

Abstract
Transdermal delivery of water-insoluble drugs via hydrogel-based microneedle (MN) arrays is crucial for improving their therapeutic efficacies. However, direct loading water-insoluble drug into hydrophilic matrices remains challenging. Here we report a biodegradable MN arrays patch that is fabricated from naturally derived polymer conjugates of gelatin methacryloyl and β-cyclodextrin (GelMA-β-CD). When curcumin, an unstable and water-insoluble anticancer drug, is loaded as a model drug, its stability and solubility are improved due to the formation of inclusion complex. The polymer-drug complex GelMA-β-CD/CUR) can be formulated into MN arrays with sufficient mechanical strength for skin penetration and tunable drug release profile. Anticancer efficacy of released curcumin is observed in three-dimensional (3D) B16F10 melanoma models. The GelMA-β-CD/CUR MN exhibits relatively higher therapeutic efficacy through more localized and deeper penetrated manner compared with a control non-transdermal patch. In vivo studies also verify biocompatibility and degradability of the GelMA-β-CD MN arrays patch.
Keywords: Gelatin Methacryloyl, β-cyclodextrin, microneedle, water insoluble drug, transdermal delivery
ToC
Hydrogel microneedle (MN) patch based on naturally derived gelatin methacryloyl and β-cyclodextrin conjugates is developed for transdermal delivery of water-insoluble drugs. Tunable drug release profiles and enhanced drug stability are demonstrated via curcumin as a model drug. The newly developed MN patch possesses good biocompatibility that can potentially be used for various pharmaceutical applications.
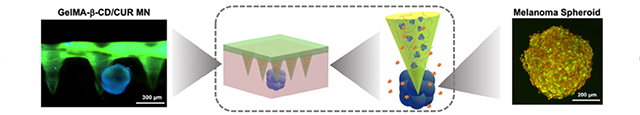
1. Introduction
Transdermal drug delivery offers advantages over non-parenteral routes such as bypassing first-pass metabolism and facile administration.[1] Mammalian skin, however, functions as a protective layer to the external environment and a fundamental barrier for transdermal delivery. Various strategies have been developed to overcome this barrier physically including ultrasound, iontophoresis, electroporation, and transdermal microneedle (MN) array patches.[2] Among those, MN arrays have been widely studied in clinical trials because of their capability of penetrating the stratum corneum that greatly enhances systemic drug delivery with minimal pain and improved patient compliance.[3] These properties enable MN arrays to be used for various biomedical applications and precision medicine tools,[4] including insulin delivery,[5] immunotherapy,[6] cancer vaccine,[7] sampling,[8] and contraceptive delivery.[9]
Natural hydrogel-MNs, such as those derived from alginate,[10] cellulose,[11] gelatin,[12] and hyaluronic acid,[13] have drawn extensive attention because of their biocompatibility and innate biodegradability. However, hydrogels are inherently composed of hydrophilic materials that is exclusively compatible with water-soluble molecules such as growth factors, chemokines, or hydrophilic drugs.[14] These gels are not suitable for many drugs as around 90% of FDA approved drugs are lipophilic.[15] Additionally, sustained release of these agents generally requires homogenous distribution of water-insoluble drugs in the matrix, which is challenging to achieve in hydrogel-based materials.[16] Hydrophobic polymers can be implemented for delivery of water-insoluble drugs though these materials generally induce stronger inflammatory responses.[16] Because of clear clinical need, development of biocompatible and biodegradable hydrogel-MN arrays that could directly function as a versatile platform for water-insoluble drug delivery is desired.
Previously, we have shown that gelatin methacryloyl (GelMA) can be used for fabrication of MN arrays and delivery of water-soluble drug with desirable release profiles.[12] GelMA is derived from the natural polymer gelatin with crosslinkable methacrylate group.[17] The incorporation of methacrylate groups in gelatin endows GelMA with tunable mechanical and drug release properties through the control of crosslinking degree by ultraviolet or visible light.[18] These properties make GelMA an ideal candidate for MN fabrication as well as various other biomedical applications.[18, 19] To harness the favorable properties of GelMA as MN arrays and extend its applications for water-insoluble drug delivery, we hypothesize to modify GelMA side chains with amphiphilic β-cyclodextrin (β-CD) and endow the resulting peptide-saccharide hybrid material capability of loading water-insoluble drug (Figure 1A). β-CD is derived from enzymatically degraded products of starch with external hydrophilic surface and stable hydrophobic core.[20] It does not change overall hydrophilicity of hydrogel matrix while being able to trap small molecules and form inclusion complexes.[21] We selected curcumin as a model for unstable and water-insoluble drugs to test functionality of newly developed biohybrid materials and potential applications as hydrogel-based MN patch (Figure 1B). The anti-cancer efficacy of curcumin delivered with MN arrays are explored on melanoma B16F10 cell culture and a 3D melanoma spheroid embedded hydrogel. Finally, in vivo biocompatibility and biodegradability of GelMA-β-CD is characterized for future in vivo applications.
Figure 1.

Schematic for GelMA-β-CD synthesis and GelMA-β-CD based microneedle fabrication and application. A. Synthesis route of GelMA-β-CD by conjugating GelMA with (I) CM-β-CD conjugation via EDC/NHS coupling. (II) Curcumin was loaded within GelMA-β-CD pre-gel solution to form curcumin inclusion complex. B. Schematic for fabrication of GelMA-β-CD/CUR MNs by (III) centrifugation and UV crosslinking; GelMA-β-CD/CUR MN arrays were tested on 3D B16F10 skin cancer model (IV).
2. Results
2.1. Characterization of GelMA-β-CD
Gelatin with a low degree of methacrylation was synthesized due to the need for further chemical modification of GelMA. The degree of MA conjugation was verified using 1H NMR by comparing the intensity of the double bond region (5.2 ppm, representative of the degree of methacrylation) with the integrated intensity of the aromatic region (7.2 ppm, representative of gelatin concentration). Synthesized GelMA was found to be approximately 20% methacrylated and was further used for β-CD modification.[22]
To optimize the conjugation of CM-β-CD onto the GelMA backbone, three different feed ratios of CM-β-CD (200, 400, 600 mg) to the GelMA (300 mg) were selected for the reaction (detailed reaction formulation shown in Table S1, Supporting Information). Because CM-β-CD is the primary factor in establishing drug release activity of the synthesized material, the dialysis process was optimized to ensure unconjugated compound was completely removed from the solution. This process was characterized by the acid-phenol assay shown in Figure S1 (Supporting Information) demonstrating that the amount of CM-β-CD in the dialysis water was undetectable relative to DI water after 3 days of dialysis. In addition, both 1H NMR and FTIR were used to confirm conjugation by examining the characteristic peaks of GelMA and the polysaccharide ring of CD. Figure 2A showed the presence of both GelMA and CM-β-CD features (Figure 2C) in the conjugated polymer GelMA-β-CD. Protons from the double bond of the methacrylate group displayed peaks at 5.3 ppm and 5.7 ppm (region I). The aromatic region from GelMA presented a peak at 7.2 ppm (region II). Characteristic peaks at 4.8 ppm and 5.7 ppm (region IV) were representative of protons in the glucose ring from CM-β-CD.[23] Similarly, in the FTIR spectra (Figure 2B) the conjugated GelMA-β-CD displayed strong absorption at 1150–1070 cm−1 in enclosed region IV corresponding to the ether group of the CM-β-CD. In addition, strong peaks at 1500–1700 cm−1 (shown in enclosed region III) came from amide bonds presented in the backbone of the GelMA polymer. Both of these confirmed the successful conjugation of CM-β-CD onto the GelMA backbone via EDC/NHS coupling. To quantify the amount of CM-β-CD conjugated, a colorimetric assay was performed where concentrated sulfuric acid and 5% phenol was used to measure the absorbance at 490 nm. The amount of CM-β-CD presented within the GelMA-β-CD was estimated by comparison with a CM-β-CD standard curve. A representative 96 well-plate after conducting the colorimetric assay and measuring the absorbance spectra between 380 nm and 550 nm for low, medium, and high GelMA-β-CD was shown in Figure S2 (Supporting Information). As shown in Figure 2D, the amount of β-CD conjugation was changed from 5 wt% to 40 wt% upon increasing the relative feed ratio. Feed ratios of 3:2, 3:4, and 3:6 altered the weight percentage of conjugated CM-β-CD to 5.4 ± 1.3%, 22.1 ± 0.6%, and 40.4 ± 1.9%, respectively. In further experiments, the formulation yielding the highest degree of CM-β-CD conjugation was used to maximize drug loading capacity.
Figure 2.

Characterization of β-CD conjugation on GelMA backbone and loading of water insoluble drug curcumin. A. NMR spectra of (a) GelMA-β-CD, (b) CM-β-CD, and (c) GelMA and B. FTIR spectra of (a) GelMA-β-CD, (b) CM-β-CD, and (c) GelMA with dash-line enclosed area corresponding to C. Chemical structures of (b) GelMA and (c) CM-β-CD. D. Modification degrees of CM-β-CD on GelMA-β-CD at different feeding weight ratios of GelMA:CM-β-CD. E. Loading capacity of curcumin in GelMA and GelMA-β-CD. F. Optical image of freeze-dried GelMA/CUR and GelMA-β-CD/CUR. (Scale bar: 1 cm).
Curcumin was selected as a model for water-insoluble drugs (Figure S3, Supporting Information) to test the loading efficacy of GelMA-β-CD. Aqueous drug suspension was used to compare the loading capacity relative to unmodified GelMA. Curcumin displayed limited solubility in GelMA (84.8 ± 13.5 μg/mL) but GelMA-β-CD had a loading capacity about 5.5 times higher (466.0 ± 31.2 μg/mL) (Figure 2E). Although the GelMA backbone contains some hydrophobic moieties that could facilitate curcumin loading inside the 3D molecular network, covalently incorporated β-CD significantly increased the solubility of the water insoluble drug through its hydrophobic cavities. The intermolecular interaction between CD and curcumin primarily dictated the desirable properties of water-insoluble drug delivery. This can be seen in Figure 2F in which the freeze-dried sample of GelMA-β-CD/CUR was observed to be much darker in color than GelMA/CUR, indicating increased curcumin loading.
2.2. Characterization of inclusion complex
Intermolecular hydrophobic interactions between the CD cavities and curcumin enhanced the loading efficiency through the guest-host interactions. The inclusion complexes GelMA-β-CD/CUR were characterized by both FTIR and DSC compared with physical mixture GelMA-β-CD+CUR. In inclusion complex, curcumin is expected to be inside the cavity of CD while physical mixture should present characteristics of both components since the lack of host-guest interactions. Figure 3A showed the FTIR spectra of GelMA-β-CD/CUR which closely resembled the spectra of pure GelMA-β-CD except for traces of peaks from curcumin. An expanded view of the same spectra is shown in which free curcumin molecules exhibit peaks at 1602 cm−1 and 1627 cm−1 (enclosed region (I)), related to the stretching of aromatic ring and C=C, C=O bonds), and a peak at 1509 cm−1 (enclosed region (II) representing stretching of C=O bond).[24] The characteristic band appearing at 1033 cm−1 was from the vibration of C-O-C of β-CD (shaded area (III)). Strong hydrophobic interactions during the formation of the inclusion complex suppressed few characteristic peaks of curcumin molecules shown in enclosed region (I) and (II) in Figure 3A. However, in the case of GelMA-β-CD+CUR, the FTIR spectrum contains peaks associated with both components to confirm the lack of host-guest interactions between two components.[25]
Figure 3.

Characterization of GelMA-β-CD/CUR inclusion complex. A. FTIR spectra (500–4000 cm−1) and zoomed-in FTIR spectra (900–1900 cm−1), and B. DSC thermograms of (a) curcumin, (b) GelMA-β-CD+CUR (c) GelMA-β-CD, (d) GelMA-β-CD/CUR C. Aqueous stability of curcumin in DPBS. Curcumin was either in free form or in GelMA-β-CD/CUR.
DSC thermograms of curcumin, GelMA-β-CD+CUR, GelMA-β-CD, and GelMA-β-CD/CUR are presented in Figure 3B. Free curcumin molecules displayed an endothermic peak at ~ 180.7 °C, which corresponded to its melting point.[26] In contrast, GelMA-β-CD did not present a melting endotherm between 100 °C and 220 °C. The GelMA-β-CD+CUR yielded the endothermic peak of curcumin, however it was broadened and shifted to 178.3 °C. This indicated that, in the physical mixture, free curcumin molecules weakly interacting with the GelMA-β-CD matrix are present. The lower peak position in the thermal profile can be explained by the lower amount of energy needed to break physical interactions between molecules in comparison to the energy required to melt the material. In contrast, the DSC curve of GelMA-β-CD/CUR displayed the features of GelMA-β-CD polymer. The difference between the DSC curves of the physical mixtures and inclusion complex mixtures indicated a strong interaction between curcumin and β-CD and is evidence of the successful inclusion of curcumin molecules inside the cavity of β-CD.[27]
Furthermore, because free curcumin was unstable in aqueous solutions,[28] the presence of the β-CD cavity that formed the inclusion complex not only improved the loading capacity of GelMA-β-CD, but also stabilizes the unstable guest molecules.[26] The stability of curcumin was monitored by measuring absorbance at 430 nm for both the inclusion complex and free curcumin dissolved in DPBS (DPBS diluted with ethanol). The percentage of remaining curcumin was estimated by measuring the absorbance compared to the initial absorbance over a 6 h period. The results (Figure 3C) demonstrated that only approximately 45% of free curcumin in DPBS remained after 1 h at 37 °C incubation, while the inclusion complex of curcumin maintained 90% of the initial concentration. This result indicates that the formation of an inclusion complex can greatly improve the aqueous stability of curcumin.
2.3. MN arrays fabrication and characterization
The GelMA-β-CD/CUR inclusion complex solution was used for the fabrication of biodegradable MN arrays (Figure 4A). Here, GelMA-β-CD/CUR inclusion complex solution was casted into a PDMS mold for an 11×11 MN array and centrifuged. After crosslinking with UV light and drying overnight, the MN arrays were easily peeled off from the PDMS mold. Because curcumin fluoresces green, successful loading of curcumin into MN arrays was observed with fluorescence microscopy (Figure 4B). In addition, MN arrays were uniformly displayed in the SEM images in Figure 4C. These images also confirmed that the dimensions of the fabricated MN arrays were approximately 600 μm long and 300 μm wide at the base of the MNs. In addition, in vivo MN arrays penetration (Figure 4D) was also studied with H&E staining of skins in which the dermal layer was disrupted by MN arrays. Tissue sections showed micro-holes from the needle tips (Figure 4E). Fabricated hydrogel-based MN arrays also demonstrated mechanical strength sufficient to penetrate mouse cadaver skin ex vivo. Trypan blue preferentially stained physically damaged cells showing that successful MN arrays penetration was achieved (Figure 4F).
Figure 4.

Characterization of MNs properties fabricated with GelMA-β-CD. A. Schematic of MN arrays fabrication process by (I) centrifugation, (II) UV crosslinking, (III) overnight dry and peeled off from PDMS mold. B. Fluorescence image of curcumin loaded MN arrays. Curcumin displays green fluorescence. (Scale Bar: 200 μm) C. SEM image of MNs. (Scale Bar: 300 μm) D. Representative image of a mouse transcutaneously administered with the MN array patch and the enclosed dash-line circle indicating the treatment area. (Scale Bar: 1 cm) E. H&E-staining image of mouse skin showing the penetration of the MN patch into the mouse skin. (Scale Bar: 200 μm) F. Skin penetration of the MN arrays patch. MN arrays patch was applied on mouse cadaver skin and treated with 0.5% trypan blue. (Scale Bar: 2 mm) G. Mechanical strength test and H. In vitro curcumin release study for MNs-0, MNs-15, and MNs-30.
In addition, we demonstrated that by controlling the crosslinking time of MN arrays the mechanical and drug release properties can be modulated. The mechanical properties were evaluated by a compression test with a stainless-steel plate. By profiling the applied compressive force and the displacement of the MN arrays, it was demonstrated that longer crosslinking times led to higher density of crosslinked networks that required larger force to achieve similar displacement (Figure 4G). In addition, we monitored in vitro curcumin release from the MNs over a period of 24 h. As shown in Figure 4H, increased crosslinking time prolonged the drug release time. For MNs-0, 90% of curcumin was released after an 8 h incubation while for MNs-30, only approximately 50% of curcumin was released after a 24 h incubation. Increased crosslinking density led to slower release of curcumin due to the density of the hydrogel network. Interestingly, even in the MNs-0 curcumin was gradually released from the needles, likely due to the physical entrapment of in β-CD. In summary, we demonstrated that GelMA-β-CD based MNs have tunable mechanical properties and drug release profiles by simply adjusting the duration of crosslinking.
2.4. In vitro anticancer efficacy of MN arrays on B16F10 cells
Next, we evaluated the anticancer efficacy of curcumin released from MNs-0, MNs-15, and MNs30.[29] The highly invasive mouse melanoma cell line B16F10 was selected as the model cell line. MNs with different crosslinking times were applied to 24 well-plates 24 h after seeding cells, and curcumin uptake was monitored after 6 h of patch application (Figure 5A). We first validated that the cells could effectively uptake the released curcumin (Figure S4, Supporting Information). By monitoring the intensity of curcumin green fluorescence, it was observed that slow release of curcumin from MNs-15 and MNs-30 displayed less uptake compared to the non-crosslinked MNs in terms of curcumin fluorescence intensity. Moreover, after 12 h of further incubation with the MN arrays, B16F10 cells viability was investigated using the CCK-8 assay. As shown in Figure 5B, the MNs-30-treated group displayed the highest cell viability while MNs-0 displayed the lowest cell viability. Furthermore, cell death was visualized by a Live/Dead assay in which live cells are stained with green fluorescent calcein and dead cells are stained with red fluorescent EthD-1. As shown in Figure 5C, cell death occurred in all wells treated with variably crosslinked MN patches. However, the degree of cell death was different, showing the same trend as the previous cell viability assay in which the MNs-30-treated group had less cell death compared to the MNs-0 group. This could be due to the faster release of curcumin from the MNs-0 patch yielding a higher local concentration of drug and inducing more cytotoxicity.
Figure 5.

In vitro anticancer efficacy of MNs on B16F10 cells. A. Timeline and schematic of 2D B16F10 melanoma model study via MN arrays patch. B.CCK-8 assay for quantitatively evaluation of cell viability treated by MNs-0, MNs-15, and MNs-30. C. Live/Dead assay for 2D B16F10 cells treated with MNs-0, MNs-15, and MNs-30; control was with no treatment (Scale bar: 100 μm).
2.5. In vitro anticancer efficacy of MN arrays on 3D B16F10 spheroids
To prove that the fabricated MNs can be used as an effective tool for transdermal delivery of water-insoluble drugs, a 3D skin cancer model was established by embedding the melanoma spheroids in the GelMA hydrogel. Employing a system with 3D tumor spheroids was better than conventional 2D cultures because it provided a more physiologically relevant microenvironment for various anti-cancer drugs.[30] Therefore, we designed a 3D skin cancer screening model by embedding B16F10 spheroids into GelMA hydrogel and applied the MNs (Figure 6A). The GelMA hydrogel functioned as both an extracellular matrix (ECM) for the spheroids as well as a tissue-mimicking substrate on which the MN arrays can be applied. As shown in Figure 6B, fabricated spheroids were embedded in a pre-polymer solution held by a PDMS mold that facilitated later MN application after the solution solidified (Figure S5, Supporting Information). The fabrication process of 3D skin model does not affect cell viability over the study period (Figure S6A, Supporting Information). B16F10 melanoma spheroids were successfully formed after initiation with 8×103 cells and 3 days of incubation. Spheroids were stained with DAPI and a 3D reconstructed image of the melanoma spheroids was visualized by confocal microscopy shown in Figure 6C indicating that the diameter of formed spheroids was ~400 μm. The thickness of the spheroid embedded system was ~ 1 mm and the spheroid was located ~ 200 μm below the surface of the hydrogel. Because height of the needles is ~600 μm, the MNs (Figure 6D) were long enough to penetrate the ECM and effectively deliver the therapeutic to the target site (Figure 6E). The efficacy of the MN arrays was evaluated by comparing responses of the spheroid to a GelMA-β-CD/CUR non-transdermal patch, where the base had the same dimension as the backing layer of the microneedle patch but without the needles (Figure 6F). After 1-day of application, cell viability was screened by Live/Dead assay. The spheroid volume change analysis after treatment indicated GelMA-β-CD/CUR MN patch treatment can inhibit spheroid growth more effectively compared to non-transdermal patch (Figure S6B, Supporting Information).[31] The MN arrays demonstrated improved anticancer efficacy relative to the flat patch due to its deeper penetration into the spheroid model.
Figure 6.

In vitro anticancer efficacy of MNs on 3D B16F10 spheroids. A. Design of curcumin delivery study on 3D skin cancer model. B. Optical image of B16F10 spheroid embedded hydrogel (Scale bar: 5 mm). C. 3D reconstructed image of B16F10 spheroid via confocal microscopy (Scale bar: 100 μm). D. 3D reconstructed image of GelMA-β-CD/CUR MNs via confocal microscopy (Scale bar: 600 μm). E. Fluorescent image of MN arrays applied on the spheroid embedded hydrogel (Scale bar: 200 μm). F. Live/Dead screening for viability of spheroid for (a) blank GelMA-β-CD MN arrays, (b) GelMA-β-CD/CUR non-transdermal patch, and (c) GelMA-β-CD/CUR MN arrays (Scale bar: 100 μm).
2.6. In vivo biocompatibility and biodegradability of GelMA-β-CD MN arrays
To investigate the biocompatibility and biodegradability of the MN arrays, we subcutaneously implanted the patches into C57BL/6J and monitored their degradation in vivo over 14 days. As shown from H&E staining results, all the MN groups tested (GelMA, GelMA mixed with β-CD (GelMA+β-CD), GelMA-β-CD MN) did not cause significant inflammation by day 7. However, it was noted that MN arrays residual area under the skin were significantly different between groups. The conjugation of β-CD onto the GelMA backbone prolonged the degradation process significantly compared to the GelMA and GelMA+β-CD. At Day 7 and Day 14, GelMA-β-CD was degraded slower than GelMA and GelMA+β-CD displayed in Figure 7A and B. The prolonged biodegradation could be result of extra crosslinking of the GelMA chains from non-specific EDC/NHS reactions and steric change of the inner gel microstructure due to the presence of β-CD that reduced enzymatic cleavage of the hybrid material.
Figure 7.

In vivo biocompatibility and biodegradability of GelMA-β-CD MN arrays. A. H&E staining of materials (GelMA, GelMA+β-CD, and GelMA-β-CD displayed within enclosed region) associated skin tissues at Day 7 and Day 14 of post-implantation (Scale bar: 1 mm). B. Plots showing quantification of residual materials within skin tissues at Day 7 and Day 14 of post-implantation (n=5). C. In vivo biocompatibility evaluation of GelMA-β-CD MN arrays with H&E staining at 1 h, Day 1, and Day 3 post-administration of MN arrays. D. H&E staining of GelMA-β-CD implanted skin showing negligible inflammatory cells at Day 7 and Day 14 of post-implantation (Scale bar: 50 μm). E. Immuno-histological staining of immune cells in the tissue. The mice were subcutaneously implanted GelMA-β-CD MN arrays showing no significant local lymphocyte infiltration (CD3) and macrophages (CD68) presence at Day 7 and Day 14 of post-implantation (Scale bar: 50 μm).
Biocompatibility was also assessed through topical application of the MN arrays to the mouse skin. As shown in Figure 7C, GelMA-β-CD MNs penetrated the epidermal layer of the skin after 1-hour of topical implantation. No MN arrays-related damage or inflammation was observed up to 3 days. In addition, CD3 (T-cell), CD68 (macrophage), and CD79A (B cell) immunofluorescence staining revealed that there was no significant immune reaction in the MN-applied dermal area (Figure S6, Supporting Information). Similar to the topical biocompatibility assay, no significant mononuclear cell (CD3+ T-cell or CD68+ macrophage) infiltration was observed around the GelMA-β-CD MN during degradation, as displayed in Figure 7D and E. Based on these results, the MN arrays was biocompatible as gelatin is naturally present within the physiological environment and the conjugation of the additional methacrylate groups and β-cyclodextrin did not induce significant inflammation within the treated area 1 h, 1 day, or 3 days post-administration. Our results indicated biosynthetic GelMA-β-CD MN arrays was biocompatible with the skin and was potentially capable of sustained drug release.
3. Discussion
Hydrogel-based drug delivery systems are particularly attractive and have been applied in multiple pharmaceutical fields for the treatment of cardiovascular diseases, cancer, wounds, and chronic pain. Materials with high water content within hydrophilic networks resemble the structure of native tissue and provide certain level of inherent biocompatibility. Crosslinking within the polymer network can tune the mesh size and porosity to further alter the mechanical and drug release properties of hydrogels.[32] Generally, these advantages can be exploited in hydrogel-based MN arrays. However, these hydrophilic networks are primarily capable of encapsulating water-soluble small molecules or proteins. For example, Lee et al. developed dissolving MN arrays based on carboxymethylcellulose for protein delivery and demonstrated that direct encapsulation of proteins maintained their activity and achieved sustained release.[11] In terms of lipophilic small molecules, it is challenging to directly load these into hydrogel matrices. Most strategies for transdermal delivery of water-insoluble drugs involve the use of liposomes, dendrimers, or microemulsions in the forms of gel or spray that are directly applied as a topical formulation to the skin.[33] In this case, the stratum corneum presents a major barrier to limit efficient drug penetration. These formulations can also cause skin irritation. Therefore, localized delivery of water-insoluble drugs with tissue penetrating MNs is an attractive strategy. Current designs of MN for delivery of water insoluble drugs often incorporate synthetic polymers with high hydrophobicity, such as polylactic acid to enhance drug loading capacity,[34] or embedding secondary nanocarriers to indirectly load water insoluble drugs. Compared with synthetic polymer-based MN patch, GelMA resembles native ECM with high biocompatibility. In terms of the use of secondary carriers, it raises concerns over the stability of them during the drying-based fabrication process.[35]
Since β-CD is a biocompatible molecule and it can camouflage undesirable physiochemical features of drugs, β-CD has been used in several FDA-approved drug delivery formulations, such as Glymesason™ for dexamethasone delivery in treating dermatitis and Indocid™ for indomethacin delivery in relieving pain from arthritis.[36] In addition to small molecules, β-CD is also capable of delivering proteins, and oligonucleotides.[37] Therefore, β-CD can be conjugated with diverse materials for delivering various therapeutics. Two main strategies for creating CD-pendant polymers are: (1) direct polymerization with CD-containing monomers or copolymerization with other monomers;[38] or (2) coupling CD to suitable functional groups on the polymers. In general, the first strategy generates synthetic polymers that are not as biocompatible as natural-derived polymers. The second strategy is mainly used for homopolymers with repeated units, such as dextran,[39] PEG,[40] or chitosan,[41] that allow random functionalization with CD through coupling bonds. In previous studies, little effort was made to conjugate CD onto gelatin even though gelatin is biocompatible and has uniformly distributed RGD sequences that are essential for cell adhesion and elongation. Previously created CD-pendant gelatin[42] achieved either limited or weak crosslinking because the crosslinking mechanism was strictly dependent on β-CD or physical interactions. In addition, both reactions involve the use of organic solvents that may induce cytotoxicity and not suitable for biomedical applications.
Our strategy of further functionalizing GelMA with β-CD in aqueous solvent maintains the tunable photo-crosslinkable properties of the material while mitigating cytotoxicity related to chemical synthesis. Moreover, controlled drug release can be achieved by tuning crosslinking degrees of GelMA-β-CD where fast release can be achieved with low degrees of crosslinking and sustained release can be achieved with high degrees of crosslinking. Prolonged in vivo biodegradation profiles of GelMA-β-CD after crosslinking are also expected to be beneficial for fabrication of sustained drug delivery MN depot. Both fast release and sustained release profiles can be adopted in transdermal drug delivery. For example, cancer immunotherapy delivered by MN patch may benefit from the fast release,[43] while prolonged release is preferred delivery of antibiotics.[44] Therefore, the GelMA-β-CD MN-based platform can be further optimized for different applications. Furthermore, the amphiphilic feature of β-CD maintains an overall apparent hydrophilicity of the hydrogel network, which is crucial for biocompatibility[45] and loading drugs with different physiochemical properties. In the case of vaccination, most vaccines are water soluble but adjuvants can be poorly water-soluble.[46] One study utilized individual MN coating-based method to load both water soluble and insoluble drugs,[47] but such method requires sophisticated microfabrication process. In contrast, GelMA-β-CD MN arrays can be easily fabricated and function as a versatile platform for transdermal delivery.
4. Conclusion
In this study, we demonstrated the feasibility of modifying GelMA with amphiphilic β-cyclodextrin to effectively load water-insoluble drugs, like curcumin, through the formation of drug-polymer inclusion complex via host-guest interactions. The host-guest system can not only carry water-insoluble drugs in aqueous solutions but also improve the stability of the compounds within the cyclodextrin cavity. Such drug-containing polymer solutions can be further fabricated into MNs. The mechanical strength of the MNs is sufficient for skin penetration, which is a prerequisite of transdermal drug delivery devices. In addition, both mechanical properties and drug release profiles can be adjusted by tuning the degree of crosslinking, making the MNs suitable for various applications. Loaded curcumin maintains its anti-cancer activity in both 2D and 3D in vitro study. In addition, the MNs demonstrate the ability to locally deliver therapeutics by penetrating the tissue. These data suggest that GelMA-β-CD based materials combine unique advantages derived from both GelMA and β-cyclodextrin. This material has great potential to improve the delivery of lipophilic compounds and to develop combination therapies of hydrophilic and hydrophobic drugs that can be used to treat a variety of medical conditions in a minimally invasive way.
5. Experimental Section
Materials
Gelatin (Type A, 300 bloom from porcine skin), methacrylic anhydride (MA), carboxymethyl-β-cyclodextrin sodium salt (CM-β-CD), N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC), N-hydroxysuccinimide (NHS), 2-hydroxy-4’-(2-hydroxyethoxy)-2-methylpropiophenone) (UV photoinitiator), curcumin, potassium bromide (KBr) and dimethyl sulfoxide-d6 were purchased from Sigma-Aldrich (Wisconsin, USA). The negative mold of the 11×11 MN (600 μm height and 300 μm base) array in polydimethylsiloxane (PDMS) was purchased from Blueacre Technology (Dundalk, Co Louth, Ireland).
GelMA synthesis
GelMA was synthesized using the established protocol.[48] Briefly, 10 g of type A porcine skin gelatin was slowly added into 100 mL of preheated Dulbecco’s phosphate buffered saline (DPBS) at 50 °C and stirred constantly with a magnetic stir bar until fully dissolved. Afterwards, MA (0.25%, v: v) was slowly added to the gelatin solution under vigorous stirring for 3 h at 50 °C. Finally, the reaction was stopped by adding warm DPBS (100 mL) to the reaction mixture. Unreacted MA and other species were removed by dialysis in distilled water (40 °C). This was continued for one week in dialysis tubing with a molecular weight cut-off of 12–14 kDa. The dialyzed product was filtered and collected in 50 mL conical tubes for lyophilization. After one-week of lyophilization, a white porous foam of GelMA was obtained and stored at −20 °C until further use.
β-CD conjugated GelMA synthesis
GelMA-β-CD was synthesized based on the previously reported method with minor modifications.[23, 49] Briefly, the synthesized GelMA (300 mg) described above was dissolved in 3 mL DPBS (pH = 7.4) and kept in an 80 °C oven to fully dissolve. Different amounts of CM-β-CD (200 mg, 400 mg, and 600 mg) were added into 3 mL MES buffer (2-(N-morpholino) ethanesulfonic acid, pH = 6) followed by the addition of EDC and NHS to activate the carboxyl group for 30 min. After adding 3 mL of the dissolved GelMA solution, the pH of the reaction system was adjusted to 8–9 by adding concentrated NaOH. The reaction was kept with magnetic stirring at 40 °C overnight. Unreacted CM-β-CD and residual salts were removed by dialysis in distilled water for 5 days using dialysis tubing with a molecular weight cut-off of 12–14 kDa. In accordance with the variable amount of CM-β-CD, three types of GelMA-β-CD were obtained and named as low GelMA-β-CD (L), medium GelMA-β-CD (M), and high GelMA-β-CD (H), respectively.
1H NMR
To perform 1H NMR analysis, GelMA, CM-β-CD, and GelMA-β-CD were dissolved in dimethyl sulfoxide-d6 respectively at 30 mg/mL concentration. The 1H NMR spectra were obtained using a Brucker AV400 broad band FT NMR spectrometer with 256 scans at room temperature for each sample. The time domain data (raw data) was processed in Topspin for peak recognition and integration.
β-CD quantization assay
The amount of conjugated β-CD on the GelMA backbone was determined by a previously reported phenol-Sulfuric acid colorimetric assay with minor modifications.[50] Briefly, 20 mg of dried GelMA and GelMA-β-CD (L, M, H) were dissolved in 4 mL of distilled water to prepare stock solutions (5 mg/mL) which were kept in an 80 °C oven until fully dissolved. The stock solutions (5 mg/mL) were further diluted to 0.5 mg/mL for the colorimetric assay. Finally, 30 μL of the diluted solutions from 4 samples were added into a 96-well plate in triplicate, followed by pipetting 150 μL of concentrated sulfuric acid and 30 μL of 5% phenol solution into each microwell sequentially. The loaded 96-well plate was put in an 80 °C oven for 15 min to ensure complete reaction. Samples were cooled to room temperature prior to absorbance measurement. The absorbance at 490 nm was measured using a plate reader (BioTek, Winooski, VT).
GelMA-β-CD and curcumin inclusion complex preparation
To prepare the inclusion complex, excess amount of curcumin (5 mg) was added to pre-dissolved GelMA-β-CD solution in 1 mL DPBS (20%, g/mL) and stirred at 37 °C for 30 min to form the inclusion complex solution where curcumin could be encapsulated within the cavity of CD and the resulting solution was named as GelMA-β-CD/CUR. For comparison of curcumin solubility, GelMA was used as a control and the resulting solution was named as GelMA/CUR. After the equilibrium was reached, the suspension was centrifuged at 12,000 rpm for 10 min; supernatants (GelMA/CUR and GelMA-β-CD/CUR) were separated and further filtered through a 0.22 μm filter. Absorbance at 430 nm for the inclusion complex solutions was measured to directly assess the amount of curcumin within GelMA and GelMA-β-CD. The prepared GelMA-β-CD/CUR solution was also used for aqueous stability study compared to free curcumin dissolved in DPBS (DPBS: ethanol as 4:1). The GelMA-β-CD/CUR solutions were freeze-dried for additional FTIR and DSC analysis.
Fourier transform infrared (FTIR) spectroscopy
To confirm the conjugation of β-CD to GelMA, the FTIR spectra of GelMA, CM-β-CD, and GelMA-β-CD were obtained using a Jasco 420 FTIR spectrophotometer recording over the range of 400–4000 cm−1. To characterize the inclusion complex GelMA-β-CD/CUR, physical mixtures of GelMA-β-CD and curcumin (named as GelMA-β-CD+CUR) were prepared by pulverizing freeze-dried GelMA-β-CD in mortars and mixing with curcumin powder at weight ratio 20:1 by spatula until the homogenous mixtures were obtained. The FTIR spectra of curcumin, GelMA-β-CD, GelMA-β-CD+CUR, and GelMA-β-CD/CUR were recorded over a range of 400–4000 cm−1 with 128 scans and 1 cm−1 resolution. The samples were prepared by mixing each sample with KBr at 1:100 weight ratio and ground into fine powder for pellet preparation.
Differential scanning calorimetry (DSC)
DSC curves of curcumin, GelMA-β-CD, GelMA-β-CD+CUR, and GelMA-β-CD/CUR were obtained using a PerkinElmer differential scanning calorimeter. Each sample (3–5 mg) was sealed within an aluminum pan and heated from 80 to 250 °C at a rate of 5 °C/min under nitrogen flow of 30 mL/min. An empty sealed aluminum pan was used as a reference.
Microneedle fabrication
GelMA-β-CD/CUR MN were prepared using a centrifugation method. Briefly, GelMA-β-CD/CUR inclusion complex solution was prepared as above and 5 mg of UV photoinitiator was added into 1 mL GelMA-β-CD/CUR solution (0.5%, g/mL) for MN fabrication. Negative PDMS MN array molds were put inside a 6 well plate and 100 μL of GelMA-β-CD/CUR solution was added on the top of the MN array mold. The well plate was centrifuged at 3500 rpm for 15 min at 37 °C in order to completely fill the mold and remove bubbles. The resulting MNs were exposed to 350 mW/cm2 UV light (360–480 nm) for predetermined durations: 0, 15, and 30s (named as MNs-0, MNs-15, MNs-30). The MN arrays were detached from the PDMS mold manually after overnight drying shielded from light at room temperature. The GelMA-β-CD/CUR non-transdermal patch was fabricated by pipetting 80 μL GelMA-β-CD/CUR inclusion complex solution into a 1 cm X 1cm PDMS mold followed by the same procedure used for the MNs described above.
Mechanical strength test
Mechanical testing of the MNs was performed using a 5943 Microtester Instron with a stress-strain gauge. The relationship between the applied force and deformation of the MN tips was recorded and profiled for MNs-5, MNs-15, and MNs-30. First, the MNs were placed on a glass slide with the tips pointing upward, facing the stainless-steel plate 1.5 mm above. The maximum loading force was set to 45.0 N and the stainless-steel plate was moved downward at a constant speed of 0.5 mm/min. The mechanical properties of MNs-5, MNs-15, and MNs-30 were profiled and analyzed.
Ex vivo Skin penetration
Cadaver skin from a mouse was used to perform skin penetration test. The MNs were pushed into the skin with 20 N of force for 30 seconds. Trypan blue (0.5%) was used to stain the penetrated skin for 10 min at room temperature. After washing the stained skin with DPBS three times, the trypan blue-stained MNs-treated skin sample was imaged.
Release of curcumin in vitro
The in vitro release of curcumin from the MNs was evaluated by immersing MNs patch in 1 mL of release media at 37 °C in Eppendorf tubes. Release media was prepared by mixing DPBS buffer and DMSO at a 4:1 volume ratio in order to facilitate the dissolution of hydrophobic curcumin. At predetermined times, 10 μL of the sample was withdrawn for absorbance measurement and 10 μL of fresh release media was replaced into the Eppendorf tube which was returned to the incubator. The absorbance of samples at each predetermined time point were detected by Nanodrop at 430 nm and the concentration was interpolated from a curcumin standard curve dissolved in the same media.
In vitro anticancer efficacy
The anticancer efficacy of the released curcumin from MNs was evaluated by using the B16F10 melanoma cell line. Briefly, cultured B16F10 cells were seeded in 24-well plates and incubated to suitable cell density. MNs with different crosslinking times were added into each well and the cells were incubated for another 18 h. The viability of B16F10 cells after treatment with MNs patches was evaluated by using the CCK-8 assay (Thermo Fisher Scientific, MA, USA) according to the manufacturer’s protocol. Live/Dead staining (Thermo Fisher Scientific, MA, USA) was utilized to investigate the viability of B16F10 cells following the manufacturer’s protocol and was subsequently imaged with a fluorescent microscope (Zeiss, Sweden).
In vitro anticancer efficacy on 3D cancer spheroids
B16F10 tumor spheroids were formed and cultured according to a previously reported strategy with minor modifications.[51] Briefly, 50 μL of hot 1.5% (g/mL) agarose solution was distributed into a 96 well plate and transferred to a 4 °C fridge to gel prior to cell seeding. Cultured B16F10 cells were suspended and diluted to a density of 8 × 104 cells mL−1. 100 μL of the cell suspension was then added to the agarose pre-coated 96 well plate. The cell seeded 96 well plate was centrifuged at 600×g for 10 min to initiate the formation of the B16F10 spheroids. After allowing 3 days for spheroid formation, the spheroids were embedded in GelMA by transferring spheroids into 10% (g/mL) GelMA prepolymer solution followed by 15 s of crosslinking under UV light. The previously fabricated MNs-15 and GelMA-β-CD/CUR non-transdermal patch were applied to the surface of the spheroid-laden hydrogels. Live/dead staining (following the manufacture’s protocol) was used to quantify the viability of the spheroids by imaging using a fluorescent microscope (Zeiss, Sweden).
In vivo biodegradation and biocompatibility
All animal experiments were approved by the UCLA Animal Research Committee. The animal experiments were conducted in alignment with relevant guidelines. Seven-week-old, C57BL/6J male mice (average weight: 20 g) were purchased from Jackson Laboratory (Sacramento, CA) and housed in an approved animal facility. For the biodegradability test, GelMA, GelMA mixed with CM-β-CD (GelMA+β-CD), GelMA-β-CD MNs were implanted in mice under inhalation anesthesia (1.5% isoflurane in 100% O2). A 1 cm incision was made on the posterior dorsal skin and a 1cm X 1cm dry patch was subcutaneously implanted for 7 and 14 days. To determine the capability of MNs to penetrate skin and to identify side effects (biocompatibility), GelMA-β-CD based MNs were also topically applied for 1 hour, 1 day, and 3 days.
Histological analysis and immunofluorescent staining
To analyze the response of the host skin tissue to the application of the MNs, animals were sacrificed using CO2. Skin tissue subject to MNs treatment was immediately collected and fixed in 10% neutral buffered formalin (Leica Biosystems, IL, USA). Fixed tissues were processed using standard methods and embedded in paraffin. 4 μm skin tissue sections were stained with hematoxylin (Leica Biosystems) and eosin (Sigma) (H&E) staining. Histology images were acquired with a Nikon inverted microscope. Quantitative data such as percentage of residual microneedle area was measured using the AmScope image analysis software (AmScope, Irvine, CA, USA). Additionally, serial formalin-fixed tissue sections were used for immunofluorescence staining. The sections were deparaffinized and underwent heat-induced antigen retrieval according to the protocol. Sections were soaked in antigen retrieval buffer, permeabilized in PBST (PBS + 0.3% Triton), and blocked with Goat serum for 30 min. Then, sections were incubated overnight at 4 °C with primary antibodies targeting CD3 (Rat, 1: 200; Abcam, Cambridge, UK), CD68 (Rat, 1: 200; Abcam), and CD79A (Rat, 1:200; Santa Cruz Biotechnology). After incubation, the sections were rinsed with PBST and incubated with 1: 1000 diluted Alexa 555-conjugated secondary antibody (Thermo Fisher Scientific, MA, USA) at room temperature for 60 min and counterstained with 4’,6-Diamidino-2-Phenylindole, Dihydrochloride (DAPI) (Thermo Fisher Scientific, MA, USA) for 5 min. The fluorescent images were collected using a Nikon Eclipse Ti-S Inverted Phase Contrast Fluorescent Microscope.
Statistical analysis
All data is presented as the mean ± standard deviation (SD). Student’s t-test were performed for statistical significance. Significance is denoted in the figures as *p < 0.05, **p < 0.01 and ***p < 0.001.
Supplementary Material
Acknowledgements
X. Zhou and Z. Luo contribute equally to this work. The authors declare no conflict of interests. This work has been supported by the National Institutes of Health (1R01EB024403–01, 1R01GM126831–01). The authors acknowledge scientific discussions with Dr. Samanvaya Srivastsva.
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Contributor Information
Xingwu Zhou, Department of Bioengineering, Henry Samueli School of Engineering and Applied Sciences, University of California, Los Angeles, Los Angeles, CA 90095, USA; Center for Minimally Invasive Therapeutics (C-MIT), University of California, Los Angeles, Los Angeles, CA 90095, USA; California NanoSystems Institute, University of California, Los Angeles, Los Angeles, CA90095, USA; Department of Chemical and Biomolecular Engineering, Henry Samueli School of Engineering and Applied Sciences, University of California, Los Angeles, Los Angeles, CA 90095, USA.
Zhimin Luo, Department of Bioengineering, Henry Samueli School of Engineering and Applied Sciences, University of California, Los Angeles, Los Angeles, CA 90095, USA; Center for Minimally Invasive Therapeutics (C-MIT), University of California, Los Angeles, Los Angeles, CA 90095, USA; California NanoSystems Institute, University of California, Los Angeles, Los Angeles, CA90095, USA; School of Pharmacy, Xi’an Jiaotong University, Xi’an, 710061, China.
Avijit Baidya, Department of Bioengineering, Henry Samueli School of Engineering and Applied Sciences, University of California, Los Angeles, Los Angeles, CA 90095, USA; Center for Minimally Invasive Therapeutics (C-MIT), University of California, Los Angeles, Los Angeles, CA 90095, USA; California NanoSystems Institute, University of California, Los Angeles, Los Angeles, CA90095, USA.
Hanjun Kim, Department of Bioengineering, Henry Samueli School of Engineering and Applied Sciences, University of California, Los Angeles, Los Angeles, CA 90095, USA; Center for Minimally Invasive Therapeutics (C-MIT), University of California, Los Angeles, Los Angeles, CA 90095, USA; California NanoSystems Institute, University of California, Los Angeles, Los Angeles, CA90095, USA.
Canran Wang, Department of Bioengineering, Henry Samueli School of Engineering and Applied Sciences, University of California, Los Angeles, Los Angeles, CA 90095, USA; Center for Minimally Invasive Therapeutics (C-MIT), University of California, Los Angeles, Los Angeles, CA 90095, USA; California NanoSystems Institute, University of California, Los Angeles, Los Angeles, CA90095, USA.
Xing Jiang, Department of Bioengineering, Henry Samueli School of Engineering and Applied Sciences, University of California, Los Angeles, Los Angeles, CA 90095, USA; Center for Minimally Invasive Therapeutics (C-MIT), University of California, Los Angeles, Los Angeles, CA 90095, USA; California NanoSystems Institute, University of California, Los Angeles, Los Angeles, CA90095, USA; School of Nursing, Nanjing University of Chinese Medicine, Nanjing, 210023, China.
Moyuan Qu, Department of Bioengineering, Henry Samueli School of Engineering and Applied Sciences, University of California, Los Angeles, Los Angeles, CA 90095, USA; Center for Minimally Invasive Therapeutics (C-MIT), University of California, Los Angeles, Los Angeles, CA 90095, USA; California NanoSystems Institute, University of California, Los Angeles, Los Angeles, CA90095, USA; State Key Laboratory of Oral Diseases, National Clinical Research Center for Oral Diseases, West China Hospital of Stomatology, Sichuan University, Chengdu, 610041, China.
Jixiang Zhu, Department of Bioengineering, Henry Samueli School of Engineering and Applied Sciences, University of California, Los Angeles, Los Angeles, CA 90095, USA; Center for Minimally Invasive Therapeutics (C-MIT), University of California, Los Angeles, Los Angeles, CA 90095, USA; California NanoSystems Institute, University of California, Los Angeles, Los Angeles, CA90095, USA; Department of Biomedical Engineering, School of Basic Medical Sciences, Guangzhou Medical University, Guangzhou, 511436, China.
Li Ren, Department of Bioengineering, Henry Samueli School of Engineering and Applied Sciences, University of California, Los Angeles, Los Angeles, CA 90095, USA; Center for Minimally Invasive Therapeutics (C-MIT), University of California, Los Angeles, Los Angeles, CA 90095, USA; California NanoSystems Institute, University of California, Los Angeles, Los Angeles, CA90095, USA.
Fereshteh Vajhadin, Department of Bioengineering, Henry Samueli School of Engineering and Applied Sciences, University of California, Los Angeles, Los Angeles, CA 90095, USA; Center for Minimally Invasive Therapeutics (C-MIT), University of California, Los Angeles, Los Angeles, CA 90095, USA; California NanoSystems Institute, University of California, Los Angeles, Los Angeles, CA90095, USA; Department of Chemistry, Yazd University, Yazd, 89195, Iran.
Peyton Tebon, Department of Bioengineering, Henry Samueli School of Engineering and Applied Sciences, University of California, Los Angeles, Los Angeles, CA 90095, USA; Center for Minimally Invasive Therapeutics (C-MIT), University of California, Los Angeles, Los Angeles, CA 90095, USA; California NanoSystems Institute, University of California, Los Angeles, Los Angeles, CA90095, USA.
Niyuan Zhang, Department of Bioengineering, Henry Samueli School of Engineering and Applied Sciences, University of California, Los Angeles, Los Angeles, CA 90095, USA; Center for Minimally Invasive Therapeutics (C-MIT), University of California, Los Angeles, Los Angeles, CA 90095, USA; California NanoSystems Institute, University of California, Los Angeles, Los Angeles, CA90095, USA.
Yumeng Xue, Department of Bioengineering, Henry Samueli School of Engineering and Applied Sciences, University of California, Los Angeles, Los Angeles, CA 90095, USA; Center for Minimally Invasive Therapeutics (C-MIT), University of California, Los Angeles, Los Angeles, CA 90095, USA; California NanoSystems Institute, University of California, Los Angeles, Los Angeles, CA90095, USA.
Yudi Feng, Department of Bioengineering, Henry Samueli School of Engineering and Applied Sciences, University of California, Los Angeles, Los Angeles, CA 90095, USA; Center for Minimally Invasive Therapeutics (C-MIT), University of California, Los Angeles, Los Angeles, CA 90095, USA; California NanoSystems Institute, University of California, Los Angeles, Los Angeles, CA90095, USA.
Chengbin Xue, Department of Bioengineering, Henry Samueli School of Engineering and Applied Sciences, University of California, Los Angeles, Los Angeles, CA 90095, USA; Center for Minimally Invasive Therapeutics (C-MIT), University of California, Los Angeles, Los Angeles, CA 90095, USA; California NanoSystems Institute, University of California, Los Angeles, Los Angeles, CA90095, USA; School of Dentistry, The University of Queensland, Herston, QLD 4006, Australia.
Yi Chen, Department of Bioengineering, Henry Samueli School of Engineering and Applied Sciences, University of California, Los Angeles, Los Angeles, CA 90095, USA; Center for Minimally Invasive Therapeutics (C-MIT), University of California, Los Angeles, Los Angeles, CA 90095, USA; California NanoSystems Institute, University of California, Los Angeles, Los Angeles, CA90095, USA.
KangJu Lee, Department of Bioengineering, Henry Samueli School of Engineering and Applied Sciences, University of California, Los Angeles, Los Angeles, CA 90095, USA; Center for Minimally Invasive Therapeutics (C-MIT), University of California, Los Angeles, Los Angeles, CA 90095, USA; California NanoSystems Institute, University of California, Los Angeles, Los Angeles, CA90095, USA.
Junmin Lee, Department of Bioengineering, Henry Samueli School of Engineering and Applied Sciences, University of California, Los Angeles, Los Angeles, CA 90095, USA; Center for Minimally Invasive Therapeutics (C-MIT), University of California, Los Angeles, Los Angeles, CA 90095, USA; California NanoSystems Institute, University of California, Los Angeles, Los Angeles, CA90095, USA.
Shiming Zhang, Department of Bioengineering, Henry Samueli School of Engineering and Applied Sciences, University of California, Los Angeles, Los Angeles, CA 90095, USA; Center for Minimally Invasive Therapeutics (C-MIT), University of California, Los Angeles, Los Angeles, CA 90095, USA; California NanoSystems Institute, University of California, Los Angeles, Los Angeles, CA90095, USA.
Chun Xu, Department of Bioengineering, Henry Samueli School of Engineering and Applied Sciences, University of California, Los Angeles, Los Angeles, CA 90095, USA; Center for Minimally Invasive Therapeutics (C-MIT), University of California, Los Angeles, Los Angeles, CA 90095, USA; California NanoSystems Institute, University of California, Los Angeles, Los Angeles, CA90095, USA.
Nureddin Ashammakhi, Department of Bioengineering, Henry Samueli School of Engineering and Applied Sciences, University of California, Los Angeles, Los Angeles, CA 90095, USA; Center for Minimally Invasive Therapeutics (C-MIT), University of California, Los Angeles, Los Angeles, CA 90095, USA; California NanoSystems Institute, University of California, Los Angeles, Los Angeles, CA90095, USA; Department of Radiology, David Geffen School of Medicine, University of California, Los Angeles, Los Angeles, CA 90095, USA; Terasaki Institute for Biomedical Innovation, Los Angeles, CA 90064, USA.
Samad Ahadian, Department of Bioengineering, Henry Samueli School of Engineering and Applied Sciences, University of California, Los Angeles, Los Angeles, CA 90095, USA; Center for Minimally Invasive Therapeutics (C-MIT), University of California, Los Angeles, Los Angeles, CA 90095, USA; California NanoSystems Institute, University of California, Los Angeles, Los Angeles, CA90095, USA.
Mehmet Remzi Dokmeci, Department of Bioengineering, Henry Samueli School of Engineering and Applied Sciences, University of California, Los Angeles, Los Angeles, CA 90095, USA; Center for Minimally Invasive Therapeutics (C-MIT), University of California, Los Angeles, Los Angeles, CA 90095, USA; California NanoSystems Institute, University of California, Los Angeles, Los Angeles, CA90095, USA; Department of Radiology, David Geffen School of Medicine, University of California, Los Angeles, Los Angeles, CA 90095, USA; Terasaki Institute for Biomedical Innovation, Los Angeles, CA 90064, USA.
Zhen Gu, Department of Bioengineering, Henry Samueli School of Engineering and Applied Sciences, University of California, Los Angeles, Los Angeles, CA 90095, USA; Center for Minimally Invasive Therapeutics (C-MIT), University of California, Los Angeles, Los Angeles, CA 90095, USA; California NanoSystems Institute, University of California, Los Angeles, Los Angeles, CA90095, USA; Jonsson Comprehensive Cancer Center, University of California, Los Angeles, Los Angeles, CA90095, USA; California NanoSystems Institute, University of California, Los Angeles, Los Angeles, CA90095, USA.
Wujin Sun, Department of Bioengineering, Henry Samueli School of Engineering and Applied Sciences, University of California, Los Angeles, Los Angeles, CA 90095, USA; Center for Minimally Invasive Therapeutics (C-MIT), University of California, Los Angeles, Los Angeles, CA 90095, USA; California NanoSystems Institute, University of California, Los Angeles, Los Angeles, CA90095, USA.
Ali Khademhosseini, Department of Bioengineering, Henry Samueli School of Engineering and Applied Sciences, University of California, Los Angeles, Los Angeles, CA 90095, USA; Center for Minimally Invasive Therapeutics (C-MIT), University of California, Los Angeles, Los Angeles, CA 90095, USA; California NanoSystems Institute, University of California, Los Angeles, Los Angeles, CA90095, USA; Jonsson Comprehensive Cancer Center, University of California, Los Angeles, Los Angeles, CA90095, USA; California NanoSystems Institute, University of California, Los Angeles, Los Angeles, CA90095, USA; Department of Radiology, David Geffen School of Medicine, University of California, Los Angeles, Los Angeles, CA 90095, USA; Terasaki Institute for Biomedical Innovation, Los Angeles, CA 90064, USA.
References
- [1].Sun W, Hu Q, Ji W, Wright G, Gu Z, Physiol. Rev 2016, 97, 189. [Google Scholar]
- [2].Anselmo AC, Gokarn Y, Mitragotri S, Nat. Rev. Drug Discovery 2018, 18, 19. [DOI] [PubMed] [Google Scholar]
- [3].a) Kim Y-C, Park J-H, Prausnitz MR, Adv. Drug Del. Rev 2012, 64, 1547; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Prausnitz MR, Mitragotri S, Langer R, Nat. Rev. Drug Discovery 2004, 3, 115. [DOI] [PubMed] [Google Scholar]
- [4].Sun W, Lee J, Zhang S, Benyshek C, Dokmeci MR, Khademhosseini A, Adv. Sci 2019, 6, 1801039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Yu J, Zhang Y, Ye Y, DiSanto R, Sun W, Ranson D, Ligler FS, Buse JB, Gu Z, Proc. Natl. Acad. Sci. USA 2015, 112, 8260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Wang C, Ye Y, Hochu GM, Sadeghifar H, Gu Z, Nano Lett. 2016, 16, 2334. [DOI] [PubMed] [Google Scholar]
- [7].Ye Y, Wang C, Zhang X, Hu Q, Zhang Y, Liu Q, Wen D, Milligan J, Bellotti A, Huang L, Dotti G, Gu Z, Sci. Immunol 2017, 2, eaan5692. [DOI] [PubMed] [Google Scholar]
- [8].a) Paul R, Saville AC, Hansel JC, Ye Y, Ball C, Williams A, Chang X, Chen G, Gu Z, Ristaino JB, Wei Q, ACS Nano 2019, 13, 6540; [DOI] [PubMed] [Google Scholar]; b) Kolluru C, Gupta R, Jiang Q, Williams M, Gholami Derami H, Cao S, Noel RK, Singamaneni S, Prausnitz MR, ACS Sensors 2019, 4, 1569; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Mandal A, Boopathy AV, Lam LKW, Moynihan KD, Welch ME, Bennett NR, Turvey ME, Thai N, Van JH, Love JC, Hammond PT, Irvine DJ, Sci. Transl. Med 2018, 10, eaar2227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Li W, Terry RN, Tang J, Feng MR, Schwendeman SP, Prausnitz MR, Nat. Biomed. Eng 2019, 3, 220. [DOI] [PubMed] [Google Scholar]
- [10].Al Sulaiman D, Chang JYH, Bennett NR, Topouzi H, Higgins CA, Irvine DJ, Ladame S, ACS Nano 2019, 13, 9620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Lee JW, Park J-H, Prausnitz MR, Biomaterials 2008, 29, 2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Luo Z, Sun W, Fang J, Lee K, Li S, Gu Z, Dokmeci MR, Khademhosseini A, Adv. Healthcare Mater 2019, 8, 1801054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Zhang Y, Feng P, Yu J, Yang J, Zhao J, Wang J, Shen Q, Gu Z, Adv. Ther 2018, 1, 1800035. [Google Scholar]
- [14].Larrañeta E, Stewart S, Ervine M, Al-Kasasbeh R, Donnelly FR, J. Funct. Biomater 2018, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Kalepu S, Nekkanti V, Acta Pharm. Sin. B 2015, 5, 442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Tanigo T, Takaoka R, Tabata Y, Control J Release 2010, 143, 201. [DOI] [PubMed] [Google Scholar]
- [17].Nichol JW, Koshy ST, Bae H, Hwang CM, Yamanlar S, Khademhosseini A, Biomaterials 2010, 31, 5536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Noshadi I, Hong S, Sullivan KE, Shirzaei Sani E, Portillo-Lara R, Tamayol A, Shin SR, Gao AE, Stoppel WL, Black Iii LD, Khademhosseini A, Annabi N, Biomater. Sci 2017, 5, 2093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].a) Yue K, Trujillo-de Santiago G, Alvarez MM, Tamayol A, Annabi N, Khademhosseini A, Biomaterials 2015, 73, 254; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Liu W, Heinrich MA, Zhou Y, Akpek A, Hu N, Liu X, Guan X, Zhong Z, Jin X, Khademhosseini A, Zhang YS, Adv. Healthcare Mater 2017, 6, 1601451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Davis ME, Brewster ME, Nat. Rev. Drug Discovery 2004, 3, 1023. [DOI] [PubMed] [Google Scholar]
- [21].a) Crini G, Chem. Rev 2014, 114, 10940; [DOI] [PubMed] [Google Scholar]; b) Mester U, Lohmann C, Pleyer U, Steinkamp G, Völcker E, Kruger H, Raj PS, Drugs in R & D 2002, 3, 143. [DOI] [PubMed] [Google Scholar]
- [22].Brinkman WT, Nagapudi K, Thomas BS, Chaikof EL, Biomacromolecules 2003, 4, 890. [DOI] [PubMed] [Google Scholar]
- [23].Li R, Zhang X, Zhang Q, Liu H, Rong J, Tu M, Zeng R, Zhao J, J. Appl. Polym. Sci 2016, 133. [Google Scholar]
- [24].Mohan PRK, Sreelakshmi G, Muraleedharan CV, Joseph R, Vib. Spectrosc 2012, 62, 77. [Google Scholar]
- [25].Rachmawati H, Edityaningrum CA, Mauludin R, AAPS PharmSciTech 2013, 14, 1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Yadav VR, Suresh S, Devi K, Yadav S, AAPS PharmSciTech 2009, 10, 752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].a) Rachmawati H, Edityaningrum CA, Mauludin R, AAPS PharmSciTech 2013, 14, 1303; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Chen J, Qin X, Zhong S, Chen S, Su W, Liu Y, Molecules (Basel, Switzerland) 2018, 23, 1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Wang Y-J, Pan M-H, Cheng A-L, Lin L-I, Ho Y-S, Hsieh C-Y, Lin J-K, J. Pharm. Biomed. Anal 1997, 15, 1867. [DOI] [PubMed] [Google Scholar]
- [29].Tomeh MA, Hadianamrei R, Zhao X, Int. J. Mol. Sci. 2019, 20, 1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Langhans SA, Front. Pharmacol 2018, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].a) Au - Chen W, Au - Wong C, Au - Vosburgh E, Au - Levine AJ, Au - Foran DJ, Au - Xu EY, JoVE 2014, e51639; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Friedrich J, Seidel C, Ebner R, Kunz-Schughart LA, Nat. Protoc 2009, 4, 309. [DOI] [PubMed] [Google Scholar]
- [32].a) Zhang YS, Khademhosseini A, Science 2017, 356, eaaf3627; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Li J, Mooney DJ, Nat. Rev. Mater 2016, 1, 16071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Morgan TM, Reed BL, Finnin BC, Journal of Pharmaceutical Sciences 1998, 87, 1213. [DOI] [PubMed] [Google Scholar]
- [34].Chen Y, Chen BZ, Wang QL, Jin X, Guo XD, J. Control. Release 2017, 265, 14. [DOI] [PubMed] [Google Scholar]
- [35].Abdelwahed W, Degobert G, Stainmesse S, Fessi H, Adv. Drug Del. Rev 2006, 58, 1688. [DOI] [PubMed] [Google Scholar]
- [36].Brewster ME, Loftsson T, Adv. Drug Del. Rev 2007, 59, 645. [DOI] [PubMed] [Google Scholar]
- [37].Zhang J, Ma PX, Adv. Drug Del. Rev 2013, 65, 1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Munteanu M, Choi S, Ritter H, Macromolecules 2008, 41, 9619. [Google Scholar]
- [39].Xiao W, Chen W-H, Zhang J, Li C, Zhuo R-X, Zhang X-Z, J. Phys. Chem. B 2011, 115, 13796. [DOI] [PubMed] [Google Scholar]
- [40].Zhang X, Zhang X, Wu Z, Gao X, Cheng C, Wang Z, Li C, Acta Biomater. 2011, 7, 585. [DOI] [PubMed] [Google Scholar]
- [41].Liu Y, Yu Z-L, Zhang Y-M, Guo D-S, Liu Y-P, J. Am. Chem. Soc 2008, 130, 10431. [DOI] [PubMed] [Google Scholar]
- [42].a) Liu C, Zhang Z, Liu X, Ni X, Li J, RSC Advances 2013, 3, 25041; [Google Scholar]; b) Xu J, Feng Q, Lin S, Yuan W, Li R, Li J, Wei K, Chen X, Zhang K, Yang Y, Wu T, Wang B, Zhu M, Guo R, Li G, Bian L, Biomaterials 2019, 210, 51. [DOI] [PubMed] [Google Scholar]
- [43].Lopez-Ramirez MA, Soto F, Wang C, Rueda R, Shukla S, Silva-Lopez C, Kupor D, McBride DA, Pokorski JK, Nourhani A, Steinmetz NF, Shah NJ, Wang J, Adv. Mater 2020, 32, 1905740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Tsioris K, Raja WK, Pritchard EM, Panilaitis B, Kaplan DL, Omenetto FG, Adv. Funct. Mater 2012, 22, 330. [Google Scholar]
- [45].Hezi-Yamit A, Sullivan C, Wong J, David L, Chen M, Cheng P, Shumaker D, Wilcox JN, Udipi K, J. Biomed. Mater. Res. A 2009, 90A, 133. [DOI] [PubMed] [Google Scholar]
- [46].Reed SG, Orr MT, Fox CB, Nat. Med 2013, 19, 1597. [DOI] [PubMed] [Google Scholar]
- [47].Li S, Li W, Prausnitz M, Drug Deliv Transl Res 2018, 8, 1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Yue K, Li X, Schrobback K, Sheikhi A, Annabi N, Leijten J, Zhang W, Zhang YS, Hutmacher DW, Klein TJ, Khademhosseini A, Biomaterials 2017, 139, 163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Nakajima N, Ikada Y, Bioconjug. Chem 1995, 6, 123. [DOI] [PubMed] [Google Scholar]
- [50].a) Masuko T, Minami A, Iwasaki N, Majima T, Nishimura S-I, Lee YC, Anal. Biochem 2005, 339, 69; [DOI] [PubMed] [Google Scholar]; b) Xu J, Li X, Sun F, Acta Biomater. 2010, 6, 486. [DOI] [PubMed] [Google Scholar]
- [51].Zanoni M, Piccinini F, Arienti C, Zamagni A, Santi S, Polico R, Bevilacqua A, Tesei A, Sci. Rep 2016, 6, 19103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.