

Abstract
Evidence has shown that certain methylation markers derived from blood can mirror corresponding methylation signatures in internal tissues. In the current study, we aimed to investigate two strong epigenetic predictors for life span, derived from blood DNA methylation data, in tissue samples of solid cancer patients. Using data from the Cancer Genome Atlas (TCGA) and the German DACHS study, we compared a mortality risk score (MRscore) and DNAmPhenoAge in paired tumor and adjacent normal tissue samples of patients with lung (N = 69), colorectal (n = 299), breast (n = 90), head/neck (n = 50), prostate (n = 50), and liver (n = 50) cancer. To explore the concordance across tissue and blood, we additionally assessed the two markers in blood samples of colorectal cancer (CRC) cases and matched controls (n = 93) in the DACHS+ study. The MRscore was significantly elevated in tumor tissues compared to normal tissues of all cancers except prostate cancer, for which an opposite pattern was observed. DNAmPhenoAge was consistently higher in all tumor tissues. The MRscore discriminated lung, colorectal, and prostate tumor tissues from normal tissues with very high accuracy [AUCs of 0.87, 0.99 (TCGA) /0.94 (DACHS), and 0.92, respectively]. DNAmPhenoAge accurately discriminated five types of tumor tissues from normal tissues (except prostate cancer), with AUCs of 0.82–0.93. The MRscore was also significantly higher in blood samples of CRC cases than in controls, with areas under the curve (AUC) of 0.74, whereas DNAmPhenoAge did not distinguish cases from controls, with AUC of 0.54. This study provides compelling evidence that blood‐derived DNAm markers could reflect methylation changes in less accessible tissues. Further research should explore the potential use of these findings for cancer diagnosis and early detection.
Keywords: blood and tissues, cancer, DNA methylation, DNAmPhenoAge, mortality risk score
In this study, methylation predictors of mortality, derived from blood DNA methylation analysis, were found to be remarkably elevated in tumor tissue, when compared to adjacent normal tissue of multiple organs. In patients with colorectal cancer, the mortality methylation predictor pattern was consistent across tumor tissue and peripheral blood. Thus, blood‐derived methylation markers could mirror tumorigenesis‐related methylation changes in less accessible tissues.

Abbreviations
- AUCs
areas under the curve
- BC
breast cancer
- CI
confidence interval
- CRC
colorectal cancer
- CRP
C‐reactive protein
- DNAm
deoxyribonucleic acid methylation
- EWAS
epigenome‐wide association study
- LC
lung cancer
- MRscore
mortality risk score
- ROC curves
receiver operating characteristic curves
- TILs
tumor‐infiltrating leukocytes
1. Introduction
DNA methylation (DNAm), one of the key epigenetic processes, plays a critical role in tissue and cellular differentiation such that different tissues are characterized by distinct DNAm patterns [1,2]. Studies, however, via either direct comparing DNAm profiles across tissues [2,3] or exploring phenotype‐related DNAm alterations across tissues (such as adipose or brain tissues vs. blood, and buccal cells vs. blood), have shown concordance or correlation of a subset of methylation signatures between tissues [4, 5, 6, 7, 8], suggesting the potential of blood DNAm as a surrogate measure of methylation at less accessible internal tissues. In the field of aging research, Teschendorff et al. identified an age‐dependent signature based on 69 CpGs mapping to promoter of polycomb group proteins, which is common not only to multiple tissues including blood and epithelial tissues, but also to the process of carcinogenesis [9]. Horvath developed a 353‐CpG‐based multitissue age predictor, known as ‘Epigenetic clock’, which can accurately predict age across a variety of tissues (e.g., whole blood, blood mononuclear cells, colon, adipose, liver, and lung) [10]. These findings support the hypothesis that robust DNAm markers derived from blood are able to mirror those in solid tissues.
Recently, two strong predictors for life span (time‐to‐death due to all‐cause mortality) have been developed using blood DNAm profiles [11,12]. We first derived a 10‐CpG‐based mortality risk score (MRscore) from a whole blood epigenome‐wide association study (EWAS) of mortality [12]. The MRscore was shown to strongly predict all‐cause and cause‐specific mortality, and to strongly correlate with other well‐established aging indicators (such as telomere length, oxidative stress, frailty index, and the epigenetic clock), while outperforming these indicators in survival prediction [13, 14, 15]. The MRscore was first derived and validated in two large German cohorts, and its strong association with mortality has subsequently been confirmed in multiple large cohort studies from the United States, such as the Framingham Heart Study, the Women's Health Initiative, and the Normative Aging Study [11,16,17]. Later, Levine et al. [11] developed another survival predictor by regressing a phenotypic measure of mortality risk on 513 CpGs, that is, DNAmPhenoAge, which showed an association with mortality as strongly as that for the MRscore. Although it was also developed based on whole blood samples, the DNAmPhenoAge correlated to various extents with chronological age in multiple tissues (r = 0.47–0.92). A question of potential high clinical interest is to what extent methylation changes identified in blood to be predictive of major health outcomes would reflect methylation changes on tissue levels in the pathogenesis of major diseases, such as various cancers. The aim of the current study was to evaluate whether and to what extent the MRscore and DNAmPhenoAge would differ between cancer tissue and adjacent normal tissue for six common cancers, including lung, colorectal, breast, head/neck, prostate, and liver cancer. In parallel, we comparatively evaluated the epigenetic clock‐derived age acceleration, that is, DNAmAge acceleration, a well‐studied epigenetic marker derived from multitissue analysis, which has been shown to correlate with a wide spectrum of health conditions [18, 19, 20]. To assess the concordance from solid tissue to peripheral blood, we additionally analyzed the three markers in blood samples of colorectal cancer (CRC) cases and matched controls.
2. Materials and methods
2.1. Study population
The analysis of paired tumor‐normal tissues of six types of cancers was based on the Cancer Genome Atlas (TCGA) data, where DNAm data for tumor/adjacent normal tissues were extracted from patients with lung (n = 833/69), colorectal (n = 367/45), breast (n = 789/90), head/neck (n = 528/50), prostate (n = 498/50), and liver (n = 377/50) cancer. To preclude the impact of interindividual variation in DNAm, our analyses focused only on corresponding matched tumor and normal tissues from the same patients.
In addition, DNAm data of paired tumor and adjacent normal tissues of CRC patients were also drawn from the German DACHS study, an ongoing large population‐based case–control study on CRC. The DACHS study enrolls patients with histologically confirmed CRC from 22 hospitals in the Rhine–Neckar–Odenwald region in southwestern Germany [21,22]. For 254 patients diagnosed between 2003 and 2007, genome‐wide DNAm assessment in paired tumor‐normal tissues was available and included in the current study. The study was approved by the ethical committees of the University of Heidelberg and of the Medical Chambers of Baden‐Württemberg and Rhineland‐Palatinate. Written informed consent was obtained from each participant.
The analysis in blood samples was carried out in the German DACHS+ study, a satellite substudy to the DACHS study. In brief, the DACHS+ study recruited 819 CRC patients (age 55–75 years) referred by general practitioners or gastroenterologists for surgery to four hospitals in and around Heidelberg after diagnosis but before initiation of treatment between October 2006 and December 2014 [23]. Blood samples were obtained before surgery. Epigenome‐wide DNAm analysis using the Infinium HumanMethylation450K platform was conducted in blood samples of 93 randomly selected DACHS+ CRC cases and 94 age‐ and sex‐matched controls randomly selected from the Blitz Study, an ongoing epidemiological study recruiting participants (age 55–75 years) of screening colonoscopy in southwestern Germany [23]. The DACHS+ study was approved by the ethics committee of the University of Heidelberg.
Methodologies in the current study conformed to the standards set by the Declaration of Helsinki.
2.2. DNA methylation profiling and data preprocessing
For the TCGA samples, IDAT format files of the Infinium 450K methylation data were extracted from the TCGA website (https://portal.gdc.cancer.gov/legacy‐archive/search/f). DNAm of paired tissue samples of the DACHS and DACHS+ study was measured using the Infinium Methylation450K BeadChip (Illumina Inc., San Diego, CA, USA) at the Genomics and Proteomics Core Facility of the German Cancer Research Center, Heidelberg, Germany, according to the manufacturer's instructions. Details of DNA isolation from the tissue samples were described in a previous study [22]. All methylation data were preprocessed following the CPACOR pipeline [24]. Probes with detection P‐value > 0.01 and missing values > 5% were removed. Quantile normalization was applied following separating the probe type into six categories, based on probe type and color channel, using the ‘limma’ R package included in the Bioconductor [25]. Methylation beta values of the 10 CpGs and 513 CpGs, respectively, included in the MRscore and DNAmPhenoAge calculation were extracted. Horvath’s DNAmAge was calculated using the online tool available at https://dnamage.genetics.ucla.edu/.
2.3. Statistical analysis
The MRscore was computed in a continuous form as in our previous study [12], that is, as the sum of weighted methylation beta values of 10 CpGs: cg01612140*(−0.38253) + cg05575921*(−0.92224) + cg06126421*(−1.70129) + cg08362785*(2.71749) + cg10321156*(−0.02073) + cg14975410* (−0.04156) + cg19572487*(−0.28069) + cg23665802*(−0.89440) + cg24704287*(−2.98637) + cg25983901*(−1.80325). The weights were taken from the original study [12]. DNAmPhenoAge was calculated according to the formula reported by Levine et al. [11]. The DNAmAge acceleration was calculated as residuals of Horvath’s DNAmAge regressed on chronological age.
The levels of MRscore, DNAmPhenoAge, and DNAmAge acceleration were first described by boxplots and compared between tumor and normal tissues among each type of cancer patients and among cancer stage‐stratified patients by nonparametric Wilcoxon signed‐rank test. The performance of the three epigenetic markers for discriminating tumor tissues from normal tissues was evaluated using receiver operating characteristic (ROC) curves, and areas under the curve (AUCs) and confidence interval (CI) derived from logistic regression. All analyses were repeated in blood samples of CRC cases and controls.
Methylation data were preprocessed and normalized in r (version 3.2.3). All statistical analyses were conducted in sas 9.4 (SAS Institute, Cary, NC, USA).
3. Results
The analyses on paired tumor‐normal tissues were based on 612 cancer patients. Characteristics of those patients are presented in Table 1. The average age of each type of cancer patients (except for breast cancer [BC]) was above 60 years. Most patients had tumor diagnosed at stage I or stage II (except head‐and‐neck cancer).
Table 1.
Characteristics of cancer patients.
| Characteristics | LC (n = 69) TCGA | CRC | BC (n = 90) TCGA | Head‐and‐neck cancer (n = 50) TCGA | Prostate cancer (n = 50) TCGA | Liver cancer (n = 50) TCGA | |
|---|---|---|---|---|---|---|---|
| (n = 45) TCGA | (n = 254) DACHS | ||||||
| Age (mean ± SD) | 67.6 ± 11.0 | 69.4 ± 12.3 | 69.7 ± 10.5 | 57.9 ± 15.3 | 62.6 ± 10.7 | 62.4 ± 6.5 | 62.7 ± 16.1 |
| Men (N/%) | 43 (62.3) | 24 (53.3) | 124 (58.2) | 38 (76.0) | 50 (100.0) | 30 (60.0) | |
| Stages (N/%) | |||||||
| I | 40 (58.8) | 5 (11.1) | 41 (19.2) | 13 (14.6) | – | – | 21 (52.5) |
| II | 11 (16.2) | 21 (46.7) | 79 (37.1) | 55 (61.8) | 8 (16.0) | – | 8 (20.0) |
| III | 14 (20.6) | 10 (22.2) | 65 (30.5) | 20 (22.5) | 10 (20.0) | – | 10 (25.0) |
| IV | 3 (4.4) | 9 (20.0) | 28 (13.2) | 1 (1.1) | 32 (64.0) | – | 1 (2.5) |
3.1. Mortality risk score (MRscore)
Figure 1 shows the levels of MRscore among tumor and normal tissues of all six types of cancer patients. Higher levels of MRscore in tumor tissue than in adjacent normal tissue were observed for five types of cancers (except prostate cancer), and the differences are most remarkable for CRC patients in both TCGA and the DACHS study (Fig. 1B). Consistently elevated MRscore in tumor tissues was also seen across stage I to stage IV tumors of each type of these cancers (Fig. S1). Among prostate cancer patients, tumor tissue showed significantly lower levels of the MRscore than normal tissues (Fig. 1E). This resulted from several CpGs that constitute the major components of the MRscore, which were hypermethylated in prostate tumor tissues (Fig. S2), such as cg06126421, cg24704287, and cg25983901, and have large absolute values of weights in the MRscore calculation, whereas these CpGs were hypomethylated in other tumor tissues, particularly for lung and CRC.
Fig. 1.
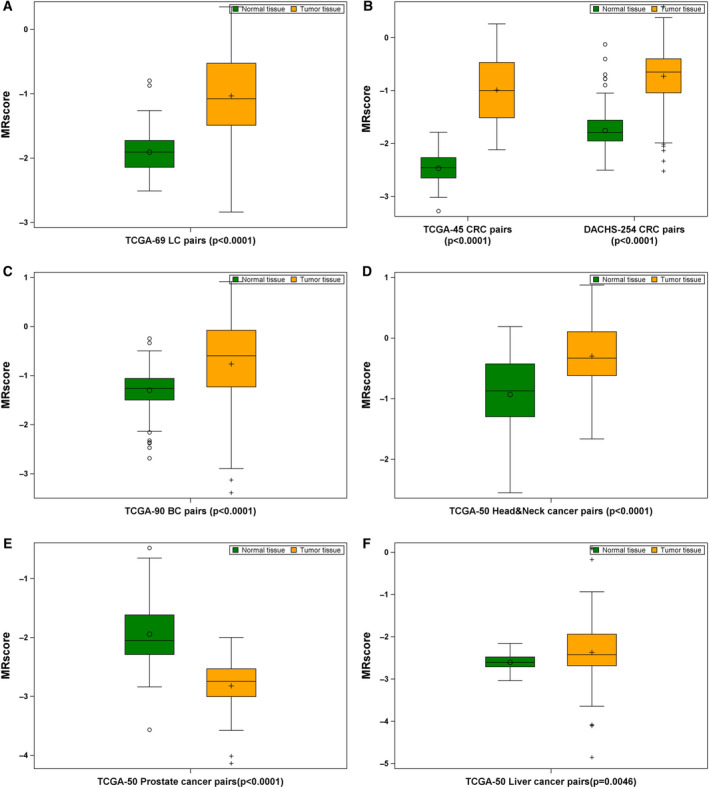
MRscore in tumor‐normal tissue pairs of patients with LC (n = 69; A), CRC (n = 299; B), BC (n = 90; C), head‐and‐neck cancer (n = 50; D), prostate cancer (n = 50; E), and liver cancer (n = 50; F). Difference between tumor and normal tissues was examined using nonparametric Wilcoxon signed‐rank test.
3.2. DNAmPhenoAge
Tumor tissues of all six types of cancers exhibited significantly higher levels of DNAmPhenoAge than the corresponding adjacent normal tissues (Fig. 2). Consistent increases of DNAmPhenoAge across all stages of tumor tissues were also observed for all six types of cancers (Fig. S3).
Fig. 2.
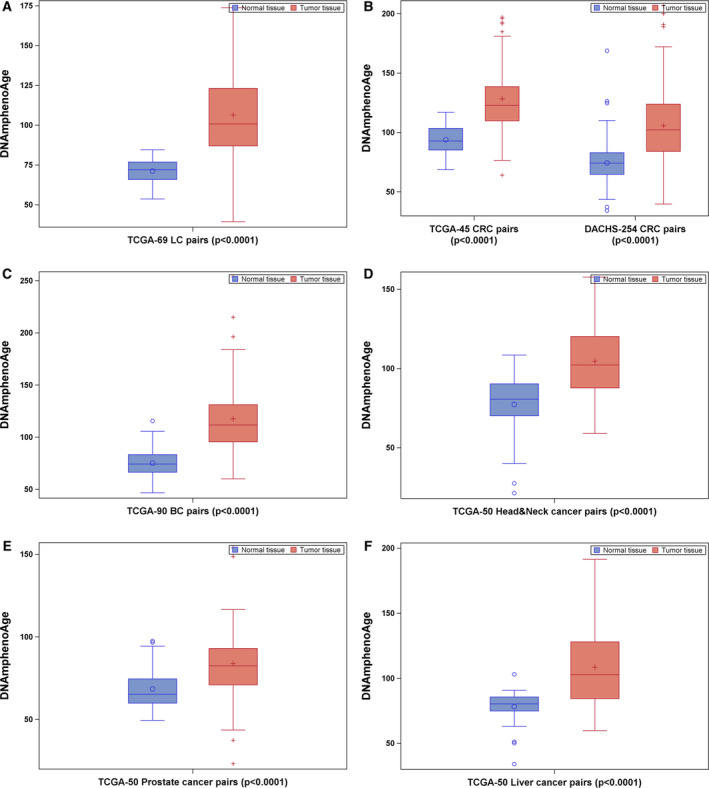
DNAmPhenoAge in tumor‐normal tissue pairs of patients with LC (n = 69; A), CRC (n = 299; B), BC (n = 90; C), head‐and‐neck cancer (n = 50; D), prostate cancer (n = 50; E), and liver cancer (n = 50; F). Difference between tumor and normal tissues was examined using nonparametric Wilcoxon signed‐rank test.
3.3. DNAmAge acceleration
Unlike the pattern of the MRscore and DNAmPhenoAge, DNAmAge acceleration was lower among tumor tissues of lung, colorectal, head‐and‐neck, and prostate cancer patients, compared to the corresponding adjacent normal tissues (Fig. 3). No difference in DNAmAge acceleration between tumor and normal tissues of breast and liver cancer patients was seen (Fig. 3C,F). These patterns were also observed when stratifying patients by cancer stages (Fig. S4).
Fig. 3.
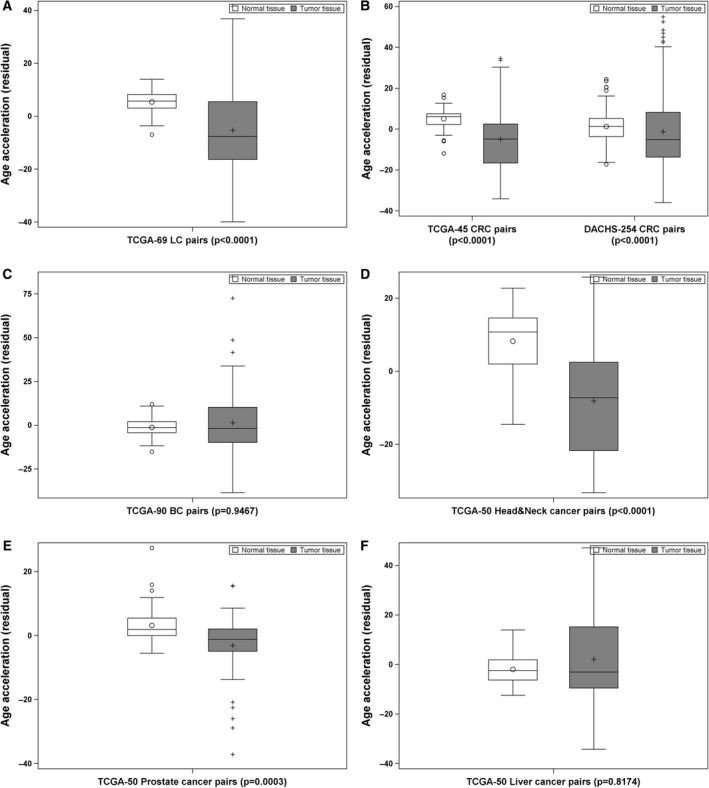
DNAmAge acceleration in tumor‐normal tissue pairs of patients with LC (n = 69; A), CRC (n = 299; B), BC (n = 90; C), head‐and‐neck cancer (n = 50; D), prostate cancer (n = 50; E), and liver cancer (n = 50; F). Difference between tumor and normal tissues was examined using nonparametric Wilcoxon signed‐rank test.
3.4. Discriminative performance of MRscore, DNAmPhenoAge, and DNAmAge acceleration
Figure 4A shows that the MRscore can discriminate lung, colorectal, and prostate tumor tissues from normal tissues with very high accuracy (AUCs of 0.87–0.99). DNAmPhenoAge can accurately discriminate five of six types of tumor tissues (except prostate tumor) from normal tissues, with AUCs of 0.82–0.93 (Fig. 4B). Compared to the MRscore and DNAmPhenoAge, DNAmAge acceleration showed relatively lower and limited accuracy for discriminating five of six types of tumor tissues (except head‐and‐neck tumor) from normal tissues (Fig. 4C). Overall, the MRscore outperformed DNAmPhenoAge for colorectal and prostate cancer tissue discrimination, and DNAmPhenoAge outperformed the MRscore for breast and liver cancer tissue discrimination. Both MRscore and DNAmPhenoAge showed similarly high accuracy for lung cancer (LC) tissue discrimination.
Fig. 4.
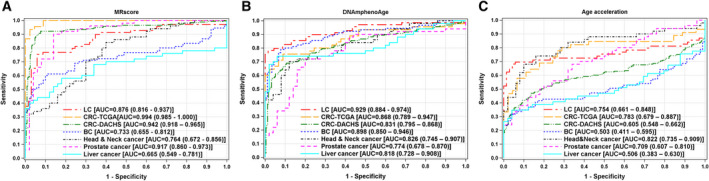
ROC curves for discriminating tumor tissues from normal tissues of common cancer patients by MRscore (A), DNAmPhenoAge (B), and DNAmAge acceleration (C).
3.5. MRscore, DNAmPhenoAge, and DNAmAge acceleration in blood samples of CRC cases and controls
The characteristics of the CRC cases and controls are presented in Table S1. Distribution of major risk factors for CRC, such as age [mean (SD), 65 (8.4)], sex, smoking, body mass index, and colonoscopy history, is comparable between cases and controls. MRscore is significantly higher in cases than in controls (Fig. 5A), whereas no difference in both DNAmPhenoAge and DNAmAge acceleration was observed between cases and controls (Fig. 5B,C). The MRscore showed modest accuracy for discrimination of CRC cases from controls (AUC of 0.74) and outperformed DNAmPhenoAge and DNAmAge acceleration (AUCs of 0.54 and 0.56, respectively).
Fig. 5.
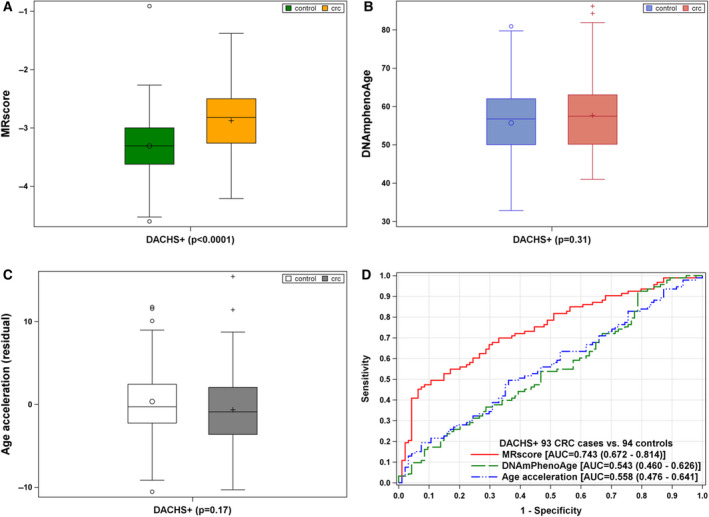
MRscore, DNAmPhenoAge, and DNAmAge acceleration in blood samples of CRC cases (n = 93) and controls (n = 94) [A‐C; difference between cases and controls was examined using nonparametric Wilcoxon signed‐rank test], and in discrimination of cases from controls in the DACHS+ study (D).
4. Discussion
Based on both publicly available and local DNAm data, the current study demonstrated distinct alterations of two strong survival predictors, that is, the MRscore and DNAmPhenoAge, in tumor tissue compared to adjacent normal tissue samples of patients with common forms of cancer. The MRscore accurately differentiated lung, colorectal, and prostate tumor tissues from normal tissues (AUCs of 0.87–0.99), and DNAmPhenoAge accurately differentiated lung, colorectal, breast, head/neck, and liver tumor tissues from adjacent normal tissues (AUCs of 0.82–0.93). Our findings thus further substantiate the evidence that the two predictors are robust epigenetic markers of health outcomes. The consistent pattern of MRscore across target tissue and blood samples of CRC patients strengthens the hypothesis that blood‐derived methylation markers could mirror tumorigenesis‐related methylation changes in less accessible tissues.
The MRscore was computed based on 10 CpGs derived from direct regression of mortality (time‐to‐death) on DNAm levels in a large EWAS, in which the majority of participants were healthy individuals at the time of blood collection and the MRscore strongly predicted their mortality during 14 years of follow‐up [12]. DNAm was quantified among participants with average age of 62 at baseline (range, 50–75 years), for whom common chronic diseases, such as malignant diseases, are the predominant causes of death [26]. The MRscore in nature thus captures and aggregates the risk of developing and surviving common chronic diseases including major cancers. The DNAmPhenoAge was developed through first estimating a phenotypic age based on clinical biomarkers and chronological age, followed by regressing this phenotypic age on DNAm levels of 513 CpGs [11]. This two‐step‐produced DNAmPhenoAge is thus in principle an estimator of biological aging, a major risk factor for cancer [27]. In line with the previous findings that high levels of MRscore and DNAmPhenoAge correlate with increased risk of adverse outcomes [11,12], we found elevated levels of the two markers in tumor tissues rather than in normal tissues. This suggests that regulation of DNAm patterns in CpGs included in the MRscore and DNAmPhenoAge could potentially be relevant for tumorigenesis in solid tissues.
Solid tumors are constituted of malignant cells as well as nonmalignant cell populations that largely overnumber tumor cells [28,29]. Tumors are thus abundantly infiltrated by leukocytes, so‐called tumor‐infiltrating leukocytes (TILs), which consist of immune cells (such as T cells, B cells, natural killer cells, macrophages, neutrophils, and eosinophils) and play a fundamental role in cancer immune surveillance [30]. Consistent methylation changes in the TILs and leukocytes in the peripheral blood might explain why the MRscore and DNAmPhenoAge, which were originally derived from blood sample analysis, showed such strong discrimination between tumor and adjacent tissues for most of the cancers studied. In the current study, we also observed variations of the performance of the MRscore and DNAmPhenoAge in tumor/normal tissue discrimination between different types of tumors, that is, the MRscore outperformed DNAmPhenoAge for colorectal and prostate cancer tissue discrimination, whereas DNAmPhenoAge outperformed the MRscore for breast and liver cancer tissue discrimination. This, on the one hand, may result from differential methylation profiles of the two markers, with a complete lack of overlap between the 10 CpGs in the MRscore and the 513 CpGs in DNAmPhenoAge. On the other hand, the differences may also be explained by heterogeneity of the TILs across various tumors, as compelling evidence from tumor immunology has shown that the abundance and composition of TILs strikingly vary with tumor type and indicate differential prognostic and predictive value [28, 29, 30]. Furthermore, pathophysiological, immune infiltrate promoted inflammation in tumor microenvironment is consistent with potential biological functions of the DNAm markers in the MRscore and DNAmPhenoAge. For example, in addition to four CpGs in the MRscore mapped to genes involved in various cancers (cg23665802 in MIR19A, cg08362785 in MKL1, cg19572487 in RARA, cg05575921 in AHRR) [31, 32, 33, 34, 35, 36, 37, 38, 39], three CpGs in the MRscore (cg05575921, cg06126421, and cg08362785) were identified to correlate with C‐reactive protein, a sensitive indicator of chronic inflammation, in a meta‐analysis of EWAS [40]. Six CpGs in the MRscore are strongly related to tobacco smoking [12,41,42], a factor with well‐established strong effects on inflammation/immune processes [43,44] and 18 types of cancer such as lung, colorectal, and liver cancer [45], [46]. For DNAmPhenoAge, Levine et al. [11] conducted GO enrichment analysis and observed enrichment for a number of pro‐inflammatory signaling pathways, including but not limited to regulation of inflammatory response, tumor necrosis factor‐mediated signaling pathway, and positive regulation of NFkappaB transcription factor activity. Taken together, notwithstanding the need to further unravel a clear picture of how the MRscore and DNAmPhenoAge are related to cancer development and progression, TILs and their involved inflammatory/immunologic responses may explain our findings.
Previous EWASs that investigated cancer‐related DNAm signatures across the whole genome through direct comparison of tumor and adjacent normal tissues have disclosed numerous differentially methylated CpG sites of tumor tissues relative to normal tissues [22,47,48]. It is thus not difficult to derive algorithms with sufficient amount of CpGs, which could reach high distinction between tumor and normal tissues when analyzing tissue samples. However, algorithms derived in such a way usually exhibit completely different performance in nontarget tissues and can hardly be transferred to blood samples for clinical application, whereas the MRscore and DNAmPhenoAge bear potential concordance in blood and target solid tissues, as illustrated by the MRscore that elevated in both tumor tissue and blood samples of CRC patients in the current study. In addition, we in parallel assessed the well‐studied Horvath's epigenetic clock, which was derived from multitissue analysis and combined far more CpGs (n = 353) than the MRscore (n = 10). Our study did not yield evidence that this epigenetic clock outperformed the MRscore in terms of tumor‐normal tissue discrimination, even though it has been linked to various chronic diseases including cancers, such as LC and BC [18, 19, 20]. This finding is also not surprising given that the effect sizes for the associations of the epigenetic clock reflected age acceleration with health conditions are typically small or moderate. Nevertheless, it is worthwhile pointing out that the MRscore and DNAmPhenoAge are indicative of the presence of multiple cancers rather than of a specific cancer. However, variations of the MRscore and DNAmPhenoAge between cancers were also found, including but not limited to the opposite pattern of MRscore in prostate cancer tissue vs. normal tissue; thus, their applications in specific cancers need to be explored by future research.
In contrast to the MRscore and DNAmPhenoAge that were developed to capture the risk and physiological dysregulation [11,12], Horvath's epigenetic clock was built upon methylation markers that are strongly correlated with chronological age and trained to be an age estimator [10]. In the current study, we found, compared to the consistent pattern of the MRscore and DNAmPhenoAge in tumor vs. normal tissues, opposite patterns for the DNAmAge acceleration based on Horvath’s epigenetic clock. This is in line with findings from previous studies that in many types of cancer tissues, age‐associated DNAm signatures hardly correlated with chronological age of the cancer patients and DNAmAge is often predicted to be younger [49,50]. A plausible explanation is that DNAmAge estimated in tumor tissue may partly reflect the state of aging in the tumor‐initiating cells, the cancer stem cells, which exhibit young biological age [10]. However, a caveat needs to be considered given the relatively small sample size of each cancer type in the current study.
In the current study, although we examined the DNAm‐based algorithms in several common forms of cancer and yielded basically consistent patterns across most cancers, the sample size for each cancer type was rather limited (< 100 pairs of tissue samples except for CRC). Another major limitation of the current study is that we assessed the DNAm‐based algorithms in paired tissues and blood samples only for CRC patients, and were not able to investigate their levels in both target tissues and blood samples of other cancer patients and healthy controls. The analyses for CRC were not conducted in tissues and blood samples from the same patients, which may bring up additional variation. Disconcordance of DNAmPhenoAge between tissue and blood samples based on the current CRC analyses thus should be interpreted with caution and should not be extrapolated to other types of cancers. Future studies with a large number of ‘tripled samples’, including blood, tumor, and adjacent normal tissues of the same patients, along with blood samples from tumor‐free participants, are needed to confirm our findings, clarify the relevant biological pathways, and evaluate the potential use of the blood‐based algorithms for cancer diagnosis and early detection.
5. Conclusions
The current study demonstrated that two DNAm‐based algorithms, which were previously shown to be strongly predictive of mortality when measured in blood samples, were also indicative of methylation changes in tissues of various common cancers, suggesting that they might reflect tumor‐related methylation changes. TILs and the underlying inflammatory process may explain potential concordance of methylation changes in both blood and solid tumor tissues, which, however, needs to be further explored by future studies with simultaneous collection and analyses of blood and tissue samples. Given the properties of easy accessibility and processing of whole blood samples, the use of DNAm‐based algorithms for cancer diagnosis and early detection should be explored in future research.
Conflict of interest
The authors declare no conflict of interest.
Author contributions
YZ and HB designed and conceived the study. YZ retrieved the data, carried out the analyses, and drafted the manuscript. HB, JC, and MH are the principal investigators of the DACHS study and carried out, led, and supervised its conduction. MS, MB, and BB contributed to processing the DACHS DNA with Illumina Human Methylation arrays. EH contributed to the provision of DACHS tissue samples. All authors contributed to the revision of the manuscript and approved the final version for submission.
Supporting information
Fig. S1. Stage‐specific levels of MRscore in paired tumor and normal tissues of 69 lung cancer (LC) patients from the TCGA (A), 45 colorectal cancer (CRC) patients from the TCGA (B), 254 CRC patients from the DACHS study (C), 90 breast cancer (BC) patients from the TCGA (D), 50 head‐and‐neck (HN) cancer patients from the TCGA (E), and 50 liver cancer patients from the TCGA (F).
Fig. S2. Methylation level of the 10 CpGs in MRscore by tumor and normal tissues of LC (A), CRC (B), BC (C), head/neck cancer (D), prostate cancer (E), and liver cancer (F) patients from the TCGA, and of CRC (G) patients from the DACHS study.
Fig. S3. Stage‐specific levels of DNAmPhenoAge in paired tumor and normal tissues of 69 LC patients from the TCGA (A), 45 CRC patients from the TCGA (B), 254 CRC from the DACHS study (C), 90 BC patients from the TCGA (D), 50 HN cancer patients from the TCGA (E), and 50 liver cancer patients from the TCGA (F).
Fig. S4. Stage‐specific levels of DNAmAge acceleration in paired tumor and normal tissues of 69 LC patients from the TCGA (A), 45 CRC patients from the TCGA (B), 254 CRC from the DACHS study (C), 90 BC patients from the TCGA (D), 50 HN cancer patients from the TCGA (E), and 50 liver cancer patients from the TCGA (F).
Table S1. Characteristics of the CRC cases and controls in the DACHS+ study.
Acknowledgement
The DACHS study was supported by the German Research Council (BR 1704/6‐1, BR 1704/6‐3, BR 1704/6‐4, CH 117/1‐1, HO 5117/2‐1, HE 5998/2‐1, KL 2354/3‐1, RO 2270/8‐1, BR 1704/17‐1), the Interdisciplinary Research Program of the National Center for Tumor Diseases (NCT), Germany, and the German Federal Ministry of Education and Research (01KH0404, 01ER0814, 01ER0815, 01ER1505A, 01ER1505B).
The sponsors had no role in the study design, in the collection, analysis, and interpretation of data and preparation, review, or approval of the manuscript.
Data accessibility
TCGA DNAm and clinical data are available in the Cancer Genome Atlas database (https://www.cancer.gov/about‐nci/organization/ccg/research/structural‐genomics/tcga). For the DACHS study, due to restrictions of informed consent we are not allowed making the dataset publicly available. However, the use of the data for collaboration projects has been and will remain the approach for data sharing.
References
- 1. Lokk K, Modhukur V, Rajashekar B, Martens K, Magi R, Kolde R, Koltsina M, Nilsson TK, Vilo J, Salumets A et al (2014) DNA methylome profiling of human tissues identifies global and tissue‐specific methylation patterns. Genome Biol 15, r54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Slieker RC, Bos SD, Goeman JJ, Bovee JV, Talens RP, van der Breggen R, Suchiman HE, Lameijer EW, Putter H, van den Akker EB et al (2013) Identification and systematic annotation of tissue‐specific differentially methylated regions using the Illumina 450k array. Epigenetics Chromatin 6, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Huang YT, Chu S, Loucks EB, Lin CL, Eaton CB, Buka SL & Kelsey KT (2016) Epigenome‐wide profiling of DNA methylation in paired samples of adipose tissue and blood. Epigenetics 11, 227–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Day K, Waite LL, Thalacker‐Mercer A, West A, Bamman MM, Brooks JD, Myers RM & Absher D (2013) Differential DNA methylation with age displays both common and dynamic features across human tissues that are influenced by CpG landscape. Genome Biol 14, R102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Horvath S, Zhang Y, Langfelder P, Kahn RS, Boks MP, van Eijk K, van den Berg LH & Ophoff RA (2012) Aging effects on DNA methylation modules in human brain and blood tissue. Genome Biol 13, R97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Houde AA, Legare C, Biron S, Lescelleur O, Biertho L, Marceau S, Tchernof A, Vohl MC, Hivert MF & Bouchard L (2015) Leptin and adiponectin DNA methylation levels in adipose tissues and blood cells are associated with BMI, waist girth and LDL‐cholesterol levels in severely obese men and women. BMC Med Genet 16, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Teschendorff AE, Yang Z, Wong A, Pipinikas CP, Jiao Y, Jones A, Anjum S, Hardy R, Salvesen HB, Thirlwell C et al (2015) Correlation of smoking‐associated DNA methylation changes in buccal cells with DNA methylation changes in epithelial cancer. JAMA Oncol 1, 476–485. [DOI] [PubMed] [Google Scholar]
- 8. Walton E, Hass J, Liu J, Roffman JL, Bernardoni F, Roessner V, Kirsch M, Schackert G, Calhoun V & Ehrlich S (2016) Correspondence of DNA methylation between blood and brain tissue and its application to schizophrenia research. Schizophr Bull 42, 406–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Teschendorff AE, Menon U, Gentry‐Maharaj A, Ramus SJ, Weisenberger DJ, Shen H, Campan M, Noushmehr H, Bell CG, Maxwell AP et al (2010) Age‐dependent DNA methylation of genes that are suppressed in stem cells is a hallmark of cancer. Genome Res 20, 440–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Horvath S (2013) DNA methylation age of human tissues and cell types. Genome Biol 14, R115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Levine ME, Lu AT, Quach A, Chen BH, Assimes TL, Bandinelli S, Hou L, Baccarelli AA, Stewart JD, Li Y et al (2018) An epigenetic biomarker of aging for lifespan and healthspan. Aging 10, 573–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhang Y, Wilson R, Heiss J, Breitling LP, Saum KU, Schottker B, Holleczek B, Waldenberger M, Peters A & Brenner H (2017) DNA methylation signatures in peripheral blood strongly predict all‐cause mortality. Nat Commun 8, 14617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gao X, Gao X, Zhang Y, Holleczek B, Schottker B & Brenner H (2019) Oxidative stress and epigenetic mortality risk score: associations with all‐cause mortality among elderly people. Eur J Epidemiol 34, 451–462. [DOI] [PubMed] [Google Scholar]
- 14. Gao X, Zhang Y, Mons U & Brenner H (2018) Leukocyte telomere length and epigenetic‐based mortality risk score: associations with all‐cause mortality among older adults. Epigenetics 13, 846–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhang Y, Saum KU, Schottker B, Holleczek B & Brenner H (2018) Methylomic survival predictors, frailty, and mortality. Aging 10, 339–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gao X, Colicino E, Shen J, Just AC, Nwanaji‐Enwerem JC, Wang C, Coull B, Lin X, Vokonas P, Zheng Y et al (2019) Comparative validation of an epigenetic mortality risk score with three aging biomarkers for predicting mortality risks among older adult males. Int J Epidemiol 48, 1958–1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lu AT, Quach A, Wilson JG, Reiner AP, Aviv A, Raj K, Hou L, Baccarelli AA, Li Y, Stewart JD et al (2019) DNA methylation GrimAge strongly predicts lifespan and healthspan. Aging 11, 303–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ambatipudi S, Horvath S, Perrier F, Cuenin C, Hernandez‐Vargas H, Le Calvez‐Kelm F, Durand G, Byrnes G, Ferrari P, Bouaoun L et al (2017) DNA methylome analysis identifies accelerated epigenetic ageing associated with postmenopausal breast cancer susceptibility. Eur J Cancer 75, 299–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chen BH, Marioni RE, Colicino E, Peters MJ, Ward‐Caviness CK, Tsai PC, Roetker NS, Just AC, Demerath EW, Guan W et al (2016) DNA methylation‐based measures of biological age: meta‐analysis predicting time to death. Aging 8, 1844–1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Levine ME, Hosgood HD, Chen B, Absher D, Assimes T & Horvath S (2015) DNA methylation age of blood predicts future onset of lung cancer in the women's health initiative. Aging 7, 690–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Brenner H, Chang‐Claude J, Jansen L, Knebel P, Stock C & Hoffmeister M (2014) Reduced risk of colorectal cancer up to 10 years after screening, surveillance, or diagnostic colonoscopy. Gastroenterology 146, 709–717. [DOI] [PubMed] [Google Scholar]
- 22. Gundert M, Edelmann D, Benner A, Jansen L, Jia M, Walter V, Knebel P, Herpel E, Chang‐Claude J, Hoffmeister M et al (2019) Genome‐wide DNA methylation analysis reveals a prognostic classifier for non‐metastatic colorectal cancer (ProMCol classifier). Gut 68, 101–110. [DOI] [PubMed] [Google Scholar]
- 23. Tao S, Haug U, Kuhn K & Brenner H (2012) Comparison and combination of blood‐based inflammatory markers with faecal occult blood tests for non‐invasive colorectal cancer screening. Br J Cancer 106, 1424–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lehne B, Drong AW, Loh M, Zhang W, Scott WR, Tan ST, Afzal U, Scott J, Jarvelin MR, Elliott P et al (2015) A coherent approach for analysis of the Illumina HumanMethylation450 BeadChip improves data quality and performance in epigenome‐wide association studies. Genome Biol 16, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Smyth G (2005) Limma: linear models for microarray data In Bioinformatics and Computational Biology Solutions Using R and Bioconductor (Gentleman R, Carey V, Dudoit S, Irizarry R. & Huber W, eds), pp. 397–420. Springer, New York. [Google Scholar]
- 26. Global Burden of Disease Cancer Collaboration , Fitzmaurice C, Allen C, Barber RM, Barregard L, Bhutta ZA, Brenner H, Dicker DJ, Chimed‐Orchir O, Dandona R et al (2017) Global, regional, and national cancer incidence, mortality, years of life lost, years lived with disability, and disability‐adjusted life‐years for 32 cancer groups, 1990 to 2015: a systematic analysis for the global burden of disease study. JAMA Oncol 3, 524–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lopez‐Otin C, Blasco MA, Partridge L, Serrano M & Kroemer G (2013) The hallmarks of aging. Cell 153, 1194–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gentles AJ, Newman AM, Liu CL, Bratman SV, Feng W, Kim D, Nair VS, Xu Y, Khuong A, Hoang CD et al (2015) The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat Med 21, 938–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Senovilla L, Vacchelli E, Galon J, Adjemian S, Eggermont A, Fridman WH, Sautes‐Fridman C, Ma Y, Tartour E, Zitvogel L et al (2012) Trial watch: prognostic and predictive value of the immune infiltrate in cancer. Oncoimmunology 1, 1323–1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lanca T & Silva‐Santos B (2012) The split nature of tumor‐infiltrating leukocytes: implications for cancer surveillance and immunotherapy. Oncoimmunology 1, 717–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Anfossi S, Giordano A, Gao H, Cohen EN, Tin S, Wu Q, Garza RJ, Debeb BG, Alvarez RH, Valero V et al (2014) High serum miR‐19a levels are associated with inflammatory breast cancer and are predictive of favorable clinical outcome in patients with metastatic HER2+ inflammatory breast cancer. PLoS One 9, e83113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cheng X, Yang Y, Fan Z, Yu L, Bai H, Zhou B, Wu X, Xu H, Fang M, Shen A et al (2015) MKL1 potentiates lung cancer cell migration and invasion by epigenetically activating MMP9 transcription. Oncogene 34, 5570–5581. [DOI] [PubMed] [Google Scholar]
- 33. Conway K, Edmiston SN, May R, Kuan PF, Chu H, Bryant C, Tse CK, Swift‐Scanlan T, Geradts J, Troester MA et al (2014) DNA methylation profiling in the Carolina Breast Cancer Study defines cancer subclasses differing in clinicopathologic characteristics and survival. Breast Cancer Res 16, 450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fasanelli F, Baglietto L, Ponzi E, Guida F, Campanella G, Johansson M, Grankvist K, Johansson M, Assumma MB, Naccarati A et al (2015) Hypomethylation of smoking‐related genes is associated with future lung cancer in four prospective cohorts. Nat Commun 6, 10192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gaillard C, Tokuyasu TA, Rosen G, Sotzen J, Vitaliano‐Prunier A, Roy R, Passegue E, de The H, Figueroa ME & Kogan SC (2015) Transcription and methylation analyses of preleukemic promyelocytes indicate a dual role for PML/RARA in leukemia initiation. Haematologica 100, 1064–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Giraldez MD, Lozano JJ, Ramirez G, Hijona E, Bujanda L, Castells A & Gironella M (2013) Circulating microRNAs as biomarkers of colorectal cancer: results from a genome‐wide profiling and validation study. Clin Gastroenterol Hepatol 11, 681–688 e683 [DOI] [PubMed] [Google Scholar]
- 37. Wang X & Chen Z (2015) MicroRNA‐19a functions as an oncogenic microRNA in non‐small cell lung cancer by targeting the suppressor of cytokine signaling 1 and mediating STAT3 activation. Int J Mol Med 35, 839–846. [DOI] [PubMed] [Google Scholar]
- 38. Zhang Y, Elgizouli M, Schottker B, Holleczek B, Nieters A & Brenner H (2016) Smoking‐associated DNA methylation markers predict lung cancer incidence. Clin Epigenetics 8, 127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhang Y, Guo X, Li Z, Li B, Li Z, Li R, Guo Q, Xiong L, Yu L, Zhao J et al (2015) A systematic investigation based on microRNA‐mediated gene regulatory network reveals that dysregulation of microRNA‐19a/Cyclin D1 axis confers an oncogenic potential and a worse prognosis in human hepatocellular carcinoma. RNA Biol 12, 643–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ligthart S, Marzi C, Aslibekyan S, Mendelson MM, Conneely KN, Tanaka T, Colicino E, Waite LL, Joehanes R, Guan W et al (2016) DNA methylation signatures of chronic low‐grade inflammation are associated with complex diseases. Genome Biol 17, 255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gao X, Jia M, Zhang Y, Breitling LP & Brenner H (2015) DNA methylation changes of whole blood cells in response to active smoking exposure in adults: a systematic review of DNA methylation studies. Clin Epigenetics 7, 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Joehanes R, Just AC, Marioni RE, Pilling LC, Reynolds LM, Mandaviya PR, Guan W, Xu T, Elks CE, Aslibekyan S et al (2016) Epigenetic signatures of cigarette smoking. Circ Cardiovasc Genet 9, 436–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lee J, Taneja V & Vassallo R (2012) Cigarette smoking and inflammation: cellular and molecular mechanisms. J Dent Res 91, 142–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Shiels MS, Katki HA, Freedman ND, Purdue MP, Wentzensen N, Trabert B, Kitahara CM, Furr M, Li Y, Kemp TJ et al (2014) Cigarette smoking and variations in systemic immune and inflammation markers. J Natl Cancer Inst 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Centers for Disease Control and Prevention (US) (2010). In How Tobacco Smoke Causes Disease: The Biology and Behavioral Basis for Smoking‐Attributable Disease: A Report of the Surgeon General. Atlanta, GA. [PubMed] [Google Scholar]
- 46. Centers for Disease Control and Prevention (US) (2014). In 2014 Surgeon General's Report: The Health Consequences of Smoking—50 Years of Progress. Atlanta, GA. [Google Scholar]
- 47. Hao X, Luo H, Krawczyk M, Wei W, Wang W, Wang J, Flagg K, Hou J, Zhang H, Yi S et al (2017) DNA methylation markers for diagnosis and prognosis of common cancers. Proc Natl Acad Sci USA 114, 7414–7419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Karlsson A, Jonsson M, Lauss M, Brunnstrom H, Jonsson P, Borg A, Jonsson G, Ringner M, Planck M & Staaf J (2014) Genome‐wide DNA methylation analysis of lung carcinoma reveals one neuroendocrine and four adenocarcinoma epitypes associated with patient outcome. Clin Cancer Res 20, 6127–6140. [DOI] [PubMed] [Google Scholar]
- 49. Horvath S (2015) Erratum to: DNA methylation age of human tissues and cell types. Genome Biol 16, 96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lin Q & Wagner W (2015) Epigenetic aging signatures are coherently modified in cancer. PLoS Genet 11, e1005334. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Stage‐specific levels of MRscore in paired tumor and normal tissues of 69 lung cancer (LC) patients from the TCGA (A), 45 colorectal cancer (CRC) patients from the TCGA (B), 254 CRC patients from the DACHS study (C), 90 breast cancer (BC) patients from the TCGA (D), 50 head‐and‐neck (HN) cancer patients from the TCGA (E), and 50 liver cancer patients from the TCGA (F).
Fig. S2. Methylation level of the 10 CpGs in MRscore by tumor and normal tissues of LC (A), CRC (B), BC (C), head/neck cancer (D), prostate cancer (E), and liver cancer (F) patients from the TCGA, and of CRC (G) patients from the DACHS study.
Fig. S3. Stage‐specific levels of DNAmPhenoAge in paired tumor and normal tissues of 69 LC patients from the TCGA (A), 45 CRC patients from the TCGA (B), 254 CRC from the DACHS study (C), 90 BC patients from the TCGA (D), 50 HN cancer patients from the TCGA (E), and 50 liver cancer patients from the TCGA (F).
Fig. S4. Stage‐specific levels of DNAmAge acceleration in paired tumor and normal tissues of 69 LC patients from the TCGA (A), 45 CRC patients from the TCGA (B), 254 CRC from the DACHS study (C), 90 BC patients from the TCGA (D), 50 HN cancer patients from the TCGA (E), and 50 liver cancer patients from the TCGA (F).
Table S1. Characteristics of the CRC cases and controls in the DACHS+ study.
Data Availability Statement
TCGA DNAm and clinical data are available in the Cancer Genome Atlas database (https://www.cancer.gov/about‐nci/organization/ccg/research/structural‐genomics/tcga). For the DACHS study, due to restrictions of informed consent we are not allowed making the dataset publicly available. However, the use of the data for collaboration projects has been and will remain the approach for data sharing.