

SUMMARY
In response to estrogens, estrogen receptor alpha (ERα), a critical regulator of homeostasis, is degraded through the 26S proteasome. However, despite the continued presence of estrogen before menopause, ERα protein levels are maintained. We discovered that ERK1/2-RSK2 activity oscillates during the estrous cycle. In response to high estrogen levels, ERK1/2 is activated and phosphorylates ERα to drive ERα degradation and estrogen-responsive gene expression. Reduction of estrogen levels results in ERK1/2 deactivation. RSK2 maintains redox homeostasis, which prevents sustained ERK1/2 activation. In juveniles, ERK1/2-RSK2 activity is not required. Mammary gland regeneration demonstrates that ERK1/2-RSK2 regulation of ERα is intrinsic to the epithelium. Reduced RSK2 and enrichment in an estrogen-regulated gene signature occur in individuals taking oral contraceptives. RSK2 loss enhances DNA damage, which may account for the elevated breast cancer risk with the use of exogenous estrogens. These findings implicate RSK2 as a critical component for the preservation of estrogen homeostasis.
In Brief
Ludwik et al. find that ERK1/2-RSK2 activity oscillates with each reproductive cycle. The estrogen surge activates ERK1/2, which phosphorylates estrogen receptor alpha to drive estrogen responsiveness. Active RSK2 acts as a brake on the estrogen response by maintaining redox homeostasis. Oral contraceptive use correlates with disruption of ERK1/2-RSK2 regulation.
Graphical Abstract
INTRODUCTION
The importance of estrogen signaling is highlighted by the numerous physiological alterations that occur during menopause, oophorectomy, or anti-estrogen therapy (Barros and Gustafsson, 2011). In the adult, human estrogen levels are highest in the follicular phase, reaching a level of ~1 nM and decrease approximately 5-fold in the luteal phase of the menstrual cycle (Lovett et al., 2017). In the mouse, the estrous cycle is divided into four stages, which are based on vaginal cytology and comprise proestrus, estrus, metestrus, and diestrus. The highest level of estrogen ~0.2 nM occurs during proestrus and then decreases by approximately 3-fold in diestrus (Walmer et al., 1992). All estrogen-receptor-positive (ER+) tissues respond to fluctuations in estrogen levels. The mammary gland undergoes extensive morphological changes as estrogen levels change (Vogel et al., 1981); therefore, it is an ideal organ in which to investigate the mechanisms that regulate estrogen homeostasis. Estrogen acts primarily through the steroid hormone receptors, estrogen receptor alpha (ERα) and ERβ (Jia et al., 2015). In the mammary gland, ERα is of particular importance for its contributions to gland development (Feng et al., 2007). Mammary gland development is normal in the absence of ERβ (Förster et al., 2002); therefore, to examine estrogen homeostasis in the mammary gland, we focused our studies on ERα.
Estrogen binding to the receptor results in ERα degradation through the 26S proteasome pathway, and both are required for activation of estrogen-responsive gene expression (Lonard et al., 2000; Nawaz et al., 1999; Reid et al., 2003; Silberstein et al., 2006; Zhang et al., 2006). However, it is puzzling how this degradation-coupled transcription is regulated to maintain ERα protein levels because, theoretically, estrogen levels are sufficient throughout the menstrual cycle to continuously drive degradation (Blair et al., 2000). Therefore, it might be expected that ERα levels would eventually drop below that required to generate a physiological response. However, both ERα levels and estrogen responsiveness are maintained to allow progression into the next menstrual cycle, but the mechanisms regulating ERα degradation are unknown (Valley et al., 2008; Zhou and Slingerland, 2014). Maintenance of responsiveness is of particular relevance to individuals taking estrogen-containing oral contraceptives in which estrogen levels do not fluctuate compared with that of the normal menstrual cycle (Lovett et al., 2017) and in transgendered individuals, in which estrogen levels can reach supra-physiological levels (Deutsch et al., 2015).
We hypothesized that a negative regulatory mechanism must exist to limit ERα degradation to preserve ERα levels and, as a result, maintain estrogen responsiveness. To identify that mechanism, we focused on estrogen and its control of the epidermal growth factor receptor (EGFR) signaling pathway because of the importance of EGFR in mammary gland development (Xie et al., 1997), cell fate specification (Pasic et al., 2011), and breast cancer (Maennling et al., 2019). Stimulation of EGFR activates the MEK-ERK1/2 signaling cascade. Activated ERK1/2 and its downstream effector, p90 ribosomal S6 kinase (RSK), directly phosphorylate ERα at Ser-118 and Ser-167, respectively (Joel et al., 1998a; Kato et al., 1995). These sites increase ERα transcriptional activity in cell-based systems. In a transgenic mouse model RSK2 nuclear accumulation in the mammary gland drives high-grade ER+ ductal carcinoma in situ (Ludwik et al., 2018). Because cancer often exploits mechanisms important in development and homeostasis, we investigated the contributions of ERK1/2-RSK2 signaling to normal ERα biology.
Unexpectedly, we discovered a novel regulatory mechanism in which the ERK1/2-RSK2 pathway acts as a developmentally regulated switch that is required for maintaining ERα protein levels in the mammary gland and uterus in the adult but not in the juvenile. ERK1/2 is activated during the estrus phase of the cycle as a consequence of an estrogen pulse that occurs in proestrus. Activated ERK1/2 phosphorylates ERα, driving the degradation of ERα and estrogen-responsive gene expression. To enable estrogen responsiveness for the subsequent cycle ERK1/2 is inactivated when estrogen levels are low. Active RSK2 limits the response to estrogen by maintaining redox homeostasis, which prevents ERK1/2 activation in response to reactive oxygen species (ROS). In the RSK2 knockout (RSK2-KO) and in individuals taking oral contraceptives decreased RSK2 levels are correlated with an enriched signature for estrogen-responsive gene expression. These observations may explain the mechanism underlying the increase in breast cancer risk that is observed for individuals taking exogenous estrogens because reduced RSK2 is correlated with increased DNA damage.
RESULTS
RSK2 Is Required to Maintain ERα Homeostasis in the Adult Mammary Gland
In the mouse, estrogen levels are highest during proestrus, akin to the follicular stage in humans (Fata et al., 2001). Analysis of ERα in the mammary gland of wild-type (WT) mice in situ using quantitative immunofluorescence (IF) revealed that ERα protein levels varied during the estrous cycle (Figures 1A and 1B). In the WT mice, the lowest ERα protein levels occurred during estrus, which is consistent with observations that ERα protein degradation increases in response to the estrogen pulse in proestrus (Table S1; Nawaz et al., 1999). Staging of the estrous cycle was determined by analysis of vaginal cytology and uterine wet weight (Wood et al., 2007; Figure S1A). WT and RSK2-KO mice moved through the estrous cycle in a similar manner (Figure S1B). In the RSK2-KO glands, ERα levels were consistently lower than in the WT glands across all estrous stages (Figures 1A, 1B, and S1C). These results were unexpected because RSK2 phosphorylation of ERα stimulates transcription (Joel et al., 1998a) and would, presumably, increase ERα degradation. Therefore, based on these observations, we would expect that loss of RSK2 would increase ERα protein levels.
Figure 1. RSK2 Regulates ERα Protein Levels in the Adult Mammary Gland throughout the Estrous Cycle.

(A) ERα protein expression in the adult mammary gland of WT and RSK2-KO mice during the estrous cycle. Scale bar: 20 μm.
(B) ERα protein levels are lower in the RSK2-KO mice at all stages of the estrous cycle in adult mammary glands as determined by IF. ERα protein levels normalized to the average level observed in the WT mice at proestrus (median ± quartile, n ≥ 3 mice/genotype and stage, one-way ANOVA with Holm-Sidak’s correction for multiple comparisons).
(C) Loss of RSK2 results in a decrease in the number of ERα cells relative to K8+ cells at all stages of the estrous cycle in adult mammary glands (median ± quartile, n ≥ 4 mice/genotype, ≥150 cells/mouse, one-way ANOVA with Holm-Sidak’s correction for multiple comparisons). See Figure S1 and Table S1.
To further investigate the decrease in ERα levels that occur in the RSK2-KO glands, we analyzed cell populations within the adult mammary glands by fluorescence-activated cell sorting (FACS). A novel FACS protocol that allowed the simultaneous analysis of WT and RSK2-KO mammary epithelial cells (MECs) was developed in which one of the genotypes was permanently marked, and equal numbers of cells from the marked and unmarked genotypes were mixed (Figure 2A). The marked genotype was varied, and live cells and lineage-negative MECs were determined (Figure S2A). The luminal and basal populations were clearly separated using epithelial cell adhesion molecule (EpCAM) and integrin alpha 6 (CD49f) (Figure 2B). The distributions were fairly similar in adult WT and RSK2-KO mice at each stage of the estrous cycle (Figure S2B). Further fractionation of the luminal cells by stem cells antigen-1 (Sca1) and integrin alpha 2 (CD49b) resulted in four populations with the gates for each experiment established using a fluorescence-minus-one strategy (Figure S2C; Shehata et al., 2012). The EpCAMhiCD49f+Sca1+CD49b− population, which consists primarily of ERα cells (Giraddi et al., 2015), is referred to as non-clonogenic luminal (NCL) because of its lack of colony-forming potential in vitro and engrafting ability in vivo. The EpCAMhiCD49f+Sca−CD49b+ and EpCAMhiCD49f+Sca+CD49b+ are luminal progenitors, which express low or high levels of luminal differentiation markers, respectively (Giraddi et al., 2015). The EpCAMhiCD49f+Sca−Cd49b− population is currently undefined. In comparison to the WT population at estrus, the NCL population was decreased in the RSK2-KO mice, with a concomitant increase in the undefined population but no change in the luminal progenitor populations (Figures 2B and S2D). These observations are consistent with those observed in situ in which fewer ERα cells were observed in the RSK2-KO population (Figures 1C and S1C). A decrease in ERα protein levels was also observed in NCL cells isolated during FACS, consistent with our in situ analysis (Figures 1A, 1B, and 2D). At each stage of the estrous cycle, a reduction in the NCL population was observed (Figures 2C and S2D).
Figure 2. RSK2 Maintains the EpCAMhiCD49f+Sca1+CD49b− (NCL) Population within the Adult Mammary Gland throughout the Estrous Cycle.

(A) Schematic of FACS protocol.
(B) FACS analysis of adult mammary glands isolated from females during estrus. Gating strategy of luminal cells by further subdivision using Sca-1 and CD49b. The percentage of NCL cells within the luminal population at estrus decreases in adult RSK2-KO mice (median ± quartile, n≥6mice/genotype, one-way ANOVA with Holm-Sidak’s correction for multiple comparisons).
(C) Loss of RSK2 results in a reduction in the percentage of NCL cells at all stages of the estrous cycle in adult mammary glands (median ± quartile, n≥3 mice/genotype and stage, one-way ANOVA with Holm-Sidak’s correction for multiple comparisons). PE, proestrus; E, estrus; ME, metestrus; DE, diestrus.
(D) ERα protein levels are decreased in cells isolated from the NCL population of RSK2-KO mice (median ± quartile, n = 3 mice/genotype, >20 cells/mouse, Student’s t test). Scale bar: 10 μm. Fn, fluorescence.
(E) RSK2 regulation of the NCL population is intrinsic to the epithelium (median ± quartile, n = 3 mice/genotype, Student’s t test).
(F) The percentage of NCL cells within the luminal population is similar between WT and RSK2-KO juvenile female mice (median ± quartile, n≥3 mice/genotype and age group, one-way ANOVA with Holm-Sidak’s correction for multiple comparisons).
(G and H) The levels of ERα protein expression (G) and the number of ERα cells (H) relative to K8+ cells are similar in WT and RSK2-KO juvenile female mice (median ± quartile, n = 3 mice/genotype, ≥5 fields/mouse, one-way ANOVA with Holm-Sidak’s correction for multiple comparisons). Scale bar: 20 μm. See Figures S1 and S2 and Table S1.
RSK2-KO is a constitutive knockout, and therefore, we evaluated the contributions of systemic and intrinsic mechanisms that facilitate RSK2 regulation of the ERα population. To perform these analyses, mammary epithelial cells from WT and RSK2-KO mice were separately introduced into the cleared fourth mammary fat pads of a WT recipient. The glands from the transplanted cells regenerated to similar extents (Figure S2E). In regenerated glands, loss of RSK2 also resulted in a decrease in the NCL population (Figure 2E), indicating that the effects on the ERα population caused by the loss of RSK2 are intrinsic to the mammary epithelial cells.
Because ERα is absolutely required for mammary gland development (Mehta et al., 2014), we analyzed the mammary gland at different ages starting at puberty. No detectable difference in the expansion of the mammary gland into the fat pad or branching during development was observed (Figure S2F). Analysis by FACS showed that all cell populations were similar between RSK2-KO and WT mice in juveniles (Figures 2F, S2B, and S2D). Consistent with these data, in situ analysis of the juvenile mammary glands showed similar ERα protein levels (Figure 2G) and numbers of ERα cells (Figure 2H). We conclude that RSK2 regulates the ERα population only in the adult, which explains the absence of a developmental defect.
ERK1/2-RSK2 Signaling Is Dependent on Estrogens
At the onset of puberty estrogen increases the levels of growth factors (Brisken and Ataca, 2015; Hynes and Watson, 2010), which, theoretically, would result in RSK activation through its upstream activator, ERK1/2 (Eisinger-Mathason et al., 2010). C57BL/6J mice initiate cycling by ~6 weeks (Pinter et al., 2007), although we observed that cycling was irregular until ~10–12 weeks old. Interestingly, ERK1/2, as shown by Thr202/Tyr204 phosphorylation (pERK1/2), was not active until the animals were ≥10 weeks old, and the levels of active ERK1/2 were similar at estrus between the WT and RSK2-KO mice at the same age (Figure 3A). A causal relationship between estrogen and ERK1/2 activity was demonstrated by the observations that ERK1/2 activation was prevented by oophorectomy at 6 weeks (Figure 3B). ERK1/2 activation in the WT mouse occurs in estrus after the estrogen burst in proestrus and then decreases during diestrus when estrogen levels are lowest (Figure 3C). The inactivation of ERK1/2 appears to be consistent with increased phosphatase activity because the protein levels of ERK1/2 do not change between estrus and diestrus (Figure S3B). We conclude that the ability of estrogen to activate ERK1/2 and regulate its cyclic activation appears as the mice sexually mature.
Figure 3. ERK1/2-RSK2 Signaling Is Activated Only in the Adult Mammary Gland.

(A) ERK1/2 activity is increased in the adult compared with juvenile animals (median ± quartile, n≥2 mice/genotype and age, ≥3 fields/mouse, one-way ANOVA with Holm-Sidak’s correction for multiple comparisons). Scale bar: 20 μm.
(B) ERK1/2 activity in the mammary gland depends on estrogen (median ± quartile, n ≥ 2 mice/genotype and procedure, ≥3 fields/mouse, Student’s t test). Scale bar: 20 μm.
(C) ERK1/2 activity varies during the estrous cycle in the WT mice adult mammary gland (median ± quartile, n ≥ 2 mice/genotype, ≥3 fields/mouse, one-way ANOVA with Tukey’s correction for multiple comparisons). Scale bar: 20 μm.
(D) Active nuclear RSK2 is the predominant RSK in adult mammary glands. Scale bar: 20 μm. See Figures S3 and Table S1.
Active ERK1/2 was primarily confined to the luminal compartment and was present in ERα cells (Figure S3C). To confirm that RSK was activated in the WT mammary gland, an anti-active RSK antibody (pRSK) was used. RSK is activated in response to coordinated inter- and intra-molecular phosphorylation events (Dalby et al., 1998), which are identical within the RSK family, and therefore, identification of the active state of a particular RSK is not possible. However, active RSK was not detectable in the adult RSK2-KO mammary glands, indicating that RSK2 is the predominant active RSK isoform (Figure 3D). These results demonstrate that, in the WT mouse, estrogen activates ERK1/2-RSK2 signaling, and that this activation corresponds with the ability of RSK2 to regulate ERα protein levels (Figures 1B and 3C).
RSK2 Negatively Regulates Proteasome-Coupled Transcription in the Adult Mammary Gland
To identify a mechanism that would explain the reduced ERα protein levels with the loss of RSK2, we performed transcriptomic analyses on the NCL population (Table S2). Estrus was chosen because changes in gene expression would be occurring in response to the estrogen pulse that happened in proestrus. We contrasted these data with those obtained in diestrus, which has the lowest estrogen levels. The transcriptomic analysis of the RSK2-KO mice showed 2,747 differentially expressed genes (DEGs) between estrus and diestrus as compared with 39 in the WT mice between estrus and diestrus (Figures 4A and S4A). The transcriptomic data of RSK2-KO mice at estrus showed a significant correlation with a signature obtained from the ERα breast cancer cell line MCF-7 at 24 h after estrogen treatment (Figures 4B and 4C; Table S3; Dutertre et al., 2010). This correlation was not driven by cell cycle genes (Figure S4B; Table S3). No significant correlation with the estrogen-responsive gene signature was obtained for the WT mice at estrus (Figures 4B and 4C). ESR1 (gene encoding ERα) mRNA levels were similar between WT and RSK2-KO (Figure S4C), eliminating the possibility that ERα mRNA expression levels accounted for the transcriptomic differences. Taken together, these data demonstrate that estrogen signaling is higher in the RSK2-KO than in the WT mice, and therefore, we conclude that RSK2 acts to inhibit estrogen-responsive gene expression.
Figure 4. RSK2 Is a Negative Regulator of ERα-Mediated Signaling.

(A) RSK2-KO mice show greater numbers of DEGs between estrus and diestrus (right panel) than do WT mice (left panel). Genes with a fold-change ≥ |1.5| (log2[fold-change]≥|0.5|) and a false discovery rate (FDR)-adjusted p < 0.05 are shown as black dots, and genes with a fold-change < |1.5| (log2[fold-change] < |0.5|) and an FDR-adjusted p value > 0.05 are shown as gray dots. The dashed line indicates the cutoff values.
(B) Heatmap illustrating that the gene expression of NCL cells isolated from RSK2-KO mice in estrus correlates with a 24-h estrogen-regulated gene signature identified from MCF-7 cells (Dutertre et al., 2010).
(C) Quantitative assessment of enrichment for estrogen-regulated genes. Cumulative Z scores were generated for each mouse by summing individual Z scores of genes upregulated in estrogen-regulated signature and subtracting individual Z scores of genes downregulated (mean ± SD, each point represents a mouse; one-way ANOVA with Holm-Sidak’s correction for multiple comparisons).
(D) Loss of RSK2 increases ERα turnover. Adult mice staged at estrus were treated with vehicle or PS-341 (5 mg/kg) intraperitoneally (i.p.) for 4 h before euthanasia and isolation of the mammary gland. ERα protein levels were normalized to those observed in the WT mice at estrus (median ± quartile, n = 3 mice/genotype and condition, ≥200 cells/mouse, one-way ANOVA with Holm-Sidak’s correction for multiple comparisons). Scale bar: 20 μm.
(E) RSK2 kinase activity is necessary to maintain ERα protein levels. Adult mice staged at estrus were treated with vehicle or C5′′-n-propyl cyclitol SL0101 (C5′′) (40 mg/kg) IP twice every 7 h before euthanasia and isolation of the mammary gland (median ± quartile, n≥3 mice/genotype, ≥3 fields/mouse, Student’s t test). See Figures S4 and S6 and Tables S1, S2, and S3.
Estrogen-responsive gene expression is interconnected with ERα destruction through the 26S proteasome pathway (Lonard et al., 2000; Nawaz et al., 1999; Reid et al., 2003; Silberstein et al., 2006; Zhang et al., 2006). Therefore, it would be expected that ERα degradation would be greater in the RSK2-KO, than the WT, mice. To investigate this possibility, the rate of in vivo ERα degradation was determined using the 26S proteasome inhibitor PS-341(Silberstein et al., 2006). In the WT gland, ERα levels did not substantially change in response to proteasome inhibition. However, in the RSK2-KO gland, ERα protein levels increased by ~5-fold in response to PS-341; therefore, ERα degradation is much higher in the absence of RSK2 (Figure 4D). This increased degradation explains our in situ observations that ERα levels are lower in the RSK2-KO, compared with those in the WT, mice (Figures 1A, 1B, and S1C).
Reduced ERα protein levels in the RSK2-KO could be the result of decreased RSK2 kinase activity or the loss of the RSK2 protein. To distinguish between these mechanisms, RSK2 activity was inhibited in vivo by the specific RSK1/2 inhibitor C5′′-n-propyl cyclitol SL0101 (C5′′) (Ludwik et al., 2016; Li et al., 2017). ERα protein levels were reduced by the RSK1/2 inhibitor (Figure 4E). To demonstrate that the inhibitor was on target, we used phosphorylation of the elongation translation factor 2 (peEF2) as a biomarker (Wang et al., 2001; Figure S4D). We conclude that RSK2 kinase activity is important in ERα degradation.
RSK2 Maintains ERα Protein Levels in Adult Reproductive Tissue
We next investigated whether RSK2 preserved ERα protein levels in other estrogen-responsive tissues. We focused on the female reproductive tract because we observed a 40% reduction in the fertility rate in crosses between RSK2-KO female and male mice (Figure 5A). RSK2-KO male mice crossed with heterozygote female mice had similar fertility rates to those of the WT mice crosses, indicating that the reduced fertility is associated with the RSK2-KO female mice. Ovaries in the RSK2-KO and WT mice showed all stages of follicular development and the presence of the corpora luteum (Figures 5B and S5A), demonstrating that hormonal signaling (Robertshaw et al., 2016) through the hypothalamic-pituitary-ovarian axis is not impaired in RSK2-KO mice. The uterus expresses high levels of ERα, which is present in stromal cells as well as in glandular and luminal epithelium. In comparison to the WT mice, the ERα protein levels were substantially decreased in the epithelial, but not in the stromal cells, in RSK2-KO mice (Figure 5C). Interestingly, ERK1/2 activity was detected in the uterine epithelium but not in the stroma cells, providing further evidence of the connection between ERK1/2-RSK2 signaling and the regulation of ERα protein levels (Figure 5D). Uterine wet weight and total uterine width were similar in the WT and RSK2-KO mice, which is consistent with the literature because stromal cells are thought to mediate uterine expansion (Figures S1A and S5B; Winuthayanon et al., 2014; Wood et al., 2007). These data indicate that RSK2 regulates ERα protein levels in multiple tissues.
Figure 5. RSK2 Maintains ERα Protein Levels in the Uterine Epithelium.

(A) RSK2-KO mice show a fertility defect (n ≥ 15 dams/genotype, Chi-square test p = 0.0299).
(B) The hypothalamic-pituitary-ovarian axis is not disrupted in RSK2-KO female mice. H&E sections of ovaries isolated from adult mice in estrus. Scale bar: 1 mm, PF, primary follicle; SF, secondary follicle; TF, tertiary follicle; CL, corpus luteum.
(C) RSK2-KO mice have reduced ERα protein levels in the glandular and luminal epithelium of the uterus (median ± quartile, n = 3 mice/genotype, >120 cells/mouse, Student’s t test). Scale bar: 40 μm. GE, glandular epithelium; S, stroma; LE, luminal epithelium.
(D) Active ERK1/2 is confined to the epithelium of the uterus. Scale bar: 40 μm. See Figures S1 and S5.
ERK1/2 Drives ERα Degradation through Phosphorylation of Ser-118 on ERα
To address the mechanism by which RSK2 regulates ERα protein levels, we initially focused on GATA3 because GATA3 and ERα regulate each other’s expression via a positive-feedback mechanism in breast cancer (Eeckhoute et al., 2007). Therefore, it is conceivable that RSK2 indirectly regulates ERα protein levels through GATA3. However, no difference in GATA3 mRNA levels was observed between WT and RSK2-KO mice (Figure S4C). Furthermore, GATA3 protein levels in the uterine glandular epithelium (Uhlen et al., 2017) are extremely low, whereas ERα protein levels are very high (Figures S6A). We conclude that RSK2 regulation of ERα through GATA3 is unlikely.
Interestingly, in contrast to that of the WT mice, ERK1/2 activity remains elevated in the RSK2-KO mice during diestrus (Figure 6A), and coincident with these observations, ERα protein levels remain lower in the RSK2-KO (Figures 1A, 1B, and S1C). Therefore, we investigated whether ERK1/2 activity was a driver of ERα degradation. In support of this hypothesis, when we prevented ERK1/2 activation by oophorectomizing RSK2-KO mice, we observed that the levels of ERα were rescued to WT levels (Figure S3A). To perform further mechanistic studies, we used the normal mouse Leydig cell line TM3, which expresses ERα but does not form tumors in vivo. Survival of the TM3 line was dependent on RSK2, which prevented knockout approaches. However, short-term treatment with two structurally distinct RSK inhibitors decreased ERα protein levels, which was prevented by the inhibition of the 26S proteasome (Figures 6B and 6C). This effect is specific because androgen receptor protein levels do not change in response to RSK1/2 inhibition (Figure 6D). ERK1/2 activity increased in response to the RSK inhibitors (Figure 6B), which is consistent with our observations at diestrus in the RSK2-KO mice. MEK inhibition by trametinib or U0126 did not decrease ERα levels. Taken together, these results indicate that ERK1/2 activity increases ERα degradation through the 26S proteasome.
Figure 6. ERK1/2 Drives ERα Degradation through Phosphorylation of Ser-118.

(A) ERK1/2 activity remains elevated during diestrus in the adult mammary gland (median ± quartile, n = 3 mice, ≥3 fields/mice, Student’s t test).
(B) RSK2 is a negative regulator of ERK1/2 activity. Serum-starved TM3 was treated for 6 h with vehicle, C5′′-n-propyl cyclitol SL0101 (C5′′) (20 μM), BI-D1870 (10 μM), trametinib (1 μM), or U0126 (10 μM). The white vertical line indicates that conditions not relevant to the manuscript were removed. ERα levels were normalized to Ran and then to the vehicle (mean, n = 3, one-way ANOVA with Dunnett’s correction for multiple comparisons).
(C) RSK1/2 inhibition stimulates ERα degradation through the 26S proteasome pathway. Serum-starved TM3 was treated for 6 h with vehicle, C5′′ (20 μM) with or without a 1 h of pre-treatment with MG132 (10 μM).
(D) RSK2 does not regulate androgen receptor (AR) degradation. Serum-starved TM3 was treated for 6 h with vehicle or C5′′ (20 μM). AR levels were normalized to Ran and then to the vehicle (mean, n = 3 in duplicate, Student’s t test).
(E) Phosphorylation of Ser-118A is required for ERα degradation. Cells transduced with WT or mutant ERα-VENUS were treated with vehicle or C5′′ (20 μM) as in (B). The range was normalized to WT ERα (mean, n = 3, >150 cells/condition/experiment, one-way ANOVA with Holm-Sidak’s correction for multiple comparisons). Scale bar: 10 μm.
(F) Loss of RSK2 increases double-stranded DNA breaks (γ-H2AX foci) in the mammary gland (median ± quartile, n ≥ 4 mice/genotype, ≥3 fields/mouse, Student’s t test).
(G) RSK1/2 inhibition increases ROS. Serum-starved TM3 was treated as in (B). The data were normalized to the range with and without C5′′ (20 μM) (mean, n = 3, >100 cells/condition/experiment, Student’s t test).
(H) RSK1/2 inhibition increases DNA damage in vitro. Cells treated for 72 h with vehicle or C5′′ (20 μM). The data were normalized to the range with and without C5′′ (mean, n = 3, >80 cells/condition/experiment, Student’s t test).
(I) Inhibition of ROS rescues ERα levels. Serum-starved TM3 was treated for 6 h with vehicle or C5′′ (20 μM) with or without ebselen (Ebs) (50 μM) or N-acetyl cysteine (NAC) (15 mM) for the final 2 h. The range was normalized to ERα levels in the absence of anti-oxidants (mean, n = 3, >50 cells/condition/experiment, one-way ANOVA with Dunnett’s correction for multiple comparisons).
(J) Inhibition of ROS inhibits ERK1/2 activation. Cells treated and analyzed as in (I). See Figures S3 and S6 and Table S1.
It is hypothesized that degradation of phosphorylated ERα occurs at a faster rate than that of the unphosphorylated (Tian et al., 2015). Therefore, we investigated whether the ERK1/2 and RSK2 phosphorylation of ERα (Joel et al., 1998a; Kato et al., 1995) regulated ERα turnover. GFP-tagged ERα mutants were generated, in which the ERK1/2 phosphorylation site, Ser-118 (S118A-ERα), or the RSK2 site, Ser-167 (S167A- ERα), was mutated to Ala. In response to ERK1/2 activation, mutation of Ser-167 did not alter ERα turnover; however, mutation of Ser-118 prevented ERα destruction (Figure 6E). An electrophoretic mobility-shift assay was used to confirm phosphorylation of Ser-118 in response to RSK1/2 inhibition because phospho-specific antibodies to human ERα do not recognize the mouse protein (Joel et al., 1998b; Figure S6B). We conclude that ERK1/2 phosphorylation of Ser-118 targets ERα for destruction and that RSK2 negatively regulates ERK1/2 activity to protect ERα from degradation.
RSK2 Negatively Regulates ERK1/2 Activity by Controlling Oxidative Stress Levels
To investigate the mechanism by which RSK2 negatively regulates ERK1/2, we determined whether a loss of RSK2 resulted in increased oxidative stress. In support of this hypothesis, an increase in ROS is associated with estrogen-regulated transcription (Perillo et al., 2008), and ROS activates ERK1/2 (Wentworth et al., 2011). Therefore, in the RSK2-KO mouse, the increased estrogen-regulated transcription could result in elevated ROS levels compared with that of the WT mouse, resulting in ERK1/2 activation. The presence of γ-H2AX provides a readout for the formation of DNA double-stranded breaks, which occur in response to oxidative stress (Tanaka et al., 2006). Consistent with our hypothesis, γ-H2AX was elevated in the RSK2-KO (Figure 6F). Analysis of the genes upregulated in the RSK2-KO at estrus compared with diestrus revealed enrichment for genes associated with oxidative stress (Figure S6C). Additionally, an over-representation of genes was associated with the glutathione metabolic process, suggesting that the cells were experiencing oxidative stress and attempting to compensate by increasing glutathione production. Consistent with the in vivo data, RSK2 inhibition in the TM3 line exhibited elevated ROS (Figure 6G) and DNA damage (Figure 6H). Importantly, reduction of ROS by two structurally distinct anti-oxidants rescued ERα levels in the presence of RSK2 inhibition (Figure 6I) and prevented ERK1/2 activation (Figure 6J). Taken together, these data demonstrate that RSK2 maintains estrogen homeostasis by preventing the activation of ERK1/2 by ROS.
RSK2 Integrates Estrogen-Mediated Transcription and Translational Responses to Maintain Homeostasis
There was no evidence of hyperplasia in the RSK2-KO glands, which was surprising because of their increased expression of cell cycle genes. In fact, the rate of proliferation was decreased in the NCL population of RSK2-KO mice (Figures 7A, S7A, and S7B), which is consistent with the reduced number of ERα cells observed in these mice (Figures 1C, 2B, and 2C). Because of this disconnect between the gene expression and proliferation data, we investigated whether RSK2 was important in translational regulation in the ERα population (Roux and Topisirovic, 2018). As a readout for translational activity in vivo, we measured eEF2 phosphorylation (peEF2). The levels of peEF2 were higher at diestrus in the RSK2-KO mice (Figure 7B), which is consistent with inhibition of protein synthesis (Wang et al., 2001). We also observed that RSK1/2 inhibition decreased protein synthesis in the TM3 line (Figure 7C). Taken together, these data support a model in which RSK2 regulation of translation contributes to the physiological responses induced by estrogen.
Figure 7. RSK2 Is Necessary for Alveolar Expansion.

(A) RSK2-KO NCL cells show a decrease in proliferation as compared with the WT cells. RSK2-KO or WT MECs were used to regenerate the mammary gland in a WT mouse. These mice were staged in proestrus and administered 5-ethynyl-2′-deoxyuridine (EdU) throughout one estrus cycle. The mammary glands were isolated and analyzed by FACS (n = 3 glands/genotype; paired Student’s t test).
(B) RSK2 regulates eEF2K activity in vivo (median ± quartile, n ≥ 3 mice/genotype, ≥5 fields/mouse, Student’s t test).
(C) Inhibition of RSK1/2 decreases translation in vitro. Serum-starved TM3 was treated for 6 h with vehicle or C5′′ (20 μM). The range was normalized to the o-propargyl-puromycin (OPP) in the absence and presence of C5′′ (mean, n ≥ 3, >150 cells/condition/experiment, one-way ANOVA with Holm-Sidak’s correction for multiple comparisons).
(D) Alveolar expansion is reduced in RSK2-KO dams as shown by H&E stains of mammary glands isolated from dams 1 d after birth (median ± quartile, n ≥ 3 mice/genotype, ≥3 fields/mouse, Student’s t test). Scale bar: 1 mm.
(E) Pups nursed by RSK2-KO dams are smaller than those nursed by WT dams. Weanling weight at 21 d nursed by a dam with the indicated genotype (median ± quartile, n = 3 litters matched for size/dam genotype, one-way ANOVA with Holm-Sidak’s correction for multiple comparisons).
(F) The estrogen-regulated signature is enriched in the luteal phase or with oral contraceptive use. Cumulative patient Z scores were generated for each individual by summing individual Z scores of genes upregulated in estrogen-regulated signature and subtracting individual Z scores of genes downregulated (mean ± SD, n = 8; F, follicular, 3 L, luteal; 4 OC, oral contraceptive; one-way ANOVA with Tukey’s correction for multiple comparisons).
(G) RSK2 mRNA levels are decreased in response to the luteal phase or oral contraceptives based on Z score analysis as in (F).
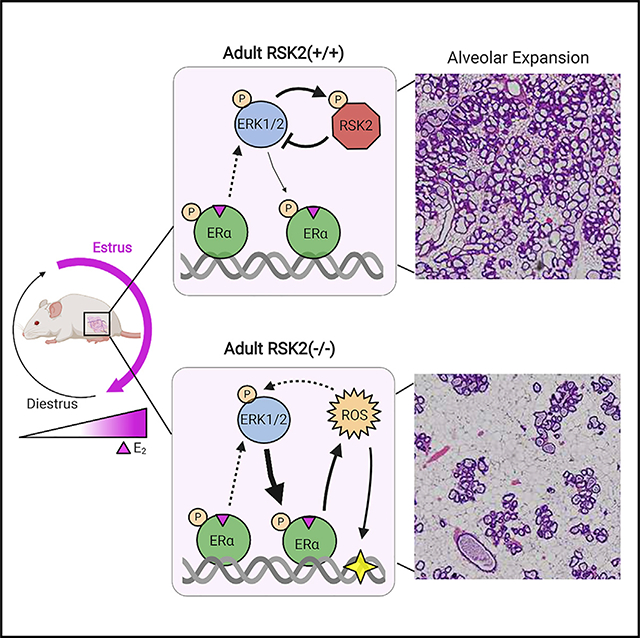
(H) Schematic illustrating maintenance of estrogen homeostasis by RSK2. See Discussion for further explanation. See Figure S7 and Table S1.
To further evaluate the physiological importance of RSK2 in estrogen responsiveness, we investigated the remodeling of the mammary gland that occurs during pregnancy. This remodeling is dependent on the ERα cells within the mammary gland, which act as sensors to facilitate alveolar expansion and lactation (Brisken and Ataca, 2015; Feng et al., 2007). Alveolar expansion in the whole-animal knockout (Figure 7D) and in glands regenerated from RSK2-KO (Figure S7C) were reduced, consistent, with the decrease in the ERα population observed in the RSK2-KO glands. Pup weight was reduced in litters arising from RSK2-KO crosses, which could be rescued by fostering RSK2-KO pups to WT dams (Figures 7E and S7D). These results argue that the reduced alveolar expansion in the RSK2-KO dams does not provide sufficient nutrition for the pups, rather than a developmental defect in the offspring. These results support our hypothesis that RSK2 is a critical regulator of estrogen responsiveness in vivo.
Estrogen Homeostasis in the Human Breast
To evaluate whether RSK2 also functions in regulating estrogen responsiveness in humans, we examined transcriptomic data obtained from normal breast tissue at different stages of the menstrual cycle or from women who were taking oral contraceptives (Pardo et al., 2014). In women taking oral contraceptives, the levels of synthetic estrogen remain elevated over the time the drugs are administered. In individuals in the luteal phase and in those taking oral contraceptives (Figure 7F), a significant correlation was observed with the estrogen-responsive gene signature obtained from the ERα breast cancer cell line MCF-7 (Dutertre et al., 2010). Interestingly, RSK2 mRNA levels were inversely correlated with the estrogen-responsive gene signature (Figures 7G and S7E), which is consistent with the RSK2-KO data. This correlation was not driven by cell cycle genes (Figure S7F). We propose that individuals who take oral contraceptives are subject to prolonged estrogen-responsive gene expression in comparison to individuals who are normally cycling.
DISCUSSION
All ER+ tissues respond to estrogen signaling and, therefore, are subject to the normal fluctuations in the levels of estrogen that occur throughout the estrous cycle. The importance of estrogen signaling is highlighted by the numerous physiological alterations, which occur during menopause, oophorectomy, or anti-estrogen therapy (Barros and Gustafsson, 2011). Here, we provide the first evidence that growth-factor signaling through the ERK1/2-RSK2 pathway is required to maintain cyclic estrogen responsiveness in vivo. In the schematic for the WT mice (Figure 7H, left) in step one, we propose that the estrogen pulse in proestrus activates growth factor pathway signaling. This hypothesis is based on observations in neuroendocrine tissues that ERK1/2 is activated after the estrogen surge (Scharfman and MacLusky, 2008). Consistent with these data, we found in the mammary gland that ERK1/2 was activated in estrus and that activation was dependent on estrogen. The second step of the schematic shows we identified that ERK1/2 phosphorylates ERα to enhance degradation through the 26S proteasome pathway because mutation of the ERK1/2 phosphorylation site Ser-118 prevents ERα degradation. The most likely mechanism for the increased ERα turnover is through creation of a phosphodegron at Ser-118, which results in E3 ligase recruitment (Filipčík et al., 2017). In step three, we determined that activated ERK1/2 drives ERα degradation to enhance estrogen-responsive gene expression. Additionally, activated RSK2, which regulates protein synthesis (Roux and Topisirovic, 2018), was identified to be important in translation of the estrogen-mediated gene program. The physiological importance of RSK2 translational regulation is demonstrated by the reduced pup size and decreased fertility in the RSK2-KO female mice. We propose that the fertility defect is most likely explained by decreased translation in the glandular epithelium because of the loss of RSK2 because estrogen-induced glandular secretions are known to be important for implantation (Kelleher et al., 2016). To reset the cycle, we propose in step four that ERK1/2 is dephosphorylated and inactivated by phosphatases. This hypothesis is based on observations in neutrophils that estrogen upregulates expression of ERK1/2 phosphatases (Zhang et al., 2019) and our data demonstrating that total ERK1/2 protein levels do not vary with estrogen levels. The cycle is then reinitiated at the next proestrus.
In contrast to temporal activation of ERK1/2 during the estrus cycle, we show in the schematic for the RSK2-KO mice (Figure 7H, right) the disruption of this homeostatic mechanism because of the loss of RSK2. We determined that the loss of RSK2 maintains activation of ERK1/2 in diestrus, which results in increased estrogen-responsive gene expression. We identified that loss of RSK2 resulted in elevated ROS levels, and we hypothesize that this increased ROS inhibits phosphatase activity. This hypothesis is supported by studies showing that oxidation of the reactive-site cysteine in ERK1/2 phosphatases results in their inactivation (Denu and Tanner, 1998; Meng et al., 2002). We speculate that the increased ROS is a result of elevated estrogen-responsive gene expression, which is known to occur (Perillo et al., 2008) and to increased energy requirements. This later hypothesis is supported by gene ontology analysis of the NCL population at estrus, which showed an over-representation of genes associated with the mitochondria. We conclude that RSK2 regulates estrogen-responsive gene expression by controlling redox homeostasis. These findings represent a previously unidentified function for RSK2. Negative regulation of estrogen-responsive gene expression by RSK2 was unexpected because its contributions to ERα breast cancer are well established (Jiang et al., 2007; Ludwik et al., 2018; Moon et al., 2012).
We show the importance of ERK1/2 in regulating ERα degradation in vivo. Phosphorylation of ERα at Ser-118 has been reported to occur by a number of different kinases and has been associated with increased ERα-mediated transcription in breast cancer cells (Bunone et al., 1996; Chen et al., 2000; González et al., 2009; Joel et al., 1998b; Kato et al., 1995; Park and Lee, 2017; Weitsman et al., 2009). Furthermore, mutation of Ser-118 to Ala in an ectopic expression system prevented degradation (Valley et al., 2005). Numerous ubiquitin ligases have been reported to regulate ERα stability, and components of the 26S proteasome are found in association with ERα on the chromatin in studies using breast cancer cells (Zhou and Slingerland, 2014). It is unclear whether the degradation mechanism differs between breast cancer and normal physiology because ERα protein levels are higher in breast cancer (Yang et al., 2013), which does suggest that the homeostatic mechanisms have been disrupted.
We also report the analysis of gene expression in the purified NCL population. Relatively few differences in DEGs were detected in the WT mice between estrus and diestrus as compared with the RSK2-KO mice. We propose that the increased gene expression is driven by the continuous ERα transcriptional activation in response to activated ERK1/2 in the RSK2-KO mice. To accurately compare gene expression in the WT and RSK2-KO mice, we developed a FACS protocol that permitted mixing the genotypes and sorting simultaneously. This approach eliminated artifacts from differences in staining among preparations.
RSK2 regulation of estrogen responsiveness occurs in the mature gland but not during puberty. It is possible that unopposed estrogen action is required to facilitate the extensive remodeling of the gland that begins at puberty. However, in the adult, this extensive proliferative response could lead to dysfunction and hyperproliferation within the gland. In human females, we observed an inverse relationship between RSK2 mRNA levels and an estrogen-responsive gene signature in the breast tissue of women in the luteal phase or on oral contraceptives. Consistent with those observations, RSK2 mRNA levels also decreased in endometrial tissue of women in the luteal, compared with the follicular, phase (Sigurgeirsson et al., 2017). ERα protein levels are known to decrease in women taking hormone-replacement therapy, suggesting increased ERα-mediated transcription-coupled degradation occurs in those individuals (Yang et al., 2013). We speculate that RSK2 levels are decreased in individuals in which normal estrogen levels are disrupted resulting in chronic activation of ERK1/2 and dysregulated estrogen-mediated transcription. This increase in estrogen-mediated signaling could lead to an increase in DNA damage as we observed in the RSK2-KO mice and may account for the higher risk of breast cancer associated with the use of hormonal contraceptives and hormone-replacement therapy (Beral, 1996; Hunter et al., 2010; Mørch et al., 2017; Romieu et al., 1989; Simin et al., 2017).
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Requests for further information and reagents should be directed to and will be fulfilled by Deborah Lannigan (deborah.lannigan@vumc.org).
Materials Availability
This study did not generate any new reagents.
Data and Code Availability
The accession number for the RNA sequencing reported in this paper is GEO: GSE113323.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
All procedures involving animals were done in accordance with current federal (NIH Guide for Care and Use of Laboratory Animals) and university guidelines and were approved by the University of Virginia and Vanderbilt University Institutional Animal Care and Use Committee.
Female WT or RSK2-KO mice (Yang et al., 2004) between six and fourteen weeks old were studied. The age of animals in specific experiments are indicated in the figures with adult animals ranging from twelve to fourteen weeks. For whole mount analysis the 4th mammary gland was fixed and stained in Carmine Alum. Ductal distance was measured from the nipple to the tip of the longest duct. The number of secondary branches along the longest primary branch were counted.
The stages of the estrous cycle were determined by cytological analysis of vaginal swabs (Wood et al., 2007; Byers et al., 2012). For all experiments requiring matched estrous stages, the cycles were monitored for 2 weeks prior to end point to ensure continuous cycling.
Mammary epithelial cells were isolated with modifications (Pasic et al., 2011). Briefly, mammary glands were isolated from donor mice, minced, and digested in DMEM/F12 supplemented with 2mg/ml Collagenase A and 100U/ml Pen/Strep for 2.5h in 37°C 5% CO2 incubator. Digested material was pelleted at 180 g for 5 min and the pellet was suspended in DNase I (1000U/ml) for 3–5 min in 37°C in 5% CO2. Fetal bovine serum (FBS) was added and the digested tissue was pelleted at 180 g for 10 min. The pellet was washed with phosphate-buffer saline, pelleted, suspended in Accumax (StemCell Technologies Inc.) and placed in Thermomixer at 37°C for 10 min. Digested material was pelleted at 180 g for 3 min, suspended in 5x trypsin for 5 min at 37°C. Trypsin was quenched with FBS and cells were pelleted and suspended in phosphate buffered saline (PBS) or DMEM/F12. The cell preparation was filtered through 70-μm mesh to obtain single cell suspensions. For mammary gland regenerations, 4×107 cells/ml of single cells in DMEM/F12 were mixed 1:1 with matrigel. 10 μL of cell suspension in matrigel was injected into the cleared 4th mammary fat pad of a recipient 3wk old mouse (Brill et al., 2008).
To inhibit the 26S proteasome pathway or RSK1/2 in vivo female mice in estrus (12 wk) were injected intraperitoneally (IP) with vehicle or PS-341 at 5 mg/kg in 2% DMSO, 30% PEG, and 68% saline or C5′′-n-propyl cyclitol SL0101 at 40mg/kg in one part DMSO and nine parts 25% hydroxypropyl-beta-cyclodextrin. Animals in the PS-341 study were euthanized 4h after injection and animals in the RSK1/2 study were injected twice at 7 h interval before euthanasia.
Cell Line Studies
TM3 cells were purchased and cultured according to ATCC. Cells were maintained in log-phase and screened for Mycoplasma by PCR. Prior to experiments, cells were serum-starved in phenol red-free media for 48 h followed by addition of vehicle, C5”-n-propyl-cyclitol SL0101 (20 μM, 6h), BI-D1870 (10 μM, 6h), trametinib (1 μM, 6h), or U0126 (10 μM, 6h). In experiments with MG-132, cells were pretreated (10 μM, 1h). For analysis of Ser-118 upshift, cells were serum-starved as above and treated with phorbol 12-myristate 13-acetate (PMA) (0.5 μM, 20 min), EGF and FGF7 cocktail (12.5 nM each, 5 min), C5” (20 μM for 2 h). In experiments with trametinib, cells were pretreated (1 μM, 2h). Cells were lysed and analyzed (Joel et al., 1998a).
METHOD DETAILS
Transduction
Constructs to generate lentivirus including psPAX2, pMD2.G, and pLVTHM were provided by D. Trono, Ph.D. (Swiss Institute of Technology, Lausanne, Switzerland). The pLV-Venus lentivirus construct was provided by Ian Macara, Ph.D. (Vanderbilt University, Nashville, TN). Lentiviral production was performed using Lipofectamine 3000 (Invitrogen) according to the manufacturer’s instructions. S118A-ERα and S167A-ERα were generated using Q5 site-directed mutagenesis.
Fluorescence Activated Cell Sorting (FACS)
For FACS, single epithelial cells (106 cells/ml) obtained from mammary glands in PBS were incubated with Cell Trace Violet (1 μM) and Zombie Yellow (1:250) for 20 min at room temperature. Cells were washed and suspended in5%FBS in PBS. Cells were blocked with 10% normal rat serum in 5% FBS for 10 min at 4°C, followed by incubation with biotin-conjugated primary antibodies against lineage markers for 10 min at 4°C. The cells were incubated with primary antibodies for 20 min at 4°C, washed and suspended in 5% FBS. Cells were analyzed using FACSCantoII or sorted using FACSAriaII. Flow cytometry data were analyzed using Cytobank version 6.2. Further reagents details are provided in the Key Resources Table.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rat anti-keratin 8 | University of Iowa | TROMA-I; RRID:AB_531826 |
| Chicken anti-keratin14 | BioLegend | SIG-3476; RRID:AB_10718041 |
| Rabbit anti-pRSK (Thr359/Ser363) (Tris) | Santa Cruz Biotechnology, Inc. | sc-12898-R; RRID:AB_2181303 |
| Mouse anti-ERα 6F11 (Citrate) | Thermo Fisher Scientific Inc. | MA5–13304; RRID:AB_11002193 |
| Mouse anti-γH2A.X (Ser139) (Tris) | EMD Millipore | JBW301; RRID:AB_568825 |
| Rabbit anit-pERK1/2 (pTEpY) (Tris) | Promega | V803A; RRID:AB_2335893 |
| Rabbit anti-peEF2 (Thr56) (Tris) | Cell Signal Technology | 2331; RRID:AB_10015204 |
| Mouse anti-GATA3 (Tris) | Thermo Fisher Scientific Inc. | 1A12–1D9; RRID:AB_2536713 |
| Rabbit anti-AR | Thermo Fisher Scientific Inc. | MA5–13426; RRID:AB_11000751 |
| Rabbit anti-E cadherin | Cell Signal Technology | 3195; RRID:AB_2291471 |
| Mouse anti-ERK | BD Biosciences | 610124; RRID:AB_397530 |
| Donkey anti-rabbit 647 | Invitrogen | A31573; RRID:AB_2536183 |
| Donkey anti-mouse 647 | Invitrogen | A31571; RRID:AB_162542 |
| Goat anti-rat 546 | Invitrogen | A11081; RRID:AB_2534125 |
| Goat anti-chicken 488 | Invitrogen | A11039; RRID:AB_2534096 |
| Biotin anti-CD140 | Biolegend | APA5; RRID:AB_11211998 |
| Biotin anti-CD31 | Biolegend | MEC13.3; RRID:AB_312910 |
| Biotin anti-Ter-119 | Biolegend | TER-119; RRID:AB_313704 |
| Biotin anti-CD45 | Biolegend | 30-F11; RRID:AB_312968 |
| Brilliant Violet 510 Streptavidin | Biolegend | 405233 |
| Anti-Sca1-PerCP | Biolegend | 108121; RRID:AB_893618 |
| Anti-Sca1-FITC | Biolegend | D7; RRID:AB_313342 |
| Anti-CD49b-APC/Cy7 | Biolegend | DX5; RRID:AB_313416 |
| Anti-EpCAM-APC | Biolegend | G8.8; RRID:AB_1134105 |
| Anti-CD49f-PE/Cy7 | Biolegend | GoH3; RRID:AB_2561704 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Cell Trace Violet | Life Technologies Corp. | C34557 |
| Zombie Yellow | Biolegend | 423104 |
| EdU (5-Ethynyl-2′-deoxyuridine) | Life Technologies Corp. | NEO87011604 |
| Bortezomib (PS-341) | Calbiochem | 50–431-40001 |
| BI-D1870 | Enzo Life Sciences | BML-EI407 |
| Trametinib | Selleck Chem | S2673 |
| U0126 | Sigma | U120 |
| MG-132 | Calbiochem | 474790 |
| 17-β estradiol (E2) | Sigma | E2758 |
| Phorbol 12-myristate 13-acetate (PMA) | Sigma | P1585 |
| EGF | Calbiochem | 324831 |
| FGF7 | R&D Systems | 251KG010CF |
| Critical Commercial Assays | ||
| Click-iT Plus EdU Alexa Fluor 488 Flow Cytometry Assay Kit | Thermo Fisher Scientific Inc. | C10632 |
| Click-iT Plus OPP Alexa Fluor 647 Protein Synthesis Assay Kit | Thermo Fisher Scientific Inc. | C10458 |
| CellROX Green Reagent | Thermo Fisher Scientific Inc. | C10444 |
| RNeasy Micro Kit | QIAGEN | 74004 |
| Deposited Data | ||
| RNA sequencing data | This paper | GEO: GSE113323 |
| Experimental Models: Cell Lines | ||
| Mouse: TM3 cell line | ATCC | CRL-1714 |
| Experimental Models: Organisms/Strains | ||
| Mouse: RSK2-KO: C57BL/6JRSK2−/− | Andre Hanauer, PhD. Institut de Genetique et Biologie Moleculaire et Cellulaire, C.U. de Strasbourg, France | N/A |
| Oligonucleotides | ||
| f-GAPDHm | AGAACATCATCCCTGCATCCA | N/A |
| r-GAPDHm | CAGATCCACGACGGACACATT | N/A |
| f-GATA3m | GATGTAAGTCGAGGCCCAAG | N/A |
| r-GATA3m | GCAGGCATTGCAAAGGTAGT | N/A |
| f-ESR1m | TTACGAAGTGGGCATGATGA | N/A |
| r-ESR1m | CCTGAAGCACCCATTTCATT | N/A |
| Recombinant DNA | ||
| pLVTHM | Wiznerowicz and Trono, 2003 | Addgene Cat #12247 |
| psPAX2 | Provided by Dr. Didier Trono | Addgene Cat #12260 |
| pMD2.G | Provided by Dr. Didier Trono | Addgene Cat #12259 |
| Software and Algorithms | ||
| LSM-FCS/ ZEN | Carl Zeiss, Inc. | N/A |
| Openlab 5.5.0 / Volocity 6.2.1 | PerkinElmer Inc. | N/A |
| GraphPad Prism 6.0a | GraphPad Spftware Inc. | N/A |
| Morpheus | Broad Institute | https://software.broadinstitute.org/morpheus/ |
| BioRender | BioRender | https://biorender.com/ |
EdU labeling was performed (Giraddi et al., 2015) in mice staged in proestrus were injected intraperitoneally with 10 mg/ml EdU in PBS (100 mg/kg) and then administered EdU in the drinking water (1mg/ml). The estrus stage was monitored, and mammary glands were isolated in metestrus (2 days after EdU injection). Mammary cells were isolated and analyzed for EdU incorporation using the Click-iT Edu Flow Cytometry Assay Kit, followed by the antibody staining as described above carried out in 1xClick-iT saponin based permeabilization buffer. Further reagents details are provided in the Key Resources Table.
Immunostaining
Mouse organs were fixed in buffered 10% formalin for 2 d and then placed in 70% ethanol. The fixed samples were paraffin-embedded, and sectioned. Sections were deparaffinized and antigen retrieval performed in tris-EDTA buffer pH 8.0 or citrate buffer pH 6.0 or pH 7.0 (Key Resources Table). The sections were blocked in 10% bovine serum albumin (BSA) in PBS and incubated with primary antibody in 3% BSA in PBS o/n at 4°C. The sections were washed and incubated with secondary antibody for 1 h in room temperature. For detection of Venus-tagged ERα in TM3 cells, 1×104 cells were seeded on laminin-coated glass coverslips. After treatment, cells were fixed in 4% PFA in PBS (pH 7.4, 15 min). Antibodies are listed in the Key Resources Table. For immunofluorescence staining, cells were fixed in 4% PFA in PBS (pH 7.4, 15 min) and permeabilized with 0.1% Triton X-100 in PBS (15 min), DNA was stained with Hoechst in PBS (10 min) and coverslips mounted using Fluoro-Gel (Electron Microscopy Sciences). Images were collected with a laser-scanning microscope (LSM 510/Meta/FCS, Carl Zeiss Inc.).
RNA Analysis
For RNA isolation, 5×104 EpCAMhiCD49f+Sca1+Cd49b− cells were FACS sorted and total RNA extraction (RNeasy Micro Kit) was performed. The RNA quality was tested using Agilent 100 Bioanalyzer (RIN 8). Libraries were constructed and sequenced by Genewiz LLC. Reads were aligned to the mm10 mouse genome with STAR, the transcripts were assembled using Gencode version 15 as gene models. Genes and transcripts were quantified with HTSeq. Two samples were clear outliers and were discarded. Batch correction was done with SVA, and differential gene expression analysis was performed with DESeq2. Gene set enrichment was done with GSEA using MSigDB and GSVA using GSKB mouse gene sets. RNASeq data is available at Gene Expression Omnibus under accession GSE113323.
For qRT-PCR RNA (1 μg) was reverse transcribed using High Capacity cDNA Reverse Transcription Kit. Analysis was performed using IQ RealTime SyberGreen PCR Supermix (BioRad Laboratories) on the C1000Thermal Cycler CFX96 Real-Time System (Ludwik et al., 2018). The ΔΔCt was calculated using GAPDH as a control. Primers are listed in the Key Resources Table.
Raw reads from the sequencing of normal breast tissue at different stages of menstrual cycle (Pardo et al., 2014) were normalized using DESeq2 according to the estimated size of the libraries. Based on unsupervised hierarchical clustering, 5 samples were rejected as outliers and Z-scores were calculated correcting for sequencing batch.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses were performed using GraphPad Prism 6. The statistical test used is reported in the figure legends. Additional ANOVA values for complex comparisons are provided (Table S1).
Supplementary Material
Highlights.
ERK1/2-RSK2 signaling maintains estrogen homeostasis in the adult mouse
ERK1/2 drives ERα degradation and estrogen-mediated transcription
RSK2 prevents sustained ERK1/2 activity by inhibiting ROS production
Oral contraceptives correlate with lowered RSK2 and an enhanced estrogen response
ACKNOWLEDGMENTS
We would like to thank Andre Hanauer (IGBMC) for providing the RSK2-KO mice (Institut de Genetique et Biologie Moleculaire et Cellulaire, C.U. de Strasbourg) (Yang et al., 2004). The cytokeratin Endo-A (K8) monoclonal developed by Philippe Brulet and Rolf Kemler was obtained from the Developmental Studies Hybridoma Bank, developed under the auspices of the NICHD, and maintained by the Department of Biology, University of Iowa, Iowa City, IA. Data generated by the TCGA Research Network: https://www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga was used. This work was supported by NIH grant DK113423 (D.A.L.), CA213201 (D.A.L.), AI144196 (G.A.O), AI142040 (G.A.O), and NSF grant CHE-1565788 (G.A.O.).
Footnotes
DECLARATION OF INTERESTS
The authors D.A.L. and G.A.O. have a patent related to this work.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.107931.
REFERENCES
- Barros RP, and Gustafsson JA (2011). Estrogen receptors and the metabolic network. Cell Metab. 14, 289–299. [DOI] [PubMed] [Google Scholar]
- Blair RM, Fang H, Branham WS, Hass BS, Dial SL, Moland CL, Tong W, Shi L, Perkins R, and Sheehan DM (2000). The estrogen receptor relative binding affinities of 188 natural and xenochemicals: structural diversity of ligands. Toxicol. Sci 54, 138–153. [DOI] [PubMed] [Google Scholar]
- Brill B, Boecher N, Groner B, and Shemanko CS (2008). A sparing procedure to clear the mouse mammary fat pad of epithelial components for transplantation analysis. Lab. Anim 42, 104–110. [DOI] [PubMed] [Google Scholar]
- Brisken C, and Ataca D (2015). Endocrine hormones and local signals during the development of the mouse mammary gland. Wiley Interdiscip. Rev. Dev. Biol 4, 181–195. [DOI] [PubMed] [Google Scholar]
- Bunone G, Briand PA, Miksicek RJ, and Picard D (1996). Activation of the unliganded estrogen receptor by EGF involves the MAP kinase pathway and direct phosphorylation. EMBO J. 15, 2174–2183. [PMC free article] [PubMed] [Google Scholar]
- Byers SL, Wiles MV, Dunn SL, and Taft RA (2012). Mouse estrous cycle identification tool and images. PLoS ONE 7, e35538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Riedl T, Washbrook E, Pace PE, Coombes RC, Egly JM, and Ali S (2000). Activation of estrogen receptor alpha by S118 phosphorylation involves a ligand-dependent interaction with TFIIH and participation of CDK7. Mol. Cell 6, 127–137. [PubMed] [Google Scholar]
- Beral V; Collaborative Group on Hormonal Factors in Breast Cancer (1996). Breast cancer and hormonal contraceptives: further results. Contraception 54 (3, Suppl), 1S–106S. [DOI] [PubMed] [Google Scholar]
- Dalby KN, Morrice N, Caudwell FB, Avruch J, and Cohen P (1998). Identification of regulatory phosphorylation sites in mitogen-activated protein kinase (MAPK)-activated protein kinase-1a/p90rsk that are inducible by MAPK. J. Biol. Chem 273, 1496–1505. [DOI] [PubMed] [Google Scholar]
- Denu JM, and Tanner KG (1998). Specific and reversible inactivation of protein tyrosine phosphatases by hydrogen peroxide: evidence for a sulfenic acid intermediate and implications for redox regulation. Biochemistry 37, 5633–5642. [DOI] [PubMed] [Google Scholar]
- Deutsch MB, Bhakri V, and Kubicek K (2015). Effects of cross-sex hormone treatment on transgender women and men. Obstet. Gynecol 125, 605–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutertre M, Gratadou L, Dardenne E, Germann S, Samaan S, Lidereau R, Driouch K, de la Grange P, and Auboeuf D (2010). Estrogen regulation and physiopathologic significance of alternative promoters in breast cancer. Cancer Res. 70, 3760–3770. [DOI] [PubMed] [Google Scholar]
- Eeckhoute J, Keeton EK, Lupien M, Krum SA, Carroll JS, and Brown M (2007). Positive cross-regulatory loop ties GATA-3 to estrogen receptor alpha expression in breast cancer. Cancer Res. 67, 6477–6483. [DOI] [PubMed] [Google Scholar]
- Eisinger-Mathason TS, Andrade J, and Lannigan DA (2010). RSK in tumorigenesis: connections to steroid signaling. Steroids 75, 191–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fata JE, Chaudhary V, and Khokha R (2001). Cellular turnover in the mammary gland is correlated with systemic levels of progesterone and not 17betaestradiol during the estrous cycle. Biol. Reprod 65, 680–688. [DOI] [PubMed] [Google Scholar]
- Feng Y, Manka D, Wagner KU, and Khan SA (2007). Estrogen receptor-alpha expression in the mammary epithelium is required for ductal and alveolar morphogenesis in mice. Proc. Natl. Acad. Sci. USA 104, 14718–14723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filipčík P, Curry JR, and Mace PD (2017). When worlds collide-mechanisms at the interface between phosphorylation and ubiquitination. J. Mol. Biol 429, 1097–1113. [DOI] [PubMed] [Google Scholar]
- Förster C, Mäkela S, Wärri A, Kietz S, Becker D, Hultenby K, Warner M, and Gustafsson JA (2002). Involvement of estrogen receptor beta in terminal differentiation of mammary gland epithelium. Proc. Natl. Acad. Sci. USA 99, 15578–15583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giraddi RR, Shehata M, Gallardo M, Blasco MA, Simons BD, and Stingl J (2015). Stem and progenitor cell division kinetics during postnatal mouse mammary gland development. Nat. Commun 6, 8487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González L, Zambrano A, Lazaro-Trueba I, Lopéz E, González JJ, Martín-Pérez J, and Aranda A (2009). Activation of the unliganded estrogen receptor by prolactin in breast cancer cells. Oncogene 28, 1298–1308. [DOI] [PubMed] [Google Scholar]
- Hunter DJ, Colditz GA, Hankinson SE, Malspeis S, Spiegelman D, Chen W, Stampfer MJ, and Willett WC (2010). Oral contraceptive use and breast cancer: a prospective study of young women. Cancer Epidemiol. Biomarkers Prev 19, 2496–2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hynes NE, and Watson CJ (2010). Mammary gland growth factors: roles in normal development and in cancer. Cold Spring Harb. Perspect. Biol 2, a003186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia M, Dahlman-Wright K, and Gustafsson JA (2015). Estrogen receptor alpha and beta in health and disease. Best Pract. Res. Clin. Endocrinol. Metab 29, 557–568. [DOI] [PubMed] [Google Scholar]
- Jiang J, Sarwar N, Peston D, Kulinskaya E, Shousha S, Coombes RC, and Ali S (2007). Phosphorylation of estrogen receptor-alpha at Ser167 is indicative of longer disease-free and overall survival in breast cancer patients. Clin. Cancer Res. 13, 5769–5776. [DOI] [PubMed] [Google Scholar]
- Joel PB, Smith J, Sturgill TW, Fisher TL, Blenis J, and Lannigan DA (1998a). pp90rsk1 regulates estrogen receptor-mediated transcription through phosphorylation of Ser-167. Mol. Cell. Biol 18, 1978–1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joel PB, Traish AM, and Lannigan DA (1998b). Estradiol-induced phosphorylation of serine 118 in the estrogen receptor is independent of p42/p44 mitogen-activated protein kinase. J. Biol. Chem 273, 13317–13323. [DOI] [PubMed] [Google Scholar]
- Kato S, Endoh H, Masuhiro Y, Kitamoto T, Uchiyama S, Sasaki H, Masushige S, Gotoh Y, Nishida E, Kawashima H, et al. (1995). Activation of the estrogen receptor through phosphorylation by mitogen-activated protein kinase. Science 270, 1491–1494. [DOI] [PubMed] [Google Scholar]
- Kelleher AM, Burns GW, Behura S, Wu G, and Spencer TE (2016). Uterine glands impact uterine receptivity, luminal fluid homeostasis and blastocyst implantation. Sci. Rep 6, 38078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Li Y, Ludwik KA, Sandusky ZM, Lannigan DA, and O’Doherty GA (2017). Stereoselective synthesis and evaluation of C6′′-substituted 5acarbasugar analogues of SL0101 as inhibitors of RSK1/2. Org. Lett 19, 2410–2413. [DOI] [PubMed] [Google Scholar]
- Lonard DM, Nawaz Z, Smith CL, and O’Malley BW (2000). The 26S proteasome is required for estrogen receptor-alpha and coactivator turnover and for efficient estrogen receptor-alpha transactivation. Mol. Cell 5, 939–948. [DOI] [PubMed] [Google Scholar]
- Lovett JL, Chima MA, Wexler JK, Arslanian KJ, Friedman AB, Yousif CB, and Strassmann BI (2017). Oral contraceptives cause evolutionarily novel increases in hormone exposure: A risk factor for breast cancer. Evol. Med. Public Health 2017, 97–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwik KA, Campbell JP, Li M, Li Y, Sandusky ZM, Pasic L, Sowder ME, Brenin DR, Pietenpol JA, O’Doherty GA, and Lannigan DA (2016). Development of a RSK inhibitor as a novel therapy for triple-negative breast cancer. Mol. Cancer Ther 15, 2598–2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwik KA, McDonald OG, Brenin DR, and Lannigan DA (2018). ERα-mediated nuclear sequestration of RSK2 is required for ER+ breast cancer tumorigenesis. Cancer Res. 78, 2014–2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maennling AE, Tur MK, Niebert M, Klockenbring T, Zeppernick F, Gattenlöhner S, Meinhold-Heerlein I, and Hussain AF (2019). Molecular targeting therapy against EGFR family in breast cancer: progress and future potentials. Cancers (Basel) 11, 1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta RG, Hawthorne M, Mehta RR, Torres KE, Peng X, McCormick DL, and Kopelovich L (2014). Differential roles of ERα and ERβ in normal and neoplastic development in the mouse mammary gland. PLoS ONE 9, e113175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng TC, Fukada T, and Tonks NK (2002). Reversible oxidation and inactivation of protein tyrosine phosphatases in vivo. Mol. Cell 9, 387–399. [DOI] [PubMed] [Google Scholar]
- Moon HG, Yi JK, Kim HS, Lee HY, Lee KM, Yi M, Ahn S, Shin HC, Ju JH, Shin I, et al. (2012). Phosphorylation of p90RSK is associated with increased response to neoadjuvant chemotherapy in ER-positive breast cancer. BMC Cancer 12, 585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mørch LS, Skovlund CW, Hannaford PC, Iversen L, Fielding S, and Lidegaard Ø (2017). Contemporary hormonal contraception and the risk of breast cancer. N. Engl. J. Med 377, 2228–2239. [DOI] [PubMed] [Google Scholar]
- Nawaz Z, Lonard DM, Dennis AP, Smith CL, and O’Malley BW (1999). Proteasome-dependent degradation of the human estrogen receptor. Proc. Natl. Acad. Sci. USA 96, 1858–1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardo I, Lillemoe HA, Blosser RJ, Choi M, Sauder CA, Doxey DK, Mathieson T, Hancock BA, Baptiste D, Atale R, et al. ; Susan G. Komen for the Cure Tissue Bank at the IU Simon Cancer Center (2014). Next-generation transcriptome sequencing of the premenopausal breast epithelium using specimens from a normal human breast tissue bank. Breast Cancer Res. 16, R26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J, and Lee Y (2017). Hypoxia induced phosphorylation of estrogen receptor at serine 118 in the absence of ligand. J. Steroid Biochem. Mol. Biol 174, 146–152. [DOI] [PubMed] [Google Scholar]
- Pasic L, Eisinger-Mathason TS, Velayudhan BT, Moskaluk CA, Brenin DR, Macara IG, and Lannigan DA (2011). Sustained activation of the HER1-ERK1/2-RSK signaling pathway controls myoepithelial cell fate in human mammary tissue. Genes Dev. 25, 1641–1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perillo B, Ombra MN, Bertoni A, Cuozzo C, Sacchetti S, Sasso A, Chiariotti L, Malorni A, Abbondanza C, and Avvedimento EV (2008). DNA oxidation as triggered by H3K9me2 demethylation drives estrogen-induced gene expression. Science 319, 202–206. [DOI] [PubMed] [Google Scholar]
- Pinter O, Beda Z, Csaba Z, and Gerendai I (2007). Differences in the onset of puberty in selected inbred mouse strains. Endocr. Abstr 14, P617. [Google Scholar]
- Reid G, H€ubner MR, Métivier R, Brand H, Denger S, Manu D, Beaudouin J, Ellenberg J, and Gannon F (2003). Cyclic, proteasome-mediated turnover of unliganded and liganded ERalpha on responsive promoters is an integral feature of estrogen signaling. Mol. Cell 11, 695–707. [DOI] [PubMed] [Google Scholar]
- Robertshaw I, Bian F, and Das SK (2016). Mechanisms of uterine estrogen signaling during early pregnancy in mice: an update. J. Mol. Endocrinol 56, R127–R138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romieu I, Hernandez-Avila M, and Liang MH (1989). Oral contraceptives and the risk of rheumatoid arthritis: a meta-analysis of a conflicting literature. Br. J. Rheumatol 28 (Suppl 1), 13–17, discussion 18–23. [DOI] [PubMed] [Google Scholar]
- Roux PP, and Topisirovic I (2018). Signaling pathways involved in the regulation of mRNA translation. Mol. Cell. Biol 38, e00070–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharfman HE, and MacLusky NJ (2008). Estrogen-growth factor interactions and their contributions to neurological disorders. Headache 48 (Suppl 2), S77–S89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shehata M, Teschendorff A, Sharp G, Novcic N, Russell IA, Avril S, Prater M, Eirew P, Caldas C, Watson CJ, and Stingl J (2012). Phenotypic and functional characterisation of the luminal cell hierarchy of the mammary gland. Breast Cancer Res. 14, R134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigurgeirsson B, Åmark H, Jemt A, Ujvari D, Westgren M, Lundeberg J, and Gidlöf S (2017). Comprehensive RNA sequencing of healthy human endometrium at two time points of the menstrual cycle. Biol. Reprod 96, 24–33. [DOI] [PubMed] [Google Scholar]
- Silberstein GB, Van Horn K, Hrabeta-Robinson E, and Compton J (2006). Estrogen-triggered delays in mammary gland gene expression during the estrous cycle: evidence for a novel timing system. J. Endocrinol 190, 225–239. [DOI] [PubMed] [Google Scholar]
- Simin J, Tamimi R, Lagergren J, Adami HO, and Brusselaers N (2017). Menopausal hormone therapy and cancer risk: An overestimated risk? Eur. J. Cancer 84, 60–68. [DOI] [PubMed] [Google Scholar]
- Tanaka T, Halicka HD, Huang X, Traganos F, and Darzynkiewicz Z (2006). Constitutive histone H2AX phosphorylation and ATM activation, the reporters of DNA damage by endogenous oxidants. Cell Cycle 5, 1940–1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian D, Solodin NM, Rajbhandari P, Bjorklund K, Alarid ET, and Kreeger PK (2015). A kinetic model identifies phosphorylated estrogen receptor-α (ERα) as a critical regulator of ERα dynamics in breast cancer. FASEB J. 29, 2022–2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlen M, Zhang C, Lee S, Sjöstedt E, Fagerberg L, Bidkhori G, Benfeitas R, Arif M, Liu Z, Edfors F, et al. (2017). A pathology atlas of the human cancer transcriptome. Science 357, eaan2507. [DOI] [PubMed] [Google Scholar]
- Valley CC, Métivier R, Solodin NM, Fowler AM, Mashek MT, Hill L, and Alarid ET (2005). Differential regulation of estrogen-inducible proteolysis and transcription by the estrogen receptor alpha Nterminus. Mol. Cell. Biol 25, 5417–5428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valley CC, Solodin NM, Powers GL, Ellison SJ, and Alarid ET (2008). Temporal variation in estrogen receptor-alpha protein turnover in the presence of estrogen. J. Mol. Endocrinol 40, 23–34. [DOI] [PubMed] [Google Scholar]
- Vogel PM, Georgiade NG, Fetter BF, Vogel FS, and McCarty KS Jr. (1981). The correlation of histologic changes in the human breast with the menstrual cycle. Am. J. Pathol 104, 23–34. [PMC free article] [PubMed] [Google Scholar]
- Walmer DK, Wrona MA, Hughes CL, and Nelson KG (1992). Lactoferrin expression in the mouse reproductive tract during the natural estrous cycle: correlation with circulating estradiol and progesterone. Endocrinology 131, 1458–1466. [DOI] [PubMed] [Google Scholar]
- Wang X, Li W, Williams M, Terada N, Alessi DR, and Proud CG (2001). Regulation of elongation factor 2 kinase by p90(RSK1) and p70 S6 kinase. EMBO J. 20, 4370–4379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weitsman GE, Weebadda W, Ung K, and Murphy LC (2009). Reactive oxygen species induce phosphorylation of serine 118 and 167 on estrogen receptor alpha. Breast Cancer Res. Treat 118, 269–279. [DOI] [PubMed] [Google Scholar]
- Wentworth CC, Alam A, Jones RM, Nusrat A, and Neish AS (2011). Enteric commensal bacteria induce extracellular signal-regulated kinase pathway signaling via formyl peptide receptor-dependent redox modulation of dual specific phosphatase 3. J. Biol. Chem 286, 38448–38455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winuthayanon W, Hewitt SC, and Korach KS (2014). Uterine epithelial cell estrogen receptor alpha-dependent and -independent genomic profiles that underlie estrogen responses in mice. Biol. Reprod 91, 110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiznerowicz M, and Trono D (2003). Conditional suppression of cellular genes: lentivirus vector-mediated drug-inducible RNA interference. J. Virol 77, 8957–8961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood GA, Fata JE, Watson KL, and Khokha R (2007). Circulating hormones and estrous stage predict cellular and stromal remodeling in murine uterus. Reproduction 133, 1035–1044. [DOI] [PubMed] [Google Scholar]
- Xie W, Paterson AJ, Chin E, Nabell LM, and Kudlow JE (1997). Targeted expression of a dominant negative epidermal growth factor receptor in the mammary gland of transgenic mice inhibits pubertal mammary duct development. Mol. Endocrinol 11, 1766–1781. [DOI] [PubMed] [Google Scholar]
- Yang X, Matsuda K, Bialek P, Jacquot S, Masuoka HC, Schinke T, Li L, Brancorsini S, Sassone-Corsi P, Townes TM, et al. (2004). ATF4 is a substrate of RSK2 and an essential regulator of osteoblast biology; implication for Coffin-Lowry Syndrome. Cell 117, 387–398. [DOI] [PubMed] [Google Scholar]
- Yang XR, Figueroa JD, Hewitt SM, Falk RT, Pfeiffer RM, Lissowska J, Peplonska B, Brinton LA, Garcia-Closas M, and Sherman ME (2013). Estrogen receptor and progesterone receptor expression in normal terminal duct lobular units surrounding invasive breast cancer. Breast Cancer Res. Treat 137, 837–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Sun L, Liang J, Yu W, Zhang Y, Wang Y, Chen Y, Li R, Sun X, and Shang Y (2006). The catalytic subunit of the proteasome is engaged in the entire process of estrogen receptor-regulated transcription. EMBO J. 25, 4223–4233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Fu Y, Ju J, Wan D, Su H, Wang Z, Rui H, Jin Q, Le Y, and Hou R (2019). Estradiol inhibits fMLP-induced neutrophil migration and superoxide production by upregulating MKP-2 and dephosphorylating ERK. Int. Immunopharmacol 75, 105787. [DOI] [PubMed] [Google Scholar]
- Zhou W, and Slingerland JM (2014). Links between oestrogen receptor activation and proteolysis: relevance to hormone-regulated cancer therapy. Nat. Rev. Cancer 14, 26–38. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the RNA sequencing reported in this paper is GEO: GSE113323.