

Abstract
Purpose
Several hundred genetic muscle diseases have been described, all of which are rare. Their clinical and genetic heterogeneity means that a genetic diagnosis is challenging. We established an international consortium, MYO-SEQ, to aid the work-ups of muscle disease patients and to better understand disease etiology.
Methods
Exome sequencing was applied to 1001 undiagnosed patients recruited from more than 40 neuromuscular disease referral centers; standardized phenotypic information was collected for each patient. Exomes were examined for variants in 429 genes associated with muscle conditions.
Results
We identified suspected pathogenic variants in 52% of patients across 87 genes. We detected 401 novel variants, 116 of which were recurrent. Variants in CAPN3, DYSF, ANO5, DMD, RYR1, TTN, COL6A2, and SGCA collectively accounted for over half of the solved cases; while variants in newer disease genes, such as BVES and POGLUT1, were also found. The remaining well-characterized unsolved patients (48%) need further investigation.
Conclusion
Using our unique infrastructure, we developed a pathway to expedite muscle disease diagnoses. Our data suggest that exome sequencing should be used for pathogenic variant detection in patients with suspected genetic muscle diseases, focusing first on the most common disease genes described here, and subsequently in rarer and newly characterized disease genes.
Keywords: next-generation sequencing, targeted exome analysis, neuromuscular disease, limb-girdle weakness, genetic diagnosis
INTRODUCTION
Genetic muscle diseases with limb-girdle weakness (LGW) are highly heterogeneous, rare neuromuscular disorders (NMD). In addition to the characteristic progressive pelvic and shoulder girdle muscle weakness, other manifestations may include respiratory insufficiency, cardiomyopathy, contractures, and gastrointestinal complications.1 Consequently, these disorders diminish an individual’s quality of life and can reduce life expectancy.2 There is a critical need to fully characterize disease etiology to achieve accurate diagnoses and offer appropriate genetic counseling and disease management.
Despite 535 genes known to be associated with 955 distinct NMDs,3 many muscle disease patients still remain genetically undiagnosed. This is partly because gene-by-gene and small panel testing strategies are usually dictated by phenotype, leaving little power for the expansion of disease associations and novel disease characterizations.4 An unbiased methodology has thus been sought to enhance standard clinical practices. Consistent with an increased affordability over the past decade, next-generation sequencing (NGS) technologies have been integral in the diagnosis of disorders such as mitochondrial disease among others.5 Large-scale NGS panels were applied to 504 Italian undiagnosed muscular dystrophy and myopathy patients,6 and more recently to 4655 limb-girdle muscular dystrophy (LGMD) patients in the United States.7 By investigating 93 and 35 NMD genes, respectively, they achieved a diagnostic rate of 43% and 27%. However, to date no study has been carried out where exome sequencing (ES) has been applied to similar limb-girdle cohorts; this allowed us not only to determine the impact that the panel size has on the detection rate, but also to interrogate the exomes beyond the known NMD genes.
We describe here the application of sequential targeted exome sequencing (TES) to a large cohort of undiagnosed patients presenting with proximal muscle weakness and/or elevated serum creatine kinase (CK) levels: the MYO-SEQ project. We established a partnership between academia, industry, and patient organizations, and developed an international network that connected over 40 specialist neuromuscular referral sites. A total of 1001 exomes were first analyzed for rare pathogenic variants in 169 common NMD genes, and then in a further 260 genes associated with rarer muscle conditions. We identify the most common muscle diseases in this population and suggest that an increased number of patients can be more rapidly tested and diagnosed through an appropriate TES approach.
MATERIALS AND METHODS
Patient recruitment
We utilized the infrastructure established by TREAT-NMD (https://treat-nmd.org/) to recruit 43 NMD referral centers from throughout Europe and the Middle East (Figure S1). Singleton patients matching the inclusion criteria of unexplained limb-girdle weakness and/or elevated CK were included in the project. Informed consent was obtained for all patients. Anonymized clinical information was entered on the PhenoTips platform (https://phenotips.org/) using NMD-specific forms. Clinical features, histological findings, electromyography results, CK levels, mode of inheritance, and disease onset and progression were recorded using Human Phenotype Ontology terms. DNA samples were submitted to the Newcastle Medical Research Council (MRC) Centre Biobank for Neuromuscular Diseases for which ethical approval was granted by the National Research Ethics Service (NRES) Committee North East–Newcastle & North Tyneside 1 (reference 08/H0906/28).
Exome sequencing, data analysis, and interpretation
ES was performed at the Broad Institute of MIT and Harvard’s Genomics Platform (Cambridge, MA, USA) using a 38-Mb targeted Illumina exome capture, as described previously.8 Data were first filtered for variants in 169 NMD-causing genes (Table S1), with a minor allele frequency in the ExAC control population9 of ≤1% and a moderate to high Ensembl Variant Effect Predictor. The resulting shortlists of single-nucleotide variants (SNVs) and small insertions/deletions (indels) were examined for pathogenicity by two independent data analysts (M.B./J.D./K.J./M.M./A.T. at the John Walton Muscular Dystrophy Research Centre [JWMDRC] and L.X./E.M.E./E.V./T.M. at the Broad Institute). We deemed SNVs and indels pathogenic if they had been previously and consistently reported as pathogenic in ClinVar (two gold stars review status; https://www.ncbi.nlm.nih.gov/clinvar/) and/or published literature. In line with American College of Medical Genetics and Genomics (ACMG) guidelines,10 pathogenicity of novel variants was evaluated based on (1) computational data, i.e., predicted to be deleterious by all, or the majority, of the in silico tools used; (2) population data, i.e., have a frequency in the control population compatible with the variant being rare disease-causing, according to its inheritance mode; and (3) allelic data, i.e., be in trans with a pathogenic variant; however, as phase could rarely be established for putative compound heterozygous cases we refer to these as double heterozygous. Variants of uncertain significance are not discussed here. To be considered solved, a patient was required to carry two known or suspected pathogenic variants in a recessive gene, or (at least) one known or suspected pathogenic variant in a dominant gene. By suspected pathogenic variants we refer to rare and damaging variants identified in a gene associated with a disease that matches the patients’ clinical presentation, disease onset and progression, magnetic resonance image (MRI) findings, and/or muscle histopathology. The findings were disclosed to the submitting centers in tailored reports, signposting the patients to relevant disease registries. Where necessary and possible, the suspected pathogenic variants were confirmed and segregated through Sanger sequencing, muscle biopsies further investigated, and clinical histories reassessed by the referring clinicians (Figure S2).
The remaining unsolved cases were subsequently analyzed for likely pathogenic variants in a further 260 genes (Table S2) including those associated with neuropathies, cardiomyopathies, and other phenotypes presenting with muscle weakness to identify rarer neuromuscular conditions. This extended gene list also included genes newly associated with muscle disease. ES data were also analyzed for copy-number variants (CNVs)11 and SMN1 exon 7 deletions12 (Supplementary Materials and Methods). For those cases carrying a single heterozygous pathogenic variant in the most common (exclusively recessive) genes found in our cohort (i.e., CAPN3, DYSF, ANO5, and SGCA) we further interrogated the noncoding regions of the gene captured in the ES data. In addition, for the DYSF carriers we Sanger sequenced two previously reported pathogenic intronic variants.13 Finally, one patient was also subjected to genome sequence (GS) and RNA sequencing (RNA-Seq), as described elsewhere.14
RESULTS
Demographics of 1001 patients with suspected genetic muscle disease
One thousand and one patients were recruited into the MYO-SEQ project, with the majority (93%) inferred to be of European or Middle Eastern ancestry. The study cohort comprised 545 (54%) males and 456 (46%) females and originated from 972 families. Disease onset for 42% of the patients was in adulthood (Figure S3A) and the mean age at the time of recruitment was 39 years (Figure S3D). Proximal muscle weakness, either in isolation or with distal muscle weakness, affected most participants (77%; Figure S3B), while 68% of the cohort had increased serum CK levels (Figure S3C). Based on family history, 46% of patients were sporadic cases, while 14%, 7%, and 1% showed recessive, dominant, and X-linked inheritance, respectively; for the remaining 32%, no indication of inheritance pattern was stated by the referring clinician.
Initial screening detected suspected pathogenic variants in 47% of participants
Using the World Muscle Society gene table (http://www.musclegenetable.fr/) we generated a list of 169 genes known to be associated with limb-girdle muscle disease (Table S1) and initially restricted our search for likely causal variants to only these genes. As a result, 468 patients (47%) had rare known or suspected pathogenic variants across 72 genes that we considered were causal; these cases were deemed solved (Table 1, Fig. 1).
Table 1.
Breakdown of genes in which suspected causal variants were identified in the MYO-SEQ cohort.
| Gene | Number of patients | Gene | Number of patients | Gene | Number of patients | Gene | Number of patients |
|---|---|---|---|---|---|---|---|
| CAPN3 | 78 | POGLUT1a | 5 | PYROXD1a | 2 | HSPB1a | 1 |
| RYR1 | 26 | SCN4A | 5 | SGCB | 2 | HSPB8a | 1 |
| DYSF | 45 | CLCN1 | 3 | ACADVL | 1 | INPP5Ka | 1 |
| ANO5 | 33 | DNM2 | 4 | BAG3 | 1 | LPIN1 | 1 |
| DMD | 31 | MYOT | 4 | BVESa | 1 | MATR3 | 1 |
| TTN | 25 | RAPSN | 4 | CACNA1Sa | 1 | MGME1a | 1 |
| COL6A2 | 17 | DNAJB6 | 3 | CHRND | 1 | MMEa | 1 |
| SGCA | 16 | GMPPB | 3 | COL12A1 | 1 | MYF6 | 1 |
| COL6A3 | 12 | MTM1 | 3 | CPT2 | 1 | MYH14 | 1 |
| FLNC | 12 | SEPN1 | 3 | CRYAB | 1 | NEB | 1 |
| DES | 10 | TCAP | 3 | DOK7 | 1 | PFKM | 1 |
| GAA | 10 | TRAPPC11 | 3 | DPM3 | 1 | PGK1 | 1 |
| LAMA2 | 10 | ACTA1 | 2 | DYNC1H1a | 1 | POLG | 1 |
| POMT2 | 10 | AGL | 2 | EGR2a | 1 | PTPLA | 1 |
| COL6A1 | 10 | ATP2A1 | 2 | ETFDH | 1 | SYNE2 | 1 |
| LMNA | 9 | COLQ | 2 | FKTN | 1 | TMEM8Ca | 1 |
| MYH7 | 9 | FHL1 | 2 | GBE1 | 1 | TNNT3 | 1 |
| FKRP | 8 | PHKA1 | 2 | GFPT1 | 1 | TPM2 | 1 |
| SGCG | 8 | POMK | 2 | GNE | 1 | TTRa | 1 |
| SMCHD1 | 8 | POMT1 | 2 | GYG1 | 1 | VCP | 1 |
| TRIM32 | 7 | PRPS1a | 2 | HNRNPA1a | 1 | VMA21 | 1 |
| CAV3 | 5 | PYGM | 2 | HNRNPDL | 1 |
Suspected causative variants were identified in 87 genes in 489 patients: 72 genes during the first round of analysis using a gene list of 169 neuromuscular disorder (NMD) genes, and 15 additional genes when using a gene list extended by a further 260 NMD genes.
aFifteen additional genes identified.
Fig. 1. Summary of the solved rate and sequential analysis in the MYO-SEQ cohort.

Suspected pathogenic variants were detected in a total of 520 patients (52%). Initially, 468 (47%) patients were solved through a screening of the MYO-SEQ gene list of 169 neuromuscular disorder (NMD) genes; of these, 450 (45%) had pathogenic variants in one of 72 genes and 18 (2%) had pathogenic variants in two genes. Pathogenic variants in eight of these genes alone accounted for half of the solved patients (26%); the most common disease detected in our cohort was limb-girdle muscular dystrophy (LGMD) R1 calpain3-related (CAPN3). Variants in 64 genes accounted for the remaining patients with one causal gene. The analysis strategy was extended by a further 260 genes, solving an additional 2% of the cohort. Copy-number variations (CNVs) were detected in 26 (3%) patients, SMN1 deletions in five (0·5%) patients and transcriptional perturbations in one patient. n = 1001. GS genome sequencing.
Frequency of NMD genes varied greatly in our cohort
The most common disease in our cohort was LGMD R1 calpain3-related, caused by autosomal recessive pathogenic variants in CAPN3. Variants in CAPN3 and seven other genes—DYSF, ANO5, DMD, RYR1, TTN, COL6A2, and SGCA—accounted for over half of the solved cases (n = 260). The other 208 solved cases had suspected pathogenic variants across 64 additional genes. Importantly, we identified a stop-gain TTN founder pathogenic variant (p.[Gln35879Ter]) in 14 patients from a Serbian subpopulation.8 Without this variant, TTN would be outnumbered by COL6A2 and COL6A3 (collagen VI–related myopathies), SGCA (LGMD R3 α-sarcoglycan–related), and FLNC (myofibrillar myopathy 5 and distal myopathy 4). Some diseases were notably absent from our cohort; for example, no POMGnT1 (LGMD R15 POMGnT1–related) or ISPD (LGMD R20 ISPD-related) patients were identified, highlighting the extreme rarity of these diseases. When the data were stratified by sex, there appeared to be a skewed ratio of males (n = 22; 69%) to females (n = 10; 31%) harboring suspected pathogenic variants in ANO5 (p < 0.0691), confirming previously reported findings.15
Homozygous variants associated with established autosomal dominant inheritance
A proband from a consanguineous pedigree harbored a previously reported dominant pathogenic variant (c.476G>A p.[Arg159His]) in VCP in homozygosity, while another proband carried a novel homozygous variant (c.1325C>G p.[Pro442Arg]) in FLNC; both genes have established autosomal dominant inheritance. In both cases, the clinical presentation was in keeping with what was expected, but at the more severe end of the spectrum with an earlier disease onset. Both sets of parents were heterozygous for the respective variants. The mother of the homozygous VCP patient remained asymptomatic at the time of examination, while the father presented with proximal weakness and atrophy of the upper and lower limbs, and rimmed vacuoles on histopathology,16 as would be expected for this previously reported dominant variant.17 On the other hand, the parents of the homozygous FLNC patient were both clinically unaffected on examination. This would suggest a different pathological mechanism for this novel FLNC variant and functional work is underway (unpublished data by University Duisburg–Essen).
Solved rate differed with geographical location of the participating center
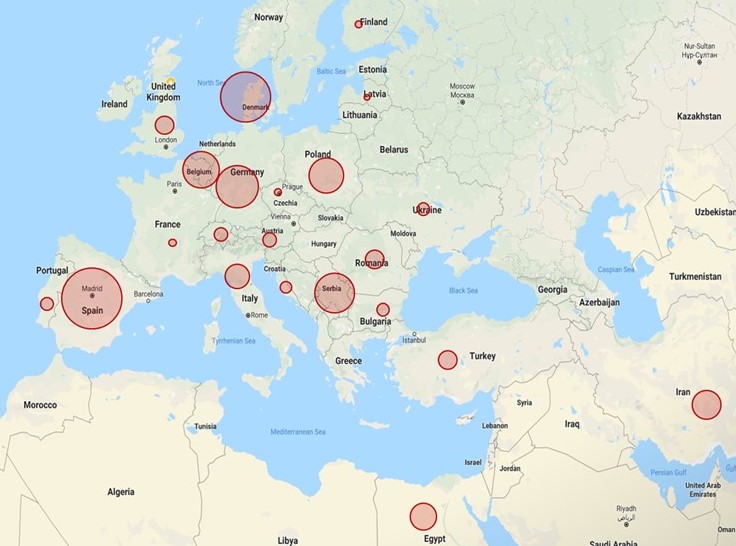
When the data were stratified by geographic location, there was a clear distinction between regions with and without the infrastructure to sufficiently prescreen patients prior to submission. The detection rate for referring centers in Eastern Europe and the Middle East was as high as 95%, including, for example, 57 LGMD R1 calpain3-related and 32 LGMD R2 dysferlin-related cases. Conversely, the detection rate for Western Europe was as low as 35%, with only 23 LGMD R1 calpain3-related (p < 0.00001) and 15 LGMD R2 dysferlin-related cases (p < 0.00001), and a larger proportion of rarer genes and phenotypes (Fig. 2).
Fig. 2. Solved rate by country of origin.

The MYO-SEQ solved rate (in dark gray) was higher in countries such as Egypt and Turkey where the infrastructure for genetic testing for prescreening is not as widely available, and lower in Western European countries where genetic prescreening of common limb-girdle muscular dystrophy (LGMD) genes is routinely performed. Calculated for referring centers submitting more than 20 samples.
Analysis of an extended gene list increased the diagnostic yield to 49%
For the remaining unsolved cases, we extended the analysis to a total of 429 disease-causing genes (Table S2). This resulted in a 2% increase in detection rate to an overall 49% (n = 488), across 87 muscle disease–associated genes; 15 genes more than the initial analysis. These genes were associated with neuropathies (EGR2, DYNC1H1, HSPB1, HSPB8, PRPS1, and MME), multisystemic disorders presenting with muscle weakness (HNRNPA1, INPP5K, TMEM8C, and TTR), channelopathies (CACNA1S), and mitochondrial disease (MGME1), all detected in patients who had been initially clinically diagnosed as LGW (n = 12). In addition, three genes only recently associated with muscular dystrophy (BVES and POGLUT1)18,19 and myofibrillar myopathy (PYROXD1)20 were found in eight patients. Thus, after the eight most common disease genes, accounting for 25% of the full cohort, the next ten genes (COL6A3, FLNC, DES, GAA, LAMA2, POMT2, COL6A1, LMNA, MYH7, and FKRP) account for an additional 10%, followed by a very small but nonetheless meaningful increase in solved rate as very rare and/or unexpected disease genes, as well as those recently described, are detected (Table 1).
Expanding exome sequencing beyond single base pair changes and small indels increased the detection yield by a further 3%
Further to point variants and small indels, we detected suspected pathogenic CNVs in 26 (3%) additional patients. Of these, 17 carried copy-number zero deletions (at exon or multiexon level) in autosomal recessive genes (DYSF, SGCA, SGCB, SGCD, SGCG, and LPIN1; n = 10) and in X-linked genes (DMD, EMD, and FHL1; n = 7, all males). Eight cases carried a heterozygous CNV in combination with a suspected pathogenic SNV in the same gene and one case, presenting with a classical dominant myofibrillar myopathy phenotype, carried a heterozygous deletion in DES. Some of these CNVs were recurring; we identified a 10.4-kb deletion in CAPN3 in five unrelated patients, and a 0.7-kb and 0.12-kb deletion in SGCD and SGCG, respectively, in two unrelated patients each (Table S3). In addition, we looked for homozygous deletions of exon 7 in SMN1, causative of spinal muscular atrophy. Such deletions were detected in five patients (0.5%), all of which were confirmed by multiplex ligation-dependent probe amplification (MLPA). Finally, through additional RNA-Seq and GS, we were able to identify an intronic change resulting in a 73-bp intron inclusion in DMD leading to a premature stop codon in one patient with a clear Becker-like phenotype and supporting family history, but with no evident causal exonic DMD variant,14 highlighting that strong phenotypic clues should always be followed up.
Over 400 novel suspected pathogenic variants were identified
In total, we identified 865 SNV and indel changes in 429 genes that were most likely to contribute to the 496 participants’ diseases. A breakdown of the genotypes and types of variants is shown in Fig. 3a, b. The 865 variants were accounted for by 520 distinct variants (Table S4); of these, 119 had been submitted to ClinVar previously, while the remaining 401 were novel and had not been yet reported at the time of the analysis (Fig. 3c). Of the novel variants, 285 were seen only once and, notably, 116 were seen multiple times in our patient population (Fig. 3d). These novel variants will be submitted to ClinVar to aid future genetic diagnoses.
Fig. 3. Breakdown of the suspected pathogenic variants and genotypes in the MYO-SEQ cohort.

(a) Zygosity of the solved patients’ variants (n = 506; 489 patients, 20 with an additional gene to report). (b) Type of variants suspected to be pathogenic. Initiation and stop loss occurred twice each (n = 865). (c) The 865 occurrences were accounted for by 520 distinct variants, of which 119 were reported as pathogenic in ClinVar and 401 were novel in their association to disease at the time of the analysis. (d) Of the 119 distinct variants reported in ClinVar, 70 were detected in only one individual (unique) while 49 were detected in multiple patients. Of the 401 distinct variants that were novel in their association to disease, 285 were detected in only individual cases, while 116 were detected in multiple families.
Single heterozygous pathogenic variants in the most common recessive genes account for 2.5% of the unsolved cases
To unravel the remaining unsolved patients, we looked at those who carried single reported pathogenic variants in the four most common exclusively recessive genes found in our cohort. We found 25 (2.5%) heterozygous carriers: 13 CAPN3, 9 ANO5, 2 SGCA, and 1 DYSF. These cases were accounted for by 16 pathogenic variants: 10 CAPN3, 4 ANO5, 1 DYSF, and 1 SGCA. These variants were all more frequent in our cohort of patients than in the control population. For example, a rare SGCA variant (c.229C>T; p.[Arg77Cys]) occurred four times more frequently in the disease population (p = 0.02), the common Eastern European CAPN3 variant (c.550delA; p.[Thr184ArgfsTer36]) over eight times more (p = 0.00028), and the European founder ANO5 variant (c.191dupA; p.[Asn63fs]) almost five times more (p = 0.00016). This suggests that these variants are likely to play a role in disease manifestation and that a second cryptic disease-causing variant might not have been detected by our analysis; we therefore interrogated these carriers further by looking into the noncoding regions present in their exome data. We thus identified one novel 3′ UTR and two rare CAPN3 intronic variants, one of which (c.1746–20C>G) occurred in three cases. Histopathological analysis showed markedly reduced calpain3 immunostaining, suggesting that this noncoding variant may affect splicing or protein stability. In addition, we identified an intronic deletion (c.585–31_585–24delTCTGCTGA) in one of the SGCA carriers, which results in partial expression of the α-sarcoglycan protein.21 Conversely, none of the reported pathogenic intronic variants in DYSF13 were found in the DYSF carrier.
Many patients harbored suspected pathogenic variants in genes associated with treatable or manageable conditions
Our study identified 64 patients (6.5%) that might benefit from treatment or specific management options related to their diagnosis. For example, variants in COLQ, DOK7, GFPT1, and RAPSN—all associated with congenital myasthenic syndrome (CMS)—were identified in eight patients (<1%). We also identified ten patients (1%) with compound heterozygous variants in GAA, associated with Pompe disease.22 Nine patients (1%) harbored variants in either SCN4A or CLCN1 and thus were suspected to have ion channel disorders that could benefit from selective drug interventions. We identified 13 female patients (1%) who were likely to be manifesting carriers of Duchenne muscular dystrophy, an X-linked disorder that often escapes clinical diagnosis in females. Finally, we identified 24 patients (2%) with heterozygous suspected pathogenic variants in RYR1, which can confer susceptibility to malignant hyperthermia.
DISCUSSION
We sought to improve the diagnostic pathway of patients with limb-girdle muscle diseases by implementing TES as a first-pass diagnostic strategy. By focusing on 429 NMD genes within the exome data, we solved 49% of our cohort, comparable with equivalent NGS panel studies.6,7 A small number of common NMD genes, namely CAPN3, DYSF, ANO5, DMD, RYR1, TTN, COL6A2, and SGCA accounted for more than half of the solved cases. Our finding that LGMD R1 calpain3-related was the most common LGMD in our cohort is in agreement with other European studies.6 Standard calpain3 immunoblot testing can only achieve a low sensitivity,23 which could explain why a tenth of the MYO-SEQ cohort was not diagnosed with this LGMD prior to enrollment. Moreover, many of the calpain3-related patients were referred from centers without the technical infrastructure to prescreen their patients.
Given the considerable phenotypic overlap and the rarity of some of these muscle conditions, it is almost impossible for clinicians, however expert, to individually diagnose every disease. Some of the LGMDs, such as R15 POMGnT1-related and R20 ISPD-related, were absent from our cohort. No SNVs and only one CNV were identified in the SGCD gene, in keeping with other sequencing projects from China (n = 756),24 Italy (n = 504),6 and the United States (n = 4656)7 where no SGCD variants were found. Interestingly, LGMD R6 δ-sarcoglycan–related patients accounted for >1% of the diagnosed cases in a South American cohort (n = 2103),25 highlighting that the geography-specific prevalence of some diseases should be considered during clinical work-ups. Per sex bias, we corroborated previous findings that ANO5 diagnosis is more frequent in males.15 This is likely due to the milder, sometimes asymptomatic, presentation of LGMD R12 anoctamin5-related in females; because of this, females can present to clinic much later in life, or not at all, skewing the diagnostic rate in favor of males.
The relatively small benefit observed when extending the gene list from 169 to 429 genes is in line with a panel study of 700 NMD genes where exome sequencing of the remaining undiagnosed cases did not improve the pick-up rate for known NMD genes.26 However, having exome data available, such as in our study, allows for both gene discovery and data analysis reiteration when new disease genes are characterized, neither of which are possible even with a large NGS panel approach. This was the case for the BVES, POGLUT1, and PYROXD1 patients who were missed in the first round of analysis, as the genes had not yet been described as disease-causing. By interrogating the whole exome of the unsolved patients, we identified and have functionally characterized two novel genes, one associated with a rare secondary dystroglycanopathy and the other, POPDC3, found in three unrelated individuals, associated with a typical LGMD phenotype.27 Two additional novel candidate genes are under investigation and we expect that this will only increase as the exome data of unsolved patients are more intricately examined.
It is possible that further causative variants have not been assigned pathogenicity due to the limitations of proband-only analysis or that they have not been detected due to nonuniform coverage and/or poorly designed capture baits that might not target relevant tissue-specific exons. For example, repeat regions in NEB or TTN are known to be difficult to map to the genome and therefore variants in these regions are poorly called.28 Somatic mosaicisms,29 only detected by deep coverage sequencing, would also have been missed. A further proportion of the remaining unsolved cases might be caused by genetic changes in intergenic, intronic, or regulatory regions not covered by ES. The overrepresentation in our disease cohort of carriers of known pathogenic variants in recessive genes likely implies that a second cryptic variant is yet to be detected. In fact, when interrogating these cases further we identified recurrent suspected pathogenic intronic variants and CNVs. Otherwise, these pathogenic heterozygous variants might act as disease modifiers, contributing to the disease severity or progression.30
Using modified pipelines, we have identified several disease-causing CNVs, accounting for 3% of the diagnoses. However, more complex structural variants such as translocations and inversions, would not have been detected.31 In a similar manner, tri- and tetranucleotide expansion repeats, such as the cause of some adult onset neuromuscular conditions (e.g., myotonic dystrophy), are typically missed in standard short-read NGS data. Interestingly, four unsolved cases were reported to present with myotonia, percussion myotonia, or myotonic discharges. Other novel late-onset myopathies might be caused by similar mechanisms and long-read NGS would be needed to identify them. Digenic or non-Mendelian inheritance, such as genomic imprinting, may also account for a proportion of the undiagnosed patients32,33 and will need a different methodological and analytical strategy.
It is also possible that some of the unsolved patients may have acquired forms of muscle disease, such as acquired immune, inflammatory, noninflammatory, or even statin-induced myopathies. In fact, during the project four patients (<1%) obtained a confirmed diagnosis of inclusion body myositis. Regular screening for anti-NT5c1A and anti-HMGCoR should provide a more realistic indication of the prevalence of these diseases in the affected population.
Over 6% of our patients received a genetic outcome that resulted in specific monitoring and tailored treatments. For example, for the CMS patients, genetic diagnoses were vital to provide adequate treatment, as this depends on the disease subtype and causative gene. Indeed, the referring clinicians reported that these patients all showed marked improvement after appropriate treatments were commenced. The pathogenicity of the RYR1 variants could not always be fully confirmed, yet it was prudent to return these potential diagnoses to physicians to manage possible risk of malignant hyperthermia. For most of the solved cases, however, who might not benefit from specific treatment, their genetic diagnosis will nevertheless allow appropriate family planning, counseling, and monitoring of their diseases.
MYO-SEQ was a worldwide partnership between academia, industry, and patient organizations. Frequent communication with the referring physicians was essential for the success of the project; to reach a meaningful diagnosis, interpretation of the genetic data was always in the context of clinical, histopathological, and MRI data. The participating centers provided consent for data sharing facilitated by the European Genome-Phenome Archive (EGA) and RD-Connect (https://platform.rd-connect.eu/), and we advocate that adopting such an approach will enable future matchmaking between extremely rare cases, such as BVES-related myopathy34 or LGMD R21 POGLUT1-related.35 In addition, thanks to the large cohort size and standardized deep phenotypic data, we were able to expand the clinical and mutational spectrum of known causative genes, such as TRIM32,36 POMK,37 DPM3,38 POMT2,39 and other dystroglycanopathies.40
Based on our findings from this large-scale international collaboration, we suggest a new diagnostic approach in the clinic and/or private health providers. Rather than implementing small NGS panel approaches, led by phenotypic clues that can often be misleading due to the high clinical heterogeneity of muscle diseases, patients should be directly referred for exome sequencing. The exome sequencing data of NMD patients of European and Middle Eastern origin should first be promptly analyzed for pathogenic variants in the eight genes most commonly associated with muscle disease in our cohort—CAPN3, DYSF, ANO5, DMD, RYR1, TTN, COL6A2, and SGCA. This is expected to diagnose over a quarter of individuals. The analysis pipeline should then be extended to include additional NMD genes, enabling a diagnosis in a further quarter of patients. For those patients who remain undiagnosed, their ES data can be further interrogated. Importantly, exome data can be retrospectively and repeatedly analyzed for pathogenic variants in novel disease genes as new discoveries are made, making it a much more cost-effective approach.
Supplementary information
{kind=link}
Online Supplementary Material and Methods
Acknowledgements
We thank the patients for donating their tissue samples. MYO-SEQ was funded by Sanofi Genzyme, Ultragenyx, LGMD2I Research Fund, Samantha J. Brazzo Foundation, LGMD2D Foundation and Kurt+Peter Foundation, Muscular Dystrophy UK, and Coalition to Cure Calpain 3. Analysis was provided by the Broad Institute of MIT and Harvard Center for Mendelian Genomics (Broad CMG) and was funded by the National Human Genome Research Institute, the National Eye Institute, and the National Heart, Lung, and Blood Institute grant UM1 HG008900, and in part by National Human Genome Research Institute grant R01 HG009141.
Disclosure
D.G.M. is a founder with equity in Goldfinch Bio, and has received research support from AbbVie, Astellas, Biogen, BioMarin, Eisai, Merck, Pfizer, and Sanofi Genzyme. V.S. is or has been a principal investigator for trials sponsored by Sanofi Genzyme, GSK, Prosensa/Biomarin, IonisPharmceuticals, and Sarepta. He has received speaker honoraria from Sanofi Genzyme. He is or has been on advisory boards for Acceleron Pharma, Audentes Therapeutics, Biomarin, Bristol-Myer Squibb, Italfarmaco S.p.A., Nicox, Pfizer, Sanofi Genzyme, Santhera, Sarepta Therapeutics, Summit Therapeutics, Tivorsan, and TrophyNOD. The other authors declare no conflicts of interest.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Ana Töpf, Katherine Johnson
Contributor Information
Volker Straub, Email: volker.straub@ncl.ac.uk.
The MYO-SEQ consortium:
Ela Akay, Jorge Alonso-Pérez, Jonathan Baets, Nina Barisic, Alexandra Bastian, Sabine Borell, Teodora Chamova, Kristl Claeys, Jaume Colomer, Sandra Coppens, Nicolas Deconinck, Willem de Ridder, Jordi Díaz-Manera, Cristina Domínguez-González, Alexis Duncan, Hacer Durmus, Nagia A. Fahmy, Maria Elena Farrugia, Roberto Fernández-Torrón, Lidia Gonzalez-Quereda, Jana Haberlova, Maja von der Hagen, Andreas Hahn, Antonia Jakovčević, Ivonne Jerico Pascual, Solange Kapetanovic, Viktorija Kenina, Janbernd Kirschner, Andrea Klein, Heike Kölbel, Anna Kostera-Pruszczyk, Richa Kulshrestha, Jaana Lähdetie, Mahsa Layegh, Cheryl Longman, Adolfo López de Munain, Wolfgang Loscher, Anna Lusakowska, Paul Maddison, Armelle Magot, Anirban Majumdar, Pilar Martí, Amaia Martínez Arroyo, Radim Mazanec, Sandra Mercier, Tiziana Mongini, Nuria Muelas, Andrés Nascimento, Shahriar Nafissi, Shirin Omidi, Carlos Ortez, Stéphanie Paquay, Yann Pereon, Stojan Perić, Valentina Ponzalino, Vidosava Rakočević Stojanović , Gauthier Remiche, Aida Rodríguez Sainz, Sabine Rudnik, Iciar Sanchez Albisua, Manuela Santos, Ulrike Schara, Andriy Shatillo, Jadranka Sertić, Ulrich Stephani, Sonja Strang-Karlsson, Yves Sznajer, Ani Tanev, Ivailo Tournev, Peter Van den Bergh, Vinciane Van Parijs, Juan Vílchez, Katharina Vill, John Vissing, Carina Wallgren-Pettersson, Julia Wanschitz, Tracey Willis, Nanna Witting, and Miren Zulaica
Supplementary information
The online version of this article (10.1038/s41436-020-0840-3) contains supplementary material, which is available to authorized users.
References
- 1.Mercuri E, Muntoni F. Muscular dystrophies. Lancet. 2013;381:845–860. doi: 10.1016/S0140-6736(12)61897-2. [DOI] [PubMed] [Google Scholar]
- 2.Bushby K, Finkel R, Birnkrant DJ, et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. Lancet Neurol. 2010;9:77–93. doi: 10.1016/s1474-4422(09)70271-6.. [DOI] [PubMed] [Google Scholar]
- 3.Bonne G, Rivier F, Hamroun D. The 2018 version of the gene table of monogenic neuromuscular disorders (nuclear genome) Neuromuscul Disord. 2017;27:1152–1183. doi: 10.1016/j.nmd.2017.10.005. [DOI] [PubMed] [Google Scholar]
- 4.Harris E, Laval S, Hudson J, et al. Undiagnosed genetic muscle disease in the north of England: an in depth phenotype analysis. PLoS Curr. 2013;5:10.1371/currents.md.37f840ca67f5e722945ecf755f40487e. doi: 10.1371/currents.md.37f840ca67f5e722945ecf755f40487e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Calvo SE, Compton AG, Hershman SG, et al. Molecular diagnosis of infantile mitochondrial disease with targeted next-generation sequencing. Sci Transl Med. 2012;4:118ra10. doi: 10.1126/scitranslmed.3003310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Savarese M, Di Fruscio G, Torella A, et al. The genetic basis of undiagnosed muscular dystrophies and myopathies: results from 504 patients. Neurology. 2016;87:71–76. doi: 10.1212/WNL.0000000000002800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nallamilli BRR, Chakravorty S, Kesari A, et al. Genetic landscape and novel disease mechanisms from a large LGMD cohort of 4656 patients. Ann Clin Transl Neurol. 2018;5:1574–1587. doi: 10.1002/acn3.649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Peric S, Glumac JN, Topf A, et al. A novel recessive TTN founder variant is a common cause of distal myopathy in the Serbian population. Eur J Hum Genet. 2017;25:572–581. doi: 10.1038/ejhg.2017.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–291. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang K, Li M, Hadley D, et al. PennCNV: an integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res. 2007;17:1665–1674. doi: 10.1101/gr.6861907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Larson JL, Silver AJ, Chan D, et al. Validation of a high resolution NGS method for detecting spinal muscular atrophy carriers among phase 3 participants in the 1000 Genomes Project. BMC Med Genet. 2015;16:100. doi: 10.1186/s12881-015-0246-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dominov JA, Uyan O, McKenna-Yasek D, et al. Correction of pseudoexon splicing caused by a novel intronic dysferlin mutation. Ann Clin Transl Neurol. 2019;6:642–654. doi: 10.1002/acn3.738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cummings BB, Marshall JL, Tukiainen T, et al. Improving genetic diagnosis in Mendelian disease with transcriptome sequencing. Sci Transl Med. 2017;9:eaal5209. doi: 10.1126/scitranslmed.aal5209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sarkozy A, Hicks D, Hudson J, et al. ANO5 gene analysis in a large cohort of patients with anoctaminopathy: confirmation of male prevalence and high occurrence of the common exon 5 gene mutation. Hum Mutat. 2013;34:1111–1118. doi: 10.1002/humu.22342. [DOI] [PubMed] [Google Scholar]
- 16.De Ridder W, Azmi A, Clemen CS, et al. Multisystem proteinopathy due to a homozygous p.Arg159His VCP mutation: a tale of the unexpected. Neurology. 2019;94:e785–e796. doi: 10.1212/WNL.0000000000008763. [DOI] [PubMed] [Google Scholar]
- 17.van der Zee J, Pirici D, Van Langenhove T, et al. Clinical heterogeneity in 3 unrelated families linked to VCP p.Arg159His. Neurology. 2009;73:626–632. doi: 10.1212/WNL.0b013e3181b389d9. [DOI] [PubMed] [Google Scholar]
- 18.Schindler RF, Scotton C, Zhang J, et al. POPDC1(S201F) causes muscular dystrophy and arrhythmia by affecting protein trafficking. J Clin Invest. 2016;126:239–253. doi: 10.1172/JCI79562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Servian-Morilla E, Takeuchi H, Lee TV, et al. A POGLUT1 mutation causes a muscular dystrophy with reduced Notch signaling and satellite cell loss. EMBO Mol Med. 2016;8:1289–1309. doi: 10.15252/emmm.201505815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.O’Grady GL, Best HA, Sztal TE, et al. Variants in the oxidoreductase PYROXD1 cause early-onset myopathy with internalized nuclei and myofibrillar disorganization. Am J Hum Genet. 2016;99:1086–1105. doi: 10.1016/j.ajhg.2016.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gonzalez-Quereda L, Gallardo E, Topf A, et al. A new mutation of the SCGA gene is the cause of a late onset mild phenotype limb girdle muscular dystrophy type 2D with axial involvement. Neuromuscul Disord. 2018;28:633–638. doi: 10.1016/j.nmd.2018.06.002. [DOI] [PubMed] [Google Scholar]
- 22.Johnson K, Topf A, Bertoli M, et al. Identification of GAA variants through whole exome sequencing targeted to a cohort of 606 patients with unexplained limb-girdle muscle weakness. Orphanet J Rare Dis. 2017;12:173. doi: 10.1186/s13023-017-0722-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fanin M, Angelini C. Protein and genetic diagnosis of limb girdle muscular dystrophy type 2A: the yield and the pitfalls. Muscle Nerve. 2015;52:163–173. doi: 10.1002/mus.24682. [DOI] [PubMed] [Google Scholar]
- 24.Xie Z, Hou Y, Yu M, et al. Clinical and genetic spectrum of sarcoglycanopathies in a large cohort of Chinese patients. Orphanet J Rare Dis. 2019;14:43. doi: 10.1186/s13023-019-1021-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bevilacqua JA, Guecaimburu Ehuletche MDR, Perna A, et al. The Latin American experience with a next generation sequencing genetic panel for recessive limb-girdle muscular weakness and Pompe disease. Orphanet J Rare Dis. 2020;15:11. doi: 10.1186/s13023-019-1291-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Monies D, Alhindi HN, Almuhaizea MA, et al. A first-line diagnostic assay for limb-girdle muscular dystrophy and other myopathies. Hum Genomics. 2016;10:32. doi: 10.1186/s40246-016-0089-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vissing J, Johnson K, Topf A, et al. POPDC3 gene variants associate with a new form of limb girdle muscular dystrophy. Ann Neurol. 2019;86:832–843. doi: 10.1002/ana.25620. [DOI] [PubMed] [Google Scholar]
- 28.Zenagui R, Lacourt D, Pegeot H, et al. A reliable targeted next-generation sequencing strategy for diagnosis of myopathies and muscular dystrophies, especially for the giant titin and nebulin genes. J Mol Diagn. 2018;20:533–549. doi: 10.1016/j.jmoldx.2018.04.001. [DOI] [PubMed] [Google Scholar]
- 29.Harris E, Topf A, Barresi R, et al. Exome sequences versus sequential gene testing in the UK highly specialised service for limb girdle muscular dystrophy. Orphanet J Rare Dis. 2017;12:151. doi: 10.1186/s13023-017-0699-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhao M, Maani N, Dowling JJ. Dynamin 2 (DNM2) as cause of, and modifier for, human neuromuscular disease. Neurotherapeutics. 2018;15:966–975. doi: 10.1007/s13311-018-00686-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pang AW, MacDonald JR, Pinto D, et al. Towards a comprehensive structural variation map of an individual human genome. Genome Biol. 2010;11:R52. doi: 10.1186/gb-2010-11-5-r52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee Y, Jonson PH, Sarparanta J, et al. TIA1 variant drives myodegeneration in multisystem proteinopathy with SQSTM1 mutations. J Clin Invest. 2018;128:1164–1177. doi: 10.1172/JCI97103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Andrade RC, Nevado J, de Faria Domingues de Lima MA, et al. Segmental uniparental isodisomy of chromosome 6 causing transient diabetes mellitus and merosin-deficient congenital muscular dystrophy. Am J Med Genet A. 2014;164A:2908–2913. doi: 10.1002/ajmg.a.36716. [DOI] [PubMed] [Google Scholar]
- 34.De Ridder W, Nelson I, Asselbergh B, et al. Muscular dystrophy with arrhythmia caused by loss-of-function mutations in BVES. Neurol Genet. 2019;5:e321. doi: 10.1212/NXG.0000000000000321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Servian-Morilla E, Cabrera-Serrano M, Johnson K, et al. POGLUT1 biallelic mutations cause myopathy with reduced satellite cells, alpha-dystroglycan hypoglycosylation and a distinctive radiological pattern. Acta Neuropathol. 2020;139:565–582. doi: 10.1007/s00401-019-02117-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Johnson K, De Ridder W, Topf A, et al. Extending the clinical and mutational spectrum of TRIM32-related myopathies in a non-Hutterite population. J Neurol Neurosurg Psychiatry. 2019;90:490–493. doi: 10.1136/jnnp-2018-318288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Strang-Karlsson S, Johnson K, Topf A, et al. A novel compound heterozygous mutation in the POMK gene causing limb-girdle muscular dystrophy-dystroglycanopathy in a sib pair. Neuromuscul Disord. 2018;28:614–618. doi: 10.1016/j.nmd.2018.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Van den Bergh PYK, Sznajer Y, Van Parys V, et al. A homozygous DPM3 mutation in a patient with alpha-dystroglycan-related limb girdle muscular dystrophy. Neuromuscul Disord. 2017;27:1043–1046. doi: 10.1016/j.nmd.2017.07.006. [DOI] [PubMed] [Google Scholar]
- 39.Ostergaard ST, Johnson K, Stojkovic T, et al. Limb girdle muscular dystrophy due to mutations in POMT2. J Neurol Neurosurg Psychiatry. 2018;89:506–512. doi: 10.1136/jnnp-2017-317018. [DOI] [PubMed] [Google Scholar]
- 40.Johnson K, Bertoli M, Phillips L, et al. Detection of variants in dystroglycanopathy-associated genes through the application of targeted whole-exome sequencing analysis to a large cohort of patients with unexplained limb-girdle muscle weakness. Skelet Muscle. 2018;8:23. doi: 10.1186/s13395-018-0170-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Online Supplementary Material and Methods