

Abstract
The regenerative capacity of the liver plays a protective role against hepatotoxins and impaired regeneration exacerbates liver dysfunction in non-alcoholic fatty liver disease (NAFLD). Mitochondrial bioenergetic and biosynthetic functions are important contributory factors in hepatic regeneration and the control of mitochondrial protein acetylation is implicated in the mitochondrial susceptibility to liver stressors. Here, we evaluated the role how GCN5L1, a mediator of mitochondrial metabolism and acetylation, in modulating murine liver regeneration in response to acute CCl4-induced hepatotoxicity. Initial metabolomic screening found that liver GCN5L1 knockout (LKO) mice have augmented glutaminolysis. The absence of GCN5L1 modified the enzyme activity of the liver enriched glutaminase enzyme (GLS2) and GCN5L1 levels modulated GLS2 oligomerization and acetylation. This metabolic remodeling resulted in the elevation of α-ketoglutarate levels, which are known to activate mTORC1. This signaling pathway was induced with increased phosphorylation of S-6-kinase in LKO hepatocytes and inhibition of glutaminolysis reversed aberrant mTORC1 signaling. At the same time, glutaminolysis, the activity of GLS2 and activation of mTORC1 signaling were reversed by the genetic reintroduction of the mitochondrial isoform of GCN5L1 into LKO primary hepatocytes. Finally, LKO mice had a more robust regenerative capacity in response to CCl4 hepatoxicity and this response was blunted by both the mTORC1 inhibitor rapamycin and by pharmacologic blunting of glutaminolysis.
Conclusion:
These data point to a central role of glutaminolysis in modulating the regenerative capacity in liver. Furthermore, inhibition of mitochondrial GCN5L1 to augment liver regeneration may be a useful strategy in disease states linked to hepatotoxicity.
The liver plays a central role in filtration, biosynthesis, excretion and metabolism. To underlie its critical role in the defense against toxins, the liver has a remarkable regenerative capacity in response to acute/subacute hepatotoxin exposure.(1) Of all the liver cells, hepatocytes are the major cell type to proliferate during liver regeneration, and to orchestrate this replicative capacity hepatocytes must augment their metabolic output to match proliferative demand.(2) Hence, it follows that liver regeneration is paired with enhanced hepatic mitochondrial oxidative phosphorylation to support this energetic demand.(3) At the same time, mitochondria also function as biosynthetic organelles where metabolic intermediates contribute to nucleotide biosynthesis, macromolecular synthesis and redox control. This role of mitochondrial metabolism in biosynthesis is most well established in cancer, where the oncogenic induction of glutaminolysis plays a central role in promoting cellular growth and proliferation.(4)
The regulation of mitochondrial function to sustain energetic and biosynthetic function in response to varying demands is complex and regulated at numerous levels. Interestingly, one such level of regulation is via post-translational modification by acetylation of lysine residues of mitochondrial proteins. These acetyl modifications alter mitochondrial protein and organelle function.(5–7) In the context of hepatotoxicity, this control node has been found to be operational where the levels and function of the mitochondrial deacetylase Sirt3 plays an important role in modulating susceptibility to hepatotoxic injury.(8–11) However, the link between mitochondrial acetylation and metabolic reprogramming during liver regeneration is less well understood.
Our laboratory has focused on the study of the GCN5L1 protein, which modulates hepatic mitochondrial protein acetylation.(12) In hepatocyte specific GCN5L1 knockout mice (LKO) we have found that hepatic lipid accumulation was diminished in response to high fat feeding and that these mice showed increased hepatocyte mitochondrial protein acetylation and fatty acid oxidation, and reduced gluconeogenesis.(13, 14) Given these phenotypes, we proposed that this model may uncover insights into the role of mitochondrial biology and acetylation-mediated remodeling during hepatoxic stress-induced hepatic regeneration.
As background, GCN5L1 has been found to reside in mitochondria and in the cytosol,(12) and it was initially identified as a putative component of a mitochondrial acetyltransferase, although this protein itself does not contain an acetyltransferase catalytic domain.(15) Moreover, no mitochondrial acetyltransferase has been identified to interact with GCN5L1.(12) Although, it is interesting that mitochondrial linked GCN5L1 is dependent on an acetyl-CoA generating enzyme, to acetylate a lysine residue on a molecular motor protein, kinesin binding protein.(16) In contrast, the cytosolic GCN5L1 has been found to bind to a canonical acetyltransferase and modulate microtubular acetylation.(17) The mitochondrial phenotype following the disruption of GCN5L1 suggests that this protein plays an integral role in fatty acid and glucose metabolism,(13, 14) in mitochondrial turnover(16, 18, 19) and in retrograde signaling from the mitochondria to nucleus for the regulation of gluconeogenesis.(14)
To better explore the mitochondrial regulatory effects of GCN5L1 and its effect on hepatic regeneration we initially employed an unbiased metabolomics approach to uncover the metabolic signatures linked to GCN5L1 depletion in primary hepatocytes extracted from littermate-control compared to LKO mice.(14) The metabolomic data implicated that the glutaminolysis pathway was induced in LKO liver. Given that glutaminolysis is a metabolic signature of proliferating cells, we hypothesized that GCN5L1 may regulate glutaminolysis to modulate the liver’s regenerative capacity. To explore this further we showed that the mitochondrial GCN5L1 regulated the activity of the liver-type glutaminase (GLS2) and that the absence of GCN5L1 resulted in GLS2 activation with the induction of glutaminolysis. This metabolic remodeling was found to activate the mammalian target of rapamycin (mTORC1), which in turn was linked to an enhanced hepatic regenerative response to the acute hepatoxicity of carbon tetrachloride (CCl4)
Materials and methods
Mouse studies
All animal protocols were in accordance with Institutional Guidelines and approved by the Institutional Animal Care and Use Committee of the NHLBI of the National Institutes of Health, USA. The mice were maintained on a 12-hour light/dark cycle and housed with free access to water and normal chow diet. GCN5L1 liver knockout mice (LKO) have been described previously(14) and compared to littermate controls.
For CCl4-induced acute liver injury/regeneration, male mice were injected with a single intraperitoneal (IP) dose of CCl4/olive oil (1:5, v/v) mixture (1 ml/kg body weight) or vehicle (olive oil). In selected experiments mTORC1 activity was inhibited by rapamycin and glutaminolysis was inhibited using epigallocatechin gallate (EGCG). Six hours following CCl4/vehicle injection and 24h. thereafter mice were administered rapamycin IP at 5mg/kg or EGCG IP at 50mg/kg. Forty-eight hours post-CCl4, mice were euthanized, livers were extracted and weighed and preserved in liquid nitrogen for further analysis. For CCl4-induced liver regeneration in GCN5L1 overexpression mice, C57BL/6 mice received a tail vein injection of adenovirus harboring either GFP or mitochondrial restricted GCN5L1 (MtG). Seven days post adenoviruses injection, mice were exposed to CCl4 IP and liver regeneration was assayed 48h. later.
Statistical analysis
Data are presented as the mean ± standard errors of the mean (s.e.m.). Statistical analysis was conducted using unpaired two‐tailed t‐test, one‐way or two‐way analysis of variance (ANOVA), followed by Bonferroni’s post hoc tests with GraphPad Prism 5.0. P<0.05 was considered statistically significant.
Results
GCN5L1 levels modulate hepatic metabolic programing
To obtain an overview of basal metabolism in the absence of hepatic GCN5L1 we employed metabolomics to quantify steady-state levels of sugars, amino acids and nucleotides by capillary electrophoresis mass spectroscopy (CE-MS) comparing littermate-control and LKO primary hepatocytes. The major differences noted were a marked reduction in glycolytic intermediates, increased lactate levels and accumulation of tricarboxylic acid cycle (TCA) intermediates. This evidence suggesting increased glycolysis was compatible with our prior finding of reduced flux through the counter-regulatory gluconeogenesis pathway, although we did not pursue this further.(14) The increase in TCA cycle intermediates suggested there was either impaired TCA flux or that excess anaplerosis was saturating the pathway (Figures 1A and 1B) in LKO hepatocytes. To begin to explore the potential effects on the TCA cycle we indirectly measured the TCA cycle activity by measuring oxygen consumption in response to the administration to TCA and electron transfer chain (ETC) metabolic intermediates, i.e. pyruvate and malate, respectively. We found no differences in oxygen consumption (supplemental Fig1A) suggesting that the TCA and ETC were not disrupted by GCN5L1 levels in primary hepatocytes. Hence, to explore this metabolic remodeling in further detail we began to measure anaplerotic flux by assaying glutamine metabolism by U-13C glutamine incubation. In keeping with the steady-state metabolite levels, 13C-incorporation was enriched into TCA intermediates downstream of isocitrate, namely alpha-ketoglutarate (α-KG), succinate, fumarate and malate in the LKO hepatocytes supporting that glutamine-fueled anaplerosis may be enhanced by GCN5L1 depletion (Fig 1C and supplemental Fig 1B). Furthermore, intracellular glutamine levels were increased in LKO, suggesting glutamine uptake may be upregulated in LKO (Fig 1D). To test this further endogenous level of α-KG were measured. α−KG levels were increased in LKO (Fig 1E), being compatible with GCN5L1-depletion driven glutaminolysis.
FIG. 1.
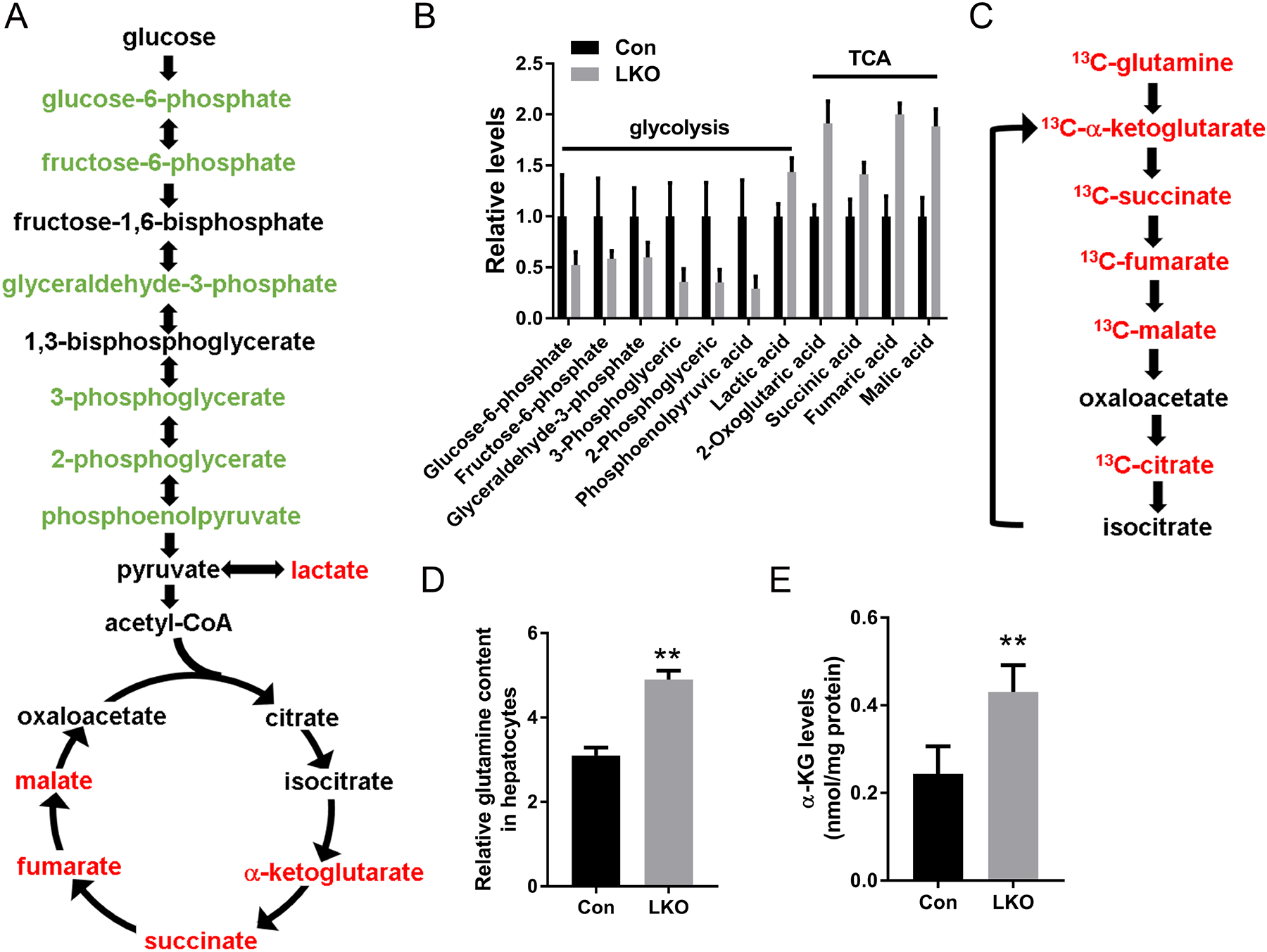
Metabolic profiles of GCN5L1 LKO primary hepatocytes reflect increases in glycolysis and glutaminolysis. (A) Schematic illustrating metabolites that are increased (red) or decreased (blue) in GCN5L1 LKO hepatocytes compared to flox/flox hepatocytes (n = 3; p < 0.05). (B) Relative levels of glycolytic and TCA cycle intermediates from metabolomic analysis in primary hepatocytes. (C) Schematic illustrating the 13C incorporated metabolites that are increased (red) in GCN5L1 LKO compared to flox/flox in primary hepatocytes isolated from 8 weeks mice after plating for 6 h on collagen-coated plates and incubation with 4 mM [U-13C] glutamine. (D) Glutamine content in primary hepatocytes of GCN5L1 LKO and control. Data is from metabolomic analysis (n=3). (E) Intracellular α-KG levels in primary hepatocytes from GCN5L1 LKO and control (n=5). Values are expressed as mean ± s.e.m. **p < 0.01 versus respective control groups by unpaired Student’s t-test.
GCN5L1 LKO enhanced glutamine metabolism by increasing glutamine transporter transcript and glutaminase 2 activity
We next investigated the molecular mechanism whereby GCN5L1 regulated glutamine metabolism by measuring the expression of the glutamine transporter SLC1A5 (also known as ASCT2) and the expression and activity of the enzymes that catabolize glutamine. Upon entry into the cell, glutamine is converted by the mitochondrial enriched enzyme GLS2 into glutamate, which is the rate limiting enzyme in glutaminolysis. Glutamate is further catabolized to α-KG by either glutamate dehydrogenase (GLUD or GDH) or two aminotransferases which are the mitochondrial form of glutamate pyruvate transferase (GPT2) and glutamate OAA transferase (GOT2). The α-KG then enters the TCA cycle to generate biosynthetic building block intermediates or reducing equivalents for oxidative phosphorylation. We firstly measured Slc1a5 transcripts in primary hepatocytes and liver and found that glutamine transporter levels were increased in GCN5L1 LKO primary hepatocytes and liver tissues (Fig 2A). We then measured the expression of glutaminolysis encoding enzymes and enzyme activity. Transcript level of GLS2 was unchanged between groups (Fig 2B). To exclude cytosolic aminotransferase contamination, we isolated mitochondria from LKO and control livers and measured enzyme activities in mitochondrial lysate. GLS2 activity was significantly increased in LKO hepatocytes and liver tissues (Fig 2C and supplemental Fig 2), conversely, GDH, GPT and GOT activities were not different (Fig 2D, 2E and 2F). To evaluate whether this change in activity was due to the levels of GLS2, immunoblot analysis was performed. However, the steady-state levels of GLS2 protein were not different between the groups (Fig 2G), suggesting that the change in GLS2 activity is not regulated at the translational level.
FIG. 2.
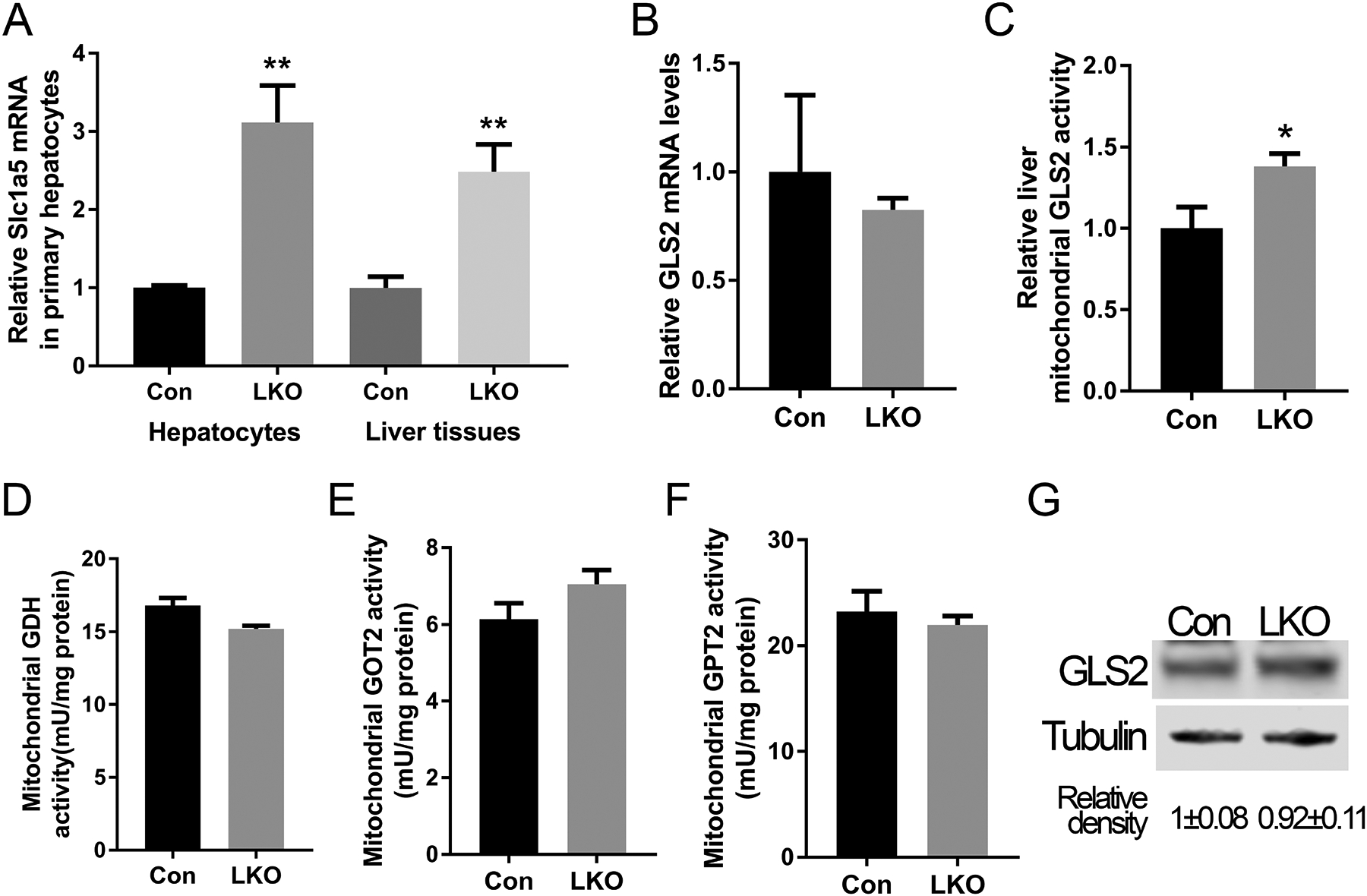
GCN5L1 LKO enhances glutamine metabolism by increasing glutamine transporter transcript levels and glutaminase 2 activity. (A) The transcript levels of glutamine transporter Slc1a5 in control and GCN5L1 LKO primary hepatocytes and liver tissues (n = 5). (B) The transcript levels of GLS2in control and GCN5L1 LKO liver tissues from 8 weeks male mice (n = 5). (C) Relative GLS2 activity in mitochondrial lysates of GCN5L1 LKO and control livers (n=5). Enzyme activities of GDH (D), GOT2 (E) and GPT2 (F) in mitochondrial lysates from GCN5L1 LKO and control livers (n=5). (G) GLS2 protein levels in GCN5L1 LKO and control primary hepatocytes. Values are expressed as mean ± s.e.m. **P < 0.01 versus respective control groups by unpaired Student’s t-test.
GCN5L1 disrupted GLS2 oligomerization and activity
The kidney-type glutaminase (GLS1) and GLS2 isoforms are encoded by different chromosomes and exhibit distinct kinetic activities.(20) Concurrently characterization of GLS1 has been more extensively studied showing that GLS1 activity is governed, in part, by enzyme oligomerization and that GLS1 acetylation modifies this oligomerization.(21) In contrast, delineation and functional characterization of GLS2 are limited. However, data suggests that GLS2 has a compromised ability to assemble into a catalytically active supra-tetrameric complexes compared to the splice variant of GLS1 designated as glutaminase C.(22) Given that GCN5L1 has been shown to modify mitochondrial protein acetylation levels, we explored whether the oligomerization and activity of overexpressed HA-tagged GLS2 were GCN5L1-level dependent. In an in-vitro assay of HA-GLS2 extracted from cells in the presence or absence of GCN5L1, cross-linked HA-GLS2 generated higher molecular weight species in the absence of GCN5L1, with reduced oligomers in HA-GLS2 extracted from GCN5L1 overexpressing cells (Fig 3A). Moreover, purified GSL2 activity was decreased when co-expressed with GCN5L1 comparing with vector controls (Fig 3B). Furthermore, we overexpressed HA-GLS2 and His-GCN5L1 in 293T cells and found that GLS2 interacted with His-GCN5L1 (Fig 3C and 3D). Considering that GCN5L1 regulate mitochondrial protein acetylation, and protein acetylation is a common post translational modification that modifies enzyme activities, we employed IP-MS to assess GLS2 acetylation in mouse livers. Total acetylated peptides were pulled down by anti-acetylated lysine antibody from liver mitochondrial lysates from GFP or MtG overexpression mice, followed by MS analysis. We found acetylated K-279 was increased by approximately 30% following MtG overexpression (Fig 3E). Interestingly based on the homologous site in the human GLS1, K-279 localizes to the putative catalytic domain of GLS2.(23) To explore this in more detail, we employed mutagenesis studies to replace K-residues on GLS2 with a glutamine, to mimic acetylation.(24) The site chosen included the sites identified in our LCMS studies i.e. K-279 and K-284 which reside in the putative catalytic domain and K-329 which resides in the putative oligomerization domain. Interestingly, compared to the WT control or acetyl mimics of K-284 and K-329, the acetylation mimic of K-279 had the greatest effect on blunting GLS2 activity in primary hepatocytes (Fig 3F). These data support that the acetylation level of GLS2 K-279 plays an important role in the modulation of GLS2 activity.
FIG. 3.
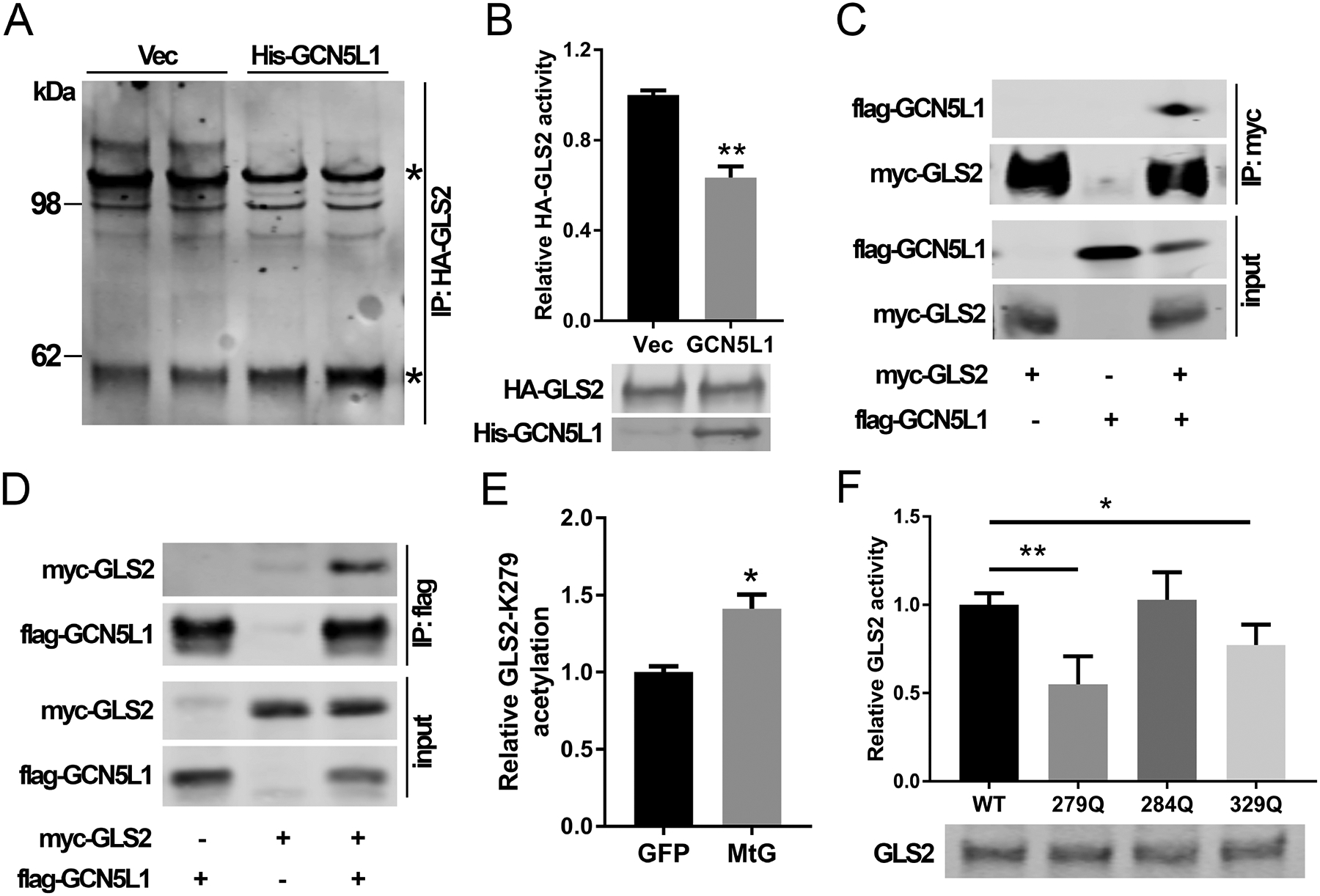
GCN5L1 disrupted GLS2 oligomerization and activity. (A) HA-GLS2 was purified with/without GCN5L1 from 293T cells by pull-down assay using an HA antibody and elution of HA peptides. HA-GLS2 was cross-linked with DSP. Stars indicate GLS2 monomer (~60kDa) and higher molecular weight superstructures. Results are representative of three independent experiments. (B) GLS2 activity was measured using purified HA-GLS2 without crosslinking from 293T cells in the presence acetate and the overexpression of GCN5L1 or a vector control (n = 3 independent experiments). (C and D) GLS2 and GCN5L1 were transfected into 293T cells. Cell lysates were immunoprecipitated with anti-myc (C) or anti-flag (D) antibodies before immunoblotting analysis using anti-flag or anti-myc antibody as indicated. (E) Total acetylated peptides from liver mitochondria of GFP or mitochondrial GCN5L1 expression mice were analyzed by LC-MS. The relative difference in the levels of the only differentiated acetylated GLS2 peptide is shown. (F) WT or mutant GLS2 constructs were transfected into 293T cells. Cell lysates were used to measure GLS2 activities Values are expressed as mean ± s.e.m. *P<0.05, **P < 0.01 versus respective control groups by unpaired Student’s t-test.
Mitochondrial GCN5L1 overexpression normalized glutamine uptake and glutaminolysis in GCN5L1 LKO hepatocytes and inhibited glutaminolysis in vivo
Given that GCN5L1 accumulates in mitochondria (supplementary Fig 3) and as glutaminolysis is predominantly catabolized in mitochondria, we tested whether mitochondrial restricted GCN5L1 (MtG)(14) was sufficient to reverse glutamine metabolism in LKO hepatocytes. Glutamine uptake was indirectly measured by HPLC assessment of glutamine consumption from the culture media of primary hepatocytes. Glutamine consumption was significantly increased in LKO hepatocytes and reversed by overexpression of MtG in KO (Fig 4A). Meanwhile, transcript of Slc1a5 was elevated in LKO and restored by MtG rescue (Fig 4B) and α-KG levels were also restored by overexpression of MtG in LKO hepatocytes (Fig 4C). To test the influence of MtG on glutaminolysis in-vivo, we introduced adenoviral expression vectors encoding MtG or GFP control through the tail vein of WT mice. In the MtG overexpression mice, but not in GFP control infection, hepatic GLS2 activity decreased (Fig 4D). Mitochondrial GDH, GOT2 or GPT2 activities did not change in response to MtG expression (Fig 4E – G). Taken together these data support that mitochondrial GCN5L1 governs GLS2 activity to regulate glutaminolysis and suggest that the regulation of Slc1a5 may be responsive to the cellular demand for glutamine.
FIG. 4.
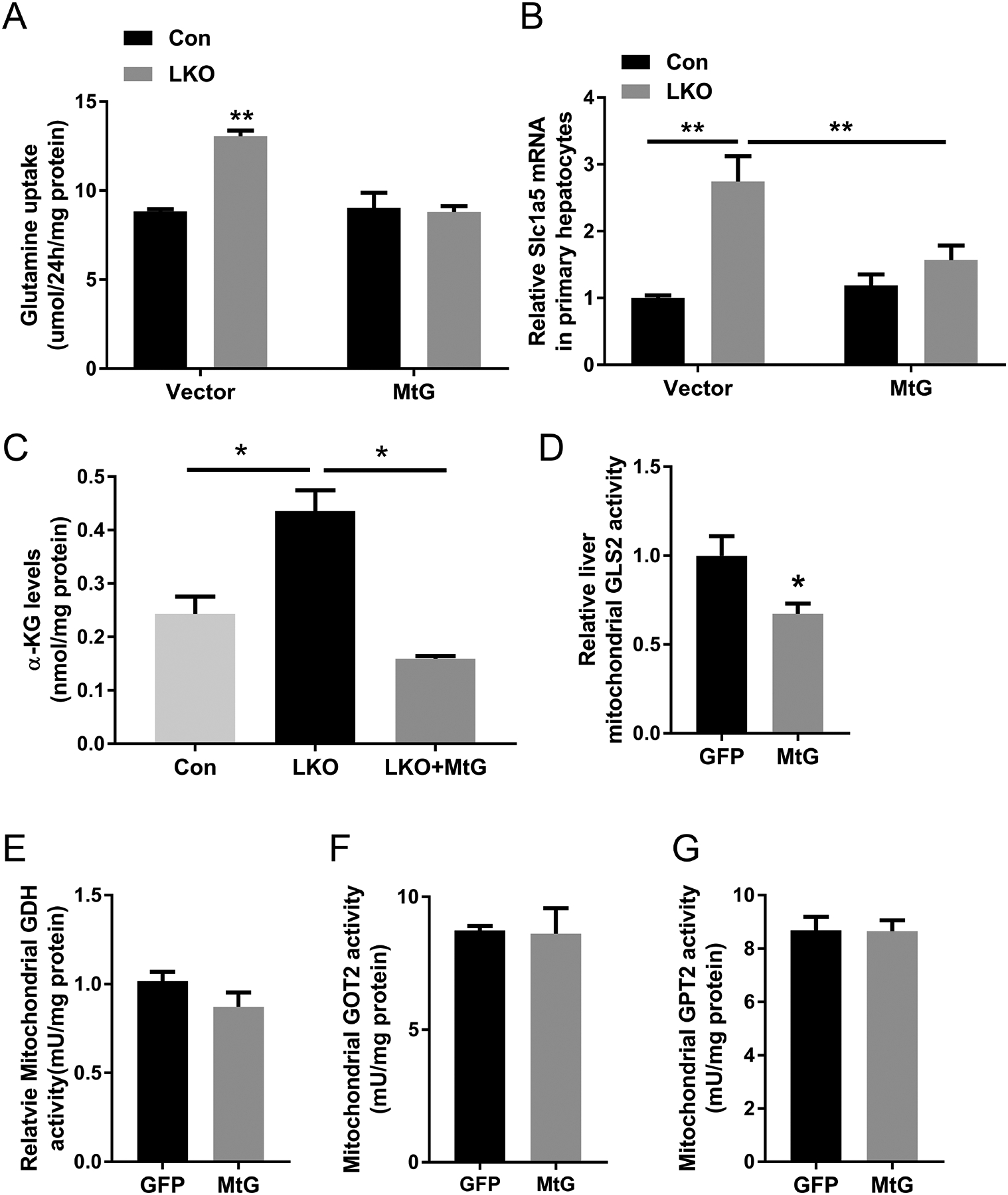
Mitochondrial GCN5L1 normalizes glutamine uptake and glutaminolysis in GCN5L1 LKO hepatocytes and inhibits glutaminolysis in vivo. (A) Glutamine uptake was measured in the culture medium of primary hepatocytes isolated from GCN5L1 LKO and littermate controls with/without mitochondrial GCN5L1 (MtG) reintroduction at 24 h after incubation with 4mM glutamine. Glutamine contents in culture medium were measured by HPLC. Glutamine uptake was calculated by subtracting glutamine content from original medium (without cells) and normalized to protein content (n = 3 independent experiments). (B) The transcript levels of Slc1a5 in primary hepatocytes of GCN5L1 LKO and controls with/without MtG reintroduction (n = 3 independent experiments). (C) α-KG levels in primary hepatocytes of GCN5L1 LKO with/without MtG overexpression and controls (n = 3 independent experiments). WT male mice were injected adenoviral expression of mitochondrial GCN5L1(MtG) or GFP control. GLS2 (D), GDH (E), GOT2 (F) and GPT2 (G) activities were measured in liver mitochondrial lysates (n=5). Values are expressed as mean ± s.e.m. *P<0.05, **P < 0.01 versus respective control groups by unpaired Student’s t-test.
GCN5L1 LKO showed increased mTORC1 activity through enhanced glutaminolysis
Glutaminolysis has been shown to activate the amino acid sensor kinase mTORC1, in part via elevating α-KG levels(25). We explored this amino acid sensing pathway, in the context of the manipulation of GCN5L1 levels as a functional biochemical validation of the role of GCN5L1 in modulating cellular amino acid content. To initially manipulate this program WT and LKO hepatocytes were deprived of glutamine for 24h., followed by incubation with glutamine or with a cell permeable α-KG (DM-KG) for 1h. The phosphorylation of a canonical mTORC1 substrate, S6K was measured. As expected, the glutamine depletion completely abolished S6K phosphorylation (p-S6K) (Fig 5A). Interestingly, and in keeping with the induction of the glutamine transporter, glutamine repletion results in greater p-S6K levels in LKO hepatocytes. In contrast, when this transporter was circumvented by supplementation with DM-KG the relative p-S6K/total S6K was induced to similar levels irrespective of GCN5L1 levels (Fig 5A). To rescue this signaling cascade we over-expressed MtG in LKO and control hepatocytes and found that restoration of mitochondrial-enriched GCN5L1 blunted phosphorylation of both mTOR and S6K (Fig 5B). To test whether increased glutaminolysis activated mTOR in GCN5L1 LKO, we employed EGCG, an allosteric inhibitor of GDH, to inhibit glutaminolysis(26) and found that the increased phosphorylation of mTOR and S6K were reversed, suggesting that induction of glutaminolysis activates mTORC1 in GCN5L1 LKO (Fig 5C). Given that GCN5L1 loss enhanced glutaminolysis by activating GLS2, we knocked down GLS2 levels by siRNA. The depletion of GLS2 in LKO hepatocytes blunted the extent of S6K phosphorylation (Fig 5D). Finally, we overexpressed the K-279-Q GLS2 expression construct in primary hepatocytes and showed that the expression of this acetyl-mimetic mutant similarly blunted the phosphorylation of the mTORC1 targets p-S6k and p-4EBP1 (Fig 5E). Together, these results demonstrated that GCN5L1 loss promotes glutamine-induced anaplerosis via the induction of GLS2 activity.
Fig. 5.
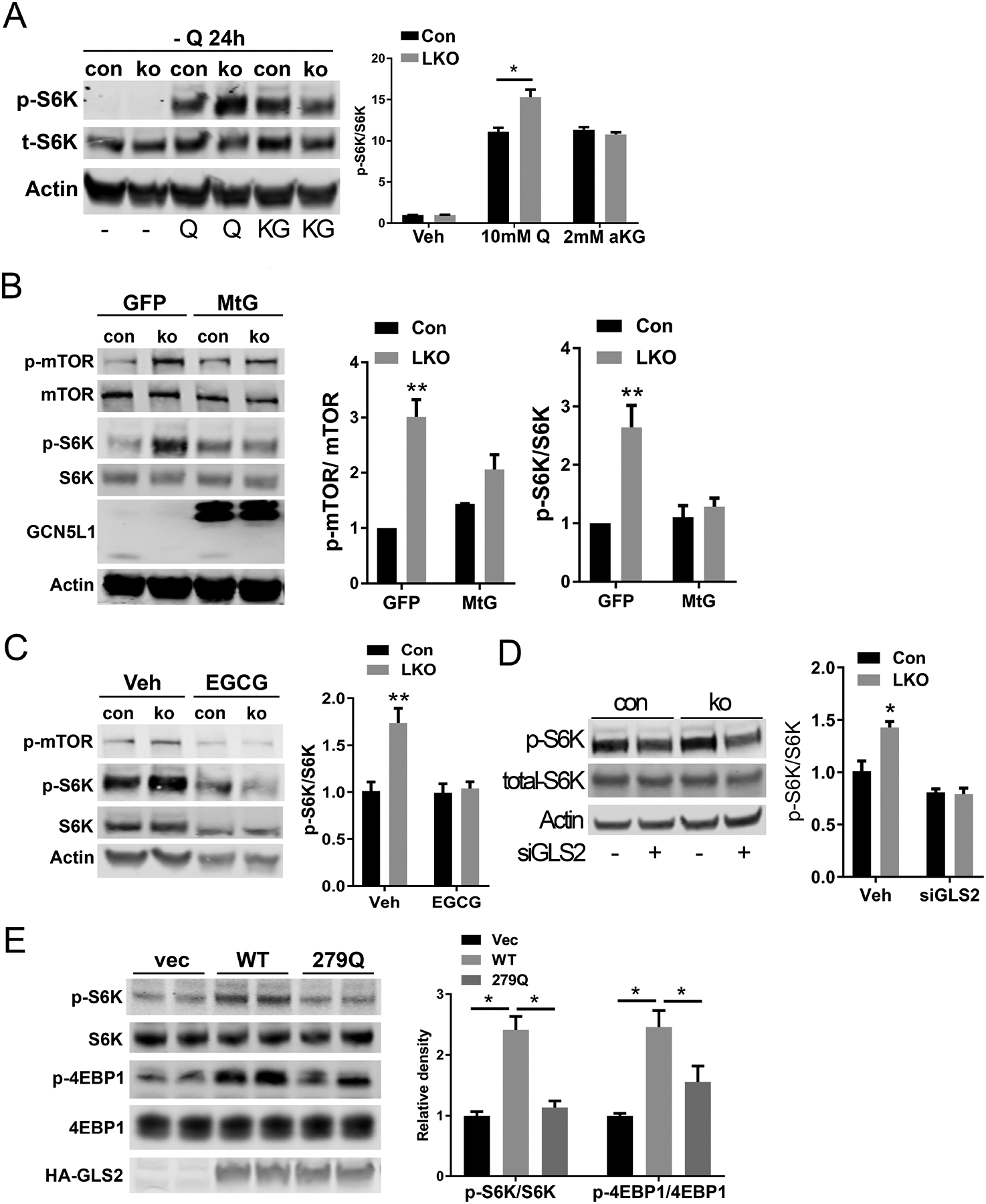
GCN5L1 LKO shows an increase of mTORC1 activity through enhanced glutaminolysis. (A) Primary hepatocytes isolated from flox/flox or LKO mice were incubated in glutamine deficient DMEM for 24h and treated with 10 mM glutamine or 4mM cell permeable α-KG for 1h. Phosphorylation levels of S6K were analyzed by immunoblotting, and results of quantification are shown after normalization to the value of total S6K (n = 3 independent experiments). (B) Primary hepatocytes from GCN5L1 LKO or control mice were infected with the adenovirus expressing GFP or MtG. At 2 days after infection, cell lysates were used for immunoblotting. Phosphorylation levels of mTOR and S6K were analyzed, and quantitative results are shown after normalization to the total proteins (n = 3 independent experiments). (C) Primary hepatocytes from GCN5L1 LKO or control mice were treated with glutaminolysis inhibitor EGCG for 15h, followed by immunoblotting analysis of mTORC1 pathway. (D) Primary hepatocytes were transfected with GLS2 siRNA, 2 days after transfection, cell lysates were analyzed by immunoblotting. (E) Primary hepatocytes from WT mice were transfected with. WT GLS2 or 279Q mutant or vector plasmids for 24h, followed by immunoblotting analysis of mTORC1 pathway (n = 3 independent experiments). Values are expressed as mean ± s.e.m. *P<0.05, **P < 0.01 versus respective control groups by unpaired Student’s t-test.
GCN5L1 LKO enabled liver regeneration following CCl4 exposure by increased glutaminolysis and mTOR activation
As mTORC1 activation promotes protein translation, and cell growth we explored whether the functional consequences of the GCN5L1-deficiency induction of mTORC1 in the LKO mouse could enhance regenerative repair of the liver in response to the hepatotoxin CCl4. We injected CCl4 or vehicle control (olive oil) into male LKO and littermate control mice. Forty-eight hours post injection, liver/ body weight ratio was assessed to measure relative liver regeneration. Upon olive oil injection, liver/ body weight ratios were similar between the groups (Fig 6A). As expected, CCl4 injection stimulated liver regeneration in both genotypes and disruption of GCN5L1 amplified liver regeneration upon CCl4 injection, as evidenced by increased liver/body weight ratio (Fig 6A). This phenotype was supported by elevated transcript levels of Cyclin D (Fig 6B) and by increased levels of the nuclear proliferation marker PCNA in LKO livers and by increased mTORC1 signaling (Fig 6C–E). At the same time, the elevation of liver associated enzymes, AST and ALT, and the transcript levels of IL-6 were induced to similar levels in response to CCl4 in both mice strains, suggesting that hepatic GCN5L1 deficiency did not differentially affect liver injury or Kupffer cell IL-6 production in response to CCl4 (Supplementary figure).
FIG. 6.
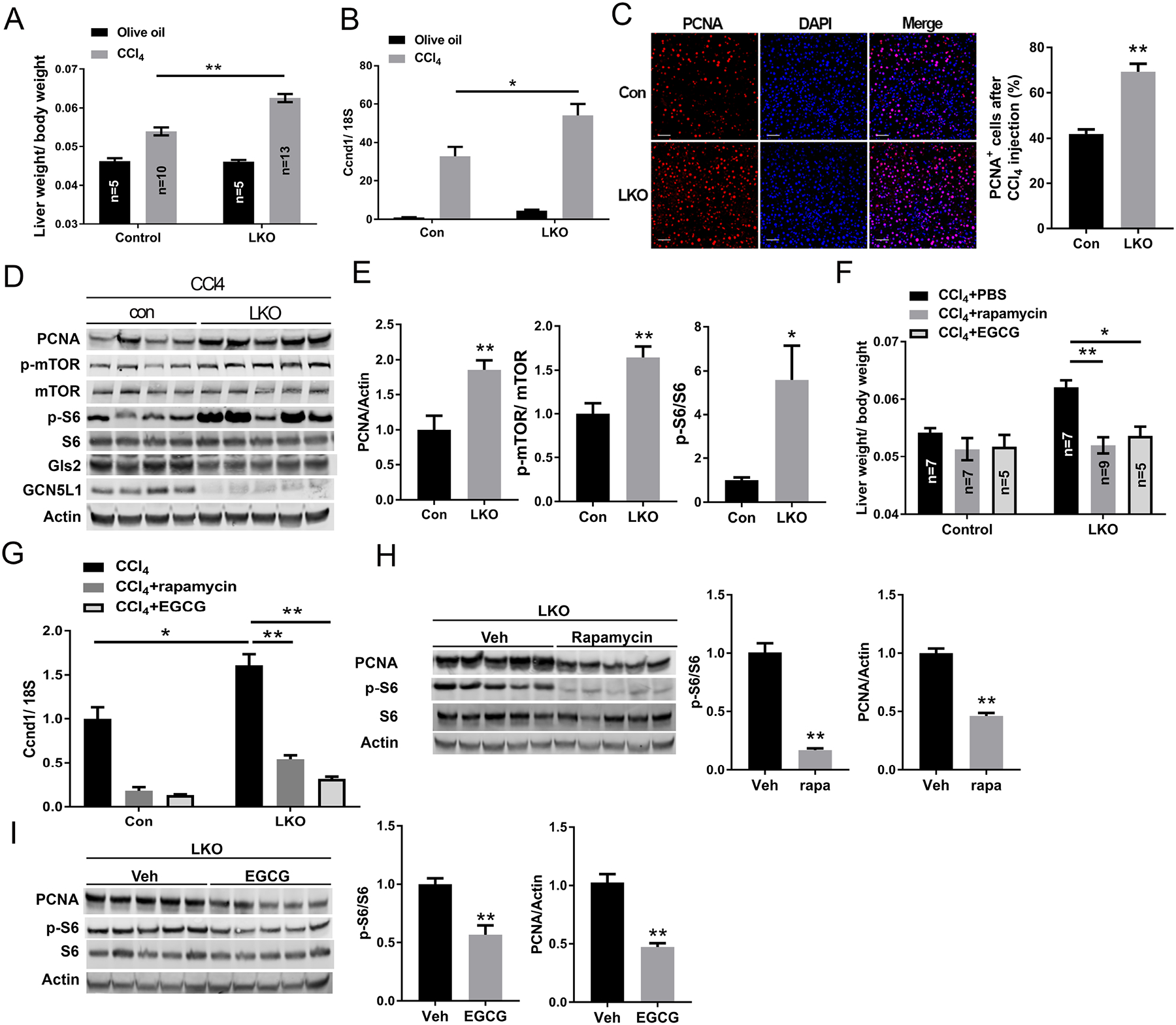
GCN5L1 LKO promotes liver regeneration following CCl4 exposure by increasing glutaminolysis and mTOR activation. (A) GCN5L1 LKO mice and flox/flox littermates at 8–9 weeks were treated for 2 days with vehicle (olive oil) or CCl4 (n = 5–13 per group). Animals were sacrificed at 48h after CCl4 injection. Liver weight was determined relative to body weight (n = 5–13 per group). (B) Quantitative RT-PCR analyses of hepatic mRNA abundance of cyclin D1 upon CCl4 or vehicle injection. (C) Proliferation marker PCNA staining was employed on liver paraffin sections with concurrent DAPI staining. The percentage of PCNA positive cells was analyzed by ImageJ. Scale bar indicates 50μm. (D and E) Immunoblotting of liver extracts using the indicated antibodies. Representative results are shown for individual mice of CCl4 treatment (D). Actin was used as loading control. Phosphorylation levels of mTOR and S6 were determined by densitometric quantification (E). (F) 6h after CCl4 injection, mice were injected i.p. with PBS, rapamycin or EGCG twice at 24h intervals. 48h after CCl4 injection mice were sacrificed and liver weight was determined relative to body weight (n = 5–9 per group). (G) Quantitative RT-PCR analyses of hepatic mRNA of cyclin D1 upon treatments in E. (H and I) Immunoblotting of liver extracts using the indicated antibodies. Representative results are shown for individual mice with CCl4 and rapamycin treatment (H) or CCl4 and EGCG treatment (I). Quantification of the immunoblots were shown on the right of each panel. Values are expressed as mean ± s.e.m. *P<0.05, **P < 0.01 versus respective control groups by unpaired Student’s t-test.
To evaluate the role of the glutaminolysis in LKO-mediated enhanced regenerative capacity we treated CCl4 exposed mice with the mTOR inhibitor Rapamycin or with the glutaminolysis inhibitor EGCG. Inhibition of glutaminolysis and mTORC1 activity both decreased the liver/body ratio and the transcript levels of Cyclin D following CCl4 injection in LKO mice livers (Fig 6F and 6G). We then evaluated the link between glutaminolysis, mTORC1 activity, and liver regeneration. As expected, Rapamycin inhibited mTORC1 activity and proliferation, and the inhibition of glutaminolysis by EGCG similarly blunted mTORC1 activity and PCNA levels (Fig 6H and 6I). Interestingly, when we assessed control mice following CCl4 exposure compared to vehicle injected controls, we found that mitochondrial localized GCN5L1 was diminished and mitochondrial protein acetylation was blunted in response to CCl4 (supplementary Fig 5). Taken together, these findings correlate with, and are supportive of the concept that GCN5L1 loss promotes cell proliferation through the induction of glutaminolysis and mTORC1 signaling.
The glutamine transporter Slc1a5 induction is not a major control node in enhanced mTORC1 signaling in GCN5L1 LKO hepatocytes
At the same time, given that mTORC1 activation targets Slc1a5 transcription,(27) we hypothesized a positive feedback loop of transcriptional activation of Slc1a5 was through GCN5L1-depletion mediated glutaminolysis. However, the induction of Slc1a5 transcript levels in response to GCN5L1 depletion may be an alternate or parallel mechanism to induce glutamine uptake, GLS2 activation, glutaminolysis and mTORC1 signaling. To explore this mechanism, we inhibited mTORC1 signaling with rapamycin in-vivo in WT and LKO mice in the CCl4 toxicity study. Rapamycin blunted Slc1a5 transcript levels in the control and LKO livers (Fig 7A). This intervention equalized the levels of glutamine but did not affect the LKO linked elevation in α−KG levels (Fig 7B) or the induction of GLS2 activity (Fig 7C). To validate that the inhibition of mTORC1 blunted glutamine levels, rapamycin was administered to WT and LKO primary hepatocytes. Here, the blunting of mTORC1, blunted intracellular glutamine levels in both the WT and LKO hepatocytes (Fig 7D). Finally, we employed siRNA to knock down GLS2 in the control and LKO hepatocytes. Despite a 50% reduction in the steady-state levels of GLS2 (Fig 7E), the transcript levels encoding Slc1a5 were still significantly elevated, albeit to a slightly lesser degree, in the LKO hepatocytes (Fig 7F). Taken together these data support that the increased activity of GLS2 is a primary consequence of GCN5L1 deficiency and that the activation of mTORC1 and the increase in Slc1a5 transcript levels are responses to GLS2-driven glutaminolysis.
Fig 7.
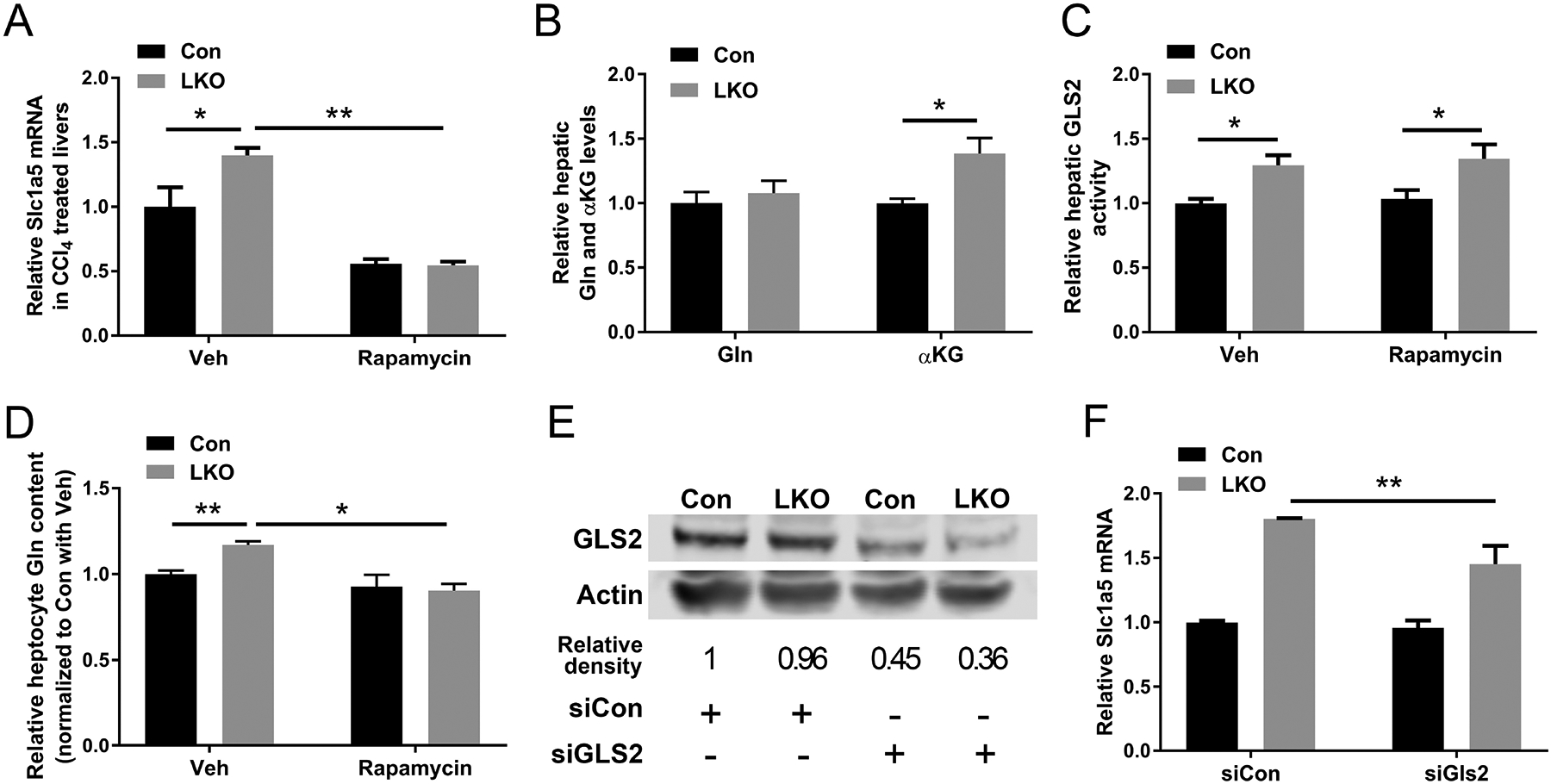
GLS2 but not Slc1a5 is the major control node in enhanced glutaminolysis in GCN5L1 LKO. (A-C) GCN5L1 LKO mice and flox/flox littermates were treated with CCl4 and rapamycin or vehicle (n = 5–7 per group). Quantitative RT-PCR analyses of hepatic mRNA abundance of Slc1a5 (A). Hepatic glutamine and α−KG levels were measured in liver samples following CCl4 and rapamycin administration (B). Mitochondrial fractions were isolated from livers and GLS2 activities were measured using mitochondrial lysates (C). (D) Primary hepatocytes were exposed to rapamycin or vehicle control. Intracellular glutamine levels were measured (n=5–6 per group). (E and F) Primary hepatocytes were transfected with siRNA to knockdown GLS2. The knockdown efficiency was showing by immunoblotting and densitometric analysis (E). Hepatic mRNA abundance of Slc1a5 was analyzed (F) (n = 3 independent experiments). Values are expressed as mean ± s.e.m. *P<0.05, **P < 0.01 versus respective control groups by unpaired Student’s t-test.
Discussion
In the current study, we present evidence that the absence of GCN5L1 promotes hepatic glutaminolysis in response to increased GLS2 activity and this metabolic remodeling increases both mTORC1 activity and the regenerative capacity of hepatocytes in response to an acute hepatotoxic stress with CCl4. Furthermore, we show that mitochondrial-enriched GCN5L1 suppresses GLS2 activity. We show that this regulation is mediated in part by the change in acetylation in the GLS2 putative catalytic domain. Although whether this is also dependent on an acetylation modification of GLS2 oligomerization and/or acetylation-dependent binding of GLS2 to its cognate substrate needs further clarification.
Given that GCN5L1 has an enriched mitochondrial isoform and that it plays a role in mitochondrial protein acetylation it is not surprising that its genetic manipulation has been found to effect mitochondrial metabolism in tissue and cell-distinct patterns. To explore this in an unbiased manner metabolomic analysis was performed in primary hepatocytes. This uncovered a role of GCN5L1 in the modulation of glutamine metabolism and further analysis shows that this regulation was controlled, in large part, by the role of GCN5L1 in controlling the activity of the liver isoform of glutaminase GLS2.
Four mammalian isoforms of glutaminase exist, with GLS1 being the most ubiquitous and major isoform, whereas GLS2 is restricted to the liver. GLS1 has been the most well characterized and it is structural regulated by oligomerization-induced augmentation of its enzymatic activity.(20) Interestingly, more recently the acetylation of the murine Lys316 (and human Lys311) was found to disrupt GLS1 tetramerization and blunt enzyme activity.(21) The role of lysine acetylation in directly modulating enzyme activity has been shown for multiple enzyme where lysine acetylation abolishes the positive charge of the lysine side chain.(28) This regulation can take the form of neutralizing the active site of an enzyme, causing allosteric changes or by recruiting a negative regulator.(28) In contrast to GLS1, the post-translational control of GLS2 has been less well characterized. Our findings of reduced GLS2 oligomerization, and diminished enzyme activity in the presence of GCN5L1 would suggest, that the modulation of enzyme activity by oligomerization may be shared with GLS1. Although the molecular structure of GLS2 has not been defined at a high resolution, K-279 appears to be in the GLS2 catalytic domain and K-329 may play a role in dimerization, when compared the to the three-dimensional structural of GLS1.(23). Our finding that GCN5L1 increases GLS2 acetylation on K-279, and that the acetyl-mimic of K-279 blunts GLS2, strongly support that the acetylation of GLS2 may also play a direct role in modulating enzyme activity. Furthermore, our lysine to glutamine substitution studies also suggest that the acetylation of K-329 effects enzyme activity, albeit to a less extent than the K-279-Q substitution. Whether this is operational in modulating GLS2 oligomerization requires further study. In parallel, it is interesting to note that acetylome analysis in the Sirt3 knockout mice also support that GLS2 may be acetylated on K-329.(29)
Increased glutaminolysis is a biological signature of cancer cell proliferation, however, GLS2 as a rate-controlling enzyme in liver glutaminolysis, has been reported to be an inhibitory factor of cell proliferation due to that it being a target of the p53 tumor suppressor, and due to its decreased expression in tumors.(30, 31) However, recent research has shown that the inhibitory function of GLS2 on proliferation is independent of its glutaminase activity.(32) As a dominant isoform of glutaminase in liver, whether GLS2 glutaminase activity and glutaminolysis regulate normal hepatocyte proliferation is not well established. Our data would support that glutaminase activity of GLS2 is necessary for liver regeneration in response to the exposure to CCl4.
Given that elevated levels of α-KG activate mTORC1, it is not surprising that in this study we find that this signaling pathway is activated by GCN5L1 deficiency induced glutaminolysis. Interestingly, previous work had shown that glutamine catabolism drives mitochondrial ROS and downstream ERK signaling,(33) and we had previously found that in the absence of mitochondrial GCN5L1 drives retrograde mitochondrial ROS signaling via the phosphorylation and activation of ERK to then control the transcription of FoxO1.(14) However, in this study the inhibition of ERK and ROS signaling did not modulate mTORC1 mediated induction of Slc1a5 expression (data not shown). Taken together, these findings suggest that distinct mitochondrial effects of GCN5L1 can, via different retrograde signaling mechanisms, modulate separate signaling pathways.
The role of mTORC1 in the activation of S6 kinase to augment hepatocyte proliferation in liver regeneration has been established.(34) Interestingly, we find that this signaling pathway is dependent on GLS2 activation and increased glutaminolysis. At the same time the distribution of hepatocytes in the liver are heterogeneous with two distinct populations, defined in part by their roles and associated metabolic demands. These are termed periportal and pericentral hepatocytes, respectively and it of interesting that the GLS2 enriched periportal hepatocytes(35) have been identified as the subtype that undergo hyperplasia during CCl4-induced regeneration.(36)
Interestingly, in this study in the control mice, we found that liver mitochondrial acetylation was decreased in response to CCl4 exposure. Although we have not expanded our studies into this regulation, mitochondrial acetylation is emerging as an important mitochondrial modification linking nutrient status and mitochondrial respiration capacity in cancer cell proliferation.(37) Furthermore, nicotinamide riboside, an NAD+ precursor, which activates sirtuin deacetylase enzymes, promoted liver regeneration following a partial hepatectomy.(38) Taken together these data suggest that the further exploration of acetylation in the regulation of hepatic regeneration is necessary.
A prior study has shown that the absence of GCN5L1 in the liver promotes fat oxidation with a concomitant reduction in high-fat feeding induced liver fatty liver.(13) At the same time, the loss of hepatocytic regenerative capacity in nonalcoholic fatty liver disease (NAFLD) is proposed to facilitate progressive liver disease with NAFLD.(39) Whether the blunting of excess fat accumulation in the liver is an additional mechanism promulgating regeneration in our study has not been evaluated. However, collectively, this prior study and our current results raise the potential that the blunting of GCN5L1 activity in the liver may confer ameliorative effects against a broader array of insults through pleotropic actions that could contribute towards a greater regenerative-capacity.
It should also be noted, that in this study we explored the role of mitochondrial localized GCN5L1. However, cytosolic GCN5L1 has been found to modulate endosome, lysosome and vesicular trafficking(12, 17) and whether cytosolic-enriched GCN5L1 plays any role in GLS2 regulation remains to be determined. At the same time, we only explored the effects of GCN5L1 depletion on acute CCl4 hepatotoxicity to explore hepatic regenerative capacity. However, more chronic models would need to be investigated to validate whether the augmentation of the glutaminolysis/mTORC1 pathway by GCN5L1 depletion could have ameliorative effects against hepatotoxicity for long term liver health.
In conclusion, this study reveals a novel function on the mitochondrial enriched GCN5L1 in the regulation of hepatic glutaminolysis via the control of the activity of GLS2. The consequences of increased glutaminolysis in the GCN5L1 liver knockout mice increases amino acid intermediate levels with the subsequent activation of mTORC1 signaling. This, in turn, was shown to enhance the hepatic regenerative capacity in response to acute exposure to CCL4. Given the myriad of metabolic consequences of hepatic GCN5L1 depletion, this model will be useful to reconcile diverse mechanism to control hepatocyte regeneration. Additionally, the pharmacologic inhibition of GCN5L1 may be a potential therapeutic target to promote hepatic regeneration.
Supplementary Material
Supplementary Figure 1. GCN5L1 deletion reprograms hepatic metabolism. (A) Basal mitochondrial respiration measured by Seahorse using isolated mitochondria from GCN5L1 LKO or control livers with pyruvate, malate and ADP. (B) primary hepatocytes were incubated with U-13C glutamine for 6h. 13C isotope incorporated into TCA intermediates (n=3). Values are expressed as mean ± s.e.m. *P<0.05, **P < 0.01 versus respective control groups by Student’s t-test.
Supplementary Figure 2. GCN5L1 deletion enhances glutaminase 2 activity in primary hepatocytes. Primary hepatocytes were isolated from GCN5L1 LKO or control mice. Mitochondrial lysates from the hepatocytes were used for GLS2 activity measurement (n=3). Values are expressed as mean ± s.e.m. **p < 0.01 versus respective control groups by Student’s t-test.
Supplementary Figure 3 GCN5L1 accumulates in liver mitochondria. Primary hepatocytes were isolated from GCN5L1 LKO or control mice, followed by adenovirus expression of GCN5L1 or mitochondrial restricted GCN5L1(MtG). 50μg cytosolic or mitochondrial fractions were analyzed by immunoblotting for GCN5L1 expression. * indicates full-length GCN5L1 expression and # indicates MtG expression.
Supplementary Figure 4 GCN5L1 deletion does not affect CCl4 induced liver injury. (A) Serum AST or ALT activities were analyzed upon CCl4 injection (n=10–13). (B) Liver IL-6 transcripts were analyzed by quantitative PCR (n=5–10). Values are expressed as mean ± s.e.m.
Supplementary Figure 5. Liver mitochondrial acetylation and mitochondrial GCN5L1 expression were decreased in CCl4 induced liver regeneration. Mitochondria were isolated from CCl4 or olive oil injected wildtype mouse livers. 50ug mitochondrial protein for each mouse was analyzed by immunoblotting, quantification of GCN5L1 expression and mitochondrial acetylation were normalized to VDAC (n=5). Values are expressed as mean ± s.e.m. *p<0.05, **p < 0.01 versus respective control groups by Student’s t-test.
Acknowledgements:
This study was funded by the NHLBI Division of Intramural Research (MNS). This study was designed by L.W. and L.Z. Experiments were performed by L.W, L. Z., K.W., Y. C, and D.Y.L. Data was analyzed and interpreted by L.W, L. Z., K.W., D.Y.L., Y.C, M.G. and M.N.S. The manuscript was written by L.W., L.Z and M.N.S. with input from all authors. We sincerely thank Dr. Zu-Xi Yu (NHLBI, Bethesda, MD, USA) for preparation of liver paraffin section and PCNA staining.
Abbreviations:
- CCl4
carbon tetrachloride
- Ccnd1
Cyclin D 1
- CE-MS
capillary electrophoresis mass spectroscopy
- DM-KG
dimethyl 2-oxoglutarate
- EGCG
epigallocatechin gallate
- GCN5L1
general control of amino acid synthesis 5 like 1
- GlS1/2
glutaminase 1/2
- GDH
glutamate dehydrogenase
- GOT2
glutamic oxaloacetic transaminase 2
- GPT2
glutamic pyruvic transaminase
- LKO
liver specific Gcn5l1 knockout
- MtG
mitochondrial restricted GCN5L1
- mTOR
mammalian target of rapamycin
- mTORC1/2
mTOR complexes 1 and 2
- NASH
nonalcoholic steatohepatitis
- p70S6K
p70 ribosomal S6 kinase
- PCNA
proliferating cell nuclear antigen
- Q
glutamine
- S6
ribosomal protein S6
- Slc1a5
solute carrier family 1 member a 5
- Sirt3
sirtuin 3
- TCA
tricarboxylic acid cycle
References
- 1.Farber JL, Gerson RJ. Mechanisms of cell injury with hepatotoxic chemicals. Pharmacol Rev 1984;36:71S–75S. [PubMed] [Google Scholar]
- 2.Fausto N, Campbell JS, Riehle KJ. Liver regeneration. Hepatology 2006;43:S45–53. [DOI] [PubMed] [Google Scholar]
- 3.Polimeno L, Capuano F, Marangi LC, Margiotta M, Lisowsky T, Ierardi E, Francavilla R, et al. The augmenter of liver regeneration induces mitochondrial gene expression in rat liver and enhances oxidative phosphorylation capacity of liver mitochondria. Dig Liver Dis 2000;32:510–517. [DOI] [PubMed] [Google Scholar]
- 4.Wise DR, Thompson CB. Glutamine addiction: a new therapeutic target in cancer. Trends Biochem Sci 2010;35:427–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lu Z, Chen Y, Aponte AM, Battaglia V, Gucek M, Sack MN. Prolonged fasting identifies heat shock protein 10 as a Sirtuin 3 substrate: elucidating a new mechanism linking mitochondrial protein acetylation to fatty acid oxidation enzyme folding and function. J Biol Chem 2015;290:2466–2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee IH, Cao L, Mostoslavsky R, Lombard DB, Liu J, Bruns NE, Tsokos M, et al. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc Natl Acad Sci U S A 2008;105:3374–3379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hirschey MD, Shimazu T, Goetzman E, Jing E, Schwer B, Lombard DB, Grueter CA, et al. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature 2010;464:121–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lu Z, Bourdi M, Li JH, Aponte AM, Chen Y, Lombard DB, Gucek M, et al. SIRT3-dependent deacetylation exacerbates acetaminophen hepatotoxicity. EMBO Rep 2011;12:840–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bao J, Scott I, Lu Z, Pang L, Dimond CC, Gius D, Sack MN. SIRT3 is regulated by nutrient excess and modulates hepatic susceptibility to lipotoxicity. Free Radic Biol Med 2010;49:1230–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kendrick AA, Choudhury M, Rahman SM, McCurdy CE, Friederich M, Van Hove JL, Watson PA, et al. Fatty liver is associated with reduced SIRT3 activity and mitochondrial protein hyperacetylation. Biochem J 2011;433:505–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hirschey MD, Shimazu T, Jing E, Grueter CA, Collins AM, Aouizerat B, Stancakova A, et al. SIRT3 deficiency and mitochondrial protein hyperacetylation accelerate the development of the metabolic syndrome. Mol Cell 2011;44:177–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Scott I, Wang L, Wu K, Thapa D, Sack MN. GCN5L1/BLOS1 Links Acetylation, Organelle Remodeling, and Metabolism. Trends Cell Biol 2018;28:346–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thapa D, Wu K, Stoner MW, Xie B, Zhang M, Manning JR, Lu Z, et al. The protein acetylase GCN5L1 modulates hepatic fatty acid oxidation activity via acetylation of the mitochondrial beta-oxidation enzyme HADHA. J Biol Chem 2018;293:17676–17684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang L, Scott I, Zhu L, Wu K, Han K, Chen Y, Gucek M, et al. GCN5L1 modulates cross-talk between mitochondria and cell signaling to regulate FoxO1 stability and gluconeogenesis. Nat Commun 2017;8:523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Scott I, Webster BR, Li JH, Sack MN. Identification of a molecular component of the mitochondrial acetyltransferase programme: a novel role for GCN5L1. Biochem J 2012;443:655–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Donato V, Bonora M, Simoneschi D, Sartini D, Kudo Y, Saraf A, Florens L, et al. The TDH-GCN5L1-Fbxo15-KBP axis limits mitochondrial biogenesis in mouse embryonic stem cells. Nat Cell Biol 2017;19:341–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu K, Wang L, Chen Y, Pirooznia M, Singh K, Walde S, Kehlenbach RH, et al. GCN5L1 interacts with alphaTAT1 and RanBP2 to regulate hepatic alpha-tubulin acetylation and lysosome trafficking. J Cell Sci 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Webster BR, Scott I, Han K, Li JH, Lu Z, Stevens MV, Malide D, et al. Restricted mitochondrial protein acetylation initiates mitochondrial autophagy. J Cell Sci 2013;126:4843–4849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Scott I, Webster BR, Chan CK, Okonkwo JU, Han K, Sack MN. GCN5-like protein 1 (GCN5L1) controls mitochondrial content through coordinated regulation of mitochondrial biogenesis and mitophagy. J Biol Chem 2014;289:2864–2872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Curthoys NP, Watford M. Regulation of glutaminase activity and glutamine metabolism. Annu Rev Nutr 1995;15:133–159. [DOI] [PubMed] [Google Scholar]
- 21.Ferreira AP, Cassago A, Goncalves Kde A, Dias MM, Adamoski D, Ascencao CF, Honorato RV, et al. Active glutaminase C self-assembles into a supratetrameric oligomer that can be disrupted by an allosteric inhibitor. J Biol Chem 2013;288:28009–28020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pasquali CC, Islam Z, Adamoski D, Ferreira IM, Righeto RD, Bettini J, Portugal RV, et al. The origin and evolution of human glutaminases and their atypical C-terminal ankyrin repeats. J Biol Chem 2017;292:11572–11585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.DeLaBarre B, Gross S, Fang C, Gao Y, Jha A, Jiang F, Song JJ, et al. Full-length human glutaminase in complex with an allosteric inhibitor. Biochemistry 2011;50:10764–10770. [DOI] [PubMed] [Google Scholar]
- 24.Traba J, Geiger SS, Kwarteng Siaw M, Han K, Ra OH, Siegel RM, Gius D, et al. Prolonged fasting suppresses mitochondrial NLRP3 inflammasome assembly and activation via SIRT3 mediated activation of Superoxide Dismutase 2. J Biol Chem 2017;292:12153–12164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Duran RV, Oppliger W, Robitaille AM, Heiserich L, Skendaj R, Gottlieb E, Hall MN. Glutaminolysis activates Rag-mTORC1 signaling. Mol Cell 2012;47:349–358. [DOI] [PubMed] [Google Scholar]
- 26.Villar VH, Merhi F, Djavaheri-Mergny M, Duran RV. Glutaminolysis and autophagy in cancer. Autophagy 2015;11:1198–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.White MA, Lin C, Rajapakshe K, Dong J, Shi Y, Tsouko E, Mukhopadhyay R, et al. Glutamine Transporters Are Targets of Multiple Oncogenic Signaling Pathways in Prostate Cancer. Mol Cancer Res 2017;15:1017–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xiong Y, Guan KL. Mechanistic insights into the regulation of metabolic enzymes by acetylation. J Cell Biol 2012;198:155–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hebert AS, Dittenhafer-Reed KE, Yu W, Bailey DJ, Selen ES, Boersma MD, Carson JJ, et al. Calorie restriction and SIRT3 trigger global reprogramming of the mitochondrial protein acetylome. Mol Cell 2013;49:186–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hu W, Zhang C, Wu R, Sun Y, Levine A, Feng Z. Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proc Natl Acad Sci U S A 2010;107:7455–7460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang J, Wang C, Chen M, Cao J, Zhong Y, Chen L, Shen HM, et al. Epigenetic silencing of glutaminase 2 in human liver and colon cancers. BMC Cancer 2013;13:601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang C, Liu J, Zhao Y, Yue X, Zhu Y, Wang X, Wu H, et al. Glutaminase 2 is a novel negative regulator of small GTPase Rac1 and mediates p53 function in suppressing metastasis. Elife 2016;5:e10727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Weinberg F, Hamanaka R, Wheaton WW, Weinberg S, Joseph J, Lopez M, Kalyanaraman B, et al. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc Natl Acad Sci U S A 2010;107:8788–8793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Espeillac C, Mitchell C, Celton-Morizur S, Chauvin C, Koka V, Gillet C, Albrecht JH, et al. S6 kinase 1 is required for rapamycin-sensitive liver proliferation after mouse hepatectomy. J Clin Invest 2011;121:2821–2832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Benhamouche S, Decaens T, Godard C, Chambrey R, Rickman DS, Moinard C, Vasseur-Cognet M, et al. Apc tumor suppressor gene is the “zonation-keeper” of mouse liver. Dev Cell 2006;10:759–770. [DOI] [PubMed] [Google Scholar]
- 36.Pu W, Zhang H, Huang X, Tian X, He L, Wang Y, Zhang L, et al. Mfsd2a+ hepatocytes repopulate the liver during injury and regeneration. Nat Commun 2016;7:13369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Corbet C, Pinto A, Martherus R, Santiago de Jesus JP, Polet F, Feron O. Acidosis Drives the Reprogramming of Fatty Acid Metabolism in Cancer Cells through Changes in Mitochondrial and Histone Acetylation. Cell Metab 2016;24:311–323. [DOI] [PubMed] [Google Scholar]
- 38.Mukherjee S, Chellappa K, Moffitt A, Ndungu J, Dellinger RW, Davis JG, Agarwal B, et al. Nicotinamide adenine dinucleotide biosynthesis promotes liver regeneration. Hepatology 2017;65:616–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Richardson MM, Jonsson JR, Powell EE, Brunt EM, Neuschwander-Tetri BA, Bhathal PS, Dixon JB, et al. Progressive fibrosis in nonalcoholic steatohepatitis: association with altered regeneration and a ductular reaction. Gastroenterology 2007;133:80–90. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. GCN5L1 deletion reprograms hepatic metabolism. (A) Basal mitochondrial respiration measured by Seahorse using isolated mitochondria from GCN5L1 LKO or control livers with pyruvate, malate and ADP. (B) primary hepatocytes were incubated with U-13C glutamine for 6h. 13C isotope incorporated into TCA intermediates (n=3). Values are expressed as mean ± s.e.m. *P<0.05, **P < 0.01 versus respective control groups by Student’s t-test.
Supplementary Figure 2. GCN5L1 deletion enhances glutaminase 2 activity in primary hepatocytes. Primary hepatocytes were isolated from GCN5L1 LKO or control mice. Mitochondrial lysates from the hepatocytes were used for GLS2 activity measurement (n=3). Values are expressed as mean ± s.e.m. **p < 0.01 versus respective control groups by Student’s t-test.
Supplementary Figure 3 GCN5L1 accumulates in liver mitochondria. Primary hepatocytes were isolated from GCN5L1 LKO or control mice, followed by adenovirus expression of GCN5L1 or mitochondrial restricted GCN5L1(MtG). 50μg cytosolic or mitochondrial fractions were analyzed by immunoblotting for GCN5L1 expression. * indicates full-length GCN5L1 expression and # indicates MtG expression.
Supplementary Figure 4 GCN5L1 deletion does not affect CCl4 induced liver injury. (A) Serum AST or ALT activities were analyzed upon CCl4 injection (n=10–13). (B) Liver IL-6 transcripts were analyzed by quantitative PCR (n=5–10). Values are expressed as mean ± s.e.m.
Supplementary Figure 5. Liver mitochondrial acetylation and mitochondrial GCN5L1 expression were decreased in CCl4 induced liver regeneration. Mitochondria were isolated from CCl4 or olive oil injected wildtype mouse livers. 50ug mitochondrial protein for each mouse was analyzed by immunoblotting, quantification of GCN5L1 expression and mitochondrial acetylation were normalized to VDAC (n=5). Values are expressed as mean ± s.e.m. *p<0.05, **p < 0.01 versus respective control groups by Student’s t-test.