; Poetz O; Haeussler RS; Maier J; Kaiser PD; Scholz AM; Nueske S; Buchfellner A; et al. Monitoring Interactions and Dynamics of Endogenous Beta-Catenin with Intracellular Nanobodies in Living Cells. Mol. Cell. Proteomics 2015, 14 (3), 707–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Buchfellner A; Yurlova L; Nüske S; Scholz AM; Bogner J; Ruf B; Zolghadr K; Drexler SE; Drexler GA; Girst S; et al. A New Nanobody-Based Biosensor to Study Endogenous PARP1 In Vitro and in Live Human Cells. PLoS One 2016, 11 (3), e0151041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Cetin M; Evenson WE; Gross GG; Jalali-Yazdi F; Krieger D; Arnold D; Takahashi TT; Roberts RW RasIns: Genetically Encoded Intrabodies of Activated Ras Proteins. J. Mol. Biol 2017, 429 (4), 562–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Burgess A; Lorca T; Castro A Quantitative Live Imaging of Endogenous DNA Replication in Mammalian Cells. PLoS One 2012, 7 (9), e45726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Maier J; Traenkle B; Rothbauer U Real-Time Analysis of Epithelial-Mesenchymal Transition Using Fluorescent Single-Domain Antibodies. Sci. Rep 2015, 5 (1), 13402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Wongso D; Dong J; Ueda H; Kitaguchi T Flashbody: A Next Generation Fluobody with Fluorescence Intensity Enhanced by Antigen Binding. Anal. Chem 2017, 89 (12), 6719–6725. [DOI] [PubMed] [Google Scholar]
- (47).Cardullo RA Theoretical Principles and Practical Considerations for Fluorescence Resonance Energy Transfer Microscopy. Methods Cell Biol 2013, 114, 441–456. [DOI] [PubMed] [Google Scholar]
- (48).Stryer L Fluorescence Energy Transfer as a Spectroscopic Ruler. Annu. Rev. Biochem 1978, 47 (1), 819–846. [DOI] [PubMed] [Google Scholar]
- (49).Ciruela F Fluorescence-Based Methods in the Study of Protein-Protein Interactions in Living Cells. Curr. Opin. Biotechnol 2008, 19 (4), 338–343. [DOI] [PubMed] [Google Scholar]
- (50).Kohnhorst CL; Kyoung M; Jeon M; Schmitt DL; Kennedy EL; Ramirez J; Bracey SM; Luu BT; Russell SJ; An S Identification of a Multienzyme Complex for Glucose Metabolism in Living Cells. J. Biol. Chem 2017, 292 (22), 9191–9203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Takagi S; Momose F; Morikawa Y FRET Analysis of HIV-1 Gag and GagPol Interactions. FEBS Open Bio 2017, 7 (11), 1815–1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Cole GB; Reichheld SE; Sharpe S FRET Analysis of the Promiscuous yet Specific Interactions of the HIV-1 Vpu Transmembrane Domain. Biophys. J 2017, 113 (9), 1992–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).McNally BA; Pendon ZD; Trudeau MC hERG1a and hERG1b Potassium Channel Subunits Directly Interact and Preferentially Form Heteromeric Channels. J. Biol. Chem 2017, 292 (52), 21548–21557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Efendiev R; Samelson BK; Nguyen BT; Phatarpekar PV; Baameur F; Scott JD; Dessauer CW AKAP79 Interacts with Multiple Adenylyl Cyclase (AC) Isoforms and Scaffolds AC5 and −6 to Alpha-Amino-3-Hydroxyl-5-Methyl-4-Isoxazole-Propionate (AMPA) Receptors. J. Biol. Chem 2010, 285 (19), 14450–14458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Willoughby D; Masada N; Wachten S; Pagano M; Halls ML; Everett KL; Ciruela A; Cooper DMF AKAP79/150 Interacts with AC8 and Regulates Ca2+-Dependent cAMP Synthesis in Pancreatic and Neuronal Systems. J. Biol. Chem 2010, 285 (26), 20328–20342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Oliveria SF; Dell’Acqua ML; Sather WA AKAP79/150 Anchoring of Calcineurin Controls Neuronal L-Type Ca2+ Channel Activity and Nuclear Signaling. Neuron 2007, 55 (2), 261–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Li H; Pink MD; Murphy JG; Stein A; Dell’Acqua ML; Hogan PG Balanced Interactions of Calcineurin with AKAP79 Regulate Ca2+-Calcineurin-NFAT Signaling. Nat. Struct. Mol. Biol 2012, 19 (3), 337–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Oliveria SF; Dittmer PJ; Youn D; Dell’Acqua ML; Sather WA Localized Calcineurin Confers Ca2+-Dependent Inactivation on Neuronal L-Type Ca2+ Channels. J. Neurosci 2012, 32 (44), 15328–15337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Murphy JG; Sanderson JL; Gorski JA; Scott JD; Catterall WA; Sather WA; Dell’Acqua ML AKAP-Anchored PKA Maintains Neuronal L-Type Calcium Channel Activity and NFAT Transcriptional Signaling. Cell Rep 2014, 7 (5), 1577–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Ullmann A; Jacob F; Monod J Characterization by in Vitro Complementation of a Peptide Corresponding to an Operator-Proximal Segment of the Beta-Galactosidase Structural Gene of Escherichia Coli. J. Mol. Biol 1967, 24 (2), 339–343. [DOI] [PubMed] [Google Scholar]
- (61).Rossi F; Charlton CA; Blau HM Monitoring Protein-Protein Interactions in Intact Eukaryotic Cells by Beta-Galactosidase Complementation. Proc. Natl. Acad. Sci. U. S. A 1997, 94 (16), 8405–8410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Pelletier JN; Campbell-Valois FX; Michnick SW Oligomerization DomainDirected Reassembly of Active Dihydrofolate Reductase from Rationally Designed Fragments. Proc. Natl. Acad. Sci. U. S. A 1998, 95 (21), 12141–12146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Galarneau A; Primeau M; Trudeau L-E; Michnick SW Beta-Lactamase Protein Fragment Complementation Assays as in Vivo and in Vitro Sensors of Protein Protein Interactions. Nat. Biotechnol 2002, 20 (6), 619–622. [DOI] [PubMed] [Google Scholar]
- (64).Wehrman T; Kleaveland B; Her J-H; Balint RF; Blau HM Protein-Protein Interactions Monitored in Mammalian Cells via Complementation of Beta -Lactamase Enzyme Fragments. Proc. Natl. Acad. Sci. U. S. A 2002, 99 (6), 3469–3474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Ghosh I; Hamilton AD; Regan L Antiparallel Leucine Zipper-Directed Protein Reassembly: Application to the Green Fluorescent Protein [12]. J. Am. Chem. Soc 2000, 122 (23), 5658–5659. [Google Scholar]
- (66).Hu C-D; Chinenov Y; Kerppola TK Visualization of Interactions among bZIP and Rel Family Proteins in Living Cells Using Bimolecular Fluorescence Complementation. Mol. Cell 2002, 9 (4), 789–798. [DOI] [PubMed] [Google Scholar]
- (67).Kerppola TK Visualization of Molecular Interactions Using Bimolecular Fluorescence Complementation Analysis: Characteristics of Protein Fragment Complementation. Chem. Soc. Rev 2009, 38 (10), 2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Kerppola TK Bimolecular Fluorescence Complementation (BiFC) Analysis as a Probe of Protein Interactions in Living Cells. Annu. Rev. Biophys 2008, 37 (1), 465–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Cabantous S; Nguyen HB; Pedelacq J-D; Koraïchi F; Chaudhary A; Ganguly K; Lockard MA; Favre G; Terwilliger TC; Waldo GS A New Protein-Protein Interaction Sensor Based on Tripartite Split-GFP Association. Sci. Rep 2013, 3 (1), 2854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Foglieni C; Papin S; Salvadè A; Afroz T; Pinton S; Pedrioli G; Ulrich G; Polymenidou M; Paganetti P Split GFP Technologies to Structurally Characterize and Quantify Functional Biomolecular Interactions of FTD-Related Proteins. Sci. Rep 2017, 7 (1), 14013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Robida AM; Kerppola TK Bimolecular Fluorescence Complementation Analysis of Inducible Protein Interactions: Effects of Factors Affecting Protein Folding on Fluorescent Protein Fragment Association. J. Mol. Biol 2009, 394 (3), 391–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Sharan A; Soni P; Nongpiur RC; Singla-Pareek SL; Pareek A Mapping the “TwoComponent System” Network in Rice. Sci. Rep 2017, 7 (1), 9287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Subotić A; Swinnen E; Demuyser L; De Keersmaecker H; Mizuno H; Tournu H; Van Dijck P A Bimolecular Fluorescence Complementation Tool for Identification of Protein-Protein Interactions inCandida Albicans. G3 (Bethesda) 2017, 7 (10), 3509–3520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Liu Z; Xing D; Su QP; Zhu Y; Zhang J; Kong X; Xue B; Wang S; Sun H; Tao Y; et al. Super-Resolution Imaging and Tracking of Protein-Protein Interactions in Sub-Diffraction Cellular Space. Nat. Commun 2014, 5, 4443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Xia P; Liu X; Wu B; Zhang S; Song X; Yao PY; Lippincott-Schwartz J; Yao X Superresolution Imaging Reveals Structural Features of EB1 in Microtubule plus-End Tracking. Mol. Biol. Cell 2014, 25 (25), 4166–4173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Hertel F; Mo GCH; Duwé S; Dedecker P; Zhang J RefSOFI for Mapping Nanoscale Organization of Protein-Protein Interactions in Living Cells. Cell Rep 2016, 14 (2), 390–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Jean-Alphonse FG; Wehbi VL; Chen J; Noda M; Taboas JM; Xiao K; Vilardaga J-P β2-Adrenergic Receptor Control of Endosomal PTH Receptor Signaling via Gβγ. Nat. Chem. Biol 2017, 13 (3), 259–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Smith FD; Esseltine JL; Nygren PJ; Veesler D; Byrne DP; Vonderach M; Strashnov I; Eyers CE; Eyers PA; Langeberg LK; et al. Local Protein Kinase A Action Proceeds through Intact Holoenzymes. Science 2017, 356 (6344), 1288–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Martin TF Phosphoinositide Lipids as Signaling Molecules: Common Themes for Signal Transduction, Cytoskeletal Regulation, and Membrane Trafficking. Annu. Rev. Cell Dev. Biol 1998, 14 (1), 231–264. [DOI] [PubMed] [Google Scholar]
- (80).Hurley JH; Meyer T Subcellular Targeting by Membrane Lipids. Curr. Opin. Cell Biol 2001, 13 (2), 146–152. [DOI] [PubMed] [Google Scholar]
- (81).Lemmon MA Phosphoinositide Recognition Domains. Traffic 2003, 4 (4), 201–213. [DOI] [PubMed] [Google Scholar]
- (82).Várnai P; Balla T Live Cell Imaging of Phosphoinositide Dynamics with Fluorescent Protein Domains. Biochim. Biophys. Acta 2006, 1761 (8), 957–967. [DOI] [PubMed] [Google Scholar]
- (83).Harlan JE; Hajduk PJ; Yoon HS; Fesik SW Pleckstrin Homology Domains Bind to Phosphatidylinositol-4,5-Bisphosphate. Nature 1994, 371 (6493), 168–170. [DOI] [PubMed] [Google Scholar]
- (84).De Camilli P; Chen H; Hyman J; Panepucci E; Bateman A; Brunger AT The ENTH Domain. FEBS Lett 2002, 513 (1), 11–18. [DOI] [PubMed] [Google Scholar]
- (85).Kutateladze TG Phosphatidylinositol 3-Phosphate Recognition and Membrane Docking by the FYVE Domain. Biochim. Biophys. Acta 2006, 1761 (8), 868–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Burd CG; Emr SD Phosphatidylinositol(3)-Phosphate Signaling Mediated by Specific Binding to RING FYVE Domains. Mol. Cell 1998, 2 (1), 157–162. [DOI] [PubMed] [Google Scholar]
- (87).Watton SJ; Downward J Akt/PKB Localisation and 3’ Phosphoinositide Generation at Sites of Epithelial Cell-Matrix and Cell-Cell Interaction. Curr. Biol 1999, 9 (8), 433–436. [DOI] [PubMed] [Google Scholar]
- (88).Várnai P; Balla T Visualization of Phosphoinositides That Bind Pleckstrin Homology Domains: Calcium- and Agonist-Induced Dynamic Changes and Relationship to Myo[3H]inositol-Labeled Phosphoinositide Pools. J. Cell Biol 1998, 143 (2), 501–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (89).Gray A; Van Der Kaay J; Downes CP The Pleckstrin Homology Domains of Protein Kinase B and GRP1 (General Receptor for Phosphoinositides-1) Are Sensitive and Selective Probes for the Cellular Detection of Phosphatidylinositol 3,4-Bisphosphate And/or Phosphatidylinositol 3,4,5-Trisphosphate in Vivo. Biochem. J 1999, 344 Pt 3, 929–936. [PMC free article] [PubMed] [Google Scholar]
- (90).Ellson CD; Anderson KE; Morgan G; Chilvers ER; Lipp P; Stephens LR; Hawkins PT Phosphatidylinositol 3-Phosphate Is Generated in Phagosomal Membranes. Curr. Biol 2001, 11 (20), 1631–1635. [DOI] [PubMed] [Google Scholar]
- (91).Yoo SK; Deng Q; Cavnar PJ; Wu YI; Hahn KM; Huttenlocher A Differential Regulation of Protrusion and Polarity by PI3K during Neutrophil Motility in Live Zebrafish. Dev. Cell 2010, 18 (2), 226–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (92).Oancea E; Teruel MN; Quest AF; Meyer T Green Fluorescent Protein (GFP)Tagged Cysteine-Rich Domains from Protein Kinase C as Fluorescent Indicators for Diacylglycerol Signaling in Living Cells. J. Cell Biol 1998, 140 (3), 485–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (93).Codazzi F; Teruel MN; Meyer T Control of Astrocyte Ca(2+) Oscillations and Waves by Oscillating Translocation and Activation of Protein Kinase C. Curr. Biol 2001, 11 (14), 1089–1097. [DOI] [PubMed] [Google Scholar]
- (94).Oancea E; Meyer T Protein Kinase C as a Molecular Machine for Decoding Calcium and Diacylglycerol Signals. Cell 1998, 95 (3), 307–318. [DOI] [PubMed] [Google Scholar]
- (95).Stahelin RV; Rafter JD; Das S; Cho W The Molecular Basis of Differential Subcellular Localization of C2 Domains of Protein Kinase C-Alpha and Group IVa Cytosolic Phospholipase A2. J. Biol. Chem 2003, 278 (14), 12452–12460. [DOI] [PubMed] [Google Scholar]
- (96).Ananthanarayanan B; Das S; Rhee SG; Murray D; Cho W Membrane Targeting of C2 Domains of Phospholipase C-δ Isoforms. J. Biol. Chem 2002, 277 (5), 3568–3575. [DOI] [PubMed] [Google Scholar]
- (97).Yeung T; Gilbert GE; Shi J; Silvius J; Kapus A; Grinstein S Membrane Phosphatidylserine Regulates Surface Charge and Protein Localization. Science (80-. ) 2008, 319 (5860), 210–213. [DOI] [PubMed] [Google Scholar]
- (98).Gonzalez-Sapienza G; Rossotti MA; Tabares-da Rosa S Single-Domain Antibodies As Versatile Affinity Reagents for Analytical and Diagnostic Applications. Front. Immunol 2017, 8, 977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (99).Rajan M; Mortusewicz O; Rothbauer U; Hastert FD; Schmidthals K; Rapp A; Leonhardt H; Cardoso MC Generation of an Alpaca-Derived Nanobody Recognizing γ-H2AX. FEBS Open Bio 2015, 5 (1), 779–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (100).Rasmussen SGF; Choi H-J; Fung JJ; Pardon E; Casarosa P; Chae PS; Devree BT; Rosenbaum DM; Thian FS; Kobilka TS; et al. Structure of a Nanobody-Stabilized Active State of the β(2) Adrenoceptor. Nature 2011, 469 (7329), 175–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (101).Ring AM; Manglik A; Kruse AC; Enos MD; Weis WI; Garcia KC; Kobilka BK Adrenaline-Activated Structure of β2-Adrenoceptor Stabilized by an Engineered Nanobody. Nature 2013, 502 (7472), 575–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (102).Kruse AC; Ring AM; Manglik A; Hu J; Hu K; Eitel K; Hübner H; Pardon E; Valant C; Sexton PM; et al. Activation and Allosteric Modulation of a Muscarinic Acetylcholine Receptor. Nature 2013, 504 (7478), 101–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (103).Huang W; Manglik A; Venkatakrishnan AJ; Laeremans T; Feinberg EN; Sanborn AL; Kato HE; Livingston KE; Thorsen TS; Kling RC; et al. Structural Insights into M-Opioid Receptor Activation. Nature 2015, 524 (7565), 315–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (104).Westfield GH; Rasmussen SGF; Su M; Dutta S; DeVree BT; Chung KY; Calinski D; Velez-Ruiz G; Oleskie AN; Pardon E; et al. Structural Flexibility of the G Alpha S Alpha-Helical Domain in the beta2-Adrenoceptor Gs Complex. Proc. Natl. Acad. Sci. U. S. A 2011, 108 (38), 16086–16091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (105).Irannejad R; Tomshine JC; Tomshine JR; Chevalier M; Mahoney JP; Steyaert J; Rasmussen SGF; Sunahara RK; El-Samad H; Huang B; et al. Conformational Biosensors Reveal GPCR Signalling from Endosomes. Nature 2013, 495 (7442), 534–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (106).Godbole A; Lyga S; Lohse MJ; Calebiro D Internalized TSH Receptors En Route to the TGN Induce Local Gs-Protein Signaling and Gene Transcription. Nat. Commun 2017, 8 (1), 443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (107).Spencer SL; Cappell SD; Tsai F-C; Overton KW; Wang CL; Meyer T The Proliferation-Quiescence Decision Is Controlled by a Bifurcation in CDK2 Activity at Mitotic Exit. Cell 2013, 155 (2), 369–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (108).Gu J; Xia X; Yan P; Liu H; Podust VN; Reynolds AB; Fanning E Cell CycleDependent Regulation of a Human DNA Helicase That Localizes in DNA Damage Foci. Mol. Biol. Cell 2004, 15 (7), 3320–3332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (109).Gross SM; Rotwein P Akt Signaling Dynamics in Individual Cells. J. Cell Sci 2015, 128 (14), 2509–2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (110).Maryu G; Matsuda M; Aoki K Multiplexed Fluorescence Imaging of ERK and Akt Activities and Cell-Cycle Progression. Cell Struct. Funct 2016, 41 (2), 81–92. [DOI] [PubMed] [Google Scholar]
- (111).Kosugi S; Hasebe M; Tomita M; Yanagawa H Nuclear Export Signal Consensus Sequences Defined Using a Localization-Based Yeast Selection System. Traffic 2008, 9 (12), 2053–2062. [DOI] [PubMed] [Google Scholar]
- (112).Regot S; Hughey JJ; Bajar BT; Carrasco S; Covert MW High-Sensitivity Measurements of Multiple Kinase Activities in Live Single Cells. Cell 2014, 157 (7), 1724–1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (113).Durandau E; Aymoz D; Pelet S Dynamic Single Cell Measurements of Kinase Activity by Synthetic Kinase Activity Relocation Sensors. BMC Biol 2015, 13, 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (114).Chattoraj M; King BA; Bublitz GU; Boxer SG Ultra-Fast Excited State Dynamics in Green Fluorescent Protein: Multiple States and Proton Transfer. Proc. Natl. Acad. Sci. U. S. A 1996, 93 (16), 8362–8367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (115).Brejc K; Sixma TK; Kitts PA; Kain SR; Tsien RY; Ormö M; Remington SJ Structural Basis for Dual Excitation and Photoisomerization of the Aequorea Victoria Green Fluorescent Protein. Proc. Natl. Acad. Sci. U. S. A 1997, 94 (6), 2306–2311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (116).Bokman SH; Ward WW Renaturation of Aequorea Gree-Fluorescent Protein. Biochem. Biophys. Res. Commun 1981, 101 (4), 1372–1380. [DOI] [PubMed] [Google Scholar]
- (117).Miesenböck G; De Angelis DA; Rothman JE Visualizing Secretion and Synaptic Transmission with pH-Sensitive Green Fluorescent Proteins. Nature 1998, 394 (6689), 192–195. [DOI] [PubMed] [Google Scholar]
- (118).Heim R; Prasher DC; Tsien RY Wavelength Mutations and Posttranslational Autoxidation of Green Fluorescent Protein. Proc. Natl. Acad. Sci. U. S. A 1994, 91 (26), 12501–12504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (119).Wachter RM; King BA; Heim R; Kallio K; Tsien RY; Boxer SG; Remington SJ Crystal Structure and Photodynamic Behavior of the Blue Emission Variant Y66H/Y145F of Green Fluorescent Protein. Biochemistry 1997, 36 (32), 9759–9765. [DOI] [PubMed] [Google Scholar]
- (120).Kneen M; Farinas J; Li Y; Verkman AS Green Fluorescent Protein as a Noninvasive Intracellular pH Indicator. Biophys. J 1998, 74 (3), 1591–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (121).Llopis J; McCaffery JM; Miyawaki A; Farquhar MG; Tsien RY Measurement of Cytosolic, Mitochondrial, and Golgi pH in Single Living Cells with Green Fluorescent Proteins. Proc. Natl. Acad. Sci. U. S. A 1998, 95 (12), 6803–6808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (122).Sankaranarayanan S; De Angelis D; Rothman JE; Ryan TA The Use of pHluorins for Optical Measurements of Presynaptic Activity. Biophys. J 2000, 79 (4), 2199–2208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (123).Li Y; Tsien RW pHTomato, a Red, Genetically Encoded Indicator That Enables Multiplex Interrogation of Synaptic Activity. Nat. Neurosci 2012, 15 (7), 1047–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (124).Wachter RM; Elsliger MA; Kallio K; Hanson GT; Remington SJ Structural Basis of Spectral Shifts in the Yellow-Emission Variants of Green Fluorescent Protein. Structure 1998, 6 (10), 1267–1277. [DOI] [PubMed] [Google Scholar]
- (125).Tojima T; Akiyama H; Itofusa R; Li Y; Katayama H; Miyawaki A; Kamiguchi H Attractive Axon Guidance Involves Asymmetric Membrane Transport and Exocytosis in the Growth Cone. Nat. Neurosci 2007, 10 (1), 58–66. [DOI] [PubMed] [Google Scholar]
- (126).Jayaraman S; Haggie P; Wachter RM; Remington SJ; Verkman AS Mechanism and Cellular Applications of a Green Fluorescent Protein-Based Halide Sensor. J. Biol. Chem 2000, 275 (9), 6047–6050. [DOI] [PubMed] [Google Scholar]
- (127).Wachter RM; Remington SJ Sensitivity of the Yellow Variant of Green Fluorescent Protein to Halides and Nitrate. Curr. Biol 1999, 9 (17), R628–9. [DOI] [PubMed] [Google Scholar]
- (128).Galietta LJ; Haggie PM; Verkman AS Green Fluorescent Protein-Based Halide Indicators with Improved Chloride and Iodide Affinities. FEBS Lett 2001, 499 (3), 220–224. [DOI] [PubMed] [Google Scholar]
- (129).Zhong S; Navaratnam D; Santos-Sacchi J A Genetically-Encoded YFP Sensor with Enhanced Chloride Sensitivity, Photostability and Reduced Ph Interference Demonstrates Augmented Transmembrane Chloride Movement by Gerbil Prestin (SLC26a5). PLoS One 2014, 9 (6), e99095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (130).Hanson GT; McAnaney TB; Park ES; Rendell MEP; Yarbrough DK; Chu S; Xi L; Boxer SG; Montrose MH; Remington SJ Green Fluorescent Protein Variants as Ratiometric Dual Emission pH Sensors. 1. Structural Characterization and Preliminary Application. Biochemistry 2002, 41 (52), 15477–15488. [DOI] [PubMed] [Google Scholar]
- (131).Grynkiewicz G; Poenie M; Tsien RY A New Generation of Ca2+ Indicators with Greatly Improved Fluorescence Properties. J. Biol. Chem 1985, 260 (6), 3440–3450. [PubMed] [Google Scholar]
- (132).Cinelli RAG; Pellegrini V; Ferrari A; Faraci P; Nifosı R; Tyagi M; Giacca M; Beltram F Green Fluorescent Proteins as Optically Controllable Elements in Bioelectronics. Appl. Phys. Lett 2001, 79 (20), 3353–3355. [Google Scholar]
- (133).Nifosì R; Ferrari A; Arcangeli C; Tozzini V; Pellegrini V; Beltram F Photoreversible Dark State in a Tristable Green Fluorescent Protein Variant. J. Phys. Chem. B 2003, 107 (7), 1679–1684. [Google Scholar]
- (134).Bizzarri R; Arcangeli C; Arosio D; Ricci F; Faraci P; Cardarelli F; Beltram F Development of a Novel GFP-Based Ratiometric Excitation and Emission pH Indicator for Intracellular Studies. Biophys. J 2006, 90 (9), 3300–3314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (135).Kuner T; Augustine GJ A Genetically Encoded Ratiometric Indicator for Chloride: Capturing Chloride Transients in Cultured Hippocampal Neurons. Neuron 2000, 27 (3), 447–459. [DOI] [PubMed] [Google Scholar]
- (136).Markova O; Mukhtarov M; Real E; Jacob Y; Bregestovski P Genetically Encoded Chloride Indicator with Improved Sensitivity. J. Neurosci. Methods 2008, 170 (1), 67–76. [DOI] [PubMed] [Google Scholar]
- (137).Arosio D; Garau G; Ricci F; Marchetti L; Bizzarri R; Nifosì R; Beltram F Spectroscopic and Structural Study of Proton and Halide Ion Cooperative Binding to Gfp. Biophys. J 2007, 93 (1), 232–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (138).Arosio D; Ricci F; Marchetti L; Gualdani R; Albertazzi L; Beltram F Simultaneous Intracellular Chloride and pH Measurements Using a GFP-Based Sensor. Nat. Methods 2010, 7 (7), 516–518. [DOI] [PubMed] [Google Scholar]
- (139).Hanson GT; Aggeler R; Oglesbee D; Cannon M; Capaldi RA; Tsien RY; Remington SJ Investigating Mitochondrial Redox Potential with Redox-Sensitive Green Fluorescent Protein Indicators. J. Biol. Chem 2004, 279 (13), 13044–13053. [DOI] [PubMed] [Google Scholar]
- (140).Dooley CT; Dore TM; Hanson GT; Jackson WC; Remington SJ; Tsien RY Imaging Dynamic Redox Changes in Mammalian Cells with Green Fluorescent Protein Indicators. J. Biol. Chem 2004, 279 (21), 22284–22293. [DOI] [PubMed] [Google Scholar]
- (141).Evers TH; Appelhof MAM; de Graaf-Heuvelmans PTHM; Meijer EW; Merkx M Ratiometric Detection of Zn(II) Using Chelating Fluorescent Protein Chimeras. J. Mol. Biol 2007, 374 (2), 411–425. [DOI] [PubMed] [Google Scholar]
- (142).Vinkenborg JL; van Duijnhoven SMJ; Merkx M Reengineering of a Fluorescent Zinc Sensor Protein Yields the First Genetically Encoded Cadmium Probe. Chem. Commun 2011, 47 (43), 11879. [DOI] [PubMed] [Google Scholar]
- (143).Heinemann U; Hahn M Circular Permutation of Polypeptide Chains: Implications for Protein Folding and Stability. Prog. Biophys. Mol. Biol 1995, 64 (2–3), 121–143. [DOI] [PubMed] [Google Scholar]
- (144).Baird GS; Zacharias DA; Tsien RY Circular Permutation and Receptor Insertion within Green Fluorescent Proteins. Proc. Natl. Acad. Sci. U. S. A 1999, 96 (20), 11241–11246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (145).Zhang J The Colorful Journey of Green Fluorescent Protein. ACS Chem. Biol 2009, 4 (2), 85–88. [DOI] [PubMed] [Google Scholar]
- (146).Klee CB; Crouch TH; Richman PG Calmodulin. Annu. Rev. Biochem 1980, 49 (1), 489–515. [DOI] [PubMed] [Google Scholar]
- (147).Chin D; Means AR Calmodulin: A Prototypical Calcium Sensor. Trends Cell Biol 2000, 10 (8), 322–328. [DOI] [PubMed] [Google Scholar]
- (148).Nagai T; Sawano A; Park ES; Miyawaki A Circularly Permuted Green Fluorescent Proteins Engineered to Sense Ca2+. Proc. Natl. Acad. Sci. U. S. A 2001, 98 (6), 3197–3202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (149).Rhoads AR; Friedberg F Sequence Motifs for Calmodulin Recognition. FASEB J 1997, 11 (5), 331–340. [DOI] [PubMed] [Google Scholar]
- (150).Nakai J; Ohkura M; Imoto K A High Signal-to-Noise Ca(2+) Probe Composed of a Single Green Fluorescent Protein. Nat. Biotechnol 2001, 19 (2), 137–141. [DOI] [PubMed] [Google Scholar]
- (151).Siemering KR; Golbik R; Sever R; Haseloff J Mutations That Suppress the Thermosensitivity of Green Fluorescent Protein. Curr. Biol 1996, 6 (12), 1653–1663. [DOI] [PubMed] [Google Scholar]
- (152).Zacharias DA; Violin JD; Newton AC; Tsien RY Partitioning of Lipid-Modified Monomeric GFPs into Membrane Microdomains of Live Cells. Science 2002, 296 (5569), 913–916. [DOI] [PubMed] [Google Scholar]
- (153).Tallini YN; Ohkura M; Choi B-R; Ji G; Imoto K; Doran R; Lee J; Plan P; Wilson J; Xin H-B; et al. Imaging Cellular Signals in the Heart in Vivo: Cardiac Expression of the High-Signal Ca2+ Indicator GCaMP2. Proc. Natl. Acad. Sci. U. S. A 2006, 103 (12), 4753–4758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (154).Tian L; Hires SA; Mao T; Huber D; Chiappe ME; Chalasani SH; Petreanu L; Akerboom J; McKinney SA; Schreiter ER; et al. Imaging Neural Activity in Worms, Flies and Mice with Improved GCaMP Calcium Indicators. Nat. Methods 2009, 6 (12), 875–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (155).Akerboom J; Chen T-W; Wardill TJ; Tian L; Marvin JS; Mutlu S; Calderón NC; Esposti F; Borghuis BG; Sun XR; et al. Optimization of a GCaMP Calcium Indicator for Neural Activity Imaging. J. Neurosci 2012, 32 (40), 13819–13840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (156).Chen TW; Wardill TJ; Sun Y; Pulver SR; Renninger SL; Baohan A; Schreiter ER; Kerr RA; Orger MB; Jayaraman V; et al. Ultrasensitive Fluorescent Proteins for Imaging Neuronal Activity. Nature 2013, 499 (7458), 295–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (157).Shaner NC; Lin MZ; McKeown MR; Steinbach PA; Hazelwood KL; Davidson MW; Tsien RY Improving the Photostability of Bright Monomeric Orange and Red Fluorescent Proteins. Nat. Methods 2008, 5 (6), 545–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (158).Zhao Y; Araki S; Wu J; Teramoto T; Chang Y-F; Nakano M; Abdelfattah AS; Fujiwara M; Ishihara T; Nagai T; et al. An Expanded Palette of Genetically Encoded Ca2+ Indicators. Science 2011, 333 (6051), 1888–1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (159).Wu J; Liu L; Matsuda T; Zhao Y; Rebane A; Drobizhev M; Chang Y-F; Araki S; Arai Y; March K; et al. Improved Orange and Red Ca2± Indicators and Photophysical Considerations for Optogenetic Applications. ACS Chem. Neurosci 2013, 4 (6), 963–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (160).Akerboom J; Carreras Calderón N; Tian L; Wabnig S; Prigge M; Tolö J; Gordus A; Orger MB; Severi KE; Macklin JJ; et al. Genetically Encoded Calcium Indicators for Multi-Color Neural Activity Imaging and Combination with Optogenetics. Front. Mol. Neurosci 2013, 6, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (161).Kredel S; Oswald F; Nienhaus K; Deuschle K; Röcker C; Wolff M; Heilker R; Nienhaus GU; Wiedenmann J mRuby, a Bright Monomeric Red Fluorescent Protein for Labeling of Subcellular Structures. PLoS One 2009, 4 (2), e4391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (162).Dana H; Mohar B; Sun Y; Narayan S; Gordus A; Hasseman JP; Tsegaye G; Holt GT; Hu A; Walpita D; et al. Sensitive Red Protein Calcium Indicators for Imaging Neural Activity. Elife 2016, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (163).Kitaguchi T; Oya M; Wada Y; Tsuboi T; Miyawaki A Extracellular Calcium Influx Activates Adenylate Cyclase 1 and Potentiates Insulin Secretion in MIN6 Cells. Biochem. J 2013, 450 (2), 365–373. [DOI] [PubMed] [Google Scholar]
- (164).Matsuda S; Harada K; Ito M; Takizawa M; Wongso D; Tsuboi T; Kitaguchi T Generation of a cGMP Indicator with an Expanded Dynamic Range by Optimization of Amino Acid Linkers between a Fluorescent Protein and PDE5α. ACS Sensors 2017, 2 (1), 46–51. [DOI] [PubMed] [Google Scholar]
- (165).Nausch LWM; Ledoux J; Bonev AD; Nelson MT; Dostmann WR Differential Patterning of cGMP in Vascular Smooth Muscle Cells Revealed by Single GFP-Linked Biosensors. Proc. Natl. Acad. Sci. U. S. A 2008, 105 (1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (166).Marvin JS; Borghuis BG; Tian L; Cichon J; Harnett MT; Akerboom J; Gordus A; Renninger SL; Chen T-W; Bargmann CI; et al. An Optimized Fluorescent Probe for Visualizing Glutamate Neurotransmission. Nat. Methods 2013, 10 (2), 162–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (167).St-Pierre F; Marshall JD; Yang Y; Gong Y; Schnitzer MJ; Lin MZ HighFidelity Optical Reporting of Neuronal Electrical Activity with an Ultrafast Fluorescent Voltage Sensor. Nat. Neurosci 2014, 17 (6), 884–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (168).Siegel MS; Isacoff EY A Genetically Encoded Optical Probe of Membrane Voltage. Neuron 1997, 19 (4), 735–741. [DOI] [PubMed] [Google Scholar]
- (169).Ataka K; Pieribone VA A Genetically Targetable Fluorescent Probe of Channel Gating with Rapid Kinetics. Biophys. J 2002, 82 (1 Pt 1), 509–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (170).Murata Y; Iwasaki H; Sasaki M; Inaba K; Okamura Y Phosphoinositide Phosphatase Activity Coupled to an Intrinsic Voltage Sensor. Nature 2005, 435 (7046), 1239–1243. [DOI] [PubMed] [Google Scholar]
- (171).Alabi AA; Bahamonde MI; Jung HJ; Kim J Il; Swartz, K. J. Portability of Paddle Motif Function and Pharmacology in Voltage Sensors. Nature 2007, 450 (7168), 370–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (172).Lundby A; Mutoh H; Dimitrov D; Akemann W; Knöpfel T Engineering of a Genetically Encodable Fluorescent Voltage Sensor Exploiting Fast Ci-VSP Voltage-Sensing Movements. PLoS One 2008, 3 (6), e2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (173).Rizzo MA; Springer GH; Granada B; Piston DW An Improved Cyan Fluorescent Protein Variant Useful for FRET. Nat. Biotechnol 2004, 22 (4), 445–449. [DOI] [PubMed] [Google Scholar]
- (174).Barnett L; Platisa J; Popovic M; Pieribone VA; Hughes T A Fluorescent, Genetically-Encoded Voltage Probe Capable of Resolving Action Potentials. PLoS One 2012, 7 (9), e43454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (175).Jensen MØ; Jogini V; Borhani DW; Leffler AE; Dror RO; Shaw DE Mechanism of Voltage Gating in Potassium Channels. Science 2012, 336 (6078), 229–233. [DOI] [PubMed] [Google Scholar]
- (176).Yang HH; St-Pierre F; Sun X; Ding X; Lin MZZ; Clandinin TRR Subcellular Imaging of Voltage and Calcium Signals Reveals Neural Processing In Vivo. Cell 2016, 166 (1), 245–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (177).Abdelfattah AS; Farhi SL; Zhao Y; Brinks D; Zou P; Ruangkittisakul A; Platisa J; Pieribone VA; Ballanyi K; Cohen AE; et al. A Bright and Fast Red Fluorescent Protein Voltage Indicator That Reports Neuronal Activity in Organotypic Brain Slices. J. Neurosci 2016, 36 (8), 2458–2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (178).Rehmann H; Prakash B; Wolf E; Rueppel A; de Rooij J; Bos JL; Wittinghofer A Structure and Regulation of the cAMP-Binding Domains of Epac2. Nat. Struct. Biol 2003, 10 (1), 26–32. [DOI] [PubMed] [Google Scholar]
- (179).Rehmann H; Rueppel A; Bos JL; Wittinghofer A Communication between the Regulatory and the Catalytic Region of the cAMP-Responsive Guanine Nucleotide Exchange Factor Epac. J. Biol. Chem 2003, 278 (26), 23508–23514. [DOI] [PubMed] [Google Scholar]
- (180).Odaka H; Arai S; Inoue T; Kitaguchi T Genetically-Encoded Yellow Fluorescent cAMP Indicator with an Expanded Dynamic Range for Dual-Color Imaging. PLoS One 2014, 9 (6), e100252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (181).Shaner NC; Lambert GG; Chammas A; Ni Y; Cranfill PJ; Baird MA; Sell BR; Allen JR; Day RN; Israelsson M; et al. A Bright Monomeric Green Fluorescent Protein Derived from Branchiostoma Lanceolatum. Nat. Methods 2013, 10 (5), 407–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (182).Tewson PH; Martinka S; Shaner NC; Hughes TE; Quinn AM New DAG and cAMP Sensors Optimized for Live-Cell Assays in Automated Laboratories. J. Biomol. Screen 2016, 21 (3), 298–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (183).Harada K; Ito M; Wang X; Tanaka M; Wongso D; Konno A; Hirai H; Hirase H; Tsuboi T; Kitaguchi T Red Fluorescent Protein-Based cAMP Indicator Applicable to Optogenetics and in Vivo Imaging. Sci. Rep 2017, 7 (1), 7351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (184).Beavo JA; Brunton LL Cyclic Nucleotide Research -- Still Expanding after Half a Century. Nat. Rev. Mol. Cell Biol 2002, 3 (9), 710–718. [DOI] [PubMed] [Google Scholar]
- (185).Zhao J; Trewhella J; Corbin J; Francis S; Mitchell R; Brushia R; Walsh D Progressive Cyclic Nucleotide-Induced Conformational Changes in the cGMP-Dependent Protein Kinase Studied by Small Angle X-Ray Scattering in Solution. J. Biol. Chem 1997, 272 (50), 31929–31936. [DOI] [PubMed] [Google Scholar]
- (186).Richie-Jannetta R; Busch JL; Higgins KA; Corbin JD; Francis SH Isolated Regulatory Domains of cGMP-Dependent Protein Kinase Ialpha and Ibeta Retain Dimerization and Native cGMP-Binding Properties and Undergo Isoform-Specific Conformational Changes. J. Biol. Chem 2006, 281 (11), 6977–6984. [DOI] [PubMed] [Google Scholar]
- (187).Ho YS; Burden LM; Hurley JH Structure of the GAF Domain, a Ubiquitous Signaling Motif and a New Class of Cyclic GMP Receptor. EMBO J 2000, 19 (20), 5288–5299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (188).Jäger R; Schwede F; Genieser H-G; Koesling D; Russwurm M Activation of PDE2 and PDE5 by Specific GAF Ligands: Delayed Activation of PDE5. Br. J. Pharmacol 2010, 161 (7), 1645–1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (189).Tam R; Saier MH Structural, Functional, and Evolutionary Relationships among Extracellular Solute-Binding Receptors of Bacteria. Microbiol. Rev 1993, 57 (2), 320–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (190).Dwyer MA; Hellinga HW Periplasmic Binding Proteins: A Versatile Superfamily for Protein Engineering. Curr. Opin. Struct. Biol 2004, 14 (4), 495–504. [DOI] [PubMed] [Google Scholar]
- (191).Sharff AJ; Rodseth LE; Spurlino JC; Quiocho FA Crystallographic Evidence of a Large Ligand-Induced Hinge-Twist Motion between the Two Domains of the Maltodextrin Binding Protein Involved in Active Transport and Chemotaxis. Biochemistry 1992, 31 (44), 10657–10663. [DOI] [PubMed] [Google Scholar]
- (192).Quiocho FA; Ledvina PS Atomic Structure and Specificity of Bacterial Periplasmic Receptors for Active Transport and Chemotaxis: Variation of Common Themes. Mol. Microbiol 1996, 20 (1), 17–25. [DOI] [PubMed] [Google Scholar]
- (193).Marvin JS; Schreiter ER; Echevarría IM; Looger LL A Genetically Encoded, High-Signal-to-Noise Maltose Sensor. Proteins Struct. Funct. Bioinforma 2011, 79 (11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (194).Alicea I; Marvin JS; Miklos AE; Ellington AD; Looger LL; Schreiter ER Structure of the Escherichia Coli Phosphonate Binding Protein PhnD and Rationally Optimized Phosphonate Biosensors. J. Mol. Biol 2011, 414 (3), 356–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (195).Dickson RM; Cubitt AB; Tsien RY; Moerner WE On/off Blinking and Switching Behaviour of Single Molecules of Green Fluorescent Protein. Nature 1997, 388 (6640), 355–358. [DOI] [PubMed] [Google Scholar]
- (196).Wu J; Abdelfattah AS; Miraucourt LS; Kutsarova E; Ruangkittisakul A; Zhou H; Ballanyi K; Wicks G; Drobizhev M; Rebane A; et al. A Long Stokes Shift Red Fluorescent Ca2+ Indicator Protein for Two-Photon and Ratiometric Imaging. Nat. Commun 2014, 5, 5262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (197).Zhao Y; Hu Q; Cheng F; Su N; Wang A; Zou Y; Hu H; Chen X; Zhou H-M; Huang X; et al. SoNar, a Highly Responsive NAD+/NADH Sensor, Allows High-Throughput Metabolic Screening of Anti-Tumor Agents. Cell Metab 2015, 21 (5), 777–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (198).Tao R; Zhao Y; Chu H; Wang A; Zhu J; Chen X; Zou Y; Shi M; Liu R; Su N; et al. Genetically Encoded Fluorescent Sensors Reveal Dynamic Regulation of NADPH Metabolism. Nat. Methods 2017, 14 (7), 720–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (199).Zapata-Hommer O; Griesbeck O Efficiently Folding and Circularly Permuted Variants of the Sapphire Mutant of GFP. BMC Biotechnol 2003, 3 (1), 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (200).Shaner NC; Campbell RE; Steinbach PA; Giepmans BNG; Palmer AE; Tsien RY Improved Monomeric Red, Orange and Yellow Fluorescent Proteins Derived from Discosoma Sp. Red Fluorescent Protein. Nat. Biotechnol 2004, 22 (12), 1567–1572. [DOI] [PubMed] [Google Scholar]
- (201).Hung YP; Albeck JG; Tantama M; Yellen G Imaging Cytosolic NADH-NAD+ Redox State with a Genetically Encoded Fluorescent Biosensor. Cell Metab 2011, 14 (4), 545–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (202).Cho J-H; Swanson CJ; Chen J; Li A; Lippert LG; Boye SE; Rose K; Sivaramakrishnan S; Chuong C-M; Chow RH The GCaMP-R Family of Genetically Encoded Ratiometric Calcium Indicators. ACS Chem. Biol 2017, 12 (4), 1066–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (203).Sivaramakrishnan S; Spudich JA Systematic Control of Protein Interaction Using a Modular ER/K α-Helix Linker. Proc. Natl. Acad. Sci. U. S. A 2011, 108 (51), 20467–20472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (204).Swanson CJ; Sivaramakrishnan S Harnessing the Unique Structural Properties of Isolated α-Helices. J. Biol. Chem 2014, 289 (37), 25460–25467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (205).Shcherbakova DM; Hink MA; Joosen L; Gadella TWJ; Verkhusha VV An Orange Fluorescent Protein with a Large Stokes Shift for Single-Excitation Multicolor FCCS and FRET Imaging. J. Am. Chem. Soc 2012, 134 (18), 7913–7923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (206).Ast C; Foret J; Oltrogge LM; De Michele R; Kleist TJ; Ho C-H; Frommer WB Ratiometric Matryoshka Biosensors from a Nested Cassette of Green- and Orange-Emitting Fluorescent Proteins. Nat. Commun 2017, 8 (1), 431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (207).Sakaue-Sawano A; Yo M; Komatsu N; Hiratsuka T; Kogure T; Hoshida T; Goshima N; Matsuda M; Miyoshi H; Miyawaki A Genetically Encoded Tools for Optical Dissection of the Mammalian Cell Cycle. Mol. Cell 2017, 68 (3), 626–640.e5. [DOI] [PubMed] [Google Scholar]
- (208).Bajar BT; Lam AJ; Badiee RK; Oh Y-H; Chu J; Zhou XX; Kim N; Kim BB; Chung M; Yablonovitch AL; et al. Fluorescent Indicators for Simultaneous Reporting of All Four Cell Cycle Phases. Nat. Methods 2016, 13 (12), 993–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (209).Rodriguez EA; Tran GN; Gross LA; Crisp JL; Shu X; Lin JY; Tsien RY A Far-Red Fluorescent Protein Evolved from a Cyanobacterial Phycobiliprotein. Nat. Methods 2016, 13 (9), 763–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (210).Shcherbakova DM; Baloban M; Emelyanov AV; Brenowitz M; Guo P; Verkhusha VV Bright Monomeric near-Infrared Fluorescent Proteins as Tags and Biosensors for Multiscale Imaging. Nat. Commun 2016, 7, 12405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (211).Nishimura K; Oki T; Kitaura J; Kuninaka S; Saya H; Sakaue-Sawano A; Miyawaki A; Kitamura T APC(CDH1) Targets MgcRacGAP for Destruction in the Late M Phase. PLoS One 2013, 8 (5), e63001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (212).Sakaue-Sawano A; Kobayashi T; Ohtawa K; Miyawaki A Drug-Induced Cell Cycle Modulation Leading to Cell-Cycle Arrest, Nuclear Mis-Segregation, or Endoreplication. BMC Cell Biol 2011, 12 (1), 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (213).Sugiyama M; Sakaue-Sawano A; Iimura T; Fukami K; Kitaguchi T; Kawakami K; Okamoto H; Higashijima S; Miyawaki A Illuminating Cell-Cycle Progression in the Developing Zebrafish Embryo. Proc. Natl. Acad. Sci. U. S. A 2009, 106 (49), 20812–20817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (214).Sakaue-Sawano A; Ohtawa K; Hama H; Kawano M; Ogawa M; Miyawaki A Tracing the Silhouette of Individual Cells in S/G2/M Phases with Fluorescence. Chem. Biol 2008, 15 (12), 1243–1248. [DOI] [PubMed] [Google Scholar]
- (215).Sakaue-Sawano A; Kurokawa H; Morimura T; Hanyu A; Hama H; Osawa H; Kashiwagi S; Fukami K; Miyata T; Miyoshi H; et al. Visualizing Spatiotemporal Dynamics of Multicellular Cell-Cycle Progression. Cell 2008, 132 (3), 487–498. [DOI] [PubMed] [Google Scholar]
- (216).Hahn AT; Jones JT; Meyer T Quantitative Analysis of Cell Cycle Phase Durations and PC12 Differentiation Using Fluorescent Biosensors. Cell Cycle 2009, 8 (7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (217).Austen K; Ringer P; Mehlich A; Chrostek-Grashoff A; Kluger C; Klingner C; Sabass B; Zent R; Rief M; Grashoff C Extracellular Rigidity Sensing by Talin Isoform-Specific Mechanical Linkages. Nat. Cell Biol 2015, 17 (12), 1597–1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (218).Iwai S; Uyeda TQP Visualizing Myosin-Actin Interaction with a Genetically-Encoded Fluorescent Strain Sensor. Proc. Natl. Acad. Sci. U. S. A 2008, 105 (44), 16882–16887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (219).Meng F; Sachs F Orientation-Based FRET Sensor for Real-Time Imaging of Cellular Forces. J. Cell Sci 2012, 125 (Pt 3), 743–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (220).Meng F; Sachs F Visualizing Dynamic Cytoplasmic Forces with a ComplianceMatched FRET Sensor. J. Cell Sci 2011, 124 (Pt 2), 261–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (221).Meng F; Suchyna TM; Sachs F A Fluorescence Energy Transfer-Based Mechanical Stress Sensor for Specific Proteins in Situ. FEBS J 2008, 275 (12), 3072–3087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (222).Brenner MD; Zhou R; Conway DE; Lanzano L; Gratton E; Schwartz MA; Ha T Spider Silk Peptide Is a Compact, Linear Nanospring Ideal for Intracellular Tension Sensing. Nano Lett 2016, 16 (3), 2096–2102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (223).Grashoff C; Hoffman BD; Brenner MD; Zhou R; Parsons M; Yang MT; McLean MA; Sligar SG; Chen CS; Ha T; et al. Measuring Mechanical Tension across Vinculin Reveals Regulation of Focal Adhesion Dynamics. Nature 2010, 466 (7303), 263–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (224).Rupprecht C; Wingen M; Potzkei J; Gensch T; Jaeger K-E; Drepper T A Novel FbFP-Based Biosensor Toolbox for Sensitive in Vivo Determination of Intracellular pH. J. Biotechnol 2017, 258, 25–32. [DOI] [PubMed] [Google Scholar]
- (225).Abad MFC; Di Benedetto G; Magalhães PJ; Filippin L; Pozzan T Mitochondrial pH Monitored by a New Engineered Green Fluorescent Protein Mutant. J. Biol. Chem 2004, 279 (12), 11521–11529. [DOI] [PubMed] [Google Scholar]
- (226).Johnson DE; Ai H; Wong P; Young JD; Campbell RE; Casey JR Red Fluorescent Protein pH Biosensor to Detect Concentrative Nucleoside Transport. J. Biol. Chem 2009, 284 (31), 20499–20511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (227).Hellwig N; Plant TD; Janson W; Schäfer M; Schultz G; Schaefer M TRPV1 Acts as Proton Channel to Induce Acidification in Nociceptive Neurons. J. Biol. Chem 2004, 279 (33), 34553–34561. [DOI] [PubMed] [Google Scholar]
- (228).Urra J; Sandoval M; Cornejo I; Barros LF; Sepúlveda FV; Cid LP A Genetically Encoded Ratiometric Sensor to Measure Extracellular pH in Microdomains Bounded by Basolateral Membranes of Epithelial Cells. Pflügers Arch. - Eur. J. Physiol 2008, 457 (1), 233–242. [DOI] [PubMed] [Google Scholar]
- (229).Mahon MJ pHluorin2: An Enhanced, Ratiometric, pH-Sensitive Green Florescent Protein. Adv. Biosci. Biotechnol 2011, 2 (3), 132–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (230).Tantama M; Hung YP; Yellen G Imaging Intracellular pH in Live Cells with a Genetically Encoded Red Fluorescent Protein Sensor. J. Am. Chem. Soc 2011, 133 (26), 10034–10037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (231).Shen Y; Rosendale M; Campbell RE; Perrais D pHuji, a pH-Sensitive Red Fluorescent Protein for Imaging of Exo- and Endocytosis. J. Cell Biol 2014, 207 (3), 419–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (232).Gjetting SK; Ytting CK; Schulz A; Fuglsang AT Live Imaging of Intra- and Extracellular pH in Plants Using pHusion, a Novel Genetically Encoded Biosensor. J. Exp. Bot 2012, 63 (8), 3207–3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (233).Poburko D; Santo-Domingo J; Demaurex N Dynamic Regulation of the Mitochondrial Proton Gradient during Cytosolic Calcium Elevations. J. Biol. Chem 2011, 286 (13), 11672–11684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (234).Awaji T; Hirasawa A; Shirakawa H; Tsujimoto G; Miyazaki S Novel Green Fluorescent Protein-Based Ratiometric Indicators for Monitoring pH in Defined Intracellular Microdomains. Biochem. Biophys. Res. Commun 2001, 289 (2), 457–462. [DOI] [PubMed] [Google Scholar]
- (235).Han Z; Jin L; Platisa J; Cohen LB; Baker BJ; Pieribone VA Fluorescent Protein Voltage Probes Derived from ArcLight That Respond to Membrane Voltage Changes with Fast Kinetics. PLoS One 2013, 8 (11), e81295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (236).Kost LA; Nikitin ES; Ivanova VO; Sung U; Putintseva EV; Chudakov DM; Balaban PM; Lukyanov KA; Bogdanov AM Insertion of the Voltage-Sensitive Domain into Circularly Permuted Red Fluorescent Protein as a Design for Genetically Encoded Voltage Sensor. PLoS One 2017, 12 (9), e0184225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (237).Inagaki S; Tsutsui H; Suzuki K; Agetsuma M; Arai Y; Jinno Y; Bai G; Daniels MJ; Okamura Y; Matsuda T; et al. Genetically Encoded Bioluminescent Voltage Indicator for Multi-Purpose Use in Wide Range of Bioimaging. Sci. Rep 2017, 7, 42398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (238).Platisa J; Vasan G; Yang A; Pieribone VA Directed Evolution of Key Residues in Fluorescent Protein Inverses the Polarity of Voltage Sensitivity in the Genetically Encoded Indicator ArcLight. ACS Chem. Neurosci 2017, 8 (3), 513–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (239).Abdelfattah AS; Rancic V; Rawal B; Ballanyi K; Campbell RE Ratiometric and Photoconvertible Fluorescent Protein-Based Voltage Indicator Prototypes. Chem. Commun 2016, 52 (98), 14153–14156. [DOI] [PubMed] [Google Scholar]
- (240).Piao HH; Rajakumar D; Kang BE; Kim EH; Baker BJ Combinatorial Mutagenesis of the Voltage-Sensing Domain Enables the Optical Resolution of Action Potentials Firing at 60 Hz by a Genetically Encoded Fluorescent Sensor of Membrane Potential. J. Neurosci 2015, 35 (1), 372–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (241).Treger JS; Priest MF; Bezanilla F Single-Molecule Fluorimetry and Gating Currents Inspire an Improved Optical Voltage Indicator. Elife 2015, 4, e10482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (242).Sung U; Sepehri-Rad M; Piao HH; Jin L; Hughes T; Cohen LB; Baker BJ Developing Fast Fluorescent Protein Voltage Sensors by Optimizing FRET Interactions. PLoS One 2015, 10 (11), e0141585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (243).Mishina Y; Mutoh H; Song C; Knöpfel T Exploration of Genetically Encoded Voltage Indicators Based on a Chimeric Voltage Sensing Domain. Front. Mol. Neurosci 2014, 7, 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (244).Tsutsui H; Jinno Y; Tomita A; Niino Y; Yamada Y; Mikoshiba K; Miyawaki A; Okamura Y Improved Detection of Electrical Activity with a Voltage Probe Based on a Voltage-Sensing Phosphatase. J. Physiol 2013, 591 (18), 4427–4437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (245).Jin L; Han Z; Platisa J; Wooltorton JRA; Cohen LB; Pieribone VA Single Action Potentials and Subthreshold Electrical Events Imaged in Neurons with a Fluorescent Protein Voltage Probe. Neuron 2012, 75 (5), 779–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (246).Mishina Y; Mutoh H; Knöpfel T Transfer of Kv3.1 Voltage Sensor Features to the Isolated Ci-VSP Voltage-Sensing Domain. Biophys. J 2012, 103 (4), 669–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (247).Lam AJ; St-Pierre F; Gong Y; Marshall JD; Cranfill PJ; Baird MA; McKeown MR; Wiedenmann J; Davidson MW; Schnitzer MJ; et al. Improving FRET Dynamic Range with Bright Green and Red Fluorescent Proteins. Nat. Methods 2012, 9 (10), 1005–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (248).Akemann W; Mutoh H; Perron A; Park YK; Iwamoto Y; Knöpfel T Imaging Neural Circuit Dynamics with a Voltage-Sensitive Fluorescent Protein. J. Neurophysiol 2012, 108 (8), 2323–2337. [DOI] [PubMed] [Google Scholar]
- (249).Lundby A; Akemann W; Knöpfel T Biophysical Characterization of the Fluorescent Protein Voltage Probe VSFP2.3 Based on the Voltage-Sensing Domain of Ci-VSP. Eur. Biophys. J 2010, 39 (12), 1625–1635. [DOI] [PubMed] [Google Scholar]
- (250).Akemann W; Mutoh H; Perron A; Rossier J; Knöpfel T Imaging Brain Electric Signals with Genetically Targeted Voltage-Sensitive Fluorescent Proteins. Nat. Methods 2010, 7 (8), 643–649. [DOI] [PubMed] [Google Scholar]
- (251).Mutoh H; Perron A; Dimitrov D; Iwamoto Y; Akemann W; Chudakov DM; Knöpfel T Spectrally-Resolved Response Properties of the Three Most Advanced FRET Based Fluorescent Protein Voltage Probes. PLoS One 2009, 4 (2), e4555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (252).Tsutsui H; Karasawa S; Okamura Y; Miyawaki A Improving Membrane Voltage Measurements Using FRET with New Fluorescent Proteins. Nat. Methods 2008, 5 (8), 683–685. [DOI] [PubMed] [Google Scholar]
- (253).Dimitrov D; He Y; Mutoh H; Baker BJ; Cohen L; Akemann W; Knöpfel T Engineering and Characterization of an Enhanced Fluorescent Protein Voltage Sensor. PLoS One 2007, 2 (5), e440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (254).Jung A; Garcia JE; Kim E; Yoon B-J; Baker BJ Linker Length and Fusion Site Composition Improve the Optical Signal of Genetically Encoded Fluorescent Voltage Sensors. Neurophotonics 2015, 2 (2), 21012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (255).Baker BJ; Jin L; Han Z; Cohen LB; Popovic M; Platisa J; Pieribone V Genetically Encoded Fluorescent Voltage Sensors Using the Voltage-Sensing Domain of Nematostella and Danio Phosphatases Exhibit Fast Kinetics. J. Neurosci. Methods 2012, 208 (2), 190–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (256).Guerrero G; Siegel MS; Roska B; Loots E; Isacoff EY Tuning FlaSh: Redesign of the Dynamics, Voltage Range, and Color of the Genetically Encoded Optical Sensor of Membrane Potential. Biophys. J 2002, 83 (6), 3607–3618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (257).Sakai R; Repunte-Canonigo V; Raj CD; Knöpfel T Design and Characterization of a DNA-Encoded, Voltage-Sensitive Fluorescent Protein. Eur. J. Neurosci 2001, 13 (12), 2314–2318. [DOI] [PubMed] [Google Scholar]
- (258).Piatkevich KD; Jung EE; Straub C; Linghu C; Park D; Suk H-J; Hochbaum DR; Goodwin D; Pnevmatikakis E; Pak N; et al. A Robotic Multidimensional Directed Evolution Approach Applied to Fluorescent Voltage Reporters. Nat. Chem. Biol 2018, 14 (4), 352–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (259).Gong Y; Huang C; Li JZ; Grewe BF; Zhang Y; Eismann S; Schnitzer MJ High-Speed Recording of Neural Spikes in Awake Mice and Flies with a Fluorescent Voltage Sensor. Science 2015, 350 (6266), 1361–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (260).Hochbaum DR; Zhao Y; Farhi SL; Klapoetke N; Werley CA; Kapoor V; Zou P; Kralj JM; Maclaurin D; Smedemark-Margulies N; et al. All-Optical Electrophysiology in Mammalian Neurons Using Engineered Microbial Rhodopsins. Nat. Methods 2014, 11 (8), 825–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (261).Flytzanis NC; Bedbrook CN; Chiu H; Engqvist MKM; Xiao C; Chan KY; Sternberg PW; Arnold FH; Gradinaru V Archaerhodopsin Variants with Enhanced Voltage-Sensitive Fluorescence in Mammalian and Caenorhabditis Elegans Neurons. Nat. Commun 2014, 5, 4894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (262).Zou P; Zhao Y; Douglass AD; Hochbaum DR; Brinks D; Werley CA; Harrison DJ; Campbell RE; Cohen AE Bright and Fast Multicoloured Voltage Reporters via Electrochromic FRET. Nat. Commun 2014, 5, 4625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (263).Gong Y; Wagner MJ; Zhong Li J; Schnitzer MJ Imaging Neural Spiking in Brain Tissue Using FRET-Opsin Protein Voltage Sensors. Nat. Commun 2014, 5, 3674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (264).Gong Y; Li JZ; Schnitzer MJ Enhanced Archaerhodopsin Fluorescent Protein Voltage Indicators. PLoS One 2013, 8 (6), e66959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (265).Kralj JM; Hochbaum DR; Douglass AD; Cohen AE Electrical Spiking in Escherichia Coli Probed with a Fluorescent Voltage-Indicating Protein. Science 2011, 333 (6040), 345–348. [DOI] [PubMed] [Google Scholar]
- (266).Kralj JM; Douglass AD; Hochbaum DR; Maclaurin D; Cohen AE Optical Recording of Action Potentials in Mammalian Neurons Using a Microbial Rhodopsin. Nat. Methods 2011, 9 (1), 90–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (267).Zhang C; Ye B-C A Single Fluorescent Protein-Based Sensor for in Vivo 2Oxogluatarate Detection in Cell. Biosens. Bioelectron 2014, 54, 15–19. [DOI] [PubMed] [Google Scholar]
- (268).De Michele R; Ast C; Loqué D; Ho C-H; Andrade SLA; Lanquar V; Grossmann G; Gehne S; Kumke MU; Frommer WB Fluorescent Sensors Reporting the Activity of Ammonium Transceptors in Live Cells. Elife 2013, 2, e00800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (269).Kaper T; Lager I; Looger LL; Chermak D; Frommer WB Fluorescence Resonance Energy Transfer Sensors for Quantitative Monitoring of Pentose and Disaccharide Accumulation in Bacteria. Biotechnol. Biofuels 2008, 1 (1), 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (270).Bogner M; Ludewig U Visualization of Arginine Influx into Plant Cells Using a Specific FRET-Sensor. J. Fluoresc 2007, 17 (4), 350–360. [DOI] [PubMed] [Google Scholar]
- (271).Yoshida T; Kakizuka A; Imamura H BTeam, a Novel BRET-Based Biosensor for the Accurate Quantification of ATP Concentration within Living Cells. Sci. Rep 2016, 6 (1), 39618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (272).Tsuyama T; Kishikawa J; Han Y-W; Harada Y; Tsubouchi A; Noji H; Kakizuka A; Yokoyama K; Uemura T; Imamura H In Vivo Fluorescent Adenosine 5’triphosphate (ATP) Imaging of Drosophila Melanogaster and Caenorhabditis Elegans by Using a Genetically Encoded Fluorescent ATP Biosensor Optimized for Low Temperatures. Anal. Chem 2013, 85 (16), 7889–7896. [DOI] [PubMed] [Google Scholar]
- (273).Nakano M; Imamura H; Nagai T; Noji H Ca2+ Regulation of Mitochondrial ATP Synthesis Visualized at the Single Cell Level. ACS Chem. Biol 2011, 6 (7), 709–715. [DOI] [PubMed] [Google Scholar]
- (274).Imamura H; Huynh Nhat KP; Togawa H; Saito K; Iino R; Kato-Yamada Y; Nagai T; Noji H Visualization of ATP Levels inside Single Living Cells with Fluorescence Resonance Energy Transfer-Based Genetically Encoded Indicators. Proc. Natl. Acad. Sci 2009, 106 (37), 15651–15656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (275).Zadran S; Sanchez D; Zadran H; Amighi A; Otiniano E; Wong K Enhanced-Acceptor Fluorescence-Based Single Cell ATP Biosensor Monitors ATP in Heterogeneous Cancer Populations in Real Time. Biotechnol. Lett 2013, 35 (2), 175–180. [DOI] [PubMed] [Google Scholar]
- (276).Saito K; Chang Y-F; Horikawa K; Hatsugai N; Higuchi Y; Hashida M; Yoshida Y; Matsuda T; Arai Y; Nagai T Luminescent Proteins for High-Speed Single-Cell and Whole-Body Imaging. Nat. Commun 2012, 3, 1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (277).Tantama M; Martínez-François JR; Mongeon R; Yellen G Imaging Energy Status in Live Cells with a Fluorescent Biosensor of the Intracellular ATP-to-ADP Ratio. Nat. Commun 2013, 4, 2550.24096541 [Google Scholar]
- (278).Berg J; Hung YP; Yellen G A Genetically Encoded Fluorescent Reporter of ATP:ADP Ratio. Nat. Methods 2009, 6 (2), 161–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (279).Yaginuma H; Kawai S; Tabata KV; Tomiyama K; Kakizuka A; Komatsuzaki T; Noji H; Imamura H Diversity in ATP Concentrations in a Single Bacterial Cell Population Revealed by Quantitative Single-Cell Imaging. Sci. Rep 2014, 4 (1), 6522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (280).Rangaraju V; Calloway N; Ryan TA Activity-Driven Local ATP Synthesis Is Required for Synaptic Function. Cell 2014, 156 (4), 825–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (281).Nakajima T; Sato M; Akaza N; Umezawa Y Cell-Based Fluorescent Indicator To Visualize Brain-Derived Neurotrophic Factor Secreted from Living Neurons. ACS Chem. Biol 2008, 3 (6). [DOI] [PubMed] [Google Scholar]
- (282).Castro LRV; Gervasi N; Guiot E; Cavellini L; Nikolaev VO; Paupardin-Tritsch D; Vincent P Type 4 Phosphodiesterase Plays Different Integrating Roles in Different Cellular Domains in Pyramidal Cortical Neurons. J. Neurosci 2010, 30 (17), 6143–6151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (283).Norris RP; Ratzan WJ; Freudzon M; Mehlmann LM; Krall J; Movsesian MA; Wang H; Ke H; Nikolaev VO; Jaffe LA Cyclic GMP from the Surrounding Somatic Cells Regulates Cyclic AMP and Meiosis in the Mouse Oocyte. Development 2009, 136 (11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (284).Nikolaev VO; Bünemann M; Schmitteckert E; Lohse MJ; Engelhardt S Cyclic AMP Imaging in Adult Cardiac Myocytes Reveals Far-Reaching beta1-Adrenergic but Locally Confined beta2-Adrenergic Receptor-Mediated Signaling. Circ. Res 2006, 99 (10), 1084–1091. [DOI] [PubMed] [Google Scholar]
- (285).Nikolaev VO; Bünemann M; Hein L; Hannawacker A; Lohse MJ Novel Single Chain cAMP Sensors for Receptor-Induced Signal Propagation. J. Biol. Chem 2004, 279 (36), 37215–37218. [DOI] [PubMed] [Google Scholar]
- (286).Jiang LI; Collins J; Davis R; Lin K-M; DeCamp D; Roach T; Hsueh R; Rebres RA; Ross EM; Taussig R; et al. Use of a cAMP BRET Sensor to Characterize a Novel Regulation of cAMP by the Sphingosine 1-phosphate/G13 Pathway. J. Biol. Chem 2007, 282 (14), 10576–10584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (287).Surdo NC; Berrera M; Koschinski A; Brescia M; Machado MR; Carr C; Wright P; Gorelik J; Morotti S; Grandi E; et al. FRET Biosensor Uncovers cAMP NanoDomains at β-Adrenergic Targets That Dictate Precise Tuning of Cardiac Contractility. Nat. Commun 2017, 8, 15031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (288).Klarenbeek J; Goedhart J; van Batenburg A; Groenewald D; Jalink K FourthGeneration Epac-Based FRET Sensors for cAMP Feature Exceptional Brightness, Photostability and Dynamic Range: Characterization of Dedicated Sensors for FLIM, for Ratiometry and with High Affinity. PLoS One 2015, 10 (4), e0122513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (289).Klarenbeek JB; Goedhart J; Hink MA; Gadella TWJ; Jalink K A mTurquoiseBased cAMP Sensor for Both FLIM and Ratiometric Read-out Has Improved Dynamic Range. PLoS One 2011, 6 (4), e19170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (290).van der Krogt GNM; Ogink J; Ponsioen B; Jalink K A Comparison of DonorAcceptor Pairs for Genetically Encoded FRET Sensors: Application to the Epac cAMP Sensor as an Example. PLoS One 2008, 3 (4), e1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (291).Salonikidis PS; Niebert M; Ullrich T; Bao G; Zeug A; Richter DW An IonInsensitive cAMP Biosensor for Long Term Quantitative Ratiometric Fluorescence Resonance Energy Transfer (FRET) Measurements under Variable Physiological Conditions. J. Biol. Chem 2011, 286 (26), 23419–23431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (292).Ponsioen B; Zhao J; Riedl J; Zwartkruis F; van der Krogt G; Zaccolo M; Moolenaar WH; Bos JL; Jalink K Detecting cAMP-Induced Epac Activation by Fluorescence Resonance Energy Transfer: Epac as a Novel cAMP Indicator. EMBO Rep 2004, 5 (12), 1176–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (293).Ding Y; Li J; Enterina JR; Shen Y; Zhang I; Tewson PH; Mo GCH; Zhang J; Quinn AM; Hughes TE; et al. Ratiometric Biosensors Based on Dimerization-Dependent Fluorescent Protein Exchange. Nat. Methods 2015, 12 (3), 195–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (294).Ni Q; Ganesan A; Aye-Han N-N; Gao X; Allen MD; Levchenko A; Zhang J Signaling Diversity of PKA Achieved via a Ca(2+)-cAMP-PKA Oscillatory Circuit. Nat. Chem. Biol 2011, 7 (1), 34–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (295).DiPilato LM; Zhang J The Role of Membrane Microdomains in Shaping beta2-Adrenergic Receptor-Mediated cAMP Dynamics. Mol. Biosyst 2009, 5 (8), 832–837. [DOI] [PubMed] [Google Scholar]
- (296).Barak LS; Salahpour A; Zhang X; Masri B; Sotnikova TD; Ramsey AJ; Violin JD; Lefkowitz RJ; Caron MG; Gainetdinov RR Pharmacological Characterization of Membrane-Expressed Human Trace Amine-Associated Receptor 1 (TAAR1) by a Bioluminescence Resonance Energy Transfer cAMP Biosensor. Mol. Pharmacol 2008, 74 (3), 585–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (297).Violin JD; DiPilato LM; Yildirim N; Elston TC; Zhang J; Lefkowitz RJ beta2-Adrenergic Receptor Signaling and Desensitization Elucidated by Quantitative Modeling of Real Time cAMP Dynamics. J. Biol. Chem 2008, 283 (5), 2949–2961. [DOI] [PubMed] [Google Scholar]
- (298).DiPilato LM; Cheng X; Zhang J Fluorescent Indicators of cAMP and Epac Activation Reveal Differential Dynamics of cAMP Signaling within Discrete Subcellular Compartments. Proc. Natl. Acad. Sci. U. S. A 2004, 101 (47), 16513–16518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (299).Krähling AM; Alvarez L; Debowski K; Van Q; Gunkel M; Irsen S; Al-Amoudi A; Strünker T; Kremmer E; Krause E; et al. CRIS-a Novel cAMP-Binding Protein Controlling Spermiogenesis and the Development of Flagellar Bending. PLoS Genet 2013, 9 (12), e1003960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (300).Mukherjee S; Jansen V; Jikeli JF; Hamzeh H; Alvarez L; Dombrowski M; Balbach M; Strünker T; Seifert R; Kaupp UB; et al. A Novel Biosensor to Study cAMP Dynamics in Cilia and Flagella. Elife 2016, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (301).Ohta Y; Kamagata T; Mukai A; Takada S; Nagai T; Horikawa K Nontrivial Effect of the Color-Exchange of a Donor/Acceptor Pair in the Engineering of Förster Resonance Energy Transfer (FRET)-Based Indicators. ACS Chem. Biol 2016, 11 (7), 1816–1822. [DOI] [PubMed] [Google Scholar]
- (302).Binkowski BF; Butler BL; Stecha PF; Eggers CT; Otto P; Zimmerman K; Vidugiris G; Wood MG; Encell LP; Fan F; et al. A Luminescent Biosensor with Increased Dynamic Range for Intracellular cAMP. ACS Chem. Biol 2011, 6 (11), 1193–1197. [DOI] [PubMed] [Google Scholar]
- (303).Fan F; Binkowski BF; Butler BL; Stecha PF; Lewis MK; Wood KV Novel Genetically Encoded Biosensors Using Firefly Luciferase. ACS Chem. Biol 2008, 3 (6), 346–351. [DOI] [PubMed] [Google Scholar]
- (304).Dyachok O; Isakov Y; Sågetorp J; Tengholm A Oscillations of Cyclic AMP in Hormone-Stimulated Insulin-Secreting β-Cells. Nature 2006, 439 (7074), 349–352. [DOI] [PubMed] [Google Scholar]
- (305).Prinz A; Diskar M; Erlbruch A; Herberg FW Novel, Isotype-Specific Sensors for Protein Kinase A Subunit Interaction Based on Bioluminescence Resonance Energy Transfer (BRET). Cell. Signal 2006, 18 (10), 1616–1625. [DOI] [PubMed] [Google Scholar]
- (306).Mongillo M; McSorley T; Evellin S; Sood A; Lissandron V; Terrin A; Huston E; Hannawacker A; Lohse MJ; Pozzan T; et al. Fluorescence Resonance Energy Transfer-Based Analysis of cAMP Dynamics in Live Neonatal Rat Cardiac Myocytes Reveals Distinct Functions of Compartmentalized Phosphodiesterases. Circ. Res 2004, 95 (1), 67–75. [DOI] [PubMed] [Google Scholar]
- (307).Zaccolo M; Pozzan T Discrete Microdomains with High Concentration of cAMP in Stimulated Rat Neonatal Cardiac Myocytes. Science 2002, 295 (5560), 1711–1715. [DOI] [PubMed] [Google Scholar]
- (308).Zaccolo M; De Giorgi F; Cho CY; Feng L; Knapp T; Negulescu PA; Taylor SS; Tsien RY; Pozzan T A Genetically Encoded, Fluorescent Indicator for Cyclic AMP in Living Cells. Nat. Cell Biol 2000, 2 (1), 25–29. [DOI] [PubMed] [Google Scholar]
- (309).Niino Y; Hotta K; Oka K Blue Fluorescent cGMP Sensor for Multiparameter Fluorescence Imaging. PLoS One 2010, 5 (2), e9164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (310).Nikolaev VO; Gambaryan S; Lohse MJ Fluorescent Sensors for Rapid Monitoring of Intracellular cGMP. Nat. Methods 2006, 3 (1), 23–25. [DOI] [PubMed] [Google Scholar]
- (311).Russwurm M; Mullershausen F; Friebe A; Jäger R; Russwurm C; Koesling D Design of Fluorescence Resonance Energy Transfer (FRET)-Based cGMP Indicators: A Systematic Approach. Biochem. J 2007, 407 (1), 69–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (312).Sato M; Hida N; Ozawa T; Umezawa Y Fluorescent Indicators for Cyclic GMP Based on Cyclic GMP-Dependent Protein Kinase Ialpha and Green Fluorescent Proteins. Anal. Chem 2000, 72 (24), 5918–5924. [DOI] [PubMed] [Google Scholar]
- (313).Honda A; Sawyer CL; Cawley SM; Dostmann WRG Cygnets: In Vivo Characterization of Novel cGMP Indicators and in Vivo Imaging of Intracellular cGMP. Methods Mol. Biol 2005, 307, 27–43. [DOI] [PubMed] [Google Scholar]
- (314).Honda A; Adams SR; Sawyer CL; Lev-Ram V; Tsien RY; Dostmann WR Spatiotemporal Dynamics of Guanosine 3’,5’-cyclic Monophosphate Revealed by a Genetically Encoded, Fluorescent Indicator. Proc. Natl. Acad. Sci. U. S. A 2001, 98 (5), 2437–2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (315).Bhargava Y; Hampden-Smith K; Chachlaki K; Wood KC; Vernon J; Allerston CK; Batchelor AM; Garthwaite J Improved Genetically-Encoded, FlincG-Type Fluorescent Biosensors for Neural cGMP Imaging. Front. Mol. Neurosci 2013, 6, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (316).Ewald JC; Reich S; Baumann S; Frommer WB; Zamboni N Engineering Genetically Encoded Nanosensors for Real-Time in Vivo Measurements of Citrate Concentrations. PLoS One 2011, 6 (12), e28245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (317).Wang J; Karpus J; Zhao BS; Luo Z; Chen PR; He C A Selective Fluorescent Probe for Carbon Monoxide Imaging in Living Cells. Angew. Chemie Int. Ed 2012, 51 (38), 9652–9656. [DOI] [PubMed] [Google Scholar]
- (318).Takanaga H; Chaudhuri B; Frommer WB GLUT1 and GLUT9 as Major Contributors to Glucose Influx in HepG2 Cells Identified by a High Sensitivity Intramolecular FRET Glucose Sensor. Biochim. Biophys. Acta 2008, 1778 (4), 1091–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (319).Deuschle K; Chaudhuri B; Okumoto S; Lager I; Lalonde S; Frommer WB Rapid Metabolism of Glucose Detected with FRET Glucose Nanosensors in Epidermal Cells and Intact Roots of Arabidopsis RNA-Silencing Mutants. Plant Cell 2006, 18 (9), 2314–2325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (320).Deuschle K; Okumoto S; Fehr M; Looger LL; Kozhukh L; Frommer WB Construction and Optimization of a Family of Genetically Encoded Metabolite Sensors by Semirational Protein Engineering. Protein Sci 2005, 14 (9), 2304–2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (321).Fehr M; Lalonde S; Lager I; Wolff MW; Frommer WB In Vivo Imaging of the Dynamics of Glucose Uptake in the Cytosol of COS-7 Cells by Fluorescent Nanosensors. J. Biol. Chem 2003, 278 (21), 19127–19133. [DOI] [PubMed] [Google Scholar]
- (322).Veetil JV; Jin S; Ye K A Glucose Sensor Protein for Continuous Glucose Monitoring. Biosens. Bioelectron 2010, 26 (4), 1650–1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (323).Garrett JR; Wu X; Jin S; Ye K pH-Insensitive Glucose Indicators. Biotechnol. Prog 2008, 24 (5), 1085–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (324).Ye K; Schultz JS Genetic Engineering of an Allosterically Based Glucose Indicator Protein for Continuous Glucose Monitoring by Fluorescence Resonance Energy Transfer. Anal. Chem 2003, 75 (14), 3451–3459. [DOI] [PubMed] [Google Scholar]
- (325).Gruenwald K; Holland JT; Stromberg V; Ahmad A; Watcharakichkorn D; Okumoto S Visualization of Glutamine Transporter Activities in Living Cells Using Genetically Encoded Glutamine Sensors. PLoS One 2012, 7 (6), e38591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (326).Abshire JR; Rowlands CJ; Ganesan SM; So PTC; Niles JC Quantification of Labile Heme in Live Malaria Parasites Using a Genetically Encoded Biosensor. Proc. Natl. Acad. Sci. U. S. A 2017, 114 (11), E2068–E2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (327).Song Y; Yang M; Wegner SV; Zhao J; Zhu R; Wu Y; He C; Chen PR A Genetically Encoded FRET Sensor for Intracellular Heme. ACS Chem. Biol 2015, 10 (7), 1610–1615. [DOI] [PubMed] [Google Scholar]
- (328).Hanna DA; Harvey RM; Martinez-Guzman O; Yuan X; Chandrasekharan B; Raju G; Outten FW; Hamza I; Reddi AR Heme Dynamics and Trafficking Factors Revealed by Genetically Encoded Fluorescent Heme Sensors. Proc. Natl. Acad. Sci. U. S. A 2016, 113 (27), 7539–7544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (329).Arpino JAJ; Czapinska H; Piasecka A; Edwards WR; Barker P; Gajda MJ; Bochtler M; Jones DD Structural Basis for Efficient Chromophore Communication and Energy Transfer in a Constructed Didomain Protein Scaffold. J. Am. Chem. Soc 2012, 134 (33), 13632–13640. [DOI] [PubMed] [Google Scholar]
- (330).Okada S; Ota K; Ito T Circular Permutation of Ligand-Binding Module Improves Dynamic Range of Genetically Encoded FRET-Based Nanosensor. Protein Sci 2009, 18 (12), 2518–2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (331).Schifferer M; Yushchenko DA; Stein F; Bolbat A; Schultz C A Ratiometric Sensor for Imaging Insulin Secretion in Single β Cells. Cell Chem. Biol 2017, 24 (4), 525–531.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (332).San Martín A; Ceballo S; Ruminot I; Lerchundi R; Frommer WB; Barros LF A Genetically Encoded FRET Lactate Sensor and Its Use To Detect the Warburg Effect in Single Cancer Cells. PLoS One 2013, 8 (2), e57712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (333).Dacres H; Michie M; Anderson A; Trowell SC Advantages of Substituting Bioluminescence for Fluorescence in a Resonance Energy Transfer-Based Periplasmic Binding Protein Biosensor. Biosens. Bioelectron 2013, 41, 459–464. [DOI] [PubMed] [Google Scholar]
- (334).Fehr M; Frommer WB; Lalonde S Visualization of Maltose Uptake in Living Yeast Cells by Fluorescent Nanosensors. Proc. Natl. Acad. Sci. U. S. A 2002, 99 (15), 9846–9851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (335).Bilan DS; Matlashov ME; Gorokhovatsky AY; Schultz C; Enikolopov G; Belousov VV Genetically Encoded Fluorescent Indicator for Imaging NAD+/NADH Ratio Changes in Different Cellular Compartments. Biochim. Biophys. Acta - Gen. Subj 2014, 1840 (3), 951–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (336).Zhao Y; Jin J; Hu Q; Zhou H-M; Yi J; Yu Z; Xu L; Wang X; Yang Y; Loscalzo J Genetically Encoded Fluorescent Sensors for Intracellular NADH Detection. Cell Metab 2011, 14 (4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (337).Cambronne XA; Stewart ML; Kim D; Jones-Brunette AM; Morgan RK; Farrens DL; Cohen MS; Goodman RH Biosensor Reveals Multiple Sources for Mitochondrial NAD+. Science (80-. ) 2016, 352 (6292), 1474–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (338).Cameron WD; Bui CV; Hutchinson A; Loppnau P; Gräslund S; Rocheleau JV Apollo-NADP(+): A Spectrally Tunable Family of Genetically Encoded Sensors for NADP(+). Nat. Methods 2016, 13 (4), 352–358. [DOI] [PubMed] [Google Scholar]
- (339).Zhao F-L; Zhang C; Zhang C; Tang Y; Ye B-C A Genetically Encoded Biosensor for in Vitro and in Vivo Detection of NADP(.). Biosens. Bioelectron 2016, 77, 901–906. [DOI] [PubMed] [Google Scholar]
- (340).Erapaneedi R; Belousov VV; Scha fers M; Kiefer F A Novel Family of Fluorescent Hypoxia Sensors Reveal Strong Heterogeneity in Tumor Hypoxia at the Cellular Level. EMBO J 2016, 35 (1), 102–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (341).Potzkei J; Kunze M; Drepper T; Gensch T; Jaeger K-E; Büchs J Real-Time Determination of Intracellular Oxygen in Bacteria Using a Genetically Encoded FRET-Based Biosensor. BMC Biol 2012, 10 (1), 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (342).San Martín A; Ceballo S; Baeza-Lehnert F; Lerchundi R; Valdebenito R; ContrerasBaeza Y; Alegría K; Barros LF Imaging Mitochondrial Flux in Single Cells with a FRET Sensor for Pyruvate. PLoS One 2014, 9 (1), e85780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (343).Lager I; Fehr M; Frommer WB; Lalonde S Development of a Fluorescent Nanosensor for Ribose. FEBS Lett 2003, 553 (1–2), 85–89. [DOI] [PubMed] [Google Scholar]
- (344).Lager I; Looger LL; Hilpert M; Lalonde S; Frommer WB Conversion of a Putative Agrobacterium Sugar-Binding Protein into a FRET Sensor with High Selectivity for Sucrose. J. Biol. Chem 2006, 281 (41), 30875–30883. [DOI] [PubMed] [Google Scholar]
- (345).Kikuta S; Hou B-H; Sato R; Frommer WB; Kikawada T FRET Sensor-Based Quantification of Intracellular Trehalose in Mammalian Cells. Biosci. Biotechnol. Biochem 2016, 80 (1), 162–165. [DOI] [PubMed] [Google Scholar]
- (346).Kaper T; Looger LL; Takanaga H; Platten M; Steinman L; Frommer WB Nanosensor Detection of an Immunoregulatory Tryptophan Influx/kynurenine Efflux Cycle. PLoS Biol 2007, 5 (10), e257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (347).Wang W; Wildes CP; Pattarabanjird T; Sanchez MI; Glober GF; Matthews GA; Tye KM; Ting AY A Light- and Calcium-Gated Transcription Factor for Imaging and Manipulating Activated Neurons. Nat. Biotechnol 2017, 35 (9), 864–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (348).Lee D; Hyun JH; Jung K; Hannan P; Kwon H-B A Calcium- and Light-Gated Switch to Induce Gene Expression in Activated Neurons. Nat. Biotechnol 2017, 35 (9), 858–863. [DOI] [PubMed] [Google Scholar]
- (349).Gulyás G; Tóth JT; Tóth DJ; Kurucz I; Hunyady L; Balla T; Várnai P Measurement of Inositol 1,4,5-Trisphosphate in Living Cells Using an Improved Set of Resonance Energy Transfer-Based Biosensors. PLoS One 2015, 10 (5), e0125601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (350).Waldeck-Weiermair M; Alam MR; Khan MJ; Deak AT; Vishnu N; Karsten F; Imamura H; Graier WF; Malli R Spatiotemporal Correlations between Cytosolic and Mitochondrial Ca2+ Signals Using a Novel Red-Shifted Mitochondrial Targeted Cameleon. PLoS One 2012, 7 (9), e45917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (351).Ding Y; Ai H; Hoi H; Campbell RE Förster Resonance Energy Transfer-Based Biosensors for Multiparameter Ratiometric Imaging of Ca2+ Dynamics and Caspase-3 Activity in Single Cells. Anal. Chem 2011, 83 (24), 9687–9693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (352).Horikawa K; Yamada Y; Matsuda T; Kobayashi K; Hashimoto M; Matsu-ura T; Miyawaki A; Michikawa T; Mikoshiba K; Nagai T Spontaneous Network Activity Visualized by Ultrasensitive Ca(2+) Indicators, Yellow Cameleon-Nano. Nat. Methods 2010, 7 (9), 729–732. [DOI] [PubMed] [Google Scholar]
- (353).Palmer AE; Giacomello M; Kortemme T; Hires SA; Lev-Ram V; Baker D; Tsien RY Ca2+ Indicators Based on Computationally Redesigned Calmodulin-Peptide Pairs. Chem. Biol 2006, 13 (5), 521–530. [DOI] [PubMed] [Google Scholar]
- (354).Palmer AE; Jin C; Reed JC; Tsien RY Bcl-2-Mediated Alterations in Endoplasmic Reticulum Ca2+ Analyzed with an Improved Genetically Encoded Fluorescent Sensor. Proc. Natl. Acad. Sci. U. S. A 2004, 101 (50), 17404–17409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (355).Nagai T; Yamada S; Tominaga T; Ichikawa M; Miyawaki A Expanded Dynamic Range of Fluorescent Indicators for Ca(2+) by Circularly Permuted Yellow Fluorescent Proteins. Proc. Natl. Acad. Sci. U. S. A 2004, 101 (29), 10554–10559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (356).Nagai T; Ibata K; Park ES; Kubota M; Mikoshiba K; Miyawaki A A Variant of Yellow Fluorescent Protein with Fast and Efficient Maturation for Cell-Biological Applications. Nat. Biotechnol 2002, 20 (1), 87–90. [DOI] [PubMed] [Google Scholar]
- (357).Truong K; Sawano A; Mizuno H; Hama H; Tong KI; Mal TK; Miyawaki A; Ikura M FRET-Based in Vivo Ca2+ Imaging by a New Calmodulin-GFP Fusion Molecule. Nat. Struct. Biol 2001, 8 (12), 1069–1073. [DOI] [PubMed] [Google Scholar]
- (358).Miyawaki A; Griesbeck O; Heim R; Tsien RY Dynamic and Quantitative Ca2+ Measurements Using Improved Cameleons. Proc. Natl. Acad. Sci. U. S. A 1999, 96 (5), 2135–2140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (359).Miyawaki A; Llopis J; Heim R; McCaffery JM; Adams JA; Ikura M; Tsien RY Fluorescent Indicators for Ca2+ Based on Green Fluorescent Proteins and Calmodulin. Nature 1997, 388 (6645), 882–887. [DOI] [PubMed] [Google Scholar]
- (360).Griesbeck O; Baird GS; Campbell RE; Zacharias DA; Tsien RY Reducing the Environmental Sensitivity of Yellow Fluorescent Protein. Mechanism and Applications. J. Biol. Chem 2001, 276 (31), 29188–29194. [DOI] [PubMed] [Google Scholar]
- (361).Fosque BF; Sun Y; Dana H; Yang C-T; Ohyama T; Tadross MR; Patel R; Zlatic M; Kim DS; Ahrens MB; et al. Neural Circuits. Labeling of Active Neural Circuits in Vivo with Designed Calcium Integrators. Science 2015, 347 (6223), 755–760. [DOI] [PubMed] [Google Scholar]
- (362).Souslova EA; Belousov VV; Lock JG; Strömblad S; Kasparov S; Bolshakov AP; Pinelis VG; Labas YA; Lukyanov S; Mayr LM; et al. Single Fluorescent Protein-Based Ca2+ Sensors with Increased Dynamic Range. BMC Biotechnol 2007, 7 (1), 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (363).Tang S; Wong H-C; Wang Z-M; Huang Y; Zou J; Zhuo Y; Pennati A; Gadda G; Delbono O; Yang JJ Design and Application of a Class of Sensors to Monitor Ca2+ Dynamics in High Ca2+ Concentration Cellular Compartments. Proc. Natl. Acad. Sci. U. S. A 2011, 108 (39), 16265–16270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (364).Alford SC; Abdelfattah AS; Ding Y; Campbell RE A Fluorogenic Red Fluorescent Protein Heterodimer. Chem. Biol 2012, 19 (3), 353–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (365).de Juan-Sanz J; Holt GT; Schreiter ER; de Juan F; Kim DS; Ryan TA Axonal Endoplasmic Reticulum Ca 2+ Content Controls Release Probability in CNS Nerve Terminals. Neuron 2017, 93 (4), 867–881.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (366).Henderson MJ; Baldwin HA; Werley CA; Boccardo S; Whitaker LR; Yan X; Holt GT; Schreiter ER; Looger LL; Cohen AE; et al. A Low Affinity GCaMP3 Variant (GCaMPer) for Imaging the Endoplasmic Reticulum Calcium Store. PLoS One 2015, 10 (10), e0139273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (367).Suzuki J; Kanemaru K; Ishii K; Ohkura M; Okubo Y; Iino M Imaging Intraorganellar Ca2+ at Subcellular Resolution Using CEPIA. Nat. Commun 2014, 5, 4153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (368).Navas-Navarro P; Rojo-Ruiz J; Rodriguez-Prados M; Ganfornina MD; Looger LL; Alonso MT; García-Sancho J GFP-Aequorin Protein Sensor for Ex Vivo and In Vivo Imaging of Ca 2+ Dynamics in High-Ca 2+ Organelles. Cell Chem. Biol 2016, 23 (6), 738–745. [DOI] [PubMed] [Google Scholar]
- (369).Rodriguez-Garcia A; Rojo-Ruiz J; Navas-Navarro P; Aulestia FJ; Gallego-Sandin S; Garcia-Sancho J; Alonso MT GAP, an Aequorin-Based Fluorescent Indicator for Imaging Ca2+ in Organelles. Proc. Natl. Acad. Sci 2014, 111 (7), 2584–2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (370).Helassa N; Zhang X; Conte I; Scaringi J; Esposito E; Bradley J; Carter T; Ogden D; Morad M; Török K Fast-Response Calmodulin-Based Fluorescent Indicators Reveal Rapid Intracellular Calcium Dynamics. Sci. Rep 2015, 5 (1), 15978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (371).Despa S; Shui B; Bossuyt J; Lang D; Kotlikoff MI; Bers DM Junctional Cleft [Ca2+]i Measurements Using Novel Cleft-Targeted Ca2+ Sensors. Circ. Res 2014, 115 (3), 339–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (372).Ohkura M; Sasaki T; Sadakari J; Gengyo-Ando K; Kagawa-Nagamura Y; Kobayashi C; Ikegaya Y; Nakai J Genetically Encoded Green Fluorescent Ca2+ Indicators with Improved Detectability for Neuronal Ca2+ Signals. PLoS One 2012, 7 (12), e51286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (373).Muto A; Ohkura M; Kotani T; Higashijima S; Nakai J; Kawakami K Genetic Visualization with an Improved GCaMP Calcium Indicator Reveals Spatiotemporal Activation of the Spinal Motor Neurons in Zebrafish. Proc. Natl. Acad. Sci. U. S. A 2011, 108 (13), 5425–5430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (374).Shen Y; Dana H; Abdelfattah AS; Patel R; Shea J; Molina RS; Rawal B; Rancic V; Chang Y-F; Wu L; et al. A Genetically Encoded Ca2+ Indicator Based on Circularly Permutated Sea Anemone Red Fluorescent Protein eqFP578. BMC Biol 2018, 16 (1), 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (375).Wu J; Prole DL; Shen Y; Lin Z; Gnanasekaran A; Liu Y; Chen L; Zhou H; Chen SRW; Usachev YM; et al. Red Fluorescent Genetically Encoded Ca 2+ Indicators for Use in Mitochondria and Endoplasmic Reticulum. Biochem. J 2014, 464 (1), 13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (376).Hoi H; Matsuda T; Nagai T; Campbell RE Highlightable Ca 2+ Indicators for Live Cell Imaging. J. Am. Chem. Soc 2013, 135 (1), 46–49. [DOI] [PubMed] [Google Scholar]
- (377).Suzuki K; Kimura T; Shinoda H; Bai G; Daniels MJ; Arai Y; Nakano M; Nagai T Five Colour Variants of Bright Luminescent Protein for Real-Time Multicolour Bioimaging. Nat. Commun 2016, 7, 13718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (378).Takai A; Nakano M; Saito K; Haruno R; Watanabe TM; Ohyanagi T; Jin T; Okada Y; Nagai T Expanded Palette of Nano-Lanterns for Real-Time Multicolor Luminescence Imaging. Proc. Natl. Acad. Sci. U. S. A 2015, 112 (14), 4352–4356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (379).Inoue M; Takeuchi A; Horigane SI; Ohkura M; Gengyo-Ando K; Fujii H; Kamijo S; Takemoto-Kimura S; Kano M; Nakai J; et al. Rational Design of a High-Affinity, Fast, Red Calcium Indicator R-CaMP2. Nat. Methods 2014, 12 (1), 64–70. [DOI] [PubMed] [Google Scholar]
- (380).Ohkura M; Sasaki T; Kobayashi C; Ikegaya Y; Nakai J An Improved Genetically Encoded Red Fluorescent Ca2+ Indicator for Detecting Optically Evoked Action Potentials. PLoS One 2012, 7 (7), e39933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (381).Yang J; Cumberbatch D; Centanni S; Shi S-Q; Winder D; Webb D; Johnson CH Coupling Optogenetic Stimulation with NanoLuc-Based Luminescence (BRET) Ca++ Sensing. Nat. Commun 2016, 7, 13268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (382).Thestrup T; Litzlbauer J; Bartholomäus I; Mues M; Russo L; Dana H; Kovalchuk Y; Liang Y; Kalamakis G; Laukat Y; et al. Optimized Ratiometric Calcium Sensors for Functional in Vivo Imaging of Neurons and T Lymphocytes. Nat. Methods 2014, 11 (2), 175–182. [DOI] [PubMed] [Google Scholar]
- (383).Su T; Zhang Z; Luo Q Ratiometric Fluorescence Imaging of Dual Bio-Molecular Events in Single Living Cells Using a New FRET Pair mVenus/mKOκ-Based Biosensor and a Single Fluorescent Protein Biosensor. Biosens. Bioelectron 2012, 31 (1), 292–298. [DOI] [PubMed] [Google Scholar]
- (384).Mank M; Santos AF; Direnberger S; Mrsic-Flogel TD; Hofer SB; Stein V; Hendel T; Reiff DF; Levelt C; Borst A; et al. A Genetically Encoded Calcium Indicator for Chronic in Vivo Two-Photon Imaging. Nat. Methods 2008, 5 (9), 805–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (385).Mank M; Reiff DF; Heim N; Friedrich MW; Borst A; Griesbeck O A FRET-Based Calcium Biosensor with Fast Signal Kinetics and High Fluorescence Change. Biophys. J 2006, 90 (5), 1790–1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (386).Heim N; Griesbeck O Genetically Encoded Indicators of Cellular Calcium Dynamics Based on Troponin C and Green Fluorescent Protein. J. Biol. Chem 2004, 279 (14), 14280–14286. [DOI] [PubMed] [Google Scholar]
- (387).Wegner SV; Sun F; Hernandez N; He C The Tightly Regulated Copper Window in Yeast. Chem. Commun. (Camb) 2011, 47 (9), 2571–2573. [DOI] [PubMed] [Google Scholar]
- (388).Wegner SV; Arslan H; Sunbul M; Yin J; He C Dynamic Copper(I) Imaging in Mammalian Cells with a Genetically Encoded Fluorescent Copper(I) Sensor. J. Am. Chem. Soc 2010, 132 (8), 2567–2569. [DOI] [PubMed] [Google Scholar]
- (389).Grimley JS; Li L; Wang W; Wen L; Beese LS; Hellinga HW; Augustine GJ Visualization of Synaptic Inhibition with an Optogenetic Sensor Developed by Cell-Free Protein Engineering Automation. J. Neurosci 2013, 33 (41), 16297–16309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (390).Chapleau RR; Blomberg R; Ford PC; Sagermann M Design of a Highly Specific and Noninvasive Biosensor Suitable for Real-Time in Vivo Imaging of Mercury (II) Uptake. Protein Sci 2008, 17 (4), 614–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (391).Gu Z; Zhao M; Sheng Y; Bentolila LA; Tang Y Detection of Mercury Ion by Infrared Fluorescent Protein and Its Hydrogel-Based Paper Assay. Anal. Chem 2011, 83 (6), 2324–2329. [DOI] [PubMed] [Google Scholar]
- (392).Gu H; Lalonde S; Okumoto S; Looger LL; Scharff-Poulsen AM; Grossman AR; Kossmann J; Jakobsen I; Frommer WB A Novel Analytical Method for in Vivo Phosphate Tracking. FEBS Lett 2006, 580 (25), 5885–5893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (393).Aper SJA; Dierickx P; Merkx M Dual Readout BRET/FRET Sensors for Measuring Intracellular Zinc. ACS Chem. Biol 2016, 11 (10), 2854–2864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (394).Lindenburg LH; Hessels AM; Ebberink EHTM; Arts R; Merkx M Robust Red FRET Sensors Using Self-Associating Fluorescent Domains. ACS Chem. Biol 2013, 8 (10), 2133–2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (395).Vinkenborg JL; Nicolson TJ; Bellomo EA; Koay MS; Rutter GA; Merkx M Genetically Encoded FRET Sensors to Monitor Intracellular Zn2+ Homeostasis. Nat. Methods 2009, 6 (10), 737–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (396).van Dongen EMWM; Evers TH; Dekkers LM; Meijer EW; Klomp LWJ; Merkx M Variation of Linker Length in Ratiometric Fluorescent Sensor Proteins Allows Rational Tuning of Zn(II) Affinity in the Picomolar to Femtomolar Range. J. Am. Chem. Soc 2007, 129 (12), 3494–3495. [DOI] [PubMed] [Google Scholar]
- (397).van Dongen EMWM; Dekkers LM; Spijker K; Meijer EW; Klomp LWJ; Merkx M Ratiometric Fluorescent Sensor Proteins with Subnanomolar Affinity for Zn(II) Based on Copper Chaperone Domains. J. Am. Chem. Soc 2006, 128 (33), 10754–10762. [DOI] [PubMed] [Google Scholar]
- (398).Park JG; Qin Y; Galati DF; Palmer AE New Sensors for Quantitative Measurement of Mitochondrial Zn(2+). ACS Chem. Biol 2012, 7 (10), 1636–1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (399).Dittmer PJ; Miranda JG; Gorski JA; Palmer AE Genetically Encoded Sensors to Elucidate Spatial Distribution of Cellular Zinc. J. Biol. Chem 2009, 284 (24), 16289–16297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (400).Qin Y; Dittmer PJ; Park JG; Jansen KB; Palmer AE Measuring Steady-State and Dynamic Endoplasmic Reticulum and Golgi Zn2+ with Genetically Encoded Sensors. Proc. Natl. Acad. Sci 2011, 108 (18), 7351–7356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (401).Qiao W; Mooney M; Bird AJ; Winge DR; Eide DJ Zinc Binding to a Regulatory Zinc-Sensing Domain Monitored in Vivo by Using FRET. Proc. Natl. Acad. Sci 2006, 103 (23), 8674–8679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (402).Hessels AM; Chabosseau P; Bakker MH; Engelen W; Rutter GA; Taylor KM; Merkx M eZinCh-2: A Versatile, Genetically Encoded FRET Sensor for Cytosolic and Intraorganelle Zn(2+) Imaging. ACS Chem. Biol 2015, 10 (9), 2126–2134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (403).Evers TH; Appelhof MAM; Meijer EW; Merkx M His-Tags as Zn(II) Binding Motifs in a Protein-Based Fluorescent Sensor. Protein Eng. Des. Sel 2008, 21 (8), 529–536. [DOI] [PubMed] [Google Scholar]
- (404).Ting AY; Kain KH; Klemke RL; Tsien RY Genetically Encoded Fluorescent Reporters of Protein Tyrosine Kinase Activities in Living Cells. Proc. Natl. Acad. Sci. U. S. A 2001, 98 (26), 15003–15008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (405).Yoshizaki H; Aoki K; Nakamura T; Matsuda M Regulation of RalA GTPase by Phosphatidylinositol 3-Kinase as Visualized by FRET Probes. Biochem. Soc. Trans 2006, 34 (Pt 5), 851–854. [DOI] [PubMed] [Google Scholar]
- (406).Zhou X; Clister TL; Lowry PR; Seldin MM; Wong GW; Zhang J Dynamic Visualization of mTORC1 Activity in Living Cells. Cell Rep 2015, 10 (10), 1767–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (407).Gao X; Zhang J Spatiotemporal Analysis of Differential Akt Regulation in Plasma Membrane Microdomains. Mol. Biol. Cell 2008, 19 (10), 4366–4373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (408).Sasaki K; Sato M; Umezawa Y Fluorescent Indicators for Akt/protein Kinase B and Dynamics of Akt Activity Visualized in Living Cells. J. Biol. Chem 2003, 278 (33), 30945–30951. [DOI] [PubMed] [Google Scholar]
- (409).Belal ASF; Sell BR; Hoi H; Davidson MW; Campbell RE Optimization of a Genetically Encoded Biosensor for Cyclin B1-Cyclin Dependent Kinase 1. Mol. Biosyst 2014, 10 (2), 191–195. [DOI] [PubMed] [Google Scholar]
- (410).Kunkel MT; Ni Q; Tsien RY; Zhang J; Newton AC Spatio-Temporal Dynamics of Protein Kinase B/Akt Signaling Revealed by a Genetically Encoded Fluorescent Reporter. J. Biol. Chem 2005, 280 (7), 5581–5587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (411).Calleja V; Alcor D; Laguerre M; Park J; Vojnovic B; Hemmings BA; Downward J; Parker PJ; Larijani B Intramolecular and Intermolecular Interactions of Protein Kinase B Define Its Activation in Vivo. PLoS Biol 2007, 5 (4), e95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (412).Calleja V; Ameer-Beg SM; Vojnovic B; Woscholski R; Downward J; Larijani B Monitoring Conformational Changes of Proteins in Cells by Fluorescence Lifetime Imaging Microscopy. Biochem. J 2003, 372 (Pt 1), 33–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (413).Miura H; Matsuda M; Aoki K Development of a FRET Biosensor with High Specificity for Akt. Cell Struct. Funct 2014, 39 (1), 9–20. [DOI] [PubMed] [Google Scholar]
- (414).Komatsu N; Aoki K; Yamada M; Yukinaga H; Fujita Y; Kamioka Y; Matsuda M Development of an Optimized Backbone of FRET Biosensors for Kinases and GTPases. Mol. Biol. Cell 2011, 22 (23), 4647–4656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (415).Ananthanarayanan B; Fosbrink M; Rahdar M; Zhang J Live-Cell Molecular Analysis of Akt Activation Reveals Roles for Activation Loop Phosphorylation. J. Biol. Chem 2007, 282 (50), 36634–36641. [DOI] [PubMed] [Google Scholar]
- (416).Depry C; Mehta S; Li R; Zhang J Visualization of Compartmentalized Kinase Activity Dynamics Using Adaptable BimKARs. Chem. Biol 2015, 22 (11), 1470–1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (417).Sample V; Ramamurthy S; Gorshkov K; Ronnett GV; Zhang J Polarized Activities of AMPK and BRSK in Primary Hippocampal Neurons. Mol. Biol. Cell 2015, 26 (10), 1935–1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (418).Tsou P; Zheng B; Hsu C-H; Sasaki AT; Cantley LC A Fluorescent Reporter of AMPK Activity and Cellular Energy Stress. Cell Metab 2011, 13 (4), 476–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (419).Johnson SA; You Z; Hunter T Monitoring ATM Kinase Activity in Living Cells. DNA Repair (Amst) 2007, 6 (9), 1277–1284. [DOI] [PubMed] [Google Scholar]
- (420).Chu Y; Yao PY; Wang W; Wang D; Wang Z; Zhang L; Huang Y; Ke Y; Ding X; Yao X Aurora B Kinase Activation Requires Survivin Priming Phosphorylation by PLK1. J. Mol. Cell Biol 2011, 3 (4), 260–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (421).Fuller BG; Lampson MA; Foley EA; Rosasco-Nitcher S; Le KV; Tobelmann P; Brautigan DL; Stukenberg PT; Kapoor TM Midzone Activation of Aurora B in Anaphase Produces an Intracellular Phosphorylation Gradient. Nature 2008, 453 (7198), 1132–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (422).Bertolin G; Sizaire F; Herbomel G; Reboutier D; Prigent C; Tramier M A FRET Biosensor Reveals Spatiotemporal Activation and Functions of Aurora Kinase A in Living Cells. Nat. Commun 2016, 7, 12674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (423).Terai K; Matsuda M The Amino-Terminal B-Raf-Specific Region Mediates Calcium-Dependent Homo- and Hetero-Dimerization of Raf. EMBO J 2006, 25 (15), 3556–3564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (424).Mizutani T; Kondo T; Darmanin S; Tsuda M; Tanaka S; Tobiume M; Asaka M; Ohba Y A Novel FRET-Based Biosensor for the Measurement of BCR-ABL Activity and Its Response to Drugs in Living Cells. Clin. Cancer Res 2010, 16 (15), 3964–3975. [DOI] [PubMed] [Google Scholar]
- (425).Terai K; Matsuda M Ras Binding Opens c-Raf to Expose the Docking Site for Mitogen-Activated Protein Kinase Kinase. EMBO Rep 2005, 6 (3), 251–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (426).Nakahata Y; Nabekura J; Murakoshi H Dual Observation of the ATP-Evoked Small GTPase Activation and Ca2+ Transient in Astrocytes Using a Dark Red Fluorescent Protein. Sci. Rep 2016, 6 (1), 39564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (427).Murakoshi H; Shibata ACE; Nakahata Y; Nabekura J A Dark Green Fluorescent Protein as an Acceptor for Measurement of Förster Resonance Energy Transfer. Sci. Rep 2015, 5 (1), 15334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (428).Lee S-JR; Escobedo-Lozoya Y; Szatmari EM; Yasuda R Activation of CaMKII in Single Dendritic Spines during Long-Term Potentiation. Nature 2009, 458 (7236), 299–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (429).Kwok S; Lee C; Sánchez SA; Hazlett TL; Gratton E; Hayashi Y Genetically Encoded Probe for Fluorescence Lifetime Imaging of CaMKII Activity. Biochem. Biophys. Res. Commun 2008, 369 (2), 519–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (430).Takao K; Okamoto K-I; Nakagawa T; Neve RL; Nagai T; Miyawaki A; Hashikawa T; Kobayashi S; Hayashi Y Visualization of Synaptic Ca2+ /Calmodulin-Dependent Protein Kinase II Activity in Living Neurons. J. Neurosci 2005, 25 (12), 3107–3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (431).Mehta S; Aye-Han N-N; Ganesan A; Oldach L; Gorshkov K; Zhang J Calmodulin-Controlled Spatial Decoding of Oscillatory Ca2+ Signals by Calcineurin. Elife 2014, 3, e03765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (432).Newman RH; Zhang J Visualization of Phosphatase Activity in Living Cells with a FRET-Based Calcineurin Activity Sensor. Mol. Biosyst 2008, 4 (6), 496–501. [DOI] [PubMed] [Google Scholar]
- (433).Bazzazi H; Sang L; Dick IE; Joshi-Mukherjee R; Yang W; Yue DT Novel Fluorescence Resonance Energy Transfer-Based Reporter Reveals Differential Calcineurin Activation in Neonatal and Adult Cardiomyocytes. J. Physiol 2015, 593 (17), 3865–3884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (434).Gavet O; Pines J Progressive Activation of CyclinB1-Cdk1 Coordinates Entry to Mitosis. Dev. Cell 2010, 18 (4), 533–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (435).Piljić A; de Diego I; Wilmanns M; Schultz C Rapid Development of Genetically Encoded FRET Reporters. ACS Chem. Biol 2011, 6 (7), 685–691. [DOI] [PubMed] [Google Scholar]
- (436).Offterdinger M; Georget V; Girod A; Bastiaens PIH Imaging Phosphorylation Dynamics of the Epidermal Growth Factor Receptor. J. Biol. Chem 2004, 279 (35), 36972–36981. [DOI] [PubMed] [Google Scholar]
- (437).Itoh RE; Kurokawa K; Fujioka A; Sharma A; Mayer BJ; Matsuda M A FRET-Based Probe for Epidermal Growth Factor Receptor Bound Non-Covalently to a Pair of Synthetic Amphipathic Helixes. Exp. Cell Res 2005, 307 (1), 142–152. [DOI] [PubMed] [Google Scholar]
- (438).Green HM; Alberola-Ila J Development of ERK Activity Sensor, an in Vitro, FRET-Based Sensor of Extracellular Regulated Kinase Activity. BMC Chem. Biol 2005, 5 (1), 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (439).Demeautis C; Sipieter F; Roul J; Chapuis C; Padilla-Parra S; Riquet FB; Tramier M Multiplexing PKA and ERK1&2 Kinases FRET Biosensors in Living Cells Using Single Excitation Wavelength Dual Colour FLIM. Sci. Rep 2017, 7, 41026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (440).Sparta B; Pargett M; Minguet M; Distor K; Bell G; Albeck JG Receptor Level Mechanisms Are Required for Epidermal Growth Factor (EGF)-Stimulated Extracellular Signal-Regulated Kinase (ERK) Activity Pulses. J. Biol. Chem 2015, 290 (41), 24784–24792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (441).Vandame P; Spriet C; Riquet F; Trinel D; Cailliau-Maggio K; Bodart J-F Optimization of ERK Activity Biosensors for Both Ratiometric and Lifetime FRET Measurements. Sensors (Basel) 2014, 14 (1), 1140–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (442).Xu C; Peter M; Bouquier N; Ollendorff V; Villamil I; Liu J; Fagni L; Perroy J REV, A BRET-Based Sensor of ERK Activity. Front. Endocrinol. (Lausanne) 2013, 4, 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (443).Fritz RD; Letzelter M; Reimann A; Martin K; Fusco L; Ritsma L; Ponsioen B; Fluri E; Schulte-Merker S; van Rheenen J; et al. A Versatile Toolkit to Produce Sensitive FRET Biosensors to Visualize Signaling in Time and Space. Sci. Signal 2013, 6 (285), rs12. [DOI] [PubMed] [Google Scholar]
- (444).Harvey CD; Ehrhardt AG; Cellurale C; Zhong H; Yasuda R; Davis RJ; Svoboda K A Genetically Encoded Fluorescent Sensor of ERK Activity. Proc. Natl. Acad. Sci. U. S. A 2008, 105 (49), 19264–19269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (445).Sato M; Kawai Y; Umezawa Y Genetically Encoded Fluorescent Indicators to Visualize Protein Phosphorylation by Extracellular Signal-Regulated Kinase in Single Living Cells. Anal. Chem 2007, 79 (6), 2570–2575. [DOI] [PubMed] [Google Scholar]
- (446).Tomida T; Oda S; Takekawa M; Iino Y; Saito H The Temporal Pattern of Stimulation Determines the Extent and Duration of MAPK Activation in a Caenorhabditis Elegans Sensory Neuron. Sci. Signal 2012, 5 (246), ra76. [DOI] [PubMed] [Google Scholar]
- (447).Mo GCHGCH; Ross B; Hertel F; Manna P; Yang X; Greenwald E; Booth C; Plummer AMAM; Tenner B; Chen Z; et al. Genetically Encoded Biosensors for Visualizing Live-Cell Biochemical Activity at Super-Resolution. Nat. Methods 2017, 14 (4), 427–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (448).Fujioka A; Terai K; Itoh RE; Aoki K; Nakamura T; Kuroda S; Nishida E; Matsuda M Dynamics of the Ras/ERK MAPK Cascade as Monitored by Fluorescent Probes. J. Biol. Chem 2006, 281 (13), 8917–8926. [DOI] [PubMed] [Google Scholar]
- (449).Cai X; Lietha D; Ceccarelli DF; Karginov AV; Rajfur Z; Jacobson K; Hahn KM; Eck MJ; Schaller MD Spatial and Temporal Regulation of Focal Adhesion Kinase Activity in Living Cells. Mol. Cell. Biol 2008, 28 (1), 201–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (450).Seong J; Ouyang M; Kim T; Sun J; Wen P-C; Lu S; Zhuo Y; Llewellyn NM; Schlaepfer DD; Guan J-L; et al. Detection of Focal Adhesion Kinase Activation at Membrane Microdomains by Fluorescence Resonance Energy Transfer. Nat. Commun 2011, 2, 406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (451).Markwardt ML; Seckinger KM; Rizzo MA Regulation of Glucokinase by Intracellular Calcium Levels in Pancreatic β Cells. J. Biol. Chem 2016, 291 (6), 3000–3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (452).Ding S-Y; Nkobena A; Kraft CA; Markwardt ML; Rizzo MA Glucagon-like Peptide 1 Stimulates Post-Translational Activation of Glucokinase in Pancreatic Beta Cells. J. Biol. Chem 2011, 286 (19), 16768–16774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (453).Rizzo MA; Magnuson MA; Drain PF; Piston DW A Functional Link between Glucokinase Binding to Insulin Granules and Conformational Alterations in Response to Glucose and Insulin. J. Biol. Chem 2002, 277 (37), 34168–34175. [DOI] [PubMed] [Google Scholar]
- (454).Lin C-W; Ting AY A Genetically Encoded Fluorescent Reporter of Histone Phosphorylation in Living Cells. Angew. Chem. Int. Ed. Engl 2004, 43 (22), 2940–2943. [DOI] [PubMed] [Google Scholar]
- (455).Boute N; Pernet K; Issad T Monitoring the Activation State of the Insulin Receptor Using Bioluminescence Resonance Energy Transfer. Mol. Pharmacol 2001, 60 (4), 640–645. [PubMed] [Google Scholar]
- (456).Sato M; Ozawa T; Inukai K; Asano T; Umezawa Y Fluorescent Indicators for Imaging Protein Phosphorylation in Single Living Cells. Nat. Biotechnol 2002, 20 (3), 287–294. [DOI] [PubMed] [Google Scholar]
- (457).Kawai Y; Sato M; Umezawa Y Single Color Fluorescent Indicators of Protein Phosphorylation for Multicolor Imaging of Intracellular Signal Flow Dynamics. Anal. Chem 2004, 76 (20), 6144–6149. [DOI] [PubMed] [Google Scholar]
- (458).Bakal C; Linding R; Llense F; Heffern E; Martin-Blanco E; Pawson T; Perrimon N Phosphorylation Networks Regulating JNK Activity in Diverse Genetic Backgrounds. Science 2008, 322 (5900), 453–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (459).Fosbrink M; Aye-Han N-N; Cheong R; Levchenko A; Zhang J Visualization of JNK Activity Dynamics with a Genetically Encoded Fluorescent Biosensor. Proc. Natl. Acad. Sci. U. S. A 2010, 107 (12), 5459–5464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (460).Suzuki H; Sato M Genetically Encoded Fluorescent Indicators to Visualise Protein Phosphorylation by c-Jun NH 2 -Terminal Kinase in Living Cells. Supramol. Chem 2010, 22 (7–8), 434–439. [Google Scholar]
- (461).Azad T; Janse van Rensburg HJ; Lightbody ED; Neveu B; Champagne A; Ghaffari A; Kay VR; Hao Y; Shen H; Yeung B; et al. A LATS Biosensor Screen Identifies VEGFR as a Regulator of the Hippo Pathway in Angiogenesis. Nat. Commun 2018, 9 (1), 1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (462).Paster W; Paar C; Eckerstorfer P; Jakober A; Drbal K; Schutz GJ; Sonnleitner A; Stockinger H Genetically Encoded Forster Resonance Energy Transfer Sensors for the Conformation of the Src Family Kinase Lck. J. Immunol 2009, 182 (4), 2160–2167. [DOI] [PubMed] [Google Scholar]
- (463).Neininger A; Thielemann H; Gaestel M FRET-Based Detection of Different Conformations of MK2. EMBO Rep 2001, 2 (8), 703–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (464).Timm T; von Kries JP; Li X; Zempel H; Mandelkow E; Mandelkow E-M Microtubule Affinity Regulating Kinase Activity in Living Neurons Was Examined by a Genetically Encoded Fluorescence Resonance Energy Transfer/fluorescence Lifetime Imaging-Based Biosensor: Inhibitors with Therapeutic Potential. J. Biol. Chem 2011, 286 (48), 41711–41722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (465).Tomida T; Takekawa M; Saito H Oscillation of p38 Activity Controls Efficient proInflammatory Gene Expression. Nat. Commun 2015, 6 (1), 8350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (466).Parrini MC; Camonis J; Matsuda M; de Gunzburg J Dissecting Activation of the PAK1 Kinase at Protrusions in Living Cells. J. Biol. Chem 2009, 284 (36), 24133–24143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (467).Seong J; Huang M; Sim KM; Kim H; Wang Y FRET-Based Visualization of PDGF Receptor Activation at Membrane Microdomains. Sci. Rep 2017, 7 (1), 1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (468).Gao X; Lowry PR; Zhou X; Depry C; Wei Z; Wong GW; Zhang J PI3K/Akt Signaling Requires Spatial Compartmentalization in Plasma Membrane Microdomains. Proc. Natl. Acad. Sci. U. S. A 2011, 108 (35), 14509–14514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (469).Tillo SE; Xiong W-H; Takahashi M; Miao S; Andrade AL; Fortin DA; Yang G; Qin M; Smoody BF; Stork PJS; et al. Liberated PKA Catalytic Subunits Associate with the Membrane via Myristoylation to Preferentially Phosphorylate Membrane Substrates. Cell Rep 2017, 19 (3), 617–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (470).Tao W; Rubart M; Ryan J; Xiao X; Qiao C; Hato T; Davidson MW; Dunn KW; Day RN A Practical Method for Monitoring FRET-Based Biosensors in Living Animals Using Two-Photon Microscopy. Am. J. Physiol. Cell Physiol 2015, 309 (11), C724–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (471).Chen Y; Saulnier JL; Yellen G; Sabatini BL A PKA Activity Sensor for Quantitative Analysis of Endogenous GPCR Signaling via 2-Photon FRET-FLIM Imaging Front. Pharmacol 2014, 5, 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (472).Herbst KJ; Allen MD; Zhang J Luminescent Kinase Activity Biosensors Based on a Versatile Bimolecular Switch. J. Am. Chem. Soc 2011, 133 (15), 5676–5679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (473).Depry C; Allen MD; Zhang J; Huang H; Zhang J; Frame MC; Wang Y; Zhang J; Ginsberg MH; Taylor SS Visualization of PKA Activity in Plasma Membrane Microdomains. Mol. Biosyst 2011, 7 (1), 52–58. [DOI] [PubMed] [Google Scholar]
- (474).Allen MD; Zhang J A Tunable FRET Circuit for Engineering Fluorescent Biosensors. Angew. Chem. Int. Ed. Engl 2008, 47 (3), 500–502. [DOI] [PubMed] [Google Scholar]
- (475).Allen MD; Zhang J Subcellular Dynamics of Protein Kinase A Activity Visualized by FRET-Based Reporters. Biochem. Biophys. Res. Commun 2006, 348 (2), 716–721. [DOI] [PubMed] [Google Scholar]
- (476).Zhang J; Hupfeld CJ; Taylor SS; Olefsky JM; Tsien RY Insulin Disrupts Beta-Adrenergic Signalling to Protein Kinase A in Adipocytes. Nature 2005, 437 (7058), 569–573. [DOI] [PubMed] [Google Scholar]
- (477).Zhang J; Ma Y; Taylor SS; Tsien RY Genetically Encoded Reporters of Protein Kinase A Activity Reveal Impact of Substrate Tethering. Proc. Natl. Acad. Sci. U. S. A 2001, 98 (26), 14997–15002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (478).Nagai Y; Miyazaki M; Aoki R; Zama T; Inouye S; Hirose K; Iino M; Hagiwara M A Fluorescent Indicator for Visualizing cAMP-Induced Phosphorylation in Vivo. Nat. Biotechnol 2000, 18 (3), 313–316. [DOI] [PubMed] [Google Scholar]
- (479).Stefan E; Aquin S; Berger N; Landry CR; Nyfeler B; Bouvier M; Michnick SW Quantification of Dynamic Protein Complexes Using Renilla Luciferase Fragment Complementation Applied to Protein Kinase A Activities in Vivo. Proc. Natl. Acad. Sci. U. S. A 2007, 104 (43), 16916–16921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (480).Bonnot A; Guiot E; Hepp R; Cavellini L; Tricoire L; Lambolez B Single-Fluorophore Biosensors Based on Conformation-Sensitive GFP Variants. FASEB J 2014, 28 (3), 1375–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (481).Violin JD; Zhang J; Tsien RY; Newton AC A Genetically Encoded Fluorescent Reporter Reveals Oscillatory Phosphorylation by Protein Kinase C. J. Cell Biol 2003, 161 (5), 899–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (482).Brumbaugh J; Schleifenbaum A; Gasch A; Sattler M; Schultz C A Dual Parameter FRET Probe for Measuring PKC and PKA Activity in Living Cells. J. Am. Chem. Soc 2006, 128 (1), 24–25. [DOI] [PubMed] [Google Scholar]
- (483).Schleifenbaum A; Stier G; Gasch A; Sattler M; Schultz C Genetically Encoded FRET Probe for PKC Activity Based on Pleckstrin. J. Am. Chem. Soc 2004, 126 (38), 11786–11787. [DOI] [PubMed] [Google Scholar]
- (484).Kunkel MT; Toker A; Tsien RY; Newton AC Calcium-Dependent Regulation of Protein Kinase D Revealed by a Genetically Encoded Kinase Activity Reporter. J. Biol. Chem 2007, 282 (9), 6733–6742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (485).Eisler SA; Fuchs YF; Pfizenmaier K; Hausser A G-PKDrep-Live, a Genetically Encoded FRET Reporter to Measure PKD Activity at the Trans-Golgi-Network. Biotechnol. J 2012, 7 (1), 148–154. [DOI] [PubMed] [Google Scholar]
- (486).Fuchs YF; Eisler SA; Link G; Schlicker O; Bunt G; Pfizenmaier K; Hausser A A Golgi PKD Activity Reporter Reveals a Crucial Role of PKD in Nocodazole-Induced Golgi Dispersal. Traffic 2009, 10 (7), 858–867. [DOI] [PubMed] [Google Scholar]
- (487).Merrins MJ; Van Dyke AR; Mapp AK; Rizzo MA; Satin LS Direct Measurements of Oscillatory Glycolysis in Pancreatic Islet β-Cells Using Novel Fluorescence Resonance Energy Transfer (FRET) Biosensors for Pyruvate Kinase M2 Activity. J. Biol. Chem 2013, 288 (46), 33312–33322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (488).Lima-Fernandes E; Misticone S; Boularan C; Paradis JS; Enslen H; Roux PP; Bouvier M; Baillie GS; Marullo S; Scott MGH. A Biosensor to Monitor Dynamic Regulation and Function of Tumour Suppressor PTEN in Living Cells. Nat. Commun 2014, 5, 4431. [DOI] [PubMed] [Google Scholar]
- (489).Li C; Imanishi A; Komatsu N; Terai K; Amano M; Kaibuchi K; Matsuda M A FRET Biosensor for ROCK Based on a Consensus Substrate Sequence Identified by KISS Technology. Cell Struct. Funct 2017, 42 (1), 1–13. [DOI] [PubMed] [Google Scholar]
- (490).Kurokawa K; Mochizuki N; Ohba Y; Mizuno H; Miyawaki A; Matsuda M A Pair of Fluorescent Resonance Energy Transfer-Based Probes for Tyrosine Phosphorylation of the CrkII Adaptor Protein in Vivo. J. Biol. Chem 2001, 276 (33), 31305–31310. [DOI] [PubMed] [Google Scholar]
- (491).Tomida T; Takekawa M; O’Grady P; Saito H Stimulus-Specific Distinctions in Spatial and Temporal Dynamics of Stress-Activated Protein Kinase Kinase Kinases Revealed by a Fluorescence Resonance Energy Transfer Biosensor. Mol. Cell. Biol 2009, 29 (22), 6117–6127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (492).Su T; Pan S; Luo Q; Zhang Z Monitoring of Dual Bio-Molecular Events Using FRET Biosensors Based on mTagBFP/sfGFP and mVenus/mKOκ Fluorescent Protein Pairs. Biosens. Bioelectron 2013, 46, 97–101. [DOI] [PubMed] [Google Scholar]
- (493).Ouyang M; Sun J; Chien S; Wang Y Determination of Hierarchical Relationship of Src and Rac at Subcellular Locations with FRET Biosensors. Proc. Natl. Acad. Sci. U. S. A 2008, 105 (38), 14353–14358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (494).Wang Y; Botvinick EL; Zhao Y; Berns MW; Usami S; Tsien RY; Chien S Visualizing the Mechanical Activation of Src. Nature 2005, 434 (7036), 1040–1045. [DOI] [PubMed] [Google Scholar]
- (495).Hitosugi T; Sasaki K; Sato M; Suzuki Y; Umezawa Y Epidermal Growth Factor Directs Sex-Specific Steroid Signaling through Src Activation. J. Biol. Chem 2007, 282 (14), 10697–10706. [DOI] [PubMed] [Google Scholar]
- (496).Randriamampita C; Mouchacca P; Malissen B; Marguet D; Trautmann A; Lellouch AC A Novel ZAP-70 Dependent FRET Based Biosensor Reveals Kinase Activity at Both the Immunological Synapse and the Antisynapse. PLoS One 2008, 3 (1), e1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (497).Yamauchi JG; Nemecz Á; Nguyen QT; Muller A; Schroeder LF; Talley TT; Lindstrom J; Kleinfeld D; Taylor P Characterizing Ligand-Gated Ion Channel Receptors with Genetically Encoded Ca2++ Sensors. PLoS One 2011, 6 (1), e16519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (498).Nguyen Q-T; Schroeder LF; Mank M; Muller A; Taylor P; Griesbeck O; Kleinfeld D An in Vivo Biosensor for Neurotransmitter Release and in Situ Receptor Activity. Nat. Neurosci 2010, 13 (1), 127–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (499).Muller A; Joseph V; Slesinger PA; Kleinfeld D Cell-Based Reporters Reveal in Vivo Dynamics of Dopamine and Norepinephrine Release in Murine Cortex. Nat. Methods 2014, 11 (12), 1245–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (500).Okumoto S; Looger LL; Micheva KD; Reimer RJ; Smith SJ; Frommer WB Detection of Glutamate Release from Neurons by Genetically Encoded Surface-Displayed FRET Nanosensors. Proc. Natl. Acad. Sci. U. S. A 2005, 102 (24), 8740–8745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (501).Hires SA; Zhu Y; Tsien RY Optical Measurement of Synaptic Glutamate Spillover and Reuptake by Linker Optimized Glutamate-Sensitive Fluorescent Reporters. Proc. Natl. Acad. Sci. U. S. A 2008, 105 (11), 4411–4416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (502).Yang H; Bogner M; Stierhof Y-D; Ludewig U H+-Independent Glutamine Transport in Plant Root Tips PLoS One 2010, 5 (1), e8917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (503).Warren S; Margineanu A; Katan M; Dunsby C; French P Homo-FRET Based Biosensors and Their Application to Multiplexed Imaging of Signalling Events in Live Cells. Int. J. Mol. Sci 2015, 16 (7), 14695–14716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (504).Ananthanarayanan B; Ni Q; Zhang J Signal Propagation from Membrane Messengers to Nuclear Effectors Revealed by Reporters of Phosphoinositide Dynamics and Akt Activity. Proc. Natl. Acad. Sci 2005, 102 (42), 15081–15086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (505).Sato M; Ueda Y; Umezawa Y Imaging Diacylglycerol Dynamics at Organelle Membranes. Nat. Methods 2006, 3 (10), 797–799. [DOI] [PubMed] [Google Scholar]
- (506).Nishioka T; Aoki K; Hikake K; Yoshizaki H; Kiyokawa E; Matsuda M Rapid Turnover Rate of Phosphoinositides at the Front of Migrating MDCK Cells. Mol. Biol. Cell 2008, 19 (10), 4213–4223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (507).Remus TP; Zima AV; Bossuyt J; Bare DJ; Martin JL; Blatter LA; Bers DM; Mignery GA Biosensors to Measure Inositol 1,4,5-Trisphosphate Concentration in Living Cells with Spatiotemporal Resolution. J. Biol. Chem 2006, 281 (1), 608–616. [DOI] [PubMed] [Google Scholar]
- (508).Sato M; Ueda Y; Shibuya M; Umezawa Y Locating Inositol 1,4,5-Trisphosphate in the Nucleus and Neuronal Dendrites with Genetically Encoded Fluorescent Indicators. Anal. Chem 2005, 77 (15), 4751–4758. [DOI] [PubMed] [Google Scholar]
- (509).Hirose K; Kadowaki S; Tanabe M; Takeshima H; Iino M Spatiotemporal Dynamics of Inositol 1,4,5-Trisphosphate That Underlies Complex Ca2+ Mobilization Patterns. Science 1999, 284 (5419), 1527–1530. [DOI] [PubMed] [Google Scholar]
- (510).Matsu-ura T; Michikawa T; Inoue T; Miyawaki A; Yoshida M; Mikoshiba K Cytosolic Inositol 1,4,5-Trisphosphate Dynamics during Intracellular Calcium Oscillations in Living Cells. J. Cell Biol 2006, 173 (5), 755–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (511).Tanimura A; Morita T; Nezu A; Shitara A; Hashimoto N; Tojyo Y Use of Fluorescence Resonance Energy Transfer-Based Biosensors for the Quantitative Analysis of Inositol 1,4,5-Trisphosphate Dynamics in Calcium Oscillations. J. Biol. Chem 2009, 284 (13), 8910–8917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (512).Tanimura A; Nezu A; Morita T; Turner RJ; Tojyo Y Fluorescent Biosensor for Quantitative Real-Time Measurements of Inositol 1,4,5-Trisphosphate in Single Living Cells. J. Biol. Chem 2004, 279 (37), 38095–38098. [DOI] [PubMed] [Google Scholar]
- (513).Nishioka T; Frohman MA; Matsuda M; Kiyokawa E Heterogeneity of Phosphatidic Acid Levels and Distribution at the Plasma Membrane in Living Cells as Visualized by a Förster Resonance Energy Transfer (FRET) Biosensor. J. Biol. Chem 2010, 285 (46), 35979–35987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (514).Tóth JT; Gulyás G; Tóth DJ; Balla A; Hammond GRV; Hunyady L; Balla T; Várnai P BRET-Monitoring of the Dynamic Changes of Inositol Lipid Pools in Living Cells Reveals a PKC-Dependent PtdIns4P Increase upon EGF and M3 Receptor Activation. Biochim. Biophys. Acta 2016, 1861 (3), 177–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (515).Li X; Wang X; Zhang X; Zhao M; Tsang WL; Zhang Y; Yau RGW; Weisman LS; Xu H Genetically Encoded Fluorescent Probe to Visualize Intracellular Phosphatidylinositol 3,5-Bisphosphate Localization and Dynamics. Proc. Natl. Acad. Sci. U. S. A 2013, 110 (52), 21165–21170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (516).Cicchetti G; Biernacki M; Farquharson J; Allen PG A Ratiometric Expressible FRET Sensor for Phosphoinositides Displays a Signal Change in Highly Dynamic Membrane Structures in Fibroblasts. Biochemistry 2004, 43 (7), 1939–1949. [DOI] [PubMed] [Google Scholar]
- (517).Santagata S; Boggon TJ; Baird CL; Gomez CA; Zhao J; Shan WS; Myszka DG; Shapiro L G-Protein Signaling through Tubby Proteins. Science 2001, 292 (5524), 2041–2050. [DOI] [PubMed] [Google Scholar]
- (518).van der Wal J; Habets R; Várnai P; Balla T; Jalink K Monitoring Agonist-Induced Phospholipase C Activation in Live Cells by Fluorescence Resonance Energy Transfer. J. Biol. Chem 2001, 276 (18), 15337–15344. [DOI] [PubMed] [Google Scholar]
- (519).Stauffer TP; Ahn S; Meyer T Receptor-Induced Transient Reduction in Plasma Membrane PtdIns(4,5)P2 Concentration Monitored in Living Cells. Curr. Biol 1998, 8 (6), 343–346. [DOI] [PubMed] [Google Scholar]
- (520).Sato M; Ueda Y; Takagi T; Umezawa Y Production of PtdInsP3 at Endomembranes Is Triggered by Receptor Endocytosis. Nat. Cell Biol 2003, 5 (11), 1016–1022. [DOI] [PubMed] [Google Scholar]
- (521).Pierre-Eugene C; Pagesy P; Nguyen TT; Neuillé M; Tschank G; Tennagels N; Hampe C; Issad T Effect of Insulin Analogues on insulin/IGF1 Hybrid Receptors: Increased Activation by Glargine but Not by Its Metabolites M1 and M2. PLoS One 2012, 7 (7), e41992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (522).Uchida Y; Hasegawa J; Chinnapen D; Inoue T; Okazaki S; Kato R; Wakatsuki S; Misaki R; Koike M; Uchiyama Y; et al. Intracellular Phosphatidylserine Is Essential for Retrograde Membrane Traffic through Endosomes. Proc. Natl. Acad. Sci 2011, 108 (38), 15846–15851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (523).Li M; Chen X; Ye Q-Z; Vogt A; Yin X-M A High-Throughput FRET-Based Assay for Determination of Atg4 Activity. Autophagy 2012, 8 (3), 401–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (524).Ai H; Hazelwood KL; Davidson MW; Campbell RE Fluorescent Protein FRET Pairs for Ratiometric Imaging of Dual Biosensors. Nat. Methods 2008, 5 (5), 401–403. [DOI] [PubMed] [Google Scholar]
- (525).Rehm M; Dussmann H; Janicke RU; Tavare JM; Kogel D; Prehn JHM Single-Cell Fluorescence Resonance Energy Transfer Analysis Demonstrates That Caspase Activation during Apoptosis Is a Rapid Process. Role of Caspase-3. J. Biol. Chem 2002, 277 (27), 24506–24514. [DOI] [PubMed] [Google Scholar]
- (526).Luo KQ; Yu VC; Pu Y; Chang DC Application of the Fluorescence Resonance Energy Transfer Method for Studying the Dynamics of Caspase-3 Activation during UVInduced Apoptosis in Living HeLa Cells. Biochem. Biophys. Res. Commun 2001, 283 (5), 1054–1060. [DOI] [PubMed] [Google Scholar]
- (527).Tyas L; Brophy VA; Pope A; Rivett AJ; Tavaré JM Rapid Caspase-3 Activation during Apoptosis Revealed Using Fluorescence-Resonance Energy Transfer. EMBO Rep 2000, 1 (3), 266–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (528).Karasawa S; Araki T; Nagai T; Mizuno H; Miyawaki A Cyan-Emitting and Orange-Emitting Fluorescent Proteins as a Donor/acceptor Pair for Fluorescence Resonance Energy Transfer. Biochem. J 2004, 381 (Pt 1), 307–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (529).Albeck JG; Burke JM; Aldridge BB; Zhang M; Lauffenburger DA; Sorger PK Quantitative Analysis of Pathways Controlling Extrinsic Apoptosis in Single Cells. Mol. Cell 2008, 30 (1), 11–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (530).Zlobovskaya OA; Sergeeva TF; Shirmanova MV; Dudenkova VV; Sharonov GV; Zagaynova EV; Lukyanov KA Genetically Encoded Far-Red Fluorescent Sensors for Caspase-3 Activity. Biotechniques 2016, 60 (2), 62–68. [DOI] [PubMed] [Google Scholar]
- (531).To T-L; Piggott BJ; Makhijani K; Yu D; Jan YN; Shu X Rationally Designed Fluorogenic Protease Reporter Visualizes Spatiotemporal Dynamics of Apoptosis in Vivo. Proc. Natl. Acad. Sci. U. S. A 2015, 112 (11), 3338–3343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (532).Takemoto K; Nagai T; Miyawaki A; Miura M Spatio-Temporal Activation of Caspase Revealed by Indicator That Is Insensitive to Environmental Effects. J. Cell Biol 2003, 160 (2), 235–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (533).Chiang JJ-H; Truong K Computational Modeling of a New Fluorescent Biosensor for Caspase Proteolytic Activity Improves Dynamic Range. IEEE Trans. Nanobioscience 2006, 5 (1), 41–45. [DOI] [PubMed] [Google Scholar]
- (534).Onuki R; Nagasaki A; Kawasaki H; Baba T; Uyeda TQP; Taira K Confirmation by FRET in Individual Living Cells of the Absence of Significant Amyloid -Mediated Caspase 8 Activation. Proc. Natl. Acad. Sci 2002, 99 (23), 14716–14721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (535).Ouyang M; Huang H; Shaner NC; Remacle AG; Shiryaev SA; Strongin AY; Tsien RY; Wang Y Simultaneous Visualization of Protumorigenic Src and MT1-MMP Activities with Fluorescence Resonance Energy Transfer. Cancer Res 2010, 70 (6), 2204–2212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (536).Ouyang M; Lu S; Li X-Y; Xu J; Seong J; Giepmans BNG; Shyy JY-J; Weiss SJ; Wang Y Visualization of Polarized Membrane Type 1 Matrix Metalloproteinase Activity in Live Cells by Fluorescence Resonance Energy Transfer Imaging. J. Biol. Chem 2008, 283 (25), 17740–17748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (537).Malik RU; Dysthe M; Ritt M; Sunahara RK; Sivaramakrishnan S ER/K Linked GPCR-G Protein Fusions Systematically Modulate Second Messenger Response in Cells. Sci. Rep 2017, 7 (1), 7749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (538).Galés C; Rebois RV; Hogue M; Trieu P; Breit A; Hébert TE; Bouvier M Real-Time Monitoring of Receptor and G-Protein Interactions in Living Cells. Nat. Methods 2005, 2 (3), 177–184. [DOI] [PubMed] [Google Scholar]
- (539).Malik RU; Ritt M; DeVree BT; Neubig RR; Sunahara RK; Sivaramakrishnan S Detection of G Protein-Selective G Protein-Coupled Receptor (GPCR) Conformations in Live Cells. J. Biol. Chem 2013, 288 (24), 17167–17178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (540).Janetopoulos C; Jin T; Devreotes P Receptor-Mediated Activation of Heterotrimeric G-Proteins in Living Cells. Science (80-. ) 2001, 291 (5512), 2408–2411. [DOI] [PubMed] [Google Scholar]
- (541).Lee D; Creed M; Jung K; Stefanelli T; Wendler DJ; Oh WC; Mignocchi NL; Lüscher C; Kwon H-B Temporally Precise Labeling and Control of Neuromodulatory Circuits in the Mammalian Brain. Nat. Methods 2017, 14 (5), 495–503. [DOI] [PubMed] [Google Scholar]
- (542).van Unen J; Stumpf AD; Schmid B; Reinhard NR; Hordijk PL; Hoffmann C; Gadella TWJ; Goedhart J A New Generation of FRET Sensors for Robust Measurement of Gαi1, Gαi2 and Gαi3 Activation Kinetics in Single Cells. PLoS One 2016, 11 (1), e0146789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (543).Gibson SK; Gilman AG Gialpha and Gbeta Subunits Both Define Selectivity of G Protein Activation by alpha2-Adrenergic Receptors. Proc. Natl. Acad. Sci. U. S. A 2006, 103 (1), 212–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (544).Bünemann M; Frank M; Lohse MJ Gi Protein Activation in Intact Cells Involves Subunit Rearrangement rather than Dissociation. Proc. Natl. Acad. Sci. U. S. A 2003, 100 (26), 16077–16082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (545).Adjobo-Hermans MJW; Goedhart J; van Weeren L; Nijmeijer S; Manders EMM; Offermanns S; Gadella TWJ. Real-Time Visualization of Heterotrimeric G Protein Gq Activation in Living Cells. BMC Biol 2011, 9 (1), 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (546).Jensen JB; Lyssand JS; Hague C; Hille B Fluorescence Changes Reveal Kinetic Steps of Muscarinic Receptor-Mediated Modulation of Phosphoinositides and Kv7.2/7.3 K+ Channels. J. Gen. Physiol 2009, 133 (4), 347–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (547).Hein P; Rochais F; Hoffmann C; Dorsch S; Nikolaev VO; Engelhardt S; Berlot CH; Lohse MJ; Bünemann M Gs Activation Is Time-Limiting in Initiating Receptor-Mediated Signaling. J. Biol. Chem 2006, 281 (44), 33345–33351. [DOI] [PubMed] [Google Scholar]
- (548).Thomsen ARB; Plouffe B; Cahill TJ; Shukla AK; Tarrasch JT; Dosey AM; Kahsai AW; Strachan RT; Pani B; Mahoney JP; et al. GPCR-G Protein-βArrestin Super-Complex Mediates Sustained G Protein Signaling. Cell 2016, 166 (4), 907–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (549).Galés C; Van Durm JJJ; Schaak S; Pontier S; Percherancier Y; Audet M; Paris H; Bouvier M Probing the Activation-Promoted Structural Rearrangements in Preassembled Receptor-G Protein Complexes. Nat. Struct. Mol. Biol 2006, 13 (9), 778–786. [DOI] [PubMed] [Google Scholar]
- (550).Hein P; Frank M; Hoffmann C; Lohse MJ; Bünemann M Dynamics of receptor/G Protein Coupling in Living Cells. EMBO J 2005, 24 (23), 4106–4114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (551).Vilardaga J-P; Bünemann M; Krasel C; Castro M; Lohse MJ Measurement of the Millisecond Activation Switch of G Protein-Coupled Receptors in Living Cells. Nat. Biotechnol 2003, 21 (7), 807–812. [DOI] [PubMed] [Google Scholar]
- (552).Dacres H; Wang J; Leitch V; Horne I; Anderson AR; Trowell SC Greatly Enhanced Detection of a Volatile Ligand at Femtomolar Levels Using Bioluminescence Resonance Energy Transfer (BRET). Biosens. Bioelectron 2011, 29 (1), 119–124. [DOI] [PubMed] [Google Scholar]
- (553).Wimmer T; Lorenz B; Stieger K Quantification of the Vascular Endothelial Growth Factor with a Bioluminescence Resonance Energy Transfer (BRET) Based Single Molecule Biosensor. Biosens. Bioelectron 2016, 86, 609–615. [DOI] [PubMed] [Google Scholar]
- (554).Ermakova YG; Bilan DS; Matlashov ME; Mishina NM; Markvicheva KN; Subach OM; Subach FV; Bogeski I; Hoth M; Enikolopov G; et al. Red Fluorescent Genetically Encoded Indicator for Intracellular Hydrogen Peroxide. Nat. Commun 2014, 5, 5222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (555).Bilan DS; Pase L; Joosen L; Gorokhovatsky AY; Ermakova YG; Gadella TWJ; Grabher C; Schultz C; Lukyanov S; Belousov VV HyPer-3: A Genetically Encoded H(2)O(2) Probe with Improved Performance for Ratiometric and Fluorescence Lifetime Imaging. ACS Chem. Biol 2013, 8 (3), 535–542. [DOI] [PubMed] [Google Scholar]
- (556).Markvicheva KN; Bilan DS; Mishina NM; Gorokhovatsky AY; Vinokurov LM; Lukyanov S; Belousov VV A Genetically Encoded Sensor for H2O2 with Expanded Dynamic Range. Bioorg. Med. Chem 2011, 19 (3), 1079–1084. [DOI] [PubMed] [Google Scholar]
- (557).Belousov VV; Fradkov AF; Lukyanov KA; Staroverov DB; Shakhbazov KS; Terskikh AV; Lukyanov S. Genetically Encoded Fluorescent Indicator for Intracellular Hydrogen Peroxide. Nat. Methods 2006, 3 (4), 281–286. [DOI] [PubMed] [Google Scholar]
- (558).Morgan B; Van Laer K; Owusu TNE; Ezeriņa D; Pastor-Flores D; Amponsah PS; Tursch A; Dick TP Real-Time Monitoring of Basal H2O2 Levels with Peroxiredoxin-Based Probes. Nat. Chem. Biol 2016, 12 (6), 437–443. [DOI] [PubMed] [Google Scholar]
- (559).Gutscher M; Sobotta MC; Wabnitz GH; Ballikaya S; Meyer AJ; Samstag Y; Dick TP Proximity-Based Protein Thiol Oxidation by H2O2-Scavenging Peroxidases. J. Biol. Chem 2009, 284 (46), 31532–31540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (560).Wang F; Niu W; Guo J; Schultz PG Unnatural Amino Acid Mutagenesis of Fluorescent Proteins. Angew. Chem. Int. Ed. Engl 2012, 51 (40), 10132–10135. [DOI] [PubMed] [Google Scholar]
- (561).Enyedi B; Zana M; Donkó Á; Geiszt M Spatial and Temporal Analysis of NADPH Oxidase-Generated Hydrogen Peroxide Signals by Novel Fluorescent Reporter Proteins. Antioxid. Redox Signal 2013, 19 (6), 523–534. [DOI] [PubMed] [Google Scholar]
- (562).Chen Z; Ai H A Highly Responsive and Selective Fluorescent Probe for Imaging Physiological Hydrogen Sulfide. Biochemistry 2014, 53 (37), 5966–5974. [DOI] [PubMed] [Google Scholar]
- (563).Chen S; Chen Z; Ren W; Ai H Reaction-Based Genetically Encoded Fluorescent Hydrogen Sulfide Sensors. J. Am. Chem. Soc 2012, 134 (23), 9589–9592. [DOI] [PubMed] [Google Scholar]
- (564).Pearce LL; Gandley RE; Han W; Wasserloos K; Stitt M; Kanai AJ; McLaughlin MK; Pitt BR; Levitan ES Role of Metallothionein in Nitric Oxide Signaling as Revealed by a Green Fluorescent Fusion Protein. Proc. Natl. Acad. Sci. U. S. A 2000, 97 (1), 477–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (565).Sato M; Nakajima T; Goto M; Umezawa Y Cell-Based Indicator to Visualize Picomolar Dynamics of Nitric Oxide Release from Living Cells. Anal. Chem 2006, 78 (24), 8175–8182. [DOI] [PubMed] [Google Scholar]
- (566).Sato M; Hida N; Umezawa Y Imaging the Nanomolar Range of Nitric Oxide with an Amplifier-Coupled Fluorescent Indicator in Living Cells. Proc. Natl. Acad. Sci 2005, 102 (41), 14515–14520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (567).Zhao BS; Liang Y; Song Y; Zheng C; Hao Z; Chen PR A Highly Selective Fluorescent Probe for Visualization of Organic Hydroperoxides in Living Cells. J. Am. Chem. Soc 2010, 132 (48). [DOI] [PubMed] [Google Scholar]
- (568).Chen Z; Ren W; Wright QE; Ai H Genetically Encoded Fluorescent Probe for the Selective Detection of Peroxynitrite. J. Am. Chem. Soc 2013, 135 (40), 14940–14943. [DOI] [PubMed] [Google Scholar]
- (569).Waypa GB; Guzy R; Mungai PT; Mack MM; Marks JD; Roe MW; Schumacker PT Increases in Mitochondrial Reactive Oxygen Species Trigger HypoxiaInduced Calcium Responses in Pulmonary Artery Smooth Muscle Cells. Circ. Res 2006, 99 (9), 970–978. [DOI] [PubMed] [Google Scholar]
- (570).Kolossov VL; Spring BQ; Clegg RM; Henry JJ; Sokolowski A; Kenis PJA; Gaskins HR Development of a High-Dynamic Range, GFP-Based FRET Probe Sensitive to Oxidative Microenvironments. Exp. Biol. Med. (Maywood) 2011, 236 (6), 681–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (571).Yano T; Oku M; Akeyama N; Itoyama A; Yurimoto H; Kuge S; Fujiki Y; Sakai Y A Novel Fluorescent Sensor Protein for Visualization of Redox States in the Cytoplasm and in Peroxisomes. Mol. Cell. Biol 2010, 30 (15), 3758–3766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (572).Kolossov VL; Spring BQ; Sokolowski A; Conour JE; Clegg RM; Kenis PJA; Gaskins HR Engineering Redox-Sensitive Linkers for Genetically Encoded FRET-Based Biosensors. Exp. Biol. Med. (Maywood) 2008, 233 (2), 238–248. [DOI] [PubMed] [Google Scholar]
- (573).Lohman JR; Remington SJ Development of a Family of Redox-Sensitive Green Fluorescent Protein Indicators for Use in Relatively Oxidizing Subcellular Environments. Biochemistry 2008, 47 (33), 8678–8688. [DOI] [PubMed] [Google Scholar]
- (574).Gutscher M; Pauleau A-L; Marty L; Brach T; Wabnitz GH; Samstag Y; Meyer AJ; Dick TP Real-Time Imaging of the Intracellular Glutathione Redox Potential. Nat. Methods 2008, 5 (6), 553–559. [DOI] [PubMed] [Google Scholar]
- (575).Cannon MB; Remington SJ Re-Engineering Redox-Sensitive Green Fluorescent Protein for Improved Response Rate. Protein Sci 2006, 15 (1), 45–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (576).Fan Y; Makar M; Wang MX; Ai H Monitoring Thioredoxin Redox with a Genetically Encoded Red Fluorescent Biosensor. Nat. Chem. Biol 2017, 13 (9), 1045–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (577).Fan Y; Ai H Development of Redox-Sensitive Red Fluorescent Proteins for Imaging Redox Dynamics in Cellular Compartments. Anal. Bioanal. Chem 2016, 408 (11), 2901–2911. [DOI] [PubMed] [Google Scholar]
- (578).Fan Y; Chen Z; Ai H Monitoring Redox Dynamics in Living Cells with a Redox-Sensitive Red Fluorescent Protein. Anal. Chem 2015, 87 (5), 2802–2810. [DOI] [PubMed] [Google Scholar]
- (579).Björnberg O; Østergaard H; Winther JR Mechanistic Insight Provided by Glutaredoxin within a Fusion to Redox-Sensitive Yellow Fluorescent Protein. Biochemistry 2006, 45 (7), 2362–2371. [DOI] [PubMed] [Google Scholar]
- (580).Ostergaard H; Henriksen A; Hansen FG; Winther JR Shedding Light on Disulfide Bond Formation: Engineering a Redox Switch in Green Fluorescent Protein. EMBO J 2001, 20 (21), 5853–5862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (581).Wang W; Fang H; Groom L; Cheng A; Zhang W; Liu J; Wang X; Li K; Han P; Zheng M; et al. Superoxide Flashes in Single Mitochondria. Cell 2008, 134 (2), 279–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (582).Seth A; Otomo T; Yin HL; Rosen MK Rational Design of Genetically Encoded Fluorescence Resonance Energy Transfer-Based Sensors of Cellular Cdc42 Signaling. Biochemistry 2003, 42 (14), 3997–4008. [DOI] [PubMed] [Google Scholar]
- (583).Hanna S; Miskolci V; Cox D; Hodgson L A New Genetically Encoded Single-Chain Biosensor for Cdc42 Based on FRET, Useful for Live-Cell Imaging. PLoS One 2014, 9 (5), e96469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (584).Itoh RE; Kurokawa K; Ohba Y; Yoshizaki H; Mochizuki N; Matsuda M Activation of Rac and cdc42 Video Imaged by Fluorescent Resonance Energy Transfer-Based Single-Molecule Probes in the Membrane of Living Cells. Mol. Cell. Biol 2002, 22 (18), 6582–6591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (585).Martin K; Reimann A; Fritz RD; Ryu H; Jeon NL; Pertz O Spatio-Temporal CoOrdination of RhoA, Rac1 and Cdc42 Activation during Prototypical Edge Protrusion and Retraction Dynamics. Sci. Rep 2016, 6 (1), 21901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (586).Hodgson L; Spiering D; Sabouri-Ghomi M; Dagliyan O; DerMardirossian C; Danuser G; Hahn KM FRET Binding Antenna Reports Spatiotemporal Dynamics of GDI-Cdc42 GTPase Interactions. Nat. Chem. Biol 2016, 12 (10), 802–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (587).Kitano M; Nakaya M; Nakamura T; Nagata S; Matsuda M Imaging of Rab5 Activity Identifies Essential Regulators for Phagosome Maturation. Nature 2008, 453 (7192), 241–245. [DOI] [PubMed] [Google Scholar]
- (588).Kraynov VS; Chamberlain C; Bokoch GM; Schwartz MA; Slabaugh S; Hahn KM Localized Rac Activation Dynamics Visualized in Living Cells. Science (80-. ) 2000, 290 (5490), 333–337. [DOI] [PubMed] [Google Scholar]
- (589).Fritz RD; Menshykau D; Martin K; Reimann A; Pontelli V; Pertz O SrGAP2Dependent Integration of Membrane Geometry and Slit-Robo-Repulsive Cues Regulates Fibroblast Contact Inhibition of Locomotion. Dev. Cell 2015, 35 (1), 78–92. [DOI] [PubMed] [Google Scholar]
- (590).Takaya A; Ohba Y; Kurokawa K; Matsuda M RalA Activation at Nascent Lamellipodia of Epidermal Growth Factor-Stimulated Cos7 Cells and Migrating MadinDarby Canine Kidney Cells. Mol. Biol. Cell 2004, 15 (6), 2549–2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (591).Kalab P; Weis K; Heald R Visualization of a Ran-GTP Gradient in Interphase and Mitotic Xenopus Egg Extracts. Science 2002, 295 (5564), 2452–2456. [DOI] [PubMed] [Google Scholar]
- (592).Mochizuki N; Yamashita S; Kurokawa K; Ohba Y; Nagai T; Miyawaki A; Matsuda M Spatio-Temporal Images of Growth-Factor-Induced Activation of Ras and Rap1. Nature 2001, 411 (6841), 1065–1068. [DOI] [PubMed] [Google Scholar]
- (593).Ng KY; Yin T; Machida K; Wu YI; Mayer BJ Phosphorylation of Dok1 by Abl Family Kinases Inhibits CrkI Transforming Activity. Oncogene 2015, 34 (20), 2650–2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (594).Murakoshi H; Shibata ACE ShadowY: A Dark Yellow Fluorescent Protein for FLIM-Based FRET Measurement. Sci. Rep 2017, 7 (1), 6791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (595).Oliveira AF; Yasuda R An Improved Ras Sensor for Highly Sensitive and Quantitative FRET-FLIM Imaging. PLoS One 2013, 8 (1), e52874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (596).Yasuda R; Harvey CD; Zhong H; Sobczyk A; van Aelst L; Svoboda K Supersensitive Ras Activation in Dendrites and Spines Revealed by Two-Photon Fluorescence Lifetime Imaging. Nat. Neurosci 2006, 9 (2), 283–291. [DOI] [PubMed] [Google Scholar]
- (597).van Unen J; Reinhard NR; Yin T; Wu YI; Postma M; Gadella TWJ; Goedhart J Plasma Membrane Restricted RhoGEF Activity Is Sufficient for RhoAMediated Actin Polymerization. Sci. Rep 2015, 5 (1), 14693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (598).Pertz O; Hodgson L; Klemke RL; Hahn KM Spatiotemporal Dynamics of RhoA Activity in Migrating Cells. Nature 2006, 440 (7087), 1069–1072. [DOI] [PubMed] [Google Scholar]
- (599).Nakamura T; Kurokawa K; Kiyokawa E; Matsuda M Analysis of the Spatiotemporal Activation of Rho GTPases Using Raichu Probes. Methods Enzymol 2006, 406, 315–332. [DOI] [PubMed] [Google Scholar]
- (600).Yoshizaki H; Ohba Y; Kurokawa K; Itoh RE; Nakamura T; Mochizuki N; Nagashima K; Matsuda M Activity of Rho-Family GTPases during Cell Division as Visualized with FRET-Based Probes. J. Cell Biol 2003, 162 (2), 223–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (601).Zawistowski JS; Sabouri-Ghomi M; Danuser G; Hahn KM; Hodgson L A RhoC Biosensor Reveals Differences in the Activation Kinetics of RhoA and RhoC in Migrating Cells. PLoS One 2013, 8 (11), e79877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (602).Kawase K; Nakamura T; Takaya A; Aoki K; Namikawa K; Kiyama H; Inagaki S; Takemoto H; Saltiel AR; Matsuda M GTP Hydrolysis by the Rho Family GTPase TC10 Promotes Exocytic Vesicle Fusion. Dev. Cell 2006, 11 (3), 411–421. [DOI] [PubMed] [Google Scholar]
- (603).Takaya A; Kamio T; Masuda M; Mochizuki N; Sawa H; Sato M; Nagashima K; Mizutani A; Matsuno A; Kiyokawa E; et al. R-Ras Regulates Exocytosis by Rgl2/RlfMediated Activation of RalA on Endosomes. Mol. Biol. Cell 2007, 18 (5), 1850–1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (604).Perroy J; Pontier S; Charest PG; Aubry M; Bouvier M Real-Time Monitoring of Ubiquitination in Living Cells by BRET. Nat. Methods 2004, 1 (3), 203–208. [DOI] [PubMed] [Google Scholar]
- (605).Nakaoka S; Sasaki K; Ito A; Nakao Y; Yoshida M A Genetically Encoded FRET Probe to Detect Intranucleosomal Histone H3K9 or H3K14 Acetylation Using BRD4, a BET Family Member. ACS Chem. Biol 2016, 11 (3), 729–733. [DOI] [PubMed] [Google Scholar]
- (606).Ito T; Umehara T; Sasaki K; Nakamura Y; Nishino N; Terada T; Shirouzu M; Padmanabhan B; Yokoyama S; Ito A; et al. Real-Time Imaging of Histone H4K12Specific Acetylation Determines the Modes of Action of Histone Deacetylase and Bromodomain Inhibitors. Chem. Biol 2011, 18 (4), 495–507. [DOI] [PubMed] [Google Scholar]
- (607).Sasaki K; Ito T; Nishino N; Khochbin S; Yoshida M Real-Time Imaging of Histone H4 Hyperacetylation in Living Cells. Proc. Natl. Acad. Sci. U. S. A 2009, 106 (38), 16257–16262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (608).Lin C-W; Jao CY; Ting AY Genetically Encoded Fluorescent Reporters of Histone Methylation in Living Cells. J. Am. Chem. Soc 2004, 126 (19), 5982–5983. [DOI] [PubMed] [Google Scholar]
- (609).Carrillo LD; Krishnamoorthy L; Mahal LK A Cellular FRET-Based Sensor for Beta-O-GlcNAc, a Dynamic Carbohydrate Modification Involved in Signaling. J. Am. Chem. Soc 2006, 128 (46), 14768–14769. [DOI] [PubMed] [Google Scholar]
- (610).Ganesan S; Ameer-Beg SM; Ng TTC; Vojnovic B; Wouters FS A Dark Yellow Fluorescent Protein (YFP)-Based Resonance Energy-Accepting Chromoprotein (REACh) for Förster Resonance Energy Transfer with GFP. Proc. Natl. Acad. Sci. U. S. A 2006, 103 (11), 4089–4094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (611).Piljic A; Schultz C Simultaneous Recording of Multiple Cellular Events by FRET. ACS Chem. Biol 2008, 3 (3), 156–160. [DOI] [PubMed] [Google Scholar]
- (612).Piljic A; Schultz C Annexin A4 Self-Association Modulates General Membrane Protein Mobility in Living Cells. Mol. Biol. Cell 2006, 17 (7), 3318–3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (613).Persechini A; Cronk B The Relationship between the Free Concentrations of Ca2+ and Ca2+-Calmodulin in Intact Cells. J. Biol. Chem 1999, 274 (11), 6827–6830. [DOI] [PubMed] [Google Scholar]
- (614).Romoser VA; Hinkle PM; Persechini A Detection in Living Cells of Ca2+-Dependent Changes in the Fluorescence Emission of an Indicator Composed of Two Green Fluorescent Protein Variants Linked by a Calmodulin-Binding Sequence. A New Class of Fluorescent Indicators. J. Biol. Chem 1997, 272 (20), 13270–13274. [DOI] [PubMed] [Google Scholar]
- (615).Chew T-L; Wolf WA; Gallagher PJ; Matsumura F; Chisholm RL A Fluorescent Resonant Energy Transfer-Based Biosensor Reveals Transient and Regional Myosin Light Chain Kinase Activation in Lamella and Cleavage Furrows. J. Cell Biol 2002, 156 (3), 543–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (616).Dormann D; Weijer G; Parent CA; Devreotes PN; Weijer CJ Visualizing PI3 Kinase-Mediated Cell-Cell Signaling during Dictyostelium Development. Curr. Biol 2002, 12 (14), 1178–1188. [DOI] [PubMed] [Google Scholar]
- (617).Friedrich MW; Aramuni G; Mank M; Mackinnon JAG; Griesbeck O Imaging CREB Activation in Living Cells. J. Biol. Chem 2010, 285 (30), 23285–23295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (618).Lorenz M; Yamaguchi H; Wang Y; Singer RH; Condeelis J Imaging Sites of NWasp Activity in Lamellipodia and Invadopodia of Carcinoma Cells. Curr. Biol 2004, 14 (8), 697–703. [DOI] [PubMed] [Google Scholar]
- (619).Corradi GR; Adamo HP Intramolecular Fluorescence Resonance Energy Transfer between Fused Autofluorescent Proteins Reveals Rearrangements of the N- and C-Terminal Segments of the Plasma Membrane Ca2+ Pump Involved in the Activation. J. Biol. Chem 2007, 282 (49), 35440–35448. [DOI] [PubMed] [Google Scholar]
- (620).Saimi Y; Kung C Calmodulin as an Ion Channel Subunit. Annu. Rev. Physiol 2002, 64 (1), 289–311. [DOI] [PubMed] [Google Scholar]
- (621).Mori MX; Erickson MG; Yue DT Functional Stoichiometry and Local Enrichment of Calmodulin Interacting with Ca2+ Channels. Science 2004, 304 (5669), 432–435. [DOI] [PubMed] [Google Scholar]
- (622).Porumb T; Yau P; Harvey TS; Ikura M A Calmodulin-Target Peptide Hybrid Molecule with Unique Calcium-Binding Properties. Protein Eng 1994, 7 (1), 109–115. [DOI] [PubMed] [Google Scholar]
- (623).Saucerman JJ; Bers DM Calmodulin Binding Proteins Provide Domains of Local Ca2+ Signaling in Cardiac Myocytes. J. Mol. Cell. Cardiol 2012, 52 (2), 312–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (624).Martin SR; Bayley PM; Brown SE; Porumb T; Zhang M; Ikura M Spectroscopic Characterization of a High-Affinity Calmodulin-Target Peptide Hybrid Molecule. Biochemistry 1996, 35 (11), 3508–3517. [DOI] [PubMed] [Google Scholar]
- (625).Vassylyev DG; Takeda S; Wakatsuki S; Maeda K; Maéda Y Crystal Structure of Troponin C in Complex with Troponin I Fragment at 2.3-A Resolution. Proc. Natl. Acad. Sci. U. S. A 1998, 95 (9), 4847–4852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (626).Potter JD; Gergely J The Calcium and Magnesium Binding Sites on Troponin and Their Role in the Regulation of Myofibrillar Adenosine Triphosphatase. J. Biol. Chem 1975, 250 (12), 4628–4633. [PubMed] [Google Scholar]
- (627).Robertson SP; Johnson JD; Potter JD The Time-Course of Ca2+ Exchange with Calmodulin, Troponin, Parvalbumin, and Myosin in Response to Transient Increases in Ca2+. Biophys. J 1981, 34 (3), 559–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (628).Finney LA; O’Halloran TV Transition Metal Speciation in the Cell: Insights from the Chemistry of Metal Ion Receptors. Science 2003, 300 (5621), 931–936. [DOI] [PubMed] [Google Scholar]
- (629).Adams SR; Harootunian AT; Buechler YJ; Taylor SS; Tsien RY Fluorescence Ratio Imaging of Cyclic AMP in Single Cells. Nature 1991, 349 (6311), 694–697. [DOI] [PubMed] [Google Scholar]
- (630).Mari SA; Pessoa J; Altieri S; Hensen U; Thomas L; Morais-Cabral JH; Müller DJ Gating of the MlotiK1 Potassium Channel Involves Large Rearrangements of the Cyclic Nucleotide-Binding Domains. Proc. Natl. Acad. Sci. U. S. A 2011, 108 (51), 20802–20807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (631).Merutka G; Shalongo W; Stellwagen E A Model Peptide with Enhanced Helicity. Biochemistry 1991, 30 (17), 4245–4248. [DOI] [PubMed] [Google Scholar]
- (632).Thomas CC; Deak M; Alessi DR; van Aalten DMF High-Resolution Structure of the Pleckstrin Homology Domain of Protein Kinase B/akt Bound to Phosphatidylinositol (3,4,5)-Trisphosphate. Curr. Biol 2002, 12 (14), 1256–1262. [DOI] [PubMed] [Google Scholar]
- (633).Lobley CMC; Ciulli A; Whitney HM; Williams G; Smith AG; Abell C; Blundell TL The Crystal Structure of Escherichia Coli Ketopantoate Reductase with NADP+ Bound. Biochemistry 2005, 44 (25), 8930–8939. [DOI] [PubMed] [Google Scholar]
- (634).Matak-Vinković D; Vinković M; Saldanha SA; Ashurst JL; von Delft F; Inoue T; Miguel RN; Smith AG; Blundell TL; Abell C Crystal Structure of Escherichia Coli Ketopantoate Reductase at 1.7 A Resolution and Insight into the Enzyme Mechanism. Biochemistry 2001, 40 (48), 14493–14500. [DOI] [PubMed] [Google Scholar]
- (635).Tsien RY Building and Breeding Molecules to Spy on Cells and Tumors. FEBS Lett 2005, 579 (4), 927–932. [DOI] [PubMed] [Google Scholar]
- (636).Sheikh SP; Vilardarga JP; Baranski TJ; Lichtarge O; Iiri T; Meng EC; Nissenson RA; Bourne HR Similar Structures and Shared Switch Mechanisms of the beta2-Adrenoceptor and the Parathyroid Hormone Receptor. Zn(II) Bridges between Helices III and VI Block Activation. J. Biol. Chem 1999, 274 (24), 17033–17041. [DOI] [PubMed] [Google Scholar]
- (637).Kataria R; Xu X; Fusetti F; Keizer-Gunnink I; Jin T; van Haastert PJM; Kortholt A Dictyostelium Ric8 Is a Nonreceptor Guanine Exchange Factor for Heterotrimeric G Proteins and Is Important for Development and Chemotaxis. Proc. Natl. Acad. Sci. U. S. A 2013, 110 (16), 6424–6429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (638).Hoffman GR; Nassar N; Cerione RA Structure of the Rho Family GTP-Binding Protein Cdc42 in Complex with the Multifunctional Regulator RhoGDI. Cell 2000, 100 (3), 345–356. [DOI] [PubMed] [Google Scholar]
- (639).Heim R; Tsien RY Engineering Green Fluorescent Protein for Improved Brightness, Longer Wavelengths and Fluorescence Resonance Energy Transfer. Curr. Biol 1996, 6 (2), 178–182. [DOI] [PubMed] [Google Scholar]
- (640).Mitra RD; Silva CM; Youvan DC Fluorescence Resonance Energy Transfer between Blue-Emitting and Red-Shifted Excitation Derivatives of the Green Fluorescent Protein. Gene 1996, 173 (1 Spec No), 13–17. [DOI] [PubMed] [Google Scholar]
- (641).Newman RH; Zhang J; Zhu H Toward a Systems-Level View of Dynamic Phosphorylation Networks. Front. Genet 2014, 5, 263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (642).Yaffe MB; Elia AE Phosphoserine/threonine-Binding Domains. Curr. Opin. Cell Biol 2001, 13 (2), 131–138. [DOI] [PubMed] [Google Scholar]
- (643).Yaffe MB; Smerdon SJ PhosphoSerine/threonine Binding Domains: You Can’t pSERious? Structure 2001, 9 (3), R33–8. [DOI] [PubMed] [Google Scholar]
- (644).Schlessinger J; Lemmon MA SH2 and PTB Domains in Tyrosine Kinase Signaling. Sci. STKE 2003, 2003 (191), RE12. [DOI] [PubMed] [Google Scholar]
- (645).Kemp BE Phosphorylation of Acyl and Dansyl Derivatives of the Peptide Leu-ArgArg-Ala-Ser-Leu-Gly by the cAMP-Dependent Protein Kinase. J. Biol. Chem 1980, 255 (7), 2914–2918. [PubMed] [Google Scholar]
- (646).Durocher D; Taylor IA; Sarbassova D; Haire LF; Westcott SL; Jackson SP; Smerdon SJ; Yaffe MB The Molecular Basis of FHA Domain:phosphopeptide Binding Specificity and Implications for Phospho-Dependent Signaling Mechanisms. Mol. Cell 2000, 6 (5), 1169–1182. [DOI] [PubMed] [Google Scholar]
- (647).Turk BE; Hutti JE; Cantley LC Determining Protein Kinase Substrate Specificity by Parallel Solution-Phase Assay of Large Numbers of Peptide Substrates. Nat. Protoc 2006, 1 (1), 375–379. [DOI] [PubMed] [Google Scholar]
- (648).Amano M; Hamaguchi T; Shohag MH; Kozawa K; Kato K; Zhang X; Yura Y; Matsuura Y; Kataoka C; Nishioka T; et al. Kinase-Interacting Substrate Screening Is a Novel Method to Identify Kinase Substrates. J. Cell Biol 2015, 209 (6), 895–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (649).Tarrant MK; Cole PA The Chemical Biology of Protein Phosphorylation. Annu. Rev. Biochem 2009, 78 (1), 797–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (650).Roy J; Cyert MS Cracking the Phosphatase Code: Docking Interactions Determine Substrate Specificity. Sci. Signal 2009, 2 (100), re9. [DOI] [PubMed] [Google Scholar]
- (651).Sacco F; Perfetto L; Castagnoli L; Cesareni G The Human Phosphatase Interactome: An Intricate Family Portrait. FEBS Lett 2012, 586 (17), 2732–2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (652).Hogan PG; Chen L; Nardone J; Rao A Transcriptional Regulation by Calcium, Calcineurin, and NFAT. Genes Dev 2003, 17 (18), 2205–2232. [DOI] [PubMed] [Google Scholar]
- (653).Rossetto D; Avvakumov N; Côté J Histone Phosphorylation: A Chromatin Modification Involved in Diverse Nuclear Events. Epigenetics 2012, 7 (10), 1098–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (654).Eissenberg JC Structural Biology of the Chromodomain: Form and Function. Gene 2012, 496 (2), 69–78. [DOI] [PubMed] [Google Scholar]
- (655).Zeng L; Zhou MM Bromodomain: An Acetyl-Lysine Binding Domain. FEBS Lett 2002, 513 (1), 124–128. [DOI] [PubMed] [Google Scholar]
- (656).Yang X; Qian K Protein O-GlcNAcylation: Emerging Mechanisms and Functions. Nat. Rev. Mol. Cell Biol 2017, 18 (7), 452–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (657).Orr AW; Helmke BP; Blackman BR; Schwartz MA Mechanisms of Mechanotransduction. Dev. Cell 2006, 10 (1), 11–20. [DOI] [PubMed] [Google Scholar]
- (658).Becker N; Oroudjev E; Mutz S; Cleveland JP; Hansma PK; Hayashi CY; Makarov DE; Hansma HG Molecular Nanosprings in Spider Capture-Silk Threads. Nat. Mater 2003, 2 (4), 278–283. [DOI] [PubMed] [Google Scholar]
- (659).Neuman KC; Nagy A Single-Molecule Force Spectroscopy: Optical Tweezers, Magnetic Tweezers and Atomic Force Microscopy. Nat. Methods 2008, 5 (6), 491–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (660).Freikamp A; Cost A-L; Grashoff C The Piconewton Force Awakens: Quantifying Mechanics in Cells. Trends Cell Biol 2016, 26 (11), 838–847. [DOI] [PubMed] [Google Scholar]
- (661).Vinkenborg JL; Evers TH; Reulen SWA; Meijer EW; Merkx M Enhanced Sensitivity of FRET-Based Protease Sensors by Redesign of the GFP Dimerization Interface. Chembiochem 2007, 8 (10), 1119–1121. [DOI] [PubMed] [Google Scholar]
- (662).Nguyen AW; Daugherty PS Evolutionary Optimization of Fluorescent Proteins for Intracellular FRET. Nat. Biotechnol 2005, 23 (3), 355–360. [DOI] [PubMed] [Google Scholar]
- (663).Kotera I; Iwasaki T; Imamura H; Noji H; Nagai T Reversible Dimerization of Aequorea Victoria Fluorescent Proteins Increases the Dynamic Range of FRET-Based Indicators. ACS Chem. Biol 2010, 5 (2), 215–222. [DOI] [PubMed] [Google Scholar]
- (664).Yang F; Moss LG; Phillips GN The Molecular Structure of Green Fluorescent Protein. Nat. Biotechnol 1996, 14 (10), 1246–1251. [DOI] [PubMed] [Google Scholar]
- (665).Campbell RE; Tour O; Palmer AE; Steinbach PA; Baird GS; Zacharias DA; Tsien RY A Monomeric Red Fluorescent Protein. Proc. Natl. Acad. Sci. U. S. A 2002, 99 (12), 7877–7882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (666).Alford SC; Ding Y; Simmen T; Campbell RE Dimerization-Dependent Green and Yellow Fluorescent Proteins. ACS Synth. Biol 2012, 1 (12), 569–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (667).Widder EA Bioluminescence in the Ocean: Origins of Biological, Chemical, and Ecological Diversity. Science 2010, 328 (5979), 704–708. [DOI] [PubMed] [Google Scholar]
- (668).Conti E; Franks NP; Brick P Crystal Structure of Firefly Luciferase Throws Light on a Superfamily of Adenylate-Forming Enzymes. Structure 1996, 4 (3), 287–298. [DOI] [PubMed] [Google Scholar]
- (669).Woo J; von Arnim AG Mutational Optimization of the Coelenterazine-Dependent Luciferase from Renilla. Plant Methods 2008, 4 (1), 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (670).Ayoub MA Resonance Energy Transfer-Based Approaches to Study GPCRs. Methods Cell Biol 2016, 132, 255–292. [DOI] [PubMed] [Google Scholar]
- (671).Ward WW; Cormier MJ In Vitro Energy Transfer in Renilla Bioluminescence. In Journal of Physical Chemistry; 1976; Vol. 80, pp 2289–2291. [Google Scholar]
- (672).Ward WW; Cormier MJ ENERGY TRANSFER VIA PROTEIN-PROTEIN INTERACTION IN RENILLA BIOLUMINESCENCE. Photochem. Photobiol 1978, 27 (4), 389–396. [Google Scholar]
- (673).Kajiyama N; Nakano E Isolation and Characterization of Mutants of Firefly Luciferase Which Produce Different Colors of Light. Protein Eng 1991, 4 (6), 691–693. [DOI] [PubMed] [Google Scholar]
- (674).Mamaev Sergey V.; Laikhter Andrei L.; Arslan Tuncer, and; Hecht SM Firefly Luciferase: Alteration of the Color of Emitted Light Resulting from Substitutions at Position 286 1996. [Google Scholar]
- (675).Branchini BR; Magyar RA; Murtiashaw MH; Anderson SM; Helgerson LC; Zimmer M Site-Directed Mutagenesis of Firefly Luciferase Active Site Amino Acids: A Proposed Model for Bioluminescence Color. Biochemistry 1999, 38 (40), 13223–13230. [DOI] [PubMed] [Google Scholar]
- (676).Loening AM; Wu AM; Gambhir SS Red-Shifted Renilla Reniformis Luciferase Variants for Imaging in Living Subjects. Nat. Methods 2007, 4 (8), 641–643. [DOI] [PubMed] [Google Scholar]
- (677).Hall MP; Unch J; Binkowski BF; Valley MP; Butler BL; Wood MG; Otto P; Zimmerman K; Vidugiris G; Machleidt T; et al. Engineered Luciferase Reporter from a Deep Sea Shrimp Utilizing a Novel Imidazopyrazinone Substrate. ACS Chem. Biol 2012, 7 (11), 1848–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (678).Chu J; Oh Y; Sens A; Ataie N; Dana H; Macklin JJ; Laviv T; Welf ES; Dean KM; Zhang F; et al. A Bright Cyan-Excitable Orange Fluorescent Protein Facilitates Dual-Emission Microscopy and Enhances Bioluminescence Imaging in Vivo. Nat. Biotechnol 2016, 34 (7), 760–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (679).Iwano S; Sugiyama M; Hama H; Watakabe A; Hasegawa N; Kuchimaru T; Tanaka KZ; Takahashi M; Ishida Y; Hata J; et al. Single-Cell Bioluminescence Imaging of Deep Tissue in Freely Moving Animals. Science 2018, 359 (6378), 935–939. [DOI] [PubMed] [Google Scholar]
- (680).Vodermaier HC APC/C and SCF: Controlling Each Other and the Cell Cycle. Curr. Biol 2004, 14 (18), R787–96. [DOI] [PubMed] [Google Scholar]
- (681).Abe T; Sakaue-Sawano A; Kiyonari H; Shioi G; Inoue K; Horiuchi T; Nakao K; Miyawaki A; Aizawa S; Fujimori T Visualization of Cell Cycle in Mouse Embryos with Fucci2 Reporter Directed by Rosa26 Promoter. Development 2013, 140 (1), 237–246. [DOI] [PubMed] [Google Scholar]
- (682).Boström J; Sramkova Z; Salašová A; Johard H; Mahdessian D; Fedr R; Marks C; Medalová J; Souček K; Lundberg E; et al. Comparative Cell Cycle Transcriptomics Reveals Synchronization of Developmental Transcription Factor Networks in Cancer Cells. PLoS One 2017, 12 (12), e0188772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (683).Barrasso AP; Tong X; Poché RA The mito::mKate2 Mouse: A Far-Red Fluorescent Reporter Mouse Line for Tracking Mitochondrial Dynamics in Vivo. Genesis 2018, 56 (2), e23087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (684).Koesling D; Russwurm M; Mergia E; Mullershausen F; Friebe A Nitric OxideSensitive Guanylyl Cyclase: Structure and Regulation. Neurochem. Int 2004, 45 (6), 813–819. [DOI] [PubMed] [Google Scholar]
- (685).Weissleder R A Clearer Vision for in Vivo Imaging. Nat. Biotechnol 2001, 19 (4), 316–317. [DOI] [PubMed] [Google Scholar]
- (686).Shcherbakova DM; Shemetov AA; Kaberniuk AA; Verkhusha VV Natural Photoreceptors as a Source of Fluorescent Proteins, Biosensors, and Optogenetic Tools. Annu. Rev. Biochem 2015, 84 (1), 519–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (687).Wagner JR; Zhang J; von Stetten D; Günther M; Murgida DH; Mroginski MA; Walker JM; Forest KT; Hildebrandt P; Vierstra RD Mutational Analysis of Deinococcus Radiodurans Bacteriophytochrome Reveals Key Amino Acids Necessary for the Photochromicity and Proton Exchange Cycle of Phytochromes. J. Biol. Chem 2008, 283 (18), 12212–12226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (688).Giraud E; Zappa S; Vuillet L; Adriano J-M; Hannibal L; Fardoux J; Berthomieu C; Bouyer P; Pignol D; Verméglio A A New Type of Bacteriophytochrome Acts in Tandem with a Classical Bacteriophytochrome to Control the Antennae Synthesis in Rhodopseudomonas Palustris. J. Biol. Chem 2005, 280 (37), 32389–32397. [DOI] [PubMed] [Google Scholar]
- (689).Yang X; Kuk J; Moffat K Crystal Structure of Pseudomonas Aeruginosa Bacteriophytochrome: Photoconversion and Signal Transduction. Proc. Natl. Acad. Sci. U. S. A 2008, 105 (38), 14715–14720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (690).Auldridge ME; Forest KT Bacterial Phytochromes: More than Meets the Light. Crit. Rev. Biochem. Mol. Biol 2011, 46 (1), 67–88. [DOI] [PubMed] [Google Scholar]
- (691).Shu X; Royant A; Lin MZ; Aguilera TA; Lev-Ram V; Steinbach PA; Tsien RY Mammalian Expression of Infrared Fluorescent Proteins Engineered from a Bacterial Phytochrome. Science 2009, 324 (5928), 804–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (692).Rockwell NC; Su Y-S; Lagarias JC Phytochrome Structure and Signaling Mechanisms. Annu. Rev. Plant Biol 2006, 57 (1), 837–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (693).Yu D; Gustafson WC; Han C; Lafaye C; Noirclerc-Savoye M; Ge W-P; Thayer DA; Huang H; Kornberg TB; Royant A; et al. An Improved Monomeric Infrared Fluorescent Protein for Neuronal and Tumour Brain Imaging. Nat. Commun 2014, 5, 3626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (694).Yu D; Baird MA; Allen JR; Howe ES; Klassen MP; Reade A; Makhijani K; Song Y; Liu S; Murthy Z; et al. A Naturally Monomeric Infrared Fluorescent Protein for Protein Labeling in Vivo. Nat. Methods 2015, 12 (8), 763–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (695).Tsien RY Very Long-Term Memories May Be Stored in the Pattern of Holes in the Perineuronal Net. Proc. Natl. Acad. Sci. U. S. A 2013, 110 (30), 12456–12461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (696).McKinney SA; Murphy CS; Hazelwood KL; Davidson MW; Looger LL A Bright and Photostable Photoconvertible Fluorescent Protein. Nat. Methods 2009, 6 (2), 131–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (697).Zolnik TA; Sha F; Johenning FW; Schreiter ER; Looger LL; Larkum ME; Sachdev RNS All-Optical Functional Synaptic Connectivity Mapping in Acute Brain Slices Using the Calcium Integrator CaMPARI. J. Physiol 2017, 595 (5), 1465–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (698).Harper SM; Neil LC; Gardner KH Structural Basis of a Phototropin Light Switch. Science 2003, 301 (5639), 1541–1544. [DOI] [PubMed] [Google Scholar]
- (699).Lungu OI; Hallett RA; Choi EJ; Aiken MJ; Hahn KM; Kuhlman B Designing Photoswitchable Peptides Using the AsLOV2 Domain. Chem. Biol 2012, 19 (4), 507–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (700).Ando R; Mizuno H; Miyawaki A Regulated Fast Nucleocytoplasmic Shuttling Observed by Reversible Protein Highlighting. Science 2004, 306 (5700), 1370–1373. [DOI] [PubMed] [Google Scholar]
- (701).Kholodenko BN Cell-Signalling Dynamics in Time and Space. Nat. Rev. Mol. Cell Biol 2006, 7 (3), 165–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (702).Santos R; Ursu O; Gaulton A; Bento AP; Donadi RS; Bologa CG; Karlsson A; Al-Lazikani B; Hersey A; Oprea TI; et al. A Comprehensive Map of Molecular Drug Targets. Nat. Rev. Drug Discov 2017, 16 (1), 19–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (703).Luttrell LM Reviews in Molecular Biology and Biotechnology: Transmembrane Signaling by G Protein-Coupled Receptors. Mol. Biotechnol 2008, 39 (3), 239–264. [DOI] [PubMed] [Google Scholar]
- (704).El-Haibi CP; Sharma P; Singh R; Gupta P; Taub DD; Singh S; Lillard JW Jr Differential G Protein Subunit Expression by Prostate Cancer Cells and Their Interaction with CXCR5. Mol. Cancer 2013, 12 (1), 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (705).Taylor SS; Kim C; Vigil D; Haste NM; Yang J; Wu J; Anand GS Dynamics of Signaling by PKA. Biochim. Biophys. Acta 2005, 1754 (1–2), 25–37. [DOI] [PubMed] [Google Scholar]
- (706).Pidoux G; Taskén K Specificity and Spatial Dynamics of Protein Kinase A Signaling Organized by A-Kinase-Anchoring Proteins. J. Mol. Endocrinol 2010, 44 (5), 271–284. [DOI] [PubMed] [Google Scholar]
- (707).Scott JD; Dessauer CW; Taskén K Creating Order from Chaos: Cellular Regulation by Kinase Anchoring. Annu. Rev. Pharmacol. Toxicol 2013, 53 (1), 187–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (708).Herbst KJ; Allen MD; Zhang J Spatiotemporally Regulated Protein Kinase A Activity Is a Critical Regulator of Growth Factor-Stimulated Extracellular Signal-Regulated Kinase Signaling in PC12 Cells. Mol. Cell. Biol 2011, 31 (19), 4063–4075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (709).Ryu H; Chung M; Dobrzyński M; Fey D; Blum Y; Lee SS; Peter M; Kholodenko BN; Jeon NL; Pertz O Frequency Modulation of ERK Activation Dynamics Rewires Cell Fate. Mol. Syst. Biol 2015, 11 (11), 838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (710).Fey D; Halasz M; Dreidax D; Kennedy SP; Hastings JF; Rauch N; Munoz AG; Pilkington R; Fischer M; Westermann F; et al. Signaling Pathway Models as Biomarkers: Patient-Specific Simulations of JNK Activity Predict the Survival of Neuroblastoma Patients. Sci. Signal 2015, 8 (408), ra130. [DOI] [PubMed] [Google Scholar]
- (711).Tyson JJ; Chen KC; Novak B Sniffers, Buzzers, Toggles and Blinkers: Dynamics of Regulatory and Signaling Pathways in the Cell. Curr. Opin. Cell Biol 2003, 15 (2), 221–231. [DOI] [PubMed] [Google Scholar]
- (712).Milo R; Shen-Orr S; Itzkovitz S; Kashtan N; Chklovskii D; Alon U Network Motifs: Simple Building Blocks of Complex Networks. Science 2002, 298 (5594), 824–827. [DOI] [PubMed] [Google Scholar]
- (713).Tiana G; Krishna S; Pigolotti S; Jensen MH; Sneppen K Oscillations and Temporal Signalling in Cells. Phys. Biol 2007, 4 (2), R1–17. [DOI] [PubMed] [Google Scholar]
- (714).Ma W; Trusina A; El-Samad H; Lim W. a; Tang C. Defining Network Topologies That Can Achieve Biochemical Adaptation. Cell 2009, 138 (4), 760–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (715).Traverse S; Gomez N; Paterson H; Marshall C; Cohen P Sustained Activation of the Mitogen-Activated Protein (MAP) Kinase Cascade May Be Required for Differentiation of PC12 Cells. Comparison of the Effects of Nerve Growth Factor and Epidermal Growth Factor. Biochem. J 1992, 288 ( Pt 2), 351–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (716).Qui MS; Green SH PC12 Cell Neuronal Differentiation Is Associated with Prolonged p21ras Activity and Consequent Prolonged ERK Activity. Neuron 1992, 9 (4), 705–717. [DOI] [PubMed] [Google Scholar]
- (717).Zhang K; Duan L; Ong Q; Lin Z; Varman PM; Sung K; Cui B Light-Mediated Kinetic Control Reveals the Temporal Effect of the Raf/MEK/ERK Pathway in PC12 Cell Neurite Outgrowth. PLoS One 2014, 9 (3), e92917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (718).Berridge MJ; Bootman MD; Roderick HL Calcium: Calcium Signalling: Dynamics, Homeostasis and Remodelling. Nat. Rev. Mol. Cell Biol 2003, 4 (7), 517–529. [DOI] [PubMed] [Google Scholar]
- (719).Novák B; Tyson JJ Design Principles of Biochemical Oscillators. Nat. Rev. Mol. Cell Biol 2008, 9 (12), 981–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (720).Dupont G; Combettes L; Bird GS; Putney JW Calcium Oscillations. Cold Spring Harb. Perspect. Biol 2011, 3 (3), a004226–a004226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (721).Saucerman JJ; McCulloch A Cardiac Beta-Adrenergic Signaling: From Subcellular Microdomains to Heart Failure. Ann. N. Y. Acad. Sci 2006, 1080 (1), 348–361. [DOI] [PubMed] [Google Scholar]
- (722).Nelson DE; Ihekwaba AEC; Elliott M; Johnson JR; Gibney CA; Foreman BE; Nelson G; See V; Horton CA; Spiller DG; et al. Oscillations in NF- B Signaling Control the Dynamics of Gene Expression. Science (80-. ) 2004, 306 (5696), 704–708. [DOI] [PubMed] [Google Scholar]
- (723).Qian Y-W; Clusin WT; Lin S-F; Han J; Sung RJ Spatial Heterogeneity of Calcium Transient Alternans During the Early Phase of Myocardial Ischemia in the Blood-Perfused Rabbit Heart. Circulation 2001, 104 (17). [DOI] [PubMed] [Google Scholar]
- (724).Lin GC; Rurangirwa JK; Koval M; Steinberg TH Gap Junctional Communication Modulates Agonist-Induced Calcium Oscillations in Transfected HeLa Cells. J. Cell Sci 2004, 117 (6). [DOI] [PubMed] [Google Scholar]
- (725).Jacob R Calcium Oscillations in Electrically Non-Excitable Cells. Biochim. Biophys. Acta - Mol. Cell Res 1990, 1052 (3), 427–438. [DOI] [PubMed] [Google Scholar]
- (726).Barbosa RM; Silva AM; Tomé AR; Stamford JA; Santos RM; Rosário LM Control of Pulsatile 5-HT/insulin Secretion from Single Mouse Pancreatic Islets by Intracellular Calcium Dynamics. J. Physiol 1998, 510 (1), 135–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (727).Keizer J; Magnus G ATP-Sensitive Potassium Channel and Bursting in the Pancreatic Beta Cell. A Theoretical Study. Biophys. J 1989, 56 (2), 229–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (728).Li J; Shuai HY; Gylfe E; Tengholm A Oscillations of Sub-Membrane ATP in Glucose-Stimulated Beta Cells Depend on Negative Feedback from Ca(2+). Diabetologia 2013, 56 (7), 1577–1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (729).Landa LR; Harbeck M; Kaihara K; Chepurny O; Kitiphongspattana K; Graf O; Nikolaev VO; Lohse MJ; Holz GG; Roe MW Interplay of Ca2+ and cAMP Signaling in the Insulin-Secreting MIN6 Beta-Cell Line. J. Biol. Chem 2005, 280 (35), 31294–31302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (730).Ferrell JE Jr. Tripping the Switch Fantastic: How a Protein Kinase Cascade Can Convert Graded Inputs into Switch-like Outputs. Trends Biochem. Sci 1996, 21 (12), 460–466. [DOI] [PubMed] [Google Scholar]
- (731).Ferrell JE; Xiong W Bistability in Cell Signaling: How to Make Continuous Processes Discontinuous, and Reversible Processes Irreversible. Chaos 2001, 11 (1), 227–236. [DOI] [PubMed] [Google Scholar]
- (732).Craciun G; Tang Y; Feinberg M Understanding Bistability in Complex EnzymeDriven Reaction Networks MATHEMATICS 2006. [DOI] [PMC free article] [PubMed]
- (733).Angeli D; Ferrell JE; Sontag ED Detection of Multistability, Bifurcations, and Hysteresis in a Large Class of Biological Positive-Feedback Systems. Proc. Natl. Acad. Sci. U. S. A 2004, 101 (7), 1822–1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (734).Zhang Q; Bhattacharya S; Andersen ME Ultrasensitive Response Motifs: Basic Amplifiers in Molecular Signalling Networks. Open Biol 2013, 3 (4), 130031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (735).Xiong W; F. JE Jr A Positive-Feedback-Based Bistable “ Memory Module ” That Governs a Cell Fate Decision. Nature 2003, 426 (November), 460–466. [DOI] [PubMed] [Google Scholar]
- (736).Qiao L; Nachbar RB; Kevrekidis IG; Shvartsman SY Bistability and Oscillations in the Huang-Ferrell Model of MAPK Signaling. PLoS Comput. Biol 2007, 3 (9), e184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (737).Chang L; Karin M Mammalian MAP Kinase Signalling Cascades. Nature 2001, 410 (6824), 37–40. [DOI] [PubMed] [Google Scholar]
- (738).Weston CR; Davis RJ The JNK Signal Transduction Pathway. Curr. Opin. Cell Biol 2007, 19 (2), 142–149. [DOI] [PubMed] [Google Scholar]
- (739).Bagowski CP; Ferrell JE Bistability in the JNK Cascade. Curr. Biol 2001, 11 (15), 1176–1182. [DOI] [PubMed] [Google Scholar]
- (740).Zaytsev AV; Segura-Peña D; Godzi M; Calderon A; Ballister ER; Stamatov R; Mayo AM; Peterson L; Black BE; Ataullakhanov FI; et al. Bistability of a Coupled Aurora B Kinase-Phosphatase System in Cell Division. Elife 2016, 5, e10644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (741).Murata-Hori M; Tatsuka M; Wang Y-L Probing the Dynamics and Functions of Aurora B Kinase in Living Cells during Mitosis and Cytokinesis. Mol. Biol. Cell 2002, 13 (4), 1099–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (742).Albeck JG; Burke JM; Spencer SL; Lauffenburger DA; Sorger PK Modeling a Snap-Action, Variable-Delay Switch Controlling Extrinsic Cell Death. PLoS Biol 2008, 6 (12), 2831–2852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (743).Aoki K; Kumagai Y; Sakurai A; Komatsu N; Fujita Y; Shionyu C; Matsuda M Stochastic ERK Activation Induced by Noise and Cell-to-Cell Propagation Regulates Cell Density-Dependent Proliferation. Mol. Cell 2013, 52 (4), 529–540. [DOI] [PubMed] [Google Scholar]
- (744).Lakowicz JR Principles of Fluorescence Spectroscopy; Springer, 2006. [Google Scholar]
- (745).Goedhart J; von Stetten D; Noirclerc-Savoye M; Lelimousin M; Joosen L; Hink MA; van Weeren L; Gadella TWJ; Royant A Structure-Guided Evolution of Cyan Fluorescent Proteins towards a Quantum Yield of 93%. Nat. Commun 2012, 3 (1), 751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (746).Bindels DS; Haarbosch L; van Weeren L; Postma M; Wiese KE; Mastop M; Aumonier S; Gotthard G; Royant A; Hink MA; et al. mScarlet: A Bright Monomeric Red Fluorescent Protein for Cellular Imaging. Nat. Methods 2017, 14 (1), 53–56. [DOI] [PubMed] [Google Scholar]
- (747).Hires SA; Tian L; Looger LL Reporting Neural Activity with Genetically Encoded Calcium Indicators. Brain Cell Biol 2008, 36 (1–4), 69–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (748).Tay LH; Griesbeck O; Yue DT Live-Cell Transforms between Ca2+ Transients and FRET Responses for a Troponin-C-Based Ca2+ Sensor. Biophys. J 2007, 93 (11), 4031–4040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (749).Bu Z; Callaway DJE. Proteins MOVE! Protein Dynamics and Long-Range Allostery in Cell Signaling; Academic Press, 2011; Vol. 83, pp 163–221. [DOI] [PubMed] [Google Scholar]
- (750).Golding I; Cox EC Physical Nature of Bacterial Cytoplasm. Phys. Rev. Lett 2006, 96 (9), 98102. [DOI] [PubMed] [Google Scholar]
- (751).Hayes JS; Brunton LL; Brown JH; Reese JB; Mayer SE Hormonally Specific Expression of Cardiac Protein Kinase Activity. Proc. Natl. Acad. Sci. U. S. A 1979, 76 (4), 1570–1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (752).Brunton LL; Hayes JS; Mayer SE Hormonally Specific Phosphorylation of Cardiac Troponin I and Activation of Glycogen Phosphorylase. Nature 1979, 280 (5717), 78–80. [DOI] [PubMed] [Google Scholar]
- (753).Corbin JD; Sugden PH; Lincoln TM; Keely SL Compartmentalization of Adenosine 3’:5’-monophosphate and Adenosine 3’:5’-monophosphate-Dependent Protein Kinase in Heart Tissue. J. Biol. Chem 1977, 252 (11), 3854–3861. [PubMed] [Google Scholar]
- (754).Hayes JS; Brunton LL; Mayer SE Selective Activation of Particulate cAMP-Dependent Protein Kinase by Isoproterenol and Prostaglandin E1. J. Biol. Chem 1980, 255 (11), 5113–5119. [PubMed] [Google Scholar]
- (755).Buxton IL; Brunton LL Compartments of Cyclic AMP and Protein Kinase in Mammalian Cardiomyocytes. J. Biol. Chem 1983, 258 (17), 10233–10239. [PubMed] [Google Scholar]
- (756).Steinberg SF; Brunton LL Compartmentation of G Protein-Coupled Signaling Pathways in Cardiac Myocytes. Annu. Rev. Pharmacol. Toxicol 2001, 41 (1), 751–773. [DOI] [PubMed] [Google Scholar]
- (757).Shamir M; Bar-On Y; Phillips R; Milo R SnapShot: Timescales in Cell Biology. Cell 2016, 164 (6), 1302–1302.e1. [DOI] [PubMed] [Google Scholar]
- (758).Dehmelt L; Bastiaens PIH Spatial Organization of Intracellular Communication: Insights from Imaging. Nat. Rev. Mol. Cell Biol 2010, 11 (6), 440–452. [DOI] [PubMed] [Google Scholar]
- (759).Good MC; Zalatan JG; Lim WA Scaffold Proteins: Hubs for Controlling the Flow of Cellular Information. Science 2011, 332 (6030), 680–686.21551057 [Google Scholar]
- (760).Shenoy SK; Lefkowitz RJ Seven-Transmembrane Receptor Signaling through BetaArrestin. Sci. STKE 2005, 2005 (308), cm10. [DOI] [PubMed] [Google Scholar]
- (761).Shenoy SK; Lefkowitz RJ β-Arrestin-Mediated Receptor Trafficking and Signal Transduction. Trends Pharmacol. Sci 2011, 32 (9), 521–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (762).DeWire SM; Ahn S; Lefkowitz RJ; Shenoy SK Beta-Arrestins and Cell Signaling. Annu. Rev. Physiol 2007, 69 (1), 483–510. [DOI] [PubMed] [Google Scholar]
- (763).O’Hayre M; Eichel K; Avino S; Zhao X; Steffen DJ; Feng X; Kawakami K; Aoki J; Messer K; Sunahara R; et al. Genetic Evidence That β-Arrestins Are Dispensable for the Initiation of β2-Adrenergic Receptor Signaling to ERK. Sci. Signal 2017, 10 (484), eaal3395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (764).Calebiro D; Nikolaev VO; Gagliani MC; de Filippis T; Dees C; Tacchetti C; Persani L; Lohse MJ Persistent cAMP-Signals Triggered by Internalized G-ProteinCoupled Receptors. PLoS Biol 2009, 7 (8), e1000172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (765).Ferrandon S; Feinstein TN; Castro M; Wang B; Bouley R; Potts JT; Gardella TJ; Vilardaga J-P Sustained Cyclic AMP Production by Parathyroid Hormone Receptor Endocytosis. Nat. Chem. Biol 2009, 5 (10), 734–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (766).Oakley RH; Laporte SA; Holt JA; Barak LS; Caron MG Association of BetaArrestin with G Protein-Coupled Receptors during Clathrin-Mediated Endocytosis Dictates the Profile of Receptor Resensitization. J. Biol. Chem 1999, 274 (45), 32248–32257. [DOI] [PubMed] [Google Scholar]
- (767).Oakley RH; Laporte SA; Holt JA; Caron MG; Barak LS Differential Affinities of Visual Arrestin, Beta arrestin1, and Beta arrestin2 for G Protein-Coupled Receptors Delineate Two Major Classes of Receptors. J. Biol. Chem 2000, 275 (22), 17201–17210. [DOI] [PubMed] [Google Scholar]
- (768).Shukla AK; Westfield GH; Xiao K; Reis RI; Huang L-Y; Tripathi-Shukla P; Qian J; Li S; Blanc A; Oleskie AN; et al. Visualization of Arrestin Recruitment by a G-Protein-Coupled Receptor. Nature 2014, 512 (7513), 218–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (769).Altarejos JY; Montminy M CREB and the CRTC Co-Activators: Sensors for Hormonal and Metabolic Signals. Nat. Rev. Mol. Cell Biol 2011, 12 (3), 141–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (770).Mayr B; Montminy M Transcriptional Regulation by the Phosphorylation-Dependent Factor CREB. Nat. Rev. Mol. Cell Biol 2001, 2 (8), 599–609. [DOI] [PubMed] [Google Scholar]
- (771).Gu Q; Jin N; Sheng H; Yin X; Zhu J Cyclic AMP-Dependent Protein Kinase A Regulates the Alternative Splicing of CaMKIIδ. PLoS One 2011, 6 (11), e25745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (772).Shi J; Qian W; Yin X; Iqbal K; Grundke-Iqbal I; Gu X; Ding F; Gong C-X; Liu F Cyclic AMP-Dependent Protein Kinase Regulates the Alternative Splicing of Tau Exon 10: A Mechanism Involved in Tau Pathology of Alzheimer Disease. J. Biol. Chem 2011, 286 (16), 14639–14648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (773).Kvissel A-K; Ørstavik S; Eikvar S; Brede G; Jahnsen T; Collas P; Akusjärvi G; Skålhegg BS Involvement of the Catalytic Subunit of Protein Kinase A and of HA95 in Pre-mRNA Splicing. Exp. Cell Res 2007, 313 (13), 2795–2809. [DOI] [PubMed] [Google Scholar]
- (774).Taskén K; Aandahl EM Localized Effects of cAMP Mediated by Distinct Routes of Protein Kinase A. Physiol. Rev 2004, 84 (1), 137–167. [DOI] [PubMed] [Google Scholar]
- (775).Harootunian AT; Adams SR; Wen W; Meinkoth JL; Taylor SS; Tsien RY Movement of the Free Catalytic Subunit of cAMP-Dependent Protein Kinase into and out of the Nucleus Can Be Explained by Diffusion. Mol. Biol. Cell 1993, 4 (10), 993–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (776).Brown RL; August SL; Williams CJ; Moss SB AKAP7gamma Is a Nuclear RIBinding AKAP. Biochem. Biophys. Res. Commun 2003, 306 (2), 394–401. [DOI] [PubMed] [Google Scholar]
- (777).Zippin JH; Farrell J; Huron D; Kamenetsky M; Hess KC; Fischman DA; Levin LR; Buck J Bicarbonate-Responsive "soluble" Adenylyl Cyclase Defines a Nuclear cAMP Microdomain. J. Cell Biol 2004, 164 (4), 527–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (778).Kubota S; Morii M; Yuki R; Yamaguchi N; Yamaguchi H; Aoyama K; Kuga T; Tomonaga T; Yamaguchi N Role for Tyrosine Phosphorylation of A-Kinase Anchoring Protein 8 (AKAP8) in Its Dissociation from Chromatin and the Nuclear Matrix. J. Biol. Chem 2015, 290 (17), 10891–10904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (779).Meoli E; Bossis I; Cazabat L; Mavrakis M; Horvath A; Stergiopoulos S; Shiferaw ML; Fumey G; Perlemoine K; Muchow M; et al. Protein Kinase A Effects of an Expressed PRKAR1A Mutation Associated with Aggressive Tumors. Cancer Res 2008, 68 (9), 3133–3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (780).Sample V; Dipilato LM; Yang JH; Ni Q; Saucerman JJ; Zhang J Regulation of Nuclear PKA Revealed by Spatiotemporal Manipulation of Cyclic AMP. Nat. Chem. Biol 2012, 8 (4), 375–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (781).Jarnaess E; Stokka AJ; Kvissel A-K; Skålhegg BS; Torgersen KM; Scott JD; Carlson CR; Taskén K Splicing Factor Arginine/serine-Rich 17A (SFRS17A) Is an A-Kinase Anchoring Protein That Targets Protein Kinase A to Splicing Factor Compartments. J. Biol. Chem 2009, 284 (50), 35154–35164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (782).Gallegos LL; Kunkel MT; Newton AC Targeting Protein Kinase C Activity Reporter to Discrete Intracellular Regions Reveals Spatiotemporal Differences in AgonistDependent Signaling. J. Biol. Chem 2006, 281 (41), 30947–30956. [DOI] [PubMed] [Google Scholar]
- (783).Kunkel MT; Newton AC Calcium Transduces Plasma Membrane Receptor Signals to Produce Diacylglycerol at Golgi Membranes. J. Biol. Chem 2010, 285 (30), 22748–22752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (784).Persechini A; Stemmer PM Calmodulin Is a Limiting Factor in the Cell. Trends Cardiovasc. Med 2002, 12 (1), 32–37. [DOI] [PubMed] [Google Scholar]
- (785).Miyamoto T; Rho E; Sample V; Akano H; Magari M; Ueno T; Gorshkov K; Chen M; Tokumitsu H; Zhang J; et al. Compartmentalized AMPK Signaling Illuminated by Genetically Encoded Molecular Sensors and Actuators. Cell Rep 2015, 11 (4), 657–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (786).Dibble CC; Manning BD Signal Integration by mTORC1 Coordinates Nutrient Input with Biosynthetic Output. Nat. Cell Biol 2013, 15 (6), 555–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (787).Betz C; Hall MN Where Is mTOR and What Is It Doing There? J. Cell Biol 2013, 203 (4), 563–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (788).Sezgin E; Levental I; Mayor S; Eggeling C The Mystery of Membrane Organization: Composition, Regulation and Roles of Lipid Rafts. Nat. Rev. Mol. Cell Biol 2017, 18 (6), 361–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (789).Simons K; Toomre D Lipid Rafts and Signal Transduction. Nat. Rev. Mol. Cell Biol 2000, 1 (1), 31–39. [DOI] [PubMed] [Google Scholar]
- (790).Edidin M The State of Lipid Rafts: From Model Membranes to Cells. Annu. Rev. Biophys. Biomol. Struct 2003, 32 (1), 257–283. [DOI] [PubMed] [Google Scholar]
- (791).Simons K; Vaz WLC Model Systems, Lipid Rafts, and Cell Membranes. Annu. Rev. Biophys. Biomol. Struct 2004, 33 (1), 269–295. [DOI] [PubMed] [Google Scholar]
- (792).Lu S; Ouyang M; Seong J; Zhang J; Chien S; Wang Y The Spatiotemporal Pattern of Src Activation at Lipid Rafts Revealed by Diffusion-Corrected FRET Imaging. PLoS Comput. Biol 2008, 4 (7), e1000127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (793).Brandman O; Meyer T Feedback Loops Shape Cellular Signals in Space and Time. Science 2008, 322 (5900), 390–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (794).Tsien R; Pozzan T Measurement of Cytosolic Free Ca2+ with quin2. Methods Enzymol 1989, 172, 230–262. [DOI] [PubMed] [Google Scholar]
- (795).Sawyer DW; Sullivan JA; Mandell GL Intracellular Free Calcium Localization in Neutrophils during Phagocytosis. Science 1985, 230 (4726), 663–666. [DOI] [PubMed] [Google Scholar]
- (796).Brundage RA; Fogarty KE; Tuft RA; Fay FS Calcium Gradients Underlying Polarization and Chemotaxis of Eosinophils. Science 1991, 254 (5032), 703–706. [DOI] [PubMed] [Google Scholar]
- (797).Smith SJ; Buchanan J; Osses LR; Charlton MP; Augustine GJ The Spatial Distribution of Calcium Signals in Squid Presynaptic Terminals. J. Physiol 1993, 472, 573–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (798).Gee KR; Brown KA; Chen WN; Bishop-Stewart J; Gray D; Johnson I Chemical and Physiological Characterization of Fluo-4 Ca(2+)-Indicator Dyes. Cell Calcium 2000, 27 (2), 97–106. [DOI] [PubMed] [Google Scholar]
- (799).Guan C-B; Xu H-T; Jin M; Yuan X-B; Poo M-M Long-Range Ca2+ Signaling from Growth Cone to Soma Mediates Reversal of Neuronal Migration Induced by Slit-2. Cell 2007, 129 (2), 385–395. [DOI] [PubMed] [Google Scholar]
- (800).Zheng JQ; Poo M-M Calcium Signaling in Neuronal Motility. Annu. Rev. Cell Dev. Biol 2007, 23 (1), 375–404. [DOI] [PubMed] [Google Scholar]
- (801).Henley J; Poo M Guiding Neuronal Growth Cones Using Ca2+ Signals. Trends Cell Biol 2004, 14 (6), 320–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (802).Komuro H; Kumada T Ca2+ Transients Control CNS Neuronal Migration. Cell Calcium 2005, 37 (5), 387–393. [DOI] [PubMed] [Google Scholar]
- (803).Wu W; Wong K; Chen J; Jiang Z; Dupuis S; Wu JY; Rao Y Directional Guidance of Neuronal Migration in the Olfactory System by the Protein Slit. Nature 1999, 400 (6742), 331–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (804).Xu H-T; Yuan X; Guan C; Duan S; Wu C; Feng L Calcium Signaling in Chemorepellant Slit2-Dependent Regulation of Neuronal Migration. Proc. Natl. Acad. Sci. U. S. A 2004, 101 (12), 4296–4301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (805).Huang Y-W; Chang S-J; Harn HI-C; Huang H-T; Lin H-H; Shen M-R; Tang M-J; Chiu W-T Mechanosensitive Store-Operated Calcium Entry Regulates the Formation of Cell Polarity. J. Cell. Physiol 2015, 230 (9), 2086–2097. [DOI] [PubMed] [Google Scholar]
- (806).Vargas ME; Yamagishi Y; Tessier-Lavigne M; Sagasti A Live Imaging of Calcium Dynamics during Axon Degeneration Reveals Two Functionally Distinct Phases of Calcium Influx. J. Neurosci 2015, 35 (45), 15026–15038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (807).Berridge MJ Elementary and Global Aspects of Calcium Signalling. J. Physiol 1997, 499 ( Pt 2), 291–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (808).Berridge MJ Calcium Microdomains: Organization and Function. Cell Calcium 2006, 40 (5–6), 405–412. [DOI] [PubMed] [Google Scholar]
- (809).Bootman M; Niggli E; Berridge M; Lipp P Imaging the Hierarchical Ca2+ Signalling System in HeLa Cells. J. Physiol 1997, 499 ( Pt 2), 307–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (810).Wei C; Wang X; Chen M; Ouyang K; Song L-S; Cheng H Calcium Flickers Steer Cell Migration. Nature 2009, 457 (7231), 901–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (811).Sonkusare SK; Bonev AD; Ledoux J; Liedtke W; Kotlikoff MI; Heppner TJ; Hill-Eubanks DC; Nelson MT Elementary Ca2+ Signals through Endothelial TRPV4 Channels Regulate Vascular Function. Science 2012, 336 (6081), 597–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (812).Tay LH; Dick IE; Yang W; Mank M; Griesbeck O; Yue DT Nanodomain Ca2+ of Ca2+ Channels Detected by a Tethered Genetically Encoded Ca2+ Sensor. Nat. Commun 2012, 3, 778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (813).Augustine GJ; Neher E Neuronal Ca2+ Signalling Takes the Local Route. Curr. Opin. Neurobiol 1992, 2 (3), 302–307. [DOI] [PubMed] [Google Scholar]
- (814).Willoughby D; Wachten S; Masada N; Cooper DMF Direct Demonstration of Discrete Ca2+ Microdomains Associated with Different Isoforms of Adenylyl Cyclase. J. Cell Sci 2010, 123 (Pt 1), 107–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (815).Lechleiter JD; Clapham DE Molecular Mechanisms of Intracellular Calcium Excitability in X. Laevis Oocytes. Cell 1992, 69 (2), 283–294. [DOI] [PubMed] [Google Scholar]
- (816).Tischer C; Bastiaens PIH Lateral Phosphorylation Propagation: An Aspect of Feedback Signalling? Nat. Rev. Mol. Cell Biol 2003, 4 (12), 971–974. [DOI] [PubMed] [Google Scholar]
- (817).Chifflet S; Justet C; Hernández JA; Nin V; Escande C; Benech JC Early and Late Calcium Waves during Wound Healing in Corneal Endothelial Cells. Wound Repair Regen 2012, 20 (1), 28–37. [DOI] [PubMed] [Google Scholar]
- (818).Sieger D; Moritz C; Ziegenhals T; Prykhozhij S; Peri F Long-Range Ca2+ Waves Transmit Brain-Damage Signals to Microglia. Dev. Cell 2012, 22 (6), 1138–1148. [DOI] [PubMed] [Google Scholar]
- (819).Gilabert JA Cytoplasmic Calcium Buffering. Adv. Exp. Med. Biol 2012, 740, 483–498. [DOI] [PubMed] [Google Scholar]
- (820).Wang Z; Tymianski M; Jones OT; Nedergaard M Impact of Cytoplasmic Calcium Buffering on the Spatial and Temporal Characteristics of Intercellular Calcium Signals in Astrocytes. J. Neurosci 1997, 17 (19), 7359–7371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (821).Roberts WM Spatial Calcium Buffering in Saccular Hair Cells. Nature 1993, 363 (6424), 74–76. [DOI] [PubMed] [Google Scholar]
- (822).Matthews EA; Schoch S; Dietrich D Tuning Local Calcium Availability: Cell-TypeSpecific Immobile Calcium Buffer Capacity in Hippocampal Neurons. J. Neurosci 2013, 33 (36), 14431–14445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (823).Minta A; Kao JP; Tsien RY Fluorescent Indicators for Cytosolic Calcium Based on Rhodamine and Fluorescein Chromophores. J. Biol. Chem 1989, 264 (14), 8171–8178. [PubMed] [Google Scholar]
- (824).Dargan SL; Parker I Buffer Kinetics Shape the Spatiotemporal Patterns of IP3-Evoked Ca2+ Signals. J. Physiol 2003, 553 (Pt 3), 775–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (825).Gorshkov K; Mehta S; Ramamurthy S; Ronnett GV; Zhou F-Q; Zhang J AKAPMediated Feedback Control of cAMP Gradients in Developing Hippocampal Neurons. Nat. Chem. Biol 2017, 13 (4), 425–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (826).Bacskai BJ; Hochner B; Mahaut-Smith M; Adams SR; Kaang BK; Kandel ER; Tsien RY Spatially Resolved Dynamics of cAMP and Protein Kinase A Subunits in Aplysia Sensory Neurons. Science 1993, 260 (5105), 222–226. [DOI] [PubMed] [Google Scholar]
- (827).Lim CJ; Kain KH; Tkachenko E; Goldfinger LE; Gutierrez E; Allen MD; Groisman A; Zhang J; Ginsberg MH Integrin-Mediated Protein Kinase A Activation at the Leading Edge of Migrating Cells. Mol. Biol. Cell 2008, 19 (11), 4930–4941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (828).Rich TC; Fagan KA; Nakata H; Schaack J; Cooper DM; Karpen JW Cyclic Nucleotide-Gated Channels Colocalize with Adenylyl Cyclase in Regions of Restricted cAMP Diffusion. J. Gen. Physiol 2000, 116 (2), 147–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (829).Hanoune J; Defer N Regulation and Role of Adenylyl Cyclase Isoforms. Annu. Rev. Pharmacol. Toxicol 2001, 41 (1), 145–174. [DOI] [PubMed] [Google Scholar]
- (830).Sunahara RK; Taussig R Isoforms of Mammalian Adenylyl Cyclase: Multiplicities of Signaling. Mol. Interv 2002, 2 (3), 168–184. [DOI] [PubMed] [Google Scholar]
- (831).Wachten S; Masada N; Ayling L-J; Ciruela A; Nikolaev VO; Lohse MJ; Cooper DMF Distinct Pools of cAMP Centre on Different Isoforms of Adenylyl Cyclase in Pituitary-Derived GH3B6 Cells. J. Cell Sci 2010, 123 (Pt 1), 95–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (832).Zaccolo M; Di Benedetto G; Lissandron V; Mancuso L; Terrin A; Zamparo I Restricted Diffusion of a Freely Diffusible Second Messenger: Mechanisms Underlying Compartmentalized cAMP Signalling. Biochem. Soc. Trans 2006, 34 (Pt 4), 495–497. [DOI] [PubMed] [Google Scholar]
- (833).Baillie GS Compartmentalized Signalling: Spatial Regulation of cAMP by the Action of Compartmentalized Phosphodiesterases. FEBS J 2009, 276 (7), 1790–1799. [DOI] [PubMed] [Google Scholar]
- (834).Maiellaro I; Lohse MJ; Kittel RJ; Calebiro D cAMP Signals in Drosophila Motor Neurons Are Confined to Single Synaptic Boutons. Cell Rep 2016, 17 (5), 1238–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (835).Lefkimmiatis K; Leronni D; Hofer AM The Inner and Outer Compartments of Mitochondria Are Sites of Distinct cAMP/PKA Signaling Dynamics. J. Cell Biol 2013, 202 (3), 453–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (836).Monterisi S; Lobo MJ; Livie C; Castle JC; Weinberger M; Baillie G; Surdo NC; Musheshe N; Stangherlin A; Gottlieb E; et al. PDE2A2 Regulates Mitochondria Morphology and Apoptotic Cell Death via Local Modulation of cAMP/PKA Signalling. Elife 2017, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (837).Lohse C; Bock A; Maiellaro I; Hannawacker A; Schad LR; Lohse MJ; Bauer WR Experimental and Mathematical Analysis of cAMP Nanodomains. PLoS One 2017, 12 (4), e0174856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (838).Chen C; Nakamura T; Koutalos Y Cyclic AMP Diffusion Coefficient in Frog Olfactory Cilia. Biophys. J 1999, 76 (5), 2861–2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (839).Cohen MH; Drage DJ; Robertson A Iontophoresis of Cyclic AMP. Biophys. J 1975, 15 (8), 753–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (840).Dworkin M; Keller KH Solubility and Diffusion Coefficient of Adenosine 3’:5’-monophosphate. J. Biol. Chem 1977, 252 (3), 864–865. [PubMed] [Google Scholar]
- (841).Huang RC; Gillette R Kinetic Analysis of cAMP-Activated Na+ Current in the Molluscan Neuron. A Diffusion-Reaction Model. J. Gen. Physiol 1991, 98 (4), 835–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (842).Oliveira RF; Terrin A; Di Benedetto G; Cannon RC; Koh W; Kim M; Zaccolo M; Blackwell KT The Role of Type 4 Phosphodiesterases in Generating Microdomains of cAMP: Large Scale Stochastic Simulations. PLoS One 2010, 5 (7), e11725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (843).Feinstein WP; Zhu B; Leavesley SJ; Sayner SL; Rich TC Assessment of Cellular Mechanisms Contributing to cAMP Compartmentalization in Pulmonary Microvascular Endothelial Cells. AJP Cell Physiol 2012, 302 (6), C839–C852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (844).Yang P-C; Boras BW; Jeng M-T; Docken SS; Lewis TJ; McCulloch AD; Harvey RD; Clancy CE A Computational Modeling and Simulation Approach to Investigate Mechanisms of Subcellular cAMP Compartmentation. PLoS Comput. Biol 2016, 12 (7), e1005005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (845).Saucerman JJ; Greenwald EC; Polanowska-Grabowska R Mechanisms of Cyclic AMP Compartmentation Revealed by Computational Models. J. Gen. Physiol 2014, 143 (1), 39–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (846).Neves SR; Tsokas P; Sarkar A; Grace EA; Rangamani P; Taubenfeld SM; Alberini CM; Schaff JC; Blitzer RD; Moraru II; et al. Cell Shape and Negative Links in Regulatory Motifs Together Control Spatial Information Flow in Signaling Networks. Cell 2008, 133 (4), 666–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (847).Richards M; Lomas O; Jalink K; Ford KL; Vaughan-Jones RD; Lefkimmiatis K; Swietach P Intracellular Tortuosity Underlies Slow cAMP Diffusion in Adult Ventricular Myocytes. Cardiovasc. Res 2016, 110 (3), 395–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (848).Agarwal SR; Clancy CE; Harvey RD Mechanisms Restricting Diffusion of Intracellular cAMP. Sci. Rep 2016, 6, 19577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (849).Saucerman JJ; Zhang J; Martin JC; Peng LX; Stenbit AE; Tsien RY; McCulloch AD Systems Analysis of PKA-Mediated Phosphorylation Gradients in Live Cardiac Myocytes. Proc. Natl. Acad. Sci. U. S. A 2006, 103 (34), 12923–12928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (850).Nikolaev VO; Moshkov A; Lyon AR; Miragoli M; Novak P; Paur H; Lohse MJ; Korchev YE; Harding SE; Gorelik J Beta2-Adrenergic Receptor Redistribution in Heart Failure Changes cAMP Compartmentation. Science 2010, 327 (5973), 1653–1657. [DOI] [PubMed] [Google Scholar]
- (851).Beavo JA; Bechtel PJ; Krebs EG Activation of Protein Kinase by Physiological Concentrations of Cyclic AMP. Proc. Natl. Acad. Sci. U. S. A 1974, 71 (9), 3580–3583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (852).Lefkimmiatis K; Moyer MP; Curci S; Hofer AM "cAMP Sponge": A Buffer for Cyclic Adenosine 3’, 5’-monophosphate. PLoS One 2009, 4 (11), e7649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (853).Devreotes PN; Zigmond SH Chemotaxis in Eukaryotic Cells: A Focus on Leukocytes and Dictyostelium. Annu. Rev. Cell Biol 1988, 4 (1), 649–686. [DOI] [PubMed] [Google Scholar]
- (854).Parent CA; Blacklock BJ; Froehlich WM; Murphy DB; Devreotes PN G Protein Signaling Events Are Activated at the Leading Edge of Chemotactic Cells. Cell 1998, 95 (1), 81–91. [DOI] [PubMed] [Google Scholar]
- (855).Meili R; Ellsworth C; Lee S; Reddy TB; Ma H; Firtel RA ChemoattractantMediated Transient Activation and Membrane Localization of Akt/PKB Is Required for Efficient Chemotaxis to cAMP in Dictyostelium. EMBO J 1999, 18 (8), 2092–2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (856).Haugh JM; Codazzi F; Teruel M; Meyer T Spatial Sensing in Fibroblasts Mediated by 3’ Phosphoinositides. J. Cell Biol 2000, 151 (6), 1269–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (857).Servant G; Weiner OD; Herzmark P; Balla T; Sedat JW; Bourne HR Polarization of Chemoattractant Receptor Signaling during Neutrophil Chemotaxis. Science 2000, 287 (5455), 1037–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (858).Iijima M; Devreotes P Tumor Suppressor PTEN Mediates Sensing of Chemoattractant Gradients. Cell 2002, 109 (5), 599–610. [DOI] [PubMed] [Google Scholar]
- (859).Funamoto S; Meili R; Lee S; Parry L; Firtel RA Spatial and Temporal Regulation of 3-Phosphoinositides by PI 3-Kinase and PTEN Mediates Chemotaxis. Cell 2002, 109 (5), 611–623. [DOI] [PubMed] [Google Scholar]
- (860).Weiner OD; Neilsen PO; Prestwich GD; Kirschner MW; Cantley LC; Bourne HR A PtdInsP(3)- and Rho GTPase-Mediated Positive Feedback Loop Regulates Neutrophil Polarity. Nat. Cell Biol 2002, 4 (7), 509–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (861).Yang HW; Shin M-G; Lee S; Kim J-R; Park WS; Cho K-H; Meyer T; Heo W Do. Cooperative Activation of PI3K by Ras and Rho Family Small GTPases. Mol. Cell 2012, 47 (2), 281–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (862).Thevathasan JV; Tan E; Zheng H; Lin Y-C; Li Y; Inoue T; Fivaz M The Small GTPase HRas Shapes Local PI3K Signals through Positive Feedback and Regulates Persistent Membrane Extension in Migrating Fibroblasts. Mol. Biol. Cell 2013, 24 (14), 2228–2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (863).Yang HW; Collins SR; Meyer T Locally Excitable Cdc42 Signals Steer Cells during Chemotaxis. Nat. Cell Biol 2016, 18 (2), 191–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (864).Wong K; Pertz O; Hahn K; Bourne H Neutrophil Polarization: Spatiotemporal Dynamics of RhoA Activity Support a Self-Organizing Mechanism. Proc. Natl. Acad. Sci. U. S. A 2006, 103 (10), 3639–3644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (865).Ridley AJ; Schwartz MA; Burridge K; Firtel RA; Ginsberg MH; Borisy G; Parsons JT; Horwitz AR Cell Migration: Integrating Signals from Front to Back. Science 2003, 302 (5651), 1704–1709. [DOI] [PubMed] [Google Scholar]
- (866).Iglesias PA; Devreotes PN Biased Excitable Networks: How Cells Direct Motion in Response to Gradients. Curr. Opin. Cell Biol 2012, 24 (2), 245–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (867).Dasso M Running on Ran: Nuclear Transport and the Mitotic Spindle. Cell 2001, 104 (3), 321–324. [DOI] [PubMed] [Google Scholar]
- (868).Kaláb P; Pralle A; Isacoff EY; Heald R; Weis K Analysis of a RanGTP-Regulated Gradient in Mitotic Somatic Cells. Nature 2006, 440 (7084), 697–701. [DOI] [PubMed] [Google Scholar]
- (869).Lee Y-P; Wong C-H; Chan K-S; Lai S-K; Koh C-G; Li H-Y In Vivo FRET Imaging Revealed a Regulatory Role of RanGTP in Kinetochore-Microtubule Attachments via Aurora B Kinase. PLoS One 2012, 7 (9), e45836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (870).Liu D; Vader G; Vromans MJM; Lampson MA; Lens SMA Sensing Chromosome Bi-Orientation by Spatial Separation of Aurora B Kinase from Kinetochore Substrates. Science 2009, 323 (5919), 1350–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (871).Pawson T; Scott JD Signaling through Scaffold, Anchoring, and Adaptor Proteins. Science 1997, 278 (5346), 2075–2080. [DOI] [PubMed] [Google Scholar]
- (872).Greenwald EC; Saucerman JJ Bigger, Better, Faster: Principles and Models of AKAP Anchoring Protein Signaling. J. Cardiovasc. Pharmacol 2011, 58 (5), 462–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (873).Wong W; Scott JD AKAP Signalling Complexes: Focal Points in Space and Time. Nat. Rev. Mol. Cell Biol 2004, 5 (12), 959–970. [DOI] [PubMed] [Google Scholar]
- (874).Terrin A; Monterisi S; Stangherlin A; Zoccarato A; Koschinski A; Surdo NC; Mongillo M; Sawa A; Jordanides NE; Mountford JC; et al. PKA and PDE4D3 Anchoring to AKAP9 Provides Distinct Regulation of cAMP Signals at the Centrosome. J. Cell Biol 2012, 198 (4), 607–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (875).Wang Y; Ho TG; Bertinetti D; Neddermann M; Franz E; Mo GCH; Schendowich LP; Sukhu A; Spelts RC; Zhang J; et al. Isoform-Selective Disruption of AKAP-Localized PKA Using Hydrocarbon Stapled Peptides. ACS Chem. Biol 2014, 9 (3), 635–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (876).Schott MB; Gonowolo F; Maliske B; Grove B FRET Biosensors Reveal AKAPMediated Shaping of Subcellular PKA Activity and a Novel Mode of Ca2+/PKA Crosstalk. Cell. Signal 2016, 28 (4), 294–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (877).Lim CJ; Han J; Yousefi N; Ma Y; Amieux PS; McKnight GS; Taylor SS; Ginsberg MH Alpha4 Integrins Are Type I cAMP-Dependent Protein KinaseAnchoring Proteins. Nat. Cell Biol 2007, 9 (4), 415–421. [DOI] [PubMed] [Google Scholar]
- (878).Gill GN; Garren LD A Cyclic-3’,5’-adenosine Monophosphate Dependent Protein Kinase from the Adrenal Cortex: Comparison with a Cyclic AMP Binding Protein. Biochem. Biophys. Res. Commun 1970, 39 (3), 335–343. [DOI] [PubMed] [Google Scholar]
- (879).Corbin JD; Brostrom CO; Alexander RL; Krebs EG Adenosine 3’,5’-monophosphate-Dependent Protein Kinase from Adipose Tissue. J. Biol. Chem 1972, 247 (12), 3736–3743. [PubMed] [Google Scholar]
- (880).Rubin CS; Erlichman J; Rosen OM Molecular Forms and Subunit Composition of a Cyclic Adenosine 3’,5’-monophosphate-Dependent Protein Kinase Purified from Bovine Heart Muscle. J. Biol. Chem 1972, 247 (1), 36–44. [PubMed] [Google Scholar]
- (881).Beavo JA; Bechtel PJ; Krebs EG Preparation of Homogeneous Cyclic AMP-Dependent Protein Kinase(s) and Its Subunits from Rabbit Skeletal Muscle. Methods Enzymol 1974, 38, 299–308. [DOI] [PubMed] [Google Scholar]
- (882).Potter RL; Taylor SS Relationships between Structural Domains and Function in the Regulatory Subunit of cAMP-Dependent Protein Kinases I and II from Porcine Skeletal Muscle. J. Biol. Chem 1979, 254 (7), 2413–2418. [PubMed] [Google Scholar]
- (883).Martin BR; Deerinck TJ; Ellisman MH; Taylor SS; Tsien RY Isoform-Specific PKA Dynamics Revealed by Dye-Triggered Aggregation and DAKAP1alphaMediated Localization in Living Cells. Chem. Biol 2007, 14 (9), 1031–1042. [DOI] [PubMed] [Google Scholar]
- (884).Smith FD; Reichow SL; Esseltine JL; Shi D; Langeberg LK; Scott JD; Gonen T Intrinsic Disorder within an AKAP-Protein Kinase A Complex Guides Local Substrate Phosphorylation. Elife 2013, 2, e01319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (885).Walker-Gray R; Stengel F; Gold MG Mechanisms for Restraining cAMP-Dependent Protein Kinase Revealed by Subunit Quantitation and Cross-Linking Approaches. Proc. Natl. Acad. Sci. U. S. A 2017, 114 (39), 10414–10419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (886).Zhang P; Ye F; Bastidas AC; Kornev AP; Wu J; Ginsberg MH; Taylor SS An Isoform-Specific Myristylation Switch Targets Type II PKA Holoenzymes to Membranes. Structure 2015, 23 (9), 1563–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (887).Dodge-Kafka KL; Soughayer J; Pare GC; Carlisle Michel JJ; Langeberg LK; Kapiloff MS; Scott JD The Protein Kinase A Anchoring Protein mAKAP Coordinates Two Integrated cAMP Effector Pathways. Nature 2005, 437 (7058), 574–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (888).Hoshi N; Langeberg LK; Gould CM; Newton AC; Scott JD Interaction with AKAP79 Modifies the Cellular Pharmacology of PKC. Mol. Cell 2010, 37 (4), 541–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (889).Greenwald EC; Redden JM; Dodge-Kafka KL; Saucerman JJ Scaffold State Switching Amplifies, Accelerates, and Insulates Protein Kinase C Signaling. J. Biol. Chem 2014, 289 (4), 2353–2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (890).Langeberg LK; Scott JD Signalling Scaffolds and Local Organization of Cellular Behaviour. Nat. Rev. Mol. Cell Biol 2015, advance on. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (891).Kolch W Coordinating ERK/MAPK Signalling through Scaffolds and Inhibitors. Nat. Rev. Mol. Cell Biol 2005, 6 (11), 827–837. [DOI] [PubMed] [Google Scholar]
- (892).Brown MD; Sacks DB Protein Scaffolds in MAP Kinase Signalling. Cell. Signal 2009, 21 (4), 462–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (893).Matsunaga-Udagawa R; Fujita Y; Yoshiki S; Terai K; Kamioka Y; Kiyokawa E; Yugi K; Aoki K; Matsuda M The Scaffold Protein Shoc2/SUR-8 Accelerates the Interaction of Ras and Raf. J. Biol. Chem 2010, 285 (10), 7818–7826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (894).Yoshiki S; Matsunaga-Udagawa R; Aoki K; Kamioka Y; Kiyokawa E; Matsuda M Ras and Calcium Signaling Pathways Converge at Raf1 via the Shoc2 Scaffold Protein. Mol. Biol. Cell 2010, 21 (6), 1088–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (895).Tobias IS; Newton AC Protein Scaffolds Control Localized Protein Kinase Cζ Activity. J. Biol. Chem 2016, 291 (26), 13809–13822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (896).Kroon J; Heemskerk N; Kalsbeek MJT; de Waard V; van Rijssel J; van Buul JD Flow-Induced Endothelial Cell Alignment Requires the RhoGEF Trio as a Scaffold Protein to Polarize Active Rac1 Distribution. Mol. Biol. Cell 2017, 28 (13), 1745–1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (897).Bhalla US Models of Cell Signaling Pathways. Curr. Opin. Genet. Dev 2004, 14 (4), 375–381. [DOI] [PubMed] [Google Scholar]
- (898).Yang JH; Saucerman JJ Computational Models Reduce Complexity and Accelerate Insight into Cardiac Signaling Networks. Circ. Res 2011, 108 (1), 85–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (899).Saucerman JJ; McCulloch AD Mechanistic Systems Models of Cell Signaling Networks: A Case Study of Myocyte Adrenergic Regulation. Prog. Biophys. Mol. Biol 2004, 85 (2–3), 261–278. [DOI] [PubMed] [Google Scholar]
- (900).Gilbert D; Fuss H; Gu X; Orton R; Robinson S; Vyshemirsky V; Kurth MJ; Downes CS; Dubitzky W Computational Methodologies for Modelling, Analysis and Simulation of Signalling Networks. Brief. Bioinform 2006, 7 (4), 339–353. [DOI] [PubMed] [Google Scholar]
- (901).Ganesan A; Zhang J How Cells Process Information: Quantification of Spatiotemporal Signaling Dynamics. Protein Sci 2012, 21 (7), 918–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (902).Bhalla US Signaling in Small Subcellular Volumes. I. Stochastic and Diffusion Effects on Individual Pathways. Biophys. J 2004, 87 (2), 733–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (903).Oliveira RF; Kim M; Blackwell KT Subcellular Location of PKA Controls Striatal Plasticity: Stochastic Simulations in Spiny Dendrites. PLoS Comput. Biol 2012, 8 (2), e1002383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (904).Cowan AE; Moraru II; Schaff JC; Slepchenko BM; Loew LM Spatial Modeling of Cell Signaling Networks. Methods Cell Biol 2012, 110, 195–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (905).Andrews SS; Bray D Stochastic Simulation of Chemical Reactions with Spatial Resolution and Single Molecule Detail. Phys. Biol 2004, 1 (3–4), 137–151. [DOI] [PubMed] [Google Scholar]
- (906).Börner S; Schwede F; Schlipp A; Berisha F; Calebiro D; Lohse MJ; Nikolaev VO FRET Measurements of Intracellular cAMP Concentrations and cAMP Analog Permeability in Intact Cells. Nat. Protoc 2011, 6 (4), 427–438. [DOI] [PubMed] [Google Scholar]
- (907).Greenwald EC; Polanowska-Grabowska RK; Saucerman JJ Integrating Fluorescent Biosensor Data Using Computational Models. Methods Mol. Biol 2014, 1071, 227–248. [DOI] [PubMed] [Google Scholar]
- (908).Ahmed S; Grant KG; Edwards LE; Rahman A; Cirit M; Goshe MB; Haugh JM Data-Driven Modeling Reconciles Kinetics of ERK Phosphorylation, Localization, and Activity States. Mol. Syst. Biol 2014, 10 (1), 718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (909).Bers DM Cardiac Excitation-Contraction Coupling. Nature 2002, 415 (6868), 198–205. [DOI] [PubMed] [Google Scholar]
- (910).Kholodenko BN Spatially Distributed Cell Signalling. FEBS Lett 2009, 583 (24), 4006–4012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (911).Duncan GE; Little KY; Koplas PA; Kirkman JA; Breese GR; Stumpf WE Beta-Adrenergic Receptor Distribution in Human and Rat Hippocampal Formation: Marked Species Differences. Brain Res 1991, 561 (1), 84–92. [DOI] [PubMed] [Google Scholar]
- (912).Ordway GA; Gambarana C; Frazer A Quantitative Autoradiography of Central Beta Adrenoceptor Subtypes: Comparison of the Effects of Chronic Treatment with Desipramine or Centrally Administered L-Isoproterenol. J. Pharmacol. Exp. Ther 1988, 247 (1), 379–389. [PubMed] [Google Scholar]
- (913).Li L; Gervasi N; Girault J-A Dendritic Geometry Shapes Neuronal cAMP Signalling to the Nucleus. Nat. Commun 2015, 6, 6319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (914).Terrin A; Di Benedetto G; Pertegato V; Cheung Y-F; Baillie G; Lynch MJ; Elvassore N; Prinz A; Herberg FW; Houslay MD; et al. PGE(1) Stimulation of HEK293 Cells Generates Multiple Contiguous Domains with Different [cAMP]: Role of Compartmentalized Phosphodiesterases. J. Cell Biol 2006, 175 (3), 441–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (915).Handly LN; Pilko A; Wollman R Paracrine Communication Maximizes Cellular Response Fidelity in Wound Signaling. Elife 2015, 4, e09652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (916).Cordeiro JV; Jacinto A The Role of Transcription-Independent Damage Signals in the Initiation of Epithelial Wound Healing. Nat. Rev. Mol. Cell Biol 2013, 14 (4), 249–262. [DOI] [PubMed] [Google Scholar]
- (917).Rhee A; Cheong R; Levchenko A The Application of Information Theory to Biochemical Signaling Systems. Phys. Biol 2012, 9 (4), 45011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (918).Cheong R; Rhee a.; Wang CJ; Nemenman I; Levchenko a. Information Transduction Capacity of Noisy Biochemical Signaling Networks. Science (80-. ) 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (919).Selimkhanov J; Taylor B; Yao J; Pilko A; Albeck J; Hoffmann A; Tsimring L; Wollman R Accurate Information Transmission through Dynamic Biochemical Signaling Networks. Science (80-. ) 2014, 346 (6215), 1370–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (920).Hell SW Far-Field Optical Nanoscopy. Science (80-. ) 2007, 316 (5828), 1153–1158. [DOI] [PubMed] [Google Scholar]
- (921).Huang B; Bates M; Zhuang X Super-Resolution Fluorescence Microscopy. Annu. Rev. Biochem 2009, 78 (1), 993–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (922).Willig KI; Kellner RR; Medda R; Hein B; Jakobs S; Hell SW Nanoscale Resolution in GFP-Based Microscopy. Nat. Methods 2006, 3 (9), 721–723. [DOI] [PubMed] [Google Scholar]
- (923).Gustafsson MGL Nonlinear Structured-Illumination Microscopy: Wide-Field Fluorescence Imaging with Theoretically Unlimited Resolution. Proc. Natl. Acad. Sci. U. S. A 2005, 102 (37), 13081–13086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (924).Mishina NM; Mishin AS; Belyaev Y; Bogdanova EA; Lukyanov S; Schultz C; Belousov VV Live-Cell STED Microscopy with Genetically Encoded Biosensor. Nano Lett 2015, 15 (5), 2928–2932. [DOI] [PubMed] [Google Scholar]
- (925).Rust MJ; Bates M; Zhuang X Sub-Diffraction-Limit Imaging by Stochastic Optical Reconstruction Microscopy (STORM). Nat. Methods 2006, 3 (10), 793–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (926).Betzig E; Patterson GH; Sougrat R; Lindwasser OW; Olenych S; Bonifacino JS; Davidson MW; Lippincott-Schwartz J; Hess HF Imaging Intracellular Fluorescent Proteins at Nanometer Resolution. Science (80-. ) 2006, 313 (5793), 1642–1645. [DOI] [PubMed] [Google Scholar]
- (927).Richardson DS; Gregor C; Winter FR; Urban NT; Sahl SJ; Willig KI; Hell SW SRpHi Ratiometric pH Biosensors for Super-Resolution Microscopy. Nat. Commun 2017, 8 (1), 577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (928).Dertinger T; Colyer R; Iyer G; Weiss S; Enderlein J Fast, Background-Free, 3D Super-Resolution Optical Fluctuation Imaging (SOFI). Proc. Natl. Acad. Sci. U. S. A 2009, 106 (52), 22287–22292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (929).Nickerson A; Huang T; Lin L-J; Nan X Photoactivated Localization Microscopy with Bimolecular Fluorescence Complementation (BiFC-PALM) for Nanoscale Imaging of Protein-Protein Interactions in Cells. PLoS One 2014, 9 (6), e100589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (930).Welch CM; Elliott H; Danuser G; Hahn KM Imaging the Coordination of Multiple Signalling Activities in Living Cells. Nat. Rev. Mol. Cell Biol 2011, 12 (11), 749–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (931).Carlson HJ; Campbell RE Genetically Encoded FRET-Based Biosensors for Multiparameter Fluorescence Imaging. Curr. Opin. Biotechnol 2009, 20 (1), 19–27. [DOI] [PubMed] [Google Scholar]
- (932).Depry C; Mehta S; Zhang J Multiplexed Visualization of Dynamic Signaling Networks Using Genetically Encoded Fluorescent Protein-Based Biosensors. Pflugers Arch 2013, 465 (3), 373–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (933).Piston DW; Rizzo MA FRET by Fluorescence Polarization Microscopy. Methods Cell Biol 2008, 85, 415–430. [DOI] [PubMed] [Google Scholar]
- (934).Chittajallu DR; Florian S; Kohler RH; Iwamoto Y; Orth JD; Weissleder R; Danuser G; Mitchison TJ In Vivo Cell-Cycle Profiling in Xenograft Tumors by Quantitative Intravital Microscopy. Nat. Methods 2015, 12 (6), 577–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (935).Isotani E; Zhi G; Lau KS; Huang J; Mizuno Y; Persechini A; Geguchadze R; Kamm KE; Stull JT Real-Time Evaluation of Myosin Light Chain Kinase Activation in Smooth Muscle Tissues from a Transgenic Calmodulin-Biosensor Mouse. Proc. Natl. Acad. Sci. U. S. A 2004, 101 (16), 6279–6284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (936).Ntziachristos V Going Deeper than Microscopy: The Optical Imaging Frontier in Biology. Nat. Methods 2010, 7 (8), 603–614. [DOI] [PubMed] [Google Scholar]
- (937).Chernov KG; Redchuk TA; Omelina ES; Verkhusha VV Near-Infrared Fluorescent Proteins, Biosensors, and Optogenetic Tools Engineered from Phytochromes. Chem. Rev 2017, 117 (9), 6423–6446. [DOI] [PubMed] [Google Scholar]
- (938).Helmchen F; Denk W Deep Tissue Two-Photon Microscopy. Nat. Methods 2005, 2 (12), 932–940. [DOI] [PubMed] [Google Scholar]
- (939).Horton NG; Wang K; Kobat D; Clark CG; Wise FW; Schaffer CB; Xu C In VivoThree-Photon Microscopy of Subcortical Structures within an Intact Mouse Brain. Nat. Photonics 2013, 7 (3), 205–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (940).Zipfel WR; Williams RM; Webb WW Nonlinear Magic: Multiphoton Microscopy in the Biosciences. Nat. Biotechnol 2003, 21 (11), 1369–1377. [DOI] [PubMed] [Google Scholar]
- (941).Hefendehl JK; LeDue J; Ko RWY; Mahler J; Murphy TH; MacVicar BA Mapping Synaptic Glutamate Transporter Dysfunction in Vivo to Regions Surrounding Aβ Plaques by iGluSnFR Two-Photon Imaging. Nat. Commun 2016, 7, 13441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (942).Wang LV; Hu S Photoacoustic Tomography: In Vivo Imaging from Organelles to Organs. Science 2012, 335 (6075), 1458–1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (943).Brunker J; Yao J; Laufer J; Bohndiek SE Photoacoustic Imaging Using Genetically Encoded Reporters: A Review. J. Biomed. Opt 2017, 22 (7), 70901. [DOI] [PubMed] [Google Scholar]
- (944).Li Y; Forbrich A; Wu J; Shao P; Campbell RE; Zemp R Engineering Dark Chromoprotein Reporters for Photoacoustic Microscopy and FRET Imaging. Sci. Rep 2016, 6 (1), 22129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (945).Hagerty JR Nobel-Winning Chemist Created a Rainbow of Colors to Illuminate Cells. The Wall Street Journal September 9, 2016. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.