

Abstract
Rationale: In cystic fibrosis (CF), the lung clearance index (LCI), derived from multiple breath washout (MBW), is more sensitive in detecting early lung disease than FEV1; MBW has been less thoroughly evaluated in young patients with primary ciliary dyskinesia (PCD).
Objectives: Our objectives were 1) to evaluate the sensitivity of MBW and spirometry for the detection of mild lung disease in young children with PCD and CF compared with healthy control (HC) subjects and 2) to compare patterns of airway obstruction between disease populations.
Methods: We used a multicenter, single-visit, observational study in children with PCD and CF with a forced expiratory volume in 1 second (FEV1) greater than 60% predicted and HC subjects, ages 3–12 years. Nitrogen MBW and spirometry were performed and overread for acceptability. χ2 and Kruskall-Wallis tests compared demographics and lung function measures between groups, linear regression evaluated the effect of disease state, and Spearman’s rank correlation coefficient compared the LCI and spirometric measurements.
Results: Twenty-five children with PCD, 49 children with CF, and 80 HC children were enrolled, among whom 17 children with PCD (68%), 36 children with CF (73%), and 53 (66%) HC children performed both acceptable spirometry and MBW; these children made up the analytic cohort. The median age was 9.0 years (interquartile range [IQR], 6.8–11.1). The LCI was abnormal (more than 7.8) in 10 of 17 (59%) patients with PCD and 21 of 36 (58%) patients with CF, whereas FEV1 was abnormal in three of 17 (18%) patients with PCD and six of 36 (17%) patients with CF. The LCI was significantly elevated in patients with PCD and CF compared with HC subjects (ratio of geometric mean vs. HC: PCD 1.27; 95% confidence interval [CI], 1.15–1.39; and CF 1.24; 95% CI, 1.15–1.33]). Children with PCD had lower midexpiratory-phase forced expiratory flow % predicted compared with children with CF (62% [IQR, 50–78%] vs. 85% [IQR, 68–99%]; P = 0.05). LCI did not correlate with FEV1.
Conclusions: The LCI is more sensitive than FEV1 in detecting lung disease in young patients with PCD, similar to CF. LCI holds promise as a sensitive endpoint for the assessment of early PCD lung disease.
Keywords: primary ciliary dyskinesia, cystic fibrosis, lung clearance index, multiple breath washout, lung function
Primary ciliary dyskinesia (PCD) and cystic fibrosis (CF) are both autosomal recessive disorders characterized by impaired mucociliary clearance, chronic endobronchial bacterial infection, and progressive obstructive lung disease (1). Pathophysiology of these two diseases differs; CF is characterized by abnormal ion and water balance in the airways, whereas patients with PCD have ineffective ciliary function (2, 3). Patients with CF and PCD show significant variability in disease severity and progression of lung disease; causes of these differences are not fully understood (4).
Although forced expiratory volume in 1 second (FEV1) measured by spirometry is the gold standard for the assessment of pulmonary function in chronic obstructive lung diseases, it has been shown to be relatively insensitive to early, mild airway obstruction in CF (5, 6). Because spirometry is driven by airway resistance dominated by medium-sized airways, it may not detect early, mild, heterogenous obstructive disease, which is typically located in the smaller, more peripheral airways (7). Because PCD is also characterized by peripheral airway obstruction, spirometry may also be relatively insensitive to early disease in this population.
The lung clearance index (LCI), derived from multiple breath washout (MBW) testing, is a measure of global ventilation and small airway dysfunction (7). MBW is performed during tidal breathing and requires only passive cooperation, making it feasible in the preschool population. Among patients with CF, the LCI has been shown to be more sensitive than FEV1 in detecting early lung disease and has become a promising outcome measure (6, 8). The evidence for the clinical utility of the LCI in PCD is much more preliminary. Five studies have shown the LCI to be more sensitive than FEV1 in detecting abnormalities in adults and children with PCD (7, 9–12), yet these studies included few young children and were not necessarily focused on those with mild disease. In addition, prior studies primarily compared the LCI with FEV1, but few have explored other outcome measures that may be obtained from either MBW or spirometry, such as moment ratios (MBW), FEV1/forced vital capacity (FVC), or midexpiratory-phase forced expiratory flow (FEF25–75) (spirometry).
Our objectives were to evaluate the sensitivity of MBW and spirometry for the detection of lung disease in children with PCD and CF with mild disease—precisely those in whom the LCI may be the most sensitive—compared with healthy control (HC) subjects and to evaluate patterns of airway obstruction between disease populations (CF and PCD). We hypothesized that the LCI is more likely to be abnormal than FEV1 in both PCD and CF, and that participants with PCD and CF will have different patterns of airways disease when MBW parameters, FEV1, FEV1/FVC, and FEF25–75 are compared, reflecting differing locations and degrees of airway obstruction within the bronchial tree.
Methods
Study Design and Study Participants
This was a cross-sectional, single-visit, observational study of participants with CF, participants with PCD, and HC participants, ages 3 through 12 years, at five sites (Washington University School of Medicine/St. Louis Children’s Hospital, Riley Hospital for Children/Indiana University, University of Washington/Seattle Children’s Hospital, Children’s Minnesota, and University of North Carolina at Chapel Hill). Participants were enrolled from March 2017 to April 2018. In addition, two sites provided data from historical research participants who underwent spirometry and MBW using identical devices and standard operating procedures between April 2012 and July 2015 (13, 14). These participants fulfilled the same eligibility criteria. Participants with PCD and CF were recruited from the clinical and research populations at each study site; HC subjects were recruited from siblings of patients with PCD and CF and with recruitment tools, including flyers and online announcements.
Inclusion criteria included age 3–12 years and gestational age of 36 weeks or more. Participants with PCD and CF were required to have an FEV1 of greater than 60% predicted on all spirometry performed during the year before enrollment. Inclusion criteria for participants with PCD included compatible clinical phenotype as well as 1) defects in ciliary ultrastructure by transmission electron microscopy and/or 2) genetic testing with identification of biallelic disease-causing variants (1). Inclusion criteria for participants with CF included compatible phenotype with elevated sweat chloride and/or the identification of two disease-causing CFTR variants (3).
Exclusion criteria for all participants included an acute intercurrent respiratory infection or change from baseline cough or wheeze during the 2 weeks preceding enrollment. Additional exclusion criteria for control subjects included any history of chronic lung disease (CF, PCD, asthma, and/or bronchopulmonary dysplasia), inhaled corticosteroid use, inhaled bronchodilator use within the past 2 years or on more than three occasions during lifetime, history of emergency room visits or hospitalizations for respiratory indications, or complex congenital heart disease.
Spirometry
Spirometry was performed according to American Thoracic Society (ATS) guidelines for preschoolers ages 2–6 years (15) and children 7 years or older (16). Participants performed spirometric maneuvers until two (preschoolers) or three (older children) reproducible maneuvers were obtained or until it was clear that acceptable spirometry could not be obtained. Spirometry overreading was performed for acceptability, per ATS guidelines (15, 16). Overreading of historical and prospective spirometry were performed simultaneously.
Multiple Breath Washout
Nitrogen (N2) MBW measurement was performed using an Exhalyzer D (EcoMedics) according to current guidelines (17–20). Historical and prospective MBW studies were analyzed simultaneously using EcoMedics Spiroware Software version 3.1.6. A facemask was used for preschool subjects, and putty was used to ensure an adequate seal and to minimize equipment dead space per ATS guidelines (20), with no additional modifications to the manufacturer’s equipment. The primary outcome was the LCI (LCI 2.5), representing the number of functional residual capacity (FRC) turnovers needed to clear the lungs of nitrogen to 1/40th of the initial concentration. Secondary outcomes included LCI 5, moment ratios (M1/M0 and M2/M0 at six and eight turnovers), ventilation heterogeneity in the convection-dependent airways (Scond), and ventilation heterogeneity in diffusion convection–dependent airways (Sacin), which reflect convection-dependent and diffusion convection–dependent ventilation inhomogeneity, respectively. To normalize for lung size, Scond and Sacin were multiplied by tidal volume. Acceptability criteria for MBW included at least two valid LCI 2.5 readings. FRC values that differed by more than 25% from the median FRC value across all valid MBW tests (within-patient median FRC) were flagged for exclusion according to European Respiratory Society/ATS guidelines (19). MBW measurements were reported as the mean of all acceptable values (two to seven observations per patient).
Statistical Analysis
The sample size was calculated to allow the detection of a 1.0 LCI unit difference between children with PCD and those with HC. Assuming a SD of 0.4 for HC and 2.4 for PCD (7), a sample size of 50 HC children, 25 children with PCD, and 25 with CF would be sufficient to detect this difference between the groups using a t test (α = 0.05 with 80% power). The sample size for patients with CF was targeted to equal that of patients with PCD, assuming a similar SD.
Demographic and clinical characteristics were summarized descriptively by disease status. Categorical data were summarized using counts and percentages and compared using a χ2 test or Fisher’s Exact test. Continuous data were summarized using medians and interquartile ranges (IQRs) and compared using Kruskall-Wallis tests because of nonnormality. Height, weight, and body mass index (BMI) z-scores were derived from U.S. Centers for Disease Control and Prevention (CDC) reference data (21). Spirometry z-scores and percentage predicted were calculated from the Global Lung Initiative equations (22). MBW parameters were reported as raw values because robust reference equations do not exist for this age range.
Linear regression was performed to compare lung function parameters (raw MBW parameters, percentage predicted, or z-scores for spirometric measures) between groups. MBW data was adjusted for age, sex, and height. Because of the nonnormal distribution in participants with PCD and CF (though there was normal distribution among control subjects), LCI and Sacin data were log-transformed and compared using the ratio of the geometric means with 95% confidence intervals (CIs). All spirometry data and the MBW measures, Scond, and moment ratios were normally distributed and therefore compared using simple linear regression and reported as regression coefficients with 95% CIs. Forest plots were used to visualize comparisons between disease groups and HC subjects. Sensitivity analyses were conducted, restricting the cohort to participants who had at least three acceptable LCI 2.5, Scond, and Sacin measurements.
For the spirometry data, a z-score of −1.645 was used as the lower limit of normal (23). The LCI upper limit of normal (ULN) was defined, based on the observed data in the healthy group, as the mean LCI + 1.645×SD (i.e., 6.97 + 1.645 × 0.5 = 7.8). This value is similar to that in the existing literature (7.9 LCI units) for the ULN of LCI in healthy young children (24). The ULN was also calculated for additional MBW parameters, including LCI 5, Scond, Sacin, and moment ratios.
Quadrant plots were created to visualize the relationship between LCI measurements and spirometry z-scores. The proportion of patients falling outside of reference parameters was summarized by disease state.
Spearman’s rank order correlation coefficients were calculated to assess the relationship between the LCI and spirometry measurements as well as the relationship between different spirometry measurements. Linear regression models with interaction were used to test for the effect of age in each disease group on the LCI 2.5 and FEV1% predicted. An α of 0.05 was considered significant. Analyses were performed using SAS version 9.4.
Ethics Approval
This study was approved by the institutional review board at each study site. Informed consent was obtained from parents and/or guardians, and assent from participants was obtained as applicable. Sites supplying historical data (University of North Carolina and Washington University School of Medicine) included a waiver of consent for retrospective data collection.
Results
Participants
The initial cohort included 25 children with PCD, 49 children with CF, and 80 HC children (Table 1). Overall, 75% and 94% had acceptable MBW and spirometry data, respectively. Among children 6 years old or younger (n = 52), 71% and 83% had acceptable MBW and spirometry data, respectively; acceptability among children more than 6 years old (n = 102) was 76% and 98%. No differences were found in measurement acceptability by disease state. The final analytic cohort comprised 17 children with PCD (68%), 36 children with CF (73%), and 53 HC children (66%) with acceptable data for both spirometry and MBW (Table 2).
Table 1.
Baseline characteristics of all participants, including children with PCD, children with CF, and healthy control subjects*
| Characteristics | Subjects with PCD (n = 25) | Subjects with CF (n = 49) | HC Subjects (n = 80) |
|---|---|---|---|
| Sex, M, n (%) | 13 (52) | 26 (53%) | 37 (46) |
| Age, median (IQR), yr | 8.6 (5.9 to 10.0) | 8.9 (6.8 to 10.8) | 8.6 (6.0 to 10.9) |
| Weight z-score†, median (IQR) | −0.34 (−1.01 to 0.53) | −0.30 (−0.67 to 0.50) | 0.21 (−0.51 to 0.89) |
| Height z-score†, median (IQR) | −0.53 (−1.18 to 0.31) | −0.27 (−1.02 to 0.25) | −0.02 (−0.67 to 0.62) |
| BMI z-score†, median (IQR) | −0.08 (−0.78 to 0.93) | 0.13 (−0.45 to 0.73) | 0.41 (−0.30 to 0.93) |
| Race/ethnicity, n (%) | |||
| White, non-Hispanic | 23 (92) | 46 (94) | 60 (75) |
| Black | 0 (0) | 1 (2) | 6 (7) |
| Asian | 0 (0) | 0 (0) | 3 (4) |
| Hispanic | 2 (8) | 2 (4) | 8 (10) |
| Other | 0 (0) | 0 (0) | 3 (4) |
| Acceptable MBW | 18 (72) | 37 (76) | 60 (75) |
| Acceptable spirometry | 23 (92) | 47 (96) | 73 (91) |
Definition of abbreviations: BMI = body mass index; CF = cystic fibrosis; HC = healthy control; IQR = interquartile range; MBW = multiple breath washout; PCD = primary ciliary dyskinesia.
The initial cohort included 45 historical study participants.
Calculated using Centers for Disease Control and Prevention reference data.
Table 2.
Baseline characteristics of participants with acceptable MBW and spirometry (final analytical cohort), including children with PCD, children with CF, and healthy control subjects*
| Characteristics | Subjects with PCD (N = 17) | Subjects with CF (n = 36) | HC Subjects (n = 53) |
|---|---|---|---|
| Sex, M, n (%) | 7 (41) | 21 (58) | 24 (45) |
| Age, median (IQR), yr | 8.6 (6.1 to 10.0) | 8.8 (6.9 to 11.1) | 9.2 (7.3 to 11.4) |
| Weight z-score†, median (IQR) | −0.35 (−0.96 to 0.47) | −0.34 (−0.69 to 0.37) | 0.21 (−0.51 to 0.78) |
| Height z-score†, median (IQR) | −0.61 (−1.25 to 0.11) | −0.43 (−1.05 to 0.38) | −0.02 (−0.65 to 0.54) |
| BMI z-score†, median (IQR) | 0.14 (−0.76 to 0.92) | 0.11 (−0.46 to 0.61) | 0.41 (−0.25 to 0.87) |
| Race/ethnicity, n (%) | |||
| White, non-Hispanic | 16 (94) | 33 (92) | 40 (75) |
| Black | 0 (0) | 1 (3) | 5 (9) |
| Asian | 0 (0) | 0 (0) | 2 (4) |
| Hispanic | 1 (6) | 2 (5) | 4 (8) |
| Other | 0 (0) | 0 (0) | 2 (4) |
| PCD ciliary defect type‡, n (%) | |||
| ODA | 6 (35%) | ||
| ODA/IDA | 4 (24%) | ||
| IDA/CA/MTD | 5 (29%) | ||
| Normal | 2 (12%) | ||
| CF genotype, n (%) | |||
| Homozygous ΔF508 | 21 (58%) | ||
| Heterozygous ΔF508 | 13 (36%) | ||
| Other | 2 (6%) |
Definition of abbreviations: BMI = body mass index; CA = central apparatus; CF = cystic fibrosis; HC = healthy control; IDA = inner dynein arm; IQR = interquartile range; MBW = multiple breath washout; MTD = microtubular disorganization; ODA = outer dynein arm; PCD = primary ciliary dyskinesia.
The final analytic cohort included 25 historical participants (see Methods), including three (18%) subjects with PCD, 10 (28%) subjects with CF, and 12 (23%) HC subjects.
Calculated using Centers for Disease Control and Prevention reference data.
PCD ciliary defects included the following PCD-causing genes: ODA (DNAH5 in four subjects, DNAI1 in one subject, and none identified in one subject), ODA/IDA (DNAAF1 in two subjects and none identified or unknown in two subjects), IDA/CA/MTD (CCDC39 in one subject and CCDC40 in four subjects), and normal (CCDC65 in one subject and RPGR in one subject).
Baseline characteristics were similar between the enrollment and final analytic cohorts (Tables 1 and 2). In the final cohort, age was similar across all three disease groups (Table 2). The number of preschool children (age ≤ 6) in the PCD, CF, and HC cohorts were five of 17 (29.4%), 10 of 36 (27.8%), and 13 of 53 (24.5%), respectively. Weight, height, and BMI z-scores were in general lower in patients with CF and PCD than in HC subjects (Tables 1 and 2). The PCD ciliary defect and associated PCD-causing genes, and the CF genotype group are summarized in Tables 1 and 2.
MBW Parameters
In both unadjusted (Table 3) and adjusted (Figure 1) analyses, LCI 2.5 was significantly and similarly elevated in children with PCD and CF compared with HC children. The median LCI in HC children was 6.94 (IQR, 6.61–7.20), compared with 8.19 (IQR, 7.35–11.38) and 8.24 (IQR, 7.30–9.68) in children with PCD and CF, respectively (Table 3). Because LCI values were not normally distributed, they were log-transformed for regression analyses, yielding estimates of the ratio of geometric means adjusted for age, sex, and height. Patients with PCD had a geometric mean LCI that was 27% higher than that of HC children (95% CI, 15–39% higher), and patients with CF had a geometric mean LCI that was 24% higher than HC children (95% CI, 15–33% higher) (Figure 1). Children with PCD and CF also had higher LCI 5, Sacin, Scond, and moment ratios compared with HC children, yet there was no difference between the two disease groups in any of these measurements (Table E1 in the online supplement). Additional MBW parameters did not appear more accurate at predicting early lung disease than the LCI 2.5 (Table E1). No significant relationship was detected between age and the LCI in any of the three disease groups in the multivariable model with disease status as an interaction term (Figure E1).
Table 3.
MBW and spirometry parameters in children with PCD, children with CF, and healthy control children*
| Parameters | Subjects with PCD (n = 17) | Subjects with CF (n = 36) | HC Subjects (n = 53) | Pairwise Comparison P Values† |
||||
|---|---|---|---|---|---|---|---|---|
| PCD vs. HC | CF vs. HC | CF vs. PCD | ||||||
| MBW parameters | ||||||||
| LCI 2.5‡, median (IQR) | 8.19 (7.35 to 11.38) | 8.24 (7.30 to 9.68) | 6.94 (6.61 to 7.20) | <0.001 |
<0.001 |
0.85 |
||
| Abnormal LCI 2.5§, n (%) | 10 (59) | 21 (58) | 4 (8) | |
|
|||
| Spirometry | ||||||||
| FVC z-score‖, median (IQR) | 0.05 (−0.37 to 0.67) | −0.05 (−0.59 to 0.50) | 0.39 (−0.11 to 0.89) | 0.46 |
0.03 |
0.80 | ||
| FVC % predicted‖, median (IQR) | 101 (95 to 108) | 99 (93 to 106) | 105 (99 to 112) | 0.49 |
0.03 |
0.79 | ||
| Abnormal FVC§, n (%) | 2 (12) | 3 (8.3) | 0 (0.0) | |
|
|||
| FEV1z-score§, median (IQR) | −0.62 (−1.57 to −0.17) | −0.38 (−1.21 to 0.17) | 0.25 (−0.38 to 0.73) |
0.01 |
0.003 |
0.73 | ||
| FEV1% predicted‖, median (IQR) | 92 (82 to 98) | 96 (85 to 102) | 103 (95 to 109) |
0.007 |
0.004 |
0.69 | ||
| Abnormal FEV1§, n (%) | 3 (18) | 6 (17) | 0 (0.0) | |
|
|||
| FEV1/FVC z-score‖, median (IQR) | −1.47 (−1.68 to −0.72) | −0.56 (−1.51 to −0.01) | −0.44 (−0.93 to 0.00) |
<0.001 |
0.29 |
0.05 | ||
| FEV1/FVC% predicted‖, median (IQR) | 90 (88 to 95) | 96 (89 to 00) | 97 (94 to 100) |
<0.001 |
0.27 |
0.06 | ||
| Abnormal FEV1/FVC§, n (%) | 5 (29) | 7 (19) | 2 (4) | |
|
|||
| FEF25–75z-score‖, median (IQR) | −1.74 (−2.03 to −1.04) | −0.67 (−1.47 to −0.06) | −0.45 (−0.91 to 0.16) |
<0.001 |
0.23 |
0.08 | ||
| FEF25–75% predicted‖, median (IQR) | 62 (50 to 78) | 85 (68 to 99) | 90 (79 to 104) |
<0.001 |
0.25 |
0.05 | ||
| Abnormal FEF25–75§, n (%) | 9 (53) | 8 (22) | 2 (4) | |||||
Definition of abbreviations: CF = cystic fibrosis; FEF25–75 = forced midexpiratory flow; FEV1 = forced expiratory volume in 1 second; FVC = forced vital capacity; HC = healthy control; IQR = interquartile range; LCI = lung clearance index; MBW = multiple breath washout; PCD = primary ciliary dyskinesia.
Ranges for each parameter are as follows: LCI (PCD, 6.80 to 12.77; CF, 6.42 to 16.45; HC, 6.12 to 8.24), FVC z-score (PCD, −2.22 to 2.20; CF, −3.43 to 2.23; HC, −1.39 to 2.72), FVC% predicted (PCD, 66 to 116; CF, 61 to 115; HC, 85 to 122), FEV1 z-score (PCD, −2.96 to 1.34; CF, −2.95 to 1.15; HC, −1.28 to 1.77), FEV1% predicted (PCD, 66 to 116; CF, 61 to 115; HC, 85 to 122), FEV1/FVC z-score (PCD, −2.43 to 0.69; CF, −2.40 to 0.91; HC, −1.77 to 1.27), FEV1/FVC% predicted (PCD, 80 to 104; CF 79 to 105; HC, 87 to 106), FEF25–75 z-score (PCD, −2.97 to 0.59; CF, −1.47 to −0.06; HC, −0.91 to 0.16), FEF25–75% predicted (PCD, 43–113; CF, 20–139; HC, 61–144).
Dwass, Steel, Critchlow-Flinger Method, significance level P < 0.05 denoted in bold typeface.
Coefficients of variation for the LCI 2.5 were 6.01% ± 3.97% for patients with PCD, 6.36% ± 5.41% for children with CF, and 4.34% ± 3.13% for HC children.
Normal cutoff for LCI 7.8, corresponding to z-score +1.645. Normal cutoff for spirometry z-score −1.645.
Calculated using Global Lung Initiative equations.
Figure 1.
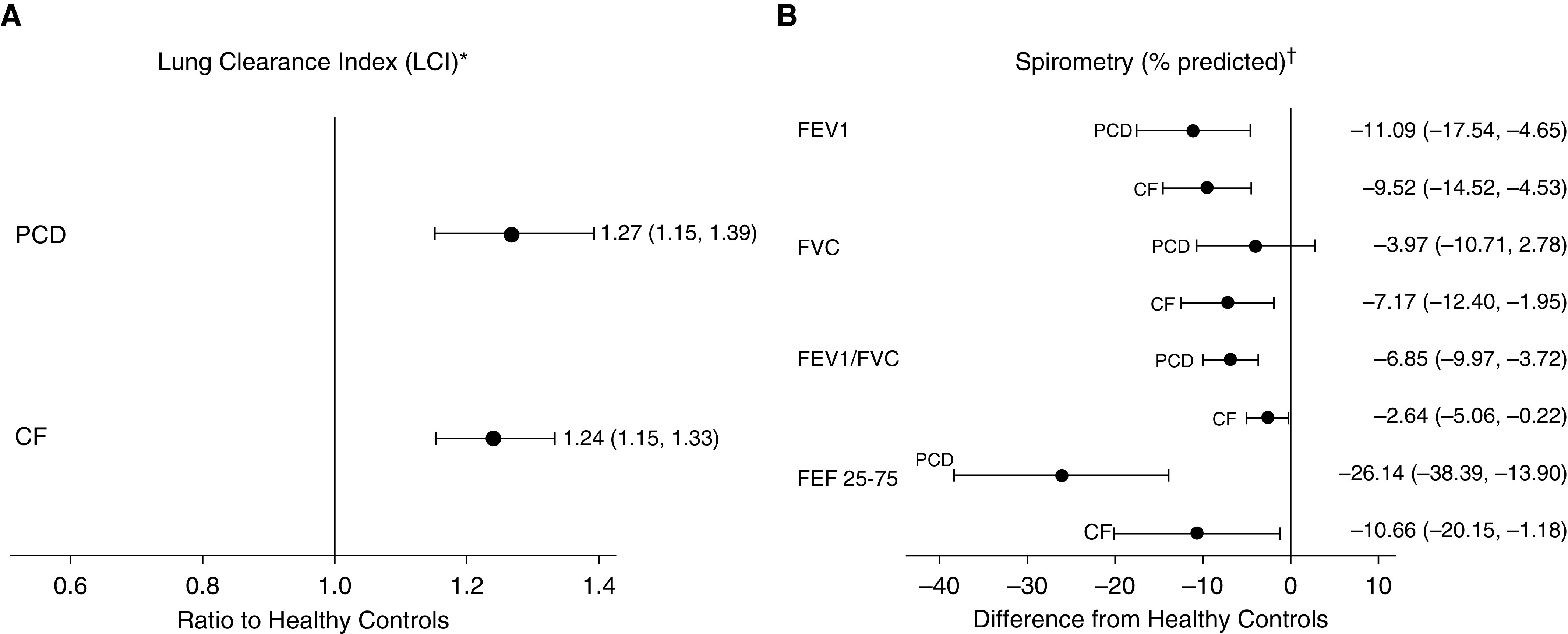
Forest plots for (A) estimated ratio of geometric mean lung clearance index (LCI) with 95% confidence interval (CI) for children with primary ciliary dyskinesia (PCD) or cystic fibrosis (CF) compared with healthy control subjects (reference), from regression analyses of log-transformed LCI data, adjusted for height, sex, and age. (B) Estimated difference in mean spirometric measures (95% CI) between children with PCD or CF and healthy control subjects from regression analyses. *LCI 2.5 were log-transformed for linear regression because of nonnormality. The resulting exponentiated parameter estimate provides the ratio of the geometric mean of each outcome by disease group to that of healthy control subjects. For example, patients with PCD have a geometric mean LCI 2.5 that is 26% higher than that of healthy control subjects of the same age, height, sex, and ethnicity (95% CI, 15–37% higher). †Calculated using Global Lung Initiative equations. The results are interpreted as the difference in the mean measure between children with PCD or CF and healthy control subjects of the same age, height, sex, and ethnicity. For example, the mean forced expiratory volume in 1 second % predicted of children with PCD is 11.1% lower than that of healthy control subjects (95% CI, −17.6 to −4.7). FEF25–75 = forced midexpiratory flow at 25–75% of the expiratory volume; FEV1 = forced expiratory volume in 1 second; FVC = forced vital capacity.
Sensitivity analyses restricted to participants with least three acceptable LCI 2.5 measurements yielded similar results. Children with PCD (n = 11) had a geometric mean LCI that was 18% (95% CI, 6–32%) higher than HC children (P = 0.003), and children with CF (n = 25) had a geometric mean LCI that was 11% (95% CI, 11–30%) higher than HC children (P < 0.001). Similarly, Sacin and Scond results did not change when restricted to those children with at least three valid measurements (Table E1).
Spirometry
In both unadjusted (Table 3) and adjusted (Figure 1) analyses, FEV1, FEV1/FVC, and FEF25–75 were significantly lower in patients with PCD and CF than in HC subjects. The pattern of spirometric abnormalities also appeared to differ slightly between participants with PCD and those with CF. Children with PCD and children with CF both had significantly lower FEV1% predicted than HC children, with a mean difference of −11.1 (95% CI, −17.5 to −4.7) and −9.5 (95% CI, −14.5 to −4.6), respectively (Figure 1), but FEV1% predicted was not significantly different between the two disease groups (P = 0.69) (Table 3). FEV1/FVC and FEF25–75 were also significantly lower in children with PCD and children with CF compared with HC children (Figure 1), but in addition, they appeared to be lower in children with PCD than in children with CF (Table 3). Linear regression, accounting for the interaction between age and disease status, showed no association between age and FEV1% predicted in our cohort (Figure E1).
Comparison of MBW and Spirometry
The LCI was more likely to be abnormal than FEV1 z-scores in both children with PCD and children with CF. Whereas the LCI was abnormal in 58% of children with CF and in 59% of children with PCD, FEV1 z-scores were abnormal in only 17% of children with CF and in 18% of children with PCD (Table 3). Importantly, 44% of children with CF and 42% of children with PCD had an abnormal LCI with a normal FEV1 z-score (Figure 2). Only one child with CF had an abnormal FEV1 z-score with a normal LCI (LCI 7.8, just at the ULN) (Figure 2). The LCI did not correlate with FEV1 z-score (Figure 2), FEV1/FVC (Figure E2A), or FEF25–75 (Figure E2B). Additional MBW parameters (Sacin/Scond and moment ratios) did not correlate with any of the spirometric indices in any of the three groups (data not shown).
Figure 2.

Lung clearance index versus forced expiratory volume in 1 second (FEV1) z-score, stratified by disease status (primary ciliary dyskinesia, cystic fibrosis, healthy control). The lung clearance index did not correlate with FEV1 (PCD, R = −0.24; P = 0.36; CF, R = −0.27, P = 0.11; and HC, R = −0.08; P = 0.57). CF = cystic fibrosis; HC = healthy control; LCI = lung clearance index; PCD = primary ciliary dyskinesia.
Discussion
Our study demonstrates that the LCI is more sensitive than FEV1 in detecting early, mild lung disease in children with PCD, which is similar to findings in the CF population. Participants with CF and PCD had similar abnormalities in LCI and FEV1; however, children with PCD had lower FEF25–75% predicted and FEV1/FVC z-scores compared with age-matched peers with CF. This finding suggests potentially worse obstructive airway disease, as detected by spirometry, in young children with PCD.
Earlier detection of lung disease in PCD could allow for intervention strategies to improve long-term outcomes. In CF, an abnormal preschool LCI is associated with concurrent measures of clinical status and later spirometry deficits, offering earlier prognostic utility of MBW testing in this age range than traditional spirometry (25). Similar to its use in CF, the LCI may also have the most clinical utility in young patients with mild lung disease, in whom FEV1 is frequently normal. The LCI correlates more strongly than FEV1 with structural changes on computed tomography scans, suggesting that ventilation heterogeneity is more sensitive than forced expiratory maneuvers in detecting early small airway damage (26, 27). As a tidal breathing maneuver, MBW can be performed in unsedated children as young as 3 years of age. We chose to focus on children ages 3–12 years with mild to moderate lung disease, enrolling younger children than previously reported (28% of children with PCD or CF were 6 yr old or younger). We found 42% of patients with PCD with a normal FEV1 to have an abnormal LCI, which is similar to our CF cohort and published studies in older patients with PCD. Boon and colleagues reported that 47% of patients with PCD ages 5–25 years (median age, 11 yr) had an abnormal LCI and a normal FEV1 (11), and Green and colleagues reported that 81% of patients with PCD ages 6–18.5 years (median age, 11 yr) had an abnormal LCI with a normal FEV1 (7). In a more recent study by Nyilas and colleagues, 83% of patients with PCD (median age, 13.4 yr; range, 5–28) had an abnormal LCI compared with 27% with an abnormal FEV1 (12). A study conducted in adults with PCD (mean age, 25 yr) with more advanced obstructive lung disease (FEV1 z-score −2.99) also demonstrated that the LCI was more likely to be abnormal compared with FEV1 (9). Thus, the LCI seems to be more sensitive than FEV1 in detecting lung disease in PCD across the age spectrum and to have a similar sensitivity in PCD and CF.
In our study, children with PCD had lower FEV1/FVC z-scores and FEF25–75% predicted compared with children with CF, whereas there was no difference in LCIs between the two groups, suggesting potentially greater limitation in airway resistance (and perhaps midsized airway obstruction) with similar degrees of peripheral airway disease in children with PCD compared with those with CF. This observation begins to add to the understanding of differences in airway pathophysiology between these two diseases. Most existing literature comparing spirometric measures in children and adults with PCD and CF have found no significant differences in FEV1, FVC, FEV1/FVC, and FEF25–75 (28–30). Only one study reports decreased FEV1, FVC, and FEF25–75 among children with PCD compared with CF at mean ages of 11.8 and 10.6, respectively (31). Our findings suggest that lung function in patients with PCD may already be reduced early in life and that airway disease may be both different from and more severe compared with that found in children with CF.
Although FEV1/FVC and FEF25–75 were lower in children with PCD than those with CF, the LCI and other MBW parameters were similar between the two groups, reinforcing the idea that MBW and spirometry measure different characteristics of these obstructive lung diseases. MBW measures the heterogeneity of ventilation, whereas spirometry measures airway obstruction as determined by airway resistance. It is therefore not surprising that we found no correlation between the LCI or other MBW indices (Sacin and Scond) and spirometric measures (FEV1, FEV1/FVC, and FEF25–75), similar to some (7, 9), but not all (11, 12, 32), prior PCD studies. Furthermore, because our study was restricted to children with mild to moderate disease, the range of FEV1 with which we are correlating LCI is much more limited—the lack of correlation here may highlight the ability of MBW to identify disease to which spirometry is less sensitive. Three studies have evaluated additional MBW parameters in children with PCD. One reported no correlation of Sacin and Scond with either FVC or FEV1 (7), whereas another demonstrated a correlation between FEV1 and both Sacin and moment ratios (33). Given the lack of consistent correlation between MBW and spirometric measures, these measurements may provide complementary information on different aspects of lung pathophysiology. More recently, Kobbernagel and colleagues performed longitudinal MBW in a cohort of 48 patients with PCD. They found that the LCI worsened significantly over time, whereas there was no significant progression of FEV1, Scond, Sacin, or moment ratios (32). In our small cohort, we had no evidence that secondary MBW measurements were more accurate than the LCI at predicting early lung disease. Therefore, at this time, it is remains unclear whether secondary LCI measurements add additional clinically relevant information to the much more widely employed LCI measurement.
Strengths of our study include the multicenter nature of the cohort, inclusion of patients with PCD only with a confirmed diagnosis, and rigorous quality control and centralized review of all MBW and spirometry data for acceptability. Our study has several limitations. First, we did not meet our target sample size for the PCD cohort because of slow accrual and lower than anticipated measurement acceptability. Feasibility of MBW testing was moderate, with only 75% of children having acceptable measurements, slightly lower than in other multicenter cohorts of a similar age range (34). The small sample size limited our ability to detect associations and correlations, so our results should be interpreted with this caution. Given our limited numbers, we were not able to investigate associations of lung function with specific CF or PCD genotypes. Second, as the objective of our study was to assess the sensitivity of MBW and spirometry for the detection of lung disease in children with PCD and CF with mild disease, we only recruited patients with an FEV1 greater than 60% predicted, so our results cannot be generalized to patients with more severe disease. We chose to report FEV1 rather than FEV0.75 for preschool children (15) for ease of interpretability of results, given the small number of young children enrolled in our study. We did not perform plethysmography or high-resolution computed tomography (HRCT) in our cohort, as has been done in prior studies (5, 34), and therefore cannot comment on the association of MBW and air trapping or structural airway abnormalities. In patients with PCD aged 5–25 years, Boon and colleagues demonstrated that LCI values had a sensitivity of 91% in detecting patients with abnormal HRCT findings, which is significantly higher than that of FEV1 (36%) (11). Studies assessing the sensitivity of LCI, FEV1, and HRCT scores in patients with PCD have been limited to older patients, and therefore, there is no information on the sensitivity of LCI and FEV1 in detecting structural damage in young children with PCD.
In summary, the LCI appears to be a promising measure of early PCD lung disease. Longitudinal studies demonstrate that airflow obstruction in PCD worsens with age (4, 35, 36), with a mean annual change in FEV1% predicted of −0.57% per year in a recent study of children and young adults (37). Future research should evaluate the longitudinal changes in LCI measurements in patients with PCD because these LCI measurements could ultimately identify early lung disease or early changes during critical windows for intervention. The evaluation of the longitudinal trajectories of these measures and response to therapeutic interventions in well-described PCD cohorts will also help to elucidate the progression of lung disease.
Acknowledgments
Acknowledgment
The authors thank the patients and their family members, Michele Manion (founder of the U.S. PCD Foundation), and the U.S. PCD Foundation. They also thank Dr. Michael Knowles, Dr. Thomas Ferkol, and the Genetic Disorders of Mucociliary Clearance for support for this project. The Genetic Disorders of Mucociliary Clearance (U54HL096458) is a part of the NCATS Rare Diseases Clinical Research Network. The Rare Diseases Clinical Research Network is an initiative of the Office of Rare Diseases Research at the NCATS and is funded through a collaboration between NCATS and NHLBI. The authors also thank Anne Mills and Elizabeth Thompson for their efforts pertaining to enrollment and data acquisition.
Footnotes
Supported by the Genetic Disorders of Mucociliary Clearance Consortium. The Genetic Disorders of Mucociliary Clearance (U54HL096458) is a part of the National Center for Advancing Translational Sciences (NCATS) Rare Diseases Clinical Research Network. The Rare Diseases Clinical Research Network is an initiative of the Office of Rare Diseases Research at the NCATS and is funded through a collaboration between the NCATS and NHLBI. Supported by a Clinical and Translational Science Awards (NIH/NCATS grant UNC ULTR000083), a NIH/NCATS Colorado Clinical and Translational Science Awards (grant UL1 TR001082), and an Intramural Research Program of NIH/National Institute of Allergy and Infectious Diseases. Funding for data analysis and multiple breath washout overreading at Washington University School of Medicine was derived, in part, from the Cystic Fibrosis Foundation Therapeutics Development Network funding for the Center for Infant and Preschool Pulmonary Function Testing (Cystic Fibrosis Foundation DAVIS08Y2).
Author Contributions: B.K. served as the site principal investigator and contributed to the conception of design, acquisition of data, analysis design, interpretation of data, and writing of the manuscript. E.S. served as the biostatistician and contributed to the analysis design, interpretation of data, and development of tables and figures. M.S. served as the study coordinator and contributed to the acquisition of data and multiple breath washout overreading. C.C. contributed to the acquisition of data and spirometry overreading. A. Griffiths served as a study site principal investigator and contributed to the editing of the manuscript. W.W. served as a study site principal investigator and contributed to the editing of the manuscript. S.D.D. served as a study site principal investigator and contributed to the editing of the manuscript. M.W.L. served as a study site principal investigator and contributed to the editing of the manuscript. M.R. served as a co–senior author and contributed to the conception of design, acquisition of data, analysis design, interpretation of data, and editing of the manuscript. J.P. served as a co–senior author and a study principal investigator and contributed to the conception of design, acquisition of data, analysis design, interpretation of data, and editing of the manuscript. S.M., A. Genatossio, M.S., I.B., R.C.J., M.D., and K.J. served as study coordinators and contributed to the acquisition of data. All authors have given approval to the final version of this manuscript.
This article has an online supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.Shapiro AJ, Zariwala MA, Ferkol T, Davis SD, Sagel SD, Dell SD, et al. Genetic Disorders of Mucociliary Clearance Consortium. Diagnosis, monitoring, and treatment of primary ciliary dyskinesia: PCD foundation consensus recommendations based on state of the art review. Pediatr Pulmonol. 2016;51:115–132. doi: 10.1002/ppul.23304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bush A, Payne D, Pike S, Jenkins G, Henke MO, Rubin BK. Mucus properties in children with primary ciliary dyskinesia: comparison with cystic fibrosis. Chest. 2006;129:118–123. doi: 10.1378/chest.129.1.118. [DOI] [PubMed] [Google Scholar]
- 3.Gibson RL, Burns JL, Ramsey BW. Pathophysiology and management of pulmonary infections in cystic fibrosis. Am J Respir Crit Care Med. 2003;168:918–951. doi: 10.1164/rccm.200304-505SO. [DOI] [PubMed] [Google Scholar]
- 4.Ellerman A, Bisgaard H. Longitudinal study of lung function in a cohort of primary ciliary dyskinesia. Eur Respir J. 1997;10:2376–2379. doi: 10.1183/09031936.97.10102376. [DOI] [PubMed] [Google Scholar]
- 5.Aurora P, Gustafsson P, Bush A, Lindblad A, Oliver C, Wallis CE, et al. Multiple breath inert gas washout as a measure of ventilation distribution in children with cystic fibrosis. Thorax. 2004;59:1068–1073. doi: 10.1136/thx.2004.022590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gustafsson PM, De Jong PA, Tiddens HA, Lindblad A. Multiple-breath inert gas washout and spirometry versus structural lung disease in cystic fibrosis. Thorax. 2008;63:129–134. doi: 10.1136/thx.2007.077784. [DOI] [PubMed] [Google Scholar]
- 7.Green K, Buchvald FF, Marthin JK, Hanel B, Gustafsson PM, Nielsen KG. Ventilation inhomogeneity in children with primary ciliary dyskinesia. Thorax. 2012;67:49–53. doi: 10.1136/thoraxjnl-2011-200726. [DOI] [PubMed] [Google Scholar]
- 8.Gustafsson PM. Peripheral airway involvement in CF and asthma compared by inert gas washout. Pediatr Pulmonol. 2007;42:168–176. doi: 10.1002/ppul.20554. [DOI] [PubMed] [Google Scholar]
- 9.Irving SJ, Ives A, Davies G, Donovan J, Edey AJ, Gill SS, et al. Lung clearance index and high-resolution computed tomography scores in primary ciliary dyskinesia. Am J Respir Crit Care Med. 2013;188:545–549. doi: 10.1164/rccm.201304-0800OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nyilas S, Schlegtendal A, Yammine S, Casaulta C, Latzin P, Koerner-Rettberg C. Further evidence for an association between LCI and FEV1 in patients with PCD. Thorax. 2015;70:896. doi: 10.1136/thoraxjnl-2015-207206. [DOI] [PubMed] [Google Scholar]
- 11.Boon M, Vermeulen FL, Gysemans W, Proesmans M, Jorissen M, De Boeck K. Lung structure-function correlation in patients with primary ciliary dyskinesia. Thorax. 2015;70:339–345. doi: 10.1136/thoraxjnl-2014-206578. [DOI] [PubMed] [Google Scholar]
- 12.Nyilas S, Bauman G, Pusterla O, Sommer G, Singer F, Stranzinger E, et al. Structural and functional lung impairment in primary ciliary dyskinesia: assessment with magnetic resonance imaging and multiple breath washout in comparison to spirometry. Ann Am Thorac Soc. 2018;15:1434–1442. doi: 10.1513/AnnalsATS.201712-967OC. [DOI] [PubMed] [Google Scholar]
- 13.Pittman JE, Jr, Laux J, Jones PW, Lin FC, Leigh MW.Nitrogen washout MBW testing in obstructive disease vs. Healthy controls: LCI vs. FEV1, age, and imaging [abstract] Am J Respir Crit Care Med 2013187:A369823668455 [Google Scholar]
- 14.Pittman JE, Detwiler C, Johnson R, Laux J, Lin FC, Leigh MW. N2 MBW testing using online analysis is more sensitive than spirometry for pediatric obstructive airways disease [abstract] Am J Respir Crit Care Med. 2014;189:A1033. [Google Scholar]
- 15.Beydon N, Davis SD, Lombardi E, Allen JL, Arets HG, Aurora P, et al. American Thoracic Society/European Respiratory Society Working Group on Infant and Young Children Pulmonary Function Testing. An official American Thoracic Society/European Respiratory Society statement: pulmonary function testing in preschool children. Am J Respir Crit Care Med. 2007;175:1304–1345. doi: 10.1164/rccm.200605-642ST. [DOI] [PubMed] [Google Scholar]
- 16.Miller MR, Hankinson J, Brusasco V, Burgos F, Casaburi R, Coates A, et al. ATS/ERS Task Force. Standardisation of spirometry. Eur Respir J. 2005;26:319–338. doi: 10.1183/09031936.05.00034805. [DOI] [PubMed] [Google Scholar]
- 17.Singer F, Houltz B, Latzin P, Robinson P, Gustafsson P. A realistic validation study of a new nitrogen multiple-breath washout system. PLoS One. 2012;7:e36083. doi: 10.1371/journal.pone.0036083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yammine S, Singer F, Gustafsson P, Latzin P. Impact of different breathing protocols on multiple-breath washout outcomes in children. J Cyst Fibros. 2014;13:190–197. doi: 10.1016/j.jcf.2013.08.010. [DOI] [PubMed] [Google Scholar]
- 19.Robinson PD, Latzin P, Verbanck S, Hall GL, Horsley A, Gappa M, et al. Consensus statement for inert gas washout measurement using multiple- and single- breath tests. Eur Respir J. 2013;41:507–522. doi: 10.1183/09031936.00069712. [Published erratum appears in Eur Respir J 42:1432.] [DOI] [PubMed] [Google Scholar]
- 20.Robinson PD, Latzin P, Ramsey KA, Stanojevic S, Aurora P, Davis SD, et al. ATS Assembly on Pediatrics. Preschool multiple-breath washout testing: an official American Thoracic Society technical statement. Am J Respir Crit Care Med. 2018;197:e1–e19. doi: 10.1164/rccm.201801-0074ST. [DOI] [PubMed] [Google Scholar]
- 21.Centers for Disease Control and Prevention. A SAS program for the 2000 CDC growth charts (ages 0 to < 20 years) 2016 [accessed 2020 Jul 21]. Available from: https://www.cdc.gov/nccdphp/dnpao/growthcharts/resources/sas.htm.
- 22.Quanjer PH, Stanojevic S, Cole TJ, Baur X, Hall GL, Culver BH, et al. ERS Global Lung Function Initiative. Multi-ethnic reference values for spirometry for the 3-95-yr age range: the global lung function 2012 equations. Eur Respir J. 2012;40:1324–1343. doi: 10.1183/09031936.00080312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Culver BH, Graham BL, Coates AL, Wanger J, Berry CE, Clarke PK, et al. ATS Committee on Proficiency Standards for Pulmonary Function Laboratories. Recommendations for a standardized pulmonary function report: an official American Thoracic Society technical statement. Am J Respir Crit Care Med. 2017;196:1463–1472. doi: 10.1164/rccm.201710-1981ST. [DOI] [PubMed] [Google Scholar]
- 24.Stanojevic S, Davis SD, Retsch-Bogart G, Webster H, Davis M, Johnson RC, et al. Progression of lung disease in preschool patients with cystic fibrosis. Am J Respir Crit Care Med. 2017;195:1216–1225. doi: 10.1164/rccm.201610-2158OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hardaker KM, Panda H, Hulme K, Wong A, Coward E, Cooper P, et al. Abnormal preschool Lung Clearance Index (LCI) reflects clinical status and predicts lower spirometry later in childhood in cystic fibrosis. J Cyst Fibros. 2019;18:721–727. doi: 10.1016/j.jcf.2019.02.007. [DOI] [PubMed] [Google Scholar]
- 26.Rowan SA, Bradley JM, Bradbury I, Lawson J, Lynch T, Gustafsson P, et al. Lung clearance index is a repeatable and sensitive indicator of radiological changes in bronchiectasis. Am J Respir Crit Care Med. 2014;189:586–592. doi: 10.1164/rccm.201310-1747OC. [DOI] [PubMed] [Google Scholar]
- 27.Horsley A. Lung clearance index in the assessment of airways disease. Respir Med. 2009;103:793–799. doi: 10.1016/j.rmed.2009.01.025. [DOI] [PubMed] [Google Scholar]
- 28.Ratjen F, Waters V, Klingel M, McDonald N, Dell S, Leahy TR, et al. Changes in airway inflammation during pulmonary exacerbations in patients with cystic fibrosis and primary ciliary dyskinesia. Eur Respir J. 2016;47:829–836. doi: 10.1183/13993003.01390-2015. [DOI] [PubMed] [Google Scholar]
- 29.Halbeisen FS, Goutaki M, Spycher BD, Amirav I, Behan L, Boon M, et al. Lung function in patients with primary ciliary dyskinesia: an iPCD Cohort study. Eur Respir J. 2018;52:1801040. doi: 10.1183/13993003.01040-2018. [DOI] [PubMed] [Google Scholar]
- 30.Maglione M, Montella S, Mollica C, Carnovale V, Iacotucci P, De Gregorio F, et al. Lung structure and function similarities between primary ciliary dyskinesia and mild cystic fibrosis: a pilot study. Ital J Pediatr. 2017;43:34. doi: 10.1186/s13052-017-0351-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ring AM, Buchvald FF, Holgersen MG, Green K, Nielsen KG. Fitness and lung function in children with primary ciliary dyskinesia and cystic fibrosis. Respir Med. 2018;139:79–85. doi: 10.1016/j.rmed.2018.05.001. [DOI] [PubMed] [Google Scholar]
- 32.Kobbernagel HE, Green K, Ring AM, Buchvald FF, Rosthøj S, Gustafsson PM, et al. One-year evolution and variability in multiple-breath washout indices in children and young adults with primary ciliary dyskinesia. Eur Clin Respir J. 2019;6:1591841. doi: 10.1080/20018525.2019.1591841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nyilas S, Schlegtendal A, Singer F, Goutaki M, Kuehni CE, Casaulta C, et al. Alternative inert gas washout outcomes in patients with primary ciliary dyskinesia. Eur Respir J. 2017;49:1600466. doi: 10.1183/13993003.00466-2016. [DOI] [PubMed] [Google Scholar]
- 34.Gustafsson PM. Inert gas washout in preschool children. Paediatr Respir Rev. 2005;6:239–245. doi: 10.1016/j.prrv.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 35.Marthin JK, Petersen N, Skovgaard LT, Nielsen KG. Lung function in patients with primary ciliary dyskinesia: a cross-sectional and 3-decade longitudinal study. Am J Respir Crit Care Med. 2010;181:1262–1268. doi: 10.1164/rccm.200811-1731OC. [DOI] [PubMed] [Google Scholar]
- 36.Hellinckx J, Demedts M, De Boeck K. Primary ciliary dyskinesia: evolution of pulmonary function. Eur J Pediatr. 1998;157:422–426. doi: 10.1007/s004310050843. [DOI] [PubMed] [Google Scholar]
- 37.Davis SD, Rosenfeld M, Lee HS, Ferkol TW, Sagel SD, Dell SD, et al. Primary ciliary dyskinesia: longitudinal study of lung disease by ultrastructure defect and genotype. Am J Respir Crit Care Med. 2019;199:190–198. doi: 10.1164/rccm.201803-0548OC. [DOI] [PMC free article] [PubMed] [Google Scholar]