

SUMMARY
Small molecule neurotensin receptor 1 (NTSR1) agonists have been pursued for more than 40 years as potential therapeutics for psychiatric disorders, including drug addiction. Clinical development of NTSR1 agonists has, however, been precluded by their severe side effects. NTSR1, a G protein-coupled receptor (GPCR), signals through the canonical activation of G proteins and engages β-arrestins to mediate distinct cellular signaling events. Here, we characterize the allosteric NTSR1 modulator SBI-553. This small molecule not only acts as a β-arrestin biased agonist, but also extends profound β-arrestin bias to the endogenous ligand by selectively antagonizing G protein signaling. SBI-553 shows efficacy in animal models of psychostimulant abuse, including cocaine self-administration, without the side effects characteristic of balanced NTSR1 agonism. These findings indicate that NTSR1 G protein and β-arrestin activation produce discrete and separable physiological effects, thus providing a strategy to develop safer GPCR-targeting therapeutics with more directed pharmacological action.
Graphical Abstract

INTRODUCTION
Drug addiction is a global public health concern for which there is a paucity of effective therapeutics. Addiction is a chronic, relapsing disorder characterized by maladaptive changes in the brain mesolimbic dopamine system. The neurotransmitter dopamine mediates reward processing and learning through action at D1 and D2 receptors (D1Rs and D2Rs). Mounting evidence suggests that dysregulation of central dopaminergic neurotransmission, particularly through D2R, is a key contributor to the abuse of psychostimulant and opioid drugs (Baicy and London, 2007; Groman et al., 2012; Parsegian and See, 2014).
A long-standing idea to combat aberrant, drug-induced dopamine signaling in the brain has been to engage modulatory neuropeptide receptors. Anatomical and functional data indicate that pharmacologically targeting the neuropeptide neurotensin (NTS) and its high affinity G protein-coupled receptor (GPCR) NTSR1 may restore homeostatic dopamine signaling (Binder et al., 2001; Ferraro et al., 2016). Indeed, NTSR1 peptide agonists directly oppose the classical effects associated with psychostimulant abuse (e.g., hyperactivity, neurotoxicity, psychotic episodes, and cognitive deficits) (Ferraro et al., 2016; St-Gelais et al., 2006). Although the precise mechanisms for these effects are not known, NTSR1 inhibits brain dopamine signaling through antagonistic functional and putative physical interactions with the D2R (Binder et al., 2001; Borroto-Escuela et al., 2013).
In addition to regulating central dopaminergic neurotransmission, NTSR1 modulates fundamental physiological outputs, including body temperature, blood pressure, and motor control. Thus, addiction therapies that engage NTSR1 could potentially produce severe, dose-limiting side effects, including hypotension (Carraway and Leeman, 1973), hypothermia (Bissette et al., 1976; Kitabgi et al., 1992), and impaired motor coordination (Osbahr et al., 1979; Pettibone et al., 2002). Overcoming this therapeutic dilemma requires a mechanistic understanding of how NTSR1 mediates its diverse physiological effects, and the identification of agents that are biased towards promoting one effect, regulating dopamine signaling, over the others.
As a GPCR, NTSR1 signals through the canonical activation of G proteins and engages β-arrestin proteins to mediate distinct cellular signaling events. The two β-arrestins, β-arrestin1 and β-arrestin2, also known as arrestin 2 and arrestin 3, are ubiquitously expressed intracellular regulators of GPCR trafficking and desensitization that, more recently, have been identified as independent mediators of GPCR signaling (Smith and Rajagopal, 2016). For the GPCRs for which this has been examined, G protein and β-arrestin signaling have been found to mediate distinct cellular and physiological functions (Lefkowitz and Shenoy, 2005; Raehal et al., 2005; Urs et al., 2016). Preclinical data have demonstrated the ability of β-arrestin2 to regulate addiction-associated behaviors in rodents (Bohn et al., 1999, 2000; Porter-Stransky and Weinshenker, 2017), and human genetic results support a role for β-arrestins, particularly β-arrestin2, in methamphetamine (Ikeda et al., 2007), nicotine (Sun et al., 2008), and opioid (Oneda et al., 2011) use disorders.
Given the evidence for a role of β-arrestin2 in addiction, we initiated a β-arrestin-based NTSR1 screening program. This program identified a series of small molecule NTSR1 ligands (Peddibhotla et al., 2013) that displayed β-arrestin bias and activity in vivo (Barak et al., 2016). The utility of these ligands, even as tool compounds, however, was limited by their low potency, poor aqueous solubility, and undesirable pharmacokinetic profiles in rodents (Hefti, 2008). A medicinal chemistry optimization of the active scaffold has now led to the current, orally available, brain-penetrant lead compound, SBI-0654553 (SBI-553, Pinkerton et al., 2019).
In the present study, we demonstrate that SBI-553 is a β-arrestin-biased allosteric NTSR1 activator with the ability to bias NTS-occupied NTSR1 against Gq protein signaling and towards β-arrestin recruitment. In addition to acting as a β-arrestin biased ligand itself, SBI-553 extends functional selectivity to NTS and, with it, more directed pharmacological action in vivo. Remarkably, SBI-553 treatment attenuates psychostimulant-associated behaviors without the concurrent hypothermia, hypotension, and motor impairment characteristic of balanced, peptide NTSR1 agonism. These findings indicate that NTSR1 G protein and β-arrestin signaling mediate distinct physiological effects and identify a distinctive class of compounds that transition the boundaries between agonist and antagonist, activator and modulator.
RESULTS
SBI-553 is an allosteric NTSR1 ligand that increases NTS affinity and receptor occupancy
SBI-553 was derived from a medicinal chemistry campaign to improve a related NTSR1 probe series. SBI-553 was selected for further study based on its potency, pharmacokinetic profile, and selectivity for NTSR1 over the neurotensin receptor 2 (Pinkerton et al. 2019). We observed that SBI-553 binds NTSR1 at an allosteric site and exhibits the binding characteristics of a positive allosteric modulator (PAM). Binding studies of the NTSR1 in HEK293T cell membranes (see Fig S1A–C for assay validation) revealed specific [3H]SBI-553 binding comprised ~50% of total binding under 400 nM (Fig 1A). Nonspecific binding at higher [3H]SBI-553 concentrations precluded an affinity (Kd) determination. [3H]SBI-553 specific binding to NTSR1 was confirmed further by comparing binding to NTSR1-containing membranes with that to membranes containing an off-target receptor (i.e., β2-adrenergic receptor) (Fig S1D–E). Notably, [3H]SBI-553 was not displaced from NTSR1 by the competitive, orthosteric NTSR1/2 antagonist SR142948A or unlabeled NTS (Fig S1G). Rather, NTS increased total [3H]SBI-553 binding, and, conversely, SBI-553 increased total [3H]NTS binding (Fig S1F–G). These behaviors are most simply explained by the simultaneous, co-operative binding of NTS to the NTSR1 orthosteric site and SBI-553 to a distinct allosteric location (Canals et al., 2012). SBI-553 behaved as an NTSR1 PAM, dose-dependently increasing both NTS affinity for NTSR1 (Kd, up to 3.9-fold) and the number of available [3H]NTS binding sites (Bmax, up to 3.3-fold) (Fig 1B; Table S1).
Figure 1. SBI-553 activates NTSR1 to stimulate β-arrestin-associated cellular responses.
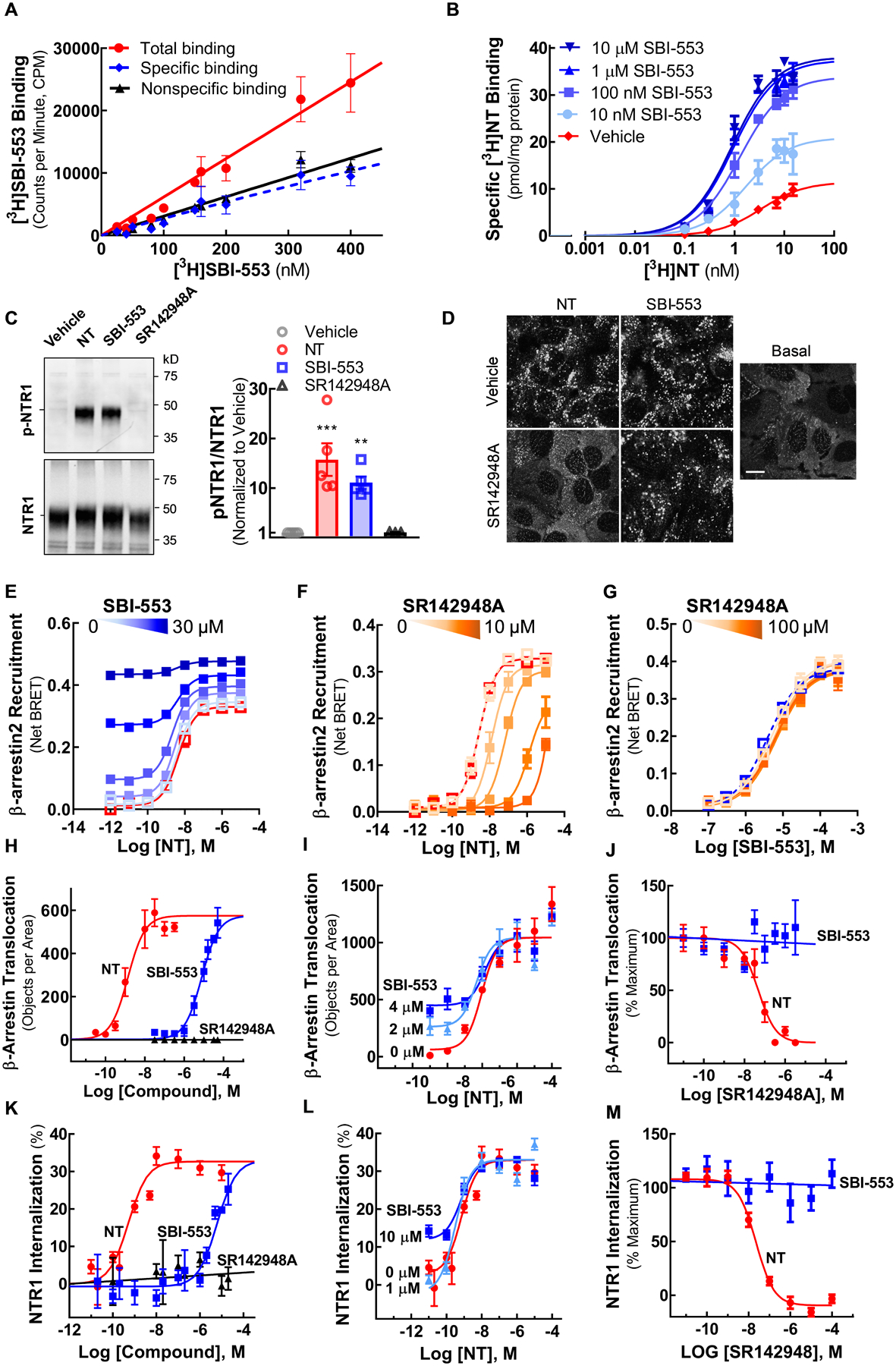
(A) [3H]SBI-553 saturation binding to NTSR1-containing HEK293T cell membranes.
(B) [3H]NTS saturation binding to NTSR1-containing HEK293T cell membranes in the absence (vehicle) and presence of SBI-553 (0.01 – 10 μM).
(C) Western blot analysis of NTSR1 phosphorylation. NTSR1-expressing HEK293T cells were treated with vehicle, NTS (10 nM), SBI-553 (10 μM) or SR142948A (10 μM). Phosphorylation of NTSR1 at threonine residues (p-NTSR1) was assessed by anti-phosphothreonine antibody.
(D) Confocal imaging of β-arrestin2 translocation to NTSR1. U2OS cells expressing NTSR1 and β-arrestin2-GFP without stimulation (Basal) and following treatment with vehicle or 30 μM SR142948A in combination with 10 nM NTS or 10 μM SBI-553. Scale bar = 10 μm.
(E-G) BRET-based assay of β-arrestin2 recruitment to NTSR1 in HEK293T cells expressing NTSR1-Rluc and β-arrestin2-Venus. (E) β-arrestin2 recruitment to NTSR1 induced by co-treatment with NTS and Vehicle or 0.03, 0.3, 1, 3, or 30 μM SBI-553. (F) NTS-induced β-arrestin2 recruitment to NTSR1 in the presence of Vehicle or 0.001, 0.01, 0.1, 1, or 10 μM SR142948A. (G) SBI-553-induced β-arrestin2 recruitment to NTSR1 in the presence of Vehicle or 0.01, 0.1, 1, 10 or 100 μM SR142948A. Increasing concentrations are indicated by increasing color intensity. NTS + Vehicle -- Red line.
(H-J) Confocal microscopy-based assay of β-arrestin2 translocation to NTSR1 in U2OS cells expressing NTSR1 and β-arrestin2-GFP. (H) Ligand induced-β-arrestin2 translocation to NTSR1 with NTS, SBI-553, or SR1425948A. (I) β-arrestin2 translocation to NTSR1 induced by co-treatment with NTS and SBI-553. (J) β-arrestin2 translocation to NTSR1 induced by NTS (5 nM) or SBI-553 (10 μM) in the presence of SR142948A.
(K-L) U2OS cells transfected with a fluorogen-activated protein (FAP, MarsCy1) tagged-NTSR1 and imaged after fluorogen treatment. (K) Dose-response of ligand-induced NTSR1 internalization. (L) NTSR1 internalization induced by co-treatment with NTS and SBI-553. Cells were pretreated with SBI-553 (0, 1 or 10 μM) prior to addition of NTS. (M) NTSR1 internalization induced by NTS (10 nM) or SBI-553 (10 μM) in the presence of SR142948A.
For curve parameters and details on statistical comparisons, see Table S1.
SBI-553 stimulates NTSR1 phosphorylation, β-arrestin recruitment and receptor internalization
Most rhodopsin family GPCRs require agonist-mediated phosphorylation by a G protein-coupled receptor kinase (GRK) prior to β-arrestin binding and subsequent β-arrestin-directed trafficking to clathrin-coated pits and endocytic vesicles (Ferguson et al., 1996; Goodman et al., 1996; Oakley et al., 2001). NTSR1 is phosphorylated by GRKs at C-terminal serine and C-terminal and intracellular loop 3 threonine residues (Inagaki et al., 2015). Here, we demonstrate that SBI-553 is as effective as NTS at stimulating NTSR1 phosphorylation, β-arrestin2 recruitment, and receptor internalization.
As shown by the phosphothreonine-NTSR1 blot (Fig 1C), the addition of either NTS or SBI-553, but not the competitive NTSR1 antagonist SR142948A, to NTSR1-expressing HEK293T cells resulted in NTSR1 phosphorylation. In NTSR1-expressing U2OS cells containing fluorescent protein-tagged β-arrestin2, addition of either NTS or SBI-553 stimulated β-arrestin2 translocation to the plasma membrane (Fig 1D,H) while SR142948A was without effect (Fig 1H). Neither NTS nor SBI-553 treatment stimulated β-arrestin2 translocation in U2OS cells lacking NTSR1 (Fig 2S). In a bioluminescence resonance energy transfer (BRET) assay assessing β-arrestin2 recruitment to NTSR1, addition of either NTS or SBI-553, but not SR142948A, stimulated β-arrestin2 recruitment (Fig 1E–G). Similarly, in NTSR1-expressing HEK293T cells, addition of either NTS or SBI-553, but not SR142948A, stimulated NTSR1 internalization (Fig 1K,L). Hence, both NTS and SBI-553 stimulate NTSR1 phosphorylation, β-arrestin2 translocation and recruitment, and receptor internalization.
Figure 2. SBI-553 does not stimulate NTSR1 Gq activation and antagonizes NT-induced Gq signaling.
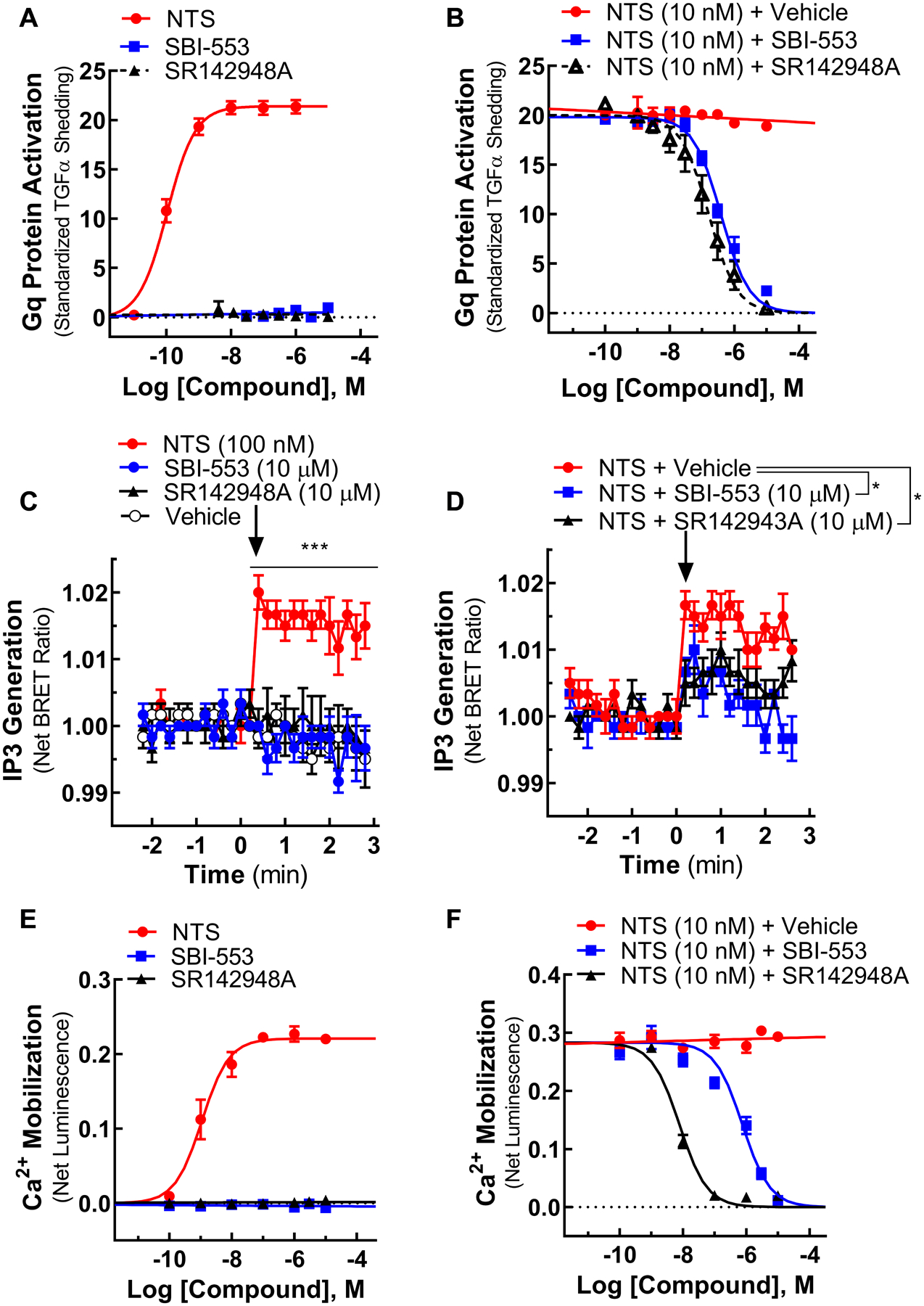
(A) Gq protein activation in the TGFα shedding assay induced by individual NTSR1 ligands.
(B) Gq activation induced by NTS in the presence of Vehicle, SBI-553, or SR142948A.
(C) Inositol 1,4,5-triphosphate (IP3) generation in an intramolecular BRET assay induced by individual NTSR1 ligands.
(D) IP3 generation induced by NTS in the presence of Vehicle, SR142948A or SBI-553.
(E) Calcium (Ca2+) mobilization in an aequorin assay induced by individual NTSR1 ligands.
(F) Ca2+ mobilization induced by NTS in the presence of Vehicle, SBI-553, or SR142948A.
For curve parameters and details on statistical comparisons, see Table S2.
To determine whether NTS-induced β-arrestin2 recruitment to and internalization of NTSR1 were influenced by SBI-553, cells were pretreated with SBI-553 prior to NTS treatment. SBI-553 enhanced low-concentration NTS-induced β-arrestin2 recruitment to NTSR1 (Fig 1E), as well as β-arrestin2 translocation (Fig 1I) and NTSR1 internalization (Fig 1L), without altering maximal levels of β-arrestin2 translocation or receptor endocytosis. These results are consistent with the action of SBI-553 at an allosteric receptor site. To confirm an allosteric mechanism of action, we evaluated the ability of SR142948A to block NTS and SBI-553-induced cellular responses. Concordant with an allosteric mechanism, SR142948A antagonized NTS-induced, but not SBI-553-induced β-arrestin2 recruitment to NTSR1, β-arrestin2 translocation, and NTSR1 internalization (Fig 1D,F,G,J,M).
SBI-553 antagonizes NTSR1 Gq protein signaling
To date the physiological effects of NTS/NTSR1 in vivo have been attributed primarily to Gq heterotrimeric G protein coupling which leads to phospholipase C activation, generation of inositol 1,4,5-triphosphate (IP3), and the mobilization of intracellular calcium (Ca2+) (Watson et al., 1992; Cathala and Paupardin-Tritsch, 1997; St-Gelais et al., 2004; Wu et al., 1995; Wu and Wang, 1995). Here, we find that SBI-553, in contrast to NTS, is incapable of stimulating Gq protein activation, IP3 generation, and Ca2+ mobilization.
In a transforming growth factor-α (TGFα) shedding assay that directly assesses G protein activation (Inoue et al., 2012), NTS increased Gq activation (EC50=106 pM), while neither SBI-553 nor the antagonist SR142948A had any effect (Fig 2A). Using an intramolecular BRET sensor to monitor IP3 generation (Gulyas et al., 2015) and an aequorin calcium reporter to monitor intracellular calcium Ca2+ levels (Evron et al., 2014; Rizzuto et al., 1992b), we observed that, unlike NTS, neither SBI-553 nor SR142948A stimulated IP3 generation (Fig 2C) or Ca2+ mobilization (Fig 2E). When applied in combination with NTS, SBI-553 and the antagonist SR142943A both attenuated NT-induced Gq activation (Fig 2B), IP3 production (Fig 2D), and Ca2+ mobilization (Fig 2F). These findings suggest SBI-553 is a β-arrestin biased ligand that stimulates receptor phosphorylation (Fig 1C) and β-arrestin-mediated processes at NTSR1 (Fig 1D,E,H,K) without activating Gq protein signaling (Fig 2A,C,E). Critically, SBI-553 extended complete β-arrestin functional selectivity to NTS: the otherwise balanced, endogenous ligand.
SBI-553 biases NTS-NTSR1 signaling towards the β-arrestin pathway
Both G proteins and β-arrestins are capable of stimulating the phosphorylation and activation of the extracellular signal-regulated kinase (ERK) (i.e., phospho-ERK or pERK). The temporal pattern of generation, subcellular localization, and phosphorylation targets of pERK mediated by these two pathways, however, differ (Cervantes et al., 2010; Lefkowitz and Shenoy, 2005; Shukla et al., 2011). G protein-mediated ERK activation manifests as a sharp peak (i.e., within 5 min) followed by a rapid decline in the pERK/total ERK ratio (Shenoy et al., 2006). By comparison, β-arrestin-mediated ERK activation produces a more blunted but long-lasting (i.e., >30 min) elevation in pERK/ERK levels (Shenoy et al., 2006). Here, we demonstrate that SBI-553 biases NTSR1 signaling, selectively permitting NTS-induced, β-arrestin-mediated ERK activation.
When pERK generation was monitored in HEK293T cells expressing human NTSR1, NTS produced both a rapid (i.e., within 5 min) and a long-lasting (i.e., >60 min) increase in pERK. This response was abolished by pretreatment with the antagonist SR142948A and was absent in cells not expressing NTSR1 (Fig 3A,B). Despite its ability to recruit β-arrestin, SBI-553 alone failed to elicit pERK generation in human NTSR1-expressing cells (Fig 3C,D). In contrast to the complete blockade of NTS-induced pERK with SR142948A pre-treatment, SBI-553 pre-treatment blunted the early sharp peak in pERK, while permitting the modest early and late NTS-induced pERK attributed to β-arrestin (Fig 3E,F). The permissiveness of SBI-553 to late NTS-induced pERK generation was re-evaluated in knock-out cells lacking either the Gq protein or both β-arrestins. In HEK293 cells lacking the Gq/11 protein (i.e., Gq-null) (Alvarez-Curto et al., 2016; Schrage et al., 2015), NTS treatment alone and NTS in combination with SBI-553 produced comparable pERK time courses (Fig 3G,H). Conversely, in HEK293 cells lacking both β-arrestin1 and β-arrestin2 (i.e., β-arrestin1/2-null) (O’Hayre et al., 2017), NTS treatment produced a transient increase in pERK that was blocked in its entirety by pre-treatment with SBI-553 (Fig 3I,J). Together, these results indicate that SBI-553 antagonizes NTS-induced, Gq-mediated ERK phosphorylation, but is permissive of NTS-induced, β-arrestin-mediated ERK phosphorylation.
Figure 3. SBI-553 antagonizes NTS-induced, Gq-mediated pERK, but permits NTS-induced, β-arrestin-mediated pERK generation.
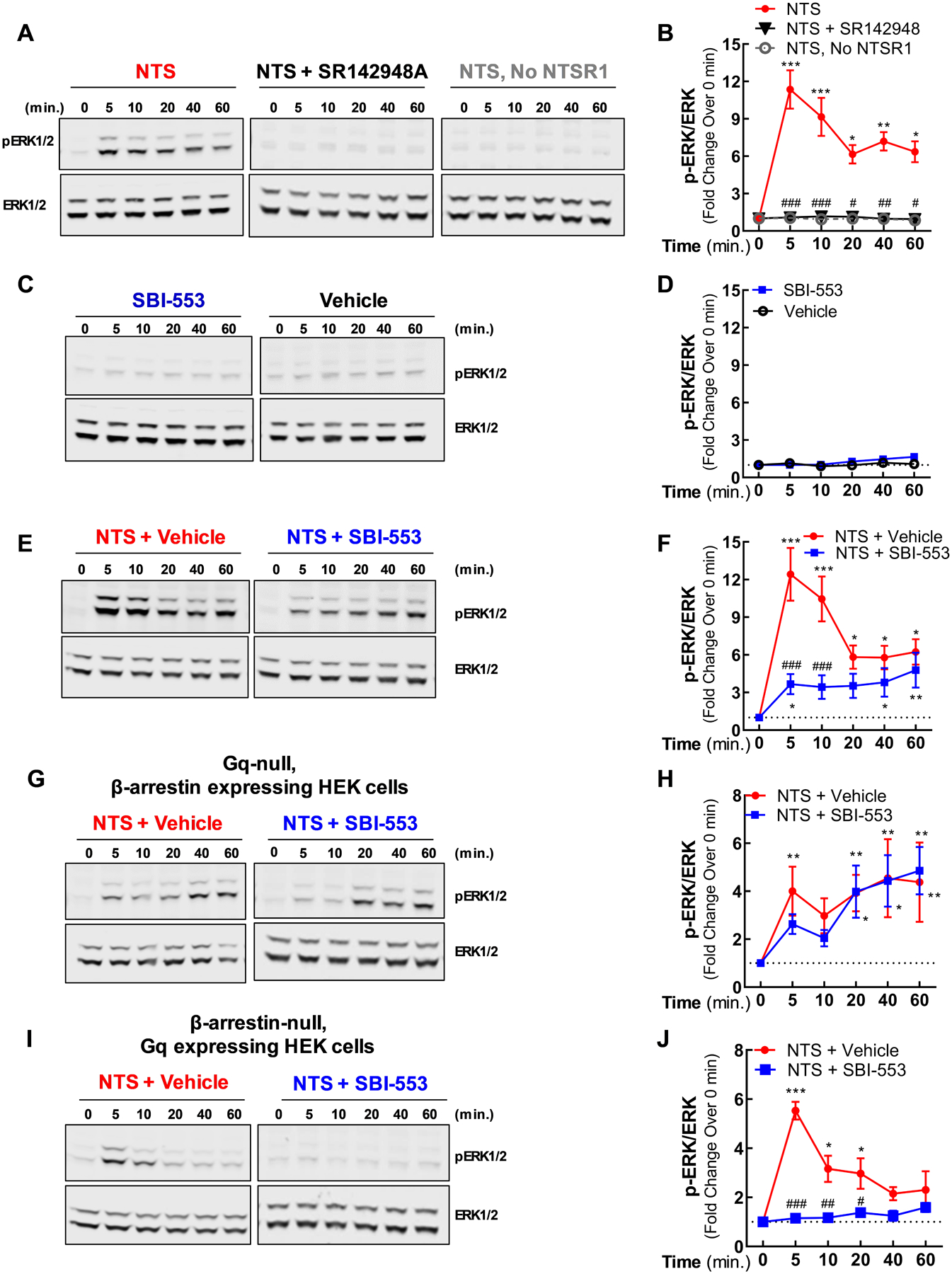
Cells expressing NTSR1 were stimulated with NTSR1 ligands. Phosphorylated ERK (pERK) and total ERK expression were assessed in whole cell lysates by Western blot analysis.
(A-B) Time course for NTS-stimulated pERK in HEK293T cells. NTSR1-expressing cells were treated with NTS in the presence or absence of the NTSR1/2 antagonist SR142948A. Controls cells lacking NTSR1 were stimulated only with NTS.
(C-D) Time course for SBI-553-stimulated pERK in HEK293T cells. NTSR1-expressing cells were treated with SBI-553 or vehicle.
(E-F) Time course for NTS and Vehicle or SBI-553 co-treatment-stimulated pERK in HEK293T cells. NTSR1-expressing cells were treated concurrently with NTS and vehicle or SBI-553.
(G-H) Time course for NTS and Vehicle or SBI-553 co-treatment-stimulated pERK in Gq-null cells. HEK293 cells genetically engineered to lack the Gq protein were treated concurrently with NTS and vehicle or SBI-553.
(I-J) Time course for NTS and Vehicle or SBI-553 co-treatment-stimulated pERK in β-arrestin1/2-null cells. HEK293 cells genetically engineered to lack β-arrestin1 and β-arrestin2 were treated concurrently with NTS and vehicle or SBI-553.
For details on statistical comparisons, see Table S3.
SBI-553 attenuates psychostimulant-like changes in regional brain metabolism
Balanced peptide NTSR1 agonists are thought to modulate motivated behavior via antagonism of aberrant drug-induced dopamine signaling at the D2R (Binder et al., 2001). Our data indicate that SBI-553 is a β-arrestin biased NTSR1 ligand. The sufficiency of NTSR1 β-arrestin signaling to modulate dopamine neurotransmission is unknown. To address this question, we determined whether systemically administered SBI-553 could modulate the brain’s metabolic response to dopamine. PET/CT with the radiolabeled glucose probe [18F]fluorodeoxyglucose ([18F]-FDG) permits visualization of brain glucose metabolism to infer neural activity and target engagement (Lancelot and Zimmer, 2010). Brain [18F]-FDG uptake is altered in a region-specific manner by psychostimulants like cocaine, which increase striatal dopamine levels (Willuhn et al., 2010). For example, acute cocaine treatment in C57BL/6J mice has been shown to reduce [18F]-FDG uptake in the frontal cortex, motor cortex, hippocampus, thalamus, cerebellum, striatum and olfactory bulb (Thanos et al., 2008). Given these established changes as well as the observed brain penetrance of SBI-553 (Pinkerton et al., 2019), the ability of SBI-553 to modulate the cellular activity of NTSR1, and the substantial overlap of dopamine-rich brain regions with the expression and activity profile of NTSR1 (Griebel and Holsboer, 2012), we investigated whether SBI-553 could alter the brain’s metabolic response to dopamine.
To maximize our ability to detect an SBI-553 modulatory effect on dopamine signaling, we used the dopamine transporter knockout (DAT KO) mouse in which dopamine can be pharmacologically raised and lowered from a zero baseline. This model produces much larger changes in brain dopamine levels and corresponding locomotor responses than moderate doses of psychostimulants (Urs et al., 2015). [18F]-FDG uptake was assessed in DAT KO mice depleted of dopamine by the tyrosine hydroxylase inhibitor α-methyl-para-tyrosine (AMPT). AMPT treatment arrest volitional movement in DAT KO mice due to dopamine depletion (Sotnikova et al., 2005). Open field motor activity was then evaluated by concurrent treatment with levodopa (L-DOPA) and vehicle (saline) or L-DOPA and SBI-553 (12 mg/kg). In this paradigm, the brain penetrant dopamine precursor L-DOPA induced robust restoration of locomotion that was attenuated with 12 mg/kg SBI-553 (Fig 4A). Note, this SBI-553 dose was selected based on pharmacokinetic data demonstrating brain SBI-553 levels in the low micromolar range following intraperitoneal (i.p.) administration (Pinkerton et al., 2019).
Figure 4. SBI-553 attenuates L-DOPA-induced changes in regional brain glucose utilization.
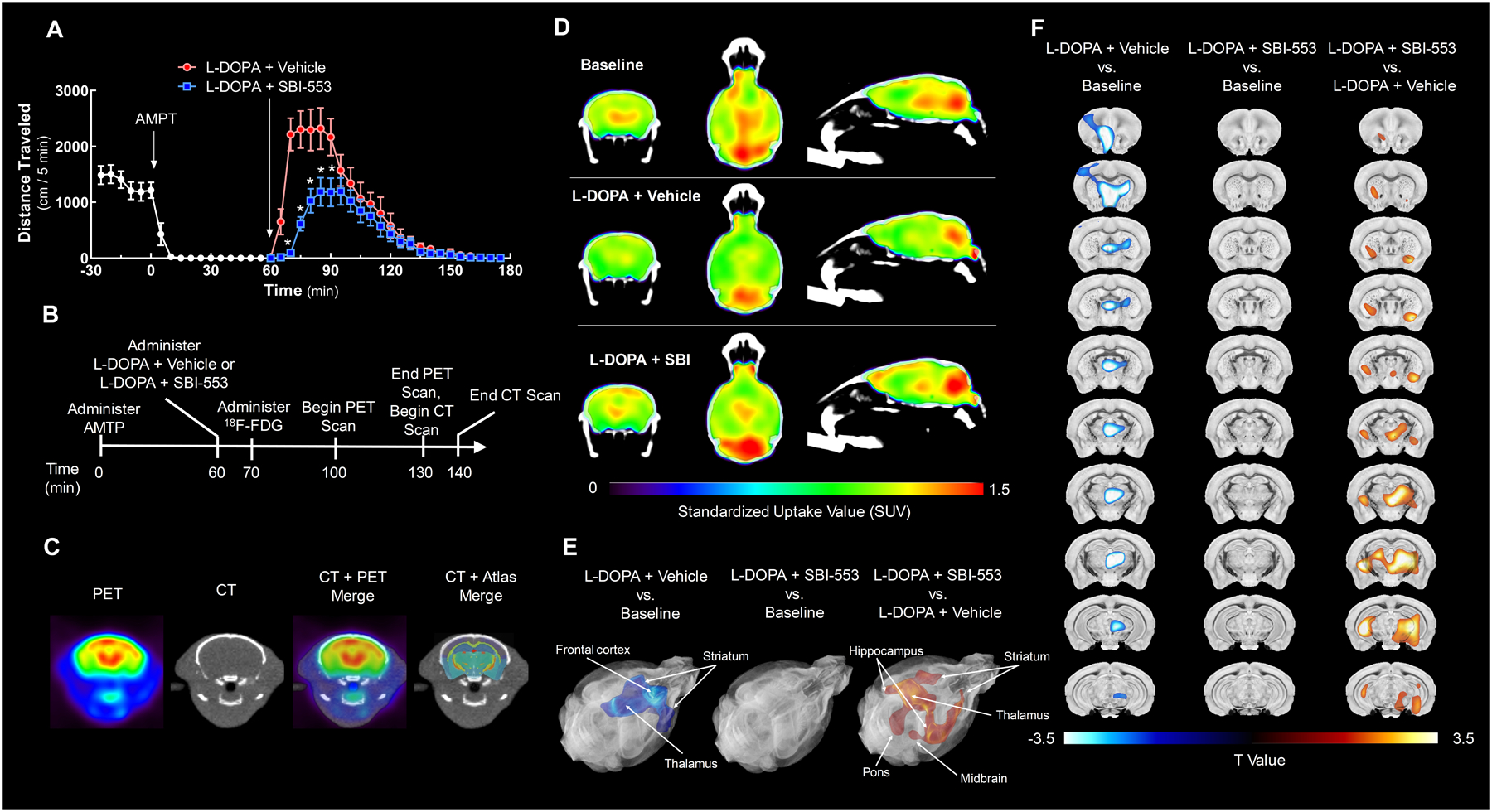
The ability of SBI-553 to modulate L-DOPA-induced changes in [18F]-FDG uptake was evaluated in dopamine-depleted dopamine transporter knockout (DAT KO) mice by PET/CT.
(A) Open field activity of DAT KO in the model of dopamine-induced locomotion used for imaging studies. DAT KO mice received the tyrosine hydroxylase inhibitor α-methyl-p-tyrosine (AMPT, 12 mg/kg, i.p.) 60 min prior to co-administration of L-DOPA (25 mg/kg, i.p.) and vehicle (saline, i.p.) or L-DOPA and SBI-553 (12 mg/kg, i.p.).
(B) Timeline indicating the schedule of treatments and scan acquisitions.
(C) Representative images showing registration of a PET to a CT scan and a CT scan to the 3-dimensional brain atlas with 332 regional labels.
(D) [18F]-FDG PET/CT scans from a representative animal acquired at baseline (top), after L-DOPA and vehicle co-treatment (middle), or after L-DOPA and SBI-553 co-treatment (bottom) are shown in coronal (left), transverse (middle) and sagittal (right) sections. Position relative to bregma: anteroposterior, −1.31 mm; mediolateral +0.05 mm; dorsoventral, +3.37 mm.
(E,F) [18F]-FDG uptake was compared among groups using statistical parametric mapping (SPM). This voxel-based analysis revealed a single cluster of significantly reduced 18F-FDG standardized uptake values (SUVs) in the L-DOPA + Vehicle versus (vs.) Baseline comparison (left; kE = 330,117 voxels, pcluster-level = 0.049; peak-level T = 7.40, peak-level Z = 3.60, peak-level <0.0001). No significant clusters were identified in the L-DOPA + SBI-553 vs. Baseline comparison (middle). A single cluster of significantly elevated SUVs was identified in the L-DOPA + SBI-553 vs. L-DOPA + Vehicle comparison (right; kE = 419,788 voxels, pcluster-level = 0.042; peak-level T = 7.74, peak-level Z = 3.67, ppeak-level <0.0001). T Maps of significantly reduced (blue) and increased (orange) voxels are shown in the brain atlas as (E) rendered 3-dimensional surfaces and (F) coronal sections. Position of coronal sections relative to bregma along anteroposterior axis: +1.51, +0.61, +0.21, −0.19, −0.63, −1.03, −1.43, −1.83, −2.23, −3.23, −3.83 mm.
For details on statistical comparisons, see Table S4. Also see Figure S3.
To validate the locomotor observations, PET/CT imaging studies were conducted using a within-subjects design. Each animal received three scans according to three treatment protocols; a baseline scan on study day 1, dopamine depletion followed by L-DOPA and vehicle co-treatment on study day 2, and dopamine depletion followed by L-DOPA and SBI-553 co-treatment on study day 3 (Fig 4B). PET/CT scans were mapped into a three-dimensional Waxholm space C57BL/6J brain atlas (Calabrese et al., 2015) (Fig 4C). To control for differences in whole brain glucose utilization, all data were standardized to mean whole brain [18F]-FDG uptake, which did not differ among groups (Fig S3). Statistical parametric mapping (SPM) was used to identify voxel clusters of standardized [18F]-FDG uptake values (SUVs) that significantly differed between treatments.
As reported for studies evaluating acute cocaine treatment in mice (Thanos et al., 2008), L-DOPA and vehicle administration in dopamine-depleted DAT KO mice reduced regional [18F]-FDG uptake as compared to baseline values (Fig 4D,E,F; L-DOPA + Vehicle vs. Baseline comparison). A cluster of significantly reduced uptake was identified in this comparison that spanned cortical areas and much of the thalamus (Fig 4E,F, left panels). SBI-553 treatment blocked L-DOPA-induced reductions in [18F]-FDG uptake, with no significant clusters identified in the L-DOPA + SBI-553 vs. Baseline comparison (Fig 4E,F, middle panels). A direct comparison of scans from SBI-553- and vehicle-treated mice revealed a large, symmetrical cluster of elevated [18F]-FDG uptake that involved the striatum, thalamus, cortical areas, hippocampus, and midbrain (Fig 4E,F, right panels; L-DOPA + SBI-553 vs. L-DOPA + Vehicle comparison). These results indicate that SBI-553 opposed the L-DOPA-induced changes in regional brain metabolism and modulated the activity of dopamine-associated structures responsible for reward processing.
SBI-553 attenuates methamphetamine and cocaine-associated behaviors in mice
Psychostimulant administration in mice has well-described behavioral consequences, including induction of hyperlocomotion, conditioned place preference, and self-administration. Balanced, peptide NTSR1 ligands can attenuate psychostimulant-induced behaviors in vivo (Ferraro et al., 2016); however, the degree to which β-arrestin agonism participates in this response has not been tested directly. To evaluate the ability of SBI-553 to modulate psychostimulant-associated behaviors in C57BL/6J mice, we assessed the effects of SBI-553 in behavioral assays of increasing complexity, including methamphetamine- and cocaine-induced hyperlocomotion, methamphetamine conditioned place preference, and cocaine self-administration.
We observed SBI-553 administration (12 mg/kg, i.p.) to attenuate cocaine-induced hyperlocomotion relative to the vehicle control (saline, 5 ml/kg, i.p.) (Fig 5A,B). By contrast, responses to the vehicle and SBI-553 were comparable in non-stimulant exposed animals (Fig 5A,B). This SBI-553 dose also reduced methamphetamine (2 mg/kg) -induced hyperlocomotion (see Fig 7B) and blocked the expression of methamphetamine conditioned place preference (Fig 5C). Similar behavioral effects have been observed with unbiased peptide NTSR1 agonists (Ferraro et al., 2016; Boules et al., 2016).
Figure 5. SBI-553 attenuates behavioral evidence of acute and chronic psychostimulant exposure.
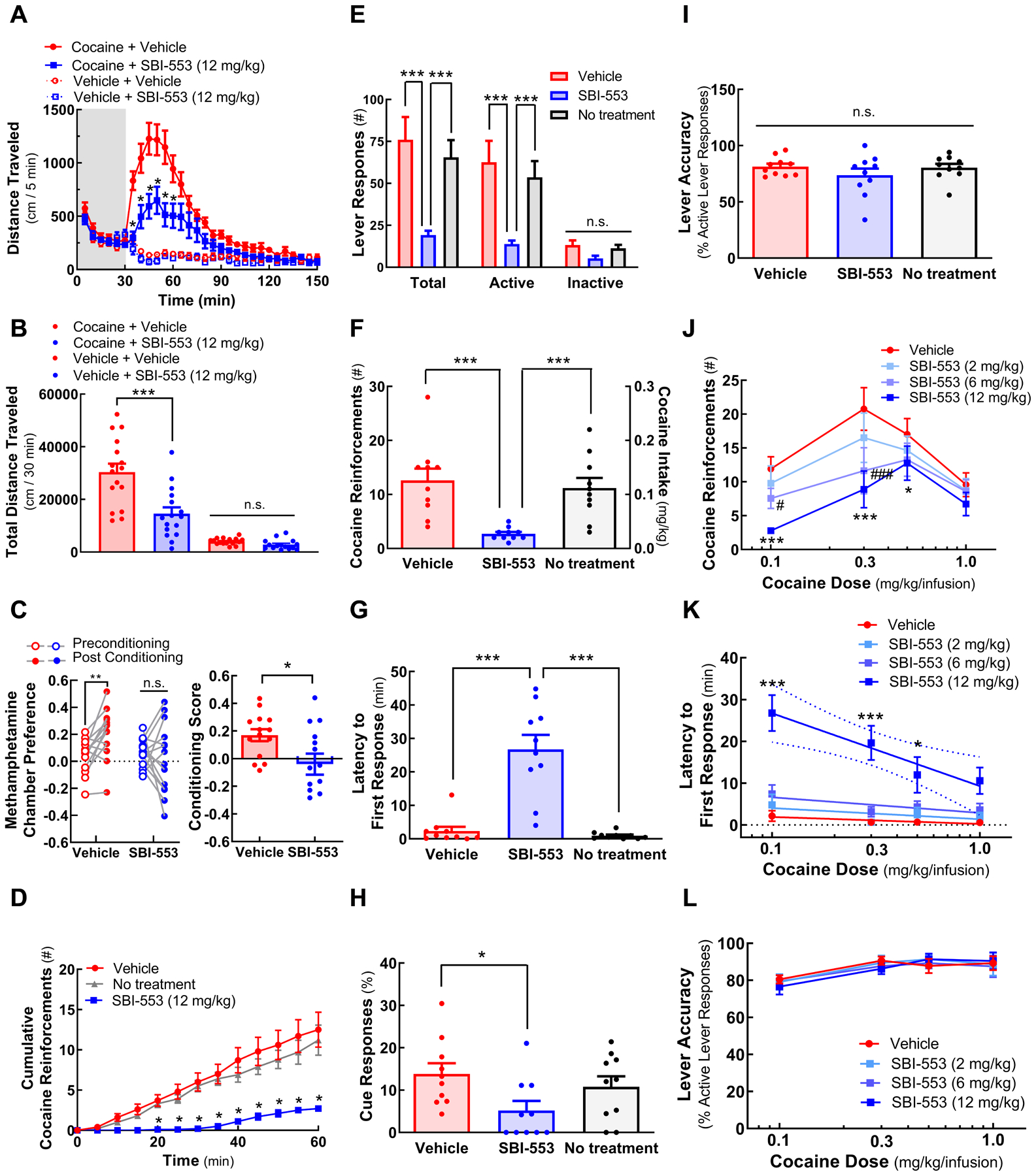
The effect of SBI-553 on cocaine and methamphetamine (meth)-associated behaviors in C57BL/6J mice.
(A,B) Cocaine-induced hyperlocomotion. Animals were acclimated to an open field for 30 min (indicated by grey box) prior to concurrent administration of cocaine (30 mg/kg, i.p.) and SBI-553 (12 mg/kg, i.p.) or vehicle (saline, i.p.). (A) Time course. (B) Cumulative distance traveled in the 30 min post-treatment period.
(C) Methamphetamine conditioned place preference (CPP). After conditioning, animals received vehicle (saline, i.p.) or SBI-553 (12 mg/kg, i.p.) 15 min prior to placement into the center chamber. Time spent in the methamphetamine- and vehicle-paired end chambers was recorded for 30 min. (Left) Pre- vs. post-conditioning chamber preference. (Right) Conditioning score.
(D-L) Cocaine self-administration. Mice were trained to self-administer cocaine intravenously via active lever responding at a fixed lever response to reinforcement schedule of 4. Once stable performance was achieved, self-administration was assessed once daily in 60 min sessions. (D-I) Results of daily 0.1 mg/kg/infusion cocaine self-administration sessions following vehicle (saline, i.p., day 1), SBI-553 (12 mg/kg, i.p., day 2) or no treatment (day 3) 5 min prior to placement into the operant chamber. (D) Cumulative cocaine reinforcement time-courses. (E) Lever responding. (F) Total cocaine reinforcements received and cocaine intake. (G) Latency to initiate the first lever response. (H) Percent of responses occurring in the post-reinforcement cue period in the absence of cocaine reinforcement. (I) Lever accuracy across treatment days. (J-L) Results of cocaine and SBI-553 dose-response studies. Animals received vehicle (saline, i.p.) or SBI-553 (2, 6 or 12 mg/kg, i.p.) immediately prior to placement into self-administration chambers with access to cocaine at doses of 0.1, 0.3, 0.5 or 1 mg/kg/infusion. (J) Cocaine reinforcement dose-response curves. (K) Latency to initiate the first lever response. (L) Lever accuracy.
For details on statistical comparisons, see Table S5.
Figure 7. SBI-553 attenuates cocaine and methamphetamine-induced locomotion via a β-arrestin2-dependent mechanism.

The effect of SBI-553 on cocaine and methamphetamine-induced hyperlocomotion was assessed in global and neuron-specific β-arrestin2 KO mice. Animals were acclimated to the open field for 30 min (indicated by grey box) prior to concurrent administration of either cocaine (30 mg/kg, i.p.) or methamphetamine (2 mg/kg, i.p.) and SBI-553 (12 mg/kg, i.p.) or vehicle (saline, i.p.).
(A) Cocaine-induced hyperlocomotion in WT and global β-arrestin2 KO mice.
(B) Methamphetamine-induced hyperlocomotion in WT and global β-arrestin2 KO mice.
(C) Diagram of the Cre-Lox breeding system used to develop region- and neuron subtype-specific β-arrestin2 KO mouse lines.
(D) Methamphetamine-induced hyperlocomotion in non-Cre expressing β-arrestin2f/f littermate control mice.
(E) Methamphetamine-induced hyperlocomotion in mice with selective deletion of β-arrestin2 in D1R-expressing neurons (D1RCre/β-arrestin2f/f).
(F) Methamphetamine-induced hyperlocomotion in mice with selective deletion of β-arrestin2 in D2R-expressing neurons (D2RCre/β-arrestin2f/f).
(G) Methamphetamine-induced hyperlocomotion in mice with selective deletion of β-arrestin2 in midbrain D2R-expressing neurons (DATCre/β-arrestin2f/f).
(H) Methamphetamine-induced hyperlocomotion in mice with selective deletion of β-arrestin2 in striatal D2R-expressing neurons (A2aCre/β-arrestin2f/f mice).
(I) Cumulative distance traveled in methamphetamine-treated mice. Distance traveled in the 50 min post-treatment in Cre negative β-arrestin2f/f control and neuron-specific β-arrestin2 KO mice.
For details on statistical comparisons, see Table S7.
To examine the effects of SBI-553 in a paradigm that more closely mimics drug taking in humans, mice were tested in cocaine self-administration, as described in Fig 5 legend. Cocaine intake (0.1 mg/kg/infusion) was assessed in animals with indwelling jugular catheters self-administering intravenous cocaine by lever press on a fixed ratio 4 schedule in 60 min sessions. SBI-553 treatment resulted in fewer cumulative self-administered reinforcements between 20 and 60 min (Fig 5D) with significantly suppressed total and active lever responses relative to Vehicle and No Treatment (Fig 5D); activity at the inactive lever was unchanged across treatments (Fig 5E). In addition to reducing cocaine intake to less than 20% of the level of the vehicle control (Fig 5F), SBI-553 increased the latency to initiate lever responding (Fig 5G): a measure inversely associated with the motivation/urgency for drug seeking (Green et al., 2015; Maccioni et al., 2015). SBI-553 also reduced the percent of lever responses that occurred during the post-reinforcement cue period (i.e., responses to the cue in the absence of cocaine reinforcement; Fig 5H), a measure that may be related to reward-cue responsivity (King et al., 2016). Critically, lever discrimination was unchanged by SBI-553, as the percent of responses at the active and inactive levers remained unchanged over the 3-day test (Fig 5I).
We further evaluated a range of SBI-553 treatments (2, 6, and 12 mg/kg, i.p.) on stable self-administration of 4 cocaine doses (0.1, 0.3, 0.5, 1 mg/kg/infusion). The effect of SBI-553 on cocaine self-administration was both cocaine- and SBI-553-dose dependent. SBI-553 exhibited a typical pattern of maintenance intake reduction (Mantsch et al., 2007), with higher SBI-553 doses (i.e., 6, 12 mg/kg) showing greater efficacy at lower cocaine infusion doses (i.e., 0.1, 0.3, 0.5 mg/kg/infusion; Fig 5J). No change in self-administration was observed with SBI-553 treatment at the highest cocaine dose (1 mg/kg/infusion). The SBI-553 (12 mg/kg)-induced increase in the latency to initiate lever responding was also cocaine dose- dependent (Fig 5K). This effect decreased linearly with the log[cocaine] dose, suggesting that the ability of SBI-553 to suppress the initiation of motivated behavior was dependent upon the magnitude of the anticipated reward. Lever accuracy increased with increasing dose of the cocaine infusion and was not altered by SBI-553 treatment (Fig 5L). Taken together, the ability of SBI-553 to attenuate methamphetamine- and cocaine-associated behaviors suggests that β-arrestin activation downstream of NTSR1 is sufficient to attenuate addiction-associated psychostimulant effects.
SBI-553 treatment is not associated with hypothermia, hypotension or motor impairment
In addition to modulating motivated behavior, NTS peptide analogues act centrally and peripherally to produce effects that may limit their utility in the treatment of CNS disorders. These effects include hypotension (Carraway and Leeman, 1973), hypothermia (Bissette et al., 1976; Kitabgi et al., 1992), muscle relaxation/impaired motor coordination (Osbahr et al., 1979; Pettibone et al., 2002), and sedation (Nemeroff, 1980). To determine whether SBI-553 shares this side effect profile, we monitored core body temperature, motor coordination, and central blood pressure in C57BL/6J mice following treatment with either SBI-553 or the unbiased, NTR1-selective, brain penetrant NTS peptide fragment 8–13 analogue PD149163 (Petrie et al., 2004; Wustrow et al., 1995). While PD149163 (0.1 – 1 mg/kg, i.p.) induced a rapid (within 30 min) and long-lasting (at least 5 hr) hypothermia (Fig 6A), SBI-553 (2–30 mg/kg, i.p.) had no effect on core body temperature (Fig 6B). Concurrent with PD149163-induced hypothermia were marked sedation and deficits in motor coordination, as evidenced by an increased latency to grasp the wire in the wire-hang test and impairment in the righting reflex (Fig 6C,E). In contrast, SBI-553, at doses 2.5 times greater than those employed in the locomotion and self-administration assays, failed to influence the wire hang or righting reflexes (Fig 6D,F).
Figure 6. SBI-553 treatment, unlike that of peptide NTSR1 ligands, is not associated with hypothermia, motor impairment or hypotension.
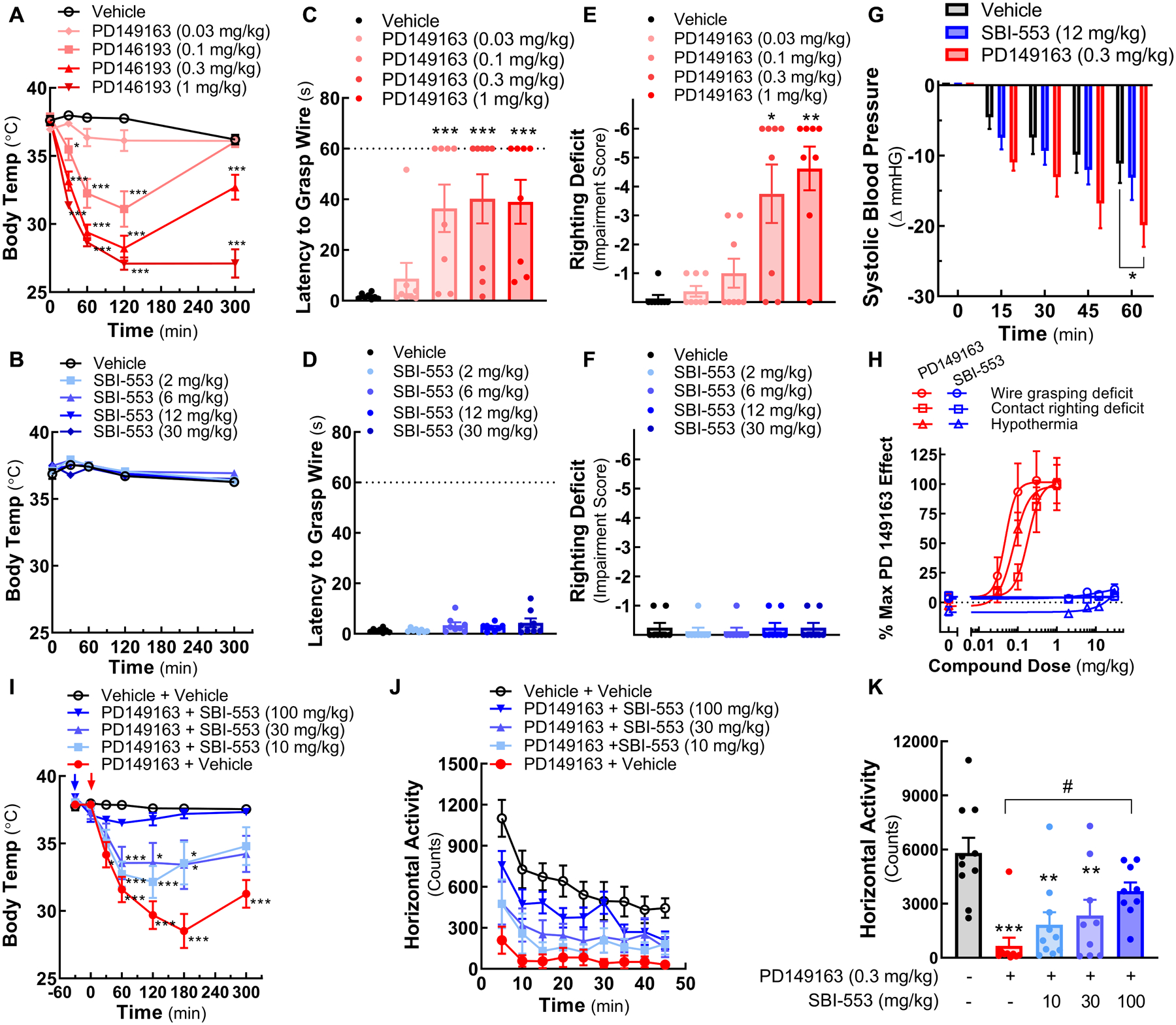
The effects of the peptide NTSR1 ligand PD149163 and SBI-553 in C57BL/6J mice.
(A, B) Core body temperature. (A) Animals received vehicle (0.2% DMSO in saline) or PD149163 (i.p.) at Time 0. (B) Animals received vehicle (saline) or SBI-553 (i.p.) at Time 0.
(C, D) Wire hang. Latency to grasp wire with hind-limbs was measured in trained animals hanging by their forepaws. (C) Animals received vehicle or PD149163 (i.p.) 120 min prior to testing. (D) Animals received vehicle or SBI-553 (i.p.) 30 min prior to testing.
(E, F) Righting reflex. Contact righting deficits were scored on a six-point scale. (E) Animals received vehicle or PD149163 (i.p.) 120 min prior to testing. (F) Animals received vehicle or SBI-553 (i.p.) 30 min prior to testing.
(G) Blood pressure. Systolic ventricular pressure was continuously recorded in anesthetized mice after treatment with vehicle (0.6% DMSO in saline), SBI-553, or PD149163. Baseline was set to 0.
(H) Side-effect dose-response curves. Data from the time of peak drug effect, 120 and 30 min post-treatment for PD149163 and SBI-553, respectively, are represented as percent PD149163 at the 1 mg/kg dose for the wire hang, contact righting, and body temperature tests.
(I,J,K) Effects of orally administered SBI-553 on PD149163-induced reductions in body temperature and motor activity. (I) Animals received vehicle (10% DMSO, 0.05% Tween-80) or 10, 30, or 100 mg/kg SBI-553 (p.o.) 30 min prior to the study (−30 min, blue arrow). Animals received vehicle (saline) or PD149163 (i.p.) at Time 0 (red arrow) and core body temperature was monitored at multiple time-points. The same administration schedule was used for quantitation of (J) horizontal motor activity (for group comparisons to Table S6) and (K) cumulative motor activity over a 45 min period starting 120 min post PD149163 or vehicle treatment.
For details on statistical comparisons, see Table S6.
To evaluate the effect of moderate doses of both compounds on blood pressure, pressure-conductance catheters were inserted retro-aortically into the left ventricle of anesthetized C57BL/6J mice. Hemodynamic parameters were recorded for 60 minutes post-treatment. While no differences between SBI-553 (12 mg/kg, i.p.) and vehicle treatments were detected, PD149163 (0.3 mg/kg, i.p.) significantly reduced peak systolic pressure at the 60-minute time point (Fig 6G). The PD149163-induced hypotension was not secondary to hypothermia, as body temperature was maintained by thermal support over the duration of the recording. Together, these findings suggest a Gq protein-dependent mechanism that is not engaged by SBI-553 may mediate NTSR1-associated hypothermia, motor coordination impairment and hypotension.
The ability of SBI-553 to modulate PD149163-induced hypothermia and suppression of motor responses was evaluated following oral (p.o.) administration. Consistent with antagonist-like action at NTSR1, SBI-553 pretreatment (p.o.) 30 minutes prior to PD149163 administration (i.p.) dose-dependently attenuated the PD149163-induced reductions in core body temperature (Fig 6I) and horizontal activity in the open field (Fig 6J,K). Remarkably, these findings suggest that while the ability of NTSR1 ligands to reduce psychostimulant-associated behaviors is preserved in the β-arrestin biased compound SBI-553, other physiological and behavioral responses are eliminated. In accordance with a Gq-mediated mechanism of NTSR1-induced hypothermia and hypoactivity, and SBI-553’s ability to antagonize NTSR1 Gq signaling, co-treatment with PD149163 and SBI-553 resulted in more restricted pharmacological action.
SBI-553 modulation of psychostimulant action requires β-arrestin2 in select neuronal population
Since cellular assays indicate that SBI-553 is a β-arrestin biased NTSR1 ligand, we hypothesized that the absence of β-arrestin2 would reduce SBI-553 activity in mice. We observed 12 mg/kg (i.p.) SBI-553 to decrease both cocaine- (30 mg/kg, i.p.; Fig 7A) and methamphetamine-induced (2 mg/kg, i.p.; Fig 7B) hyperlocomotion in WT mice on a C57BL/6J background. In contrast, and consistent with this hypothesis, a significant reduction in locomotion was absent in global β-arrestin2 knockout mice (Fig 7A,B). To identify the neuronal subpopulation involved in SBI-553-induced locomotor attenuation, we evaluated region- and neuron subtype-selective β-arrestin2 KO mouse lines generated and validated previously in our laboratory using Cre-Lox recombination (Urs et al., 2016). Here, β-arrestin2 “floxed” mice (β-arrestin2f/f) were crossed with selective Cre driver mice, resulting in β-arrestin2 deletion in select neuronal populations: D1R-Cre (deletion in all D1R-expressing neurons), D2R-Cre (deletion in all D2R-expressing neurons), DAT-Cre (deletion in midbrain presynaptic D2R-expressing neurons, characterized by dopamine transporter expression (Toth et al., 2018; Bertonlino et al., 2009)), or A2aR-Cre (deletion in adenosine A2a receptor expressing-neurons, predominantly striatal postsynaptic D2R-expressing spiny projection neurons (Schiffmann et al., 2007)) (Fig 7C). Methamphetamine was selected for further study because it produced a robust and protracted increase in locomotor activity in global β-arrestin2 knockout mice (Fig 7B). We found that 12 mg/kg (i.p.) SBI-553 effectively reduced methamphetamine (2 mg/kg, i.p.)-induced hyperlocomotion in β-arrestin2f/f Cre negative littermate controls (Fig. 7D) and D1RCre animals (Fig 7E). However, SBI-553 failed to affect locomotion in D2R-Cre mice (Fig 7F).
These results suggest that SBI-553 locomotor modulation requires β-arrestin2 expression in D2R- but not in D1R- expressing neurons, a finding consistent with the observation that NTSR1 and D2R are co-expressed within populations of midbrain and striatal neurons (Binder et al., 2001). To determine whether midbrain presynaptic D2R-Cre neurons, striatal postsynaptic D2R-Cre neurons or some combination of both populations were responsible for driving the effects in the D2R-Cre animals, we assessed SBI-553 effects on methamphetamine-stimulated hyperlocomotion in DAT-Cre and A2aR-Cre animals. SBI-553 reduced the methamphetamine-induced hyperlocomotion in DAT-Cre mice (Fig 7G,I). By contrast, no SBI-553 effects were observed in A2aR-Cre animals, in which striatal neurons were selectively affected (Fig 7H,I). Together, these results suggest that SBI-553’s effects on methamphetamine-induced hyperlocomotion may be mediated primarily through striatal D2R-expressing medium spiny neurons.
DISCUSSION
Allosteric and functionally selective GPCR ligands provide a means of developing therapeutics with more directed pharmacological action and fewer side-effects. Here, we characterize the small molecule SBI-553 as an allosteric, β-arrestin biased NTR1 agonist that confers profound β-arrestin bias to the endogenous ligand. Remarkably, SBI-553 shows efficacy in mouse models of psychostimulant abuse without the side effects characteristic of unbiased NTSR1 agonism. Studies with global and neuron-selective β-arrestin2 knockout mice reveal that SBI-553 locomotor modulation requires β-arrestin2 in striatal D2R-expressing spiny projection neurons. As the ventral striatum is a major locus for reward in the brain, SBI-553 may have broad utility in the treatment of conditions involving dysregulated reward mechanisms.
SBI-553 increases NTS binding affinity and Bmax,, and its binding affinity is reciprocally increased by NTS. These behaviors are characteristic of positive allosteric modulation and are concordant with the Monod-Wyman-Changeux model of allosteric transitions (Canals et al., 2012). At NTSR1, SBI-553 exhibited behaviors characteristic of both a β-arrestin biased agonist and a biasing modulator. Alone, SBI-553 stimulated NTSR1 phosphorylation, β-arrestin2 recruitment, and internalization. In combination with NTS, SBI-553 enhanced low-concentration NTS-induced NTSR1 β-arrestin2 recruitment and internalization and antagonized NTS-induced Gq activation. These findings indicate that SBI-553 is not only an allosteric, β-arrestin biased ligand itself but also extends β-arrestin bias to the endogenous ligand.
Sustained pERK generation following NTS stimulation was lost in β-arrestin1/2-null cells, suggesting that β-arrestins contribute to pERK downstream NTSR1 activation. This finding contrasts with a recent report for the β2-adrengeric receptor (O’Hayre et al., 2017). The inability of SBI-553 alone to stimulate pERK generation suggests the SBI-553-NTSR1-β-arrestin2 complex may not be fully signaling competent and raises the possibility that interactions with other signaling partners may be required for pERK generation (Grundmann et al., 2018; Smith et al., 2019). SBI-553 antagonized NTS-induced, G protein-mediated pERK, but was permissive of NTS-induced, β-arrestin-mediated pERK. Such action may dramatically alter NTS-NTSR1 signaling, as the subcellular localization and phosphorylation targets of G protein-mediated and β-arrestin-mediated pERK differ (Cervantes et al., 2010; Lefkowitz and Shenoy, 2005; Shukla et al., 2011). Future studies will determine the full spectrum of SBI-553 functions, which may include activation of other kinases associated with β-arrestin scaffolding (e.g., glycogen synthase kinase 3 β, protein kinase B) (Urs et al., 2012).
Together, the binding and signaling results suggest that SBI-553 locks NTSR1 into a high affinity, β-arrestin-selective conformation. While biased allosteric modulators of other GPCRs have been reported, most notably for the metabotropic glutamate receptors (for review see Stansley and Conn, 2019), SBI-553 is set apart by its ability to induce β-arrestin recruitment independent of the endogenous agonist (i.e., ago-allosteric characteristics) and by the degree to which bias is conferred. In the presence of micromolar concentrations of SBI-553, NTS is transformed from a balanced Gq/β-arrestin agonist into a fully β-arrestin biased agonist. As brain NTS expression is increased following psychostimulant exposure (Betancur et al., 2001; Betancur et al., 1997) and SBI-553 reaches micromolar concentrations in the brain after systemic (i.p. or p.o.) administration (Pinkerton et al., 2019), SBI-553’s in vivo effects may be the result of the action of endogenous NTS at SBI-553-activated receptors.
The [18F]-FDG PET/CT studies in dopamine-depleted DAT KO mice demonstrate that SBI-553 reaches central NTSR1 receptors at sufficient concentrations to modulate brain dopaminergic neurotransmission. SBI-553’s pharmacodynamic profile provides an opportunity to distinguish between β-arrestin and Gq protein NTSR1-mediated actions in vivo. In murine models of psychostimulant use and abuse, SBI-553 displays actions analogous to those of balanced peptide NTSR1 agonists (Ferraro et al., 2016) by reducing stimulant-induced hyperlocomotion, blocking expression of methamphetamine place preference, and attenuating cocaine self-administration. Together these data suggest that NTSR1/β-arrestin signaling is sufficient to reduce the behavioral consequences of psychostimulant exposure in mice. Moreover, the increase in latency to initiate lever responding in the cocaine self-administration paradigm suggests that SBI-553 may reduce the motivation to initiate drug-taking, an effect consistent with the reported actions of the PD149163 agonist (Sharpe et al., 2017). NTSR1 exerts its effects on brain dopamine signaling through antagonistic functional and putative physical interactions with the D2R (Binder et al., 2001; Borroto-Escuela et al., 2013). The ability of SBI-553 to modulate dopamine-associated brain metabolic activity and locomotor, CPP, and self-administration behaviors suggests that selective induction of the NTSR1-β-arrestin interaction may be sufficient to modulate D2R function.
Due to their dependence on the endogenous ligand and their ability to segregate G protein from β-arrestin signaling, biased modulators may confer greater spatial and temporal precision and elicit more directed physiological actions than balanced agonists (Khoury et al., 2014). While PD149163 produces hypothermia, hypotension, and deficits in motor coordination at doses that generate desired anxiolytic (Prus et al., 2014), antidepressant-like (Carey et al., 2017), and anti-addictive effects (i.e., 0.1 and 1 mg/kg) (Sharpe et al., 2017), SBI-553 alone exerts no effect on these indices. This profile indicates that NTSR1 may be one of a growing list of GPCRs for which it is now known that G protein and β-arrestin signaling mediate distinct physiological effects. Critically, selective activation of β-arrestin by NTSR1 may be sufficient to attenuate psychostimulant-associated behaviors without the undesirable side effects attributed to balanced NTSR1 activation. Although SBI-553 promotes more directed action at NTSR1 than balanced agonists, higher doses are necessary to definitively exclude hypothermia or central blood pressure changes. Nevertheless, the β-arrestin bias induced by SBI-553 creates a therapeutic window between effects on motivated behavior and thermo-/hemodynamic- regulation where one had not existed previously. This action profile may enable the safe and simultaneous administration of SBI-553 with existing NTSR1 peptide agonists.
While NTSR1 interacts with both β-arrestin1 and β-arrestin2 (Oakley et al., 2000), the current work focuses on β-arrestin2 because of the noted roles of this protein in addiction. In addition to blunted responses to stimulants, β-arrestin2 KO mice exhibit altered responses to cannabinoids, alcohol, nicotine and hallucinogens (as reviewed in Porter-Stransky and Weinshenker, 2017). Critically, human genetic studies of drug use and dependence implicate β-arrestin2 in these activities (Porter-Stransky and Weinshenker, 2017). To evaluate the role of β-arrestin2 in SBI-553’s locomotor effects in vivo, we used global and neuron-subtype specific β-arrestin2 KO mice. SBI-553’s effects on both cocaine- and methamphetamine-induced hyperlocomotion were lost in global β-arrestin2 KO mice. Our finding that β-arrestin2 expression in striatal D2R-expressing spiny projection neurons is necessary for SBI-553 modulation of methamphetamine-induced hyperlocomotion suggests that, as reported for PD149163 (Vadnie et al., 2014) and NTS itself (Binder et al., 2001), SBI-553 may act, directly or indirectly, on this population of neurons to reduce stimulant-induced motor activity.
SBI-553 typifies a distinctive class of biased allosteric modulators that stabilize GPCRs in a conformation that more selectively engages β-arrestin. The present study indicates that functionally selective modulation of NTSR1 signaling may be superior to balanced receptor activation for the treatment of substance use disorders. NTSR1-mediated β-arrestin activation may act as a general brake on dopamine-associated behavioral responses, thereby broadening SBI-553’s therapeutic utility beyond psychostimulant use disorders to the treatment of other chemical and behavioral addictions.
Capitalizing on allostery and functional selectivity in GPCR drug development is in its infancy, but these strategies present exciting new opportunities to more precisely modulate receptor signaling and develop safer, more efficacious therapeutics. Biased allosteric modulators will be especially useful for those GPCRs for which it has been demonstrated that one signaling arm produces therapeutic and the other clinically undesirable effects (McCorvy et al., 2018). The resolution of the structure of many GPCRs, which permits the use of structure-based, computer-assisted docking, has resulted in the identification of several new allosteric probes (Shoichet and Kobilka, 2012; McCorvy et al., 2018). With SBI-553’s distinctive pharmacological profile and the wealth of structural information available for the NTSR1, similar compounds may now be rationally pursued for other GPCRs. In sum, our findings reveal a class of biased allosteric modulators that permit more precise targeting of GPCR-second messenger signaling and identify an agent whose continued development may directly translate to improved clinical outcomes for patients.
RESOURCE AVAILABILITY
Lead Contact
Requests for further information or reagents should be directed to and will be fulfilled by the Lead Contact, Marc G. Caron (marc.caron@duke.edu).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
This study did not generate datasets or employ previously unreported custom computer code.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines.
U2OS (Cat# HTB-96, RRID:CVCL_0042) and HEK293T/17 (Cat# CRL-11268, RRID:CVCL_1926) cells were obtained from the American Type Culture Collection (ATCC, Manassas, VA). The U2OS cell line stably expressing NTSR1 and green fluorescent protein (GFP) tagged-βarrestin2, and the HEK293 cell line stably expressing a mitochondrially-targeted apoaequorin have been described (Barak et al., 2016). HEK293 cells that lack Gq (i.e., ΔGNAQ and GNA11 HEK293 Clone 1) or β-arrestin1 and β-arrestin2 (i.e., ΔArrb1 and Arrb2 HEK293 Clone 4), and the control parental line from which they were derived, were previously characterized (Alvarez-Curto et al., 2016) and generous gifts from Dr. Asuka Inoue (Tohoku University, Miyagi, Japan). Also provided by Dr. Inoue were the G protein-deficient cells (i.e., ΔGNAS, GNAL, GNAQ, GNA11, GNA12, and GNA13 HEK293 Clone 38) used in TGFα shedding assays of G protein activation (Inoue et al., 2012). All cells were cultured in minimum essential medium (DMEM) with 10% fetal bovine serum (FBS) and 1x antibiotic antimycotic solution (100 IU−1 penicillin, 100 μg/mL streptomycin and 250 ng/ml amphotericin B; MilliporeSigma). Cells were grown exponentially in an incubator at 37°C under 5% CO2 and subcultured at ratios of 1:5–1:10 every 2 to 4 days using 0.05% trypsin-EDTA. HEK293T/17 cells were authenticated by the Duke University DNA Analysis Facility using Short Tandem Repeat (STR) DNA profiling and confirmed mycoplasma-free by the Duke University Cell Culture Facility (CCF).
Animals.
All mouse studies were conducted in accordance with the National Institutes of Health Guidelines for Animal Care and Use of Laboratory Animals and with approved animal protocols from the Duke University Animal Care and Use Committee. The mice studied in the embodied work include: C57BL/6J (The Jackson Laboratory, Bar Harbor, ME, Cat# 000664), DAT KO (Slc6a3tm1; Giros et al., 1996), global β-arrestin2 KO (Arrb2tm1Rjl; Bohn et al., 1999; The Jackson Laboratory Cat# 011130) and WT littermates, D1R-Cre/β-arrestin2f/f (Drd1tm1(Cre)/Arrb2tm1(flox)nu; Urs et al., 2016), D2R-Cre/β-arrestin2f/f (Drd2tm1(Cre)/Arrb2tm1(flox)nu; Urs et al., 2016), A2aR-Cre/β-arrestin2f/f (Adora2tm1(Cre)/Arrb2tm1(flox)nu; Urs et al., 2016), DAT-Cre/β-arrestin2f/f (Slc6a3tm1(cre)/Arrb2tm1(flox)nu; Toth et al., 2018), and respective Cre-negative β-arrestin2f/f littermates (Arrb2tm1(flox)nu). All mouse lines were backcrossed onto a C57BL/6J genetic background for ≥ 10 generations prior to use. At study initiation, all animals were treatment-naïve adults. Mice were 8–16 weeks old, weighed 19–30 g, and were age-matched across experimental groups. Locomotor, self-administration, temperature, wire hang, and contact righting studies employed both male and female mice, and experimental groups were sex-matched. No effect of sex on drug or SBI-553 responsivity were identified in these assays. PET/CT, CPP, and hemodynamic studies utilized male C57BL/6J mice. In all studies, littermates of the same sex and genotype were randomly assigned to experimental groups. Mice for open field, PET/CT, CPP, temperature, wire hang, contact righting, and hemodynamic studies were group housed in 75 JAG micro-VENT system cages (Allentown Inc., Allentown, NJ) with Enrich-O’Cob bedding (The Anderson Inc., Maumee, Ohio) and Nestlets nesting material (Ancare, Bellmore, NY) and maintained on a 12:12 hour light:dark cycle. Mice for self-administration studies were singly housed following implantation of jugular catheters in BCU-2 (Allentown Inc., Allentown, NJ) filtertop cages with Enrich-O’Cob bedding and Enviro-Dri nesting material (Shepherd Specialty Papers, Watertown, TN) and maintained on a 14:10 light:dark cycle. Experiments were conducted at the beginning of the light cycle, with the exception of extended access self-administration training. Tap water and standard laboratory chow were supplied ad libitum, except during testing.
METHOD DETAILS
Chemicals.
All chemicals were obtained from MilliporeSigma (St. Louis, MO) unless otherwise noted. Method-specific compounds are described with their respective procedures. SBI-0654553 HCl (SBI-553) was synthesized by the Conrad Prebys Center for Chemical Genomics at Sanford Burnham Prebys Medical Discovery Institute (La Jolla, CA, USA). NTS was maintained as a 2 mM stock in 80% glycerol. SBI-553 and SR142948A were maintained as 10 mM or 50 mM stocks in DMSO.
Recombinant DNA plasmids.
The 3xHA-NTSR1 plasmid consists of N-terminal 3xHA-tagged wild-type human NTSR1 cloned into the pcDNA3.1+ vector (Invitrogen) at KpnI (5’) and XbaI (3’). This construct was purchased from the University of Missouri (cDNA Bank cDNA.org, CAT# NTSR10TN00). The 3xHA-NTSR1-GFP construct contains an N-terminal 3xHA-tagged wild-type human NTSR1 cloned into the eGFPN3 vector (Clontech, Mountain View, CA) (Barak et al., 2016). The 3xHA-NTSR1-FAP plasmid consists of N-terminal MarsCy1-tagged wild-type human NTSR1 in the pcDNA3.1+ vector (Invitrogen) (Barak et al., 2016). The β-arrestin2-Venus plasmid contains C-terminal Venus-tagged mus muluscus β-arrestin2 in the N1-mVenus vector (Addgene plasmid #54640) (Pack et al., 2018). The GRK2-Flag plasmid contains C-terminal Flag-tagged Bos taurus GRK2 in the pcDNA1/Amp vector (Pitcher et al., 1995). The NTSR1-Rluc plasmid was made previously in the Caron Laboratory by inserting wild-type human NTSR1 into a codon humanized pRluc-N vector (BioSignal Packard, Montréal, Québec, Canada). The mitochondrial-targeting apoaequorin expression vector (Rizzuto et al., 1992) was a gift from Dr. Stanley Thayer (University of Minnesota). The constructs used for the TGFα shedding assay, including the Gαq chimeric protein plasmid and AP-TGFα plasmid (Inoue et al., 2012), were generously provided by Dr. Asuka Inoue (Tohoku University, Sendai, Japan). The IP3 intramolecular BRET sensor construct was a gift from Dr. Peter Varnai (Semmelweis University, Budapest, Hungary).
Radioligand binding.
Binding assays were carried out using cell membranes from HEK293T cells transiently expressing 3xHA-NTSR1. Briefly, NTSR1-expressing HEK293T cells were grown to confluence in 150 mm dishes, washed once with cold PBS, and detached using a cell lifter in 3 ml of assay buffer (50 mM Tris, 0.1% BSA and Roche protease inhibitors, pH 7.4). The cell suspensions were disrupted over ice using a Teflon® glass homogenizer. To remove tissue debris and nuclei, the resulting homogenate was centrifuged at 1000 rpm for 10 min at 4°C. Membrane protein was collected by subsequent centrifugation at 40,000 g for 30 min at 4°C and used immediately or stored at −80 °C until use. On the day of the experiment, cell membranes were further sheared by passing them through a 25G needle 3–4 times. For experiments employing a traditional harvest system approach: [3H]NTS binding was assessed in 96-well round bottom polypropylene plates. Membranes (1–10 μg/well) were first incubated with NTS, SR142948A or SBI-553 for 10 min on a shaker at room temperature. [3H]NTS was added at indicated concentrations in each well for an additional 1 hr incubation at room temperature. Binding reactions were terminated by filtration of membranes onto glass filters (GF/B – Whatman, Brandel, Gaithersburg, MD, USA, Cat# FPD-196) using a 96 channel Brandel plate harvester. Filtration was followed by three rapid washes with cold 50 mM Tris-HCl buffer (pH 7.4). Filter disks were transferred to scintillation vials, and 4 ml Bio-Safe II scintillation cocktail was added to each vial. CPM of [3H]NTS were determined using a Packard 1900TR liquid scintillation analyzer. Specific binding was obtained by calculating the difference between the total [3H]NTS and nonspecific [3H]NTS binding curves. Nonspecific binding was determined using the competitive NTSR1 antagonist SR142948 or unlabeled NTS at a concentration of 10 μM. For experiments employing a YOx on-bead assay: 1 mg wheat germ agglutinin-coated yttrium oxide (YOx) beads (PerkinElmer, Inc., Waltham, MA, USA, Cat# RPNQ0272) were incubated overnight at 4°C with 20 μg NTSR1-HEK membrane in assay buffer. On the day of the experiment, beads were washed once with assay buffer to remove unbound membrane. Membrane coated YOx beads were incubated for 1 hr at room temperature with NTS, SR142948A, SBI-553 and [3H]SBI-553 or [3H]NTS in 1.5 ml microcentrifuge tubes. Incubation was followed by three rapid washes of the beads with cold assay buffer. Beads were transferred to scintillation vials, and 4 ml Bio-Safe II scintillation cocktail was added to each vial. Specific binding was obtained by calculating the difference between the total [3H]SBI-553 and nonspecific [3H]SBI-553 binding curves. Nonspecific binding was determined using unlabeled SBI-553 at a concentration 100x that of [3H]SBI-553. [3H]NTS (Neurotensin, [3,11-Tyrosyl-3,5–3H(N)]) was purchased from PerkinElmer, Inc. [3H]SBI-553 (concentration: 1.0 mCi/ml; specific activity: 16.9 Ci/mmol) was obtained as a custom order from Moravek, Inc. (Brea, CA, USA).
Phosphorylated NTSR1 detection by western blot.
1 × 106 HEK293T cells/well were plated onto 6-well plates. The following day, cells were transfected with 4 μg 3xHA-NTSR1-GFP and 8 μl Lipofectamine 2000 per well in 2 ml of freshly changed, antibiotic-free media. 24 hr post-transfection, cells are treated with drugs (100 nM NTS, 10 μM SBI-553 or 10 μM SR142948A) in 1 ml media for 30 minutes at 37°C. Following treatment, cells were washed once with ice-cold Hank’s Balanced Salt Solution (HBSS) and lysed in 200 μl ice-cold lysis buffer: 1% Anatrace detergent (Dodecyl maltoside (D310) and cholesteryl hemisuccinate (CH210) solution, Anatrace, Maumee, OH, USA) in TBS with protease and phosphatase inhibitors). Cell lysates were incubated for 20 min on ice with vortexing every 5 minutes and centrifuged (21,000 rcf at 4°C for 10 min). Supernatants were transferred to pre-chilled tubes and 300 μl of cold wash buffer (0.1% Anantrace detergent in TBS) was added. 15 μl of GFP-TRAP Magnetic Agarose beads (ChromoTek, Munich, Germany, Cat# gtma-20) per sample were washed 3x in wash buffer and re-suspended in 40 μl cold wash buffer. 40 μl of bead solution was added per sample and rotated overnight at 4°C. The following day, beads were washed 3x in ice-cold wash buffer. Protein was eluted with 40 μl elution buffer: 2x NuPAGE LDS sample buffer (Invitrogen NP0007), 1% Anatrace detergent and 10% 2-Mercaptoethanol (BME) with protease and phosphatase inhibitors) and heated at 95°C for 10 min. 10 μl of samples were run on Nu-PAGE 4–12% Bis-Tris gels and transferred to 0.45uM nitrocellulose membrane at 35 V for 2 hr in the cold. Blots were blocked with Li-Cor blocking buffer and probed with 1:3,000 rabbit anti-phosphothreonine antibody (Cell Signaling Technology, Cat# 9386, RRID:AB_331239) and 1:3,000 mouse anti-GFP (Abgent, Cat# AM1009a, RRID:AB_352468) overnight at 4°C. Blots were probed with secondary 1:10,000 anti-rabbit 680 (Invitrogen, Cat# A-21109, RRID:AB_2535758) and 1:10,000 anti-mouse 800 (LI-COR, Cat# 926–32212, RRID:AB_621847) antibodies. Membranes were imaged on LI-COR Biosciences Odyssey imaging system (Lincoln, NE, USA). Bands were quantitated using Image Studio Lite v. 5.2 (LI-COR Biosciences). In sample blot images, brightness and contrast were increased uniformly across all blots to optimize band visualization. All pNTSR1 data were normalized to total NTSR1-GFP signal in each lane. pNTSR1 to total NTSR1 ratios are represented as average fold change over vehicle-treated control ± SEM. Individual replicates are displayed as like-colored symbols.
Confocal β-arrestin 2 translocation assay.
The translocation of β-arrestin2 to the membrane by NTSR1 was assessed using a U2OS cell line permanently expressing the NTSR1 and green fluorescent protein (GFP) tagged-β-arrestin2, as described (Barak et al., 2016). Briefly, on day 1, stable cells were split into MGB101-1-2-LG glass-bottom 384-well plates (MatriCal, Spokane, WA, USA). Each well contained 30 μL aliquots of 8,000 cells in Minimum Eagle’s medium (MEM) containing 10% fetal bovine serum (FBS) and 100 U/mL penicillin/streptomycin (Life Technologies, Grand Island, NY, USA). The plates were incubated overnight at 37°C in 5% CO2, and on the following day the media was changed to 30 μL serum- and phenol red-free MEM. Increasing concentrations of 10x NTS, SBI-553 and SR142948A stocks were added to the wells and diluted 10-fold to reach final concentrations. To test the additive/antagonist property of NTSR1 ligands, cells were pretreated with SBI-553 (2 or 4 μM) or SR142948A (100 pM – 10 μM) 10 min prior to treatment with NTS (100 pM – 100 μM) or SBI-553 (10 μM). After treatment, plates were returned to the incubator for 40 min. The cells were then fixed by adding 30 μL of 2% paraformaldehyde-phosphate buffered saline (PBS) to each well. Plates were stored at 4 °C until analysis at 488 nm on a robotic imager (ImageXpress Ultra, Molecular Devices, Sunnyvale, CA, USA). Images were analyzed using a wavelet algorithm to measure formation of fluorescence aggregates (Evron et al., 2014). Data are represented as mean aggregates per unit cell area ± SEM. Analyzed image results were visually confirmed. In sample images, brightness and contrast were increased uniformly across all images and all conditions to optimize visualization of GFP signal. To determine whether observed β-arrestin2 translocation was NTSR1-dependent, representative images were captured from U2OS cells transiently expressing GFP tagged β-arrestin2 and human NTSR1 or empty pcDNA vector. For these experiments, cells were plated at a density of (4–8) × 104/well in 35 mm MatTek (Ashland, MA, USA) glass coverslip dishes. Cells were left unstimulated or treated with 200 nM NTS or 7.5 μM of SBI-553 for 40 min, fixed in 2% paraformaldehyde, and examined on a Zeiss Axiovert 200 fluorescence microscope using a plan-apochromat 40x/0.95 N.A. air objective.
BRET β-arrestin2 recruitment assay.
Recruitment of Venus-tagged β-arrestin2 to Renilla luciferase (Rluc)-tagged NTSR1 was assessed in HEK293T cells using a bioluminescence resonance energy transfer (BRET) assay, as described (Pack et al., 2018). On day 1, HEK293T cells were plated in 6-well plates in growth media. On day 2, cells were transiently transfected with Venus-tagged β-arrestin2 (2 ug/well), Rluc-tagged NTSR1 (100 ng/well), and flag-tagged GRK2 (500 ng/well) using a standard calcium phosphate transfection protocol. On day 3, cells were plated onto poly-D-lysine-coated, clear bottom, white-walled 96-well plates in Opti-MEM containing 2% FBS and 1x antibiotic antimycotic solution. On day 4, cells were incubated in 70 μl/well Hanks’ Balanced Salt solution (HBSS) containing calcium and magnesium for 2–4 hr prior to treatment. Treatments were freshly prepared in HBSS from 50 mM DMSO (SBI-553, SR142948A) or 2 mM 50% glycercol (NTS) stocks. A white vinyl sticker was placed on the bottom of the plate. 10 μl of 10x SBI-553, SR142948A or vehicle pretreatments were added 10 min prior to reading. 10 μl/well of a 10x concentration of coelenterazine h (final concentration ~4.7 μM; Cayman Chemical Co., Ann Arbor, MI, USA) was added 5 min prior to reading. 10 μl of 10x NTS, SBI-553, or vehicle treatments were added, and plates read with a Berthold Mithras LB 940 plate reader (Bad Wildbad, Germany) set at 35°C 5, 10, 15, and 30 min post treatment. The NET BRET ratio was calculated by subtracting the stimulated Venus/Rluc ratios from vehicle Venus/Rluc ratios at every time point. The maximum NET BRET ratio over the time course was averaged within treatments and combined between experiments. Data are presented as Mean NET BRET ratio ± SEM.
NTSR1 internalization assay.
Internalization of NTSR1 was assessed using a MarsCy1 tagged-human NTSR1 receptor, as described (Barak et al., 2016). The cell surface expression of MarsCy1 epitope-tagged receptors can be visualized by the addition of the fluorogen SCi1 (Snyder et al., 2015). SCi1 was obtained from Dr. Alan S. Waggoner (Carnegie Mellon University, Pittsburgh, PA, USA). For this assay, U2OS cells in 100 mm plates were transiently transfected on day 1 with the MarsCy1 tagged-NTSR1 plasmid using Lipofectamine 2000 via its standard protocol (Life Technologies, Carlsbad CA). On day 2, cells were split into 96 well plates in 100 μL serum free media (MEM) supplemented with 10% FBS (ThermoFisher/GIBCO, Waltham MA). On day 3, media was replaced with serum-free MEM containing 10 mM HEPES and 1% Glutamax (ThermoFisher/GIBCO). On day 4, for agonist assays, the cells were treated for 1 hr at 37°C in 5% CO2 by replacing the media with 75 μL of an equivalent solution also containing NTS (100 pm – 10 μM), SBI-553 (100 pm – 30 μM) or SR142948A (100 pm – 30 μM). Post-treatment, Sci1 was added and plates were immediately read. For additive/antagonist assays, the cells were pre-treated for 10 min with SBI-553 (1 or 10 μM) or SR142948A (100 pm – 100 μM) prior to 1 hr incubation with SBI-553 (10 μM) or NTS (10 nM). A LI-COR Odyssey® (LI-COR Biosciences, Lincoln, NE, USA) system in the 700 nm channel was used for scanning MarsCy1 tagged-NTSR1 stained with the plasma membrane impermeant fluorescent compound SCi1 with focal offsets adjusted to match the focal plane of the plate. SCi1 was reconstituted in ethanol with 5% acetic acid at 78 μM and utilized at approximately 1:4,000 dilution. Live cells were incubated with 20 nM SCi1 for 5 minutes prior to imaging. No washing was performed. % internalization was determined by expressing signal in compound-stimulated wells as a percent of signal in corresponding vehicle control wells. Data are represented as % internalization ± SEM.
Gq protein activation by TGFα shedding assay.
TGFα shedding assays were performed as originally described (Inoue et al., 2012) with previously detailed modifications (Pack et al., 2018). We used G protein-deficient (ΔGNAS, GNAL, GNAQ, GNA11, GNA12, and GNA13) HEK293 cells (Inoue et al., 2012) and the fluorescent substrate, 4-methylumbelliferyl phosphate (MilliporeSigma), instead of p-Nitrophenyl Phosphate (p-NPP). The overexpression of Gq protein as performed in the assay here in Figure 2 has been observed by Inoue and colleagues to enhance the response of the shedding assay compared to the characteristic response observed with an endogenous complement of G proteins such that the amplitude and/or EC50/IC50 is changed. Alkaline phosphatase activity was measured using a ClarioStar plate reader (BMG Labtech, Ortenberg, Germany) equipped with a fluorescent reader. Excitation was set at 360 nm (±10 nm) and emission at 450 nm (±15 nm). Data were collected over the course of 60 minutes. Shedding activity was calculated by dividing the amount of phosphatase activity present in the conditioned media by the amount present on the cells plus the conditioned media. All values were standardized to background shedding activity, defined as compound-induced shedding activity in non-NTSR1 expressing HEK293T cells, and represented at mean standardized shedding ± SEM.
IP3 BRET assay.
IP3 generation was measured using an intramolecular BRET sensor (Gulyas et al., 2015). HEK293T cells were transfected to express both NTSR1 and an IP3 biosensor: the ligand-binding domain of the human type-I IP3-receptor tagged on the N-terminus with super Renilla luciferase and on the C-terminus with Venus. Treatment compounds were injected into a cell-containing assay plates and plates were read at 485 and 530 nm every 15 sec for 3 min. Cells were stimulated with NTS (100 nM), SBI-553 (10 μM), SR142948A (10 μM) or vehicle (0.1% DMSO) at time 0. Data are represented as mean net BRET ratio ± SEM.
Apo-aequorin calcium assay.
Assessment of intracellular calcium mobilization was performed using a functional bioluminescence-based aequorin assay (Rizzuto et al., 1992), as previously described (Evron et al., 2014). Briefly, HEK293 cells stably expressing a mitochondrial membrane localized apoaequorin calcium reporter were transiently transfected with 3xHA-NTSR1 (3 μg/well on a 6-well plate) using a standard calcium phosphate transfection protocol. On the day of the experiment, cells were incubated in serum-, antibiotic- and phenol red-free Opti-MEM reduced serum media (Thermofisher) for 2 hr at 37 °C. Cells were then treated with Opti-MEM containing 2.5 μM coelenterazine h (Cayman Chemical Co.) for an additional 1–2 hr in a 37 °C incubator. Following this incubation, cells were washed from the plate and dispensed (30 μl, 20,000 cells/well) into increasing concentrations of NTS, SBI-553, SR142948A or vehicle (0.0002%−0.2% DMSO) that were predispensed into a 96-well white flat-bottom microplate at 2x concentrations in volumes of 30 μl. For antagonist assays, 10 min after the cells were dispensed into treatment-containing plates, they were treated with a 7x concentration of NTS in 10 μl/well to obtain a final concentration of 10 nM NTS. Luminescence was recorded using a Mithras LB 940 instrument (Berthold Technologies, Oak Ridge, TN) for 20 s/well immediately after dispensing cells for agonist assays and immediately after dispensing NTS for antagonist assays. After all wells had been read, 80 μl of a calcium lysis buffer (100 mM CaCl and 0.2% Triton-X) was dispensed into each well, and luminescence was recorded for 5 s/well. To control for variations in cell numbers, stimulated responses were normalized by dividing the stimulated response by the total response value (i.e., sum of responses following stimulation and detergent cell lysis) to obtain net luminescence. Background signal, defined as normalized signal following vehicle treatment, was subtracted from stimulated responses to obtain the presented average net luminescence values ± SEM.
Phosphorylated ERK detection by western blot.
Western blotting of phosphorylated (p-ERK) and total ERK was performed as previously described (Peterson et al., 2015). On day 1, HEK293T, Gq-null HEK293 (Alvarez-Curto et al., 2016), and β-arrestin1/2-null (Alvarez-Curto et al., 2016) HEK293 cells were plated at 750,000 cells per well onto 6-well plates. On day 2, cells were transiently transfected with 3xHA human NTSR1 pcDNA (1.5 μg/well) or empty pcDNA (1.5 μg/well) using a standard calcium phosphate transfection protocol. On day 3, cells were incubated overnight in serum-, phenol red- and antibiotic-free media (MEM with HEPES and Glutamax). On day 4, drug treatments were prepared as 20x stocks in Hanks’ Balanced Salt solution (HBSS) with calcium and magnesium. Cells were treated with NTS (10 nM, final concentration), SBI-553 (10 μM), SR142948A (10 μM) or vehicle (0.2% DMSO) for 0, 5, 15, 30 or 60 min. During treatment incubations cells were maintained at culture conditions (e.g., 37°C and 5% CO2). At designated treatment end points, cells were lysed in ice-cold RIPA buffer containing protease and phosphatase inhibitor mixtures (Sigma). Whole-cell lysates were analyzed for expression of p-ERK and total ERK. Cell protein samples (10 μg) were resolved on 10% SDS–polyacrylamide gels (NuPAGE, Bis-Tris; Thermo Fisher Scientific, Waltham, MA) and transferred to nitrocellulose membranes (0.45 μm pore size; Thermo Fisher Scientific). Membranes were incubated in Odyssey Blocking Buffer, TBS (Li-Cor Biosciences, Lincoln, NE) for 1 hr at room temperature. Blocked membranes were incubated overnight at 4°C with rabbit anti-pERK (Cell Signaling Technology, #4370, RRID:AB_2315112) and mouse anti-ERK (Cell Signaling Technology, #9107S, RRID:AB_10695739) in 50% blocking buffer and 50% tris-buffered saline containing 0.1% Tween-20 (vol/vol; TBST). Membranes were washed with TBST prior to and following incubation with infrared secondary antibodies Alexa Fluor® goat anti-rabbit 680 (Invitrogen, Carlsbad, CA, #A-21109, RRID:AB_2535758) and IRDye donkey anti-mouse 800 (Li-Cor Biosciences, #926–32212, RRID:AB_621847) for 1 hr at room temperature. Membranes were imaged on LI-COR Biosciences Odyssey imaging system (Lincoln, NE). Bands were quantitated using Image Studio Lite v. 5.2 (LI-COR Biosciences). In sample blot images, brightness and contrast were increased uniformly across all blots to optimize band visualization. All p-ERK data were normalized to total ERK in each lane, and pERK to total ERK ratios are reported as average fold change over untreated control (Time 0) ± SEM.
In vivo drug treatment.
SBI-553 (2, 6, 12 or 30 mg/kg), cocaine HCl (30 mg/kg, for open field studies), methamphetamine HCl (2 mg/kg), α-methyl-p-tyrosine (250 mg/kg), L-DOPA (25 mg/kg), and benserazide (12.5 mg/kg) were dissolved in sterile physiological saline (Henry Schein, Raleigh, NC) and injected intraperitoneally (i.p.) according to the experimental schedule at a volume of 5 or 10 ml/kg body weight. All compounds except for SBI-553 (provided by Sanford-Burnham-Prebys) were purchased from MilliporeSigma (Burlington, MA). Saline (at 5 or 10 ml/kg, as appropriate) was administered as a vehicle control. For intravenous cocaine self-administration studies, cocaine HCl (0.1–1 mg/kg/infusion) was administered intravenously (iv) to animals with indwelling jugular catheters according to the test protocol and the mouse’s lever responding.
Locomotor activity.
The Accuscan open field (Omnitech Electronics, Inc.) was used to assess locomotor activity, as previously described (Toth et al., 2018). For evaluating stimulant-induced hyperlocomotion, drug-naïve male and female mice were acclimated to the open field for 30 min (indicated by grey box) prior to concurrent administration (i.p.) of vehicle (saline, 5 or 10 ml/kg, i.p.) or cocaine (30 mg/kg, i.p., 5 ml/kg) or methamphetamine (2 mg/kg, i.p.) and vehicle (saline, 5 or 10 ml/kg, i.p.) or SBI-553 (12 mg/kg, i.p.). Animals were immediately returned to the open field, and locomotion was monitored over the next 2 hr. For evaluating L-DOPA-induced restoration of locomotion in dopamine depleted DAT KO mice, DAT KO mice were acclimated to the chambers for 30 min prior to administration of α-methyl-p-tyrosine (AMPT, 250 mg/kg, i.p., 5 μl/g). One hr post AMPT treatment animals received L-DOPA (25 mg/kg, i.p.) and the DOPA decarboxylase inhibitor benserazide (12.5 mg/kg, prepared with L-DOPA, i.p.) in combination with vehicle (saline, 5 ml/kg, i.p.) or SBI-553 (12 mg/kg, i.p.). Animals were immediately returned to the open field, and locomotion was monitored over the next 2 hr.
PET/CT imaging.
Studies were conducted at the Small Animal Imaging Facility at the University of North Carolina, Chapel Hill Biomedical Research Imaging Center (BRIC). Study design: On study Day 1 baseline PET/CT imaging was performed on DAT KO mice without any drug treatment. On Day 2, DAT KO mice received the tyrosine hydroxylase inhibitor α-methyl-p-tyrosine (AMPT, 250 mg/kg, i.p., 5 μl/g) 1 hr prior to administration of L-DOPA (25 mg/kg, i.p., 5 μl/g) and the DOPA decarboxylase inhibitor benserazide (12.5 mg/kg, prepared with L-DOPA). The PET probe [18F]-FDG was delivered via tail vein injection 10 min after L-DOPA treatment. On Day 3 DAT KO mice were treated with AMPT followed by an injection of L-DOPA, benserazide and SBI-533 (12 mg/kg, i.p., 5 μl/g) at 1 hr. [18F]-FDG, was administered 10 min after the co-injection of L-DOPA, benserazide and SBI-533. Scan acquisition: PET imaging with [18F]-FDG was performed using a small animal PET/CT scanner (eXplore VISTA model, GE Healthcare, Waukesha, WI). Mice were fasted for at least 3 hr prior to the PET imaging. Before imaging, mice were anesthetized briefly by inhalation of isoflurane mixed with oxygen gas (3% isoflurane for induction and 1–2% for maintenance) and a tail vein catheter was placed on the mouse tail. Approximately 0.3 mCi (11 MBq) of [18F]-FDG in saline was administered through the tail vein catheter as a bolus injection. PET imaging (30 min in duration) was acquired at 30 min post [18F]-FDG injection followed by a CT scan for attenuation correction and anatomical colocalization. PET images were reconstructed using 2D ordered subset expectation maximization (OSEM) algorithms with random, scatter, and attenuation corrections. The standardized uptake value (SUV) was calculated voxel-wise with normalization to the injected dose and animal body weight (SUV = radioactivity concentration / (Injected-dose / body weight)). Registration and image processing: PET images were aligned with corresponding CT images based on the CT imaging contrast using Advanced Normalization Tools (ANTs) (Avants et al., 2011; Kim et al., 2008), with corrections made by hand based on anatomical landmarks (i.e., heart, bladder, eyes/Harderian glands) using Amide’s a Medical Imaging Data Examiner software (AMIDE) (Loening and Gambhir, 2003). PET and CT images were cropped around the head of the animal using a rectangular bounding box. An average CT scan was generated using a minimum deformation template strategy, as in (Anderson et al., 2018). An MR-based 3-dimensional Waxholm space C57BL/6J brain atlas (Calabrese et al., 2015) was interactively aligned/mapped using an affine transform to fit the intracranial space of the average DAT CT template, using 3D Slicer (Fedorov et al., 2012) (https://www.slicer.org/). PET images were resampled to the same voxel dimensions of the atlas (0.43 μm, isotropic) and transformed according to the transformations determined by their CT corresponding scans. CT scans were co-registered based on a mutual information metric with 16 bins of the histogram, a 0.25 gradient step, using a multi resolution scheme with 150×50×50 iterations, at each level from 8×4×1 downsampling, and with 4-voxel smoothing at each level. PET and CT images were thus mapped into the space of an MR-based 3-dimensional Waxholm space C57BL/6J brain atlas through concatenating the above-mentioned image transformations. A 3-dimensional binary mask image was produced from the MR atlas and all values outside the brain were set to zero to isolate PET signal originating in the brain. Brain PET signal was smoothed (1 voxel radius kernel) to enhance signal-to-noise ratio (Reimold et al., 2006). Analysis: See Quantification and Analysis Section below.
Conditioned place preference.
Preference to conditioning was assessed in a three-chamber system (model ENV-3013, Med Associates, St. Albans, VT, USA) with the two larger end chambers used for conditioning sessions (see Barak et al., 2016). One of the two end chambers had bar flooring and black walls while the other had grid flooring and white walls. The gray center chamber had solid flooring. On pretest day 0, male mice were placed in the center chamber with the doors connecting the two end chambers open. The time spent in chamber was recorded for 30 minutes. Mice were treated with methamphetamine (2 mg/kg, i.p.) on days 1, 3, and 5 and vehicle (saline, 10 ml/kg, i.p.) on days 2, 4, and 6. Mice were confined to an assigned end chamber post-injection for 30 minutes. Each mouse was confined to either the black or white chamber when receiving methamphetamine and to the opposite chamber when given saline the following day. To ensure that the assignment was unbiased, those mice that spent more than 60% of the 30 min preconditioning period in one end chamber were assigned to the alternate chamber after methamphetamine injection. For the mice showing no marked preference for either side, methamphetamine treatment was randomly paired with end chambers such that equal numbers of mice received methamphetamine in each end chamber. On the test day (day 7), mice were grouped for vehicle (saline, 10 ml/kg, i.p.) or SBI-553 treatment (12 mg/kg, i.p.) such that each treatment group had one-half of the mice methamphetamine conditioned to the left and the other half to the right chambers. 15 minutes after injection the grouped mice were given free access to the 3 chambers for 30 min. Time spent in the vehicle- and methamphetamine-paired chambers were recorded. Preference scores were calculated as the time spent in the vehicle chamber subtracted from the time spent in the methamphetamine chamber divided by the total time spent in both chambers. Conditioning scores were calculated by subtracting the pre-conditioning preference score from the post-conditioning preference score.
Self-administration.
Indwelling catheters (Instech Laboratories, Inc., Plymouth Meeting, PA, USA, Cat# C20PU-MJV1301) were implanted in the right jugular vein of adult male and female C57BL/6J mice fitted with vascular access buttons (Instech Laboratories, Inc., Cat# VAB62BS/25), as described (Kmiotek et al., 2012). To maintain patency and prevent infection, catheters were flushed 1–2x daily with a heparin/gentamicin solution. Following a 7–10 day post-surgical recovery period, animals that passed a ketamine test for catheter patency (Kmiotek et al., 2012) were trained to self-administer iv cocaine (0.5 mg/kg/infusion) paired with a cue light via lever responding in operant chambers (Med Associates, Inc., Fairfax, VT; Instech tethers Cat# KVAH62T/MED). Active, reinforced lever(s) were denoted by a solid cue light. Reinforcements were delivered by a single-speed syringe pump (Med Associates) in a volume of 18 μl over a 4 sec period. Each infusion was followed by a 40-sec-time-out period during which no additional reinforcements were awarded. For the first 20-sec of this time-out period the stimulus light blinked and levers remained available, but lever responses (i.e., cue responses) had no programed consequences. For the second 20-sec of this time-out period levers were retracted. Training began with a single 3 hr session in which responding at either of 2 levers resulted in a cocaine reinforcement at a fixed lever response to reinforcement ratio (fixed ratio, FR) schedule of 1. In this session, mice that did not self-administer a reinforcement every 6 min received a timed non-contingent infusion, such that all animals received a total of 30 reinforcements. The following day, mice completed an extended access (12 hr, over dark cycle) training session in which cocaine infusions were contingent on pressing either of 2 levers at FR1. Animals progressed sequentially through once daily, 1 hr, 2 lever training at FR1 to 1 hr, 1 lever training at FR1, FR2 and then FR4. During 1 lever training, animals discriminated between an active, reinforced lever and an inactive, non-reinforced lever at which responding has no programmed consequences. Training advancement was contingent upon meeting of performance criteria. To progress from 1 hr, 2 lever training to 1 hr, 1 lever training, an animal had to self-administer ≥ 5 mg/kg cocaine (10 reinforcements) in three consecutive sessions. To progress from 1 hr, 1 lever training at FR1 to higher FR sessions, an animal had to self-administer ≥ 5 mg/kg cocaine with ≥ 70% of total responses occurring at the active lever in two consecutive sessions. Stable lever responding at FR4 was assessed at 4 cocaine doses (i.e., 0.1, 0.3, 0.5, and 1.0 mg/kg/infusion). Dose order was varied between animals. Stable responding was considered achieved when self-administered reinforcements in two consecutive sessions varied by ≤ 20% and ≥ 70% of total responses occurring at the active lever. Once stable responding at a given cocaine concentration was achieved with vehicle (sterile saline; 10 ml/kg, i.p.) treatment, SBI-553 dose-response testing was initiated. The effect of 3 SBI-553 doses (2, 6, and 12 mg/kg; 10 ml/kg, i.p.) were assessed in once daily sessions in which animals received either vehicle or SBI-553 immediately prior to placement in the self-administration chamber. Dose order was varied between animals and cocaine doses, with animals receiving either 2, 6, and then 12 mg/kg or 12, 6 and then 2 mg/kg SBI-553. In the sessions immediately following SBI-553 treatment, animals were placed into the self-administration chambers either without treatment or immediately following vehicle treatment. Between each SBI-553 dose, animals were required to demonstrate stable responding following vehicle treatment. Animals that failed to achieve stable responding and/or ≥ 70% active lever discrimination in 4 consecutive sessions were subjected to a test of catheter patency. Animals with non-patent catheters were removed from the study.
Core body temperature measurement.
Core body temperatures were recorded from age-matched male and female C57BL/6J mice prior to treatment (Time 0) and 30, 60, 120, and 300 min post-treatment using a rectal probe thermometer for mice (Thermalert Model TH-8, Physitemp Instruments, Inc., Clifton, NJ). On three days of the week leading up to the study, animals were handled and temperatures were taken to acclimate the animals to the procedure. For single compound dosing studies, baseline temperatures were recorded and animals received either PD149163 (0.03–1 mg/kg), PD149163’s vehicle (0.2% DMSO in saline), SBI-553 (2–30 mg/kg), or SBI-553’s vehicle (physiological saline). All treatments were administered i.p. in a volume of 10 ml/kg. For multiple compound dosing studies, animals received SBI-553 at 10, 30 or 100 mg/kg or vehicle (10% DMSO, 0.05% Tween 80) p.o. via a metal feeding tube in a volume of 10 ml/kg 30 min prior to the start of the study (−0.5 hr). Following temperature recording at Time 0, animals received PD149163 (2 mg/kg) or vehicle (saline) in volumes of 10 ml/kg i.p. Open field activity was monitored as described above between 120 and 165 min post PD149163 treatment.
Horizontal wire and contact righting tests.
Wire hang and contact righting studies were conducted with age-matched male and female C57BL/6J mice as described (Nehrenberg et al., 2009). On the day of the study, animals received either PD149163 (0.03–1 mg/kg), PD149163’s vehicle (0.2% DMSO in saline), SBI-553 (2–30 mg/kg), or SBI-553’s vehicle (physiological saline). Behavioral testing was conducted at the times of peak drug effect: 120 min post-treatment for the PD149163 group and 30 min post-treatment for the SBI-553 group. To detect drug-induced myorelaxant effects and/or coordination changes, the horizontal wire test was utilized (Bonetti et al., 1982). This assay was selected because its successful performance requires both muscle strength and coordination (Deacon, 2013). A metal wire (2 mm in diameter, 70 cm in length) was secured 33 cm over a padded surface. Mice were placed on the wire such that they were supported only by their forepaws and trained to grasp a horizontal wire with their hind-limbs. Training was conducted in once daily 60 sec sessions over 5 days. On the day of the test, latency was recorded from placement of forepaws on the wire to when two hind limbs grasped the wire. For animals that failed to grasp the wire with two hind-limbs during the test, a 60 sec latency was recorded. To detect drug-induced sedation, changes in the righting reflex were assessed (Corrigan et al., 2015). Mice were placed on their feet in a glass tube (6.5 cm in diameter × 43 cm long), and the tube was rapidly rotated 180° for 6 consecutive trials. Latency to right was recorded over the trials. Responses were scored as: −6 for failure to right, −5 for >5 sec delay in righting, −4 for >2 sec delay in righting, −3 for >1 sec delay in righting, −2 for righting from back <1 sec, −1 for immediately rolling to the side and righting within 1 sec, and 0 for no delay in righting.
Blood pressure measurement.
To assess the effect of SBI-553 and PD146193 on blood pressure, hemodynamic measurements were performed on male WT C57BL/6J mice anesthetized with 3% isoflurane. A 1.4-Fr pressure-conductance catheter (Millar Instruments, Houston, TX) was inserted retroaortically into the left ventricle to record hemodynamics. Subsequently, parallel conductance (Vp) was determined by a 10 μl injection of 15% saline into the right jugular vein to establish the parallel conductance of the blood pool. The derived Vp was used to correct the pressure-volume loop data. Data were recorded digitally at 1,000 Hz and analyzed with pressure volume analysis software (PVAN data analysis software version 3.3; Millar Instruments). Temperature measurements were taken using a rectal probe thermometer for mice, and a heating pad was used to maintain the animal’s core temperature between 35–38°C. After baseline hemodynamic measures and temperature were established, each mouse received either Vehicle (0.6% DMSO in saline, n = 6), SBI-553 (12 mg/kg, n = 7) or PD146193 (0.3 mg/kg, n = 8) in a volume of 10 ml/kg (i.p.), and systolic pressure was monitored over 60 min. Average peak systolic pressure in 15 min bins was normalized to vehicle baseline values and represented as mean ± SEM change from Time 0 pressure. To be included in the analysis, animals had to survive the entire duration of the 60 min reading. Data from 1 animal was excluded from the analysis for failing to meet study inclusion criteria. Five animals instrumented on a single day were excluded from the study due to equipment malfunction.
Graphical abstract and figure illustrations.
The graphical abstract was created using Scientific Illustration Toolkits for Presentations and Publications (Motifolio, Ellicott City, MD) and BioRender (Toronto, ON). The diagram of the sagittal section of a mouse brain is credited to Jonas Töle, Creative Commons License. In Fig 7C, the mouse and neuron images were modified from content available from somersault18:24 (Diest, Belgium), Creative Commons License.
QUANTIFICATION AND STATISTICAL ANALYSIS
Biochemical and behavioral data analysis.
All data are represented as mean ± SEM. Individuals replicates are represented as symbols and provided for all single time-point, single-dose assessments. Data were analyzed and plotted using the software GraphPad Prism version 7.0. Information on curve fitting, statistical tests, and N are provided in supporting information Tables 1–7. In supporting information Tables 1, 2, and 3, N represents the number of biological replicates. For binding and signaling assays, individual experiments also included technical replicates. No statistical methods were used to predetermine sample size in biochemical assays. In supporting information Tables 4, 5, 6, and 7, N represents the number of individual mice in each drug treatment and genotype group. All behavioral experiments were replicated in independent cohorts on different experimental days. Experimental group size (N) for mouse studies was determined based on power analyses using GraphPad StatMate version 2. In these analyses, α (two-tailed) was 0.05, the desired power (1-β) was 0.7, and anticipated effect size and SD were based on preliminary or historical data. Assay-specific inclusion and/or exclusion criteria are addressed by procedure in Method Details. Parametric statistics were used for data that met normal (gaussian) distribution assumptions, as determined by a Shapiro-Wilk test. Wire hang and contact righting data, which were not normally distributed, were analyzed using nonparametric statistics (i.e., one-way ANOVA on ranks) (see supporting information Table 7). A p value of < 0.05 was accepted as statistically significant.
PET/CT image analysis.
Differences in [18F]-FDG uptake were assessed voxel-wise using statistical parametric mapping with SPM12 (Worsley et al., 1992). As scans of the same animals were acquired on different treatment days, analyses were based on paired t-tests. Significant variability in global brain [18F]-FDG uptake has been reported in mice (Welch et al., 2013). To address this, scans were standardized for whole brain uptake by entering average whole brain uptake, determined using the whole brain as a region of interest (ROI), as a covariant in the analysis. A single animal that did not respond to L-DOPA treatment on study day 2 was removed from the study, as the objective was to examine SBI-553 modulation of the L-DOPA treatment. To control for type I errors, a cluster-level false discovery rate correction was set at α = 0.05 and an initial cluster forming threshold for p values was set at 0.05.
Supplementary Material
Table S1. Curve parameters and statistical comparisons presented in Figure 1 – Supporting Figure 1.
Table S2. Curve parameters and statistical comparisons presented in Figure 2 – Supporting Figure 2.
Table S3. Statistical comparisons presented in Figure 3 – Supporting Figure 3.
Table S4. Statistical comparisons presented in Figure 4 – Supporting Figure 4.
Table S5. Curve parameters and statistical comparisons presented in Figure 5 – Supporting Figure 5.
Table S6. Curve parameters and statistical comparisons presented in Figure 6 –Supporting Figure 6.
Table S7. Statistical comparisons presented in Figure 7 – Supporting Figure 7.
Figure S1. Validation of on-bead binding assay with [3H]NTS and confirmation of [3H]SBI-553 NTSR1 specific binding at an allosteric site – Supporting Fig 1. (A) Nonspecific [3H]SBI-553 binding to different bead substrates. SBI-553 is lipophilic, and NTSR1 binding studies required substituting wheat germ agglutinin (WGA) coated yttrium oxide (YOx) beads for glass membranes to minimize non-specific binding. [3H]SBI-553 binding to a panel of beads was evaluated to minimize nonspecific binding. Data (mean ± SEM) were normalized to L-lysine bead determinations. WGA-YOx beads were selected for the on-bead binding assays. Type A, WGA-PEI A; Type B, WGA-PEI B; WGA, wheat germ agglutinin; PEI, polyethyleneimine; PS, polystyrene; YOx, yttrium oxide. N = 2 independent experiments.
(B,C) On-bead binding assay validation. [3H]NTS saturation binding to NTSR1-expressing membranes was compared using the novel on-bead assay and the classic glass microfiber filter paper/Brandel Harvester assay. (B) [3H]NTS saturation binding to NTSR1 using glass microfiber filter paper and Brandel Harvester: Use of Brandel Harvester. Data points are presented as mean CPM ± SEM. Calculated [3H]NTS 95% confidence intervals: (Bmax = 3,651 to 4,693 CPM, Kd = 1.8 to 4.0 nM). N = 4 independent experiments. (C) [3H]NTS saturation binding to NTSR1 using YOx bead assay platform: Use of YOx bead assay platform. Data points are presented as mean CPM ± SEM. Calculated [3H]NTS 95% confidence intervals: (Bmax = 2,748 to 5,720 CPM, Kd =1.5 to 18.0 nM). N = 3 independent experiments.
(D,E) Total [3H]SBI-553 and total [3H]NTS binding with substitution of β2-adrenergic receptor (β2AR)-containing membranes for NTSR1-containing membranes. To independently demonstrate specific [3H]SBI-553 binding to NTSR1, membrane competition studies were conducted in which the ratio of membranes containing NTSR1 to membranes containing an off-target GPCR (i.e., β2AR) was varied. Cell membranes from NTSR1-expressing and β2AR-expressing HEK293T cells were conjugated together at the indicated ratios to wheat-germ agglutin (WGA) coated yttrium oxide (YOx) beads. The beads were then incubated at room temperature in the presence of (D) 100 nM [3H]SBI-553 or (E) 5 nM [3H]NTS. Data were analyzed by one-way ANOVA followed by Bonferroni’s multiple comparisons test. Total [3H]SBI-553 binding was reduced with β2AR membrane substitution. *p<0.05, **p<0.01, compared to NTSR1 membranes alone (2:0) (F(3,14)=8.0, p=0.0024). Total [3H]NTS binding was reduced with β2AR membrane substitution. **p<0.01, ***p<0.001, as compared to NTSR1 membranes alone (2:0) using a one-way ANOVA (F(3,8)=49.1, p<0.001) followed by Bonferroni’s multiple comparisons test. N = 3 independent experiments.
(F) Competitive displacement of [3H]SBI-553 from NTSR1. NTSR1 membrane-coated YOx beads were incubated at room temperature with 100 nM [3H]SBI-553 in the presence of vehicle (0.1% DMSO in Opti-MEM),10 μM unlabeled SBI-553, neurotensin (NTS), or the NTSR1 antagonist SR142948A. Data are normalized to vehicle binding and presented as mean ± SEM. ***p<0.001, compared to vehicle using a one-way ANOVA (F(4,55)=26.0, p<0.001) followed by Bonferroni’s multiple comparisons test. N = 4 independent experiments.
(G) Competitive displacement of [3H]NTS from NTSR1. NTSR1 membrane coated YOx beads were incubated at room temperature with 5 nM [3H]NTS in the presence of vehicle (0.1% DMSO in Opti-MEM),10 μM unlabeled SBI-553, NTS, or SR142948A. Data are normalized to vehicle binding and presented as mean ± SEM. ***p<0.001, compared to vehicle using a one-way ANOVA (F(4,23)=220.1, p<0.001) followed by Bonferroni’s multiple comparisons test. N = 3 independent experiments.
Figure S2. NTS and SBI-553 fail to activate β-arrestin2 in absence of NTSR1 – Supporting Fig 1. U2OS cells stably expressing β-arrestin2-GFP were transiently transfected with human NTSR1-containing (top panels) or empty (bottom panels) pcDNA vectors. Cells remained unstimulated (left) or treated with NTS (200 nM, middle) or SBI-553 (7.5 μM, right) for 40 min. Following treatment cells were fixed and imaged using a fluorescein filter set and examined at 40×, Plan Apo objective, NA/0.85, on a Zeiss Axiovert 200 microscope. β-arrestin2-GFP aggregates are apparent with NTS and SBI-553 stimulation only in NTSR1-expressing cells and some dissimilarities between these aggregates may reflect compartmentalization differences. Scale bar = 10 μm.
Figure S3. Whole brain [18F]-FDG uptake did not differ among treatment groups – Supporting Fig 4. [18F]-FDG PET/CT scans of DAT KO mice (n = 8) were acquired prior to treatment (Baseline, Day 1), following dopamine depletion and stimulation with L-DOPA and vehicle (L-DOPA + Vehicle, Day 2) and following dopamine depletion and stimulation with L-DOPA and SBI-553 (L-DOPA + SBI-553, Day 3). Standardized uptake values (SUVs) were normalized to the injected [18F]-FDG dose and mouse’s body weight. PET/CT scans were co-registered with a three-dimensional Waxholm space C57BL/6J brain atlas (Calabrese et al., 2015). Whole brain SUVs were calculated using the region of interest (ROI) feature of AMIDE: a Medical Imaging Data Examiner software (Loening and Gambhir, 2003). No differences in whole brain uptake among treatment days were identified using a one-way ANOVA (F(2,21)= 0.22, p = 0.80.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) rabbit monoclonal antibody (Clone D13.14.4E) | Cell Signaling Technology | Cat# 4370, RRID:AB_2315112 |
| p44/42 MAPK (Erk1/2) mouse monoclonal antibody (Clone 3A7) | Cell Signaling Technology | Cat# 9107, RRID:AB_10695739 |
| Goat anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 680 | Invitrogen | Cat# A-21109, RRID:AB_2535758 |
| IRDye 800CW Donkey anti-Mouse IgG antibody | LI-COR | Cat# 926–32212, RRID:AB_621847 |
| Phospho-threonine mouse monoclonal antibody (Clone 42H4) | Cell Signaling Technology | Cat# 9386, RRID:AB_331239 |
| Anti-GFP Tag mouse monoclonal antibody (Clone 6AT316) | Abgent | Cat# AM1009a, RRID:AB_352468 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| SBI-553 HCl (SBI-553) | Sanford Burnham Prebys Medical Discovery Institute | SBI-0654553 |
| SR142948A | MilliporeSigma | Cat# SML0015 |
| [3H]SBI-553, Concentration 1.0 mCi/ml, Specific Activity 16.9 Ci/mmol | Moravek | Custom order |
| [3H]Neurotensin, [3,11-Tyrosyl-3,5–3H(N)] ([3H]NTS) | PerkinElmer | Cat# NET605025UC |
| Coelenterazine h | Cayman Chemical | Cat# 16894 |
| Neurotensin (NTS) | MilliporeSigma | Cat# N6383 |
| α-Methyl-DL-tyrosine methyl ester hydrochloride (α-methyl-p-tyrosine) | MilliporeSigma | Cat# M3281 |
| L-3,4-Dihydroxyphenylalanine methyl ester hydrochloride (L-DOPA) | MilliporeSigma | Cat# D1507 |
| Benserazide hydrochloride (Benserazide) | MilliporeSigma | Cat# B7283 |
| [18F]fluorodeoxyglucose ([18F]-FDG) | Biomedical Research Imaging Center Cyclotron Facility, University of North Carolina Chapel Hill | N/A |
| Cocaine hydrochloride (Cocaine) | MilliporeSigma | Cat# C5776 |
| (+)-Methamphetamine hydrochloride (Methamphetamine) | MilliporeSigma | Cat# M8750 |
| PD 149163 tetrahydrochloride hydrate (PD149163) | MilliporeSigma | Cat# PZ0175 |
| 4-Methylumbelliferyl phosphate disodium salt | MilliporeSigma | Cat# M8168 |
| Dodecyl Maltoside (D310) and Cholesteryl Hemisuccinate (CH210) Solution | Anatrace | Cat# D310-CH210 |
| SCi1 fluorogen | Dr. Alan S. Waggoner, Carnegie Mellon University, Pittsburgh, Pennsylvania, USA | N/A |
| Experimental Models: Cell Lines | ||
| HEK293T/17 | American Type Culture Collection (ATCC) | Cat# CRL-11268, RRID:CVCL_1926 |
| U2OS | American Type Culture Collection (ATCC) | Cat# HTB-96, RRID:CVCL_0042 |
| β-arrestin1/2-deficient / arrestin 2/3-deficient (ΔArrb1/2) HEK293 - Clone 4 | Alvarez-Curto et al., 2016 | N/A |
| Gq/11-deficient (ΔGNAQ and GNA11) HEK293 - Clone 1 | Alvarez-Curto et al., 2016 | N/A |
| Parental HEK293 line from which β-arrestin and Gq null lines were generated | Alvarez-Curto et al., 2016 | N/A |
| G protein-deficient (ΔGNAS, GNAL, GNAQ, GNA11, GNA12, and GNA13) HEK293 - Clone 38 | Inoue et al., 2012 | N/A |
| U2OS cells stably expressing β-arrestin2-GFP and 3xHA-NTSR1 | Barak et al., 2016 | N/A |
| HEK293 cells stably expressing mitochondrially-targeted apoaequorin | Barak et al., 2016 | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6J: WT | The Jackson Laboratory | Cat# 000664 |
| Mouse: DAT KO: Slc6a3tm1 | Giros et al., 1996 | N/A |
| Mouse: β-arrestin2 KO: Arrb2tm1Rjl | Bohn et al., 1999; The Jackson Laboratory | Cat# 011130 |
| Mouse:β-arrestin2f/f: Arrb2tm1(flox)nu | Urs et al., 2016 | N/A |
| Mouse:D1R-Cre/β-arrestin2f/f:Drd1tm1(Cre)/Arrb2tm1(flox)nu | Urs et al., 2016 | N/A |
| Mouse:D2R-Cre/β-arrestin2f/f:Drd2tm1(Cre)/Arrb2tm1(flox)nu | Urs et al., 2016 | N/A |
| Mouse:A2aR-Cre/β-arrestin2f/f: Adora2tm1(Cre)/Arrb2tm1(flox)nu | Urs et al., 2016 | N/A |
| Mouse: DAT-Cre/β-arrestin2f/f: Slc6a3tm1(cre)/Arrb2tm1(flox)nu | Toth et al., 2018 | N/A |
| Recombinant DNA | ||
| NTSR1: N-terminal 3xHA-tagged human neurotensin receptor 1 (NTSR1) (wild-type) cloned into pcDNA3.1+ (Invitrogen) at KpnI (5’) and XbaI (3’) | cDNA Resource Center | Cat# NTSR10TN00 |
| NTSR1-GFP: N-terminal 3xHA-tagged human neurotensin receptor 1 (NTSR1) (wild-type) cloned into eGFPN3 (Clontech) | Barak et al., 2016 | N/A |
| NTSR1-FAP: N-terminal MarsCy1-tagged human neurotensin receptor 1 (NTSR1) (wild-type) in pcDNA3.1+ (Invitrogen) | Barak et al., 2016 | N/A |
| Mitochondrial-targeted apoaequorin expression vector | Rizzuto et al., 1992 | N/A |
| Inositol-1,4,5-trisphosphate BRET sensor | Gulyas et al., 2015 | N/A |
| Gαq chimeric protein plasmid | Inoue et al., 2012 | N/A |
| NTSR1-Rluc: C-terminal Rluc-tagged human NTSR1 in the pRluc-N vector (BioSignal Packard Cat. #6310220) | Caron Laboratory | N/A |
| β-arrestin2-Venus: C-terminal Venus-tagged mus muluscus β-arrestin2 in the N1-mVenus vector (Addgene plasmid #54640) | Pack et al., 2018 | N/A |
| GRK2-FLG: C-terminal Flag-tagged Bos taurus GRK2 in pcDNA1/Amp vector | Pitcher et al., 1995 | N/A |
| AP-TGFα plasmid | Inoue et al., 2012 | N/A |
| Software and Algorithms | ||
| Image Studio Lite version 5.2 | LI-COR | https://www.licor.com/bio/image-studio-lite/ |
| Prism, version 7 | GraphPad | https://www.graphpad.com/ |
| Statmate, version 2 | GraphPad | https://www.graphpad.com/ |
| Wavelet algorithm for confocal β-arrestin2 translocation assay | Kapur et al., 2009 | N/A |
| SPM12 | Worsley et al., 1992 | https://www.fil.ion.ucl.ac.uk/spm/software/spm12/ |
| MATLAB R2016a | MathWorks | https://www.mathworks.com/ |
| Advanced Normalization Tools (ANTs) | Kim et al., 2008 | https://github.com/ANTsX/ANTs |
| AMIDE: A Medical Imaging Data Examiner | Loening and Gambhir, 2003 | http://amide.sourceforge.net/ |
| 3DSlicer | Fedorov et al., 2012 | https://www.slicer.org/ |
| Other | ||
| Waxholm space C57BL/6J brain atlas | Calabrese et al., 2015 | N/A |
| Vascular Access Button for mice, 25ga, injector, sterile | Instech | Cat# VAB62BS/25 |
| Catheter for mouse jugular vein, PU 2Fr 10cm, collar @ 1.3cm. Fits 25ga. | Instech | Cat# C20PU-MJV1301 |
| Mouse VAH/VAB tether kit for Med Assoc cage: 7in VAH62, 375/25PS, 60in PU, LS25 | Instech | Cat# KVAH62T/MED |
| Wheat germ agglutinin-coated yttrium oxide (YOx) beads | PerkinElmer | Cat# RPNQ0272 |
ACKNOWLEDGEMENTS
We thank X. Zhang and W. Roberts for managing our mouse colony, Dr. A. Inoue for providing TGFα shedding assay plasmids and cell lines, the UNC Biomedical Research Imaging Center (BRIC) (P30CA016086) for providing PET/CT imaging services, and the Duke Center for In Vivo Microscopy (P41EB01589a, U24CA220245) for supporting PET/CT image analysis. Equipment for studies conducted at the Duke Mouse Behavioral and Neuroendocrine Analysis Core Facility were supported by a grant from the North Carolina Biotechnology Center. Hemodynamic studies at the Duke Cardiovascular Research Center occurred with the support of the Edna and Fred L. Mandel Jr. Foundation. For these studies, LMS was supported by F32 DA043931 and K99 DA048970, AB by K01 AG041211, DMA by K08 HL125905, ABP by R21/33 DA038019, LSB and MGC by P30 DA029925, and MGC by R37 MH073853 as well as funds from the David Pall family part of the Long Island Community Foundation, a division of The New York Community Trust. We are grateful to Dr. Brigid Hogan for the critical reading of our manuscript.
Footnotes
DECLARATION OF INTERESTS
US Patents 9,868,707 and 10,118,902 relating to the chemistry of ML314, SBI-553, and their derivatives have been issued to the Sanford Burnham Prebys Medical Research Institute and Duke University (ABP, MPH, PM, SP, PM, LSB, MGC).
REFERENCES
- Alvarez-Curto E, Inoue A, Jenkins L, Raihan SZ, Prihandoko R, Tobin AB, and Milligan G (2016). Targeted Elimination of G Proteins and Arrestins Defines Their Specific Contributions to Both Intensity and Duration of G Protein-coupled Receptor Signaling. J Biol Chem 291, 27147–27159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson RJ, Cook JJ, Delpratt N, Nouls JC, Gu B, McNamara JO, Avants BB, Johnson GA, and Badea A (2018). Small Animal Multivariate Brain Analysis (SAMBA) - a High Throughput Pipeline with a Validation Framework. Neuroinformatics. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avants BB, Tustison NJ, Song G, Cook PA, Klein A, and Gee JC (2011). A reproducible evaluation of ANTs similarity metric performance in brain image registration. Neuroimage 54, 2033–2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baicy K, and London ED (2007). Corticolimbic dysregulation and chronic methamphetamine abuse. Addiction 102 Suppl 1, 5–15. [DOI] [PubMed] [Google Scholar]
- Barak LS, Bai Y, Peterson S, Evron T, Urs NM, Peddibhotla S, Hedrick MP, Hershberger P, Maloney PR, Chung TD, et al. (2016). ML314: A Biased Neurotensin Receptor Ligand for Methamphetamine Abuse. ACS Chem Biol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betancur C, Lepee-Lorgeoux I, Cazillis M, Accili D, Fuchs S, and Rostene W (2001). Neurotensin gene expression and behavioral responses following administration of psychostimulants and antipsychotic drugs in dopamine D-3 receptor deficient mice. Neuropsychopharmacology 24, 170–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betancur C, Rostene W, and Berod A (1997). Chronic cocaine increases neurotensin gene expression in the shell of the nucleus accumbens and in discrete regions of the striatum. Mol Brain Res 44, 334–340. [DOI] [PubMed] [Google Scholar]
- Binder EB, Kinkead B, Owens MJ, and Nemeroff CB (2001). Neurotensin and dopamine interactions. Pharmacological reviews 53, 453–486. [PubMed] [Google Scholar]
- Bissette G, Nemeroff CB, Loosen PT, Prange AJ Jr., and Lipton MA (1976). Hypothermia and intolerance to cold induced by intracisternal administration of the hypothalamic peptide neurotensin. Nature 262, 607–609. [DOI] [PubMed] [Google Scholar]
- Bohn LM, Lefkowitz RJ, Gainetdinov RR, Peppel K, Caron MG, and Lin FT (1999). Enhanced morphine analgesia in mice lacking beta-arrestin 2. Science 286, 2495–2498. [DOI] [PubMed] [Google Scholar]
- Bohn LM, Gainetdinov RR, Lin FT, Lefkowitz RJ, and Caron MG (2000). Mu-opioid receptor desensitization by beta-arrestin-2 determines morphine tolerance but not dependence. Nature 408, 720–723. [DOI] [PubMed] [Google Scholar]
- Bonetti EP, Pieri L, Cumin R, Schaffner R, Pieri M, Gamzu ER, Muller RK, and Haefely W (1982). Benzodiazepine antagonist Ro 15–1788: neurological and behavioral effects. Psychopharmacology (Berl) 78, 8–18. [DOI] [PubMed] [Google Scholar]
- Borroto-Escuela DO, Ravani A, Tarakanov AO, Brito I, Narvaez M, Romero-Fernandez W, Corrales F, Agnati LF, Tanganelli S, Ferraro L, et al. (2013). Dopamine D2 receptor signaling dynamics of dopamine D2-neurotensin 1 receptor heteromers. Biochem Biophys Res Commun 435, 140–146. [DOI] [PubMed] [Google Scholar]
- Boules M, Netz R, Fredrickson PA, and Richelson E (2016). A neurotensin analog blocks cocaine-conditioned place preference and reinstatement. Behav Pharmacol 27, 236–239. [DOI] [PubMed] [Google Scholar]
- Calabrese E, Badea A, Cofer G, Qi Y, and Johnson GA (2015). A Diffusion MRI Tractography Connectome of the Mouse Brain and Comparison with Neuronal Tracer Data. Cereb Cortex 25, 4628–4637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canals M, Lane JR, Wen A, Scammells PJ, Sexton PM, and Christopoulos A (2012). A Monod-Wyman-Changeux Mechanism Can Explain G Protein-coupled Receptor (GPCR) Allosteric Modulation. J Biol Chem 287, 650–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey LM, Rice RJ, and Prus AJ (2017). The Neurotensin NTS1 Receptor Agonist PD149163 Produces Antidepressant-Like Effects in the Forced Swim Test: Further Support for Neurotensin as a Novel Pharmacologic Strategy for Antidepressant Drugs. Drug Dev Res 78, 196–202. [DOI] [PubMed] [Google Scholar]
- Carraway R, and Leeman SE (1973). The isolation of a new hypotensive peptide, neurotensin, from bovine hypothalami. J Biol Chem 248, 6854–6861. [PubMed] [Google Scholar]
- Cathala L, and Paupardin-Tritsch D (1997). Neurotensin inhibition of the hyperpolarization-activated cation current (Ih) in the rat substantia nigra pars compacta implicates the protein kinase C pathway. J Physiol 503 (Pt 1), 87–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cervantes D, Crosby C, and Xiang Y (2010). Arrestin orchestrates crosstalk between G protein-coupled receptors to modulate the spatiotemporal activation of ERK MAPK. Circ Res 106, 79–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corrigan F, Wu Y, Tuke J, Coller JK, Rice KC, Diener KR, Hayball JD, Watkins LR, Somogyi AA, and Hutchinson MR (2015). Alcohol-induced sedation and synergistic interactions between alcohol and morphine: A key mechanistic role for Toll-like receptors and MyD88-dependent signaling. Brain Behavior and Immunity 45, 245–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deacon RM (2013). Measuring motor coordination in mice. J Vis Exp, e2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evron T, Peterson SM, Urs NM, Bai YS, Rochelle LK, Caron MG, and Barak LS (2014). G Protein and beta-Arrestin Signaling Bias at the Ghrelin Receptor. J Biol Chem 289, 33442–33455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedorov A, Beichel R, Kalpathy-Cramer J, Finet J, Fillion-Robin JC, Pujol S, Bauer C, Jennings D, Fennessy F, Sonka M, et al. (2012). 3D Slicer as an image computing platform for the Quantitative Imaging Network. Magn Reson Imaging 30, 1323–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson SS, Downey WE 3rd, Colapietro AM, Barak LS, Menard L, and Caron MG (1996). Role of beta-arrestin in mediating agonist-promoted G protein-coupled receptor internalization. Science 271, 363–366. [DOI] [PubMed] [Google Scholar]
- Ferraro L, Tiozzo Fasiolo L, Beggiato S, Borelli AC, Pomierny-Chamiolo L, Frankowska M, Antonelli T, Tomasini MC, Fuxe K, and Filip M (2016). Neurotensin: A role in substance use disorder? J Psychopharmacol 30, 112–127. [DOI] [PubMed] [Google Scholar]
- Giros B, Jaber M, Jones SR, Wightman RM, and Caron MG (1996). Hyperlocomotion and indifference to cocaine and amphetamine in mice lacking the dopamine transporter. Nature 379, 606–612. [DOI] [PubMed] [Google Scholar]
- Goodman OB Jr., Krupnick JG, Santini F, Gurevich VV, Penn RB, Gagnon AW, Keen JH, and Benovic JL (1996). Beta-arrestin acts as a clathrin adaptor in endocytosis of the beta2-adrenergic receptor. Nature 383, 447–450. [DOI] [PubMed] [Google Scholar]
- Green JL, Dykstra LA, and Carelli RM (2015). Examination of cocaine dose in a preclinical model of natural reward devaluation by cocaine. Behav Pharmacol 26, 398–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griebel G, and Holsboer F (2012). Neuropeptide receptor ligands as drugs for psychiatric diseases: the end of the beginning? Nature Reviews Drug Discovery 11, 462–478. [DOI] [PubMed] [Google Scholar]
- Groman SM, Lee B, Seu E, James AS, Feiler K, Mandelkern MA, London ED, and Jentsch JD (2012). Dysregulation of D(2)-mediated dopamine transmission in monkeys after chronic escalating methamphetamine exposure. The Journal of neuroscience: the official journal of the Society for Neuroscience 32, 5843–5852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundmann M, Merten N, Malfacini D, Inoue A, Preis P, Simon K, Ruttiger N, Ziegler N, Benkel T, Schmitt NK, et al. (2018). Lack of beta-arrestin signaling in the absence of active G proteins. Nat Commun 9, 341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulyas G, Toth JT, Toth DJ, Kurucz I, Hunyady L, Balla T, and Varnai P (2015). Measurement of Inositol 1,4,5-Trisphosphate in Living Cells Using an Improved Set of Resonance Energy Transfer-Based Biosensors. PloS one 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hefti FF (2008). Requirements for a lead compound to become a clinical candidate. Bmc Neurosci 9 Suppl 3, S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda M, Ozaki N, Suzuki T, Kitajima T, Yamanouchi Y, Kinoshita Y, Kishi T, Sekine Y, Iyo M, Harano M, et al. (2007). Possible association of beta-arrestin 2 gene with methamphetamine use disorder, but not schizophrenia. Genes Brain Behav 6, 107–112. [DOI] [PubMed] [Google Scholar]
- Inagaki S, Ghirlando R, Vishnivetskiy SA, Homan KT, White JF, Tesmer JJ, Gurevich VV, and Grisshammer R (2015). G Protein-Coupled Receptor Kinase 2 (GRK2) and 5 (GRK5) Exhibit Selective Phosphorylation of the Neurotensin Receptor in Vitro. Biochemistry 54, 4320–4329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue A, Ishiguro J, Kitamura H, Arima N, Okutani M, Shuto A, Higashiyama S, Ohwada T, Arai H, Makide K, et al. (2012). TGFalpha shedding assay: an accurate and versatile method for detecting GPCR activation. Nat Methods 9, 1021–1029. [DOI] [PubMed] [Google Scholar]
- Khoury E, Clement S, and Laporte SA (2014). Allosteric and biased g protein-coupled receptor signaling regulation: potentials for new therapeutics. Front Endocrinol (Lausanne) 5, 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Avants B, Patel S, Whyte J, Coslett BH, Pluta J, Detre JA, and Gee JC (2008). Structural consequences of diffuse traumatic brain injury: A large deformation tensor-based morphometry study. Neuroimage 39, 1014–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King CP, Palmer AA, Woods LCS, Hawk LW, Richards JB, and Meyer PJ (2016). Premature responding is associated with approach to a food cue in male and female heterogeneous stock rats. Psychopharmacology 233, 2593–2605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitabgi P, De Nadai F, Labbe-Jullie C, Dubuc I, Nouel D, Costentin J, Masuo Y, Rostene W, Woulfe J, Lafortune L, et al. (1992). Functional and pharmacological aspects of central neuropeptidergic transmission mediated by neurotensin and neuromedin n. Clin Neuropharmacol 15 Suppl 1 Pt A, 313A–314A. [DOI] [PubMed] [Google Scholar]
- Kmiotek EK, Baimel C, and Gill KJ (2012). Methods for intravenous self administration in a mouse model. J Vis Exp, e3739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancelot S, and Zimmer L (2010). Small-animal positron emission tomography as a tool for neuropharmacology. Trends Pharmacol Sci 31, 411–417. [DOI] [PubMed] [Google Scholar]
- Lefkowitz RJ, and Shenoy SK (2005). Transduction of receptor signals by beta-arrestins. Science 308, 512–517. [DOI] [PubMed] [Google Scholar]
- Loening AM, and Gambhir SS (2003). AMIDE: a free software tool for multimodality medical image analysis. Mol Imaging 2, 131–137. [DOI] [PubMed] [Google Scholar]
- Maccioni P, Lorrai I, Marras MF, Contini A, Capra A, Piras G, Caboni P, Gessa GL, and Colombo G (2015). Elevated reinforcing and motivational properties of alcohol at the end of the nocturnal period in sP rats. Psychopharmacology 232, 3585–3595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantsch JR, Li SJ, Risinger R, Awad S, Katz E, Baker DA, and Yang Z (2007). Levo-tetrahydropalmatine attenuates cocaine self-administration and cocaine-induced reinstatement in rats. Psychopharmacology 192, 581–591. [DOI] [PubMed] [Google Scholar]
- McCorvy JD, Wacker D, Wang S, Agegnehu B, Liu J, Lansu K, Tribo AR, Olsen RHJ, Che T, Jin J, et al. (2018). Structural determinants of 5-HT2B receptor activation and biased agonism. Nat Struct Mol Biol 25, 787–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nehrenberg DL, Rodriguiz RM, Cyr M, Zhang X, Lauder JM, Gariepy JL, and Wetsel WC (2009). An anxiety-like phenotype in mice selectively bred for aggression. Behav Brain Res 201, 179–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemeroff CB (1980). Neurotensin: perchance an endogenous neuroleptic? Biol Psychiatry 15, 283–302. [PubMed] [Google Scholar]
- O’Hayre M, Eichel K, Avino S, Zhao XF, Steffen DJ, Feng XD, Kawakami K, Aoki J, Messer K, Sunahara R, et al. (2017). Genetic evidence that beta-arrestins are dispensable for the initiation of beta(2)-adrenergic receptor signaling to ERK. Science Signaling 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakley RH, Laporte SA, Holt JA, Caron MG, and Barak LS (2000). Differential affinities of visual arrestin, beta arrestin1, and beta arrestin2 for G protein-coupled receptors delineate two major classes of receptors. J Biol Chem 275, 17201–17210. [DOI] [PubMed] [Google Scholar]
- Oakley RH, Laporte SA, Holt JA, Barak LS, Caron MG (2001). Molecular determinants underlying the formation of stable intracellular G protein-coupled receptor-beta-arrestin complexes after receptor endocytosis. J Biol Chem 276, 19452–60. [DOI] [PubMed] [Google Scholar]
- Oneda B, Crettol S, Bochud M, Besson J, Croquette-Krokar M, Hammig R, Monnat M, Preisig M, and Eap CB (2011). beta-Arrestin2 influences the response to methadone in opioid-dependent patients. Pharmacogenomics J 11, 258–266. [DOI] [PubMed] [Google Scholar]
- Osbahr AJ 3rd, Nemeroff CB, Manberg PJ, and Prange AJ Jr. (1979). Centrally administered neurotensin: activity in the Julou-Courvoisier muscle relaxation test in mice. Eur J Pharmacol 54, 299–302. [DOI] [PubMed] [Google Scholar]
- Pack TF, Orlen MI, Ray C, Peterson SM, and Caron MG (2018). The dopamine D2 receptor can directly recruit and activate GRK2 without G protein activation. J Biol Chem 293, 6161–6171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsegian A, and See RE (2014). Dysregulation of dopamine and glutamate release in the prefrontal cortex and nucleus accumbens following methamphetamine self-administration and during reinstatement in rats. Neuropsychopharmacology 39, 811–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peddibhotla S, Hedrick MP, Hershberger P, Maloney PR, Li Y, Milewski M, Gosalia P, Gray W, Mehta A, Sugarman E, et al. (2013). Discovery of ML314, a Brain Penetrant Non-Peptidic beta-Arrestin Biased Agonist of the Neurotensin NTSR1 Receptor. ACS Med Chem Lett 4, 846–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson SM, Pack TF, Wilkins AD, Urs NM, Urban DJ, Bass CE, Lichtarge O, and Caron MG (2015). Elucidation of G-protein and beta-arrestin functional selectivity at the dopamine D2 receptor. Proceedings of the National Academy of Sciences of the United States of America 112, 7097–7102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrie KA, Bubser M, Casey CD, Davis MD, Roth BL, and Deutch AY (2004). The neurotensin agonist PD149163 increases Fos expression in the prefrontal cortex of the rat. Neuropsychopharmacology 29, 1878–1888. [DOI] [PubMed] [Google Scholar]
- Pettibone DJ, Hess JF, Hey PJ, Jacobson MA, Leviten M, Lis EV, Mallorga PJ, Pascarella DM, Snyder MA, Williams JB, et al. (2002). The effects of deleting the mouse neurotensin receptor NTSR1 on central and peripheral responses to neurotensin. The Journal of pharmacology and experimental therapeutics 300, 305–313. [DOI] [PubMed] [Google Scholar]
- Pitcher JA, Hall RA, Daaka Y, Zhang J, Ferguson SSG, Hester S, Miller S, Caron MG, Lefkowitz RJ, and Barak LS (1998). The G protein-coupled receptor kinase 2 is a microtubule-associated protein kinase that phosphorylates tubulin. J Biol Chem 273, 12316–12324. [DOI] [PubMed] [Google Scholar]
- Porter-Stransky KA, and Weinshenker D (2017). Arresting the Development of Addiction: The Role of beta-Arrestin 2 in Drug Abuse. The Journal of pharmacology and experimental therapeutics 361, 341–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prus AJ, Hillhouse TM, and LaCrosse AL (2014). Acute, but not repeated, administration of the neurotensin NTS1 receptor agonist PD149163 decreases conditioned footshock-induced ultrasonic vocalizations in rats. Prog Neuropsychopharmacol Biol Psychiatry 49, 78–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raehal KM, Walker JK, and Bohn LM (2005). Morphine side effects in beta-arrestin 2 knockout mice. The Journal of pharmacology and experimental therapeutics 314, 1195–1201. [DOI] [PubMed] [Google Scholar]
- Reimold M, Slifstein M, Heinz A, Mueller-Schauenburg W, and Bares R (2006). Effect of spatial smoothing on t-maps: arguments for going back from t-maps to masked contrast images. J Cereb Blood Flow Metab 26, 751–759. [DOI] [PubMed] [Google Scholar]
- Rizzuto R, Simpson AW, Brini M, and Pozzan T (1992). Rapid changes of mitochondrial Ca2+ revealed by specifically targeted recombinant aequorin. Nature 358, 325–327. [DOI] [PubMed] [Google Scholar]
- Schiffmann SN, Fisone G, Moresco R, Cunha RA, and Ferre S (2007). Adenosine A2A receptors and basal ganglia physiology. Prog Neurobiol 83, 277–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrage R, Schmitz AL, Gaffal E, Annala S, Kehraus S, Wenzel D, Bullesbach KM, Bald T, Inoue A, Shinjo Y, et al. (2015). The experimental power of FR900359 to study Gq-regulated biological processes. Nature Communications 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharpe AL, Varela E, and Beckstead MJ (2017). Systemic PD149163, a neurotensin receptor 1 agonist, decreases methamphetamine self-administration in DBA/2J mice without causing excessive sedation. PloS one 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenoy SK, Drake MT, Nelson CD, Houtz DA, Xiao K, Madabushi S, Reiter E, Premont RT, Lichtarge O, and Lefkowitz RJ (2006). beta-arrestin-dependent, G protein-independent ERK1/2 activation by the beta2 adrenergic receptor. J Biol Chem 281, 1261–1273. [DOI] [PubMed] [Google Scholar]
- Shoichet BK, and Kobilka BK (2012). Structure-based drug screening for G-protein-coupled receptors. Trends Pharmacol Sci 33, 268–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla AK, Xiao K, and Lefkowitz RJ (2011). Emerging paradigms of beta-arrestin-dependent seven transmembrane receptor signaling. Trends Biochem Sci 36, 457–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JS, and Rajagopal S (2016). The beta-Arrestins: Multifunctional Regulators of G Protein-coupled Receptors. J Biol Chem 291, 8969–8977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JS, Pack TF, Inoue A, Lee C, Xiong X, Zheng K, Kahsai AW, Choi I, Ma Z, Levitan IM, Rochelle LK, Staus DP, Snyder JC, Caron MG, and Rajagopal S (2019) Noncanonical scaffolding of Gαi and β-arrestin by G protein-coupled receptors. bioRxiv [Preprint] May 7, 2019; doi: 10.1101/629576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder JC, Pack TF, Rochelle LK, Chakraborty SK, Zhang M, Eaton AW, Bai Y, Ernst LA, Barak LS, Waggoner AS, et al. (2015). A rapid and affordable screening platform for membrane protein trafficking. BMC Biol 13, 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotnikova TD, Beaulieu JM, Barak LS, Wetsel WC, Caron MG, and Gainetdinov RR (2005). Dopamine-independent locomotor actions of amphetamines in a novel acute mouse model of Parkinson disease. PLoS Biol 3, e271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St-Gelais F, Jomphe C, and Trudeau LE (2006). The role of neurotensin in central nervous system pathophysiology: what is the evidence? J Psychiatry Neurosci 31, 229–245. [PMC free article] [PubMed] [Google Scholar]
- St-Gelais F, Legault M, Bourque MJ, Rompre PP, and Trudeau LE (2004). Role of calcium in neurotensin-evoked enhancement in firing in mesencephalic dopamine neurons. Journal of Neuroscience 24, 2566–2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stansley BJ, and Conn PJ (2019). Neuropharmacological Insight from Allosteric Modulation of mGlu Receptors. Trends Pharmacol Sci 40, 240–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun D, Ma JZ, Payne TJ, and Li MD (2008). beta-Arrestins 1 and 2 are associated with nicotine dependence in European American smokers. Mol Psychiatr 13, 398–406. [DOI] [PubMed] [Google Scholar]
- Thanos PK, Michaelides M, Benveniste H, Wang GJ, and Volkow ND (2008). The effects of cocaine on regional brain glucose metabolism is attenuated in dopamine transporter knockout mice. Synapse 62, 319–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toth K, Slosky LM, Pack TF, Urs NM, Boone P, Mao L, Abraham D, Caron MG, and Barak LS (2018). Ghrelin receptor antagonism of hyperlocomotion in cocaine-sensitized mice requires betaarrestin-2. Synapse 72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urs NM, Bido S, Peterson SM, Daigle TL, Bass CE, Gainetdinov RR, Bezard E, and Caron MG (2015). Targeting beta-arrestin2 in the treatment of L-DOPA-induced dyskinesia in Parkinson’s disease. Proceedings of the National Academy of Sciences of the United States of America 112, E2517–2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urs NM, Gee SM, Pack TF, McCorvy JD, Evron T, Snyder JC, Yang X, Rodriguiz RM, Borrelli E, Wetsel WC, et al. (2016). Distinct cortical and striatal actions of a beta-arrestin-biased dopamine D2 receptor ligand reveal unique antipsychotic-like properties. Proceedings of the National Academy of Sciences of the United States of America 113, E8178–E8186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urs NM, Snyder JC, Jacobsen JPR, Peterson SM, and Caron MG (2012). Deletion of GSK3 beta in D2R-expressing neurons reveals distinct roles for beta-arrestin signaling in antipsychotic and lithium action. Proceedings of the National Academy of Sciences of the United States of America 109, 20732–20737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vadnie CA, Hinton DJ, Choi S, Choi Y, Ruby CL, Oliveros A, Prieto ML, Park JH, and Choi DS (2014). Activation of neurotensin receptor type 1 attenuates locomotor activity. Neuropharmacology 85, 482–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson MA, Yamada M, Yamada M, Cusack B, Veverka K, Boldenwatson C, and Richelson E (1992). The Rat Neurotensin Receptor Expressed in Chinese-Hamster Ovary Cells Mediates the Release of Inositol Phosphates. Journal of Neurochemistry 59, 1967–1970. [DOI] [PubMed] [Google Scholar]
- Welch A, Mingarelli M, Riedel G, and Platt B (2013). Mapping changes in mouse brain metabolism with PET/CT. Journal of nuclear medicine: official publication, Society of Nuclear Medicine 54, 1946–1953. [DOI] [PubMed] [Google Scholar]
- Willuhn I, Wanat MJ, Clark JJ, and Phillips PEM (2010). Dopamine Signaling in the Nucleus Accumbens of Animals Self-Administering Drugs of Abuse. Curr Top Behav Neuro 3, 29–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worsley KJ, Evans AC, Marrett S, and Neelin P (1992). A three-dimensional statistical analysis for CBF activation studies in human brain. J Cereb Blood Flow Metab 12, 900–918. [DOI] [PubMed] [Google Scholar]
- Wu T, Li A, and Wang HL (1995). Neurotensin Increases the Cationic Conductance of Rat Substantia-Nigra Dopaminergic-Neurons through the Inositol 1,4,5-Trisphosphate-Calcium Pathway. Brain research 683, 242–250. [DOI] [PubMed] [Google Scholar]
- Wu T, and Wang HL (1995). Protein-Kinase-C Mediates Neurotensin Inhibition of Inwardly Rectifying Potassium Currents in Rat Substantia-Nigra Dopaminergic-Neurons. Neurosci Lett 184, 121–124. [DOI] [PubMed] [Google Scholar]
- Wustrow DJ, Davis MD, Akunne HC, Corbin AE, Wiley JN, Wise LD, and Heffner TG (1995). Reduced Amide Bond Neurotensin-8–13 Mimetics with Potent in-Vivo Activity. Bioorg Med Chem Lett 5, 997–1002. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Curve parameters and statistical comparisons presented in Figure 1 – Supporting Figure 1.
Table S2. Curve parameters and statistical comparisons presented in Figure 2 – Supporting Figure 2.
Table S3. Statistical comparisons presented in Figure 3 – Supporting Figure 3.
Table S4. Statistical comparisons presented in Figure 4 – Supporting Figure 4.
Table S5. Curve parameters and statistical comparisons presented in Figure 5 – Supporting Figure 5.
Table S6. Curve parameters and statistical comparisons presented in Figure 6 –Supporting Figure 6.
Table S7. Statistical comparisons presented in Figure 7 – Supporting Figure 7.
Figure S1. Validation of on-bead binding assay with [3H]NTS and confirmation of [3H]SBI-553 NTSR1 specific binding at an allosteric site – Supporting Fig 1. (A) Nonspecific [3H]SBI-553 binding to different bead substrates. SBI-553 is lipophilic, and NTSR1 binding studies required substituting wheat germ agglutinin (WGA) coated yttrium oxide (YOx) beads for glass membranes to minimize non-specific binding. [3H]SBI-553 binding to a panel of beads was evaluated to minimize nonspecific binding. Data (mean ± SEM) were normalized to L-lysine bead determinations. WGA-YOx beads were selected for the on-bead binding assays. Type A, WGA-PEI A; Type B, WGA-PEI B; WGA, wheat germ agglutinin; PEI, polyethyleneimine; PS, polystyrene; YOx, yttrium oxide. N = 2 independent experiments.
(B,C) On-bead binding assay validation. [3H]NTS saturation binding to NTSR1-expressing membranes was compared using the novel on-bead assay and the classic glass microfiber filter paper/Brandel Harvester assay. (B) [3H]NTS saturation binding to NTSR1 using glass microfiber filter paper and Brandel Harvester: Use of Brandel Harvester. Data points are presented as mean CPM ± SEM. Calculated [3H]NTS 95% confidence intervals: (Bmax = 3,651 to 4,693 CPM, Kd = 1.8 to 4.0 nM). N = 4 independent experiments. (C) [3H]NTS saturation binding to NTSR1 using YOx bead assay platform: Use of YOx bead assay platform. Data points are presented as mean CPM ± SEM. Calculated [3H]NTS 95% confidence intervals: (Bmax = 2,748 to 5,720 CPM, Kd =1.5 to 18.0 nM). N = 3 independent experiments.
(D,E) Total [3H]SBI-553 and total [3H]NTS binding with substitution of β2-adrenergic receptor (β2AR)-containing membranes for NTSR1-containing membranes. To independently demonstrate specific [3H]SBI-553 binding to NTSR1, membrane competition studies were conducted in which the ratio of membranes containing NTSR1 to membranes containing an off-target GPCR (i.e., β2AR) was varied. Cell membranes from NTSR1-expressing and β2AR-expressing HEK293T cells were conjugated together at the indicated ratios to wheat-germ agglutin (WGA) coated yttrium oxide (YOx) beads. The beads were then incubated at room temperature in the presence of (D) 100 nM [3H]SBI-553 or (E) 5 nM [3H]NTS. Data were analyzed by one-way ANOVA followed by Bonferroni’s multiple comparisons test. Total [3H]SBI-553 binding was reduced with β2AR membrane substitution. *p<0.05, **p<0.01, compared to NTSR1 membranes alone (2:0) (F(3,14)=8.0, p=0.0024). Total [3H]NTS binding was reduced with β2AR membrane substitution. **p<0.01, ***p<0.001, as compared to NTSR1 membranes alone (2:0) using a one-way ANOVA (F(3,8)=49.1, p<0.001) followed by Bonferroni’s multiple comparisons test. N = 3 independent experiments.
(F) Competitive displacement of [3H]SBI-553 from NTSR1. NTSR1 membrane-coated YOx beads were incubated at room temperature with 100 nM [3H]SBI-553 in the presence of vehicle (0.1% DMSO in Opti-MEM),10 μM unlabeled SBI-553, neurotensin (NTS), or the NTSR1 antagonist SR142948A. Data are normalized to vehicle binding and presented as mean ± SEM. ***p<0.001, compared to vehicle using a one-way ANOVA (F(4,55)=26.0, p<0.001) followed by Bonferroni’s multiple comparisons test. N = 4 independent experiments.
(G) Competitive displacement of [3H]NTS from NTSR1. NTSR1 membrane coated YOx beads were incubated at room temperature with 5 nM [3H]NTS in the presence of vehicle (0.1% DMSO in Opti-MEM),10 μM unlabeled SBI-553, NTS, or SR142948A. Data are normalized to vehicle binding and presented as mean ± SEM. ***p<0.001, compared to vehicle using a one-way ANOVA (F(4,23)=220.1, p<0.001) followed by Bonferroni’s multiple comparisons test. N = 3 independent experiments.
Figure S2. NTS and SBI-553 fail to activate β-arrestin2 in absence of NTSR1 – Supporting Fig 1. U2OS cells stably expressing β-arrestin2-GFP were transiently transfected with human NTSR1-containing (top panels) or empty (bottom panels) pcDNA vectors. Cells remained unstimulated (left) or treated with NTS (200 nM, middle) or SBI-553 (7.5 μM, right) for 40 min. Following treatment cells were fixed and imaged using a fluorescein filter set and examined at 40×, Plan Apo objective, NA/0.85, on a Zeiss Axiovert 200 microscope. β-arrestin2-GFP aggregates are apparent with NTS and SBI-553 stimulation only in NTSR1-expressing cells and some dissimilarities between these aggregates may reflect compartmentalization differences. Scale bar = 10 μm.
Figure S3. Whole brain [18F]-FDG uptake did not differ among treatment groups – Supporting Fig 4. [18F]-FDG PET/CT scans of DAT KO mice (n = 8) were acquired prior to treatment (Baseline, Day 1), following dopamine depletion and stimulation with L-DOPA and vehicle (L-DOPA + Vehicle, Day 2) and following dopamine depletion and stimulation with L-DOPA and SBI-553 (L-DOPA + SBI-553, Day 3). Standardized uptake values (SUVs) were normalized to the injected [18F]-FDG dose and mouse’s body weight. PET/CT scans were co-registered with a three-dimensional Waxholm space C57BL/6J brain atlas (Calabrese et al., 2015). Whole brain SUVs were calculated using the region of interest (ROI) feature of AMIDE: a Medical Imaging Data Examiner software (Loening and Gambhir, 2003). No differences in whole brain uptake among treatment days were identified using a one-way ANOVA (F(2,21)= 0.22, p = 0.80.
Data Availability Statement
This study did not generate datasets or employ previously unreported custom computer code.