

Abstract
This review covers the main synthetic routes to and the corresponding mechanisms of phosphoramidate formation. The synthetic routes can be separated into six categories: salt elimination, oxidative cross-coupling, azide, reduction, hydrophosphinylation, and phosphoramidate-aldehyde-dienophile (PAD). Examples of some important compounds synthesized through these routes are provided. As an important class of organophosphorus compounds, the applications of phosphoramidate compounds, are also briefly introduced.
Keywords: phosphoramidate, synthetic routes, mechanism, applications
1. Introduction to Phosphoramidates and their Applications
Phosphoramidates (P-N) are a class of organophosphorus compounds known for the presence of a single covalent bond between the tetracoordinate P(V) atom and N(III) atom. There are generally three types of phosphoroamidates, which are distinguished according to the substitution on the P and N atoms (Figure 1) [1].
Figure 1.

Three types of P-N based on the substituents directly attached to the P and N.
Another feature of these organophosphorous compounds, defined as (RO)2P(O)NR’2 (R, R’ = H, alkyl, aryl, heteroaryl), is a stable phosphoryl bond (P=O). Both P and N atoms are key physiological elements present in genetic material, energy transfer, enzymes, and other biomolecules and are required for various life processes. As such, molecules containing P-N linkages are found in a large array of biologically active natural products (1–7) (Figure 2) [2,3]. For example, Microcin C7 (1) (Figure 2) is an antibiotic produced by Escherichia coli [4]. Dinogunellin (2) (Figure 2), which is produced in the roe of some fishes, is a natural toxin [5]. Phosphoarginine and phosphocreatine (3 and 6) (Figure 2) are important biological molecules used as sources of stored energies in invertebrates and vertebrates, respectively [6]. Phosphoramidon (4) (Figure 2), derived from Streptomyces tanashiensis, inhibits the thermolysin enzyme, which is a key factor in the development of various diseases [7,8]. Lastly, phosmidosine and agrocin 84 (5 and 7) (Figure 2) are nucleotide antibiotics isolated from Streptomyces durhameusis and Agrobacterium radiobacter and are used to control gray mold disease and crown gall disease in plants, respectively [9,10].
Figure 2.

Phosphoramidate motifs in natural products: Microcin C7 (1), Dinogunellin (R = residue of fatty acid) (2), Phosphoarginine (3), Phosphoramidon (4), Phosmidosine (5), Phosphocreatine (6), Agrocin 84 (7).
Due to the inherent physiochemical properties of the phosphorus atom including its polarizability, multivalency, varying oxidation states, and low coordination number, a diverse range of compounds containing the P-N motifs have been synthesized [11]. These P-Ns are widely used in medicine for the control and treatment of various diseases, in agriculture as pesticides for crop protection, in industry as novel fire-retardant compounds to delay the flammability of polymeric compounds, and in the fields of analytical and coordination chemistry. Therefore, synthetic P-N compounds are not only researched out of academic interest, but they also have commercial applications. Uses for P-Ns can be separated into 5 main classes: agriculture, industry, analytical chemistry, synthetic chemistry, and medicinal chemistry (Figure 3) [12,13,14,15,16,17,18].
Figure 3.

Applications of phosphoramidates.
1.1. Phosphoramidates in Agriculture
This class of organophosphates (with common names such as cruformate, fenamiphos, and fosthietan) has been widely used in pest management for crop protection (Figure 3) [19,20,21]. P-Ns for pest control affect the nervous system of pests/insects by inhibiting acetylcholinesterase (AChE), a major neurotransmitter [22,23,24,25]. In addition, these P-N compounds have been utilized as urease inhibitors [26,27]. Urease is a nickel-based enzyme that catalyzes the hydrolysis of urea on soil surfaces into volatile ammonia and carbon dioxide (Scheme 1) [28,29]. Thus, on inhibiting the activity of this metalloenzyme, the availability and performance of urea is enhanced, which is a vital nitrogenous fertilizer for plant growth.
Scheme 1.

Hydrolysis of urea on soil in the presence of urease.
1.2. Phosphoramidates in Industry
P-Ns have been used as additives in both oil and water-based lubricants (Figure 3). It has been proposed that phosphorus acts as a key element in enhancing the tribological properties of water/oil-based fluids by forming a protective layer containing phosphates and/or polyphosphates. Thus, they possess anti-wear and friction-reducing properties [18,30]. Furthermore, these P-Ns have been found to be a promising alternative to halogenated flame retardants (FRs), which are being phased out as they have been recognized as global contaminants due to their ill effects on health and the environment [31]. P-N fire retardants are less volatile and relatively thermally stable with enhanced char formation, and they are more efficient and sustainable than their halogenated counterparts [32,33]. Recently, compounds containing both P and N atoms in combination have been reported to have a synergistic effect, as they can act in both vapor phase and condensed phase, thus offering improved flame retardancy on various substrates such as polyurethane foams, cotton, cellulose, epoxy resins, textiles, etc. [34,35,36,37,38,39,40]. For example, novel P-N-based intumescent flame retardants have been incorporated into polycarbonates (PC) to impart flame retardancy to the composite by forming an expandable protective char [41].
1.3. Phosphoramidates in Analytical Chemistry
MALDI-TOF MS is a prominent tool for analyzing biomolecules (Figure 3) [42]. For the detection of low molecular weight such as amino acids and peptides, conventional matrices yield a lot of matrix-related ions in the low m/z range of the spectrum, thus interfering with analyte signals. To tackle this, various approaches such as using additives in the organic matrix, high mass matrix molecules, or matrices based on inorganic compounds have been employed [43]. Furthermore, to improve analysis, synthesizing derivatives of small analyte molecules by N-phosphorylation labeling has been done. The neutral phosphoryl group attached at the N-terminal of amino acids suppresses the matrix signals and favors the transfer of energy from matrix to analyte for ionization by secondary ion-molecule reactions. Therefore, P-Ns have been used for analysis of biomolecules to improve the ionization efficiency and suppress matrix background signals [44]. In addition to this, P-Ns have found novel applications as task-specific ionic liquids (TSILs), which are tailored ionic liquids having a specific functionality to complex metal ions. Recently, these TSILs have been used for the efficient extraction of uranium metal, which is an important element in nuclear fuel, and the extraction of uranium metal from the spent/used nuclear fuel is vital for the sustainable economy [45]. The enhanced extraction capabilities using TSILs in contrast to liquid–liquid extraction could be attributed to synergistic effects as a result of complexation and H-bonding [17]. To this end, these tailored ionic liquids have been found to be selective for the micellar extraction of uranium metal from aqueous waste samples as even the ultra-trace levels of uranium are toxic [46].
1.4. Phosphoramidates in Synthetic Chemistry
P-Ns act as 1,3-N,O-chelating ligands (e.g., Figure 3, Ligand) and have diverse applications in the field of coordination chemistry for bond activation, catalysis, and metal ligand cooperativity [47,48,49]. For example, a P-N-tantalum complex has been developed as a pre-catalyst for hydroaminoalkylation reactions [50]. In addition, the synthesis of medium and large N-based heterocycles has been achieved with the aid of P-Ns, as the phosphoryl group improves intramolecular cyclization, and it can also be easily removed afterwards [51]. Furthermore, P-Ns have been used as directing groups for arylation reactions, as they selectively activate C-H bonds, and thus, desired functionality can be achieved [52].
1.5. Phosphoramidates in Pharmaceutical and Medicinal Chemistry
P-Ns have garnered considerable interest in the field of biomedicine due to their potential applications as antimicrobial [53,54,55], antioxidant [56], anticancer [57], antimalarial [58], antiviral [59,60], and anti-HIV [61] drugs and prodrugs against various ailments (Figure 3). The ProTide approach, pioneered by the McGuigan group in 1992 for drug delivery, is a powerful tool to enhance the efficacy, intracellular delivery, and therapeutic potential of the parent drugs [62,63]. Recently, remdesivir (Figure 3, drug candidate), which has a P-N moiety, has been evaluated as a therapeutic option for the treatment of COVID-19-affected patients and has attracted a lot of attention in the clinical field [64]. Additionally, these P-N compounds have been used in gene therapy and nucleoside treatment [65,66,67]. Moreover, many P-Ns have been found to be hydrolytically degradable under acidic conditions and are comparatively stable at neutral and higher pHs. The acid labile P-N bond is selectively cleaved due to protonation at N, which then undergoes nucleophilic attack by a molecule of water to eliminate the corresponding phosphates and amines [68,69]. Thus, these pH responsive P-Ns have notable applications in controlled drug release [70,71]. For example, the P-N-based prodrug of l-Dopa has been synthesized for its controlled release for the treatment of Parkinson’s disease [72]. Similarly, P-N derivatives of the antiherpetic drug, acyclovir (ACV), hydrolyses at pH 2 into ACV monophosphate and can be a promising antiviral drug due to increased cell penetration and therapeutic potential relative to the parent drug [73].
2. Synthetic Routes to Phosphoramidates
Although P-N biomolecules such as phosphocreatine have been known since the work of Fiske and Subbarow in 1927, synthetic routes to P-N compounds prior to 1988 have been limited [74,75]. These early routes involve the preparation of and use of toxic reagents over multiple synthetic steps and often resulted in the elimination of stoichiometric waste. However, with an increase in the number of uses of P-Ns, the need to develop one-pot synthetic routes that are more environmentally benign has driven researchers to discover catalytic strategies. Accordingly, all the known synthetic routes to P-Ns can be separated into six categories: salt elimination, oxidative cross-coupling, azide, reduction, hydrophosphinylation, and phosphoramidate-aldehyde-dienophile (PAD). Each of the categories will be discussed in detail in the coming sections, with an emphasis on conditions, scope, and mechanism. This discussion will be preceded by a brief introduction to the early synthetic routes.
2.1. A Brief Historical Timeline of Phosphoramidate Synthesis
Based on the available literature, one of the earliest syntheses of P-Ns involved the works of Audrieth and Toy [76,77]. Audrieth and Toy treated phosphoryl trichloride with phenol in the presence of pyridine to obtain a mixture of diphenylchlorophosphate (dPCP) and phenyl dichlorophosphate (PdCP). Upon reacting the mixture with gaseous or aqueous NH3 in 1941 or primary amine in 1942, P-N and N-P-N were formed (Scheme 2 route A, R = Ph). In 1945, P-Ns were made by Atherton, Openshaw, and Todd through a salt elimination process using disubstituted H-phosphonates (H-P), amines, and chlorinating agents (Scheme 2 route B) [78]. Subsequently, the Atherton-Todd group synthesized P-Ns via the same method using different polyhalogen solvents in 1947 [79] and using alternative phosphorylating agents in 1948 [80]. In 1951, Wagner-Jauregg et al. synthesized P-N compounds from diisopropylchlorophosphate (dPrCP) or tetraethylpyrophosphate (tEPyP) and amines (Scheme 2 route A, R = i-Pr and route C, respectively) [81]. Sodium anilide or sodium diphenylamide was refluxed with triphenylphosphate (tPPO) to eliminate phenol and give diphenyl N-phenylphosphoramidate or diphenyl N,N-diphenylphosphoramidate in 1964 (Scheme 2 route D) [82]. In 1965, Sundberg reacted 1-ethyl-2-nitrobenzene and triethyl phosphite to obtain a phosphorimidate-product (N=P) that was converted to P-N on silica or alumina columns [83]. Cadogan and coworkers reportedly synthesized and investigated the nucleophilicity of various P-Ns in 1967 [84]. Two years later, a mixture of N-arylphosphoramidates, dialkyl phosphoramidates, and phosphonates were obtained when nitroso- and nitro-compounds were reduced in the presence of (RO)3P (R = alkyl) [85]. In 1975, Appel and Einig [86] reported a new synthesis to P-Ns by reacting phosphoric acid and an amine in the presence of triphenylphosphine (tPP) and CCl4. The same year, phase-transfer catalysis was introduced by Zwierzak to synthesize P-Ns through the phosphorylation of amines [87]. The selective alkylation of (EtO)2P(O)NH2 via silylation was achieved by Zwierzak in 1982 using [n-Bu4N]Br catalyst (Scheme 2 route E) [88]. In 1984, Zwierzak and Osowska-Pacewicka reported the synthesis of various P-Ns via inorganic salt elimination [89] and the direct conversion of phosphoric acid [90]. In 1985, Riesel et al. reacted primary amines or NH3 with (RO)3P (R = alkyl) and CCl4 as an effective route to P-Ns [91]. Nielsen and Caruthers reported the first iodine-mediated reaction of phosphite and n-butylamine to synthesize P-Ns in 1988 (Scheme 2 route F) [92]. At about the same time, P-Ns were synthesized from a reaction of azide and (RO)3P (R = alkyl) in a one-pot process [93]. A magnesium chloride-catalyzed reaction of 2,6-dimethylphenol and phosphorus oxychloride was used to generate phosphorochloridate, which was used for the phosphorylation of amines to synthesize P-Ns [94].
Scheme 2.

Selected early synthetic routes to phosphoramidates.
The aforementioned syntheses demonstrate the diversity of organophosphorus synthons available for P-N chemistry up until 1988 and highlight the main pioneers in the field (Figure 4). The research in these initial studies has focused on reducing waste, limiting toxic reagents, improving selectivity, expanding scope, and searching for effective and efficient catalytic P-N bond-making solutions.
Figure 4.

Timeline illustrating the pioneers who discovered the first syntheses of phosphoramidates.
2.2. Salt Elimination Route
2.2.1. Modifications to the Atherton–Todd Reaction
A synthetic P-N was fortuitously prepared by Atherton, Openshaw, and Todd in 1945 by the salt elimination method using H-phosphonates (H-P) ((RO)2P(O)H; R = Et, i-Pr, Bn) and ammonia in the presence of a base (Scheme 2 route B) [78]. The scope of this method was investigated using different amines (e.g., morpholine, cyclohexylamine, 1-phenylethan-1-amine and benzylamine, 4-methylmorpholine) or a mixture of aniline and N,N-dimethylcyclohexanamine, as well as different solvents (e.g., CCl4, Cl3CCCl3, and HCl2CCCl3). Isolated yields of 62–92% were obtained. The Atherton–Todd method was prominent for its in situ generation of (RO)2P(O)Cl (dACP; R = alkyl, benzyl), which eliminates the use of moisture- and air-sensitive dACP. However, the major drawbacks of the method include the use of halogen sources such as CCl4, Cl3CCCl3, and HCl2CCCl3 and extensive work-up/purification of the products. In 2020, Chen et al. reported a modified Atherton–Todd reaction for the synthesis of P-Ns using air as a radical initiator (Scheme 3) [95]. The modified method explored P-N synthesis using (H-P) ((RO)2P(O)H; R = Et, i-Pr, Bu, i-Bu) and benzylamine with yields in the range of 30–75%. Although this method offered advantages such as using air as a radical initiator at room temperature, the use of relatively expensive 2,3,4,6,7,8,9,10-octahydropyrimido[1,2-a]azepine (DBU) as a base in the process, the elimination of large amounts of stoichiometric waste, and comparatively lower yields of products are some of the limitations.
Scheme 3.

A modified Atherton–Todd reaction for the synthesis of phosphoramidates.
2.2.2. Direct Conversion of Diethyl Hydrogen Phosphate
With reference to the limitations (such as low yield and the formation of organophosphorus anhydrides side products) in the method reported by Appel and Einig [86], Zwierzak and Osowska-Pacewicka [90] proposed an alternative method that uses −OH group activator, hexamethyltriaminodibromophosphorane ((Me2N)3PBr2). In the reported method, (Me2N)3PBr2 was prepared in situ in a one-pot two-step synthesis of P-Ns from (EtO)2P(O)OH and amines (Scheme 4) [90]. The synthesis was explored using various amines such as aniline, phenylmethanamine, dibutylamine, butan-1-amine, dipropylamine, and hept-1-en-2-amine. This method was favorable with improved yields of P-N products (59–91%) after workup and eliminated the route(s) for anhydride formation. Nevertheless, the method used potentially harmful bromine in a multi-step tedious process with low atom economy.
Scheme 4.

One-pot two-step synthesis of phosphoramidates via (Me2N)3PBr2.
2.2.3. Inorganic Salt Elimination
In another salt elimination route, this time using inorganic bases and [n-Bu4N]Br as a catalyst, Zwierzak and Osowska-Pacewicka synthesized P-Ns from (EtO)2P(O)H (diethyl H-P) and various amines (Scheme 5a) [89]. Aromatic amines such as aniline and 4-chloroaniline, aliphatic amines such as cyclohexylamine, n-butylamine, and ethanamine, and alkanolamines such as 2-aminoethan-1-ol, 3-aminopropan-1-ol, and bis(2-(-ethoxy)ethyl)amine were explored to demonstrate the broad scope of this synthetic transformation. The P-Ns were isolated in 83–100% yield after recrystallization. In a similar synthesis, using [BnEt3N]Cl as the catalyst instead of [n-Bu4N]Br, Zwierzak accomplished the phosphorylation of amines using dialkyl H-phosphonate (RO)2P(O)H (R = Et, Bn, t-Bu; Scheme 5b) [87]. A range of amines, such as aniline, benzylamine, cyclohexylamine, ethylamine, and diethylamine were used. The crude P-Ns were recrystallized to afford pure products in 35–93% yield. Due to the difficulty encountered in phosphorylating aromatic amines, as mentioned by Zwierzak [87,89], Lukanov et al. proposed a similar synthetic route using N-phenylformamide or 2-chloro-N-phenylacetamide as the N-source, which offered less steric hindrance and better N-H acidity (Scheme 5c) [96]. Derivatives of N-phenylformamide or 2-chloro-N-phenylacetamide (R’R’’NC(O)R’’’) were used containing a wide range of aryl-substituents (e.g., R’’ = Ph, 4-MeOPh, 4-ClPh, 2-MeOPh, 2,3-Cl2Ph, 2-MePh, and 2,6-(C2H5)2Ph and 2,6-(CH3)2Ph). Either column chromatography (on silica) or recrystallization were used to purify the P-Ns to obtain isolated yields in the range of 30–80%.
Scheme 5.

Phosphoramidate syntheses via the inorganic salt elimination route. (a) Using [n-Bu4N]Br as a catalyst, (b) using [BnEt3N]Cl as a catalyst and (c) using [BnEt3N]Br as a catalyst.
Inorganic salt elimination methods stimulated an era of catalytic transformations of dialkyl H-P ((RO)2P(O)H; R = alkyl) to (RO)2P(O)Cl (dACP; R = alkyl) in the presence of readily available inorganic bases, thereby eliminating the use of Et3N. However, this method has limitations including a pre-functionalization step, the production of large amounts of stoichiometric waste, and the use of hazardous halogenating reagents.
2.3. Oxidative Cross-Coupling Route
2.3.1. Using Chlorinating Agents
A one-pot synthesis of P-Ns was reported by Gupta et al. (Scheme 6a) [97]. According to the authors, the chlorination of (RO)2P(O)H (R = Me, Et, Pr, i-Pr; 3 eq.) to afford (RO)2P(O)Cl (dACP; R = alkyl) was possible upon reacting with trichloroisocyanuric acid (tCiC-A; 1 eq.), which was then treated with an equal mixture of amines, R’R’’NH (R’ = Me, Et, Pr; R’’ = H) and Et3N, to form the P-N in 82–92% yield, which was determined as conversions by 31P NMR spectroscopy. However, the isolation of (RO)2P(O)NR’R” (R = Et, R’, R” = Et, Pr) product gave 79% yield. A similar method, under base-free reaction conditions, was achieved by Kaboudin et al. (Scheme 6b) [98]. The scope of the synthesis was explored with (H-P) ((RO)2P(O)H; R = Et, i-Pr) and amine (e.g., N-methylaniline, naphthalen-1-amine, 2-phenylethan-1-amine, 3-chloro-2-methylaniline, 3-nitroaniline, benzylamine, or aniline). The crude product was purified by column chromatography (on silica) to yield isolated pure products in the range of 22–92%.
Scheme 6.

Phosphoramidate synthesis via oxidative cross-coupling route using different chlorinating agents, (a) trichloroisocyanuric acid and a base, (b) trichloroisocyanuric acid under base-free conditions, (c) Cl3CCN, and (d) CCl4.
Jang and coworkers reported a facile synthetic route to form P-Ns using diphenyl phosphoric acid (dPPA) and an amine in the presence of both a chlorinating agent (Cl3CCN) and a base (Et3N), which was added to sequester the acid formed during the reaction (Scheme 6c) [99]. The applicability of the method was explored using prop-2-en-1-amine, diethylamine, 2-methylpropan-2-amine, cyclohexylamine, piperidine, morpholine, and aniline. The crude product was purified by column chromatography (on silica) to obtain isolated yields of P-N products in the range of 53–93%. A similar chlorination method reacting an amine (e.g., aniline, hexylamine, diethylamine, and methanamine) with phosphoric acid ((R2P(O)OH; R = OEt, OBu, OBn) in the presence of triphenylphosphine and CCl4 was also used to synthesize P-Ns (Scheme 6d) [86]. The isolated yields of the products in this case were in the range of 30–75%.
P-N synthesis via oxidative cross-coupling using chlorinating agents offers a short reaction time, low temperature synthesis, and a diverse choice of phosphorylating reagents. Nevertheless, the methods use hazardous chlorinating agents such as CCl4 and Cl3CCN, generate large stoichiometric wastes, and involve a pre-functionalization step in the synthetic process.
2.3.2. Using Iodinating Agents
The synthesis of P-Ns from amines (aromatic and aliphatic) and symmetrical trialkyl phosphite (P(OR)3; R= Me, Et, i-Pr) in the presence I2 was reported by Chen et al. [100]. Crude products were purified by column chromatography (on silica) and resulted in yields in the range of 0–95%. For the low yielding derivatives, the reaction was more selective to three identified by-products, H-phosphonate, trialkyl phosphate, and tetraalkyl diphosphate, as well as some other unidentified by-products. Important compounds such as phosphoryl amino acid esters (8–11) were synthesized using the iodine-mediated method (Figure 5).
Figure 5.

Structures of benzyl (diethoxyphosphoryl)-d-prolinate (8), ethyl (diethoxyphosphoryl)-l-phenylalaninate (9), tetraethyl ((1S,3S)-cyclohexane-1,3-diyl)bis(phosphoramidate) (10), diethyl ((1S,3S)-3-aminocyclohexyl)phosphoramidate (11) synthesized by iodinating agents.
Iodine (I2) has also been used as a catalyst under solvent-free conditions to couple aryl amines (PhNH2 and derivatives containing OH, OEt, CN, OMe, NO2, F, Cl, or Br substituents on Ph) with (RO)2P(O)H (dialkyl H-P; R = Me, Et) affording dialkyl P-Ns (Scheme 7b) [101]. Then, the crude products were purified by column chromatography (on silica) to obtain yields of product in 0–88%. The best yielding conditions were shorter reaction times without solvent. Reactions performed in polar non-protic solvents (e.g., CH2Cl2), non-polar solvents (e.g., hexane), or protic solvents (e.g., MeOH) all suffered from reduced efficiency and selectivity. Furthermore, aniline with substituents at ortho- and meta-positions were observed to have reacted faster and were higher yielding than aniline containing substituents at the para-position.
Scheme 7.

Iodine-mediated synthesis of phosphoramidates using iodine as a catalyst (a) with a solvent, (b) under a solvent-free conditions, and (c) in the presence of H2O2.
I2 was used in the presence of H2O2 as a catalyst at 20 °C for the dehydrogenative cross-coupling of diethyl H-P and amine or sulfoximines to form P-Ns (Scheme 7c) [102]. Purification of the products by column chromatography (on silica) afforded P-Ns with isolated yields of 31–96%, with the lower yielding products reported from sulfoximine. Both alkyl and aryl H-P ((RO)2P(O)H, R = Et, i-Pr, Ph) were used as the substrate along with cyclic aromatic secondary amines (e.g., indole), derivatives of heterocyclic amines (e.g., furfurylamine), aliphatic cyclic secondary amines (e.g., 1-benzhydrylpiperazine), tertiary amines (e.g., N-methylhomopiperazine), or sulfoximine. According to the article, using other sources of I2 such as KI or N-bromosuccinimide (NBS) for the reaction resulted in decreased yield and trace quantities of the desired products, respectively.
Iodine-mediated oxidative cross-coupling methods offer better routes to P-N compounds by eliminating toxic halogenating agents such as CCl4 in the synthetic processes. The method also uses inexpensive oxidants without additives and generates water or alkanol as stoichiometric waste. However, the synthetic routes are prone to the formation of many undesired side products, suffer from low yields, and sometimes have limited applicability.
2.3.3. Using Transition Metal Catalysts
The aerobic oxidative coupling of amines and disubstituted H-phosphonates (H-P) (RO)2P(O)H (R = Me, Et, i-Pr; 1 eq.) in the presence of copper catalysts to synthesize P-Ns was reported by Hayes and coworkers (Table 1, route a) [103]. The authors reported that the ligands on the copper catalyst were important in the reaction, but not the initial oxidation state of copper catalyst, and that the primary amines coupled better than the secondary and tertiary amines due to the reduced steric hindrance. The method was slightly modified for the synthesis of some important phosphoryl amino compounds such as ethyl (diethoxyphosphoryl)glycinate, etc. (12–15) (Figure 6).
Table 1.
Oxidative cross-coupling using transition metal catalysts to form phosphoramidates.
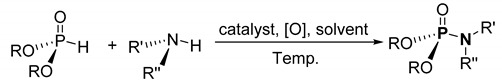
| ||||||
|---|---|---|---|---|---|---|
| Route | Catalyst | Reaction Conditions | R | R’, R’’ | Yield Range/% | Ref. |
| a | 20 mol% CuI | MeCN, 55 °C | Me, Et, i-Pr | H, alkyl | 16–98 | [103] |
| b | 5 mol% CuBr | EtOAc, 20 °C | Et, i-Pr, Bu | H, Ph, functional Ph | 20–94 | [104] |
| c | 15 mol% Fe3O4@MgO | CCl4, 20 °C | Et, i-Pr | H, Ph, Bn, cycloalkyl | 52–85 | [105] |
| d | 200 mol% CuCl2 | Acetone, Cs2CO3, 20 °C | Me, Et, Pr, i-Pr | H, alkyl | 25–93 | [106] |
| e | 10 mol% Cu(OAc)2 | Toluene, K2CO3, mol. sieve, 80 °C | Me, Et, i-Pr, Bu | RCOR a | 52–99 | [107] |
| f | 2 mol% CuBr | EtOAc, 25 °C | Me, Et, i-Pr, Bu, Ph | H, alkyl, cycloalkyl, R b | 86–96 | [108] |
| g | 5 mol% Cu(OAc)2 | MeOH, NaN3, 20 °C | Et c | R-B(OH)2 d | 67–93 | [109] |
a urea, oxazolidinone, indole, pyrrolidinone, lactam, and sulfonamide derivatives; b methyl alaninate; c triethyl phosphite; d phenylboronic acid and phenylboronic ester derivatives.
Figure 6.

Structures of ethyl (diethoxyphosphoryl)glycinate (12), methyl ((((2R,3S,5S)-5-(((tert-butyldimethylsilyl)oxy)methyl)-2-(5-methyl-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)tetrahydrofuran-3-yl)oxy)(methoxy)phosphoryl)-D-phenylalaninate (13), methyl (diethoxyphosphoryl)-l-phenylalaninate (14), (2R,3S,5S)-5-(((tert-butyldimethylsilyl)oxy)methyl)-2-(5-methyl-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)tetrahydrofuran-3-yl methyl benzylphosphoramidate (15) synthesized by aerobic oxidative cross coupling.
Another copper-catalyzed aerobic oxidative cross-coupling reaction reported by Wang et al. in 2013 involved the coupling of aryl amines (1.5 eq.) and dialkyl H-phosphonates (RO)2P(O)H (R = Et, i-Pr, Bu; 1 eq.) in the presence of CuBr (5 mol%) to give P-Ns (Table 1, route b) [104]. Although para- or meta-substituted aryl amines were very effective substrates in this reaction, the conversions observed with ortho-substituted aryl amines or aryl amines with strong electron-withdrawing substituents (e.g., 4-nitroaniline) as well as alkylamines were very poor. The effectiveness of the reaction with diaryl H-phosphonates was not reported.
In 2015, Fe3O4@MgO nanoparticles were used as a catalyst to synthesize P-N from primary amines and H-P via the Atherton–Todd coupling reaction with CCl4 (Table 1, route c) [105]. After column chromatography (on silica), P-Ns were isolated in 52–85% yield. The Fe3O4@MgO nanoparticles were recycled by washing successively with H2O and MeOH and drying before reuse. Although the scope of the transformation was not challenged by investigating aromatic H-Ps, this route offers some advantages, such as the use of a low-cost reusable catalyst with proven activity for up to four consecutive reactions.
Purohit et al. reported a one-pot synthesis of P-Ns from (RO)2P(O)H (R = Et, Pr, i-Pr; 1 eq.) and amine in the presence of CuCl2 and Cs2CO3 (Table 1, route d) [106]. The reported yields for the variety of desired products were in the range of 25–93% as determined by 31P NMR spectroscopy.
A Cu(II) catalyst, a base, and air as an oxidant were used to synthesize N-acylphosphoramidates (AcP-N) from (RO)2P(O)H (R = Me, Et, i-Pr, Bu) and an amide via an oxidative cross-coupling reaction according to Mizuno et al. (Table 1, route e) [107]. The scope of the reaction was investigated by changing the R substituent as well as the nitrogen nucleophiles (e.g., urea, oxazolidinone, indole, pyrrolidinone, lactam, and sulfonamide derivatives). Column chromatography (on silica) using different solvent mixtures was used to purify the desired P-Ns in 52–99% yield. Through the reagent screening, Mizuno et al. [107] found that Cu(OTf)2 gave similar cross-coupling results as Cu(OAc)2, while other Cu catalysts such as CuCl2 and CuSO4 and other transition metal catalysts (e.g., Co(OAc)2.4H2O, Fe(OAc)2) were not selective or completely ineffective.
Another copper-catalyzed synthesis of P-N at 20 °C in the presence of air, reported by Zhou et al., involved reacting (RO)2P(O)H (R = Me, Et, i-Pr, Ph) with primary or secondary amines (butylamine, dibutylamine, hexylamine, cylohexylamine, and methyl alaninate; Table 1, route f) [108]. The crude P-Ns were extracted and then concentrated under vacuum to obtain pure products with excellent isolated yields (86–96%). Similar to other copper-catalyzed P-N synthesis, the authors reported a low yield (21%) when dibutylamine and diisopropyl H-P were used as substrates, which could be attributed to steric effects. In addition, the method was not effective when diphenylphosphine oxide (dPPO) was reacted with butylamine (13% isolated yield) due to oxidative conversion to phosphonic acid (PP-A). Using this aerobic copper-catalyzed dehydrocoupling method, Zhou et al. synthesized a phosphoryl amino compound (16) (Figure 7).
Figure 7.

Structure of methyl (isopropoxy(((2S,5R)-5-(5-methyl-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2,5-dihydrofuran-2-yl)oxy)phosphoryl)-d-alaninate (16) synthesized by aerobic copper-catalyzed oxidative cross coupling.
Phenylboronic acid or ester-based substrates were converted in situ to an organic azide using a Cu-based catalyst and subsequently used for the synthesis of P-Ns via a coupling reaction, according to Dangroo et al. (Table 1, route g) [109]. Triethyl and trimethyl phosphate were used as the P-sources while phenylboronic acids or esters with substituents such as CN, NO2, OH, OMe and halides were used for azide conversion. The reaction was very tolerant to compounds with unsaturated boron–carbon bonds giving isolated yields of 67–93%. However, this reaction is not effective for compounds containing saturated boron–carbon bonds such as butyl boronic acid or cyclopentyl boronic acid.
Transition metal-catalyzed oxidative cross-coupling methods for P-N synthesis have become attractive in recent years because of the advantages of using readily available inexpensive catalysts, for example CuXn (X = Cl, Br, (OAc), etc., n = 1, 2), and air as an oxidant. In addition, this route uses environmentally friendly H-P (RO)2P(O)H (R = alkyl, Ph) and amines as starting materials in a one-pot single-step process without any need for purification of the starting material, thereby making the process cost- and time-effective. Apart from eliminating hazardous starting materials, the method uses fewer chemical additives and generates mainly water as a stoichiometric waste. Furthermore, the methods offer a possibility of catalyst recycling, which is an important attribute for any green chemical process in terms of mass production. However, a notable limitation of the synthetic route is the formation of various undesired by-products from the reactivities of H-P and amine with the catalysts under oxidative conditions [103].
2.3.4. Using an Organophotocatalyst
Guo et al. used visible light and organic dyes as catalysts for the dehydrogenative cross-coupling reaction of (EtO)2P(O)H (H-P) and amines (aniline, aniline derivatives with CN, X, Me, OMe and 1-naphthylamine) to form the corresponding P-Ns (Scheme 8) [110]. After an extraction, the pure product was formed with isolated yield in the range of 0–99%. It is noteworthy that the method was not effective when vinylaniline and 2-aminothiazole were used because of their radical sensitivity making them prone to either polymerization or decomposition in the reaction media, respectively. Although no product was formed when secondary aliphatic amines were used, moderate yields of up to 59% were obtained from primary or tertiary aliphatic amines, while a complex mixture of products was formed when an aromatic H-P was used.
Scheme 8.

Visible light and organic dye-catalyzed synthesis of phosphoramidates.
2.3.5. Using Alkali–Metal Catalyst
An oxidative cross-coupling reaction of amines and (EtO)2P(O)H (H-P) in the presence of lithium iodide-tert-butyl hydroperoxide (LiI/TBHP) was used for the synthesis of P-N and other compounds by Reddy et al. (Scheme 9) [111]. The crude products were purified by column chromatography (on silica) with isolated yields of 65% and 31% for the use of benzylamine and 4-chloroaniline, respectively. Other amines were not investigated. Although the method uses an inexpensive catalyst and oxidant, the applicability of the process was not explored using diverse H-P and amines.
Scheme 9.

Oxidative cross-coupling reaction of amines and H-phosphonates to form phosphoramidates in the presence of LiI/TBHP.
2.4. Azide Route
Many conventional routes to P-N synthesis as discussed above have used H-P (RO)2P(O)H (R = alkyl, Bn, Ph) or (RO)2P(O)Cl (dACP; R = alkyl, Bn, Ph) and amine as the P- and N-sources, respectively. As a result, derivatives of P-N compounds synthesized through these routes are limited mainly to substituents on the amine starting materials. Due to increased applicability, it has become necessary to synthesize P-N compounds with diverse functionalities. In order to achieve this, nitrene insertion from organic azides is becoming a popular one because the route uses phosphoryl azide, which acts a dual source of P and N. In this case, it is possible to add (or insert) diverse molecules on the P-N moiety through C-N bond formation.
2.4.1. Nitrene Insertion from Organic Azide
A route to P-N formation through C-N bond formation was explored by Chang, Kim, and coworkers (Scheme 10a) [112]. The P-N was synthesized in the presence of an Ir(III)-based catalyst using an amide or a ketone and a phosphoryl azide. The product was purified by column chromatography (on silica) with isolated yields in the range 41–99%. The scope of the synthesis was explored using phosphoryl azide substituted with binaphthyl, aryl, and alkyl groups, while the phenone had substituents such as phenyl, methyl, and isobutane groups. Furthermore, 4-methoxy, 4-CF3, and tert-butyl were some of the substituents on the benzamide. Biologically active phosphoramidates (17 and 18) were synthesized through this method (Figure 8).
Scheme 10.

Phosphoramidate synthesis via nitrene insertion using, (a) 5 mol% Ir(III)-based catalyst, (b) 5 mol% RuIV-porphyrin-catalyst, and (c) 4 mol% Ir(III)-based catalyst.
Figure 8.

Structures of (1R,2R,5R)-5-isopropyl-2-methylcyclohexyl phenyl (2-(tert-butylcarbamoyl)phenyl)phosphoramidate (17), (3S,5S,8R,9S,10S,13R,14S,17R)-10,13-dimethyl-17-((R)-6-methylheptan-2-yl)hexadecahydro-1H-cyclopenta[a]phenanthren-3-yl phenyl (2-(tert-butylcarbamoyl)phenyl)phosphoramidate (18) synthesized by nitrene insertion from organic azide.
Che and coworkers later introduced a RuIV-porphyrin-catalyst to synthesize N-acylphosphoramidates (AcP-N) from aldehydes and phosphoryl azide through nitrene insertion (Scheme 10b) [113]. The product was isolated by column chromatography (on silica). The observed product conversions were in the range 56–99%. Aromatic and aliphatic aldehydes such as 2-naphthaldehyde, benzo[d][1,3]dioxole-5-carbaldehyde, 5,6-dihydro-2H-pyran-3-carbaldehyde and 3-methylbut-2-enal were used in the reaction along with aromatic and aliphatic phosphoryl azide such as 3-nitrophenyl (4-nitrophenyl) phosphorazidate, bis(2,2,2-trichloroethyl) phosphorazidate, and diethyl phosphorazidate. However, lower yields were obtained from aliphatic aldehydes when compared with the aromatic aldehydes, suggesting that the synthetic procedure favors electron-rich aromatic aldehydes.
A nitrene insertion route in the presence of Ir(III)-based catalyst was also used to synthesize P-Ns by Zhu et al. (Scheme 10c) [114]. The P-Ns were synthesized under an inert environment and the isolated yield of the products was in the range of 52–77%. The scope of the synthesis was investigated using diethyl phosphorazidate and diphenyl phosphorazidate along with 2-phenylpyridine or pyrazole derivatives with substituents such as Me, OMe, CF3, Cl, F, Ph, and COOMe at ortho-, meta-, and para-positions as well as benzo[h]quinoline.
Although nitrene insertion provides a viable route to P-N synthesis and generates N2 gas as the only stoichiometric by-product, the process uses expensive catalysts in addition to many chemical additives. Moreover, apart from the hazards associated with the handling of phosphoryl azide, the route requires a long reaction time, high temperature, and does not favor electron-deficient starting materials.
2.4.2. Via In Situ Azide Generation
An organic azide was generated in situ from organic halides and was coupled with (RO)3P (R = alkyl) for the synthesis of P-N by Sangwan et al. (Scheme 11) The crude product was purified by column chromatography (on silica) to afford isolated products in the range of 52–96% yield. A broad scope of the reaction was tested; 3-bromoprop-1-yne, 3-bromoprop-1-ene, 1-bromopropane alkyl or (un)substituted aromatic halide such as (bromomethyl)benzene, 1-(bromomethyl)-2-nitrobenzene, 1-(chloromethyl)-4-nitrobenzene, (chloromethyl)benzene, 1-(bromomethyl)-3,5-dimethoxybenzene, or 3-(4-(bromomethyl)-1H-1,2,3-triazol-1-yl)benzyl acetate were reacted with dimethyl or diethyl H-P.
Scheme 11.

Phosphoramidate synthesis from organic azide and (RO)3P.
A similar method of P-N synthesis via in situ azide generation from organic halides was reported by Dar [115], following the procedure previously reported by Sangwan et al. (Scheme 11) [116]. The yield range of the isolated P-Ns, in this case, was 60–96%. Phosphoramidate-derived bioactive compound (19) was synthesized using the proposed method (Figure 9).
Figure 9.
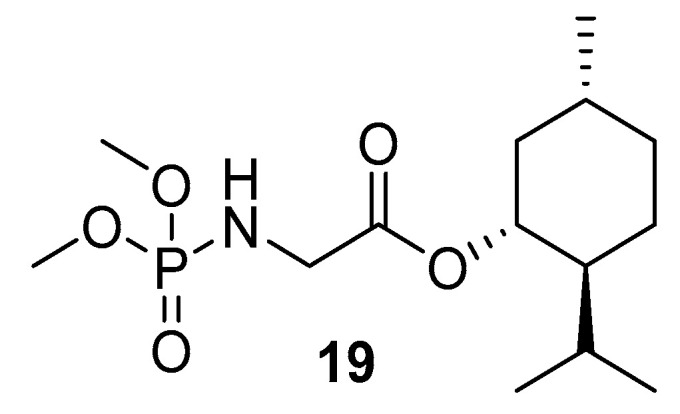
Structure of (1R,2S,5R)-2-isopropyl-5-methylcyclohexyl (dimethoxyphosphoryl)glycinate (19) synthesized via in situ azide generation.
2.4.3. Via Two-Step Organic Azide Generation
In 1988, Koziara reported a two-step P-N synthetic route via organic azide generation using a [n-Bu4N]Br catalyst (Scheme 12) [93]. The crude product was purified by crystallization or vacuum distillation with 50–93% isolated yield. The organic halides used were (3-bromopropyl)benzene, (2-bromoethyl)benzene, 1-bromohexane, bromocyclohexane, 1-bromobutane, or bromocyclopentane.
Scheme 12.

Phosphoramidate synthesis via two-step organic azide generation.
Routes via organic azide generation use readily available, less toxic organic halides and (RO)3P (R = alkyl) relative to other routes and generate N2 as stoichiometric waste. However, the method relies on the pre-functionalization and preparation of hazardous azide precursors in multi-step processes.
2.4.4. Transition Metal-Free Synthesis from Organic Azide
Transition metal-free synthesis of P-N from phosphoryl azide and amines was accomplished by Chen et al. [117]. The synthetic process uses the nitrogen atom available from the amine rather than from the azide (Scheme 13). The residue was purified by column chromatography (on silica) to afford the isolated product in 52–92% yield. The scope of the reaction was examined by using diphenyl and diisopropyl phosphoryl azide along with primary amines (e.g., isopropylamine and t-butylamine), substituted primary amines (e.g., thiophen-2-ylmethylamine and 4-chlorobenzylamine), and heterocyclic amines (e.g., pyrrolidine and morpholine). Although the method circumvents the use of metal catalysts in the synthesis, the use of a hazardous azide and high temperature are its limitations.
Scheme 13.

Phosphoramidate synthesis from organic azide and amine.
2.5. Reduction Route
2.5.1. Via Nitro-Group Reduction
Beifuss et al. reported a microwave-assisted synthesis of N-arylphosphoramidates from nitrobenzene and (RO)3P (R = alkyl) (Scheme 14) [118]. The crude residue product was purified by column chromatography (on silica) resulting in pure P-Ns with 52–79% isolated yield. The scope of the synthesis was investigated using trimethyl or triethyl phosphite along with nitrobenzene, and mono- or di-substituted nitrobenzenes with substitution such as Me, OMe, Cl, Br, or CN groups. This method is attractive because it provides a different route to P-Ns using nitro compounds instead of the conventional amines. Nonetheless, the high temperature and amount of energy required during the synthesis are the obvious limitations.
Scheme 14.

Synthesis of N-arylphosphoramidates from nitrobenzene and (RO)3P (R = alkyl).
2.5.2. Via Catalyst-Free Staudinger Reduction and Lewis-Acid Catalyzed Rearrangement
Hackenberger et al. synthesized P-N compounds from organic azide derivatives and (RO)3P (R = alkyl) after Lewis acid-catalyzed rearrangement (Scheme 15) [119]. The crude product was concentrated under reduced pressure, and the desired product was isolated (63–98% yield) through column chromatography (on silica). Triethyl or trimethyl phosphite were used as the P-source along with organic azide derivatives such as (azidomethyl)benzene, azidocyclohexane, (2-azidopropan-2-yl)benzene, azidobenzene, ethyl 2-azidoacetate, 1-azidodecane, (azidomethylene)dibenzene, and (3-azidoprop-1-en-1-yl)benzene.
Scheme 15.

Synthesis of phosphoramidates from organic azide derivatives and (RO)3P after Lewis acid-catalyzed rearrangement.
2.6. Hydrophosphinylation Route
Zimin et al. reported the 1,2-hydrophosphinylation of alkyl nitriles (R’-CN, R’ = alkyl, Ph, Bn) using sodium dialkyl phosphite ((RO)2PONa, R = Et, Pr) to achieve the formation of a P-N featuring a P-C-N-P linkage in which the dialkyl phosphite group of (RO)2PONa was added to the carbon and nitrogen atoms of the nitriles (Scheme 16) [120].
Scheme 16.

A 1,2-hydrophosphinylation of nitriles to form phosphoramidates.
2.7. Phosphoramidate-Aldehyde-Dienophile (PAD) Route
Chabour et al. reported a diastereoselective P-N synthesis via the PAD process using a TsOH acid catalyst and a dienophile, maleimide (Scheme 17) [1]. Flash chromatography was used to isolate the desired product in 40–88% yield. The scope of the synthesis was explored with maleimide derivatives with substituents such as 4-FC6H4CH2, C6H4CH2, C6H4COCH3, 4-BrC6H4, phenyl, and methyl. The unsaturated aldehyde derivatives that were used were 3,7-dimethylocta-2,6-dienal, 3-methylbut-2-enal, pent-2-enal, and hex-2-enal. This newest route for P-N synthesis uses inexpensive catalysts, generates only H2O as a by-product in stoichiometric amounts, and demonstrates an important route to the synthesis of polyfunctionalized P-N compounds.
Scheme 17.

Phosphoramidate synthesis via the PAD process.
3. Mechanistic Considerations
A wealth of information on the mechanisms for the synthetic routes to P-Ns is available. For each of the main categories highlighted above, researchers have provided mechanistic insight into the reaction by determining the main sources of oxidation as well as electronic and steric factors provided from the substituents that influence the reaction rate. Several examples of postulated catalytic cycles will be provided below, illustrating the key steps, similarities, and differences between them.
3.1. Salt Elimination Route
3.1.1. Atherton–Todd Reaction
Atherton, Openshaw and Todd suggested that the two-step mechanistic process for the salt elimination could involve the formation of dialkyl or dibenzyl (trichloromethyl)phosphonate in the first step, from a reaction of (RO)2P(O)H (H-P; R = alkyl) and CCl4 in the presence of Et3N [78]. The desired product is formed in the second step when the (trichloromethyl)phosphonate reacts with amine, forming also CHCl3 in the process (Scheme 18). Although this mechanism is plausible, evidence by NMR spectroscopy suggests that in the first step, a reaction of the base and CCl4 results in salt formation [Et3NCl]+[CCl3]− [121]. The H-P is deprotonated by [CCl3]− to form CHCl3 and anionic phosphite, which reacts further with [Et3NCl]+ to form (RO)2P(O)Cl (dACP; R = alkyl). In the second step, dACP reacts with amine to give the product.
Scheme 18.

Atherton–Todd mechanism proposed for the formation of phosphoramidates.
For the modified Atherton–Todd reaction, the two-step mechanistic process involves the deprotonation of H-P by DBU, and the radical formed is autoxidized by O2 before it abstracts chlorine from CHCl3 to form dACP (Scheme 19) [95]. The nucleophilic substitution of dACP by the amine affords the P-N product.
Scheme 19.

Modified Atherton–Todd mechanism proposed in the formation of phosphoramidates.
3.1.2. Direct Conversion of Diethyl Hydrogen Phosphate
In the plausible mechanism for the direct conversion of diethyl hydrogen phosphate, hexamethyltriaminophosphine P(NMe2)3 is oxidized to hexamethyltriaminodibromophosphorane ((Me2N)3PBr2) which is ionized in solution (Scheme 20) [90]. Subsequently, the ionized (Me2N)3PBr2 and diethyl hydrogen phosphate undergo ligand exchange in the presence of Et3N to form the intermediate, diethyl (phosphaneyloxy)phosphonate amino-hydrazine-bromide salt complex. Nucleophilic attack from the amine onto the positively charged P results in the diethyl phosphoramidate bromide salt, which reacts with Et3N to give the desired product with a concomitant elimination of triethylammonium bromide. Apart from the stoichiometric Et3N salt waste generated, the synthetic process is prone to the formation of significant quantities of pyrophosphate or phosphanetetraamine bromide salt side products, resulting in a low yield.
Scheme 20.

Proposed mechanism of direct conversion of diethyl hydrogen phosphate to form phosphoramidates.3.1.3. Inorganic Salt Elimination.
The mechanistic steps for the synthesis of P-N via a salt elimination route were not proposed by Zwierzak et al. [89]; however, based on the roles of KHCO3, K2CO3, and [n-Bu4N]Br as outlined in the paper and a recently proposed mechanism for an Atherton–Todd type reaction, a probable mechanism can be postulated (Scheme 21) [121]. In the reacting system, the HCO3− migrates to the organic phase in the presence of [n-Bu4N]Br as the catalyst, where it is protonated and reacts with CCl4 to form anionic HCO3ClH− and cationic Cl3C+. The dialkyl H-phosphonate ((RO)2P(O)H, R = alkyl) is deprotonated by the cationic Cl3C+ to yield CHCl3 and anionic phosphate, (RO)2P(O)−. Next, the anionic phosphate reacts with HCO3ClH− to form (RO)2P(O)Cl (dACP; R = alkyl) and H2CO3. In the following step, a nucleophilic substitution between the amine and dACP results in the formation of the desired product. Meanwhile, K2CO3 serves as the dehydrating agent in the reacting system. The plausible mechanism is also similar when NaOH and [BnEt3N]Cl are used in place of KHCO3 and [n-Bu4N]Br, respectively.
Scheme 21.

Potential mechanism for the formation of phosphoramidates using a salt elimination route.
Lukanov et al. suggested that the mechanistic cycle for their synthesis involves the deprotonation of dialkyl H-phosphonate ((RO)2P(O)H, R = alkyl) to form (RO)2P(O)Cl (dACP, R = alkyl) [96]. Then, the dACP reacts with N-phenylformamide or the 2-chloro-N-phenylacetamide derivative to form the product. Alternatively, the mechanism could proceed via a dialkyl (acetyl)(phenyl)phosphoramidate intermediate, which subsequently undergoes hydrolysis to afford the product.
3.2. Oxidative Cross-Coupling Route
3.2.1. Using Chlorinating Agents
The mechanisms of P-N formation from Gupta et al. [97] and Kaboudin et al. [98] suggest that the dialkyl H-phosphonate ((RO)2P(O)H, R = alkyl) reacts with trichloroisocyanuric acid (tCiC-A) to form the intermediate (RO)2P(O)Cl (dACP; R = alkyl) and 1,3,5-triazinane-2,4,6-trione (tAtO) (Scheme 22). Following, a nucleophilic substitution between the dACP and the amine result in the P-N with HCl as the major by-product.
Scheme 22.

Proposed mechanism for the base-free synthesis of phosphoramidates using a chlorinating agent.
The mechanism of the reaction by Jang et al. suggests that diphenyl phosphoric acid (dPPA) undergoes chlorination in the presence of a base, which is subsequently attacked by the amine to afford the P-N product [99]. Although the authors did not propose any mechanism, they suggested that nucleophilic attack on the P atom was consistent with aliphatic amines resulting in a higher yield of products than the aromatic amines because aliphatic amines are more nucleophilic.
The mechanistic step suggested by Appel and Einig involves the formation of oxo-triphenyl-diphosphoxanium salt intermediate from a deprotonation of phosphoric acid by CCl4 in the presence of phosphine (Scheme 23) [86]. Nucleophilic substitution from amine results in the desired product and triphenylphosphate as the by-product.
Scheme 23.

Proposed mechanism for the synthesis of phosphoramidates from phosphoric acid and amine in the presence of phosphine and CCl4.
3.2.2. Using Iodinating Agents
In the suggested reaction mechanism by [100], iodine reacts with (RO)3P (R = alkyl) to form trialkoxyiodophosphonium intermediate, which is further attacked by iodide ion to form dialkyl phosphoriodidate (dAPPI) (Arbuzov reaction, Scheme 24). Then, the dAPPI is attacked by the amine to give the desired P-N with an elimination of HI. N-phosphoryl amino acid esters and P-N with a free amino group were also synthesized using the iodine-mediated synthesis method. The challenges with this iodine-mediated P-N formation is that many side products are formed, and the reaction procedure does not generate the desired product when N-iodosuccinimide (NIS) is used as the iodine source. In addition, the reaction process is ineffective with aromatic H-phosphonates because the reaction is affected by steric effects on the substituents.
Scheme 24.

Proposed mechanism for the iodine-mediated synthesis of phosphoramidates.
In a mechanism of the I2-mediated P-N formation, according to Singh et al., H-P reacts with I2 to form an intermediate, which undergoes nucleophilic attack from the amine to generate a hydroxylated-dialkyl phenylphosphoramidate complex with an elimination of I2 (Scheme 25) [101]. The phosphoramidate complex further undergoes dehydrogenation to eliminate H2 in form of H2O and afford the desired products.
Scheme 25.

Proposed mechanism for the iodine-mediated synthesis of phosphoramidates.
The suggested mechanism of a similar reaction reported by Prabhu et al. instead involves the nucleophilic substitution of phosphoriodate intermediate after the electrophilic iodination of the H-P starting material (Scheme 26) [102]. Then, the iodide ion is oxidized to active catalyst, hypoiodous acid, in the presence of H2O2.
Scheme 26.

Proposed mechanism for the hypoiodous acid-mediated synthesis of phosphoramidates.
3.2.3. Using Transition Metal Catalyst
In a copper-catalyzed oxidative cross-coupling reaction, Hayes et al. proposed a reaction mechanism involving single-electron transfer oxidation of the copper catalyst, which favored the formation of the Cu(II)-amine complex (Scheme 27) [103]. The Cu(II)-amine complex is further oxidized to form the Cu(III)-amine-phosphonate complex, which undergoes reductive elimination of the copper catalyst to yield the desired P-N. Although this mechanism seems plausible, it could not easily explain the formation of the side products.
Scheme 27.

Proposed mechanism for the copper catalyzed oxidative cross-coupling of disubstituted H-phosphonates and amines to produce phosphoramidates.
There were no suggested mechanisms for the reaction in the report by Wang et al. [104]. However, the results of the reagent screening for the reaction had suggested that other transition metal catalysts (e.g., CoBr2) and other Cu catalysts (e.g., Cu(OAc)2·H2O) were inactive or inefficient for the catalysis. It was further suggested that although a CuCl catalyst enabled reaction, it was not as effective as CuBr and conducting the reaction under anaerobic conditions (e.g., under N2) did not lead to the desired product.
In the Fe3O4@MgO nanoparticle-mediated synthesis of P-Ns as per the procedure reported by Habibi et al. [105], the mechanism involves the formation of the (RO)2P(O)Cl (dACP; R = alkyl) intermediate from the reaction of dialkyl H-phosphonate ((OR)2P(O)H, R = alkyl) with CCl4 (Scheme 28). Following, the amine in the presence of Fe3O4@MgO nanoparticles couples with the dACP by nucleophilic substitution to give the P-N product, eliminating HCl.
Scheme 28.

Proposed mechanism for the Fe3O4@MgO nanoparticle-mediated synthesis of phosphoramidates.
According the plausible two-step mechanism for the reaction by Purohit et al., (RO)2P(O)Cl (dACP; R = alkyl) is first generated from the electrophilic displacement of hydrogen on the dialkyl H-phosphonate ((OR)2P(O)H, R = alkyl) upon reacting with CuCl2 (Scheme 29a) [106]. The phosphoramidate is formed in the second step by coupling dACP and amine. However, this reported mechanism could not properly explain the role of Cs2CO3 in the coupling of alkyl chlorophosphate and the amine to give the P-N. This is because the coupling reaction was expected to eliminate other products along with CsCl such as a highly reactive CsH and a carbonate. However, it is to be understood that the use of Cs2CO3 as the inorganic base was based on the poorly understood “caesium effect” in which the highly soluble caesium base selectively favors the formation of one functional group in preference to another [122,123].
Scheme 29.

Proposed mechanism for the Cu-mediated synthesis of phosphoramidates.
Mizuno et al. proposed a mechanism for the reaction in which K2CO3 first deprotonates the amide, affording unpaired electrons on a Cu-amide complex [107]. Furthermore, it was suggested that it is possible that K2CO3 deprotonates H-P or provides a medium for the H-P tautomer to further complex with the Cu-amide complex. The Cu-amide-phosphite intermediate undergoes reductive elimination to afford the desired product, while O2 re-oxidizes the reduced Cu species to regenerate the active catalyst. Although the oxidation state of Cu in the reaction mechanism was not explained, the authors suggested that it could be +2, and the fact that the reaction produced only stoichiometric amounts of the desired product (relative to the Cu catalyst) under an inert atmosphere was a support to the re-oxidation of the Cu species in air.
From their experimental observations, Zhou et al. strongly suggested that the oxidative dehydrocoupling reaction for P-N synthesis using Cu-catalysts proceeds stereospecifically through an inversion of the configuration at P [108]. This stereospecific inversion chemistry was used as an argument in the proposed mechanism to support a formation of halogen–phosphate intermediate when Cu catalyst is reacted with H-phosphonate (H-P). The amine (as a nucleophile) subsequently attacks the intermediate from behind to give the product, while the Cu(I) catalyst is then reoxidized by O2 to Cu(II) in the presence of hydrogen halide formed from the process. Water is also formed as a stoichiometric waste (Scheme 29b).
The mechanism as reported by Dangroo et al. involved transmetalation to form an azidocopper(II) species in the first step (Scheme 30) [109]. Similar transmetalation reactions involving the phenylboronic acid-based substrates and the azidocopper(II) species in the second step result in the formation of a phenyl–azidocopper(II) complex. The complex is oxidized in the presence of Cu(II) to a phenyl–azidocopper(III) complex, which results in the formation of the organic azide and forms Cu(I) after reductive elimination. From this, a reaction between the (RO)3P (R = alkyl) and the organic azide gives phosphorimidate (with loss of N2), which undergoes hydrolysis to afford the desired product, while the Cu(I) is reoxidized to Cu(II) in the catalytic cycle. Although the mechanism satisfies much of the observed results, the proposed second step, transmetalation, would have resulted in borazidic acid (i.e., N3-B(OH)2) and Cu(II)-phenyl species or Cu(OH)2 species when the azidocopper(II) species reacted with phenylboronic acid-based substrates, based on the exchange of ligands as in the first step.
Scheme 30.

Proposed mechanism of the Cu-catalyzed synthesis of phosphoramidates from phenylboronic acid/ester-based substrates and trialkyl phosphite.
3.2.4. Using an Organophotocatalyst
In a suggested mechanism for the organophotocatalytic synthesis of P-N, a photon from the visible light source is absorbed by the organic dye to become an excited radical (Scheme 31) [110]. By a single-electron transfer process, the excited dye-radical abstracts an electron from the N atom of the amine to form an anionic dye radical and cationic amine radical. In the presence of O2, the anionic dye radical is reoxidized to the organic dye, while the anionic superoxide that is formed in the process reacts with diethyl H-phosphonate (H-P) to generate anionic hydrogen peroxide and a diethyl phosphoryl radical. Then, the radical reacts with the cationic-amine radical to form a protonated P-N, which is then deprotonated by the anionic hydrogen peroxide. The authors reported that after an initial 74% isolated product, the organic dye catalyst was recycled a second time using an acid–base extraction, and 67% isolated product was obtained the second time.
Scheme 31.

Proposed mechanism for the visible light and organic dye-catalyzed synthesis of phosphoramidates.
3.2.5. Using Alkali–Metal Catalyst
In the overall mechanism suggested for the alkali–metal catalyzed oxidative cross-coupling reaction, a phosphoryl iodide intermediate and LiOH are generated in the first step from a reaction of LiI and the H-phosphonate (H-P) in the presence of aq. TBHP (Scheme 32) [111]. The second step involves the removal of a proton from the amine by the LiOH to form lithium amide and H2O, and the desired product is formed in the final step from the reaction of phosphoryl iodide and lithium amide.
Scheme 32.

Proposed mechanism for the synthesis of phosphoramidates using alkali–metal catalyst.
3.3. Azide Route
3.3.1. Nitrene Insertion from Organic Azide
The mechanism of the reaction of nitrene insertion according to Chang, Kim, and coworkers follows activation of the C-H bond cleavage on the amide or ketone substrate by the Ir(III)-based catalyst to generate an iridacyclic intermediate, which then reacts with the phosphoryl azide to produce a coordinated phosphoryl azide–iridacyclic adduct (Scheme 33) [112]. By intramolecular migratory insertion of C-N into the Ir-C bond, with removal of N2, or by nucleophilic attack from the azide group on the Ir-C bond to form the C-N bond with the elimination of N2, a complex of Ir-N-C bond is formed. Proto-demetallation of the Ir(III)-based catalyst affords the desired product while regenerating the catalyst.
Scheme 33.

Proposed mechanism for the Ir-catalyzed synthesis of phosphoramidates.
According to Zhu et al., the mechanistic steps involve activation of the Ir(III)-based catalyst in the presence of AgOAc and AgSbF6, which abstracts an H atom from the 2-phenylpyridine derivative to cyclo-(2-phenylpyridine)iridium(III) complex [114]. The complex further coordinates with phosphoryl azide to form a cyclo-(2-phenylpyridine)iridium(III)-phosphoryl azide complex, which undergoes migratory insertion to give a complex containing a C-N-Ir(III) bond, releasing N2. The Ir(III)-based catalyst is released through proto-demetallation to generate the desired product.
For the nitrene insertion using RuIV-porphyrin-based catalyst, Che and coworkers proposed that a reactive intermediate RuIV-porphyrin-phosphorylimide species is first formed from the interaction of phosphoryl azide and the RuIV–porphyrin catalyst [113]. Then, the intermediate reacts with the aldehyde C-H bond through nitrene insertion to form a C-N bonded RuIV-porphyrin-phosphorylimide-aldehyde complex with the removal of N2. The desired N-acylphosphoramidates (AcP-N) product is formed after proto-demetallation of the RuIV-porphyrin catalyst, and the catalyst is regenerated in the process.
3.3.2. Organic Azide Generation Route
In the reported mechanisms of the reaction for P-N synthesis via azide generation, organic azide is formed in the first step from nucleophilic substitution of the organic halide with the elimination of N2 gas. In the second step, the organic azide reacts with (RO)3P (R = alkyl) to form the P-N compound after rearrangement or hydrolysis (Scheme 34) [93,115,116].
Scheme 34.

Proposed mechanism to phosphoramidates via azide generation.
According to the mechanism suggested by Chen et al., the amine reacts with phosphoryl azide through a nucleophilic substitution reaction to generate an azido (hydroxy)-phosphanamine complex intermediate in the first step [117]. In the second step, the azide anion departs as the leaving group to give a stable P-N product (Scheme 35). Although the mechanism looks highly plausible, it is noteworthy that N3 is highly nucleophilic because in its canonical structure, one of the nitrogen atoms carries a double negative charge (N≡N+ − N2−) [124], which makes it stable and a poor leaving group as N3 in the presence of amine [125]. In addition, the mechanism does account for the potentially explosive HN3 in a closed flask at 120 °C. However, it is possible that in the reaction process, the azide is first reduced or decomposed to a nitrene phosphonate radical ((RO)2P(O)N; R = alkyl, Ph), while N2 is liberated. In the second step, the electrophilic nitrene phosphonate radical reacts with or is substituted by the nucleophile amine to give the product.
Scheme 35.

Proposed mechanism for an organic azide route to phosphoramidates.
3.4. Reduction Route
3.4.1. Via Nitro-Group Reduction
The proposed mechanism of the reaction for the nitro-group reduction route involves the reduction of nitrobenzene in the presence of (RO)3P (R = alkyl) to give nitrosobenzene with a phosphate side product (Scheme 36) [118]. In the second step, the nitrosobenzene is reduced further in the presence of another equivalent of (RO)3P, generating phenylnitride with another phosphate side product. A further reaction of (RO)3P (R = alkyl) with the basic phenylnitride generates a trialkyl phenylphosphorimidate intermediate, which undergoes hydrolysis to give the desired N-arylphosphoramidate product. Experiments to verify the proposed mechanism using aniline in the presence of excess triethyl phosphite or triethyl phosphate did not yield the desired product under the optimized conditions, suggesting that the reaction proceeds via reduction of the nitro group to N2− and that the product is not formed from phosphate.
Scheme 36.

Proposed mechanism for reduction of the nitro-group route to phosphoramidates.
3.4.2. Via Catalyst-Free Staudinger Reduction and Lewis-Acid Catalyzed Rearrangement
The plausible two-step mechanistic process suggested by Hackenberger’s group for the synthesis involves the formation of phosphorimidate from the reduction of azide in the presence of (RO)3P (R = alkyl), eliminating N2, in the first step (Scheme 37) [119]. In the second step, BF3 activates the nitrogen site of the phosphorimidate by forming a coordinated complex. Electrophilic attack by a neighbouring alkyl group of phosphorimidate on the negatively charged N atom of the phosphorimidate-BF3 complex results in an ((alkyl)amino)trialkoxyphosphonium complex, cleaving off BF3. Nucleophilic attack on the trialkoxyphosphonium complex from a neighboring phosphorimidate completes the catalytic cycle, affording a P=O bond of the desired P-N product.
Scheme 37.

Proposed mechanism for the catalyst-free Staudinger reduction and Lewis-acid catalyzed rearrangement route to phosphoramidates.
3.5. Hydrophosphinylation Route
For the hydrophosphinylation route to P-Ns, Zimin et al. proposed that in the first step, sodium dialkyl phosphite ((RO)2PONa, R = Et, Ph) reacts with alkyl nitrile to form an imine (C=N), which reacts further with a second equivalent of (RO)2PONa to form the 1,1-addition product, sodium (1,1-bis(dialkoxyphosphoryl)alkyl)amide (Scheme 38) [120]. In the second step, sodium (1,1-bis(dialkyoxyphosphoryl)alkyl)amide undergoes hydrolysis and isomerization to give the 1,2-addition phosphoramidate product containing a P-C-N-P bond, eliminating a sodium cation in the process.
Scheme 38.

Proposed mechanism for hydrophosphinylation route to phosphoramidates.
3.6. Phosphoramidate-Aldehyde-Dienophile (PAD) Route
Chabour et al. proposed a mechanism of the reaction in which (EtO)2P(O)NH2 reacts with (or is activated by) acetic anhydride to form diethyl acetylphosphoramidate and acetic acid (Scheme 39) [1]. Meanwhile, the aldehyde protonated in the presence of an acid catalyst, TsOH, reacts with the diethyl acetylphosphoramidate complex to give the cationic diethyl (alkene-acetamido)phosphonate complex intermediate. The unstable cationic complex undergoes rearrangement with a hydride shift and deprotonation in presence of TsO− to give a more stable diethyl acetyl(alk-1,3-diene)phosphoramidate. The subsequent direct addition of the maleimide derivative results in a P-N adduct intermediate, which is deacetylated in the presence of acetic acid to afford the desired product.
Scheme 39.

Proposed mechanism for the synthesis of phosphoramidates via the phosphoamidate-aldehyde-dienophile (PAD) route.
4. Summary
In this review, the applications, syntheses, and corresponding mechanisms of phosphoramidates have been discussed in depth. As demonstrated from the many categories for the uses of synthetic P-Ns, it is clear that the P-N bond motif has structural importance in diverse fields. Early synthetic routes to P-Ns are limited due to harsh reaction conditions, air and moisture-sensitive reagents, multi-step synthesis, and undesirable side reactions. The separation and purification of P-Ns is also a challenge. In particular, most of the synthetic strategies involve extraction and/or column chromatography, which not only requires large amounts of solvent, but also takes a significant amount of time. Therefore, unsurprisingly, extensive studies have been undertaken (with more to come in the future) for more sustainable, efficient, atom economic, selective, and milder synthetic procedures toward P-Ns. In addition, the discussed mechanisms require a deeper understanding of the associated phosphorus chemistry in order to have better synthetic control and to optimize the yield and selectivity toward P-N products. To overcome these challenges, it is predicted that better catalysts, improved reaction conditions, and new solvent solutions for separating the product from the by-products and potentially improving selectivity are in the forecast. For example, there is a sharp rise into synthetic research under solvent-free conditions or using ionic liquids (ILs) and deep eutectic solvents (DES). These can be environmentally benign, highly versatile, selective, and recoverable solvents for use in catalysis.
Acknowledgments
We gratefully acknowledge Ebonyi State University Abakaliki and Tertiary Education Trust Fund (TETFund) Nigeria for E.J.I’s PhD Scholarship and The MacDiarmid Institute for Advanced Materials and Nanotechnology, Wellington, New Zealand for financial support.
List of Abbreviations
Dialkyl chlorophosphate (dACP), dialkyl phosphoriodidate (dAPPI), H-phosphonate (H-P), phosphoramidate (P-N), trichloroisocyanuric acid (tCiC-A), 1,3,5-triazinane-2,4,6-trione (tAtO), N-acylphosphoramidates (AcP-N), diphenylphosphine oxide (dPPO), phosphonic acid (PP-A), N-bromosuccinimide (NBS), diphenyl phosphoric acid (dPPA), triphenylphosphine (tPP), diisopropylchlorophosphate (dPrCP), tetraethylpyrophosphate (tEPyP), triphenylphosphate (tPPO), 2,3,4,6,7,8,9,10-octahydropyrimido[1,2-a]azepine (DBU), matrix-assisted laser desorption-ionization time-of-flight mass spectrometry (MALDI-TOF MS), task specific ionic liquid (TSIL), phosphorodiamidate (N-P-N), phenyl dichlorophosphate (PdCP), diphenylchlorophosphate (dPCP), tert-butyl hydroperoxide (TBHP).
Author Contributions
Conceptualization, E.J.I. and E.M.L.; writing—original draft preparation, E.J.I. and S.D.; writing—review and editing, E.M.L.; supervision, E.M.L.; funding acquisition, E.M.L and E.J.I. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Tertiary Education Trust Fund (TETFund) Nigeria administered by Ebonyi State University Abakaliki, and The MacDiarmid Institute for Advanced Materials and Nanotechnology, Wellington, New Zealand.
Conflicts of Interest
The authors declare no conflict of interest.
References
- 1.Chabour I., Nájera C., Sansano J.M. Diastereoselective multicomponent phosphoramidate-aldehyde-dienophile (PAD) process for the synthesis of polysubstituted cyclohex-2-enyl-amine derivatives. Tetrahedron. 2020;76:130801. doi: 10.1016/j.tet.2019.130801. [DOI] [Google Scholar]
- 2.Sekine M., Moriguchi T., Wada T., Seio K. Total Synthesis of Agrocin 84 and Phosmidosine as Naturally Occurring Nucleotidic Antibiotics Having P-N Bond Linkages. J. Syn. Org. Chem. Jpn. 2001;59:1109–1120. doi: 10.5059/yukigoseikyokaishi.59.1109. [DOI] [Google Scholar]
- 3.Petkowski J.J., Bains W., Seager S. Natural Products Containing ‘Rare’ Organophosphorus Functional Groups. Molecules. 2019;24:866. doi: 10.3390/molecules24050866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guijarro J.I., González-Pastor J.E., Baleux F., Millán J.L.S., Castilla M.A., Rico M., Moreno F., Delepierre M. Chemical Structure and Translation Inhibition Studies of the Antibiotic Microcin C7. J. Biol. Chem. 1995;270:23520–23532. doi: 10.1074/jbc.270.40.23520. [DOI] [PubMed] [Google Scholar]
- 5.Hatano M., Hashimoto Y. Properties of a toxic phospholipid in the northern blenny roe. Toxicon. 1974;12:231–236. doi: 10.1016/0041-0101(74)90063-4. [DOI] [PubMed] [Google Scholar]
- 6.Besant P.G., Attwood P.V., Piggott M.J. Focus on Phosphoarginine and Phospholysine. Curr. Protein Pept. Sci. 2009;10:536–550. doi: 10.2174/138920309789630598. [DOI] [PubMed] [Google Scholar]
- 7.Adekoya O.A., Sylte I. Thermolysin. In: Kretsinger R.H., Uversky V.N., Permyakov E.A., editors. Encyclopedia of Metalloproteins. Springer New York; New York, NY, USA: 2013. pp. 2213–2221. [DOI] [Google Scholar]
- 8.Van den Burg B., Eijsink V. Chapter 111-Thermolysin and Related Bacillus Metallopeptidases. In: Rawlings N.D., Salvesen G., editors. Handbook of Proteolytic Enzymes. 3rd ed. Academic Press; Cambridge, MA, USA: 2013. pp. 540–553. [DOI] [Google Scholar]
- 9.Moriguchi T., Asai N., Wada T., Sekine M. Synthesis of nucleotide antibiotics having N-acyl phosphoramidate linkages. Nucleic Acids Symp. Ser. 1999;42:15–16. doi: 10.1093/nass/42.1.15. [DOI] [PubMed] [Google Scholar]
- 10.Phillips D.R., Uramoto M., Isono K., McCloskey J.A. Structure of the antifungal nucleotide antibiotic phosmidosine. J. Org. Chem. 1993;58:854–859. doi: 10.1021/jo00056a017. [DOI] [Google Scholar]
- 11.Oliveira F.M., Barbosa L.C.A., Ismail F.M.D. The diverse pharmacology and medicinal chemistry of phosphoramidates—A review. RSC Adv. 2014;4:18998–19012. doi: 10.1039/C4RA01454E. [DOI] [Google Scholar]
- 12.Cao Y.-C., Deng Q.-X., Dai S.-X. Remdesivir for severe acute respiratory syndrome coronavirus 2 causing COVID-19: An evaluation of the evidence. Travel Med. Infect. Dis. 2020 doi: 10.1016/j.tmaid.2020.101647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gao X., Bi X., Wei J., Peng Z., Liu H., Jiang Y., Wei W., Cai Z. N-phosphorylation labeling for analysis of twenty natural amino acids and small peptides by using matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Analyst. 2013;138:2632–2639. doi: 10.1039/c3an00036b. [DOI] [PubMed] [Google Scholar]
- 14.Nguyen T.-M., Chang S., Condon B., Slopek R., Graves E., Yoshioka-Tarver M. Structural Effect of Phosphoramidate Derivatives on the Thermal and Flame Retardant Behaviors of Treated Cotton Cellulose. Ind. Eng. Chem. Res. 2013;52:4715–4724. doi: 10.1021/ie400180f. [DOI] [Google Scholar]
- 15.Qader B., Baron M.G., Hussain I., Johnson R.P., Gonzalez-Rodriguez J. Electrochemical determination of the organophosphate compound Fenamiphos and its main metabolite, Fenamiphos sulfoxide. Mon. Chem. 2019;150:411–417. doi: 10.1007/s00706-018-2334-4. [DOI] [Google Scholar]
- 16.Rodrigues Furtado Medeiros A.C., Gouvêa M.M., Felipe T.V., Marques F.F.d.C., Bernardino A.M.R., López Ortiz F., de Souza M.C. New o-substituted diphenylphosphinic amide ligands: Synthesis, characterization and complexation with Zn2+, Cu2+ and Y3+ New J. Chem. 2019;43:13881–13890. doi: 10.1039/C9NJ02829C. [DOI] [Google Scholar]
- 17.Xie X., Qin Z., He Y., Xiong P., Huang Z., Mao Y., Wei H., Zhuo L. Significant enhanced uranyl ions extraction efficiency with phosphoramidate-functionalized ionic liquids via synergistic effect of coordination and hydrogen bond. Sci. Rep. 2017;7:15735. doi: 10.1038/s41598-017-15899-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yan J., Bai X., Li J., Ren T., Zhao Y. The tribochemical study of novel phosphorous-nitrogen (P-N) type phosphoramidate additives in water. Ind. Lubr. Tribol. 2014;66:346–352. doi: 10.1108/ilt-12-2011-0111. [DOI] [Google Scholar]
- 19.Kang J., Zettel V.H., Ward N.I. The Organophosphate Pesticides. J. Nutr. Environ. Med. 1995;5:325–339. doi: 10.3109/13590849509007239. [DOI] [Google Scholar]
- 20.Oliveira F.M., Barbosa L.C.A., Teixeira R.R., Demuner A.J., Maltha C.R.Á., Picanço M.C., Silva G.A., de Paula V.F. Synthesis and insecticidal activity of new phosphoramidates. J. Pestic. Sci. 2012;37:85–88. doi: 10.1584/jpestics.D11-014. [DOI] [Google Scholar]
- 21.Rezg R., Mornagui B., El-Fazaa S., Gharbi N. Organophosphorus pesticides as food chain contaminants and type 2 diabetes: A review. Trends Food Sci. Technol. 2010;21:345–357. doi: 10.1016/j.tifs.2010.04.006. [DOI] [Google Scholar]
- 22.Costa L.G. Current issues in organophosphate toxicology. Clin. Chim. Acta. 2006;366:1–13. doi: 10.1016/j.cca.2005.10.008. [DOI] [PubMed] [Google Scholar]
- 23.Eto M., Kobayashi K., Kato T., Kojima K., Oshima Y. Saligenin Cyclic Phosphoramidates and Phosphoramidothionates as Pesticides. Agric. Biol. Chem. 1965;29:243–248. doi: 10.1080/00021369.1965.10858381. [DOI] [Google Scholar]
- 24.Paula V.F., Barbosa L.C.A., Teixeira R.R., Picanço M.C., Silva G.A. Synthesis and insecticidal activity of new 3-benzylfuran-2-yl N,N,N′,N′-tetraethyldiamidophosphate derivatives. Pest. Manag. Sci. 2008;64:863–872. doi: 10.1002/ps.1559. [DOI] [PubMed] [Google Scholar]
- 25.Uesugi Y., Katagiri M., Noda O. Negatively Correlated Cross-resistance and Synergism between Phosphoramidates and Phosphorothiolates in Their Fungicidal Actions on Rice Blast Fungi. Agric. Biol. Chem. 1974;38:907–912. doi: 10.1080/00021369.1974.10861282. [DOI] [Google Scholar]
- 26.Modolo L.V., da-Silva C.J., Brandão D.S., Chaves I.S. A minireview on what we have learned about urease inhibitors of agricultural interest since mid-2000s. J. Adv. Res. 2018;13:29–37. doi: 10.1016/j.jare.2018.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Domínguez M.J., Sanmartín C., Font M., Palop J.A., San Francisco S., Urrutia O., Houdusse F., García-Mina J.M. Design, Synthesis, and Biological Evaluation of Phosphoramide Derivatives as Urease Inhibitors. J. Agric. Food Chem. 2008;56:3721–3731. doi: 10.1021/jf072901y. [DOI] [PubMed] [Google Scholar]
- 28.Kafarski P., Talma M. Recent advances in design of new urease inhibitors: A review. J. Adv. Res. 2018;13:101–112. doi: 10.1016/j.jare.2018.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rego Y.F., Queiroz M.P., Brito T.O., Carvalho P.G., de Queiroz V.T., de Fátima Â., Macedo F., Jr. A review on the development of urease inhibitors as antimicrobial agents against pathogenic bacteria. J. Adv. Res. 2018;13:69–100. doi: 10.1016/j.jare.2018.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yan J., Bu J., Bai X., Li J., Ren T., Zhao Y. The tribological study of novel phosphorous–nitrogen type phosphoramidate additives in rapeseed oil. Proc. Inst. Mech. Eng. Part J: J. Eng. 2012;226:377–388. doi: 10.1177/1350650111435034. [DOI] [Google Scholar]
- 31.Shaw S., Blum A., Weber R., Rich D., Lucas D., Koshland C., Dobraca D., Hanson S., Birnbaum L. Halogenated flame retardants: Do the fire safety benefits justify the risks? Rev. Environ. Health. 2010;25:261–305. doi: 10.1515/REVEH.2010.25.4.261. [DOI] [PubMed] [Google Scholar]
- 32.Gaan S., Mauclaire L., Rupper P., Salimova V., Tran T.-T., Heuberger M. Thermal degradation of cellulose acetate in presence of bis-phosphoramidates. J. Anal. Appl. Pyrolysis. 2011;90:33–41. doi: 10.1016/j.jaap.2010.10.005. [DOI] [Google Scholar]
- 33.Hirsch C., Striegl B., Mathes S., Adlhart C., Edelmann M., Bono E., Gaan S., Salmeia K.A., Hoelting L., Krebs A., et al. Multiparameter toxicity assessment of novel DOPO-derived organophosphorus flame retardants. Arch. Toxicol. 2017;91:407–425. doi: 10.1007/s00204-016-1680-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Neisius M., Liang S., Mispreuve H., Gaan S. Phosphoramidate-Containing Flame-Retardant Flexible Polyurethane Foams. Ind. Eng. Chem. Res. 2013;52:9752–9762. doi: 10.1021/ie400914u. [DOI] [Google Scholar]
- 35.Salmeia K.A., Gaan S., Malucelli G. Recent Advances for Flame Retardancy of Textiles Based on Phosphorus Chemistry. Polymers. 2016;8:319. doi: 10.3390/polym8090319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.SeChin C., Brian C., Jade S. Microwave Assisted Preparation of Self-Extinguishing Cotton Fabrics by Small Molecules Containing Phosphorous and Nitrogen. Curr. Microw. Chem. 2019;6:3–12. doi: 10.2174/2213335606666190301160053. [DOI] [Google Scholar]
- 37.Velencoso M.M., Battig A., Markwart J.C., Schartel B., Wurm F.R. Molecular Firefighting—How Modern Phosphorus Chemistry Can Help Solve the Challenge of Flame Retardancy. Angew. Chem. Int. Ed. 2018;57:10450–10467. doi: 10.1002/anie.201711735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Edwards B., Rudolf S., Hauser P., El-Shafei A. Preparation, Polymerization, and Performance Evaluation of Halogen-Free Radiation Curable Flame Retardant Monomers for Cotton Substrates. Ind. Eng. Chem. Res. 2015;54:577–584. doi: 10.1021/ie502915t. [DOI] [Google Scholar]
- 39.Markwart J.C., Battig A., Zimmermann L., Wagner M., Fischer J., Schartel B., Wurm F.R. Systematically Controlled Decomposition Mechanism in Phosphorus Flame Retardants by Precise Molecular Architecture: P–O vs P–N. ACS Appl. Polym. Mater. 2019;1:1118–1128. doi: 10.1021/acsapm.9b00129. [DOI] [Google Scholar]
- 40.Rupper P., Gaan S., Salimova V., Heuberger M. Characterization of chars obtained from cellulose treated with phosphoramidate flame retardants. J. Anal. Appl. Pyrolysis. 2010;87:93–98. doi: 10.1016/j.jaap.2009.10.011. [DOI] [Google Scholar]
- 41.Zhao W., Li B., Xu M., Yang K., Lin L. Novel intumescent flame retardants: Synthesis and application in polycarbonate. Fire Mater. 2013;37:530–546. doi: 10.1002/fam.2145. [DOI] [Google Scholar]
- 42.Jurinke C., Oeth P., van den Boom D. MALDI-TOF mass spectrometry. Mol. Biotechnol. 2004;26:147–163. doi: 10.1385/MB:26:2:147. [DOI] [PubMed] [Google Scholar]
- 43.Guo Z., Zhang Q., Zou H., Guo B., Ni J. A Method for the Analysis of Low-Mass Molecules by MALDI-TOF Mass Spectrometry. Anal. Chem. 2002;74:1637–1641. doi: 10.1021/ac010979m. [DOI] [PubMed] [Google Scholar]
- 44.Gao X., Tang Z., Lu M., Liu H., Jiang Y., Zhao Y., Cai Z. Suppression of matrix ions by N-phosphorylation labeling using matrix-assisted laser desorption–ionization time-of-flight mass spectrometry. Chem. Commun. 2012;48:10198–10200. doi: 10.1039/c2cc36091h. [DOI] [PubMed] [Google Scholar]
- 45.Saha A., Tiwari N., Deb S., Saxena M. Selective Liquid-Liquid Extraction of Uranium by Phosphoramidate-Group Bearing Ionic Liquid. ChemistrySelect. 2019;4:7691–7697. doi: 10.1002/slct.201901933. [DOI] [Google Scholar]
- 46.Saha A., Sanyal K., Rawat N., Deb S.B., Saxena M.K., Tomar B.S. Selective Micellar Extraction of Ultratrace Levels of Uranium in Aqueous Samples by Task Specific Ionic Liquid Followed by Its Detection Employing Total Reflection X-ray Fluorescence Spectrometry. Anal. Chem. 2017;89:10422–10430. doi: 10.1021/acs.analchem.7b02427. [DOI] [PubMed] [Google Scholar]
- 47.Drover M.W., Christopherson C.J., Schafer L.L., Love J.A. C(sp3)–H Bond Activation Induced by Monohydroborane Coordination at an Iridium(III)–Phosphoramidate Complex. Eur. J. Inorg. Chem. 2017;2017:2639–2642. doi: 10.1002/ejic.201601518. [DOI] [Google Scholar]
- 48.Drover M.W., Love J.A., Schafer L.L. Toward anti-Markovnikov 1-Alkyne O-Phosphoramidation: Exploiting Metal–Ligand Cooperativity in a 1,3-N,O-Chelated Cp*Ir(III) Complex. J. Am. Chem. Soc. 2016;138:8396–8399. doi: 10.1021/jacs.6b05143. [DOI] [PubMed] [Google Scholar]
- 49.Drover M.W., Love J.A., Schafer L.L. 1,3-N,O-Complexes of late transition metals. Ligands with flexible bonding modes and reaction profiles. Chem. Soc. Rev. 2017;46:2913–2940. doi: 10.1039/C6CS00715E. [DOI] [PubMed] [Google Scholar]
- 50.Garcia P., Lau Y.Y., Perry M.R., Schafer L.L. Phosphoramidate tantalum complexes for room-temperature C-H functionalization: Hydroaminoalkylation catalysis. Angew. Chem. Int. Ed. 2013;52:9144–9148. doi: 10.1002/anie.201304153. [DOI] [PubMed] [Google Scholar]
- 51.Yang T., Lin C., Fu H., Jiang Y., Zhao Y. Copper-Catalyzed Synthesis of Medium- and Large-Sized Nitrogen Heterocycles via N-Arylation of Phosphoramidates and Carbamates. Org. Lett. 2005;7:4781–4784. doi: 10.1021/ol052126c. [DOI] [PubMed] [Google Scholar]
- 52.Chary B.C., Kim S., Park Y., Kim J., Lee P.H. Palladium-Catalyzed C–H Arylation Using Phosphoramidate as a Directing Group at Room Temperature. Org. Lett. 2013;15:2692–2695. doi: 10.1021/ol4009987. [DOI] [PubMed] [Google Scholar]
- 53.Reddy S.M., Rao D.S., Madhu K., Subramanyam C., Kumari P.G., Raju C.N. 2-(Benzo[d]thiazol-2-yl) phenyl-4-nitrophenyl alkyl/aryl substituted phosphoramidates: Synthesis, characterization and antimicrobial activity evaluation. Phosphorus Sulfur Silicon Relat. Elem. 2018;193:136–140. doi: 10.1080/10426507.2017.1392960. [DOI] [Google Scholar]
- 54.Ji Q., Ge Z., Ge Z., Chen K., Wu H., Liu X., Huang Y., Yuan L., Yang X., Liao F. Synthesis and biological evaluation of novel phosphoramidate derivatives of coumarin as chitin synthase inhibitors and antifungal agents. Eur. J. Med. Chem. 2016;108:166–176. doi: 10.1016/j.ejmech.2015.11.027. [DOI] [PubMed] [Google Scholar]
- 55.Rasool N., Chennamsetty S., Janakiramudu D., Perikala S., Usha R., Raju C. Convenient one-pot synthesis and biological evaluation of phosphoramidates and phosphonates containing heterocycles. Phosphorus Sulfur Silicon Relat. Elem. 2018;193:1–14. doi: 10.1080/10426507.2018.1452229. [DOI] [Google Scholar]
- 56.Rasheed S., Madhava G., Basha S.T., Fareeda G., Raju C.N. Synthesis, spectral characterization, and pro- and antioxidant activity of phosphorylated derivatives of cis-tramadol. Phosphorus Sulfur Silicon Relat. Elem. 2017;192:1247–1251. doi: 10.1080/10426507.2015.1119138. [DOI] [Google Scholar]
- 57.McGuigan C., Thiery J.-C., Daverio F., Jiang W.G., Davies G., Mason M. Anti-cancer ProTides: Tuning the activity of BVDU phosphoramidates related to thymectacin. Bioorg. Med. Chem. 2005;13:3219–3227. doi: 10.1016/j.bmc.2005.02.041. [DOI] [PubMed] [Google Scholar]
- 58.Mara C., Dempsey E., Bell A., Barlow J.W. Synthesis and evaluation of phosphoramidate and phosphorothioamidate analogues of amiprophos methyl as potential antimalarial agents. Bioorg. Med. Chem. Lett. 2011;21:6180–6183. doi: 10.1016/j.bmcl.2011.07.088. [DOI] [PubMed] [Google Scholar]
- 59.Slusarczyk M., Serpi M., Pertusati F. Phosphoramidates and phosphonamidates (ProTides) with antiviral activity. Antivir. Chem. Chemother. 2018;26 doi: 10.1177/2040206618775243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Okon A., Matos de Souza M.R., Shah R., Amorim R., da Costa L.J., Wagner C.R. Anchimerically Activatable Antiviral ProTides. ACS Med. Chem. Lett. 2017;8:958–962. doi: 10.1021/acsmedchemlett.7b00277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li X., Fu H.-H., Jiang Y., Zhao Y., Jin J. New and Efficient Approach to Aryl Phosphoramidate Derivatives of AZT/d4T as Anti-HIV Prodrugs. Synlett. 2004;14:2600–2602. doi: 10.1055/s-2004-834800. [DOI] [Google Scholar]
- 62.Mehellou Y., Balzarini J., McGuigan C. Aryloxy Phosphoramidate Triesters: A Technology for Delivering Monophosphorylated Nucleosides and Sugars into Cells. ChemMedChem. 2009;4:1779–1791. doi: 10.1002/cmdc.200900289. [DOI] [PubMed] [Google Scholar]
- 63.Mehellou Y., Rattan H.S., Balzarini J. The ProTide Prodrug Technology: From the Concept to the Clinic. J. Med. Chem. 2018;61:2211–2226. doi: 10.1021/acs.jmedchem.7b00734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Amirian E.S., Levy J.K. Current knowledge about the antivirals remdesivir (GS-5734) and GS-441524 as therapeutic options for coronaviruses. One Health. 2020;9:100128. doi: 10.1016/j.onehlt.2020.100128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang J., Gao S.J., Zhang P.C., Wang S., Mao H.Q., Leong K.W. Polyphosphoramidate gene carriers: Effect of charge group on gene transfer efficiency. Gene Ther. 2004;11:1001–1010. doi: 10.1038/sj.gt.3302248. [DOI] [PubMed] [Google Scholar]
- 66.Zhang P.-C., Wang J., Leong K.W., Mao H.-Q. Ternary Complexes Comprising Polyphosphoramidate Gene Carriers with Different Types of Charge Groups Improve Transfection Efficiency. Biomacromolecules. 2005;6:54–60. doi: 10.1021/bm040010i. [DOI] [PubMed] [Google Scholar]
- 67.Siegel D., Hui H.C., Doerffler E., Clarke M.O., Chun K., Zhang L., Neville S., Carra E., Lew W., Ross B., et al. Discovery and Synthesis of a Phosphoramidate Prodrug of a Pyrrolo[2,1-f][triazin-4-amino] Adenine C-Nucleoside (GS-5734) for the Treatment of Ebola and Emerging Viruses. J. Med. Chem. 2017;60:1648–1661. doi: 10.1021/acs.jmedchem.6b01594. [DOI] [PubMed] [Google Scholar]
- 68.Benkovic S.J., Sampson E.J. Structure-reactivity correlation for the hydrolysis of phosphoramidate monoanions. J. Am. Chem. Soc. 1971;93:4009–4016. doi: 10.1021/ja00745a032. [DOI] [Google Scholar]
- 69.Oney I., Caplow M. Phosphoramidate solvolysis. J. Am. Chem. Soc. 1967;89:6972–6980. doi: 10.1021/ja01002a027. [DOI] [Google Scholar]
- 70.Choy C.J., Geruntho J.J., Davis A.L., Berkman C.E. Tunable pH-Sensitive Linker for Controlled Release. Bioconjug. Chem. 2016;27:824–830. doi: 10.1021/acs.bioconjchem.6b00027. [DOI] [PubMed] [Google Scholar]
- 71.Choy C.J., Ley C.R., Davis A.L., Backer B.S., Geruntho J.J., Clowers B.H., Berkman C.E. Second-Generation Tunable pH-Sensitive Phosphoramidate-Based Linkers for Controlled Release. Bioconjug. Chem. 2016;27:2206–2213. doi: 10.1021/acs.bioconjchem.6b00422. [DOI] [PubMed] [Google Scholar]
- 72.Olatunji F.P., Kesic B.N., Choy C.J., Berkman C.E. Phosphoramidate derivates as controlled-release prodrugs of l-Dopa. Bioorg. Med. Chem. Lett. 2019;29:2571–2574. doi: 10.1016/j.bmcl.2019.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zakirova N.F., Shipitsyn A.V., Jasko M.V., Prokofjeva M.M., Andronova V.L., Galegov G.A., Prassolov V.S., Kochetkov S.N. Phosphoramidate derivatives of acyclovir: Synthesis and antiviral activity in HIV-1 and HSV-1 models in vitro. Bioorg. Med. Chem. 2012;20:5802–5809. doi: 10.1016/j.bmc.2012.08.008. [DOI] [PubMed] [Google Scholar]
- 74.Fiske C.H., Subbarow Y. The nature of the "inorganic phosphate" in voluntary muscle. Science. 1927;65:401–403. doi: 10.1126/science.65.1686.401. [DOI] [PubMed] [Google Scholar]
- 75.Fiske C.H., Subbarow Y. Phosphorus compounds of muscle and liver. Science. 1929;70:381–382. doi: 10.1126/science.70.1816.381-a. [DOI] [PubMed] [Google Scholar]
- 76.Audrieth L.F., Toy A.D.F. The Aquo Ammono Phosphoric Acids. I. Preparation of Phenyl Esters of Amido- and Diamidophosphoric Acids. J. Am. Chem. Soc. 1941;63:2117–2119. doi: 10.1021/ja01853a025. [DOI] [Google Scholar]
- 77.Audrieth L.F., Toy A.D.F. The Aquo Ammono Phosphoric Acids. II. Preparation of N-Substituted Derivatives of the Phenyl Esters of Amido- and Diamidophosphoric Acids. J. Am. Chem. Soc. 1942;64:1337–1339. doi: 10.1021/ja01258a029. [DOI] [Google Scholar]
- 78.Atherton F.R., Openshaw H.T., Todd A.R. 174. Studies on phosphorylation. Part II. The reaction of dialkyl phosphites with polyhalogen compounds in presence of bases. A new method for the phosphorylation of amines. J. Chem. Soc. 1945:660–663. doi: 10.1039/jr9450000660. [DOI] [PubMed] [Google Scholar]
- 79.Atherton F.R., Todd A.R. 129. Studies on phosphorylation. Part III. Further observations on the reaction of phosphites with polyhalogen compounds in presence of bases and its application to the phosphorylation of alcohols. J. Chem. Soc. 1947:674–678. doi: 10.1039/jr9470000674. [DOI] [PubMed] [Google Scholar]
- 80.Atherton F.R., Howard H.T., Todd A.R. 220. Studies on phosphorylation. Part IV. Further studies on the use of dibenzyl chlorophosphonate and the examination of certain alternative phosphorylation methods. J. Chem. Soc. 1948:1106–1111. doi: 10.1039/jr9480001106. [DOI] [Google Scholar]
- 81.Wagner-Jauregg T., O’Neill J.J., Summerson W.H. The Reaction of Phosphorus-containing Enzyme Inhibitors with Amines and Amino Acid Derivatives1. J. Am. Chem. Soc. 1951;73:5202–5206. doi: 10.1021/ja01155a059. [DOI] [Google Scholar]
- 82.Nielsen M.L. The Formation of P-N and P-N-P Bonds by Elimination of Phenol in a Basic Condensation. Inorg. Chem. 1964;3:1760–1767. doi: 10.1021/ic50022a024. [DOI] [Google Scholar]
- 83.Sundberg R.J. Deoxygenation of Nitro Groups by Trivalent Phosphorus. Indoles from o-Nitrostyrenes. J. Org. Chem. 1965;30:3604–3610. doi: 10.1021/jo01022a006. [DOI] [Google Scholar]
- 84.Cadogan J.I.G., Mackie R.K., Maynard J.A. The Reactivity of Organophosphorus Compounds. Part XX1. P=O Nucleophilicity in Phosphonates and Phosphoramidates. J. Chem. Soc. C. 1967:1356–1360. doi: 10.1039/J39670001356. [DOI] [Google Scholar]
- 85.Cadogan J.I.G., Sears D.J., Smith D.M., Todd M.J. Reduction of nitro- and nitroso-compounds by tervalent phosphorus reagents. Part V. Reduction of alkyl- and methoxy-nitrobenzenes, and nitrobenzene by trialkyl phosphites. J. Chem. Soc. C. 1969:2813–2819. doi: 10.1039/j39690002813. [DOI] [Google Scholar]
- 86.Zwierzak A. Phase-Transfer-Catalysed Phosphorylation of Amines in an Aqueous System. Synthesis. 1975;1975:507–509. doi: 10.1055/s-1975-23821. [DOI] [Google Scholar]
- 87.Zwierzak A. Selective N-Alkylation of Diethyl Phosphoramidate: Tetrabutylaminium Bromide as Catalyst for Nucleophilic Substitution in a Homogeneous Medium. Synthesis. 1982;1982:920–922. doi: 10.1055/s-1982-29996. [DOI] [Google Scholar]
- 88.Zwierzak A., Osowska K. A Convenient Method for the Protection of Amino Groups as Diethyl Phosphoramidates. Synthesis. 1984;1984:223–224. doi: 10.1055/s-1984-30778. [DOI] [Google Scholar]
- 89.Zwierzak A., Osowska-Pacewicka K. Direct conversion of diethyl hydrogen phosphate into diethyl phosphoramides. Mon. Chem. 1984;115:117–120. doi: 10.1007/BF00798428. [DOI] [Google Scholar]
- 90.Steinbach J., Herrmann E., Riesel L. Zur Umsetzung des Zweikomponentensystems Trialkylphosphit/Tetrachlorkohlenstoff mit wasserstoffhaltigen Nucleophilen 2. Die Reaktion mit Ammoniak und Aminen. Z. Anorg. Aallg. Chem. 1985;523:180–186. doi: 10.1002/zaac.19855230422. [DOI] [Google Scholar]
- 91.Nielsen J., Caruthers M.H. Directed Arbuzov-type reactions of 2-cyano-1,1-dimethylethyl deoxynucleoside phosphites. J. Am. Chem. Soc. 1988;110:6275–6276. doi: 10.1021/ja00226a069. [DOI] [PubMed] [Google Scholar]
- 92.Koziara A. A facile one-pot transformation of alkyl bromides into Diethyl N-Alkylphosphoroamidates. J. Praktische Chem. 1988;330:473–475. doi: 10.1002/prac.19883300320. [DOI] [Google Scholar]
- 93.Talley J.J. Preparation of sterically hindered phosphoramidates. J. Chem. Eng. Data. 1988;33:221–222. doi: 10.1021/je00052a048. [DOI] [Google Scholar]
- 94.Ou Y., Huang Y., He Z., Yu G., Huo Y., Li X., Gao Y., Chen Q. A phosphoryl radical-initiated Atherton–Todd-type reaction under open air. Chem. Commun. 2020;56:1357–1360. doi: 10.1039/C9CC09407E. [DOI] [PubMed] [Google Scholar]
- 95.Lukanov L.K., Venkov A.P., Mollov N.M. A New Approach to the Atherton-Todd Reaction. Synthesis. 1985;1985:971–973. doi: 10.1055/s-1985-31409. [DOI] [Google Scholar]
- 96.Gupta A.K., Acharya J., Dubey D.K., Kaushik M.P. Efficient and Convenient One-Pot Synthesis of Phosphoramidates and Phosphates. Synth. Commun. 2007;37:3403–3407. doi: 10.1080/00397910701483662. [DOI] [Google Scholar]
- 97.Kaboudin B., Donyavi A., Kazemi F. Trichloroisocyanuric Acid as an Efficient Reagent for the Synthesis of Phosphoroamidates via Atherton–Todd Reaction under Base-Free Conditions. Synthesis. 2018;50:170–174. doi: 10.1055/s-0036-1589111. [DOI] [Google Scholar]
- 98.Jang D.O., Kasemsuknimit A., Satyender A., Chavasiri W. Efficient Amidation and Esterification of Phosphoric Acid Using Cl3CCN/Ph3P. Bull. Korean Chem. Soc. 2011;32:3486–3488. doi: 10.5012/BKCS.2011.32.9.3486. [DOI] [Google Scholar]
- 99.Appel R., Einig H. Neues Verfahren zur Darstellung von Phosphorsäureesteramiden, Phosphinsäureamiden, Phosphinsäureestern und gemischt substituierten Phosphorsäuretriestern. Z. Anorg. Aallg. Chem. 1975;414:241–246. doi: 10.1002/zaac.19754140308. [DOI] [Google Scholar]
- 100.Chen X., Xiao Z., Chu H., Wang B., Peng A.-Y. Reinvestigation of the iodine-mediated phosphoramidation reaction of amines and P(OR)3 and its synthetic applications. Org. Biomol. Chem. 2018;16:6783–6790. doi: 10.1039/C8OB01840E. [DOI] [PubMed] [Google Scholar]
- 101.Dar B.A., Dangroo N.A., Gupta A., Wali A., Khuroo M.A., Vishwakarma R.A., Singh B. Iodine catalyzed solvent-free cross-dehydrogenative coupling of arylamines and H-phosphonates for the synthesis of N-arylphosphoramidates under atmospheric conditions. Tetrahedron Lett. 2014;55:1544–1548. doi: 10.1016/j.tetlet.2014.01.064. [DOI] [Google Scholar]
- 102.Dhineshkumar J., Prabhu K.R. Cross-Hetero-Dehydrogenative Coupling Reaction of Phosphites: A Catalytic Metal-Free Phosphorylation of Amines and Alcohols. Org. Lett. 2013;15:6062–6065. doi: 10.1021/ol402956b. [DOI] [PubMed] [Google Scholar]
- 103.Fraser J., Wilson L.J., Blundell R.K., Hayes C.J. Phosphoramidate synthesis via copper-catalysed aerobic oxidative coupling of amines and H-phosphonates. Chem. Commun. 2013;49:8919–8921. doi: 10.1039/c3cc45680c. [DOI] [PubMed] [Google Scholar]
- 104.Wang G., Yu Q.-Y., Chen S.-Y., Yu X.-Q. Copper-catalyzed aerobic oxidative cross-coupling of arylamines and dialkylphosphites leading to N-arylphosphoramidates. Tetrahedron Lett. 2013;54:6230–6232. doi: 10.1016/j.tetlet.2013.09.006. [DOI] [Google Scholar]
- 105.Kaboudin B., Kazemi F., Habibi F. Fe3O4@MgO nanoparticles as an efficient recyclable catalyst for the synthesis of phosphoroamidates via the Atherton–Todd reaction. Tetrahedron Lett. 2015;56:6364–6367. doi: 10.1016/j.tetlet.2015.09.129. [DOI] [Google Scholar]
- 106.Purohit A.K., Pardasani D., Kumar A., Goud D.R., Jain R., Dubey D.K. A single-step one pot synthesis of O,O′-dialkyl N,N-dialkylphosphoramidates from dialkylphosphites. Tetrahedron Lett. 2016;57:3754–3756. doi: 10.1016/j.tetlet.2016.07.019. [DOI] [Google Scholar]
- 107.Jin X., Yamaguchi K., Mizuno N. Copper-Catalyzed Oxidative Cross-Coupling of H-Phosphonates and Amides to N-Acylphosphoramidates. Org. Lett. 2013;15:418–421. doi: 10.1021/ol303420g. [DOI] [PubMed] [Google Scholar]
- 108.Zhou Y., Yang J., Chen T., Yin S.-F., Han D., Han L.-B. Stereospecific Aerobic Oxidative Dehydrocoupling of P(O)–H Bonds with Amines Catalyzed by Copper. Bull. Chem. Soc. Jpn. 2014;87:400–402. doi: 10.1246/bcsj.20130310. [DOI] [Google Scholar]
- 109.Dangroo N.A., Ara T., Dar B.A., Khuroo M.A. Copper catalyzed tandem Chan–Lam type C—N and Staudinger-phosphite N—P coupling for the synthesis of N-arylphosphoramidates. Catal. Commun. 2019;118:76–80. doi: 10.1016/j.catcom.2018.10.006. [DOI] [Google Scholar]
- 110.Meazza M., Kowalczuk A., Shirley L., Yang J.W., Guo H., Rios R. Organophotocatalytic Synthesis of Phosphoramidates. Adv. Synth. Catal. 2016;358:719–723. doi: 10.1002/adsc.201501068. [DOI] [Google Scholar]
- 111.Anitha T., Ashalu K.C., Sandeep M., Mohd A., Wencel-Delord J., Colobert F., Reddy K.R. LiI/TBHP Mediated Oxidative Cross-Coupling of P(O)–H Compounds with Phenols and Various Nucleophiles: Direct Access to the Synthesis of Organophosphates. Eur. J. Org. Chem. 2019;2019:7463–7474. doi: 10.1002/ejoc.201901362. [DOI] [Google Scholar]
- 112.Kim H., Park J., Kim J.G., Chang S. Synthesis of Phosphoramidates: A Facile Approach Based on the C-N Bond Formation via Ir-Catalyzed Direct C–H Amidation. Org. Lett. 2014;16:5466–5469. doi: 10.1021/ol502722j. [DOI] [PubMed] [Google Scholar]
- 113.Xiao W., Zhou C.-Y., Che C.-M. Ruthenium(iv) porphyrin catalyzed phosphoramidation of aldehydes with phosphoryl azides as a nitrene source. Chem. Commun. 2012;48:5871–5873. doi: 10.1039/c2cc31686b. [DOI] [PubMed] [Google Scholar]
- 114.Pan C., Jin N., Zhang H., Han J., Zhu C. Iridium-Catalyzed Phosphoramidation of Arene C–H Bonds with Phosphoryl Azide. J. Org. Chem. 2014;79:9427–9432. doi: 10.1021/jo5018052. [DOI] [PubMed] [Google Scholar]
- 115.Dar B.A. Catalyst free, one pot synthesis of phosphoramidates under environment friendly conditions. J. Ind. Eng. Chem. 2016;36:194–197. doi: 10.1016/j.jiec.2016.01.041. [DOI] [Google Scholar]
- 116.Dangroo N.A., Dar A.A., Shankar R., Khuroo M.A., Sangwan P.L. An efficient synthesis of phosphoramidates from halides in aqueous ethanol. Tetrahedron Lett. 2016;57:2717–2722. doi: 10.1016/j.tetlet.2016.05.003. [DOI] [Google Scholar]
- 117.Li Q., Sun X., Yang X., Wu M., Sun S., Chen X. Transition-metal-free amination phosphoryl azide for the synthesis of phosphoramidates. RSC Adv. 2019;9:16040–16043. doi: 10.1039/C9RA03389K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Haggam R., Conrad J., Beifuss U. Practical and reliable synthesis of dialkyl N-arylphosphoramidates with nitroarenes as substrates. Tetrahedron Lett. 2009;50:6627–6630. doi: 10.1016/j.tetlet.2009.09.037. [DOI] [Google Scholar]
- 119.Wilkening I., del Signore G., Hackenberger C.P.R. Synthesis of N,N-disubstituted phosphoramidatesvia a Lewis acid-catalyzed phosphorimidate rearrangement. Chem. Commun. 2008:2932–2934. doi: 10.1039/b802030b. [DOI] [PubMed] [Google Scholar]
- 120.Zimin M.G., Dvoinishnikova T.A., Konovalova I.V., Pudovik A.N. Reaction of sodium salts of dialkylphosphorous acids with carboxylic acid nitriles. Bull. Acad. Sci. USSR Div. Chem. Sci. 1978;27:436–437. doi: 10.1007/BF00923903. [DOI] [Google Scholar]
- 121.Mitova V., Koseva N., Troev K. Study on the Atherton–Todd reaction mechanism. RSC Adv. 2014;4:64733–64736. doi: 10.1039/C4RA10228B. [DOI] [Google Scholar]
- 122.Salvatore R.N., Nagle A.S., Jung K.W. Cesium Effect: High Chemoselectivity in Direct N-Alkylation of Amines. J. Org. Chem. 2002;67:674–683. doi: 10.1021/jo010643c. [DOI] [PubMed] [Google Scholar]
- 123.Parrish J.P., Dueno E.E., Kim S.-I., Jung K.W. Improved Cs2CO3 Promoted O-Alkylation of Acids. Synth. Commun. 2000;30:2687–2700. doi: 10.1080/00397910008086893. [DOI] [Google Scholar]
- 124.Bunton C.A., Moffatt J.R., Rodenas E. Abnormally high nucleophilicity of micellar-bound azide ion. J. Am. Chem. Soc. 1982;104:2653–2655. doi: 10.1021/ja00373a061. [DOI] [Google Scholar]
- 125.Beckmann H.S.G., Wittmann V. One-Pot Procedure for Diazo Transfer and Azide−Alkyne Cycloaddition: Triazole Linkages from Amines. Org. Lett. 2007;9:1–4. doi: 10.1021/ol0621506. [DOI] [PubMed] [Google Scholar]