

Abstract
Bronchopulmonary dysplasia (BPD) is a common and serious complication associated with preterm birth. The pathogenesis of BPD is incompletely understood, and there is an unmet clinical need for effective treatments. The role of autophagy as a potential cytoprotective mechanism in BPD remains to be fully elucidated. In the present study, we investigated the role and regulation of autophagy in experimental models of BPD. Regulation and cellular distribution of autophagic activity during postnatal lung development and in neonatal hyperoxia-induced lung injury (nHILI) were assessed in the autophagy reporter transgenic GFP-LC3 (GFP–microtubule-associated protein 1A/1B-light chain 3) mouse model. Autophagic activity and its regulation were also examined in a baboon model of BPD. The role of autophagy in nHILI was determined by assessing lung morphometry, injury, and inflammation in autophagy-deficient Beclin 1 heterozygous knockout mice (Becn1+/−). Autophagic activity was induced during alveolarization in control murine lungs and localized primarily to alveolar type II cells and macrophages. Hyperoxia exposure of neonatal murine lungs and BPD in baboon lungs resulted in impaired autophagic activity in association with insufficient AMPK (5′-AMP–activated protein kinase) and increased mTORC1 (mTOR complex 1) activation. Becn1+/− lungs displayed impaired alveolarization, increased alveolar septal thickness, greater neutrophil accumulation, and increased IL-1β concentrations when exposed to nHILI. Becn1+/− alveolar macrophages isolated from nHILI-exposed mice displayed increased expression of proinflammatory genes. In conclusion, basal autophagy is induced during alveolarization and disrupted during progression of nHILI in mice and BPD in baboons. Becn1+/− mice are more susceptible to nHILI, suggesting that preservation of autophagic activity may be an effective protective strategy in BPD.
Keywords: bronchopulmonary dysplasia, autophagy, macrophage, GFP-LC3, beclin
Clinical Relevance
This research demonstrates that autophagic activity is impaired in experimental models of bronchopulmonary dysplasia. It also demonstrates that dysregulation of autophagy plays a role in the pathogenesis of neonatal hyperoxia-induced lung injury and inflammation through its effects in lung epithelial cells and macrophages. These findings suggest that autophagy-inducing drugs can be exploited in clinical trials to ameliorate bronchopulmonary dysplasia.
Bronchopulmonary dysplasia (BPD) is a serious and common complication of preterm birth (1–3). BPD results from the interplay between immature lungs and various antenatal and postnatal exposures, including elements necessary to support the survival of preterm infants, such as supplemental oxygen and mechanical ventilation. However, the molecular mechanisms by which these factors lead to interrupted alveolarization and dysregulated vascularization, the two pathologic hallmarks of BPD, remain to be fully elucidated because there is an unmet clinical need for effective preventive and therapeutic strategies for BPD (1, 4–6).
Macroautophagy (hereafter referred to as “autophagy”) is a highly conserved and regulated intracellular process that plays a key role in the lysosomal turnover of cytoplasmic material, including macromolecules and damaged organelles (7–9). Autophagy is induced by a broad range of stimuli, including environmental stress, and is cytoprotective in many conditions (10, 11). The initiation and progression of autophagy are regulated by a complex network of proteins, some of which are known as autophagy-related genes (Atg). Targeted homozygous deletion of several Atgs leads to either embryonic or perinatal lethality in mice (12–16). The cause of death in these mouse models may be multifactorial, but there is a growing body of evidence to suggest that respiratory failure due to aberrant in utero lung development plays a role in this outcome. For example, both Atg5−/− and Ulk1/2 (unc-51–like autophagy-activating kinase) double-knockout mice display a defect in lung development, manifested by reduced airspaces with thickened septa and glycogen-laden alveolar type II cells (AEC2s) (16). In a recent study, conditional deletion of Becn1 (Beclin 1), a key regulator of autophagosome formation, in mouse lung epithelial cells during gestation resulted in lethal respiratory distress perinatally due to reduced airway branching and impaired terminal air sac formation (17).
To date, the role of autophagy in BPD has been addressed in a limited number of studies with somewhat conflicting results. Sureshbabu and colleagues employed a murine model of BPD induced by 100% oxygen exposure between Postnatal Day (P) 1 and P7 and found that autophagy was induced during hyperoxia exposure (18). Further augmentation of autophagic activity by inhibition of RPTOR (regulatory-associated protein of mechanistic target of rapamycin) improved lung architecture and increased survival in association with decreased apoptosis. However, mechanical ventilation of newborn rats triggered autophagy and subsequent apoptosis of lung epithelial cells via increased ceramide production (19). Thus, although these studies have brought well-deserved attention to autophagy as a potential target in BPD, a better understanding of the role of autophagy in BPD is needed to be able to pursue this hypothesis.
In this study, we took advantage of the autophagy reporter GFP-LC3 (GFP–microtubule-associated protein 1A/1B-light chain 3) mouse model to investigate the temporal and spatial regulation of autophagy during progression of neonatal hyperoxia-induced lung injury (nHILI) (20). We also used lungs from a baboon model of BPD to validate the translational significance of our findings and investigated the impact of autophagy deficiency induced by heterozygous deletion of Becn1 in murine nHILI.
Methods
Additional details on the methods used for making these measurements is provided in the data supplement.
Murine Model of Neonatal Hyperoxia-induced Lung Injury
Becn1+/− mice were obtained from Dr. Beth Levine (University of Texas Southwestern Medical Center at Dallas, Dallas, TX), and GFP-LC3 mice were obtained from Dr. Noboru Mizushima (University of Tokyo, Tokyo, Japan) (21, 22). Wild-type (WT) C57BL/6 littermates were used as control animals. All procedures performed on mice were approved by the animal care and use committee at Brigham and Women’s Hospital. Neonatal hyperoxia exposure was performed as previously described (23).
Baboon Model of BPD
The frozen baboon lung tissue samples were provided by the Southwest Foundation for Biomedical Research (23, 24). All procedures performed on baboons were reviewed and approved by the animal care and use committees of the Southwest Foundation for Biomedical Research and the University of Texas Southwestern Medical Center. Briefly, baboons were delivered by hysterotomy at 125 days (∼27 wk human gestation). They were intubated, treated with surfactant, and maintained on mechanical ventilation and pro re nata (PRN) oxygen for 14 days to induce pathologic and biochemical findings that are characteristic of the “new BPD” seen in human infants (24, 25).
Human Tracheal Aspirate Samples
Human tracheal aspirate samples were obtained during routine care of intubated preterm infants with an exempt protocol that was approved by the Brigham and Women’s Hospital Institutional Review Board.
BAL, Isolation, and Culture of Murine Alveolar Macrophages
BAL was performed for isolation of alveolar macrophages as previously described (26).
Morphometric Analysis
Paraffin-embedded lung sections were stained with modified Gill’s stain for measurements of mean linear intercept as a surrogate for alveolar diameter or with hematoxylin and eosin for assessment of general architecture and measurements of alveolar septal thickness, as previously described (27).
Immunostaining of Mouse Lung Sections
Double immunofluorescence staining was performed as previously described (28).
Western Blotting
Lung tissues were homogenized in radioimmunoprecipitation assay lysis buffer (Boston BioProducts) containing protease inhibitors. Equal amounts of protein samples were subjected to SDS-PAGE followed by IB as previously described (26).
RNA Extraction and qRT-PCR
Total RNA was extracted from adhesion-purified alveolar macrophages using the RNeasy Mini Kit (Qiagen). qRT-PCR amplification assays were performed using Green-2-Go qPCR Mastermix-ROX (Bio Basic).
Quantification of IL-1β Concentrations in Whole-Lung Homogenates
IL-1β concentrations in mouse lung homogenates were quantified with a commercially available ELISA kit (Invitrogen).
Detection of Cell Death In Situ Using TUNEL Assay
For in situ detection of apoptosis in paraffin-embedded lung sections, an in situ cell death detection kit was used (TMR red; Roche Applied Science).
Caspase-1 Fluorometric Activity Assay
Enzymatic activity of caspase 1 was quantified using a caspase 1 fluorometric assay kit (R&D Systems).
Statistical Analyses
All data are presented as mean ± SEM. Statistical significance was determined by two-tailed Student’s t test for comparisons of two groups or one-way ANOVA followed by Tukey’s post hoc test for more than two groups using Prism 7 software (GraphPad Software). P < 0.05 was considered significant.
Results
Basal Autophagic Activity in the Postnatal Murine Lung Is Induced during Alveolarization
We first aimed to characterize baseline autophagic activity during postnatal lung development. In a previous study, basal autophagic activity in autophagy reporter GFP-LC3 mouse lungs was determined using GFP puncta counts and transient EM between Embryonic Day (E) 18.5 and P2.5, and it was found to peak on P1 between 3 and 12 hours of life, followed by a gradual decrease to very low concentrations by P2.5 (12). In our analysis of lung cryosections from GFP-LC3 mice, we also observed GFP puncta in lung samples harvested from 6-hour-old mice, but these were difficult to discern in older mice, owing to low degrees of basal autophagic activity in the lung (Figure E1 in the data supplement). During autophagy, LC3-I is converted to LC3-II, and p62 is degraded by autolysosomes. Therefore, an increase in LC3-II/LC3-I ratio in association with decreased concentrations of p62 indicates increased autophagic flux (29). Despite many attempts with different LC3 antibodies, we could not quantify LC3-I/II concentrations on immunoblots of neonatal murine lungs in a satisfactory manner. Therefore, we took advantage of GFP-LC3 mice to optimize and validate another method to quantify autophagic flux. Previous studies have demonstrated that when GFP-LC3 is delivered to the lysosome, the LC3 part of the fusion protein is sensitive to degradation, whereas GFP is relatively resistant to hydrolysis (30). Therefore, autophagic flux can be measured by the detection of the free GFP fragment (referred to as “GFP” hereafter) that is generated in the autolysosome by IB with an anti-GFP antibody (31). Although this assay has been widely used in yeast cells, experience is more limited in mammalian cells and tissues (31). In validation studies, we first analyzed GFP concentrations in conjunction with p62 and LC3 concentrations at P0.5, P1, and P3 by IB and found the previously reported trend in autophagic activity at these time points based on GFP concentrations alone (Figure 1A) (12). In contrast, LC3-II/LC3-I ratio did not show a good correlation with the data previously published by Kuma and colleagues (12). We further validated this method by IB of whole-lung homogenates from GFP-LC3+/−/Becn1+/− and GFP-LC3+/− mice for GFP and p62. As expected, autophagy-deficient GFP-LC3+/−/Becn1+/− lungs had lower GFP and higher p62 concentrations than did GFP-LC3+/− lungs (Figure 1B). Thus, we confirmed that GFP concentrations on immunoblots constitute a sensitive and reliable marker for quantification of autophagic activity in whole-lung tissues from neonatal GFP-LC3 mice. Next, using this method, we analyzed the time course of autophagic activity in lungs harvested from mice at P1 (24 h), P3, P5, P7, P10, and P14. We found very low degrees of basal autophagic activity in whole-lung homogenates at P1 and P3 (Figures 1C and 1D). This was followed by a marked increase in autophagic activity first detected at P5, coinciding with the beginning of alveolarization in mouse lungs (32). Autophagic activity reached its peak at P7, started declining at P10, and finally reached prealveolarization levels at P14, when the majority of alveoli have already formed in mouse lungs. As expected, the concentrations of the autophagy adapter p62 demonstrated a trend for an inverse relationship with GFP concentrations with a progressive decline between P1 and P5, but concentrations remained similar between P7 and P14 (Figures 1C and 1E). Beclin-1 protein concentrations also were developmentally regulated, with the highest concentrations at P10 (Figure E2). These results indicate that basal autophagic activity in the postnatal murine lung is developmentally regulated and peaks during alveolarization after an earlier and transient spike soon after birth (12).
Figure 1.

Autophagic activity is induced in the lung during alveolarization. (A) Representative immunoblot analysis for GFP, LC3-I and -II, and p62 using GFP-LC3 (GFP–microtubule-associated protein 1A/1B-light chain 3) whole-lung homogenates harvested at Postnatal Day (P) 0.5, P1, and P3. Relative protein concentrations normalized to ACTB (β-actin) are indicated under the GFP and p62 blots and LC3-II/LC3-I ratio is indicated under the LC3 blot. (B) Representative immunoblot analysis for GFP and p62 using GFP-LC3+/− (GFP+/−) and GFP-LC3+/−/Becn1+/− (GFP+/−/Becn1+/−) whole-lung homogenates at P7. ACTB was used as a loading control. Relative protein concentrations are indicated under the blots. (C) GFP-LC3 mice were exposed to normoxia (21% O2) starting at birth and killed on P1, 3, 5, 7, 10, or 14. (D and E) Left lungs were homogenized and used for IB and densitometric analysis of GFP (D) and p62 protein (E). Data are shown as mean ± SEM; n = 3–4 per group. ****P < 0.0001 versus all groups except for P10.
Autophagic Activity Is Impaired in Murine Lungs during Progression of nHILI
To assess the temporal regulation of autophagy during nHILI, GFP-LC3 mice were exposed to normoxia or hyperoxia (75% O2) between P1 and P3, P5, or P7, and autophagic activity was assessed in whole-lung homogenates by IB and densitometry (Figures 2A–2D). Autophagic activity as determined by GFP concentrations was significantly increased at P3 in nHILI-exposed mouse lungs as compared with control animal lungs. With ongoing hyperoxia exposure, autophagic activity was decreased to the same amounts as in control mice at P5 and further decreased compared with control animals at P7. As expected, p62 concentrations demonstrated an inverse correlation with GFP concentrations, with a slight decrease at P3 and significant increases at P5 and P7 compared with control animals during progression of nHILI. Thus, alterations in p62 concentrations were also in line with GFP concentrations, although the kinetics were slightly different, and, taken together, both GFP and p62 expression indicated that the initial induction in autophagic activity was not sustained with ongoing hyperoxia exposure beyond P3. The concentrations of Becn1, which is an upstream regulator of LC3-I to LC3-II conversion, demonstrated parallel changes to GFP, with an initial increase at P3 followed by a decrease at P7. There were no alterations noted in the expression amounts of ATG5–12 and the lysosomal marker LAMP1 (lysosomal-associated membrane protein 1) at P7 (Figure E3). We also assessed the activity of mTORC1 (mTOR complex 1) and AMPK (5′-AMP–activated protein kinase) signaling pathways as the two major upstream regulators of autophagy during progression of nHILI (33, 34). The concentrations of phospho-S6 (p-S6), a marker of mTORC1 activation and a negative regulator of autophagy, were significantly increased at P5 in nHILI mice compared with in control animals. In contrast, the protein concentrations of phospho-AMPK (p-AMPK), which positively regulates autophagy, were significantly higher in the hyperoxia group at P3 without any significant differences at P5 and P7 between the nHILI and control groups.
Figure 2.

Autophagic activity is initially induced and then impaired during progression of neonatal hyperoxia-induced lung injury. GFP-LC3 mice were randomized to normoxia (N; 21% O2) or hyperoxia (H; 75% O2) exposure within 12 hours of birth and killed on P3, 5, or 7. (A) Left lung homogenates were analyzed for GFP, p62, Becn1 (Beclin 1), AMPK (5′-AMP–activated protein kinase), phospho-AMPK (p-AMPK), and phospho-S6 (p-S6) expression by IB. Relative protein concentrations of P3, P5, and P7 samples, normalized to ACTB, were determined by densitometry and are presented in B–D, respectively. Data are shown as mean ± SEM; n = 4 per group. *P < 0.05, **P < 0.01, and ***P < 0.001.
Autophagic Activity Is Impaired in Baboon Lungs with BPD
To assess the translational significance of our autophagy-related findings in the murine model of nHILI, we analyzed the autophagy pathway in a baboon model of “new BPD,” which was induced by subjecting preterm baboons delivered at 125 days of gestation, the equivalent of 27 weeks of human gestation, to mechanical ventilation and PRN O2 after antenatal steroid and postnatal surfactant treatment. BPD group lungs displayed impaired autophagic activity, as indicated by significantly decreased LC3-II/LC3-I ratio and significantly increased concentrations of p62 compared with gestational control animals (Figure 3). Decreased autophagy was associated with significantly increased apoptotic activity, as indicated by increased cleaved caspase 3 concentrations. Becn1 and ATG5–12 (not shown) concentrations were similar in the BPD and control groups, whereas the concentrations of Parkin and PINK1 (phosphatase and tensin homolog–induced kinase 1), two key regulators of mitophagy (35), were decreased in the BPD group lungs compared with control lungs. Furthermore, there was a significant increase in mTORC1 signaling, as indicated by p-S6 concentrations in the BPD group lungs. In three of seven BPD lungs, p-AMPK concentrations were similar to the concentrations detected in control lungs and increased in the other four BPD lungs compared with control lungs. Notably, within the BPD group, p-AMPK concentrations demonstrated a positive correlation with LC3-II concentrations (Pearson r = 0.94; P < 0.01), whereas there was no correlation between p-S6 and LC3-II concentrations (Figure E4). These results indicate that, similar to the murine model, autophagy is impaired in the baboon model of “new BPD,” which recapitulates the human BPD very well. We also analyzed LC3, p62, and p-AMPK expression in baboon lungs delivered at 140 days of gestation (∼29–30 wk of human gestation) and exposed to mechanical ventilation and PRN or 100% O2 for 10 days (the “old BPD” model). Because of the limited availability of samples from this model, we were not able to perform statistical analyses, but we also found a trend for impaired autophagic activity with a marked accumulation of p62 in lungs exposed to 100% O2 as compared with PRN O2 and gestational control samples (Figure E5).
Figure 3.
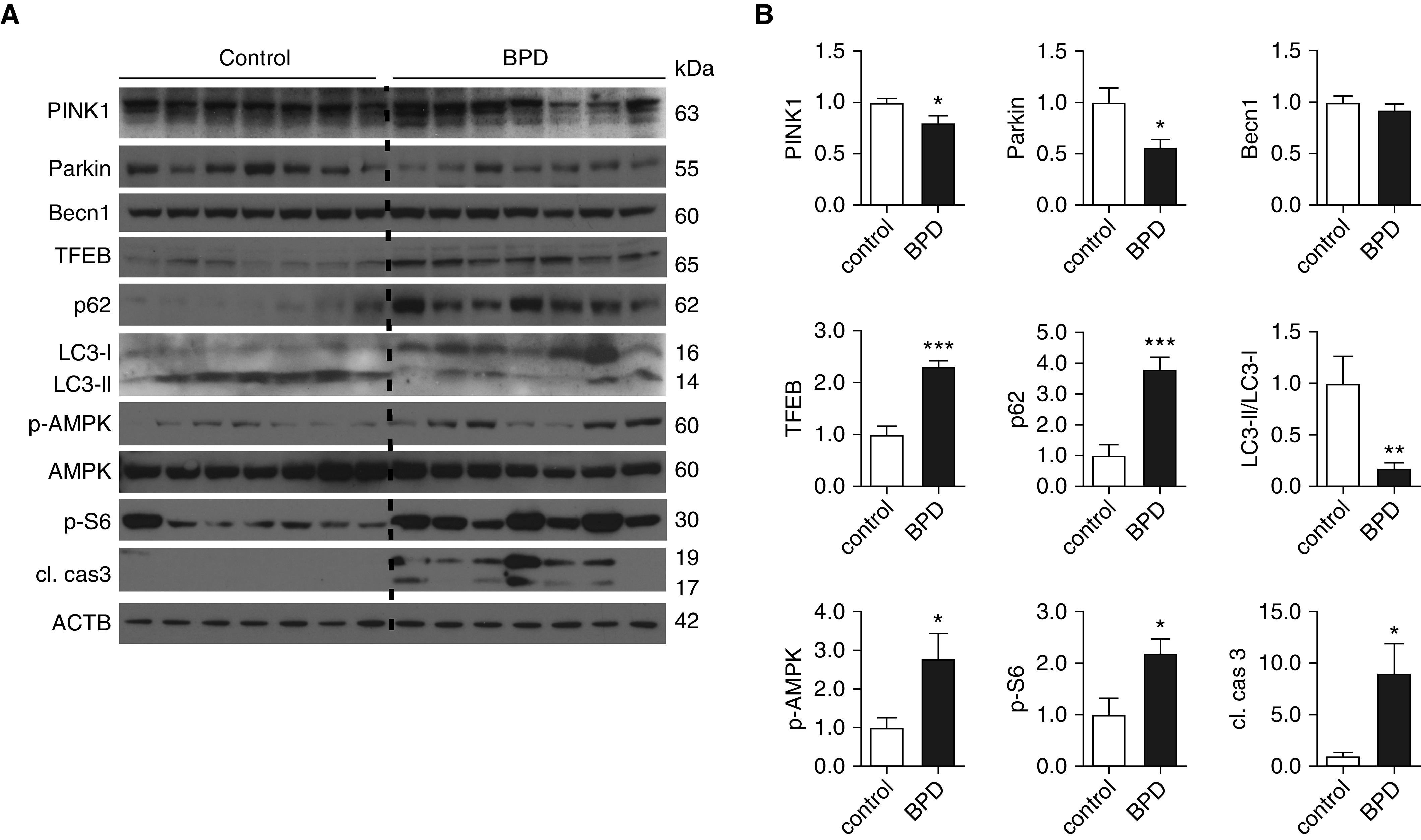
Autophagic activity is impaired in baboon lungs with bronchopulmonary dysplasia (BPD). Baboons were delivered by cesarean section at 125 days of gestation (∼27 wk of human gestation) and treated with exogenous surfactant, pro re nata O2, and mechanical ventilation for 14 days. Control baboons were delivered at 140 days of gestation and killed immediately before their first breath. (A) Lung homogenates were prepared and analyzed by IB with antibodies against cleaved caspase 3 (cl. cas3), p-S6, AMPK, p-AMPK, LC3-I and -II, p62, TFEB (transcription factor EB), BECN1, Parkin, and PINK1 (phosphatase and tensin homolog–induced kinase 1). (B) Relative protein expression levels, normalized to ACTB (except for LC3-II, which was normalized to LC3-I), were determined by densitometry. Data are shown as mean ± SEM; n = 7 per group. *P < 0.05, **P < 0.01, and ***P < 0.001.
Cellular Localization of Autophagic Activity in the Neonatal Lung
To determine the cell types in which autophagy is functional, we examined GFP-LC3 expression in GFP-LC3 lung cryosections using immunofluorescence analysis. The cellular distribution of GFP-LC3 signal was similar in normoxia- and hyperoxia-exposed lungs at all ages examined (P1, 3, 5, 7, 14, 28) and was predominantly localized to epithelial cells surrounding air saccules and alveoli. The majority of GFP-LC3 signal was colocalized with pro-SPC (pro–surfactant protein C), a marker of AEC2s and club cells (Figure 4, top row, and Figure E6). GFP-LC3 was also detected in smooth muscle cells around large airways (Figure 4, second row) and some macrophages (Figure 4, third row). Double immunofluorescence analyses revealed colocalization of p-AMPK (Figure 4, fourth row) and p-S6 (Figure 4, fifth row) with GFP-LC3 in some AEC2s in hyperoxia-exposed GFP-LC3 lungs. p-S6 was also colocalized with some alveolar macrophages in both hyperoxia-exposed GFP-LC3 lungs and baboon lungs with BPD (Figure E7). To confirm the presence of autophagic activity in alveolar macrophages, we pooled BAL cells from GFP-LC3 mice and analyzed GFP expression by IB. A GFP-LC3 band was detected in both normoxia and hyperoxia samples, whereas a GFP band was visible in the control sample and very faint in the hyperoxia sample (Figure E8A). Of note, there was no GFP-LC3 expression in neutrophils on the basis of colocalization studies with myeloperoxidase as a neutrophil marker on lung cryosections (Figure E8B). Furthermore, we detected LC3 puncta in some macrophages, but not neutrophils, in tracheal aspirate cytospins from intubated preterm infants with evolving BPD (Figure E9). Taken together, these results indicate that autophagy is functional in neonatal alveolar macrophages.
Figure 4.
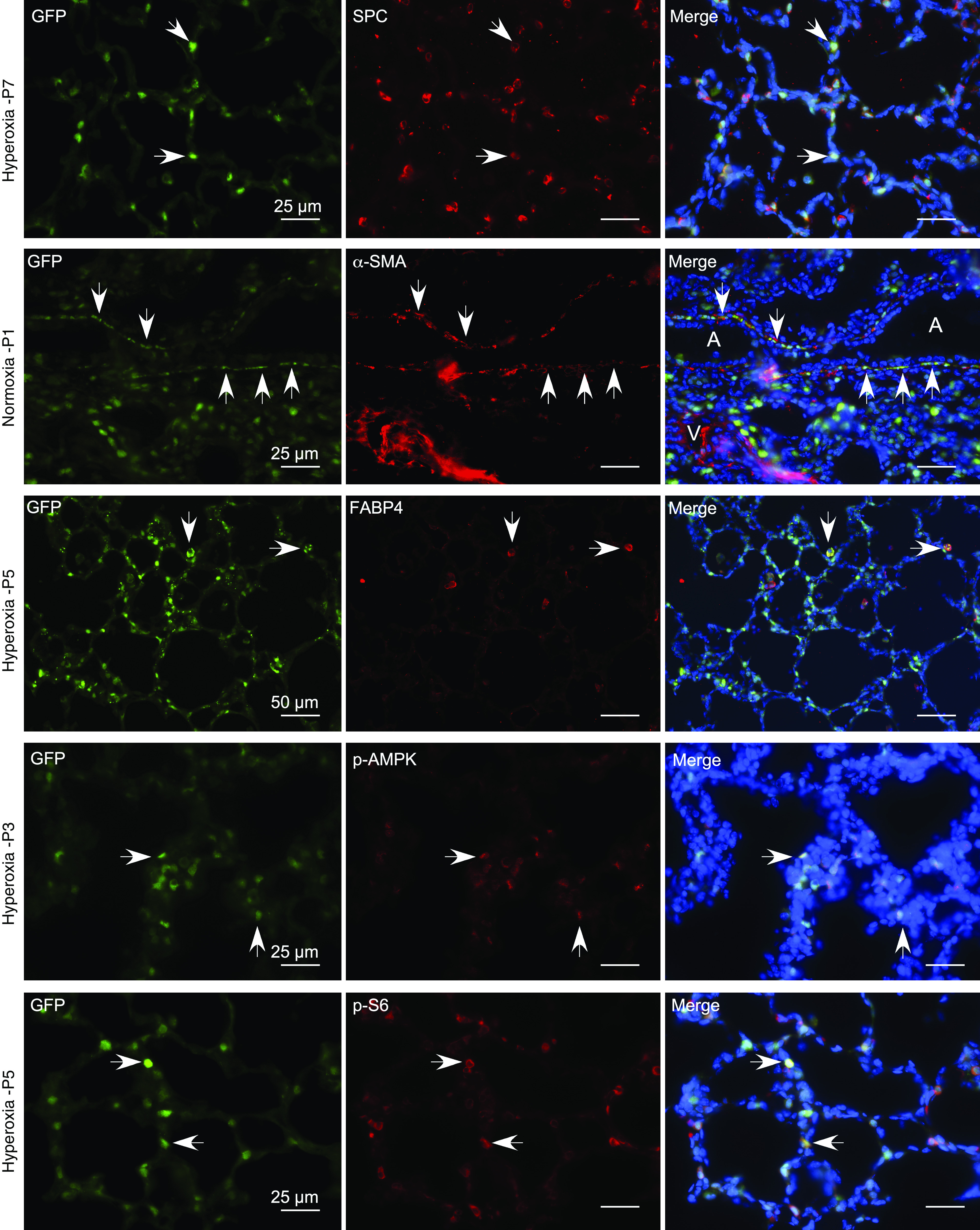
GFP-LC3 expression in neonatal murine lungs. Immunofluorescence staining was performed on cryosections of GFP-LC3 lungs with primary antibodies against SPC (surfactant protein C) (top row), ACTA2 (α-SMA) (second row), FABP4 (fatty acid-binding protein, adipocyte) (third row), p-AMPK (fourth row), and p-S6 (fifth row). The secondary antibody was Alexa Fluor 594 goat antirabbit IgG (red). Representative images are shown. White arrows indicate examples of co-localization of GFP-LC3 (green) with each of the primary antibody targets. Scale bars, 25 μm and 50 μm. A = airway; V = vessel.
Autophagy-Deficient Becn1+/− Mouse Lungs Exhibit Increased Susceptibility to Neonatal Hyperoxia Exposure
Our data summarized above demonstrate that autophagic activity is induced as an initial response to hyperoxia-exposed neonatal lungs with subsequent attenuation over time. To determine whether autophagy deficiency increases susceptibility to nHILI, we subjected autophagy-deficient Becn1+/− and littermate WT mice to nHILI between P1 and P7. After hyperoxia exposure, histologic examination of Becn1+/− lungs revealed increased interstitial cellularity in the alveolar septa and larger-appearing alveoli compared with WT lungs, without any discernible differences between the two groups under normoxic conditions (Figure 5A). Morphometric analyses confirmed these initial observations of more severe lung injury with impaired alveolarization and increased alveolar septal thickness in Becn1+/− mice compared with WT mice in response to n2eonatal hyperoxia exposure (Figure 5B). Immunoblot analysis of whole-lung homogenates demonstrated significantly decreased concentrations of p21, Parkin, SPC, and CD31 and significantly increased concentrations of cleaved caspase 3 in Becn1+/− lungs compared with WT lungs at P7 after hyperoxia exposure (Figures 5C and 5D). There were no significant differences in p-S6, p-AMPK, or Pink1 concentrations between the two groups. Decreased p21 and increased cleaved caspase 3 concentrations suggested an increase in the number of cells undergoing apoptotic cell death in Becn1+/− lungs compared with WT lungs, which was confirmed by TUNEL assay (Figures 5E and 5F).
Figure 5.
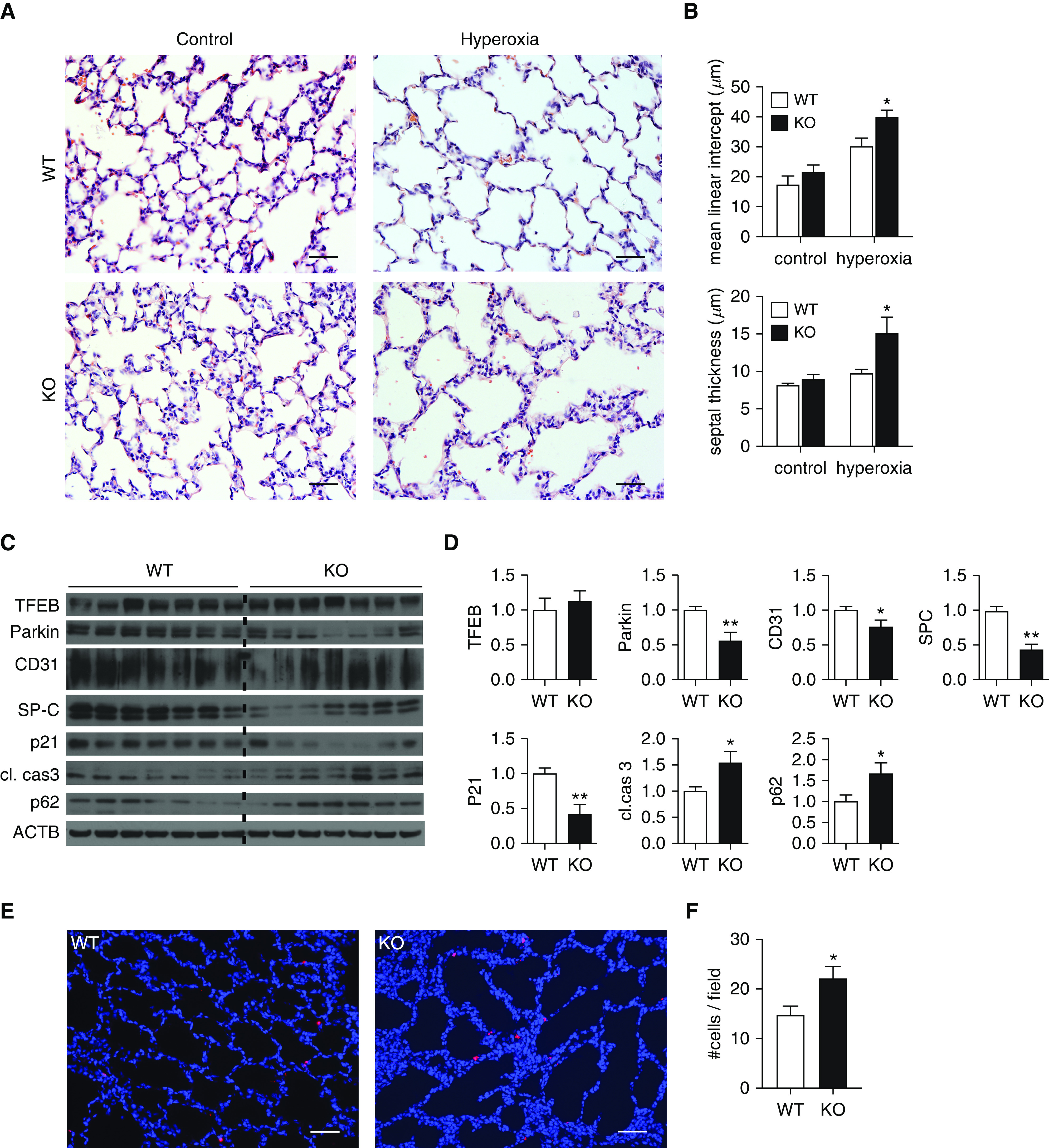
Becn1+/− mice are more susceptible to neonatal hyperoxia-induced lung injury. Becn1+/− (knockout [KO]) and wild-type (WT) littermate control mice (both on C57BL/6 background) were exposed to normoxia (21% O2) or hyperoxia (75% O2) between P1 and P7. At P7, mice were killed, and their lungs were inflation fixed with 10% formalin to 25 cm H2O and embedded in paraffin. (A) Sections were stained with hematoxylin and eosin. (B) Mean linear intercept as a surrogate for alveolar diameter and alveolar septal thickness were quantified using ImageJ and NIS-Elements Basic Research (Nikon Instruments) software, respectively. (C and D) In other cohorts of mice, left lungs were snap frozen, homogenized in radioimmunoprecipitation assay buffer containing protease inhibitors, and analyzed by IB (C) and densitometry (D) for the indicated proteins. (E and F) A TUNEL assay was performed on paraffin-embedded mouse lung tissues (E), and TUNEL-positive cells were quantified (F). Data were obtained from a minimum of three separate litters and are shown as mean ± SEM; n = 7–10 per group. *P < 0.05 and **P < 0.01. Scale bars, 50 μm.
Because autophagy is implicated in regulation of inflammatory responses in lung injury models, we next characterized the effect of Becn1 deficiency on inflammatory cell influx to the airways in nHILI and found a significantly higher number of neutrophils in Becn1+/− BAL samples than in control samples at P7. There was also a trend for an increase in total number of alveolar macrophages in Becn1+/− mice without a statistically significant difference. To determine whether Becn1 deficiency modulated inflammatory signaling in alveolar macrophages, we first assessed expression levels of a panel of genes that regulate inflammatory responses in adhesion-purified alveolar macrophages harvested from BAL of hyperoxia-exposed mice between P1 and P7. We found significant increases (approximately threefold) in the relative mRNA expression levels of the M1 markers pro–IL-1β, pro–IL-18, and CD68 in Becn1+/− alveolar macrophages compared with WT alveolar macrophages (Figure 6C). Because the NLRP3 (NACHT, LRR and PYD domains-containing protein 3) inflammasome catalyzes proteolytic cleavage of pro–IL-1β and pro–IL-18 to their active forms (36) and excessive activation of the NLRP3 inflammasome has been implicated in BPD (37), we next measured caspase 1 activity and IL-1β protein concentrations in whole-lung homogenates as markers of NLRP3 inflammasome activation and found significant increases in both measurements in Becn1+/− mice compared with WT mice. Thus, Becn1+/− alveolar macrophages and lungs display increased expression of proinflammatory genes and enhanced NLRP3 inflammasome activation, respectively, during progression of nHILI.
Figure 6.
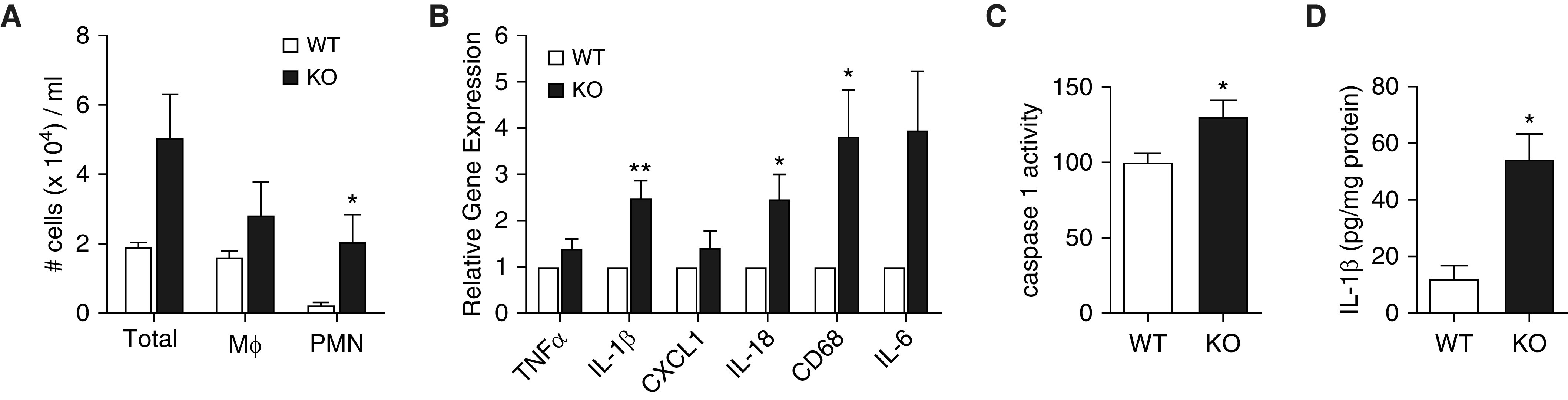
Becn1+/− lungs and alveolar macrophages exhibit enhanced inflammatory signaling. (A) BAL fluid (BALF) was obtained from Becn1+/− (KO) and WT littermates after hyperoxia exposure between P1 and P7. Macrophages (Mϕ) and polymorphonuclear neutrophils (PMN) were enumerated on cytospins stained with Diff-Quik. (B) BALF was obtained from other cohorts of hyperoxia-exposed Becn1+/− (KO) and WT littermate mice, and alveolar macrophages were isolated by adhesion purification. Total RNA was isolated, and qRT-PCR was performed for a panel of genes indicated on the figure. (C) Caspase 1 activity was measured in right lung homogenates from hyperoxia-exposed Becn1+/− (KO) and WT littermates using a fluorescence-based activity assay. Relative caspase 1 activity level was expressed as a percentage of activity detected in WT samples. (D) IL-1β protein was measured in whole-lung lysates from hyperoxia-exposed Becn1+/− (KO) and WT littermates using ELISA. All data were derived from a minimum of three different experiments with n = 6–14 per group. Data are shown as mean ± SEM; *P < 0.05 and **P < 0.01.
Discussion
Despite the availability of several methods for analysis of autophagic flux, the ideal methodology for in vivo monitoring of autophagy remains controversial and needs to be optimized for each experimental setting (29). During the onset of this study, we found it technically challenging to precisely assess autophagic activity by immunoblot detection of LC3-I and -II proteins or by autophagosome quantification in neonatal murine lung tissues, where basal autophagy levels are low compared with other organs. Therefore, we first optimized and validated a practical and sensitive method that involved quantification of GFP concentrations by IB and densitometry of whole-lung homogenates from GFP-LC3 mice. GFP is more resistant to the acidic pH of the lysosomes than LC3, and, although changes in the lysosomal acidity can affect formation of GFP fragments (38), we found GFP concentrations to correlate well with other indicators of autophagic activity, such as the LC3 puncta and p62 concentrations. Nevertheless, we simultaneously analyzed GFP and p62 concentrations in all of our experiments.
On the basis of GFP concentrations, autophagic activity peaked during alveolarization at P7 during postnatal lung development. At this time point, p62 concentrations also reached a nadir, although this did not reach statistical significance, most likely because of a few outliers. It should be noted, however, that other factors besides autophagic degradation, such as transcriptional regulation and availability of amino acids, also regulate p62 protein concentrations, and therefore p62 concentrations do not always demonstrate an inverse correlation with autophagic activity (39, 40). Markers of autophagy were initially increased during short-term hyperoxia exposure up to P3 but then significantly decreased with prolonged hyperoxia exposure at P7. p62 concentrations demonstrated an inverse correlation with GFP concentrations with a slight decrease at P3 and significant increases at P5 and P7 in hyperoxia-exposed lungs as compared with control lungs during progression of nHILI. Other investigators have reported decreased autophagy in hyperoxia-exposed neonatal rat lungs based on increased LC3-II and p62 concentrations and suggested that this might have been caused by impaired fusion of autophagosomes with lysosomes (41). A correlation between endoplasmic reticulum stress and increased LC3-II concentrations during hyperoxia exposure of neonatal rat lungs was also reported (42). Our finding of decreased autophagy in murine nHILI is consistent with these two studies in rat models, despite differences in methods employed for autophagy detection. Our results, however, differ from those of Sureshbabu and colleagues, who reported upregulation of the autophagy pathway in hyperoxia-exposed neonatal murine lungs with use of 100% O2 for 7 days (18).
Our study also provided some novel insights into upstream regulation of autophagic activity in neonatal murine lungs. The initial induction of autophagic activity was accompanied by a significant increase in p-AMPK expression at P3, which subsequently decreased to levels similar to those of normoxic control animals at P5 and P7. AMPK plays a key role in induction of autophagy through direct phosphorylation and activation of ULK1 (33). On the one hand, our findings suggest that insufficient AMPK activation beyond P3 may be an important factor in attenuation of autophagic activity during hyperoxia exposure. On the other hand, the mTORC1 pathway, which is inhibited by AMPK activation and serves as a negative regulator of autophagy through phosphorylation and inactivation of the ULK1–ATG13–FIP200 (focal adhesion kinase family kinase-interacting protein of 200 kDa) kinase complex, showed increased activity at P5 during nHILI. Thus, our studies revealed a novel and distinct pattern of time-dependent, dysregulated autophagic activity during hyperoxia exposure in murine lungs with insufficient activation of AMPK and hyperactivity of mTORC1 pathways. The translational significance of these findings was unequivocally confirmed in the baboon model of “new BPD,” with significantly decreased LC3-II/LC3-I ratios, impaired AMPK activation, and increased p62 protein concentrations. A notable alteration in the baboon model of BPD was the markedly decreased protein concentrations of Parkin, which plays a key role in mitophagy to target the damaged mitochondria for elimination by autophagosomes (43). Interestingly, Parkin concentrations were also decreased in Becn1+/− lungs compared with WT lungs in the nHILI model. Becn1 regulation of Parkin expression was previously described in a murine model of cigarette smoke–induced kidney injury (44). Collectively, our findings suggest that impaired mitophagy contributes to the decreased autophagy signal in experimental models of BPD.
Becn1+/− alveolar macrophages demonstrated increased expression of proinflammatory cytokine genes, such as pro–IL-1β and pro–IL-18, in response to neonatal hyperoxia exposure. There were also increased caspase 1 activity and IL-1β protein concentrations in whole-lung homogenates, consistent with enhanced activation of the NLRP3 inflammasome, which has been implicated in the pathogenesis of BPD (37). Previously, Nakahira and colleagues reported that in vitro treatment of Becn1+/− macrophages with LPS and ATP resulted in increased production of active caspase 1, IL-1β, and IL-18 (45). In contrast, Zhang and colleagues reported that activation of autophagy in macrophages mediates early lung inflammation during mechanical ventilation via NLRP3 inflammasome signaling (46). Thus, our findings are in line with those reported by Nakahira and colleagues and extend their observations to in vivo conditions to strongly suggest that loss of autophagic activity in alveolar macrophages contributes to neonatal hyperoxia-induced lung inflammation.
Becn1 has an antiapoptotic role in several settings, including nutrient deprivation and hypoxia; however, the precise mechanism by which Becn1 inhibits apoptosis is not yet clear (47). In previous studies, siRNA-mediated knockdown of Becn1 increased apoptotic cell death in hyperoxia-exposed MLE-12 cells in a TRP-53 (tumor protein p53)-dependent manner (18), and conditional deletion of Becn1 in mouse lung epithelial cells increased apoptotic cell death in late-gestation mouse lungs (17). We also found an increased number of apoptotic cells in hyperoxia-exposed Becn1+/− lungs in association with decreased p21 concentrations. In previous studies, neonatal hyperoxia exposure of p21−/− mice disrupted alveolarization (48), and overexpression of p21 delayed loss of antiapoptotic proteins during hyperoxia (49). Thus, decreased p21 concentrations in Becn1+/− mouse lungs, at least in part, may account for the increased amounts of apoptosis. The cross-talk between autophagy and apoptosis is known to be highly complex and context dependent (50, 51). Our findings in both murine and baboon models suggest that neonatal hyperoxia sequentially induces autophagy at early time points and low doses of stress and induces apoptosis at later time points and higher doses of stress. Further studies are needed to delineate the precise regulation and time course of the interplay between these two critical processes and inflammatory signaling in evolving BPD with an eye toward rapid translation. In this regard, it is tempting to speculate that the beneficial effects of caffeine, a widely used drug in the neonatal ICU, on BPD prevention (52) may be mediated, at least in part, through stimulation of autophagy (53). Conversely, some caution may be warranted in use of chloroquine, a strong inhibitor of autophagy, for empirical treatment of severe BPD.
It is intriguing that, among lung-resident cells, the expression of autophagy proteins is primarily restricted to AEC2s and that, despite such a restricted expression pattern and the incomplete autophagy deficiency in Becn1+/− mice, these lungs displayed significant structural perturbations involving not just epithelial but also mesenchymal cells, as evidenced by increased alveolar septal thickness and decreased CD31 expression levels, when exposed to nHILI. These findings suggest that the effects of autophagy may be mediated by not just autocrine but also paracrine mechanisms in the developing lung.
In conclusion, autophagic activity is regulated in a time-dependent manner during nHILI and is impaired in two different experimental models of BPD. Autophagy-deficient mice demonstrate greater lung injury and increased inflammation in nHILI. The extent of lung pathology in these animals suggests that combined autocrine and paracrine effects of autophagy deficiency in AEC2s and alveolar macrophages contribute to this phenotype. Further studies are needed to delineate the cell-type–specific effects of autophagy and its key upstream regulator AMPK as they emerge as attractive novel therapeutic targets in BPD pathogenesis.
Supplementary Material
Acknowledgments
Acknowledgment
The authors thank Joanne Du for technical assistance, Dr. Beth Levine for Becn1+/− mice, and Dr. Noboru Mizushima for GFP-LC3–transgenic mice.
Footnotes
Supported by U.S. National Institutes of Health grants 5R21HD092934 (S.Ç.), U01HL075904 (S.Ç.), and P51RR13986 (facility support).
Author Contributions: Conception/design: L.Z. and S.Ç. Collection and assembly of data: L.Z., S.S., E.H., and S.Ç. Experimental work: L.Z., S.S., E.H., and S.Ç. Data analysis and interpretation: L.Z., S.B., and S.Ç. Manuscript writing: L.Z., S.S., S.B., and S.Ç.
This article has a data supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1165/rcmb.2019-0445OC on May 6, 2020
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.Abman SH, Bancalari E, Jobe A. The evolution of bronchopulmonary dysplasia after 50 years. Am J Respir Crit Care Med. 2017;195:421–424. doi: 10.1164/rccm.201611-2386ED. [DOI] [PubMed] [Google Scholar]
- 2.Higgins RD, Jobe AH, Koso-Thomas M, Bancalari E, Viscardi RM, Hartert TV, et al. Bronchopulmonary dysplasia: executive summary of a workshop. J Pediatr. 2018;197:300–308. doi: 10.1016/j.jpeds.2018.01.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stoll BJ, Hansen NI, Bell EF, Walsh MC, Carlo WA, Shankaran S, et al. Eunice Kennedy Shriver National Institute of Child Health and Human Development Neonatal Research Network. Trends in care practices, morbidity, and mortality of extremely preterm neonates, 1993–2012. JAMA. 2015;314:1039–1051. doi: 10.1001/jama.2015.10244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Abman SH. The dysmorphic pulmonary circulation in bronchopulmonary dysplasia: a growing story. Am J Respir Crit Care Med. 2008;178:114–115. doi: 10.1164/rccm.200804-629ED. [DOI] [PubMed] [Google Scholar]
- 5.Jobe AH. The new bronchopulmonary dysplasia. Curr Opin Pediatr. 2011;23:167–172. doi: 10.1097/MOP.0b013e3283423e6b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mandell EW, Kratimenos P, Abman SH, Steinhorn RH. Drugs for the prevention and treatment of bronchopulmonary dysplasia. Clin Perinatol. 2019;46:291–310. doi: 10.1016/j.clp.2019.02.011. [DOI] [PubMed] [Google Scholar]
- 7.Yang Z, Klionsky DJ. Eaten alive: a history of macroautophagy. Nat Cell Biol. 2010;12:814–822. doi: 10.1038/ncb0910-814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Parzych KR, Klionsky DJ. An overview of autophagy: morphology, mechanism, and regulation. Antioxid Redox Signal. 2014;20:460–473. doi: 10.1089/ars.2013.5371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med. 2013;368:1845–1846. doi: 10.1056/NEJMc1303158. [DOI] [PubMed] [Google Scholar]
- 11.Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature. 2011;469:323–335. doi: 10.1038/nature09782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, et al. The role of autophagy during the early neonatal starvation period. Nature. 2004;432:1032–1036. doi: 10.1038/nature03029. [DOI] [PubMed] [Google Scholar]
- 13.Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol. 2005;169:425–434. doi: 10.1083/jcb.200412022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sou YS, Waguri S, Iwata J, Ueno T, Fujimura T, Hara T, et al. The Atg8 conjugation system is indispensable for proper development of autophagic isolation membranes in mice. Mol Biol Cell. 2008;19:4762–4775. doi: 10.1091/mbc.E08-03-0309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yue Z, Jin S, Yang C, Levine AJ, Heintz N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci USA. 2003;100:15077–15082. doi: 10.1073/pnas.2436255100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cheong H, Wu J, Gonzales LK, Guttentag SH, Thompson CB, Lindsten T. Analysis of a lung defect in autophagy-deficient mouse strains. Autophagy. 2014;10:45–56. doi: 10.4161/auto.26505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yeganeh B, Lee J, Ermini L, Lok I, Ackerley C, Post M. Autophagy is required for lung development and morphogenesis. J Clin Invest. 2019;129:2904–2919. doi: 10.1172/JCI127307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sureshbabu A, Syed M, Das P, Janér C, Pryhuber G, Rahman A, et al. Inhibition of regulatory-associated protein of mechanistic target of rapamycin prevents hyperoxia-induced lung injury by enhancing autophagy and reducing apoptosis in neonatal mice. Am J Respir Cell Mol Biol. 2016;55:722–735. doi: 10.1165/rcmb.2015-0349OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yeganeh B, Lee J, Bilodeau C, Lok I, Ermini L, Ackerley C, et al. Acid sphingomyelinase inhibition attenuates cell death in mechanically ventilated newborn rat lung. Am J Respir Crit Care Med. 2019;199:760–772. doi: 10.1164/rccm.201803-0583OC. [DOI] [PubMed] [Google Scholar]
- 20.Mizushima N. Methods for monitoring autophagy using GFP-LC3 transgenic mice. Methods Enzymol. 2009;452:13–23. doi: 10.1016/S0076-6879(08)03602-1. [DOI] [PubMed] [Google Scholar]
- 21.Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell. 2004;15:1101–1111. doi: 10.1091/mbc.E03-09-0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A, et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest. 2003;112:1809–1820. doi: 10.1172/JCI20039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Altiok O, Yasumatsu R, Bingol-Karakoc G, Riese RJ, Stahlman MT, Dwyer W, et al. Imbalance between cysteine proteases and inhibitors in a baboon model of bronchopulmonary dysplasia. Am J Respir Crit Care Med. 2006;173:318–326. doi: 10.1164/rccm.200503-425OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Coalson JJ, Winter VT, Siler-Khodr T, Yoder BA. Neonatal chronic lung disease in extremely immature baboons. Am J Respir Crit Care Med. 1999;160:1333–1346. doi: 10.1164/ajrccm.160.4.9810071. [DOI] [PubMed] [Google Scholar]
- 25.Yoder BA, Coalson JJ. Animal models of bronchopulmonary dysplasia: the preterm baboon models. Am J Physiol Lung Cell Mol Physiol. 2014;307:L970–L977. doi: 10.1152/ajplung.00171.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liang X, Gupta K, Quintero JR, Cernadas M, Kobzik L, Christou H, et al. Macrophage FABP4 is required for neutrophil recruitment and bacterial clearance in Pseudomonas aeruginosa pneumonia. FASEB J. 2019;33:3562–3574. doi: 10.1096/fj.201802002R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Harijith A, Choo-Wing R, Cataltepe S, Yasumatsu R, Aghai ZH, Janér J, et al. A role for matrix metalloproteinase 9 in IFNγ-mediated injury in developing lungs: relevance to bronchopulmonary dysplasia. Am J Respir Cell Mol Biol. 2011;44:621–630. doi: 10.1165/rcmb.2010-0058OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ghelfi E, Karaaslan C, Berkelhamer S, Akar S, Kozakewich H, Cataltepe S. Fatty acid-binding proteins and peribronchial angiogenesis in bronchopulmonary dysplasia. Am J Respir Cell Mol Biol. 2011;45:550–556. doi: 10.1165/rcmb.2010-0376OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition) Autophagy. 2016;12:1–222. doi: 10.1080/15548627.2015.1100356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhou B, Kreuzer J, Kumsta C, Wu L, Kamer KJ, Cedillo L, et al. Mitochondrial permeability uncouples elevated autophagy and lifespan extension. Cell. 2019;177:299–314, e16. doi: 10.1016/j.cell.2019.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140:313–326. doi: 10.1016/j.cell.2010.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mund SI, Stampanoni M, Schittny JC. Developmental alveolarization of the mouse lung. Dev Dyn. 2008;237:2108–2116. doi: 10.1002/dvdy.21633. [DOI] [PubMed] [Google Scholar]
- 33.Mihaylova MM, Shaw RJ. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol. 2011;13:1016–1023. doi: 10.1038/ncb2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim YC, Guan KL. mTOR: a pharmacologic target for autophagy regulation. J Clin Invest. 2015;125:25–32. doi: 10.1172/JCI73939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rodger CE, McWilliams TG, Ganley IG. Mammalian mitophagy: from in vitro molecules to in vivo models. FEBS J. 2018;285:1185–1202. doi: 10.1111/febs.14336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Man SM, Kanneganti TD. Regulation of inflammasome activation. Immunol Rev. 2015;265:6–21. doi: 10.1111/imr.12296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liao J, Kapadia VS, Brown LS, Cheong N, Longoria C, Mija D, et al. The NLRP3 inflammasome is critically involved in the development of bronchopulmonary dysplasia. Nat Commun. 2015;6:8977. doi: 10.1038/ncomms9977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ni HM, Bockus A, Wozniak AL, Jones K, Weinman S, Yin XM, et al. Dissecting the dynamic turnover of GFP-LC3 in the autolysosome. Autophagy. 2011;7:188–204. doi: 10.4161/auto.7.2.14181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sahani MH, Itakura E, Mizushima N. Expression of the autophagy substrate SQSTM1/p62 is restored during prolonged starvation depending on transcriptional upregulation and autophagy-derived amino acids. Autophagy. 2014;10:431–441. doi: 10.4161/auto.27344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu WJ, Ye L, Huang WF, Guo LJ, Xu ZG, Wu HL, et al. p62 links the autophagy pathway and the ubiqutin-proteasome system upon ubiquitinated protein degradation. Cell Mol Biol Lett. 2016;21:29. doi: 10.1186/s11658-016-0031-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang D, Wu L, Du Y, Zhu Y, Pan B, Xue X, et al. Autophagy inducers restore impaired autophagy, reduce apoptosis, and attenuate blunted alveolarization in hyperoxia-exposed newborn rats. Pediatr Pulmonol. 2018;53:1053–1066. doi: 10.1002/ppul.24047. [DOI] [PubMed] [Google Scholar]
- 42.Li M, Pan B, Shi Y, Fu J, Xue X. Increased expression of CHOP and LC3B in newborn rats with bronchopulmonary dysplasia. Int J Mol Med. 2018;42:1653–1665. doi: 10.3892/ijmm.2018.3724. [DOI] [PubMed] [Google Scholar]
- 43.Pickles S, Vigié P, Youle RJ. Mitophagy and quality control mechanisms in mitochondrial maintenance. Curr Biol. 2018;28:R170–R185. doi: 10.1016/j.cub.2018.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pabón MA, Patino E, Bhatia D, Rojas-Quintero J, Ma KC, Finkelsztein EJ, et al. Beclin-1 regulates cigarette smoke-induced kidney injury in a murine model of chronic obstructive pulmonary disease. JCI Insight. 2018;3:99592. doi: 10.1172/jci.insight.99592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol. 2011;12:222–230. doi: 10.1038/ni.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang Y, Liu G, Dull RO, Schwartz DE, Hu G. Autophagy in pulmonary macrophages mediates lung inflammatory injury via NLRP3 inflammasome activation during mechanical ventilation. Am J Physiol Lung Cell Mol Physiol. 2014;307:L173–L185. doi: 10.1152/ajplung.00083.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kang R, Zeh HJ, Lotze MT, Tang D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011;18:571–580. doi: 10.1038/cdd.2010.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McGrath-Morrow SA, Cho C, Soutiere S, Mitzner W, Tuder R. The effect of neonatal hyperoxia on the lung of p21Waf1/Cip1/Sdi1-deficient mice. Am J Respir Cell Mol Biol. 2004;30:635–640. doi: 10.1165/rcmb.2003-0049OC. [DOI] [PubMed] [Google Scholar]
- 49.Wu YC, O’Reilly MA. Bcl-XL is the primary mediator of p21 protection against hyperoxia-induced cell death. Exp Lung Res. 2011;37:82–91. doi: 10.3109/01902148.2010.521617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mariño G, Niso-Santano M, Baehrecke EH, Kroemer G. Self-consumption: the interplay of autophagy and apoptosis. Nat Rev Mol Cell Biol. 2014;15:81–94. doi: 10.1038/nrm3735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu B, Oltvai ZN, Bayır H, Silverman GA, Pak SC, Perlmutter DH, et al. Quantitative assessment of cell fate decision between autophagy and apoptosis. Sci Rep. 2017;7:17605. doi: 10.1038/s41598-017-18001-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schmidt B, Roberts RS, Davis P, Doyle LW, Barrington KJ, Ohlsson A, et al. Caffeine for Apnea of Prematurity Trial Group. Caffeine therapy for apnea of prematurity. N Engl J Med. 2006;354:2112–2121. doi: 10.1056/NEJMoa054065. [DOI] [PubMed] [Google Scholar]
- 53.Pietrocola F, Malik SA, Mariño G, Vacchelli E, Senovilla L, Chaba K, et al. Coffee induces autophagy in vivo. Cell Cycle. 2014;13:1987–1994. doi: 10.4161/cc.28929. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.