

Abstract
In a proportion of patients with hypersensitivity pneumonitis, the biological and environmental factors that sustain inflammation are ill defined, resulting in no effective treatment option. Bioaerosols found in occupational settings are complex and often include Toll-like receptor ligands, such as endotoxins. How Toll-like receptor ligands contribute to the persistence of hypersensitivity pneumonitis, however, remains poorly understood. In a previous study, we found that an S1P1 (sphingosine-1-phosphate receptor 1) agonist prevented the reactivation of antigen-driven B-cell responses in the lung. Here, we assessed the impact of endotoxins on B-cell activation in preexisting hypersensitivity pneumonitis and the role of S1P1 in this phenomenon. The impact of endotoxins on pre-established hypersensitivity pneumonitis was studied in vivo. S1P1 levels were tracked on B cells in the course of the disease using S1P1-eGFP knockin mice, and the role of S1P1 on B-cell functions was assessed using pharmacological tools. S1P1 was found on B cells in experimental hypersensitivity pneumonitis. Endotoxin exposure enhanced neutrophil accumulation in the BAL of mice with experimental hypersensitivity pneumonitis. This was associated with enhanced CD69 cell-surface expression on lymphocytes in the BAL. In isolated B cells, endotoxins increased cell-surface levels of costimulatory molecules and CD69, which was prevented by an S1P1 agonist. S1P1 modulators also reduced TNF production by B cells and their capacity to trigger T-cell cooperation ex vivo. An S1P1 ligand directly inhibited endotoxin-induced B-cell activation.
Keywords: extrinsic allergic alveolitis, TLR4, antibodies, TNF, ozanimod
Hypersensitivity pneumonitis (HP) is an interstitial lung disease caused by repetitive exposure to a wide variety of airborne antigens (1). In patients with HP, the lung features B-cell–enriched tertiary lymphoid tissues (TLTs) that are rapidly activated upon antigen reencounter (2). This reactivation leads to local release of antigen-specific antibodies and cytokines, which are instrumental in disease exacerbation and persistence. The identification of causal agents and improvements in working procedures have decreased the incidence of HP flare-ups in agricultural workers (3, 4). Despite these advances, the causative agent cannot be identified in nearly 30% of patients with HP (3, 5). In these patients, uninterrupted exposure often leads to a chronic form of the disease, which can evolve into pulmonary fibrosis (6). The current corticosteroid-based therapy does not impact HP progression (7, 8), and fundamental strides are required to broaden the spectrum of targets to stop the immunopathogenic mechanisms that sustain progression of the disease.
Bioaerosols found in environments associated with HP feature multiple components. However, little is known regarding the interplay between T-cell–dependent and TLR (Toll-like receptor) ligands in HP. This is surprising because potent TLR activators, such as endotoxins, are found in environments that are known to favor hypersensitivity lung diseases (9, 10). Moreover, TLR signaling is well recognized as an early immunological event that is essential for effective activation of naive B cells (11, 12). Although TLR signaling is required to mount an efficient antigen-specific B-cell response (13), TLR agonists only reactivate B-cell memory in vitro (14), and not in vivo (15). These findings suggest that environmental TLR exposure contributes to the induction, but not the reactivation, persistence, or amplification, of preestablished HP. Considering the growing body of evidence supporting deleterious interactions among the multiple components of bioaerosols (16, 17), we postulated that addressing the interplay between TLR ligands and HP-causing antigens might reveal novel aspects of HP pathogenesis that could be targeted for therapeutic benefit.
Pharmacological modulators of S1P1 (sphingosine-1-phosphate receptor 1) are believed to reduce inflammation in pathological conditions by inducing lymphocyte sequestration in lymphoid organs (18–20). However, lymphopenia induced by FTY720 (an S1P receptor agonist prodrug) is a poor predictor of clinical responses in patients with relapsing multiple sclerosis, suggesting that alternative mechanisms are in play (21). Our previous study showed that S1P1 agonism inhibited antibody accumulation in experimental HP (22), in contrast to observations in studies of T-cell–dependent antigens that the humoral response was poorly impacted by S1P1 pharmacological modulators (20, 23, 24). Moreover, several lines of evidence argue that S1P1 is involved in the immune response induced by TLR4 ligands (25, 26). Thus, we hypothesized that S1P1 regulates B-cell activation and functions in HP in response to endotoxins.
Accordingly, we investigated the contribution of endotoxins to HP induction and progression, as well as the modulation of B-cell activation in response to the picomolar S1P1 agonist, RP001 (27). We reveal a role for endotoxins in the exacerbation of HP, and show that an S1P1 ligand prevents endotoxin-induced B-cell activation, cytokine release, and B-cell–T-cell cooperation.
Methods
Additional information regarding the methods used in this work is provided in the data supplement.
Preparation of Methanosphaera stadtmanae cultures
Methanosphaera stadtmanae (MSS) cultures (strain #3091; DMSZ) were grown in MCB-3 medium enriched with ruminal fluid (graciously provided by Centre de Recherche en Science Animale de Deschambault, Québec, Canada) under strict anaerobic conditions as previously described (28).
Animals
Pathogen-free female C57Bl/6j and OT-II mice were obtained from The Jackson Laboratory. S1P1-eGFP mice were graciously provided by Hugh Rosen (27) and backcrossed at least six times on the C57Bl/6j background. The animals were kept in a specific pathogen-free environment (Centre de Recherche de l’Institut Universitaire de Cardiologie et de Pneumologie de Québec). Experiments were reviewed and approved by the Animal Care Committee of Université Laval in accordance with the Canadian Council on Animal Care guidelines (approval Nos. 2010-050 and 2018-063).
Experimental HP
Mice were exposed to 50 μl of saline (mock procedure) or to 50 μl of saline containing 100 μg of MSS by intranasal instillation, 3 days a week for 3 weeks, and euthanized at the indicated time points. In some experiments, mice were rechallenged intranasally with 50 μl of saline or 50 μl of saline containing 5 μg of endotoxins (LPS isolated from Escherichia coli O55:B5 [Sigma-Aldrich]).
B-Cell Culture
Negatively isolated splenic B cells (StemCell Technologies) were incubated for 24 hours or 72 hours with LPS 10 μg/ml (Sigma-Aldrich) or a T-cell–dependent mimic constituted of 20 μg/ml of anti-CD40 (BioLegend) and 8 μg/ml Igκ chain (BioLegend) with increasing concentrations of RP001 (Tocris). Cell-free supernatants were stored at −80°C and pellets were resuspended in staining buffer (PBS, 1% FBS, and 0.2% sodium azide) for flow-cytometric analyses.
MSS-Specific IgG1 and Cytokine Quantification
MSS-specific antibodies were quantified by indirect ELISA in the BAL supernatant harvested as described in the data supplement. Plates were coated with 50 μg lyophilized MSS/ml as described previously (29). IgG1s in BAL fluid dilutions were detected using horseradish peroxidase–coupled goat anti-mouse IgG1 (Abcam). ELISAs were also used to quantify TNF (BioLegend) in cell supernatants.
Flow Cytometry
Single-cell suspensions were prepared from tissues harvested from mice whose vasculature was perfused with PBS. Antibody labeling (Table E1 in the online supplement) was conducted for 20 minutes at 4°C in the dark (22). Data were acquired using a FACS Diva–driven customized LSR Fortessa (BD Biosciences) and analyzed with FlowJo software (Tree Star). Fluorescence Minus One controls were used to determine the staining threshold, and experimental condition–matched wild-type mice were used to determine the threshold of S1P1-eGFP fluorescence.
OT-II T-Cell Activation Assay
Ovalbumin peptide–pulsed B cells preincubated with S1P1 modulators were incubated with OT-II T cells for 12 hours. After 8 hours, Brefeldin A (Sigma-Aldrich) was added for intracellular cytokine staining.
Statistical Analyses
Data are presented using the averages ± SEM. Statistical analyses were performed using one-way or two-way ANOVA, when appropriate, with Tukey’s multiple comparison post hoc test. Log transformation of the data was used to homogenize intergroup variances when they were statistically different according to the Brown-Forsythe test. The significance threshold was set to P < 0.05.
Results
A TLR4 Ligand Exacerbates HP Signs
Exposure to archaeal species mimics HP signs in mice (28). However, bioaerosols are composed of numerous microbial components (LPS, peptidoglycan, bacterial DNA, etc.), whose proportions vary in time, and interactions can modify the nature of the immune response (30). In the first series of experiments, we determined the impact of LPS on cardinal signs of experimental HP (Figure 1A). Compared with the mock-saline group, all groups exhibited a significant accumulation of leukocytes in the BAL. LPS challenge in MSS-sensitized mice resulted in the highest accumulation of BAL leukocytes, reaching 13.7 ± 1.5 × 105 total cells (Figure 1B). Neutrophils were absent from the BAL of both mock-saline and MSS-saline mice, but LPS enhanced by 38% the accumulation of neutrophils in the BAL of MSS-sensitized mice (10.6 ± 1.3 × 105 cells) compared with mock-LPS mice (6.6 ± 0.7 × 105 cells; Figure 1C).
Figure 1.
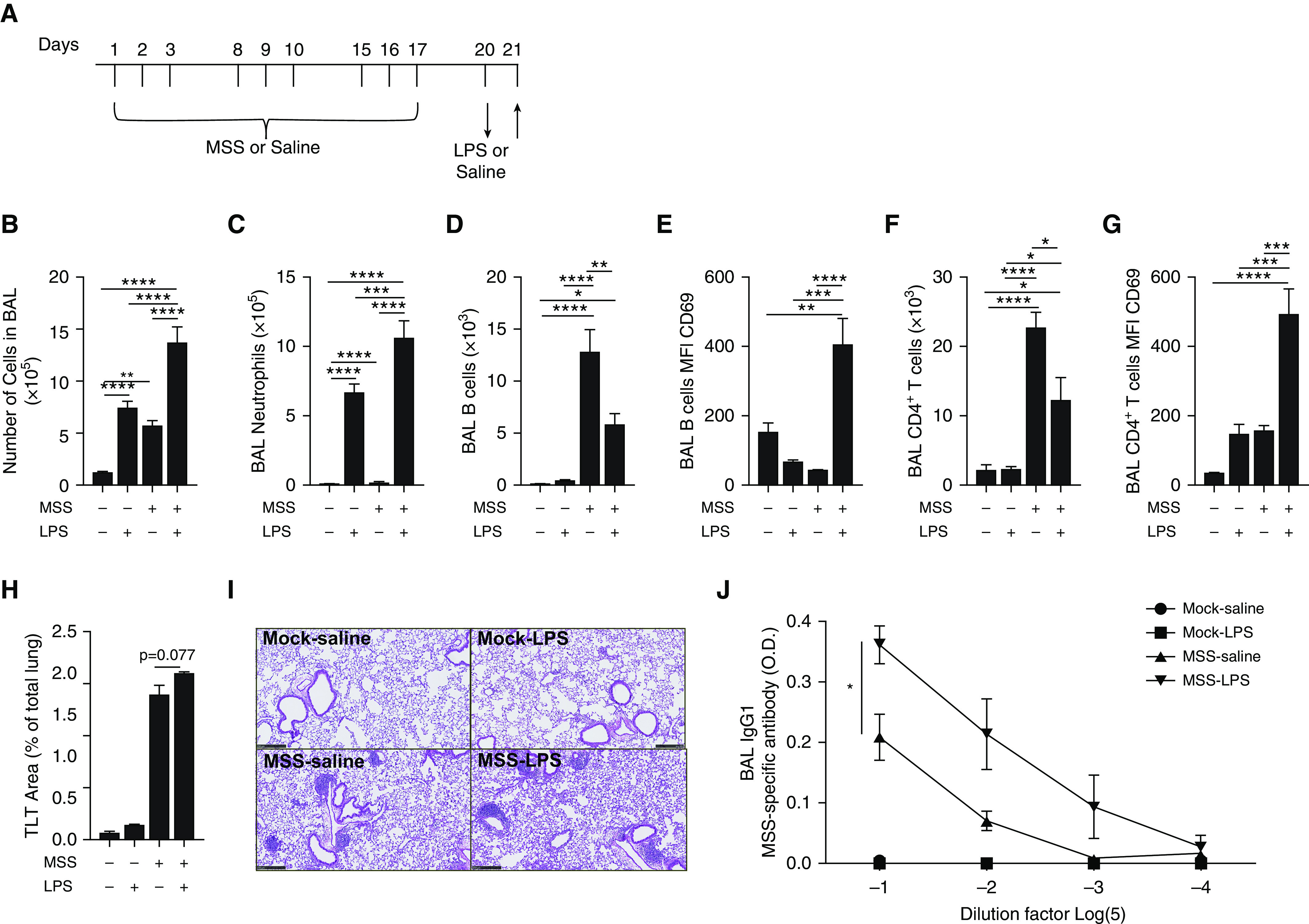
LPS challenge exacerbates preexisting experimental hypersensitivity pneumonitis (HP). (A) Mice were exposed to saline (Mock) or 100 μg of Methanosphaera stadtmanae (MSS) three times a week for 3 weeks (Days 1–3, 8–10, and 15–17) and challenged with saline or 5 μg of LPS (Day 20) 16 hours before euthanasia. Upward arrow indicates the time point when analyses were performed. (B and C) Total cell number (B) and neutrophil number (C) in the BAL were determined with hemocytometer count and differential staining. (D–G) Using flow cytometry, we quantified B cells (D), CD69 median fluorescence intensity (MFI) at the B-cell surface (E), CD4+ T cells (F), and CD69 MFI at the CD4+ T-cell surface (G). (H) The percentage of the lung area occupied by tertiary lymphoid tissues (TLTs) was determined on paraffin-embedded lung slices after hematoxylin and eosin staining. (I) Histology representative of the different experimental groups are shown. Scale bars, 250 μM. (J) MSS-specific IgG1s were quantified by indirect ELISA in BAL supernatant dilutions. Averages ± SEM. n = 6. *P < 0.05, **P < 0.01, ***P < 0.005, and ****P < 0.001. O.D. = optical density.
MSS sensitization induced B-cell accumulation in BAL in both the MSS-saline and MSS-LPS groups (Figure 1D). MSS-LPS mice had lower B-cell numbers in the BAL than MSS-saline mice (Figure 1D). Lower B-cell accumulation in the BAL was associated with a strong increase in the activation and tissue retention marker CD69 in the MSS-sensitized group (34.7 ± 3 in MSS-saline vs. 414.4 ± 70 in MSS-LPS) (Figure 1E). By themselves, neither LPS nor MSS led to an increase of CD69 on B cells found in the BAL (Figure 1E). MSS sensitization also induced CD4+ T-cell accumulation in the BAL of the MSS-saline and MSS-LPS groups (Figure 1F). Similar to the observations made with B cells, the number of CD4+ T cells was reduced in the MSS-LPS group compared with MSS-saline mice (Figure 1F), and CD69 was strongly increased on CD4+ T cells in mice that received both MSS and LPS (Figure 1G). TLTs were rare in mock-saline and mock-LPS mice, but were widespread in MSS-sensitized mice (Figures 1H and 1I). Sixteen hours after the challenges, we noted trends for the TLT area to increase in the MSS-LPS group compared with the MSS-saline group (Figures 1H and 1I). Notably, LPS also increased the MSS-specific IgG1 titers in the BAL of MSS-sensitized mice (Figure 1J).
Mediastinal lymph nodes (mLNs) featured low total cell numbers in the mock-saline and mock-LPS groups (Figure 2A). MSS sensitization induced both B-cell and CD4+ T-cell accumulation in mLNs independently of LPS challenge (Figures 2B and 2D). CD69 remained at baseline on B cells in the mock-LPS and MSS-saline groups, but was increased in the MSS-LPS group (109.4 ± 18 in the MSS-LPS group compared with 34.7 ± 3 in the MSS-saline group) (Figure 2C). In contrast to what was observed for B cells, CD69 was increased on CD4+ T cells in mice exposed to LPS, regardless of MSS sensitization (Figure 2E). Moreover, TLR4-deficient mice displayed reduced inflammation and antigen-specific antibodies in response to LPS-containing MSS (Figure E1). Together, these findings suggest that B-cell responses are modified in the context of HP and involve TLR ligands in the amplification of hypersensitivity.
Figure 2.
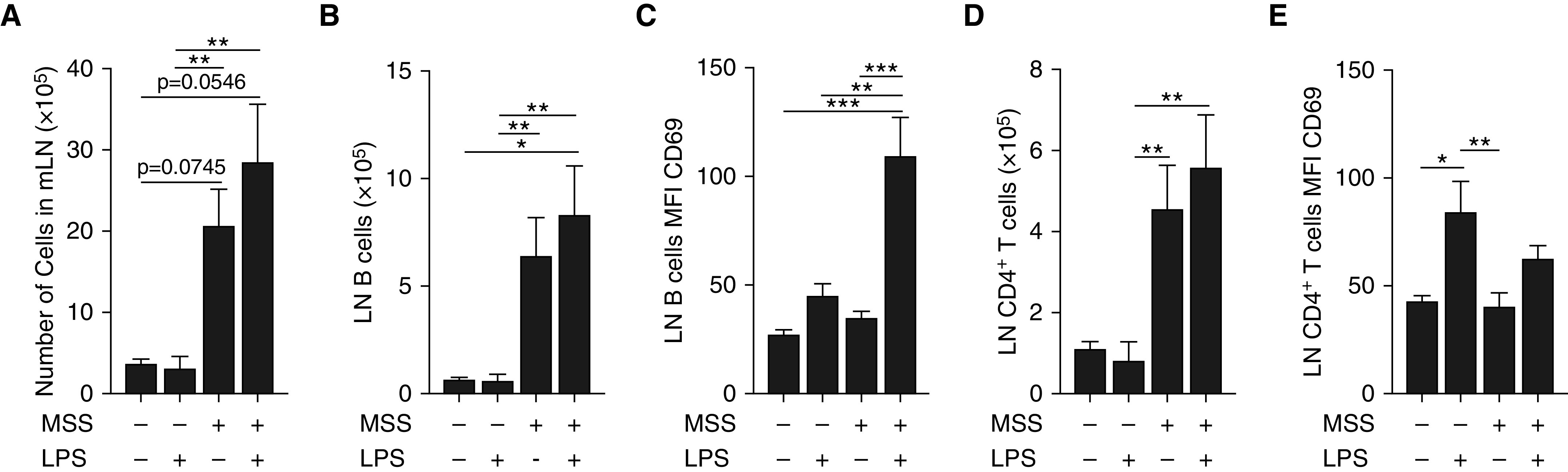
Impact of LPS challenge on lymphocyte subsets in mediastinal lymph nodes (mLNs). Mice were exposed to saline (Mock) or 100 μg of MSS three times a week for 3 weeks (Days 1–3, 8–10, and 15–17) and challenged with saline or 5 μg of LPS (Day 20) 16 hours before euthanasia. (A) Total cell numbers in mLNs were determined by direct cell count with a hemocytometer. (B–E) Using flow cytometry, we quantified B cells (B), CD69 MFI at the B-cell surface (C), CD4+ T cells (D), and CD69 MFI at the CD4+ T-cell surface (E). Averages ± SEM. n = 6. *P < 0.05, **P < 0.01, and ***P < 0.005.
S1P1 Is Increased in B Cells in the Course of Experimental HP
Given that an S1P1 agonist alleviates experimental HP (22) and that S1P1 levels can change in leukocytes under pathological conditions (31), we investigated S1P1 levels in T and B cells during HP development. To monitor this response, we took advantage of an elegant mouse strain, S1P1-eGFP knockin mice, that permits real-time monitoring of S1P1 levels (27). Compared with naive S1P1-eGFP knockin mice, acute exposure to MSS (Days 2 and 4) did not modify S1P1-eGFP fluorescence in CD19+ cells (B cells) and CD90+ cells (T cells) (Figures 3C–3E). However, subchronic exposure to MSS (Day 18) increased both the percentage of S1P1-eGFP+ B cells (from 20% ± 0.8% to 28% ± 1.0%) and the median fluorescence intensity (MFI) of S1P1-eGFP (from 84 ± 6.5 to 101 ± 6.1) (Figures 3C and 3D). S1P1-eGFP fluorescence in B cells returned to baseline 3 weeks after the last MSS exposure (Day 38) (Figures 3C and 3D). In contrast, MSS exposure caused a decrease from baseline in both the percentage of S1P1-eGFP+ T cells (from 49% ± 1.8% to 26% ± 2.2%) and the MFI of S1P1-eGFP in T cells (from 161 ± 10.3 to 42 ± 6.1), which was lowest on Day 24 (Figures 3C and 3E). Upon arrest of MSS exposure, the S1P1-eGFP signal slowly resolved, but failed to return to baseline in T cells, within the course of this study (Figures 3C and 3E).
Figure 3.

S1P1-eGFP (sphingosine-1-phosphate receptor 1–eGFP) is increased on B cells during experimental HP. (A) S1P1-eGFP mice were exposed to 100 μg MSS (Days 1–3, 8–10, and 15–17) and euthanized on Days 2, 4, 18, 24, and 38 (indicated by upward arrows). (B) Gating strategy for CD45+CD90−CD19+ B cells and CD45+CD90+CD19− T cells. (C) Representative flow cytometry scatterplots and histograms of S1P1-eGFP signal in B cells (left columns) and T cells (right columns) in naive S1P1-eGFP mice and on Days 18 and 38 in S1P1-eGFP mice exposed to MSS. In histograms, the gray area represents the signal distribution in wild-type (WT) mice and the black dotted line represents the peak of fluorescence in naive S1P1-eGFP mice. (D and E) The frequency of S1P1-eGFP+ cells and the MFI of S1P1-eGFP were quantified in both B cells (D) and T cells (E). For MFI, baseline fluorescence was removed using values from experimental condition-matched WT mice. Averages ± SEM. n = 3–7 mice per point. *P < 0.05, **P < 0.01, and ****P < 0.001. FSC-A = forward scatter area; SSC-A = side scatter area.
The S1P1 Ligand RP001 Downregulates S1P1 in B Cells during Experimental HP
The S1P1 ligand RP001 can decrease S1P1 levels on lymphocytes under noninflammatory conditions (27). However, its ability to downregulate S1P1 in the course of inflammation and to directly impact B cells remained to be determined. Subchronic exposure to MSS upregulated S1P1 in B cells found in the lung (Figure 3) and the mLNs, but not the spleen (Figures 4A and 4B). Compared with vehicle, RP001 caused a rapid (within 4 h) and generalized downregulation of S1P1 on B cells in the lung, spleen, and mLNs regardless of antigenic exposure (Figures 4A and 4B). To confirm that alteration of S1P1-eGFP levels could result from a direct effect of RP001 on B cells, in vitro experiments were conducted. S1P1 was detected on B cells in vitro until 72 hours in culture, and it remained sensitive to RP001-induced downregulation (Figure 4C). We also found that LPS and a T-cell–dependent mimic (α-CD40/Igκ chain) mitigated S1P1 levels on B cells, and that sensitivity to RP001-induced S1P1 downregulation was retained under these conditions. Thus, S1P1 is found on B cells under inflammatory conditions and RP001 reduces S1P1-eGFP MFI on B cells in vivo and in vitro, preferentially in response to a TLR ligand.
Figure 4.
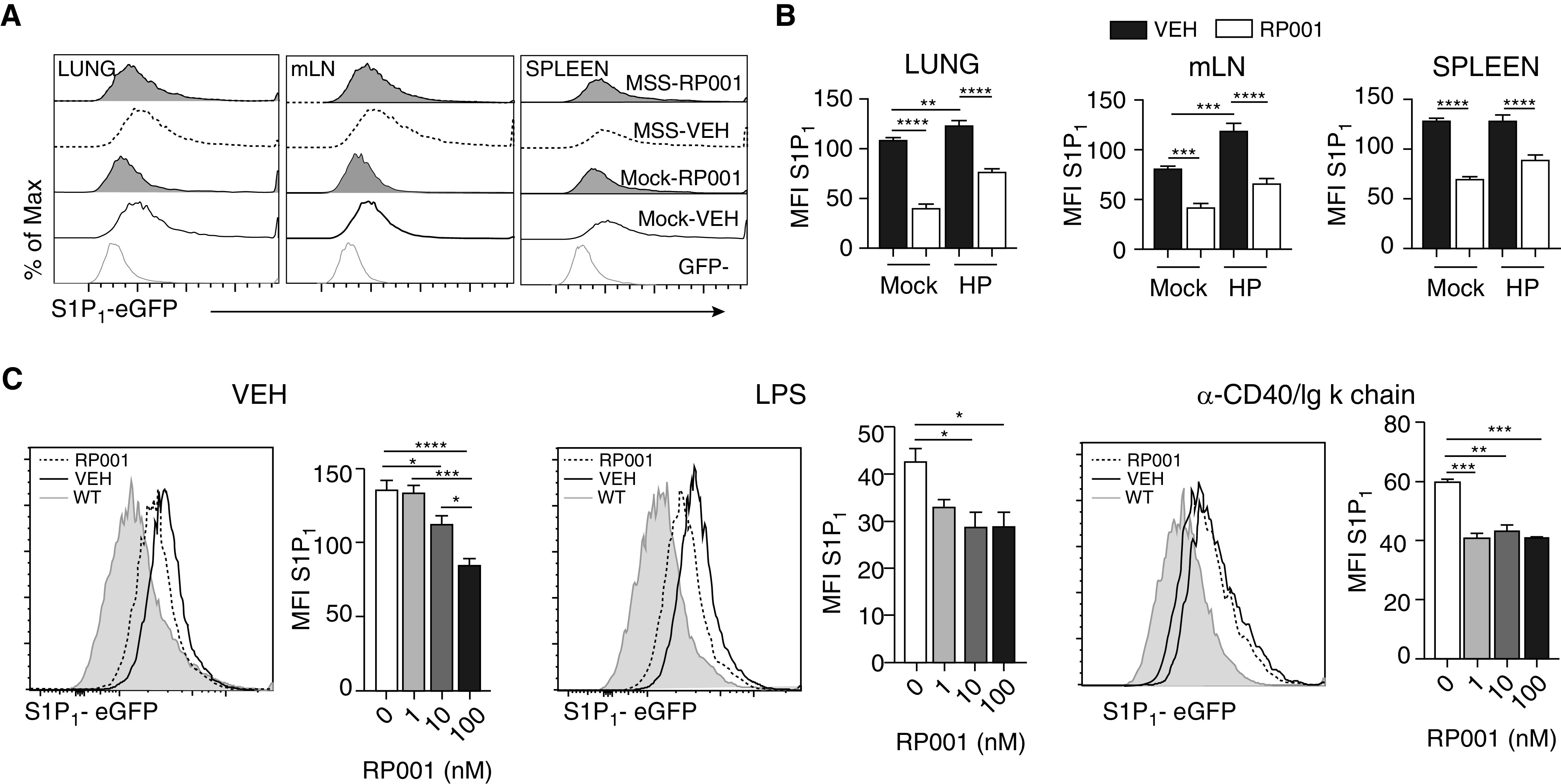
RP001 induces S1P1 downregulation in B cells. WT and S1P1-eGFP mice were exposed to saline (Mock) or MSS 100 μg three times a week for 3 weeks before intratracheal administration of RP001 1 mg/kg. The mice were euthanized 4 hours later. (A) Representative histograms of S1P1 on B cells are shown. (B) Quantification of S1P1-eGFP MFI on B cells from the lung, mLN, and spleen. (C) Isolated splenic B cells from naive S1P1-eGFP mice were incubated with vehicle (VEH), LPS, or an α-CD40/Igκ-chain cocktail for 72 hours, with increased concentrations of RP001. Representative histograms (left panel; 100 nM) and quantification of S1P1-eGFP MFI (right panel) are presented. Averages ± SEM. (A and B) n = 4–5 mice per group. (C) n = 6–12 replicates from two different experiments performed with similar results. For bar graphs, background fluorescence was removed using experimental conditions–matched WT mice. Averages ± SEM. *P < 0.05, **P < 0.01, ***P < 0.005, and ****P < 0.001.
RP001 Inhibits B-Cell Activation and TNF Release in Response to LPS
Considering that RP001 prevents reactivation of the antigen-specific antibody response (22), we next addressed the hypothesis that S1P1 directly regulates B-cell activation and functions in response to a T-cell–dependent stimulus, or in response to a TLR ligand. B-cell stimulation with a T-cell–dependent mimic (α-CD40 and Igκ chain) or LPS increased cell-surface CD86, CD69, CD40, and MHCII compared with the unstimulated cells (Figures 5A–5C). RP001 did not reduce CD86 or MHCII in B cells at baseline (Figure 5A) or activated using α-CD40 and Igκ chain (Figure 5B). However, RP001 inhibited the LPS-induced upregulation of CD86, CD69, CD40, and MHCII in a concentration-dependent manner, which reached maximal inhibition of 36%, 47%, 39%, and 39%, respectively (Figure 5C).
Figure 5.
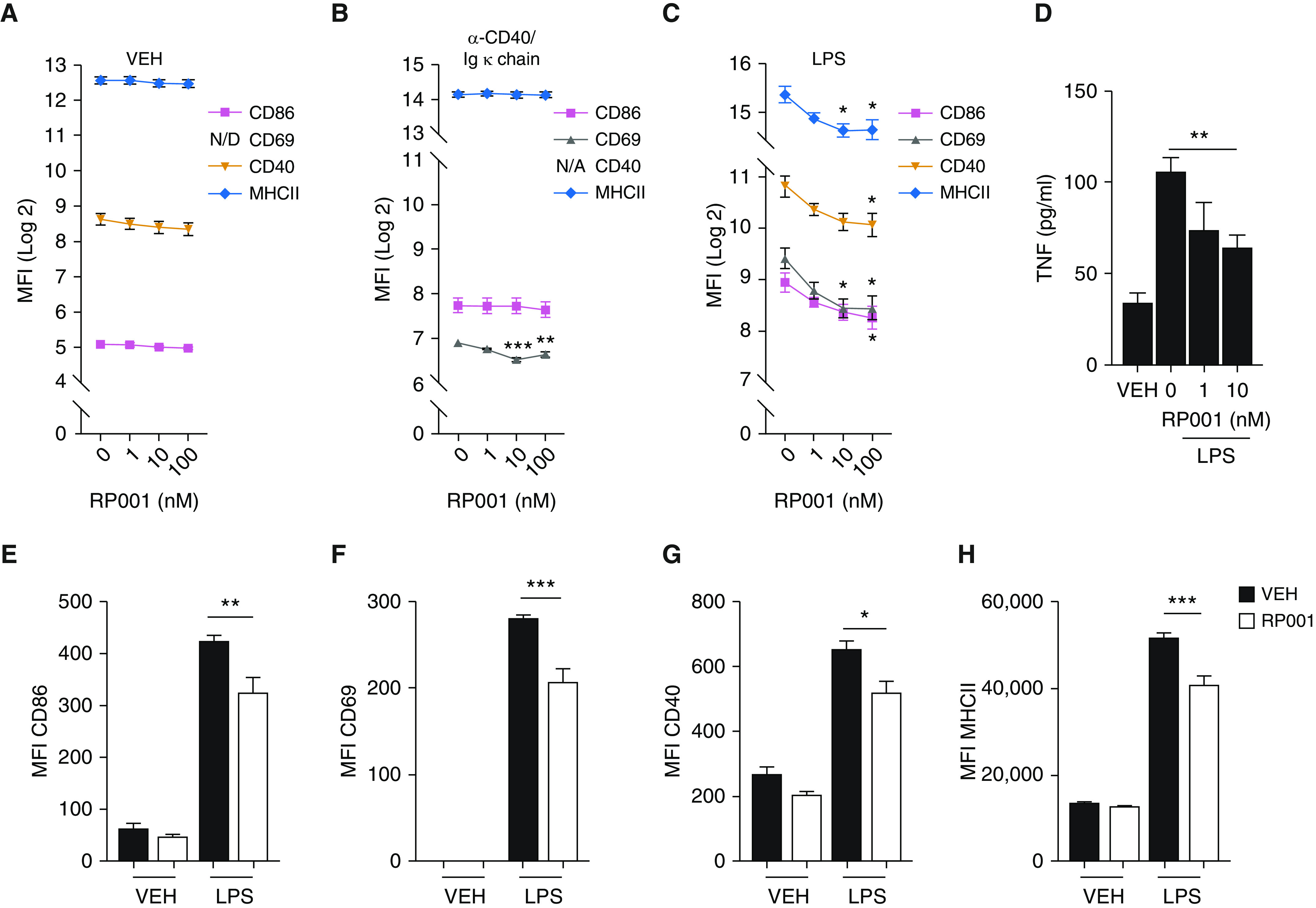
RP001 reduces LPS-induced activation of B cells. (A–C) Splenic B cells were incubated with VEH (A), α-CD40 20 μg/ml and Igκ chain 8 μg/ml (B), or LPS 10 μg/ml (C) in the presence of increasing RP001 concentrations for 24 hours. MFI of CD86, CD69, CD40, and MHCII was determined by flow cytometry. CD69 was below the detection limit in the VEH group, and CD40 was not assessed in the group incubated with the α-CD40/Igκ chain. (D) TNF concentration was quantified by ELISA in culture supernatants after a 72-hour incubation with LPS. (E–H) Pulmonary B cells from MSS-sensitized mice euthanized 4 days after the last MSS exposure (Day 21) were isolated and incubated with LPS 10 μg/ml and RP001 (10 nM), and the MFI of CD86 (E), CD69 (F), CD40 (G), and MHCII (H) was assessed. Averages ± SEM. Shown is one experiment representative of three to five independent experiments performed with similar results. Each point was minimally assessed in triplicate for each individual experiment. *P < 0.05, **P < 0.01, and ***P < 0.005. N/A = not applicable; N/D = not detected.
LPS caused a significant accumulation of TNF in the supernatant of splenic B cells compared with unstimulated cells (Figure 5D). Compared with vehicle, RP001 (10 nM) reduced LPS-induced TNF accumulation (from 106 ± 8 pg/ml to 64 ± 7 pg/ml) (Figure 5D). IL-10, IFN-γ, and IgGs were below the limit of detection (data not shown). We also found that the impact of S1P1 agonism on B-cell activation was not restricted to splenic B cells. Indeed, RP001 prevented LPS-induced upregulation of CD86, CD69, CD40, and MHCII in pulmonary B cells isolated from mice in the aforementioned experimental HP model (Figures 5E–5H), indicating that RP001 likely has direct functional impacts on B cells in the context of HP.
RP001 Blunts LPS-enhanced Cooperation between B Cells and T Cells
B-cell–T-cell cooperation mediated by MHCII and costimulatory molecules amplifies the antigen-specific response (32). To test the concept that RP001 impacts TLR-enhanced B-cell–T-cell cooperation, we used the OT-II model, as no immunodominant peptides or TCR-transgenic mice are available for HP-inducing agents. First, B cells without LPS priming induced a weak antigen-specific activation of T cells compared with LPS-primed B cells (Figure 6). Preincubation of LPS-primed B cells with RP001 decreased by 27% and 23% the frequency of CD69+ (Figure 6B) and TNF+ (Figure 6D) OT-II CD4+ T cells, respectively, compared with preincubation with medium only (vehicle). Both the S1P1 receptor antagonist W146 (10 μM) and the S1P1 agonist that is known to induce a mild degradation of S1P1, SEW2871 (1 μM) (33), failed to dampen B-cell–induced T-cell activation (Figure 6). Thus, RP001 dampens LPS-mediated B-cell–driven T-cell cooperation, resulting in a reduced production of TNF by T cells. This effect appears to relate to its ability to downregulate S1P1.
Figure 6.
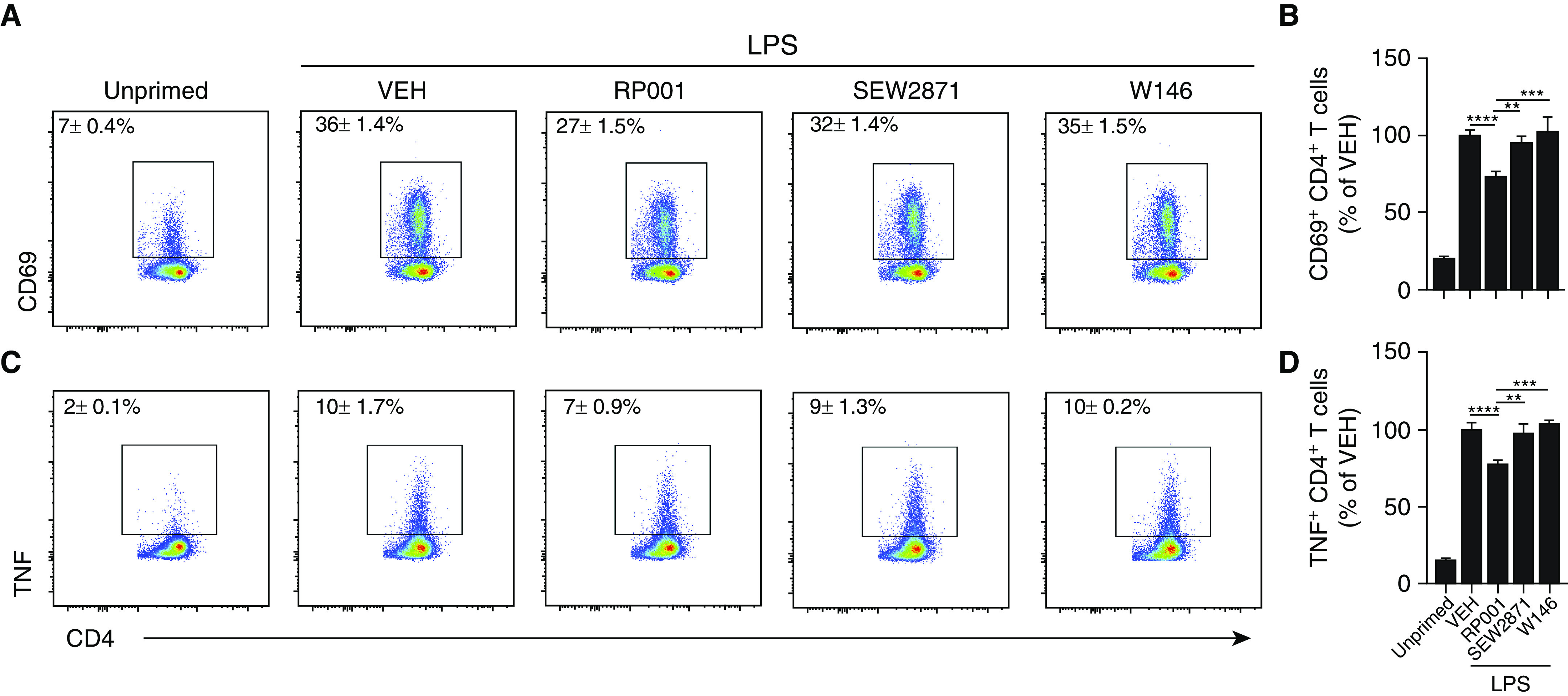
RP001 reduces the capacity of B cells to trigger antigen-specific T-cell activation in vitro. B cells isolated from the spleen of WT C57Bl/6j mice were primed overnight with LPS (10 μg/ml) or left unprimed. LPS-primed B cells were coincubated with VEH, RP001 (100 nM), SEW2871 (1 μM), or W146 (10 μM). Cells were then washed, incubated with ovalbumin-peptide (10 μg/ml) for 1 hour, and coincubated with OT-II CD4+ T cells for 12 hours. (A) Representative scatterplots of CD69+ T cells are presented. (B) Bar graph represents the normalized frequency of CD69+ T cells. (C) Representative scatterplots of TNF+ T cells. (D) Bar graph represents the normalized frequency of TNF+ T cells. Averages ± SEM. n = 3 independent experiments. Each point was minimally assessed in triplicate for each individual experiment. **P < 0.01, ***P < 0.005 and ****P < 0.001.
Discussion
Our study reveals a new role for S1P1 in TLR4-mediated B-cell functions that contribute to HP pathogenesis. We found that LPS-induced neutrophil accumulation in the airways was enhanced in HP mice, which was accompanied by increased anti-MSS IgG1 titers in the BAL. Our previous finding that S1P1 agonism prevented the accumulation of antigen-specific antibodies in HP (22) required a deeper investigation because it contrasted with the lack of impact of S1P receptor modulators on the humoral response in models involving primarily T-cell–dependent antigens (20, 23, 24). Here, we demonstrate that S1P1 modulators can directly impact B-cell activation in response to a TLR ligand, resulting in impaired B-cell–T-cell cooperation ex vivo. This study supports the concept that TLR ligands contribute to shaping the pathophysiological mechanisms underlying pulmonary hypersensitivity, and reveal S1P1 as a putative regulator of B cells upon exposure to complex bioaerosols.
The mechanisms of HP persistence are poorly understood, as evidenced by the fact that none of the current clinical approaches can reverse hypersensitivity or prevent the immunopathological consequences of antigen reencounter (7). Our study suggests that TLR ligands could contribute to sustaining or exacerbating the deleterious response to T-cell–dependent HP-amplifying antigens. Given the nature of the agents classically used to induce experimental HP (mainly Saccharopolyspora rectivirgula), most studies have addressed the involvement of TLR2, TLR6, and TLR9 in HP (34–36). However, causative HP agents are diverse (37) and rarely seen as pure entities. It is thus likely that a diversity of TLR ligands cooperate in the context of HP pathogenesis. For that matter, Thorne and colleagues suggested an involvement of TLR4 in the exacerbation of experimental HP (17). In addition, we found that TLR4 deficiency potently alleviated the antigen-specific response to LPS-containing MSS, suggesting that TLR activators are likely involved at different stages of HP pathogenesis (Figure E1).
Our observation that LPS alone increased MSS-specific antibody titers in the BAL of mice with preestablished HP is surprising given that bolstering of an antigen-specific response is commonly performed by exposure to the specific antigen (28, 38). Although our results do not prove or disprove that LPS directly enhances the release of antigen-specific antibodies by B cells, our observations are compatible with the fact that TLR ligands enhance several mechanisms that contribute to increase MSS-specific antibodies in the airways. In the current context where granulocytic but not lymphocytic accumulation is resolved, it is expected that antigen-specific B cells already in the lung are impacted by TLR4 ligands. Further supporting this contention, we found that LPS tended to induce enlargement of both secondary lymphoid tissues and TLTs, which are privileged sites for amplification of the adaptative response. On a technical note, it is likely that we underestimated the magnitude of TLT reactivation considering the exponential relationship between areas and volumes, and the fact that we perfused the vasculature before harvesting the tissue (39). Nonetheless, we also determined that LPS increased the surface expression of activation and retention markers on B cells. It is also likely that the plasma leakage associated with enhanced neutrophilic inflammation in the HP lung could have contributed to increase the anti-MSS IgG1 titers in the BAL. Altogether, these results suggest that exposure to TLR ligands after antigenic exposure ceases, but before the effector phase is resolved, promotes B-cell–associated mechanisms involved in the exacerbation or persistence of HP.
In vivo profiling of S1P1 on lymphocyte subsets provides additional insights regarding its putative involvement, and potential as a target, in the course of HP. Although we cannot discern their exact location (parenchymal vs. capillary-adhered) (40), we found that B cells retained in the lung after vascular perfusion had increased S1P1 levels during the chronic phase of the model. Intriguingly, and in contrast to previous findings regarding chronic inflammation in the gut (31), we determined that S1P1 was decreased on T cells during the chronic stage of the model, which was driven by a strong decrease of S1P1 in CD4+, but not CD8+, T cells (Figure E2). Although S1P1 downregulation in CD4+ T cells will require further investigation, it is tempting to speculate that the different profiles of S1P1 levels on B cells versus T cells might relate to the accelerating recruitment of activated B cells in parallel with the establishment of a local memory T-cell population in the parenchyma, as S1P1 is downregulated upon transition toward a tissue-homing phenotype (41, 42).
Regardless of these considerations, there is evidence that B cells are key targets in the context of HP, including the facts that mice with impaired B-cell maturation display an alleviated hypersensitivity response to MSS (43) and that anti-CD20 immunotherapy can alleviate HP (44). In the HP lung, B cells outnumber T cells by more than twofold (22). As such, they contribute to HP not only by producing antigen-specific antibodies but also by releasing cytokines (45), among which TNF is critical (46, 47). The current study demonstrates the ability of an S1P1 agonist to inhibit the release of TNF by B cells. We confirmed that the decrease of TNF was not the result of lower B-cell proliferation in response to RP001 (Figure E3). Upon activation, B cells upregulate key costimulatory factors that are critical for B-cell–T-cell cooperation, which leads to a bidirectional signaling that ultimately bolsters the antigen-specific response (32, 48). As a critical point supporting the concept that TLR ligands contribute to shaping immune dysregulation and amenability to therapeutics, we observed that the phenotype of B cells was differentially modulated by LPS in the lung and mLNs of MSS-sensitized mice. Moreover, we found that S1P1 modulators only interfered with TLR4-driven B-cell activation, and had no impact in response to the T-cell–dependent mimic ex vivo (Figure 5). Importantly, RP001 hampered LPS-induced B-cell activation to a degree that is sufficient for functional impairment of B-cell–dependent T-cell activation. Thus, RP001 does not appear to directly impact the BCR (B-cell receptor)-driven activation. Instead, it operates through dampening of TLR-induced B-cell activation, which is compatible with the pathogenesis of HP (36).
Pharmacologically, we observed a strong downregulation of S1P1 in B cells in response to RP001 in vivo, suggesting that receptor internalization and/or degradation are required for immune regulation. This idea was confirmed by the fact that neither SEW2871 (an S1P1 agonist that does not potently induce receptor degradation [33]) nor W146 (an S1P1 antagonist) interfered with B-cell–induced T-cell activation in vitro. Our findings align with the notion that mutations hampering S1P1 internalization leads to enhanced autoimmune inflammation (49). Based on these observations, we conclude that RP001-induced downregulation of S1P1 is required to interfere with HP-promoting events in B cells.
Our results challenge the concept that the role of S1P1 on B cells is limited to regulation of their recirculation. This concept is based on previous observations that S1P receptor agonists sequestered B cells in the LNs, whereas circulating antigen-specific immunoglobulins remained unaffected (20, 23, 24). Although we acknowledge the likely contribution of impaired B-cell accumulation in the lung as a supplementary means of dampening the local antigen-specific antibody release, our in vitro experiments reveal that the peculiar context of HP, where several TLR ligands are in play, renders B cells sensitive to S1P1-derived modulation of their function. Therefore, we suspect that the efficacy of RP001’s impact on B-cell functions depends on the immunological context, and that conditions in which TLR ligands are prevalent might be amenable to S1P1 pharmacological modulation. Of note, we also demonstrate that RP001 does not cause S1P1 degradation on blood endothelial cells of the lung (Figure E4). Functionally, this finding agrees with our previous observation that RP001 dampens vascular leakage into the lung (22), which in turn is supported by the endothelial barrier–enforcing effect of S1P1 (50).
In conclusion, we show that experimental HP modifies the response of B cells to endotoxins, a phenomenon associated with enhanced airway inflammation. B cells isolated from the lung express S1P1 in the chronic phase of the model, and the S1P1 ligand RP001 inhibits LPS-induced release of TNF from B cells, as well as their ability to cooperate with T cells ex vivo. Altogether, our findings implicate S1P1 in B-cell activation and HP amplification by TLR ligands.
Acknowledgments
Acknowledgment
The authors thank Marc Veillette at the Centre de Recherche de l’institut Universitaire de Cardiologie et de Pneumologie de Québec Flow Cytometry Core facility for his technical assistance.
Footnotes
Supported by grants from the Natural Sciences and Engineering Research Council of Canada (402151), the Quebec Respiratory Health Research Network, and the Fonds sur les Maladies Respiratoires J.D. Bégin–P.H. Lavoie. C.-A.H. received a scholarship from the Canadian Institutes of Health Research and P.B.-L. received a postdoctoral scholarship from the Apogee program. M.C.M. holds a Fonds de Recherche du Québec–Santé Junior 1 Research Scholar Award. D.M. held a Fonds de Recherche du Québec–Santé Junior 2 Research Scholar Award.
Author Contributions: Study conception and design: C.-A.H., P.B.-L., C.D., G.D., M.C.M., and D.M. Acquisition of data: C.-A.H., P.B.-L., and E.B. Analysis and interpretation of data: C.-A.H., P.B.L., E.B., K.M.M., M.-R.B., M.C.M., and D.M. Drafting of the manuscript for important intellectual content: C.-A.H., P.B.L., J.-F.L.-J., M.C.M., and D.M. Substantial involvement in revision of the manuscript: C.-A.H., P.B.-L., E.B., J.-F.L.-J., C.D., H.R., G.D., K.M.M., M.-R.B., M.C.M., and D.M.
This article has a data supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1165/rcmb.2019-0339OC on April 14, 2020
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1. Lacasse Y, Girard M, Cormier Y. Recent advances in hypersensitivity pneumonitis. Chest. 2012;142:208–217. doi: 10.1378/chest.11-2479. [DOI] [PubMed] [Google Scholar]
- 2. Suda T, Chida K, Hayakawa H, Imokawa S, Iwata M, Nakamura H, et al. Development of bronchus-associated lymphoid tissue in chronic hypersensitivity pneumonitis. Chest. 1999;115:357–363. doi: 10.1378/chest.115.2.357. [DOI] [PubMed] [Google Scholar]
- 3. Hanak V, Golbin JM, Ryu JH. Causes and presenting features in 85 consecutive patients with hypersensitivity pneumonitis. Mayo Clin Proc. 2007;82:812–816. doi: 10.4065/82.7.812. [DOI] [PubMed] [Google Scholar]
- 4. Fernández Pérez ER, Kong AM, Raimundo K, Koelsch TL, Kulkarni R, Cole AL. Epidemiology of hypersensitivity pneumonitis among an insured population in the United States: a claims-based cohort analysis. Ann Am Thorac Soc. 2018;15:460–469. doi: 10.1513/AnnalsATS.201704-288OC. [DOI] [PubMed] [Google Scholar]
- 5. Walters GI, Mokhlis JM, Moore VC, Robertson AS, Burge GA, Bhomra PS, et al. Characteristics of hypersensitivity pneumonitis diagnosed by interstitial and occupational lung disease multi-disciplinary team consensus. Respir Med. 2019;155:19–25. doi: 10.1016/j.rmed.2019.06.026. [DOI] [PubMed] [Google Scholar]
- 6. Morell F, Villar A, Montero MÁ, Muñoz X, Colby TV, Pipvath S, et al. Chronic hypersensitivity pneumonitis in patients diagnosed with idiopathic pulmonary fibrosis: a prospective case-cohort study. Lancet Respir Med. 2013;1:685–694. doi: 10.1016/S2213-2600(13)70191-7. [DOI] [PubMed] [Google Scholar]
- 7. De Sadeleer LJ, Hermans F, De Dycker E, Yserbyt J, Verschakelen JA, Verbeken EK, et al. Effects of corticosteroid treatment and antigen avoidance in a large hypersensitivity pneumonitis cohort: a single-centre cohort study. J Clin Med. 2018;8:E14. doi: 10.3390/jcm8010014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kokkarinen JI, Tukiainen HO, Terho EO. Effect of corticosteroid treatment on the recovery of pulmonary function in farmer’s lung. Am Rev Respir Dis. 1992;145:3–5. doi: 10.1164/ajrccm/145.1.3. [DOI] [PubMed] [Google Scholar]
- 9. Charavaryamath C, Juneau V, Suri SS, Janardhan KS, Townsend H, Singh B. Role of Toll-like receptor 4 in lung inflammation following exposure to swine barn air. Exp Lung Res. 2008;34:19–35. doi: 10.1080/01902140701807779. [DOI] [PubMed] [Google Scholar]
- 10. Duquenne P, Marchand G, Duchaine C. Measurement of endotoxins in bioaerosols at workplace: a critical review of literature and a standardization issue. Ann Occup Hyg. 2013;57:137–172. doi: 10.1093/annhyg/mes051. [DOI] [PubMed] [Google Scholar]
- 11. Ruprecht CR, Lanzavecchia A. Toll-like receptor stimulation as a third signal required for activation of human naive B cells. Eur J Immunol. 2006;36:810–816. doi: 10.1002/eji.200535744. [DOI] [PubMed] [Google Scholar]
- 12. Jegerlehner A, Maurer P, Bessa J, Hinton HJ, Kopf M, Bachmann MF. TLR9 signaling in B cells determines class switch recombination to IgG2a. J Immunol. 2007;178:2415–2420. doi: 10.4049/jimmunol.178.4.2415. [DOI] [PubMed] [Google Scholar]
- 13. Pasare C, Medzhitov R. Control of B-cell responses by Toll-like receptors. Nature. 2005;438:364–368. doi: 10.1038/nature04267. [DOI] [PubMed] [Google Scholar]
- 14. Bernasconi NL, Traggiai E, Lanzavecchia A. Maintenance of serological memory by polyclonal activation of human memory B cells. Science. 2002;298:2199–2202. doi: 10.1126/science.1076071. [DOI] [PubMed] [Google Scholar]
- 15. Richard K, Pierce SK, Song W. The agonists of TLR4 and 9 are sufficient to activate memory B cells to differentiate into plasma cells in vitro but not in vivo. J Immunol. 2008;181:1746–1752. doi: 10.4049/jimmunol.181.3.1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Boucher M, Blais Lecours P, Létourneau V, Veillette M, Duchaine C, Marsolais D. Organic components of airborne dust influence the magnitude and kinetics of dendritic cell activation. Toxicol In Vitro. 2018;50:391–398. doi: 10.1016/j.tiv.2018.04.011. [DOI] [PubMed] [Google Scholar]
- 17. Thorne PS, Adamcakova-Dodd A, Kelly KM, O’neill ME, Duchaine C. Metalworking fluid with mycobacteria and endotoxin induces hypersensitivity pneumonitis in mice. Am J Respir Crit Care Med. 2006;173:759–768. doi: 10.1164/rccm.200405-627OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Brinkmann V, Chen S, Feng L, Pinschewer D, Nikolova Z, Hof R. FTY720 alters lymphocyte homing and protects allografts without inducing general immunosuppression. Transplant Proc. 2001;33:530–531. doi: 10.1016/s0041-1345(00)02126-6. [DOI] [PubMed] [Google Scholar]
- 19. Kataoka H, Sugahara K, Shimano K, Teshima K, Koyama M, Fukunari A, et al. FTY720, sphingosine 1-phosphate receptor modulator, ameliorates experimental autoimmune encephalomyelitis by inhibition of T cell infiltration. Cell Mol Immunol. 2005;2:439–448. [PubMed] [Google Scholar]
- 20. Pinschewer DD, Ochsenbein AF, Odermatt B, Brinkmann V, Hengartner H, Zinkernagel RM. FTY720 immunosuppression impairs effector T cell peripheral homing without affecting induction, expansion, and memory. J Immunol. 2000;164:5761–5770. doi: 10.4049/jimmunol.164.11.5761. [DOI] [PubMed] [Google Scholar]
- 21. Fragoso YD, Spelman T, Boz C, Alroughani R, Lugaresi A, Vucic S, et al. Lymphopenia and Efficacy of Fingolimod MSBase sub-study Investigators. Lymphocyte count in peripheral blood is not associated with the level of clinical response to treatment with fingolimod. Mult Scler Relat Disord. 2018;19:105–108. doi: 10.1016/j.msard.2017.11.018. [DOI] [PubMed] [Google Scholar]
- 22. Huppé CA, Blais Lecours P, Lechasseur A, Gendron DR, Lemay AM, Bissonnette EY, et al. A sphingosine-1-phosphate receptor 1 agonist inhibits tertiary lymphoid tissue reactivation and hypersensitivity in the lung. Mucosal Immunol. 2018;11:112–119. doi: 10.1038/mi.2017.37. [DOI] [PubMed] [Google Scholar]
- 23. Mehling M, Hilbert P, Fritz S, Durovic B, Eichin D, Gasser O, et al. Antigen-specific adaptive immune responses in fingolimod-treated multiple sclerosis patients. Ann Neurol. 2011;69:408–413. doi: 10.1002/ana.22352. [DOI] [PubMed] [Google Scholar]
- 24. Koch M, Poehnert D, Nashan B. Effects of FTY720 on peripheral blood lymphocytes and graft infiltrating cells in a rat model of chronic renal allograft rejection. Transpl Immunol. 2016;35:12–17. doi: 10.1016/j.trim.2016.01.003. [DOI] [PubMed] [Google Scholar]
- 25. Roviezzo F, Sorrentino R, Terlizzi M, Riemma MA, Iacono VM, Rossi A, et al. Toll-like receptor 4 is essential for the expression of sphingosine-1-phosphate-dependent asthma-like disease in mice. Front Immunol. 2017;8:1336. doi: 10.3389/fimmu.2017.01336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kolahdooz Z, Nasoohi S, Asle-Rousta M, Ahmadiani A, Dargahi L. Sphingosin-1-phosphate receptor 1: a potential target to inhibit neuroinflammation and restore the sphingosin-1-phosphate metabolism. Can J Neurol Sci. 2015;42:195–202. doi: 10.1017/cjn.2015.19. [DOI] [PubMed] [Google Scholar]
- 27. Cahalan SM, Gonzalez-Cabrera PJ, Sarkisyan G, Nguyen N, Schaeffer MT, Huang L, et al. Actions of a picomolar short-acting S1P1 agonist in S1P1-eGFP knock-in mice. Nat Chem Biol. 2011;7:254–256. doi: 10.1038/nchembio.547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Blais Lecours P, Duchaine C, Taillefer M, Tremblay C, Veillette M, Cormier Y, et al. Immunogenic properties of archaeal species found in bioaerosols. PLoS One. 2011;6:e23326. doi: 10.1371/journal.pone.0023326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cayer MP, Veillette M, Pageau P, Hamelin R, Bergeron MJ, Mériaux A, et al. Identification of mycobacteria in peat moss processing plants: application of molecular biology approaches. Can J Microbiol. 2007;53:92–99. doi: 10.1139/w06-105. [DOI] [PubMed] [Google Scholar]
- 30.Delort A-M, Amato P. Microbiology of aerosols. Hoboken, NJ: John Wiley & Sons; 2018. [Google Scholar]
- 31. Karuppuchamy T, Behrens EH, González-Cabrera P, Sarkisyan G, Gima L, Boyer JD, et al. Sphingosine-1-phosphate receptor-1 (S1P1) is expressed by lymphocytes, dendritic cells, and endothelium and modulated during inflammatory bowel disease. Mucosal Immunol. 2017;10:162–171. doi: 10.1038/mi.2016.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Duchez S, Rodrigues M, Bertrand F, Valitutti S. Reciprocal polarization of T and B cells at the immunological synapse. J Immunol. 2011;187:4571–4580. doi: 10.4049/jimmunol.1100600. [DOI] [PubMed] [Google Scholar]
- 33. Gonzalez-Cabrera PJ, Hla T, Rosen H. Mapping pathways downstream of sphingosine 1-phosphate subtype 1 by differential chemical perturbation and proteomics. J Biol Chem. 2007;282:7254–7264. doi: 10.1074/jbc.M610581200. [DOI] [PubMed] [Google Scholar]
- 34. Andrews K, Abdelsamed H, Yi AK, Miller MA, Fitzpatrick EA. TLR2 regulates neutrophil recruitment and cytokine production with minor contributions from TLR9 during hypersensitivity pneumonitis. PLoS One. 2013;8:e73143. doi: 10.1371/journal.pone.0073143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Fong DJ, Hogaboam CM, Matsuno Y, Akira S, Uematsu S, Joshi AD. Toll-like receptor 6 drives interleukin-17A expression during experimental hypersensitivity pneumonitis. Immunology. 2010;130:125–136. doi: 10.1111/j.1365-2567.2009.03219.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Vasakova M, Selman M, Morell F, Sterclova M, Molina-Molina M, Raghu G. Hypersensitivity pneumonitis: current concepts of pathogenesis and potential targets for treatment. Am J Respir Crit Care Med. 2019;200:301–308. doi: 10.1164/rccm.201903-0541PP. [DOI] [PubMed] [Google Scholar]
- 37. Riario Sforza GG, Marinou A. Hypersensitivity pneumonitis: a complex lung disease. Clin Mol Allergy. 2017;15:6. doi: 10.1186/s12948-017-0062-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Waserman S, Olivenstein R, Renzi P, Xu LJ, Martin JG. The relationship between late asthmatic responses and antigen-specific immunoglobulin. J Allergy Clin Immunol. 1992;90:661–669. doi: 10.1016/0091-6749(92)90140-w. [DOI] [PubMed] [Google Scholar]
- 39. Werahera PN, Miller GJ, Taylor GD, Brubaker T, Daneshgari F, Crawford EDA. A 3-D reconstruction algorithm for interpolation and extrapolation of planar cross sectional data. IEEE Trans Med Imaging. 1995;14:765–771. doi: 10.1109/42.476120. [DOI] [PubMed] [Google Scholar]
- 40. Anderson KG, Sung H, Skon CN, Lefrancois L, Deisinger A, Vezys V, et al. Cutting edge: intravascular staining redefines lung CD8 T cell responses. J Immunol. 2012;189:2702–2706. doi: 10.4049/jimmunol.1201682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Shiow LR, Rosen DB, Brdicková N, Xu Y, An J, Lanier LL, et al. CD69 acts downstream of interferon-alpha/beta to inhibit S1P1 and lymphocyte egress from lymphoid organs. Nature. 2006;440:540–544. doi: 10.1038/nature04606. [DOI] [PubMed] [Google Scholar]
- 42. Feng C, Woodside KJ, Vance BA, El-Khoury D, Canelles M, Lee J, et al. A potential role for CD69 in thymocyte emigration. Int Immunol. 2002;14:535–544. doi: 10.1093/intimm/dxf020. [DOI] [PubMed] [Google Scholar]
- 43. Carter S, Miard S, Caron A, Sallé-Lefort S, St-Pierre P, Anhê FF, et al. Loss of OcaB prevents age-induced fat accretion and insulin resistance by altering B-lymphocyte transition and promoting energy expenditure. Diabetes. 2018;67:1285–1296. doi: 10.2337/db17-0558. [DOI] [PubMed] [Google Scholar]
- 44. Lota HK, Keir GJ, Hansell DM, Nicholson AG, Maher TM, Wells AU, et al. Novel use of rituximab in hypersensitivity pneumonitis refractory to conventional treatment. Thorax. 2013;68:780–781. doi: 10.1136/thoraxjnl-2013-203265. [DOI] [PubMed] [Google Scholar]
- 45. Vazquez MI, Catalan-Dibene J, Zlotnik A. B cells responses and cytokine production are regulated by their immune microenvironment. Cytokine. 2015;74:318–326. doi: 10.1016/j.cyto.2015.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Falfán-Valencia R, Camarena A, Pineda CL, Montaño M, Juárez A, Buendía-Roldán I, et al. Genetic susceptibility to multicase hypersensitivity pneumonitis is associated with the TNF-238 GG genotype of the promoter region and HLA-DRB1*04 bearing HLA haplotypes. Respir Med. 2014;108:211–217. doi: 10.1016/j.rmed.2013.11.004. [DOI] [PubMed] [Google Scholar]
- 47. Schaaf BM, Seitzer U, Pravica V, Aries SP, Zabel P. Tumor necrosis factor-alpha -308 promoter gene polymorphism and increased tumor necrosis factor serum bioactivity in farmer’s lung patients. Am J Respir Crit Care Med. 2001;163:379–382. doi: 10.1164/ajrccm.163.2.2002062. [DOI] [PubMed] [Google Scholar]
- 48. Constant SL. B lymphocytes as antigen-presenting cells for CD4+ T cell priming in vivo. J Immunol. 1999;162:5695–5703. [PubMed] [Google Scholar]
- 49. Garris CS, Wu L, Acharya S, Arac A, Blaho VA, Huang Y, et al. Defective sphingosine 1-phosphate receptor 1 (S1P1) phosphorylation exacerbates TH17-mediated autoimmune neuroinflammation. Nat Immunol. 2013;14:1166–1172. doi: 10.1038/ni.2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sanna MG, Wang SK, Gonzalez-Cabrera PJ, Don A, Marsolais D, Matheu MP, et al. Enhancement of capillary leakage and restoration of lymphocyte egress by a chiral S1P1 antagonist in vivo. Nat Chem Biol. 2006;2:434–441. doi: 10.1038/nchembio804. [DOI] [PubMed] [Google Scholar]