

To the Editor:
Coronavirus disease (COVID-19), the pandemic caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection, can lead to sepsis and acute respiratory distress syndrome (ARDS), resulting in an extraordinary level of ICU use and considerable mortality. Several pathophysiological features of COVID-19–associated ARDS appear to be overrepresented in comparison with non-COVID etiologies. Whether COVID-19–induced lung injury is truly unique or represents one end of the ARDS spectrum remains unclear at this time. With the caveat that studies are ongoing, and appropriately powered studies are needed, the observations discussed here implicate vascular dysfunction in the pathogenesis of COVID-19–induced ARDS, leading to the hypothesis that COVID-ARDS is a distinct vascular endotype of ARDS (Figure 1).
Figure 1.
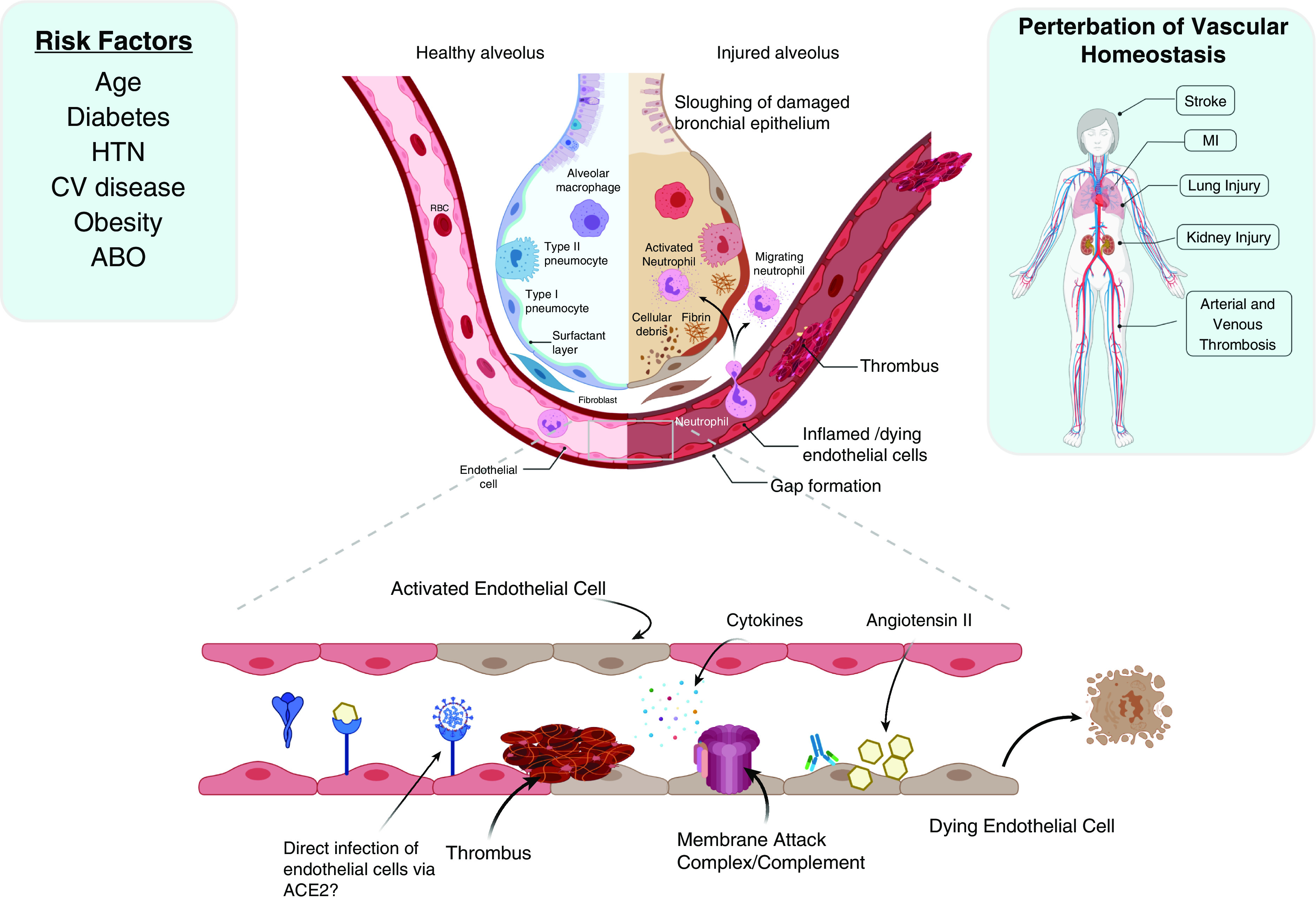
Risk factors and disease manifestations of coronavirus disease (COVID-19) suggest the central involvement of the pulmonary vasculature. Although some risk factors (age and obesity) are common for undifferentiated acute respiratory distress syndrome, others, including cardiovascular disease and diabetes, are overrepresented with regard to severe COVID-19. Regarding viral pathogenesis at the tissue level, the epithelium represents the primary cell type affected as with most respiratory viruses. However, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection is notable for the induction of microthrombi. Multiple mechanisms are likely to promote endothelial injury, including viral competition for ACE2 (angiotensin-converting enzyme 2) binding (thus increasing angiotensin II) and direct activation of complement by SARS-CoV-2 structural proteins, in addition to elevated cytokines and complement activation and cell death observed in acute respiratory distress syndrome and sepsis. These vascular perturbations likely contribute to systemic thrombosis and organ injury in susceptible hosts. CV = cardiovascular; HTN = hypertension; MI = myocardial infarction; RBC = red blood cell.
In recently published reports, ARDS related to COVID-19 often presents with relatively preserved compliance (1). In these cases, the static compliance has been 30–40 ml/cm H2O despite FiO2 > 70% and positive end-expiratory pressure >15 cm H2O. Thus, although the compliance is abnormal and consistent with the ARDS conceptual model articulated in the Berlin definition (2), compliance is less impacted than oxygenation and dead space in COVID-ARDS.
Early reports of COVID-19 patient cohorts from China and Italy identified several risk factors for severe illness and death. Some of these risk factors are unsurprising as they are also risk factors for non-COVID ARDS, including age and obesity. However, others appear to be overrepresented in patients who develop COVID-ARDS, suggesting a distinct ARDS endotype. Strikingly, diabetes is a risk factor for COVID-ARDS whereas it is a negative predictor in the Lung Injury Prediction Score for non-COVID ARDS (3). Cardiovascular disease (including hypertension and hyperlipidemia) are common among critically ill patients with COVID-19 ARDS, although they have not previously been reported as ARDS risk factors, highlighting what may be a prominent role for underlying vascular dysfunction in this subtype.
ABO blood group antigens are expressed on vascular endothelium. Blood type A has been associated with increased risk of vascular disease and possibly ARDS. Recently, the first published genome-wide association study of COVID-19 identified an association between genetic variation that determines ABO blood type and severe COVID-19 disease (4). Specifically, ABO blood type A was overrepresented and blood type O was underrepresented in patients with COVID-19 relative to blood donors. Findings were inconclusive for types B and AB given the smaller population prevalence of these blood types. This is consistent with reports of an association between blood type A and increased risk of infection with SARS-CoV-1 (5). Although the mechanism underlying this association is unknown, ABO blood type A is also associated with a higher risk of multiple thrombotic diseases including myocardial infarction, stroke, and venous thromboembolism, as well as higher plasma concentrations of endothelial-derived proteins important in microvascular coagulation and cell adhesion. Collectively, these observations suggest that blood type A and chronic conditions such as diabetes and cardiovascular disease may prime the endothelium for injury when faced with SARS-CoV-2, thereby lowering the threshold for infection to progress to organ failure including ARDS, kidney injury, and shock.
In addition to facilitating gas exchange and performing critical barrier functions, the endothelium regulates leukocyte trafficking, hemostasis, and vascular tone. The pulmonary microvascular endothelium is unique in that it filters the entire systemic circulation and is routinely exposed to noxious stimuli including bloodborne pathogens, toxins, and endogenous inflammatory mediators. Maintenance of endothelial quiescence under basal conditions is essential to lung homeostasis, and endothelial protective mechanisms promote this antiinflammatory phenotype. Although most respiratory viruses do not infect endothelial cells directly, the inflammatory response induced by these pathogens can cause significant injury to the vasculature. Inflammation-induced disruption of homeostatic endothelial functions can result in impaired diffusion, disrupted barrier function, aberrant coagulation, and increased permeability. Perturbation of endothelial homeostasis in patients with chronic diseases may predispose these susceptible populations to organ failure in response to vascular injury induced by SARS-CoV-2. This is consistent with the finding that thrombosis and kidney injury are predominant features of COVID-19 in susceptible populations. Multiple autopsy studies now confirm the involvement of the endothelium in COVID-ARDS, demonstrating microvascular thrombi, vascular complement deposition, possible direct endothelial infection, and endothelial cell death (Table 1) (6, 7). Furthermore, aberrant endothelial cell death and dysregulated angiogenesis are observed in COVID-ARDS when compared with influenza-associated ARDS (8).
Table 1.
Potential Vascular Complications in Critically Ill Patients with COVID-19
| Complication | Organ Affected | |
|---|---|---|
| Macrovascular | ||
| Venous | Deep vein thrombosis | Extremities, pelvic |
| Pulmonary embolism | Lung | |
| In situ pulmonary thrombosis* | Lung | |
| Arterial | Stroke | CNS |
| Myocardial infarction | Heart | |
| Mesenteric ischemia | Gut | |
| Limb Ischemia | Extremities | |
| Microvascular | Thrombosis | Lung, heart, intestines, kidneys, and skin |
| Extracorporeal | ECMO oxygenator clotting | N/A |
| Renal replacement filter clotting | N/A |
Definition of abbreviations: CNS = central nervous system; COVID-19 = coronavirus disease; ECMO = extracorporeal membrane oxygenation; N/A = not applicable.
Vascular complications in patients with COVID-19 that have been reported in the literature and organs involved (both reported and extrapolated).
Postulated mechanism that is not reported in the literature as of this writing.
One possible contributing factor to this vascular ARDS phenotype may be the SARS-CoV and CoV-2 receptor, ACE2 (angiotensin-converting enzyme 2). ACE2 is a key player in the renin–angiotensin system responsible for regulating vascular tone. Angiotensin II acts on a variety of target cells to produce acute and long-term physiological effects, including vasoconstriction, sympathetic nervous stimulation, smooth muscle and fibroblast proliferation, and inflammation. ACE2 counteracts angiotensin II activity by catalyzing its proteolytic cleavage into angiotensin (1–7), which counteracts acute lung injury.
As the viral receptor, it might be expected that higher levels of ACE2 would result in more severe disease. However, studies after the original SARS outbreak indicate the opposite, as ACE2 knockout mice exhibit much more severe lung injury after acid aspiration, whereas administration of recombinant ACE2 is protective. Moreover, binding of SARS-CoV Spike protein to ACE2 resulted in a loss of ACE2 protein, and administration of recombinant Spike-Fc protein worsened lung injury by increasing angiotensin II activity (9), presumably owing to competition for available ACE2. These studies were performed with the original SARS-CoV Spike protein, so it is not certain whether CoV-2 Spike would have similar effects. Nonetheless, they provide significant rationale for some of the pathophysiological differences observed with COVID-19 ARDS, and clinical trials using recombinant ACE2 and angiotensin (1–7) to treat COVID-19 are ongoing. Loss of ACE2 repression of angiotensin II activity promotes microvascular thrombosis through direct and indirect means (10), and prolonged vasoconstriction and hypertension are well known to induce endothelial injury. Recent autopsy reports demonstrating direct endothelial injury may be mediated by this dysregulation of the renin–angiotensin system.
Another potential mechanism of vascular injury contributing to ARDS and kidney injury involves dysregulated complement activation. The complement system serves as a first-line defense against pathogens and is essential for the removal of dead cells. Although the effector functions of opsonization, inflammation, chemotaxis, and cytolysis promote pathogen clearance, dysregulated or excessive complement activation can lead to tissue injury and organ failure, one of the clearest examples being the prothrombotic and anaphylatoxic effects of activated complement component 5.
Cytokine release and complement activation have long been implicated in organ failure and ARDS in sepsis (11). Although cytokine levels are comparable with non-COVID ARDS (medRxiv preprint DOI: https://doi.org/10.1101/2020.05.15.20103549), complement-mediated damage to the lung microvascular endothelial cells appears to be a predominant feature of COVID-ARDS, whereas direct comparisons with non-COVID ARDS have not been published as of this writing (6). Preclinical studies demonstrate that the nucleocapsid protein of several coronaviruses, including SARS-CoV-2, binds directly to and activates MASP-2, a key protease in the lectin pathway of complement. In murine studies of SARS-CoV–induced lung injury, mice deficient in C3 were relatively protected from lung injury following SARS-CoV infection and exhibited less lung neutrophil recruitment and lower levels of cytokines in the lungs and circulation (12).
The alternative pathway of complement activation is always “on,” requiring tight regulation by soluble and membrane-bound complement regulatory proteins to protect the endothelium. Medical conditions such as diabetes, among others identified as risk factors for SARS-CoV-2 mortality, leads to dysfunctional endothelial complement regulatory proteins, thereby increasing susceptibility to complement-induced endothelial damage. Complement activation and dysregulation of the renin–angiotensin system may be most severe within viral damaged lung vasculature but may also contribute to the pathogenesis of strokes, myocardial and mesenteric ischemia, and cutaneous lesions owing to limb ischemia. Given the atypical vascular-centric risk factors for COVID-ARDS, it is plausible that complement activation and dysregulated ACE2-angiotensin repression in susceptible hosts might lead to widespread endothelial dysfunction.
Despite the best care and implementation of lung protective strategies, the mortality for COVID-ARDS remains high. Given the many indications pointing toward vascular involvement, vascular-centric, endothelial protective therapies should be considered as adjuncts in the treatment of COVID-ARDS. Although no previous medical therapy has improved sepsis or ARDS mortality, there is reason to believe COVID-ARDS may be unique. Unlike most sepsis-associated ARDS, both the timing and pathogen are known in COVID-ARDS. Additionally, the higher incidence of vascular manifestations should justify consideration of COVID-ARDS as a distinct endotype with prominent vascular dysfunction (13). Specific vascular targeting may present a unique opportunity to intervene. In view of the potential for targeted therapies of the complement pathway, supplements, or alternatives to heparin as an antithrombotic, and endothelial protective therapies such as nitric oxide, corticosteroids, and statins to restore endothelial homeostasis, a comprehensive molecular understanding of vascular endothelial dysfunction in COVID-ARDS is urgently needed. Although we still do not know enough to definitively classify COVID-ARDS as a vascular endotype, COVID-ARDS may be an extreme example of a phenotype present in the more general population of ARDS, and investigations into the dysregulated immune response in the vasculature may advance the understanding and treatment of all forms of ARDS.
Some of the results of these studies have been previously reported in the form of a preprint (OSFPrePrints, 24 April 2020 https://osf.io/ckdpe/).
Supplementary Material
Footnotes
Supported by NHLBI 126788 (N.S.M.), NHLBI 131817 (A.E.V.), NHLBI 137006 and NHLBI 137915 (N.J.M.), and NHLBI 125723 (J.P.R.).
Originally Published in Press as DOI: 10.1164/rccm.202006-2598LE on July 6, 2020
Author disclosures are available with the text of this letter at www.atsjournals.org.
References
- 1.Ziehr DR, Alladina J, Petri CR, Maley JH, Moskowitz A, Medoff BD, et al. Respiratory pathophysiology of mechanically ventilated patients with COVID-19: a cohort study. Am J Respir Crit Care Med. 2020;201:1560–1564. doi: 10.1164/rccm.202004-1163LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ranieri VM, Rubenfeld GD, Thompson BT, Ferguson ND, Caldwell E, Fan E, et al. ARDS Definition Task Force. Acute respiratory distress syndrome: the Berlin definition. JAMA. 2012;307:2526–2533. doi: 10.1001/jama.2012.5669. [DOI] [PubMed] [Google Scholar]
- 3.Soto GJ, Kor DJ, Park PK, Hou PC, Kaufman DA, Kim M, et al. Lung injury prediction score in hospitalized patients at risk of acute respiratory distress syndrome. Crit Care Med. 2016;44:2182–2191. doi: 10.1097/CCM.0000000000002001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ellinghaus D, Degenhardt F, Bujanda L, Buti M, Albillos A, Invernizzi P, et al. Severe Covid-19 GWAS Group. Genomewide association study of severe Covid-19 with respiratory failure. N Engl J Med. doi: 10.1056/NEJMoa2020283. [online ahead of print] 17 Jun 2020; DOI: 10.1056/NEJMoa2020283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cheng Y, Cheng G, Chui CH, Lau FY, Chan PK, Ng MH, et al. ABO blood group and susceptibility to severe acute respiratory syndrome. JAMA. 2005;293:1450–1451. doi: 10.1001/jama.293.12.1450-c. [Published erratum appears in JAMA 294:794.] [DOI] [PubMed] [Google Scholar]
- 6.Magro C, Mulvey JJ, Berlin D, Nuovo G, Salvatore S, Harp J, et al. Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: a report of five cases. Transl Res. 2020;220:1–13. doi: 10.1016/j.trsl.2020.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Varga Z, Flammer AJ, Steiger P, Haberecker M, Andermatt R, Zinkernagel AS, et al. Endothelial cell infection and endotheliitis in COVID-19. Lancet. 2020;395:1417–1418. doi: 10.1016/S0140-6736(20)30937-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ackermann M, Verleden SE, Kuehnel M, Haverich A, Welte T, Laenger F, et al. Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in Covid-19. N Engl J Med. 2020;383:120–128. doi: 10.1056/NEJMoa2015432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kuba K, Imai Y, Rao S, Gao H, Guo F, Guan B, et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat Med. 2005;11:875–879. doi: 10.1038/nm1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brown NJ, Vaughan DE. Role of angiotensin ii in coagulation and fibrinolysis. Heart Fail Rev. 1999;3:193–198. [Google Scholar]
- 11.Bain W, Li H, van der Geest R, Moore SR, Olonisakin TF, Ahn B, et al. Increased alternative complement pathway function and improved survival during critical illness. Am J Respir Crit Care Med. 2020;202:230–240. doi: 10.1164/rccm.201910-2083OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gralinski LE, Sheahan TP, Morrison TE, Menachery VD, Jensen K, Leist SR, et al. Complement activation contributes to severe acute respiratory syndrome coronavirus pathogenesis. mBio. 2018;9:e01753-18. doi: 10.1128/mBio.01753-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Helms J, Tacquard C, Severac F, Leonard-Lorant I, Ohana M, Delabranche X, et al. CRICS TRIGGERSEP Group (Clinical Research in Intensive Care and Sepsis Trial Group for Global Evaluation and Research in Sepsis) High risk of thrombosis in patients with severe SARS-CoV-2 infection: a multicenter prospective cohort study. Intensive Care Med. 2020;46:1089–1098. doi: 10.1007/s00134-020-06062-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.