

Abstract
Predictive biomarkers of response to immune checkpoint inhibitors (ICI) help to identify cancer patients who will benefit from immunotherapy. Protein kinase, DNA‐activated, catalytic subunit (PRKDC) is an important gene for DNA double‐strand break (DSB) repair and central T‐cell tolerance. We aimed to investigate the association between PRKDC mutations and tumor mutation burden (TMB), tumor microenvironment (TME), and response to ICI. Whole‐exome sequencing data of 4023 solid tumor samples from the Cancer Genome Atlas (TCGA) and panel‐based sequencing data of 3877 solid tumor samples from Geneplus‐Beijing, China, were used to analyze the TMB. The mRNA expression data of 3541 solid tumor samples from TCGA were used to explore the effect of PRKDC mutations on the TME. Four ICI‐treated cohorts were analyzed for verifying the correlation between PRKDC mutations and the response to ICI. In both the TCGA and Geneplus datasets, we found that the TMB in PRKDC mutation samples was significantly higher than in PRKDC wild‐type samples (P < 0.05 and P < 0.0001, respectively). Further, TCGA datasets showed that PRKDC mutation samples were associated with a significantly increased expression of CD8+ T cells, NK cells, immune checkpoint, chemokines, etc. compared to PRKDC wild‐type samples (P < 0.05). In ICI‐treated cohorts, we also found the PRKDC mutations were associated with increased survival (median PFS, not reached vs. 6.8 months, HR, 0.2893; 95% CI, 0.1255–0.6672; P = 0.0650, Hellmann cohort; median OS, 1184 days vs. 250 days, HR, 0.5126; 95% CI, 0.2715–0.9679; P = 0.1020, Allen cohort), and the increase was significant in multivariate analysis (HR, 0.361; 95% CI, 0.155–0.841; P = 0.018, Allen cohort; HR, 0.240 95% CI, 0.058–0.998; P = 0.050, Hellmann cohort). In summary, we found that PRKDC mutation often appeared to co‐exist with deficiency in some other DNA damage repair mechanism and is nonetheless one of the important factors associated with increased TMB, inflamed TME, and better response to ICI.
Keywords: DNA‐PKcs, immune checkpoint inhibitors, PRKDC, tumor microenvironment, tumor mutation burden
Protein kinase, DNA‐activated, catalytic subunit (PRKDC) is a gene related to DNA damage response (DDR). Here, we found that PRKDC mutation often co‐exists with other DNA damage repair deficiencies and is associated with an increased tumor mutation burden, an inflamed tumor microenvironment, and a good response to immune checkpoint inhibitors.

Abbreviations
- DDR
DNA damage repair
- DNA‐PK
DNA‐dependent serine/threonine protein kinase
- DNA‐PKcs
DNA‐dependent serine/threonine protein kinase catalytic subunit
- DSB
DNA double‐strand break
- GSEA
Gene set enrichment analysis
- HR
homologous recombination
- ICIs
immune checkpoint inhibitors
- MMR
mismatch repair
- MSI
microsatellite instability
- NHEJ
nonhomologous end‐joining
- PRKDC
protein kinase, DNA‐activated, catalytic subunit
- TCGA
The Cancer Genome Atlas
- TMB
tumor mutation burden
- TME
tumor microenvironment
1. Introduction
New immune checkpoint inhibitors (ICI) have changed the therapeutic landscape for many types of cancer [1, 2, 3]. However, only a subset of cancers are responsive to ICI [4]. Several biomarkers, including PD‐L1 expression [5], RNA expression signatures [6], tumor mutational burden (TMB) [7], and lymphocyte infiltration [8], have been reported as possible biomarkers to identify patients who may benefit from ICI.
Gene mutations in the tumor DNA damage repair (DDR) pathway have also been recently reported as an important predictor for the response to ICI therapy. Mutations in DNA polymerase ɛ (POLE) and polymerase ƌ (POLD1), mismatch repair (MMR) genes, and BRCA1/BRCA2 reduce genomic stability and can lead to hypermutations [9, 10, 11]. The protein kinase, DNA‐activated, catalytic subunit (PRKDC) that encodes the DNA‐dependent serine/threonine protein kinase catalytic subunit (DNA‐PKcs) is a member of the DDR pathway. DNA‐dependent serine/threonine protein kinase (DNA‐PK) is composed of DNA‐PKcs and a heterodimer of Ku proteins (Ku70/Ku80). DNA‐PK is a critical component of the nonhomologous end‐joining (NHEJ) pathway that is mainly involved in DNA double‐strand break (DSB) repair and maintaining genomic integrity [11, 12, 13]. In this study, we performed a comprehensive analysis of PRKDC mutations by reviewing the Cancer Genome Atlas (TCGA) database, the Geneplus database, and four available clinical cohorts treated with ICI [7, 14, 15, 16]. Notably, we uncovered that PRKDC mutations were significantly associated with a high TMB similar with other DDR pathway‐related gene mutations, including POLE/D1, MMR genes, BRCA1/2, and the presence of PRKDC mutations predicted a superior response to ICI therapy compared with patients with wild‐type PRKDC. We highlight the importance of validation of PRKDC mutations for the delivery of precise immunotherapy.
2. Materials and methods
This study had been informed and approved by the ethics committee of the Fujian Medical University Cancer Hospital. All procedures in the study were conducted conformed to the standards set by the Declaration of Helsinki.
2.1. Patients and specimens
From August 12, 2016, to March 4, 2019, 3877 solid tumor samples collected from 3877 patients underwent a panel‐based next‐generation sequencing assays at Geneplus‐Beijing, Beijing, China. The cancer types and number of samples of each cancer type included in Geneplus are displayed in Table S1. All patients signed a written consent. We also analyzed 4023 samples from TCGA (is a cancer research project established by the National Cancer Institute (NCI) and the National Human Genome Research Institute (NHGRI), Bethesda, MD, USA), 110 samples from the Allen cohort [16], 75 samples from the Hellmann cohort [14], 34 samples from the Rizvi cohort [7], and 64 samples from the Snyder cohort [15].
2.2. Panel‐based sequencing
2.2.1. Specimen processing and DNA extraction
The genomic DNA from frozen tissue samples was extracted by using the Tissue gDNA exaction Kit (Qiagen, Hilden, Germany). DNA from formalin‐fixed, paraffin‐embedded specimens (FFPE) was isolated by using a commercially available kit (Maxwell® 16 FFPE Plus LEV DNA Purification; Qiagen. catalog: AS1135). The DNA concentration was measured using a Qubit fluorometer and the Qubit dsDNA HS (High Sensitivity) Assay Kit (Invitrogen, Carlsbad, CA, USA).
2.2.2. Library preparation, target capture, and next‐generation sequencing
Sequencing was carried out using Illumina 2 × 75‐bp paired‐end reads on an Illumina HiSeq 3000 instrument according to the manufacturer's recommendations using the KAPA DNA Library Preparation Kit (Kapa Biosystems, Wilmington, MA, USA). Barcoded libraries were hybridized to a customized panel of 1021 genes containing whole exons and selected introns of 288 genes and selected regions of 733 genes, and another panel of 430 genes most frequently mutated in solid tumors. The libraries were sequenced to a uniform median depth (> 500×) and assessed for somatic variants including single nucleotide variants (SNVs), small insertions and deletions (InDels), copy number alterations (CNA), and gene fusions/rearrangements.
2.2.3. Somatic mutation calling
MuTect2 (3.4‐46‐gbc02625) [17] was employed to identify somatic small InDels and SNVs.
2.3. Data source
2.3.1. The cancer Genome Atlas dataset
We obtained the WES data of 4023 solid tumors and the mRNA expression data of 3541 solid tumors across 10 tumor types from TCGA. The experimental procedures for DNA and RNA extraction from tumors, library preparation, sequencing, quality control, and subsequent data processing were published previously by TCGA [18]. The mRNA expression was quantified by RSEM (RNA‐seq by expectation‐maximization) [19]. The data were log2(x + 1)‐transformed before analysis.
2.3.2. Genome and MSI status data in four datasets
Gastric, colorectal, and endometrial tumors are the three types of cancer known to include microsatellite stability (MSS) and microsatellite instability (MSI) subtypes. To explore the association between PRKDC mutations and MSI status and their effects on TMB, we obtained the WES data from 246 endometrial carcinoma, 223 colorectal adenocarcinoma, and 295 gastric adenocarcinoma samples with MSS/MSI subtype information from three studies [20, 21, 22] in TCGA Network, and the whole genome sequencing (WGS) data of 100 gastric adenocarcinoma samples from the Kai Wang cohort [23].
2.3.3. Available clinical cohorts
To further explore the association between PRKDC mutations and the clinical benefits of ICI, we analyzed the genomic and clinical data from four clinical cohorts treated with ICI and predicted the neoantigen data from the Hellmann cohort. The first cohort consisted of 75 patients with non‐small‐cell lung cancer (NSCLC) treated with anti‐PD‐1 therapy and anti‐CTLA‐4 therapy (Hellmann cohort) [14]. The second cohort consisted of 110 patients with advanced‐stage melanoma treated with anti‐CTLA‐4 therapy (Allen cohort) [16]. The third cohort was comprised of 64 patients with advanced‐stage melanoma treated with anti‐CTLA‐4 therapy (Snyder cohort) [15]. The last cohort was comprised of 34 patients with NSCLC treated with anti‐PD‐1 therapy (Rizvi cohort) [7].
2.4. Biomarker analysis
We defined any nonsynonymous mutations in the gene of interest as ‘mut+’, including missense, nonsense, frameshift indels, in‐frame indels, and splice site mutations.
2.4.1. Tumor mutation burden analysis
Tumor mutation burden was defined as the total somatic nonsynonymous mutation counts in coding regions. TMB was classified into high or low taking the top quartile as the cutoff value.
2.4.2. mRNA expression analysis in immune‐related gene set
The response to ICI has been reported to be related to cytotoxic T cells, NK cells, chemokines, and checkpoints [7, 24]. The immune gene list is based on published articles [25] and [26] that summarized the genes related to CD8 T cells, NK cells, cytotoxic lymphocyte, chemokines, plasmacytoid dendritic cell precursors (pDCs), Th1, macrophages, CD4 Treg, CD4 T cells, neutrophils, etc. The mRNA expression of a gene set was defined as the arithmetic mean of transcripts per million values of genes in this gene set.
2.4.3. Gene set enrichment analysis
Gene set enrichment analysis (GSEA) was performed using the javagsea 3.0 Desktop Application (http://software.broadinstitute.org/gsea/index.jsp). The gene sets used for the enrichment analysis were downloaded from the Molecular Signatures Database (MsigDB, http://software.broadinstitute.org/gsea/index.jsp). The gene sets with a false discovery rate (FDR) < 0.05 were considered as significantly enriched. The normalized enrichment score (NES) is the primary statistic for examining gene set enrichment results.
2.4.4. Predicted neoantigen burden
Neoantigens in 75 samples from the Hellmann cohort were estimated [14]. The mutated DNA sequences were virtually translated into corresponding mutated peptide sequences by using Topiary (https://github.com/hammerlab/topiary/) [27]. Topiary was used to run netmhccons (v. 1.1) [28] in order to predict MHC class I binding affinity for all 8–11 mer peptide sequences containing the mutated amino acid. For variants longer than a single residue, all 8–11 mers downstream of the variant were considered. Candidate neoantigens were those peptides with a binding affinity IC50 of % 500 nm to one (or more) of the patient‐specific HLA alleles.
2.5. Prediction of the functional impact of mutation
We used the functional impact predicting tools SIFT and PolyPhen‐2 HumVar to predict the effects of PRKDC mutations on protein function in a clinical case we presented. Mutations with a SIFT score < 0.05 predicted to be deleterious or PolyPhen‐2 HumVar score > 0.5–0.9 were considered possibly damaging and probably damaging (score > 0.9).
2.6. Statistical analysis
Statistical analyses were conducted using graphpad prism (version 8.0.1; GraphPad Software, San Diego, CA, USA) and spss version 25.0 (SPSS, Inc., International Business Machines Corporation (IBM), Armonk, NY, USA). If TMB, TNB, and mRNA were normally distributed, a Student t test was used to determine the differences between two groups; otherwise, the Mann–Whitney U test was used. Logistic regression was used to analyze the influencing factors of TMB‐high. Pearson's correlation was used to analyze the correlation between the length of exons and the mean number of somatic mutations in the exon region for 800 genes with long transcripts including PRKDC, and the fitness lines in scatter plots were plotted with Loess regression which fitted by (weighted) least squares. Kaplan–Meier survival and multivariate Cox regression analyses were used to analyze associations between mutation type and survival, with a P value determined by a log‐rank test. HR was determined through Cox regression. The factors associated with survival with P < 0.15 in univariable analysis were included in Cox proportion hazard model multivariable analysis. All reported P values were two‐tailed, and P < 0.05 was considered significant.
3. Results
3.1. PRKDC mutation profile landscape in TCGA and Geneplus
We explored the prevalence of PRKDC mutations in the TCGA cohort and in the Geneplus cohort (a cohort of a Chinese pan‐cancer population). In the TCGA cohort, colorectal adenocarcinoma had the highest PRKDC mutation frequency of 9.66% (51/528), followed by gastric adenocarcinoma with 9.63% (42/436), endometrial cancer 9.27% (23/248), and non‐small‐cell lung cancer 7.86% (81/1031; Fig. 1A) In the Geneplus cohort, melanoma had the highest mutation frequency of 5.88% (1/17), followed by small‐cell lung cancer with 5.45% (3/55) and cervical squamous cell carcinoma 3.33% (1/30; Fig. 1A). PRKDC mutation sites were scattered throughout the genes analyzed, and no hotspot mutation was detected in either of the two cohorts (Fig. 1B).
Fig. 1.
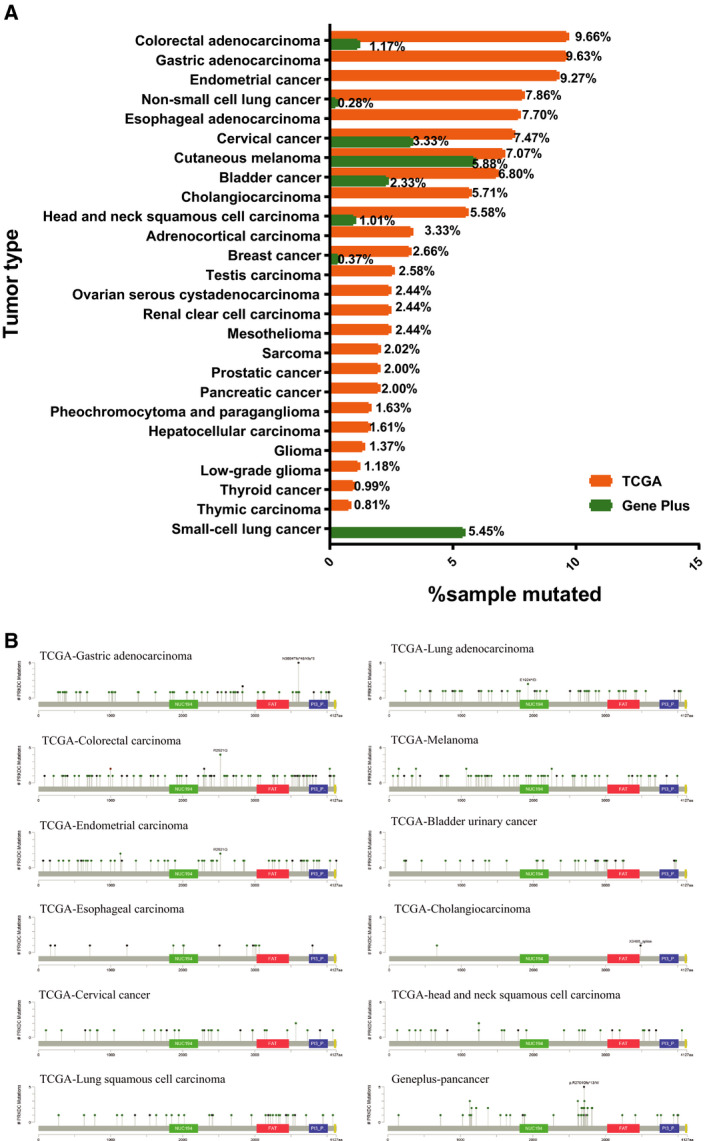
Prevalence of PRKDC Mutations. (A) Frequency of PRKDC mutations across different tumor types in TCGA and Chinese population (Geneplus). (B) Mutation sites and mutation type of the PRKDC gene in TCGA and Geneplus.
3.2. PRKDC mutation is associated with increased tumor mutation burden in pan‐cancer
To validate the association of PRKDC mutations with TMB, we compared the mutation burden of samples with PRKDC mutations and samples PRKDC wild‐type. In the top 10 cancers with the highest PRKDC mutation frequency from the TCGA dataset, the TMB in samples with PRKDC mutations was significantly higher than in those without PRKDC mutations (median nonsynonymous mutations 1278 vs. 109, P < 0.0001 for stomach adenocarcinoma; 1450 vs. 99, P < 0.0001 for colorectal adenocarcinoma; 1529 vs. 45, P < 0.0001 for uterine corpus endometrioid carcinoma). Other results are shown in (Fig. 2A). Previous studies had shown that mutations in some pivotal DDR pathway genes are associated with genomic instability and increased TMB, including MMR (PMS2/MLH1/MSH2/MSH6), POLE/POLD1, and BRCA1/BRCA2 [11]. Our study showed that there was no significant difference in TMB among PRKDC mutation group and MMR gene, POLE/D1, and BRCA1/2 mutation groups. The aforementioned groups all had a higher TMB than the nonmutation group: The nonmutation group was defined as patients without any mutations in the above genes (Fig. 2B). The TMB levels of the top quartile are commonly considered TMB‐high [29], and according to the cutoff value, we found that 96% of PRKDC mutation samples were TMB‐H in the uterine corpus endometrial carcinoma cohort, followed by 82% of the PRKDC mutation samples being TMB‐H in the colorectal adenocarcinoma cohort and 81% in the gastric adenocarcinoma group (Fig. 2C). We then conducted a logistic regression to analyze the influencing factors of TMB‐H. The results showed that the independent risk factors of TMB‐H were PRKDC mutation, BRCA1/2 mutation, POLE/D1 mutation, MMR gene mutation, and age (all P < 0.05; Table S2).
Fig. 2.
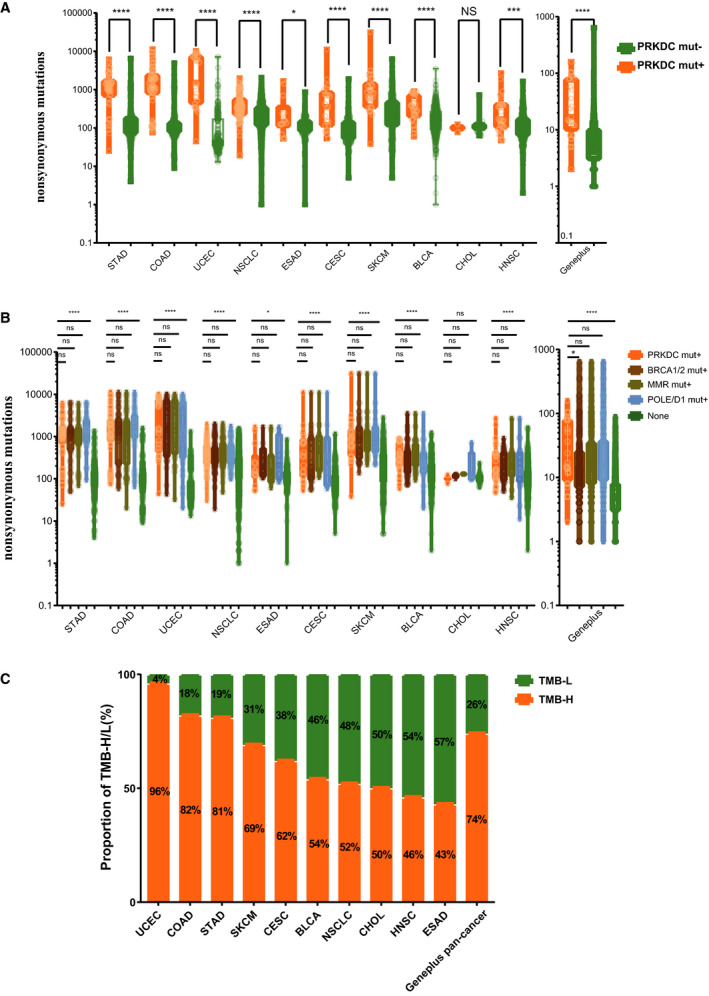
Relationships between TMB and PRKDC mutation status. (A) Comparison of TMB between PRKDC mutations and PRKDC wild‐type samples in TCGA top10 cancers and Geneplus pan‐cancer. TMB, defined as the sum of somatic nonsynonymous mutations. (B) Comparison of TMB between PRKDC mutations and other DDR‐gene (including BRCA1/BRCA2, PMS2/MSH2/MSH6/MLH1, POLE/POLD1) mutations in TCGA top10 cancers and Geneplus pan‐cancer. The none group was the referent group, defined as the absence of any of the aforementioned mutations. (C) The proportion of TMB‐high/low status in PRKDC mutation samples in TCGA top10 cancers and Geneplus pan‐cancer. TMB‐H, defined as the upper quartile of tumor all samples' TMB in each cancer type. Statistical significance was calculated using the Mann–Whitney U test. ****P < 0.0001; ***P < 0.001; **P < 0.01; *P < 0.05; ns P > 0.05. STAD, stomach adenocarcinoma (N = 436); COAD, colorectal adenocarcinoma (N = 528); UCEC, uterine corpus endometrial carcinoma (N = 248); NSCLC, non‐small‐cell lung cancer (N = 1031); ESAD, esophageal adenocarcinoma (N = 182); CESC, cervical squamous cell carcinoma (N = 281); SKCM, human skin cutaneous melanoma (N = 368); BLCA, bladder urothelial carcinoma (N = 412); CHOL, cholangiocarcinoma (N = 35); HNSC, head and neck squamous cell carcinoma (N = 502); Geneplus, pan‐cancer samples in Geneplus‐Beijing Institute (N =。 3877). (N: the number of samples).
Similar results were found in the Geneplus cohort. Patients with PRKDC mutations showed significantly higher TMB than those with PRKDC wild‐type (median nonsynonymous mutations 17.0 vs. 6.0, P < 0.0001). When compared with other DDR genes, the PRKDC mutation group had a higher TMB than the BRCA1/2 mutation group (P < 0.05) but was not different from the MMR mutation group or the POLE/D1 mutation group. The proportion of TMB‐high in the PRKDC mutation group was 74% over a pan‐cancer analysis (Fig. 2A–C).
To further explore the association among PRKDC mutation, MSI status, and TMB, we included four clinical cohorts covering three cancer types [20, 21, 22]. It showed that 63.89% (46/72) of PRKDC mutations subgroups are MSI‐H samples, but PRKDC mutation and MSI are not completely overlapped, and the majority of PRKDC mutations which did not overlap with MSI‐H group are still TMB‐H (Fig. S1). Among the nonoverlapped 26 PRKDC mut+/MSS MSI‐L patients, it also presented a large proportion (20/26) of patients with other DDR genes (BRCA1/2, POLE/D1, or MMR genes) mutation, but four of the rest six PRKDC mut+/MSS MSI‐L/other DDR mut− patients were still TMB‐H. Then, we compared the TMB among PRKDC mut+/MSI‐H, PRKDC mut−/MSI‐H, PRKDC mut+/MSS MSI‐L, and PRKDC mut−/MSS MSI‐L subgroups in the four cohorts. In the Lander's endometrial carcinoma cohort [20, 21, 22], the TMB of the PRKDC mut+ samples is significantly higher than in the PRKDC mut− samples in both MSI‐H and MSI‐L/MSS subtypes (median nonsynonymous mutations 558 vs. 220, P < 0.001 in MSI‐H subgroup, 5764 vs. 31, P < 0.0001 in MSS/MSI‐L subgroup). In the Muzny's colorectal adenocarcinoma cohort [20, 21, 22] and the Vesteinn's gastric adenocarcinoma cohort [20, 21, 22], the TMB of PRKDC mut+ samples is significantly higher than in the PRKDC mut− samples only in the MSS/MSS‐L subtypes (median nonsynonymous mutations 3209 vs. 69.50, P < 0.0001 for colorectal adenocarcinoma, 234 vs. 87, P < 0.01 for gastric adenocarcinoma). In the Kai Wang's gastric adenocarcinoma cohort [20, 21, 22], the TMB of PRKDC mut+ samples is significantly higher than in the PRKDC mut− samples only in the MSI‐H subtypes (median nonsynonymous mutations 1467 vs. 553, P < 0.01; Fig. S2). Furthermore, a logistic regression was performed to analyze the influencing factors of TMB‐H in the combined four cohorts. It showed that the independent risk factors of TMB‐H were PRKDC mutation, MSI‐H, BRCA1/2 mutation, MMR gene mutation, and POLE/D1 mutation (odds ratio, 19.428, 95% CI, 5.525–68.317, P = 0.000, PRKDC mutation; Table S3).
The PRKDC gene has a very long transcript with 12 784 bp, and genes with longer transcripts are generally considered more likely to accumulate somatic mutations by chance. In order to show that the large number of mutations that occur in PRKDC is not due to its long transcript, we analyzed whether the length of the transcript was correlated with the average number of somatic mutations in a gene set, which including 800 genes with the length of transcripts more than 8100 bp (8100–104 301 bp). In most of the genes, we found that the length of the transcript correlated with the average number of somatic mutations within them when we analyzed four TCGA patient cohorts (r = 0.7537, ****P < 0.0001, Bladder Carcinoma cohort; r = 0.7276, ****P < 0.0001, Colorectal Cancer cohort; r = 0.7679, ****P < 0.0001, Lung Adenocarcinoma cohort; and r = 0.7824, ****P < 0.0001, Head and Neck Squamous Cell Carcinoma cohort; Fig. S3). For example, TTN, SYNE2, and RSF1 genes showed a short‐weighted distance from the fitness lines, which indicated according to the trend well. But the PRKDC gene was not consistent with the fitness lines (Fig. S3). Thus, the mutations that occur in PRKDC are not only due to the longer transcript length.
3.3. The correlation of PRKDC mutations with signatures of CTL, NK cell infiltration, and inflamed tumor microenvironment
To further explore the distinct phenotypic and immunologic states caused by PRKDC mutations, we performed GSEA with the Hallmark gene set in the PRKDC mutation group and the PRKDC wild‐type group based on the TCGA top 10 cancers dataset. Notably, we found that sixteen gene sets were significantly upregulated, including five immune‐related signaling pathways: IFN‐γ response, IFN‐α response, allograft rejection, complement, and IL‐6/JAK/STAT signaling. Nine gene sets were significantly downregulated, including two immune‐related negative signaling pathways (TGF‐β signaling and Wnt/β‐catenin signaling) in the PRKDC mutation group (FDR q < 0.05; Fig. 3A and Table S4). Previous studies [30] have confirmed that the IFN‐γ pathway is one of the key pathways to induce PD‐L1 expression. TGF‐β plays an important role in promoting tumor immune escape and immunotherapy resistance [24, 31]. The upregulation of the IFN‐γ response and other immune‐related pathways along with downregulation of TGF‐β may contribute to the improvement of ICI therapy in patients with PRKDC mutations.
Fig. 3.
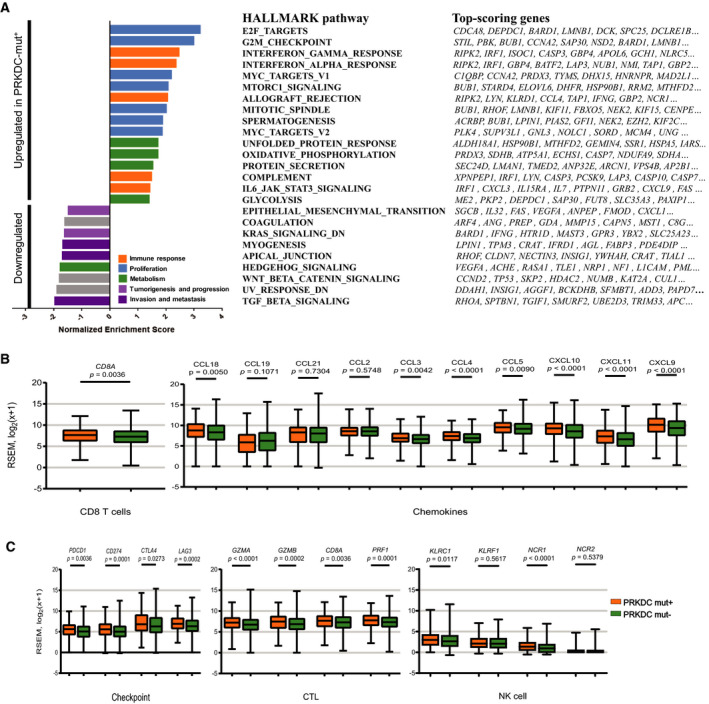
Transcriptome analysis by PRKDC mutation status in TCGA top10 cancers. (A) GSEA of hallmark gene sets downloaded from MSigDB database. Hallmark pathways significantly associated with PRKDC mutation (FDR. q < 0.05; comparing 285 PRKDC mutation samples to 3256 PRKDC wild‐type samples), and the top 10 genes per set are shown; complete lists are given in Table S1. (B, C) Comparison of the mRNA expression of genes related to immune checkpoints, cytotoxic lymphocyte, NK cells, chemokines, and Th1 cells signature between PRKDC mutations and PRKDC wild‐type groups in the TCGA top10 cancers analysis. CTL, cytotoxic lymphocyte. Statistical significance was calculated using the Mann–Whitney U test in B and C.
Previous reports showed that the presence of immune cells, especial tumor‐specific T‐cell and NK cell infiltration, which can be estimated using transcriptome signatures, had a significant association with a superior response to ICI [24]. We analyzed 3541 samples of 10 solid tumors from TCGA with a PRKDC mutation frequency in the top 10 with both RNA‐seq and WES data. The mRNA expression levels of immune‐related gene clusters were analyzed. Among the 14 selected immune‐related gene clusters (Table S5), the mRNA expression of eight gene clusters was significantly higher in the subgroup with PRKDC mutations than in the subgroup having the PRKDC wild‐type (P < 0.05; Table S6). Specifically, the PRKDC mutation subgroup demonstrated dramatically higher levels of mRNA expression than did the PRKDC wild‐type subgroup in the following gene clusters: CD8 T cells (CD8A, Fig. 3B), chemokines (CCL18, CCL3, CCL4, CCL5, CXCL10, CXCL11, and CXCL9, Fig. 3B), immune checkpoint (PD‐L1, PD‐1, CTLA‐4, and LAG3, Fig. 3C), cytotoxic T cells (GZMA, GZMB, CD8A, and PRF1, Fig. 3C), and NK cells (KLRC1 and NCR1, Fig. 3C).
3.4. PRKDC mutations predict favorable clinical benefit to ICI
We next examined four independent cohorts to investigate whether patients with PRKDC mutations could benefit from ICI therapy. In the first two cohorts [14, 16], a total of 110 patients with melanoma from the Allen cohort who received anti‐CTLA4 therapy and 75 patients with non‐small‐cell lung cancer (NSCLC) from the Hellmann cohort who received combined anti‐PD‐1 and anti‐CTLA4 therapy were analyzed. Patients with PRKDC mutations had higher TMB than patients with wild‐type PRKDC in both cohorts (Fig. 4A), and the TMB status in the PRKDC mutation group was similar to the BRCA1/2, POLE/D1, and MMR gene mutation groups (Fig. 4B). A higher neoantigen load was found in the PRKDC mutation patients in the Hellmann cohort (Fig. 4C).
Fig. 4.
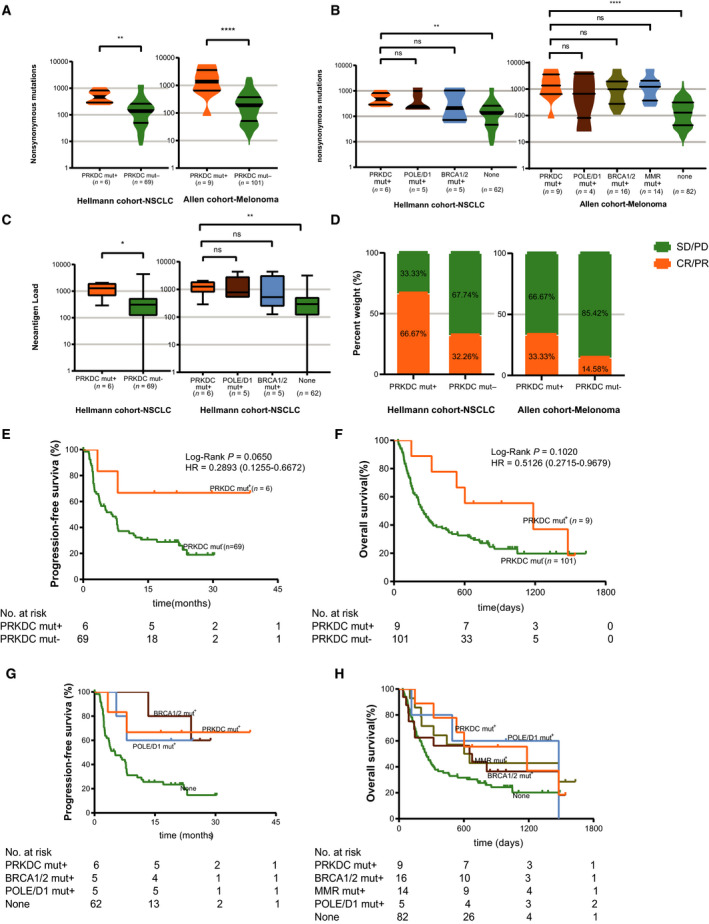
Patients with PRKDC mutations showed a favorable clinical benefit from immune checkpoint blockades. (A) Comparison of TMB between PRKDC mutations and PRKDC wild‐type in two different IO cohorts. (B) Comparison of TMB between PRKDC mutation and other DDR‐gene mutations in cohorts. (C) Comparison of neoantigen load between PRKDC mutation and PRKDC wild‐type in cohorts. (D) Comparison of the ORR between the PRKDC mutations and PRKDC wild‐type groups from cohorts. (E) Kaplan–Meier survival curves of PFS comparing the PRKDC mutations and PRKDC wild‐type groups in patients with NSCLC treated with combined PD‐1 and CTLA‐4 blockade from the Hellmann cohort. (F) Kaplan–Meier survival curves of OS comparing the PRKDC mutations and PRKDC wild‐type groups in patients with melanoma treated with CTLA‐4 blockade from the Allen cohort. (G) Kaplan–Meier survival curves of PFS comparing the PRKDC mutations and other DDR‐gene mutation groups in patients with NSCLC treated with combined PD‐1 and CTLA‐4 blockade from the Hellmann cohort. (H) Kaplan–Meier survival curves of OS comparing the PRKDC mutations and other DDR‐gene mutation groups in patients with melanoma treated with anti‐CTLA‐4 therapy from the Allen cohort. Statistical significance was calculated using the Mann–Whitney U test in comparison of TMB and neoantigen load in A–C. ****P < 0.0001; ***P < 0.001; **P < 0.01; *P < 0.05; ns P > 0.05. The P value in Kaplan–Meier survival curves was determined by a log‐rank test.
The Hellmann cohort included 75 patients with NSCLC, including six patients with PRKDC mutations for whom progression‐free survival (PFS) was superior to that of PRKDC wild‐type patients [median, not reached (NR) vs. 6.8 months; HR, 0.2893; 95% CI, 0.1255–0.6672; P = 0.0650, Fig. 4E]. The objective response rates (ORR) of patients with a PRKDC mutation and those with a PRKDC wild‐type were 66.67% and 32.26%, respectively.
The Allen cohort enrolled 110 patients with metastatic melanoma, and a total of nine patients were identified with PRKDC mutations. Compared with the patients with the PRKDC wild‐type, the PRKDC mutation patients in this cohort also trended toward a longer overall survival (OS; median, 1184 days vs. 250 days; HR, 0.5126; 95% CI, 0.2715–0.9679; P = 0.1020, Fig. 4F) and a higher ORR (33.33% vs. 14.58%, Fig. 4D).
We compared the survival of patients with PRKDC mutations, MMR gene mutations, POLE/D1 mutations, and BRCA1/2 mutations with patients with no mutations in these genes (none mutation subgroup) in the two cohorts. In the Hellmann cohort, we found that PRKDC mutations were significantly associated with a longer PFS than in the none mutation subgroup (median, NR vs. 5.092 months; HR, 0.2541; 95% CI, 0.1138–0.5674; P = 0.0382, Fig. 4G) and had similar PFS with the BRCA1/2 mutation and POLE/D1 mutation subgroups (Fig. 4G). In the Allen cohort, patients with a PRKDC mutation showed a trend toward a longer OS than those with no mutation in these DDR genes (median, 1184 days vs. 250 days, HR = 0.5051, 95% CI, 0.2645–0.9646; P = 0.0939, Fig. 4H), and also a similar OS to patients with BRCA1/2 mutations, POLE/D1 mutations, and MMR gene mutations (Fig. 4H). In multivariate analysis, it showed a significant longer survival in PRKDC mutation samples in the two cohorts, after adjustment for clinicopathological characteristics, MMR genes, POLE/D1, and BRCA1/2 (hazard ratio, 0.240 95% CI, 0.058–0.998, P = 0.050, Hellmann cohort, hazard ratio, 0.361; 95% CI, 0.155–0.841; P = 0.018, Allen cohort; Tables 1 and 2). It suggests that a PRKDC mutation is an independent predictive factor of better clinical outcome in ICI‐treated patients.
Table 1.
Univariable and multivariable analyses of progression‐free survival in the Hellmann Cohort [14]
| Parameters | N | Univariable analysis | Multivariable analysis | |||||
|---|---|---|---|---|---|---|---|---|
| HR | 95% CI | P | HR | 95% CI | P | |||
| Age | < 60 | 27 | 0.9937 | 0.5538–1.783 | 0.9830 | |||
| ≥ 60 | 48 | |||||||
| Sex | Male | 37 | 1.030 | 0.5915–1.793 | 0.9166 | |||
| Female | 38 | |||||||
| ECOG | 0 | 30 | 0.6851 | 0.3935–1.193 | 0.1837 | |||
| 1 | 45 | |||||||
| Smoking | Current/former | 60 | 0.6979 | 0.3304–1.474 | 0.2870 | |||
| Never | 15 | |||||||
| Histology | Squamous | 16 | 1.177 | 0.5678–2.441 | 0.6424 | |||
| Nonsquamous | 59 | |||||||
| %PD‐L1 expression | > 1 | 43 | 0.7307 | 0.3921–1.362 | 0.2931 | |||
| ≤ 1 | 27 | |||||||
| BRCA1/2 | Mut+ | 5 | 0.2663 | 0.1184–0.5988 | 0.0420 | 0.212 | 0.050–0.899 | 0.035 |
| Mut− | 70 | |||||||
| POLE/D1 | Mut+ | 4 | 0.4772 | 0.1721–1.323 | 0.2921 | |||
| Mut− | 71 | |||||||
| PRKDC | Mut+ | 6 | 0.2893 | 0.1255–0.6672 | 0.0650 | 0.240 | 0.058–0.998 | 0.050 |
| Mut− | 69 | |||||||
Table 2.
Univariable and multivariable analyses of overall survival in the Allen Cohort [16]
| Parameters | N | Univariable analysis | Multivariable analysis | |||||
|---|---|---|---|---|---|---|---|---|
| HR | 95% CI | P | HR | 95% CI | P | |||
| Age | < 60 | 59 | 1.073 | 0.6971–1.652 | 0.7484 | |||
| ≥ 60 | 51 | |||||||
| Sex | Male | 78 | 0.7806 | 0.4787–1.273 | 0.2924 | |||
| Female | 32 | |||||||
| Stage | IV | 100 | 4.504 | 2.443–8.306 | 0.0045 | 4.912 | 1.539–15.673 | 0.007 |
| III | 10 | |||||||
| LDH | LDH‐high | 48 | 2.030 | 1.289–3.197 | 0.0010 | 2.307 | 1.474–3.611 | 0.000 |
| LDH‐low | 58 | |||||||
| BRCA1/2 | Mut+ | 16 | 0.7409 | 0.4184–1.312 | 0.3475 | |||
| Mut− | 94 | |||||||
| MMR | Mut+ | 14 | 0.05722 | 0.3249–1.008 | 0.1020 | 0.680 | 0.327–1.417 | 0.301 |
| Mut− | 96 | |||||||
| POLE/D1 | Mut+ | 4 | 0.6708 | 0.2566–1.753 | 0.4906 | |||
| Mut− | 106 | |||||||
| PRKDC | Mut+ | 9 | 0.5126 | 0.2715–0.9679 | 0.1020 | 0.361 | 0.155–0.841 | 0.018 |
| Mut− | 101 | |||||||
In another two cohorts [7, 15], a total of two patients (study IDs were CR4880 and PR4092, [15]) with melanoma who received anti‐CTLA4 therapy and one patient with lung adenocarcinoma (study ID was DI6359, [7]) who received anti‐PD‐1 therapy were verified to harbor PRKDC mutations. The numbers of nonsynonymous mutations in patients CR4880, PR4092, and DI6359 were 527, 1108, and 228, respectively. One patient achieved a complete response, and the other two patients achieved major partial responses. The overall survival for the two melanoma patients was 5.4 and 6.1 years, and the PFS for the NSCLC patient was 9.8 months (Table S7).
In our clinic, we treated a patient (NPC_Y) with stage IVA nasopharyngeal carcinoma that harbored a PRKDC mutation. The functional impact evaluation of the PRKDC mutation in this patient showed that it can be considered as a functional mutation (SIFT score, 0.05; PolyPhen‐2 HumVar score, 0.57). The PD‐L1 expression by IHC staining in both tumor cells and immune cells was positive (tumor proportion score > 95% and > 80%, respectively; Fig. S4). Moreover, a large number of infiltrating CD8+ T cells was found in the tumor center and at the margins of all lesions (Fig. S4). The ctDNA analysis expectedly displayed a high mutation load with 26 nonsynonymous mutations (Table S8). When disease rapidly progressed with systemic metastases after first‐line therapy (left clavicle area, right lobe of liver, sternum, and T11/L4 centrum), the patient accepted nivolumab as the next‐step treatment. Treatment resulted in a complete response based on the criteria in the Response Evaluation Criteria In Solid Tumors (RECIST) 1.1, after three cycles of nivolumab. Finally, this patient had 17 months of the progression‐free survival with nivolumab (Fig. S5).
4. Discussion
In our study, the prevalence of PRKDC mutations in two large cohorts was identified in multiple solid tumor types, including common neoplasms such as gastrointestinal cancers, NSCLC, and bladder carcinoma. We are the first to comprehensively describe that PRKDC mutations, regardless of the status of other DDR‐related genes, and found an association with an increased TMB, an increased mRNA expression of immune‐related genes, and a superior response to ICI in pan‐cancer patients.
Double‐stranded DNA breaks are the most serious DNA lesions. The two major pathways for repair of DSB are homologous recombination (HR) and NHEJ [11, 32]. DNA‐PKcs is the key component of the NHEJ pathway involved in DSB repair [12]. PRKDC mutations lead to a deficiency in the DNA‐PKcs and NHEJ pathway, so DSB fail to repair and mutations tend to accumulate. Furthermore, DNA‐PKcs has been verified as essential for induction of apoptosis after massive DSB formation [33]. When PRKDC mutations are present, cancer cells are resistant to apoptosis and there is an increase in the accumulation of DNA damage that promotes genome instability. Moreover, such excess DNA damage may not only increase mutations due to error‐prone translesion synthesis, but also increase epigenetic alterations due to errors during DNA repair [34, 35]. In our study, we comprehensively reviewed PRKDC mutations and found they were significantly associated with high TMB scores in two large independent cohorts, similar to POLE/POLD1, MMR gene, and BRCA1/BRCA2 mutation patients. This finding implicates PRKDC mutations as a valuable biomarker in clinical practice.
When performing transcriptome analysis we found that, compared to the PRKDC wild‐type, the patients with PRKDC mutations tend to have an inflamed tumor microenvironment (TME) that includes higher numbers of CD8+ T cells, NK cells, Th1, and pDCs, and higher PD‐L1 expression, other immune checkpoints, and chemokine expression. The GSEA analysis also showed a remarkably upregulated expression of the IFN‐γ and IFN‐α response, and IL‐6/JAK/STAT signaling along with downregulation of TGF‐β and Wnt/β‐catenin signaling. The high TMB might be one of the reasons contributing to this inflammatory microenvironment. A previous study has shown that DNA‐PK also interacts with the transcription factor autoimmune regulator (AIRE) to promote central T‐cell tolerance [36]. Deficiency of DNA‐PK can present as an inflammatory disease with organ‐specific autoimmunity, suggesting a role of DNA‐PK in regulating autoimmune responses and maintaining AIRE‐dependent autoimmune tolerance [35]. These results support our findings that PRKDC mutations induced inflamed TME was caused by an increased TMB level together with impaired central immune tolerance. In the four independent cohorts, we also found that patients with PRKDC mutations were more likely to benefit from ICI.
However, our study has limitations. First, due to the relatively low incidence and limited data, we can only conclude that PRKDC mutation is one of the important factors which affect TMB, TME, and the prognosis of ICI therapy, but cannot confirm the extent to which PRKDC mutation sites contribute to the inflamed TME and prognosis improvements. Second, the sample size of the validation cohort was small and only melanoma and NSCLC were included, and so, the association between PRKDC mutation‐induced changes in the immune microenvironment and improvements of ICI treatment efficacy needs to be confirmed and verified in a larger population with multiple cancer types, such as MSS colorectal cancer and pancreatic cancer.
5. Conclusion
Our findings suggest that PRKDC mutations occur in a subset of solid tumor patients, and they often appeared to co‐exist with deficiency in some other DDR mechanism. But anyway, PRKDC mutation is still one of the important factors significantly associated with an increased TMB, an increased expression of immune‐related genes, and an improved response to ICI. Additional prospective studies are needed to validate this finding and to determine whether routine testing for this alteration is warranted. This work may have important implications for clinical practice and provide a potential predictive biomarker for guiding ICI therapy.
Conflict of interest
The authors declare no conflict of interest.
Author contributions
YC, CC, and YL conceived and designed the study. YG, JL, GC, LC, and XY made the collection of clinical data and sample. YG, JL, LC, and XY performed sequencing. YH, JL, LC, LP, ZG, NX, and JP analyzed the clinical data and performed statistical analysis. All authors wrote the manuscript. All authors read and approved the final manuscript.
Supporting information
Fig. S1. Venn diagram of the relationship among PRKDC mutation subgroup, MSI‐H subgroup and TMB‐H subgroup. (Red: PRKDC mutation subgroup; Green: MSI‐H subgroup; Purple: TMB‐H subgroup).
Fig. S2. Comparison of TMB in different combinations of PRKDC mutations and MSI status groups. (ns, P > 0.05, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001).
Fig. S3. Length of transcript versus the mean number of somatic mutations in coding region for each gene in the TCGA cohorts of Bladder cancer, Colorectal cancer, Lung Adenocarcinoma and Head/Neck Squamous cell carcinoma. R, Pearson's correlation; curve, fitness lines. The statistics used in this analysis is Loess regression.
Fig. S4. The hematoxylin‐eosin (HE) staining and immunohistochemistry (IHC) staining of CD8 and PD‐L1 with the recurrent nasopharyngeal lesion of patient NPC_Y. The representative microscopic findings are shown.
Fig. S5. Disease course and clinical response in an advanced nasopharyngeal carcinoma patient (NPC_Y) with PRKDC mutation treated with Nivolumab‐based multi‐combination strategies.
Table S1. The cancer types and number of samples included in Geneplus pan‐cancer cohort.
Table S2. The logistic regression of factors influencing TMB‐high in TCGA top 10 cancers.
Table S3. The logistic regression of factors influencing TMB‐high in the combined four cohorts which with MSI/MSS data.
Table S4. GSEA between PRKDC mutation group and non‐mutation group with the Hallmark gene set in TCGA top 10 cancers dataset.
Table S5. Gene list of immune‐related gene set.
Table S6. Immune‐related gene set mRNA expression analysis.
Table S7. The clinical data of three patients with PRKDC mutation from two clinical cohorts treated with ICIs.
Table S8. The mutation list of an advanced nasopharyngeal carcinoma patient (NPC_Y) by ctDNA analysis.
Appendix S1. Case report.
Appendix S2. Immunohistochemical staining and analysis for CD8, PD‐L1.
Acknowledgements
We appreciate all subjects who participated in this study and wish to thank Dr. Jian‐ping Lu and Dr. Wei‐feng Zhu (Department of Pathology, Fujian Medical University Cancer Hospital & Fujian Cancer Hospital, Fuzhou, Fujian Province, China) and all colleagues (Cancer Bio‐immunotherapy Center, Fujian Medical University Cancer Hospital) for technical support. The work was supported by Fujian provincial health and family planning research talent training program (Grant No. 2018‐ZQN‐13, 2018‐CX‐11) and Joint Funds for the innovation of science and Technology, Fujian province (Grant No. 2017Y9077, 2018Y9107).
Yu Chen, Yi Li, Yanfang Guan and Yingying Huang contributed equally to this work
Data accessibility
The TCGA datasets analyzed in the study are available at the cBioPortal (www.cbioportal.org/). Relevant data in this study were provided in the supplementary information. Other data could be obtained from the corresponding authors of this study.
Data accessibility
The TCGA datasets analyzed in the study are available at the cBioPortal (www.cbioportal.org/). Relevant data in this study were provided in the supplementary information. Other data could be obtained from the corresponding authors of this study.
References
- 1. Gandhi L, Rodriguez‐Abreu D, Gadgeel S, Esteban E, Felip E, De Angelis F, Domine M, Clingan P, Hochmair MJ, Powell SF et al (2018) Pembrolizumab plus chemotherapy in metastatic non‐small‐cell lung cancer. N Engl J Med 378, 2078–2092. [DOI] [PubMed] [Google Scholar]
- 2. Rosenberg JE, Hoffman‐Censits J, Powles T, van der Heijden MS, Balar AV, Necchi A, Dawson N, O'Donnell PH, Balmanoukian A, Loriot Y et al (2016) Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum‐based chemotherapy: a single‐arm, multicentre, phase 2 trial. Lancet (London, England) 387, 1909–1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, Segal NH, Ariyan CE, Gordon RA, Reed K et al (2013) Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med 369, 122–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Havel JJ, Chowell D & Chan TA (2019) The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat Rev Cancer 19, 133–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Taube JM, Klein A, Brahmer JR, Xu H, Pan X, Kim JH, Chen L, Pardoll DM, Topalian SL & Anders RA (2014) Association of PD‐1, PD‐1 ligands, and other features of the tumor immune microenvironment with response to anti‐PD‐1 therapy. Clin Cancer Res 20, 5064–5074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu‐Lieskovan S, Berent‐Maoz B, Pang J, Chmielowski B, Cherry G et al (2017) Genomic and transcriptomic features of response to anti‐PD‐1 therapy in metastatic melanoma. Cell 168, 542. [DOI] [PubMed] [Google Scholar]
- 7. Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, Lee W, Yuan J, Wong P, Ho TS et al (2015) Cancer immunology. Mutational landscape determines sensitivity to PD‐1 blockade in non‐small cell lung cancer. Science (New York, NY) 348, 124–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V et al (2014) PD‐1 blockade induces responses by inhibiting adaptive immune resistance. Nature 515, 568–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Germano G, Lamba S, Rospo G, Barault L, Magri A, Maione F, Russo M, Crisafulli G, Bartolini A, Lerda G et al (2017) Inactivation of DNA repair triggers neoantigen generation and impairs tumour growth. Nature 552, 116–120. [DOI] [PubMed] [Google Scholar]
- 10. Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, Lu S, Kemberling H, Wilt C, Luber BS et al (2017) Mismatch repair deficiency predicts response of solid tumors to PD‐1 blockade. Science 357, 409–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mouw KW, Goldberg MS, Konstantinopoulos PA & D'Andrea AD (2017) DNA damage and repair biomarkers of immunotherapy response. Cancer Discov 7, 675–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Goodwin JF & Knudsen KE (2014) Beyond DNA repair: DNA‐PK function in cancer. Cancer Discov 4, 1126–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hsu FM, Zhang S & Chen BP (2012) Role of DNA‐dependent protein kinase catalytic subunit in cancer development and treatment. Transl Cancer Res 1, 22–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hellmann MD, Nathanson T, Rizvi H, Creelan BC, Sanchez‐Vega F, Ahuja A, Ni A, Novik JB, Mangarin LMB, Abu‐Akeel M et al (2018) Genomic features of response to combination immunotherapy in patients with advanced non‐small‐cell lung cancer. Cancer Cell 33, 843–852.e844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, Walsh LA, Postow MA, Wong P, Ho TS et al (2014) Genetic basis for clinical response to CTLA‐4 blockade in melanoma. N Engl J Med 371, 2189–2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, Sucker A, Hillen U, Foppen MHG, Goldinger SM et al (2015) Genomic correlates of response to CTLA‐4 blockade in metastatic melanoma. Science (New York, NY) 350, 207–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Lander ES & Getz G (2013) Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nature Biotechnol 31, 213–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tomczak K, Czerwinska P & Wiznerowicz M (2015) The Cancer Genome Atlas (TCGA): an immeasurable source of knowledge. Contemporary oncology (Poznan, Poland) 19, A68–A77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Li B & Dewey CN (2011) RSEM: accurate transcript quantification from RNA‐Seq data with or without a reference genome. BMC Bioinform 12, 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Muzny DMBM, Chang K, Dinh HH, Drummond JA, Fowler G, Kovar CL, Lewis LR, Morgan MB, Newsham IF, Reid JG et al (2012) Comprehensive molecular characterization of human colon and rectal cancer. Nature 487, 330–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lander ES & Park PJ (2013) Integrated genomic characterization of endometrial carcinoma. Nature 497, 67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bass AJ, Thorsson V, Shmulevich I, Reynolds SM, Miller M, Bernard B, Hinoue T, Laird PW, Curtis C, Shen H et al (2014) Comprehensive molecular characterization of gastric adenocarcinoma. Nature 513, 202–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wang K, Yuen ST, Xu J, Lee SP, Yan HHN, Shi ST, Siu H C, Deng S, Chu KM, Law S et al (2014) Whole‐genome sequencing and comprehensive molecular profiling identify new driver mutations in gastric cancer. Nat Genet 46, 573–582. [DOI] [PubMed] [Google Scholar]
- 24. Jiang P, Gu S, Pan D, Fu J, Sahu A, Hu X, Li Z, Traugh N, Bu X, Li B et al (2018) Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat Med 24, 1550–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sade‐Feldman M, Jiao YJ, Chen JH, Rooney MS, Barzily‐Rokni M, Eliane JP, Bjorgaard SL, Hammond MR, Vitzthum H, Blackmon SM et al (2017) Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat Commun 8, 1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wang Z, Zhao J, Wang G, Zhang F, Zhang Z, Zhang F, Zhang Y, Dong H, Zhao X, Duan J et al (2018) Comutations in DNA damage response pathways serve as potential biomarkers for immune checkpoint blockade. Can Res 78, 6486–6496. [DOI] [PubMed] [Google Scholar]
- 27. Nathanson T, Ahuja A, Rubinsteyn A, Aksoy BA, Hellmann MD, Miao D, Van Allen E, Merghoub T, Wolchok JD, Snyder A et al (2017) Somatic mutations and neoepitope homology in melanomas treated with CTLA‐4 blockade. Cancer Immunol Res 5, 84–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Karosiene E, Lundegaard C, Lund O & Nielsen M (2012) NetMHCcons: a consensus method for the major histocompatibility complex class I predictions. Immunogenetics 64, 177–186. [DOI] [PubMed] [Google Scholar]
- 29. Balar AV, Galsky MD, Rosenberg JE, Powles T, Petrylak DP, Bellmunt J, Loriot Y, Necchi A, Hoffman‐Censits J, Perez‐Gracia JL et al (2017) Atezolizumab as first‐line treatment in cisplatin‐ineligible patients with locally advanced and metastatic urothelial carcinoma: a single‐arm, multicentre, phase 2 trial. Lancet (London, England) 389, 67–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Li N, Wang J, Zhang N, Zhuang M, Zong Z, Zou J, Li G, Wang X, Zhou H, Zhang L et al (2018) Cross‐talk between TNF‐alpha and IFN‐gamma signaling in induction of B7–H1 expression in hepatocellular carcinoma cells. Cancer Immunol Immunother 67, 271–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, Kadel EE III, Koeppen H, Astarita JL, Cubas R et al (2018) TGFbeta attenuates tumour response to PD‐L1 blockade by contributing to exclusion of T cells. Nature 554, 544–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dietlein F, Thelen L, Jokic M, Jachimowicz RD, Ivan L, Knittel G, Leeser U, van Oers J, Edelmann W, Heukamp LC et al (2014) A functional cancer genomics screen identifies a druggable synthetic lethal interaction between MSH3 and PRKDC. Cancer Discov 4, 592–605. [DOI] [PubMed] [Google Scholar]
- 33. Abe T, Ishiai M, Hosono Y, Yoshimura A, Tada S, Adachi N, Koyama H, Takata M, Takeda S, Enomoto T et al (2008) KU70/80, DNA‐PKcs, and Artemis are essential for the rapid induction of apoptosis after massive DSB formation. Cell Signal 20, 1978–1985. [DOI] [PubMed] [Google Scholar]
- 34. Cuozzo C, Porcellini A, Angrisano T, Morano A, Lee B, Di Pardo A, Messina S, Iuliano R, Fusco A, Santillo MR et al (2007) DNA damage, homology‐directed repair, and DNA methylation. PLoS Genet 3, e110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. O'Hagan HM, Mohammad HP & Baylin SB (2008) Double strand breaks can initiate gene silencing and SIRT1‐dependent onset of DNA methylation in an exogenous promoter CpG island. PLoS Genet 4, e1000155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mathieu AL, Verronese E, Rice GI, Fouyssac F, Bertrand Y, Picard C, Chansel M, Walter JE, Notarangelo LD, Butte MJ et al (2015) PRKDC mutations associated with immunodeficiency, granuloma, and autoimmune regulator‐dependent autoimmunity. J Allergy Clin Immunol 135, 1578–1588.e1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Venn diagram of the relationship among PRKDC mutation subgroup, MSI‐H subgroup and TMB‐H subgroup. (Red: PRKDC mutation subgroup; Green: MSI‐H subgroup; Purple: TMB‐H subgroup).
Fig. S2. Comparison of TMB in different combinations of PRKDC mutations and MSI status groups. (ns, P > 0.05, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001).
Fig. S3. Length of transcript versus the mean number of somatic mutations in coding region for each gene in the TCGA cohorts of Bladder cancer, Colorectal cancer, Lung Adenocarcinoma and Head/Neck Squamous cell carcinoma. R, Pearson's correlation; curve, fitness lines. The statistics used in this analysis is Loess regression.
Fig. S4. The hematoxylin‐eosin (HE) staining and immunohistochemistry (IHC) staining of CD8 and PD‐L1 with the recurrent nasopharyngeal lesion of patient NPC_Y. The representative microscopic findings are shown.
Fig. S5. Disease course and clinical response in an advanced nasopharyngeal carcinoma patient (NPC_Y) with PRKDC mutation treated with Nivolumab‐based multi‐combination strategies.
Table S1. The cancer types and number of samples included in Geneplus pan‐cancer cohort.
Table S2. The logistic regression of factors influencing TMB‐high in TCGA top 10 cancers.
Table S3. The logistic regression of factors influencing TMB‐high in the combined four cohorts which with MSI/MSS data.
Table S4. GSEA between PRKDC mutation group and non‐mutation group with the Hallmark gene set in TCGA top 10 cancers dataset.
Table S5. Gene list of immune‐related gene set.
Table S6. Immune‐related gene set mRNA expression analysis.
Table S7. The clinical data of three patients with PRKDC mutation from two clinical cohorts treated with ICIs.
Table S8. The mutation list of an advanced nasopharyngeal carcinoma patient (NPC_Y) by ctDNA analysis.
Appendix S1. Case report.
Appendix S2. Immunohistochemical staining and analysis for CD8, PD‐L1.
Data Availability Statement
The TCGA datasets analyzed in the study are available at the cBioPortal (www.cbioportal.org/). Relevant data in this study were provided in the supplementary information. Other data could be obtained from the corresponding authors of this study.
The TCGA datasets analyzed in the study are available at the cBioPortal (www.cbioportal.org/). Relevant data in this study were provided in the supplementary information. Other data could be obtained from the corresponding authors of this study.