

Summary
Originally discovered as an inducer of apoptosis, the TNF-family receptor Fas (CD95, APO-1, TNFRSF6) has more recently been found to have functions beyond cell death, including T cell co-stimulation and promoting terminal differentiation of CD4+ and CD8+ T cells. Other TNF-family members also discovered as apoptosis-inducers, such as TRAIL (APO-2L, TNFSF10), can promote inflammation through caspase-8. Surprisingly, non-apoptotic signaling through Fas can protect from the autoimmunity seen in Fas deficiency independently from the cell-death inducing functions of the receptor. Non-apoptotic Fas signaling can induce tumor cell growth and migration, and impair the efficacy of T cell adoptive immunotherapy. Blocking of non-apoptotic functions of these receptors may be a novel strategy to regulate autoimmunity, inflammation and enhance anti-tumor immunity.
Diverse functions of TNF-family cytokines
The 19 cytokines in the tumor necrosis factor (TNF) superfamily induce diverse cellular responses through cognate receptors and two main signaling pathways: induction of apoptosis through activation of caspases, and activation of inflammatory and cell differentiation programs [1, 2]. These cytokines have also been successful targets for therapy in a number of autoimmune diseases such as rheumatoid arthritis, inflammatory bowel disease and systemic lupus [1, 2]. Although all TNF-family cytokines share structural similarities, each cytokine only binds one or a restricted number of receptors. TNF receptors can be grouped based on their signal transduction pathways (Figure 1). One sub-family, consisting of TNFR1/TNFRSF1A, Fas/CD95/TNFRSF6, DR3/TNFRSF25, TRAIL receptor 1/TNFRSF10A, TRAIL receptor 2/TNFRSF10B, DR6/TNFRSF1 and EDAR, share an intracellular domain with a characteristic six alpha helical fold termed a ‘death domain’. The other larger sub-family of TNF receptors lack a death domain and instead recruit TRAF (TNF-Receptor associated Factor) proteins through peptide motifs in their intracellular tails. TRAF proteins in turn can recruit ubiquitin ligase complexes and kinases through protein-protein interactions and activate NF-κB and MAP Kinase signaling cascades. The receptors sharing a death domain were initially termed ‘death receptors’ due to their ability to trigger apoptosis or other forms of cell death upon overexpression or ligation with agonistic antibodies or their cognate cytokine ligands [3, 4]. However, it then emerged that TNFR1, DR3, and possibly DR6 recruit the adapter protein TRADD [5], and EDAR binds a related protein EDARADD [6]. These proteins have a death domain in their C termini, and TRAF recruitment domains at the N-termini, endowing them with the ability to activate NF-κB as do TNF-family receptors without a death domain. In most circumstances, these receptors activate inflammatory responses, although NF-κB activation is blocked, secondary signaling complexes can form, intracellularly triggering cell death [7]. This third subfamily is termed ‘dual-signaling’ receptors (Figure 1).
Figure 1: Signaling pathways initiated by the TNF-Receptor superfamily.
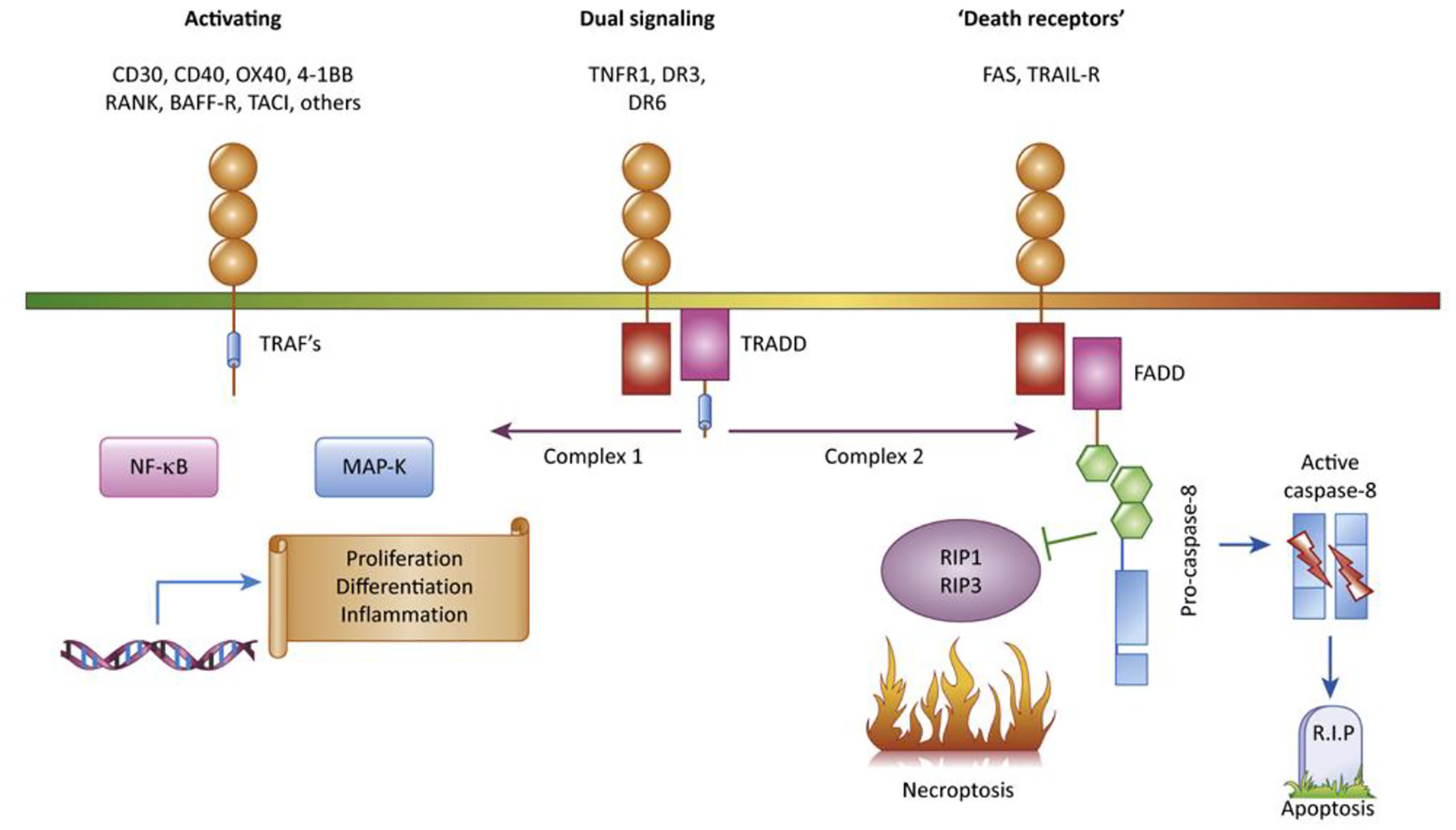
The 29 TNF receptors in the human genome can be divided into three subtypes based on the adapter proteins recruited to their cytoplasmic tails, in addition to ‘decoy receptors;’ that are soluble or lack functional intracellular signaling domains. The largest group of receptors contain peptide motifs in their cytoplasmic domains which recruit TRAF adapter proteins, which activate intracellular signaling pathways culminating in transcriptional responses and production of inflammatory cytokines, proliferation and cellular differentiation. Fas and TRAIL receptors recruit FADD, an adapter protein that mediates recruitment of caspase-8 and can activate apoptosis, with inhibition of necrosis and inflammatory and differentiation pathways as more newly recognized functions. TNFR1, DR3 and DR6 recruit the TRADD adapter protein, which can alternatively activate inflammatory or caspase-mediated signaling.
Fas and TRAIL receptors bind the adapter protein FADD, which has a C-terminal Death Domain and an N-terminal structurally related domain termed a Death Effector Domain (DED). The FADD DED recruits DED’s in the pro-enzyme form of caspase-8, an initiator caspase in the apoptotic protease cascade. Oligomerization of caspase-8 activates its protease function, resulting in cleavage of the enzymatic subunits from the DED domains and dissociation of the active enzyme complex from the Fas-induced signaling complex. Collectively, this is referred to as the ‘Death inducing Signaling Complex or DISC (see glossary) [8]. Cytoplasmic Caspase-8 can trigger apoptosis through cleavage of downstream substrates such as caspase-3 and the BH3-domain containing protein BiD, which when cleaved interacts with other Bcl-2 family proteins on mitochondria to trigger mitochondrial depolarization and release of apoptosis-promoting molecules such as cytochrome-c and SMAC[9–11]. These molecules activate the cytoplasmic ‘apoptosome’ and caspase-9, potently amplifying the apoptotic protease cascade. Ligation of Fas and TRAIL by agonistic antibodies or cognate ligands potently induces cell death in vitro, and the functions of Fas and TRAIL receptors and were thought for many years to be restricted to cell death. Critically, antigen-induced death of CD4+ T cells in vitro and in vivo was shown to be dependent on autocrine Fas-FasL interactions [12–15]. However, it has more recently emerged that both of these receptors can induce responses other than cell death both in vitro and in vivo, including inflammatory responses in innate immune cells and differentiation of CD4+ and CD8+ T cells. Here we will review these developments, which have significant implications for the biology of these receptors and pathophysiology and therapies for a number of diseases.
Potentiating innate immunity by Fas and related TNF-family receptors
Recent investigations have shown that Fas is involved in a number of immune system roles beyond apoptosis, including potentiating the innate immune system to respond to pathogens. One common pathway in activation of the innate immune system is the NF-κB pathway. NF-κB transcription factors are central to immune system activation and immune response upregulation to combat both foreign pathogens and cancers [16, 17]. Notably, in a number of contexts, Fas has been found to activate NF-κB, potentiating innate immunity [18]. Dendritic cells (DCs) express surface Fas, but are generally resistant to Fas ligand induced killing ex vivo [19]. Instead, Fas crosslinking can activate DC maturation and the production of cytokines and chemokines NF-κB was required for these responses, in that chemical inhibition of NF-κB reverted the DC’s to apoptosis in response to Fas crossinking [20, 21]. In addition to activation of NF-κB, Fas can promote IL-1β production through a pathway dependent on caspase-8 but independent of caspase-1 and the NLRP3 inflammasome [22]. Rather, Caspase-8 activated through the Fas DISC directly cleaves IL-1 and the related cytokine IL-18 [23, 24].
Fas-mediated innate immune cell activation can affect host defense and immunopathology in the intact immune system. For instance, infection with L. monocytogenes led to an upregulation of membrane bound Fas ligand expression on murine natural killer (NK) cells in vitro. Membrane bound Fas ligand, in turn, stimulated infected macrophages to begin producing IL-1β [25]. Even when FasL induces both apoptotic and non-apoptotic responses, secretion of pro-inflammatory cytokines contributes to inflammatory responses which can abrogate tolerance in immune privileged sites such as the eye [21, 26–28]. In rodent transplantation experiments, FasL expression on hematopoeitc cells can enhance engraftment and donor-specific tolerance [29, 30], but expression of FasL on graft tissues can trigger graft rejection and tumor cell killing through activation of host innate immune cells, resulting in acute neutrophilic inflammation [31–33]. In a spinal-cord injury mouse model, FasL on myeloid cells also promoted infiltration of macrophages and neutrophils, ultimately contributing to tissue repair [34, 35]. While these findings dampened hope that FasL expression on transplanted tissue could induce immunological tolerance through apoptosis, they opened up a new area of study of Fas-induced innate inflammatory responses. While the signaling cascade triggering Fas-induced inflammatory cytokine production is still unclear, a wide variety of pro-inflammatory cytokines and chemokines can be detected in the supernatant of FasL stimulated cells in vitro, including TNF, IL-6, IL-8, IL-17, and IL-18, CXCL1, MIP2, MCP-1 [36–38], which recruit and activate infiltrating leukocytes.
Inflammatory signaling by Fas and related receptors does appear to involve components shared with the apoptotic signal transduction. The Caspase-8 related protein FLICE-like inhibitory protein (FLIP), capable of binding Fas associated death domain protein (FADD) and Caspase-8, can activate the ERK1/2 and NF-κB pathways [39]. in response to TRAIL, Caspase-8 has been shown to function in a non-enzymatic fashion for the assembly of a novel inflammatory complex termed the ‘FADDosome’ [40]. RIPK1, NEMO, IKK are recruited via caspase-8 to induce the NF-κB inflammatory gene expression pathway [41]. This response is inhibited by knockdown or genetic deficiency of caspase-8, but not inhibitors of caspase-8 protease function or an enzymatically disabled caspase-8 mutant, suggesting that caspase-8 functions in a scaffolding role, rather than through cleaving specific protease substrates, to activate NF-κB.
In mouse models, constitutive expression of membrane-bound FasL in the eye, an organ with in which antigen exposure typically leads to immune tolerance rather than a pathogenic immune response, initiated a strong inflammatory response including neutrophil and macrophage infiltration and inflammatory cytokine production. In an ocular tumor model, this led to termination of ocular immune privilege and rapid rejection of the tumors, with other cell types including lymphocytes contributing to tumor rejection as well [28]. Notably, soluble FasL did not trigger the same response, suggesting that only membrane-bound FasL can initiate an inflammatory signaling cascade [27], results concordant with mouse models showing that membrane bound, rather than soluble Fas ligand is the principal inducer of apoptosis [42]. These results suggest that mFasL may be used to potentiate anti-tumor immune responses, but the other functions of FasL in lymphocytes and the inflammatory responses provoked by FasL may limit this therapeutic application (see Clinician’s corner, box 1).
Clinician’s Corner (Box 1).
Harnessing the ability of cell surface receptors to induce apoptosis, which is a generally non-inflammatory form of cell death, has long been a therapeutic goal [112], and ‘death receptors’ of TNF-family cytokines such as Fas/CD95 and TRAIL receptors have this ability.
However, results of clinical trials of agonistic antibodies against TRAIL receptors in cancer have been disappointing [113, 114]. This may be due to alternative functions of TRAIL and related receptors, including induction of inflammatory cytokine production [40].
Conversely, blocking cell death induction by these receptors may be a way to keep lymphocytes that are important for host defense or cancer immunotherapy alive, and potentiate the effects of vaccines or adoptive cellular immunotherapy.
Fas co-stimulates T cell activation and differentiation
In addition to its functions in innate immunity, Fas also plays an important role in adaptive immunity beyond its role in cell death. The description of loss-of-function mutations in Fas in mice harboring the Lymphoproliferation (lpr) mutation and dominant interfering mutations in autoimmune lymphoproliferative syndrome (ALPS) [43, 44], coupled with the demonstration that TCR-induced apoptosis is in CD4+ T cells is dependent on Fas-FasL interactions [14, 15, 45], led to a model in which defective apoptosis of autoreactive T and B cells underlies the pathogenesis of autoimmune disease in ALPS patients and lpr mice [46].
Rather than promoting apoptosis, Fas co-stimulation during T cell activation can also co-stimulate proliferation [47]. Subsequent work investigated the signaling pathways by which Fas co-stimulates CD4+ and CD8+ T cells, finding that FasL triggers recruitment of the adapter protein cFLIP to the Fas signaling complex through FADD, increasing NF-κB and Erk activation and promoting IL-2 expression [39, 48]. Activation of caspase-8 is also apparently required for Fas-induced T cell proliferation, as T cells treated with caspase inhibitors or deficient in caspase-8 show reduced proliferation [49–51]. CD4+ and CD8+ T cells co-stimulated with FasL show only caspase-8 cleavage but no evidence of downstream caspase-3 cleavage, suggesting that full caspase-8 activation may not be needed for its co-stimulatory function [49]. In accordance with these results, CD4+ and CD8+ T cells from patients homozygous for a rare loss-of-function caspase-8 mutation are resistant to Fas-mediated lymphocyte apoptosis, but also have activation defects. T B, natural killer cells from these patients have functional defects and the patients developed clinical immunodeficiency [52]. These results suggest that Fas signaling in CD4+ and CD8+ T lymphocyte promotes cell proliferation by recruiting Caspase-8 and activating NF-κB and Erk pathways. More recently, the discovery that caspase-8 can also inhibit necroptosis through cleavage of RIP3 has suggested that the reduced proliferation and dysfunction in lymphocytes lacking caspase-8 may stem from enhanced necroptosis, since these defects and the lethality caspase-8 deficiency are reversed in the absence of RIP3 [53, 54]. The finding that caspase-8 can promote apoptosis while inhibiting necroptosis placed it in a central role in deciding T cell fate after activation and the inflammatory or non-inflammatory consequences of T cell death after activation.
It has been known for some time that recently activated and naïve T cells are resistant to Fas and TCR-induced apoptosis, [55, 56], but what non-apoptotic functional changes Fas can trigger beyond proliferation was not clear. A recent insight into this question came from the study of interactions between naïve and memory T cells during T cell activation. Co-culture with memory T cells promoted more rapid differentiation of human and mouse naïve cells into effector memory T cells producing high-levels of effector cytokines and cytotoxic granules [57]. This process, termed precocious differentiation, is driven by interactions between FasL on the memory T cells and Fas expressed on recently activated naïve T cells [57]. Precocious T cell differentiation depended on Akt signaling, but was independent of apoptosis [57]. These observations provide a novel function for Fas signaling in T cells independent of apoptosis, and, as detailed below, have significant implications for autoimmunity and cellular cancer immunotherapy.
Despite these advances in understanding the alternative functions of Fas and caspase 8, dissecting the in vivo functions of apoptotic vs. non-apoptotic Fas signaling have been challenging due to the high degree of overlap between their biochemical mechanisms. Recent studies of the localization of Fas on the cellular plasma membrane have allowed new insights into this receptor’s divergent apoptotic and non-apoptotic functions. A conserved palmitoylation site on Fas at a cysteine residue just inside of the plasma membrane (C199 in human and C194 in mouse) was found to be essential for assembly and full activation of caspase 8 in the Fas receptor signaling complex and efficient initiation of Fas-induced apoptosis in human B-lymphoblastoid and T cell lines [58, 59]. Fas palmitoylation enables association of Fas with lipid raft microdomains and efficient receptor clustering on the plasma membrane [58–60]. To investigate the role of Fas membrane localization in an intact immunological setting, our group developed transgenic mice engineered to express the Fas palmitoylation mutation via BAC recombineering, and crossed these to the C57BL/6 Fas-deficient lpr mouse, termed FasC194Vlpr/lpr mice. Fas-induced apoptosis is defective in CD4+ and CD8+ T cells, total B cells and dendritic cells in these mice, but lymphocyte activation is still intact and necroptosis not enhanced. Further, Fas-induced precocious differentiation is still intact [60]. These findings highlight the role of Fas in determining cell fate not only by inducing apoptosis but more importantly by promoting cell differentiation.
Non-apoptotic Fas signaling in autoimmunity, tumor biology and cancer immunotherapy
If Fas-mediated apoptosis mediates protection from autoimmunity, then mice expressing the FasC194V mutant receptor that is defective in triggering cell death should develop the autoimmune and other features of Fas-deficiency. Cells from lpr mice and ALPS patients harbor loss of function or dominant-negative Fas mutations that impair both apoptosis and differentiation, not allowing determination of which function of Fas prevents autoimmunity. Notably, a FasC194V BAC transgene unable to mediate apoptosis almost completely reversed the B cell hyper-activation, accumulation of ‘double-negative’ CD4−/CD8−/B220+ T cells, and autoantibody production in Fas-deficient mice, even though the transgenic mutant Fas expression was somewhat lower than the endogenous receptor [60]. Because the FasC194V mutant still supports Fas-mediated precocious CD4+ and CD8+ T cell differentiation, these results suggest that Fas-mediated T cell differentiation into short-lived effector-memory cells may prevent autoimmunity through limiting the lifespan of autoimmune cells independently of the ability of Fas to induce apoptosis. Because effector memory T cells have a short in vivo half-life and are more vulnerable to Fas-induced apoptosis and other forms of cell death such as cytokine withdrawal, these two functions of Fas — promotion of differentiation into effector memory cells and triggering apoptosis — ultimately may work together to prevent expansion of autoreactive T cells and autoimmunity. Whether Fas signaling has similar functions promoting B cell differentiation in addition to triggering apoptosis of germinal center B cells, is an important question for future study, as conditional Fas knockout mice have shown a B cell-intrinsic function for Fas in preventing autoantibody production [61, 62].
Although one of the ways Fas was originally identified was through the ability of anti-Fas antibodies to trigger cell death of tumor cells, a role for endogenous Fas in promoting tumor growth and metastasis has more recently emerged (reviewed in [63]). Systematic studies of mouse and human tumor cell lines revealed that many lines express Fas but are resistant to Fas-induced apoptosis, and stimulation of these cells with FasL induced NF-κB and MAP-Kinase signaling in addition to caspae-8 activation, and promoted tumor cell motility and invasiveness [64]. Loss of Fas mRNA and protein expression on ovarian and liver tumor cells consistently resulted in lower growth rates and tumor size and incidence in animal models [65], confirming earlier studies in which genetic overexpression of Fas promoted growth of Lewis lung cancer cells in a mouse model despite conferring apoptosis sensitivity to these same cells in vitro [66]. In a glioblastoma model, tumor infiltration induced FasL expression in surrounding stromal cells, which promoted increased tumor invasiveness through activation of the AKT signaling pathway through Fas on the glioblastoma cells [67]. These findings have been translated into a clinical trial of blocking Fas signaling using a Fas-Fc fusion protein in patients with progressive glioblastoma, which did significantly increase progression-free survival when added to radiation therapy [68].
Adoptive cell transfer (ACT) is a potentially curative form of cancer immunotherapy in which tumor-antigen specific CD8+ and/or CD4+ T cells are grown outside a patient’s body to therapeutic numbers prior to re-infusion. ACT has recently entered the standard of care for selected B-cell malignancies [69, 70] and has also shown promising efficacy in early phase clinical trials for patients with selected solid cancers [71–81]. Two sources of T cells are currently used for ACT: 1) tumor infiltrating lymphocytes (TIL) obtained through the surgical resection of a metastatic deposit and, 2) peripheral blood T cells genetically engineered with an anti-tumor receptor. Regardless of source, therapeutic T cells must undergo a period of ex vivo activation and expansion to generate sufficient numbers of cells for treatment (typically 108 to 1011 cells). In the case of T cells genetically redirected to recognize cancer-associated antigens with either a T cell receptor (TCR) or chimeric antigen receptor (CAR), ex vivo activation is also required for efficient integration of the receptor transgene. During expansion, T cells experience progressive changes in epigenetic marks [82, 83], gene expression profiles [84, 85], metabolic processes [85–87], and cell-surface markers [78, 88]. The end result is the formation of a heterogenous population of antigen-experienced T-cell subsets which differ in their relative capacities to expand, persist, and mediate tumor regression following adoptive transfer [89]. Despite this diversity, expression of Fas is held in common across all T cells used for adoptive immunotherapy [90].
Pre-clinical studies in mice [71, 84, 91–96] and retrospective analyses of human clinical trials [72, 97–99] have established a correlation between the frequency of less-differentiated T cell subsets infused and improved clinical outcomes. Specifically, infusion of either T stem cell memory (TSCM) or T central memory (TCM) was associated with superior tumor regression relative to cells with a T effector memory (TEM) or effector T cell (TEFF) phenotype. Based on these findings, strategies that generate minimally-differentiated T cell products have become a major translational goal. The composition of circulating lymphocytes in cancer patients is skewed towards Fas-expressing memory T-cell subsets likely as a consequence of prior chemotherapy and immunotherapy [71, 84, 95, 100, 101]. Nevertheless, most cancer patients have at least some representation of the highly potent TSCM and TCM subsets in the peripheral blood that could be used to generate therapeutic cells. This observation raised the question of whether enrichment for minimally-differentiated T cell subsets prior to adoptive immunotherapy is required to deliver maximal in vivo antitumor efficacy.
The discovery of precocious differentiation through T cell-T cell interactions whereby TMem directly promote the functional differentiation of naïve T cells identified a novel mechanism which leads the attrition of TSCM and TCM cells and an accumulation of TEM cells during generation of T cells for ACT [57, 102, 103]. This process results in the compromised antitumor efficacy following ACT [57]. Mechanistically, precocious differentiation occurs in a cell-contact and cell-activation dependent manner [71, 102]. Further, the process is prevented when activated TMem and TN are separated by a semi-permeable membrane that allows passage of soluble factors, such as cytokines, but limits direct cell contact [57]. These observations suggested that an activation induced co-stimulatory ligand expressed on the surface of TMem may be responsible for the phenomenon.
Many co-stimulatory ligands are members of the TNF super family (TNFSF), including 4–1BBL, OX40L, CD70, CD40L, GITRL, and TL1A [2]. By comparing the gene expression of memory T cells at rest and following stimulation, it was discovered that the only TNFSF ligand differentially expressed with acute activation was Fasl, the gene encoding Fas ligand. This finding correlates with the poised epigenetic landscape of the Fasl locus in TMem, which contains activating trimethylation epigenetic modifications on histone 3 at lysine 4 (H3K4me3) and a paucity of repressive H3K27me3 marks [57, 104]. By contrast, the Fasl locus is heavily deposited with repressive H3K27me3 modifications in TN, limiting gene expression. Induction of Fas-signaling in the absence of TMem using a recombinant form of FasL oligomerized through a leucine zipper domain [56] caused augmented mouse and human T cell differentiation [57, 60]. Conversely, loss of Fas function using CD4+ or CD8+ T cells from Fas-deficient lpr mice, human T cells derived from ALPS patients with Fas mutations, or an anti-FasL antibody all blocked the accelerated maturation of T cells. Taken together, these data indicate that a FasL-Fas interaction between memory and naïve T cells are both necessary and sufficient to skew T cell differentiation.
At least three lines of evidence support the conclusion that skewing towards TEM and away from TSCM/TCM caused by Fas-signaling results from enhanced differentiation and not selective subset killing. First, Fas-signaling in naïve T cells results in increased phosphorylation of Akt at both the activating T308 and S473 residues as well as downstream phosphorylation of ribosomal protein S6 [57, 60]. Signaling through the PI3K/AKT/mTOR pathway is a common feature among a number of co-stimulatory TNFSF members and is a key driver of T cell-differentiation [105]. Second, both TSCM and TCM are relatively resistant to FasL-mediated apoptosis compared with TEM [56, 57, 106]. Thus, the accumulation of TEM when Fas-signaling is augmented with lz-FasL or co-culture of naïve with memory CD8+ T cells cannot be attributed to the selective apoptosis of TSCM/ TCM. Finally, enhanced cellular differentiation and cell death can be genetically uncoupled. Primary murine T cells expressing the FasC variant, which cannot undergo post-translation palmitoylation required for partitioning of Fas into lipid rafts [59, 60, 107], are protected from lz-FasL mediated apoptosis [57, 60]. However, FasC194V-expressing T cells remain capable of undergoing augmented FasL differentiation similar to wild type T cells.
Based on these mechanistic data, the negative influence of precocious differentiation on the function of T cell used for adoptive immunotherapy can be minimized using several translatable strategies. First, for adoptive immunotherapies using genetically engineered peripheral blood T cells, TMem can be physically separated away from TN prior to cell activation and expansion [57]. Practically, this can be accomplished using clinical cell isolation techniques such as magnetic microbeads [108] or streptamers [109]. Second, Fas-signaling can be limited using either an anti-FasL antibody or a recombinant Fas-Fc decoy to neutralize FasL [57]. Finally, bulk populations of T cells, whether derived from TIL or the peripheral blood, can be grown in the presence of pharmacological inhibitors of the PI3K/AKT pathway to block downstream signals associated with non-apoptotic Fas signaling [85, 88, 110, 111]. Pre-clinical proof of principal experiments using each of these strategies has established that blockade of signals emanating from non-apoptotic Fas signaling can enhance adoptive immunotherapy. The latter strategy using pharmacological pathway inhibitors has already been incorporated into T-cell based immunotherapy clinical trials using both a CAR (NCT03274219) and TCR (NCT03139370).
Concluding remarks
Much has been learned about the functions of Fas and TRAIL receptors beyond their originally described roles as inducers of cell death, yet many outstanding questions remain. More understanding of these functions will help unravel the complexity of how these receptors can trigger such alternative cell fates as cell death and immune cell stimulation. Greater knowledge of alternative signaling pathways by these receptors will aid development of therapeutic strategies to block or stimulate signaling in the context of specific cell types and disease states.
Figure 2: Apoptotic and non-apoptotic signaling induced by Fas.
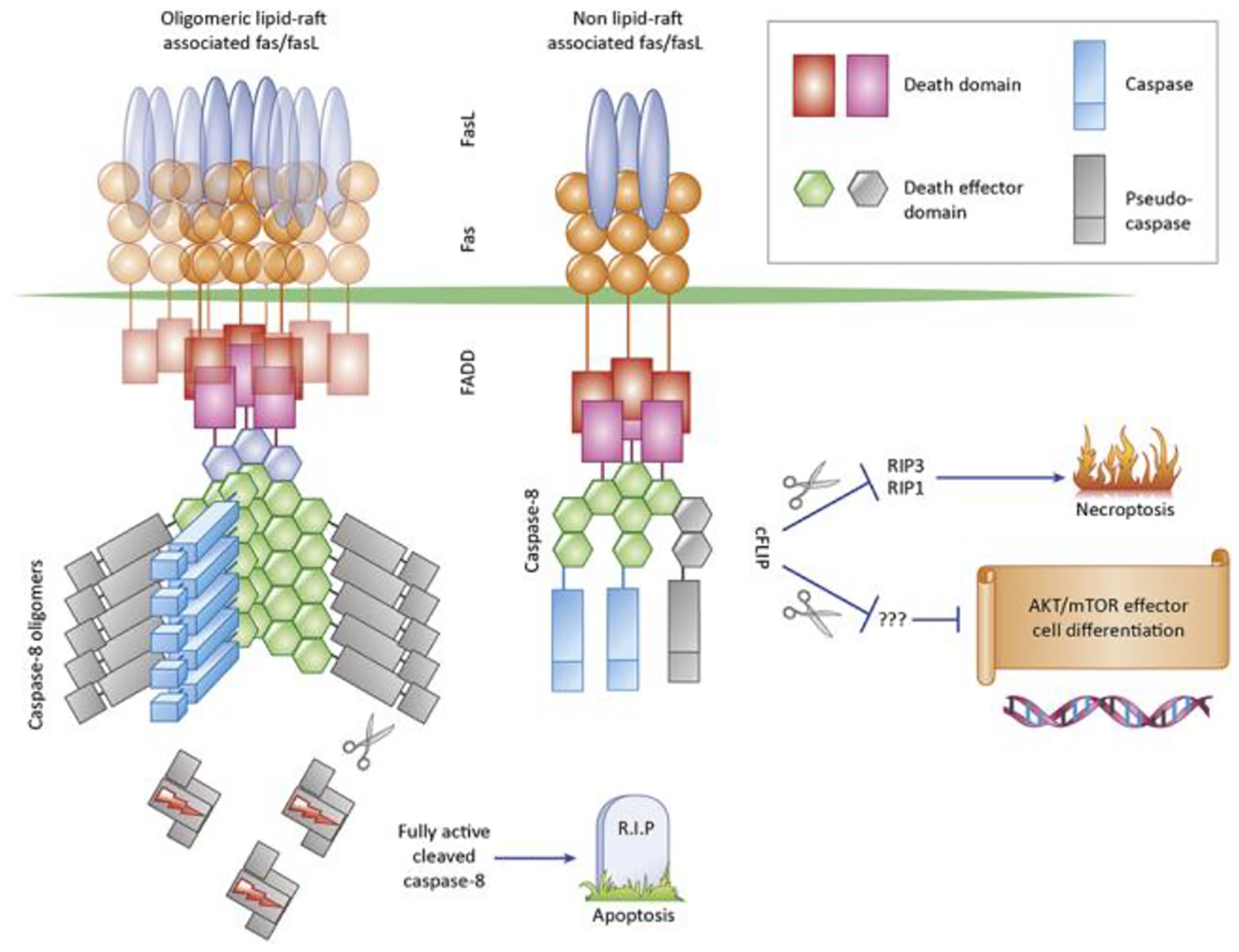
Fas is aggregated on the cell surface by cell-bound Fas ligand, which initiates formation of the Death-Inducing-Signaling Complex (DISC), consisting of Fas, the adapter protein FADD, Pro-caspase-8 and the regulatory protein c-FLIP, which shares the dual-DED and caspase domains with caspase-8 but is enzymatically inactive. In Caspase-8 is known to be able to oligomerize into large filamentous structures which facilitate autocatalytic activation and release of the enzymatically active caspase-8 tetramer into the cytoplasm, where substrates which promote apoptosis are cleaved. Fas signaling in lipid rafts promotes efficient formation of the DISC and high caspase-8 activity and release into the cytoplasm. In Fas complexes assembling outside of lipid rafts or when FasL oligomerization is low, the DISC does not become fully active, but Caspase-8 and c-FLIP form an alternate signaling complex which cleaves RIP1 and RIP3 to prevent necroptosis. This alternate signaling complex may also mediate non-apoptotic signaling by cleaving alternate substrates, most likely at the plasma membrane
Figure 3: Dual roles of Fas in promoting differentiation vs. cell death in T cells.
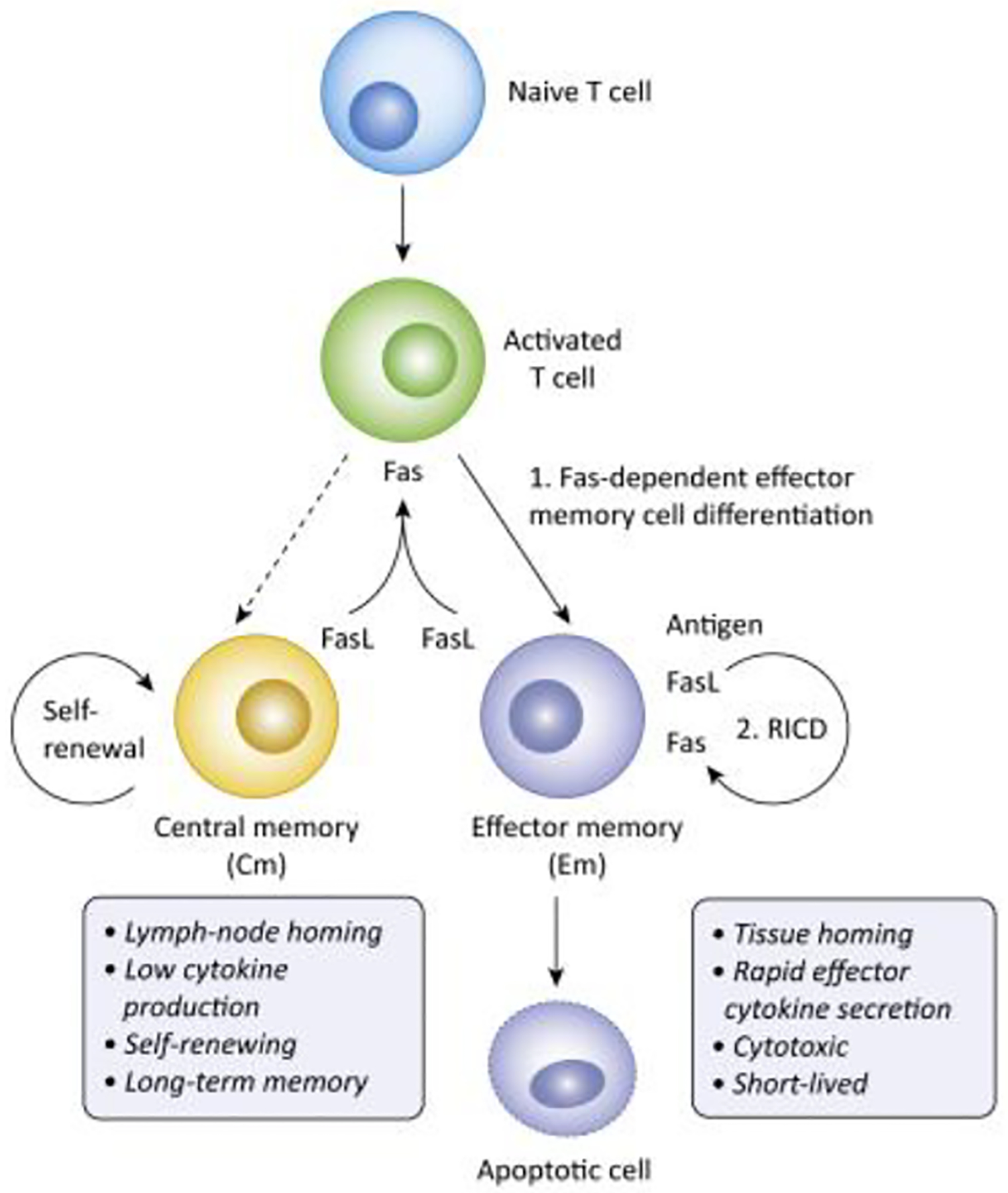
1. FasL expression on activated memory T cells, higher in effector than central memory, feeds back on early activated cells to skew differentiation towards effector memory cells, which have higher cytotoxic function and effector cytokine production than central memory cells. 2. TEM are also more susceptible to Fas-mediated apoptosis, which can be triggered through TCR stimulation and autocrine or paracrine FasL production, a process termed Restimulation-Induced Cell Death (RICD). These two mechanisms work together to limit the survival of effector memory T cells, which reduces the frequency of chronically stimulated autoreactive T cells, but also limits the efficacy of T cell adoptive cancer immunotherapy.
Highlights Box.
TNF family cytokine receptors containing a death domain, known informally as ‘death receptors’, trigger a diverse range of functions beyond inducing programmed cell death including cell growth and proliferation
The TNFR Fas, or CD95, can co-stimulate CD4+ and CD8+ T cell activation and accelerate T cell differentiation into short-lived effector memory T cells
Fas-induced effector memory T cell differentiation limits the efficacy of adoptive T cell immunotherapy and also is important for preventing the autoimmune syndrome that arises in the absence of a functional Fas receptor
TRAIL receptors can also trigger inflammatory responses through a scaffolding function of caspase-8 and activation of caspase-1 and interleukin 1β activation and release
These novel functions of TNF receptors may constitute targets for approaches to improve cancer immunotherapy and treat autoimmune diseases
Acknowledgements:
F.Y., N.F. A.C., and R.M.S. were supported by Intramural Research Funding from NIAMS and NCI. C.A.K. was supported by the Parker Institute for Cancer Immunotherapy, the MSKCC Core grant P30 CA008748, and the Damon Runyon Clinical Investigator Award. The authors thank Vera Siegel for editing.
Glossary
- ALPS
autoimmune lymphoproliferative syndrome, caused by Fas gene mutations and characterized by hypergammaglobulinema, autoimmunity (mainly hematologic) and accumulation of CD4−CD8− T cells in multiple organs
- Apoptosome
A cytoplasmic protein complex consisting of caspase-9, the scaffolding protein APAF-1, and cytochrome-c, a small protein normally present in the mitochondrial membrane. The complex efficiently cleaves the effector caspases 3 and 7, amplifying the apoptotic protease cascade
- DISC
death-inducing signaling complex. A protein complex containing Fas, the adapter protein FADD, the cysteine protease caspase 8 formed after Fas stimulation
- FADDosome
a TRAIL induced protein complex assembled by caspase 8, FADD and RIPK1 that initiates pro-inflammatory signaling
- Lpr mice
lymphproliferation mutation mouse model. lpr mice carry functional mutation in the Fas gene which causes autoimmune lymphocytes accumulation
- necroptosis
a form of necrotic programmed cell death dependent on oligomerization of RIP1/RIP3 that that results in release of cellular contents and an acute inflammatory responseanti-
- NLRP3 inflammasome
protein complex formed following TLR or cytokine stimulation. NLRP3 inflammasome converts pro-caspase 1 to active caspase 1
- precocious differentiation
accelerated differentiation of activated naïve T cell into effector memory T cells following FasL stimulation
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Aggarwal BB (2003) Signalling pathways of the TNF superfamily: a double-edged sword. Nat Rev Immunol 3 (9), 745–56. [DOI] [PubMed] [Google Scholar]
- 2.Croft M and Siegel RM (2017) Beyond TNF: TNF superfamily cytokines as targets for the treatment of rheumatic diseases. Nat Rev Rheumatol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Itoh N et al. (1991) The polypeptide encoded by the cDNA for human cell surface antigen Fas can mediate apoptosis. Cell 66 (2), 233–43. [DOI] [PubMed] [Google Scholar]
- 4.Sheridan JP et al. (1997) Control of TRAIL-induced apoptosis by a family of signaling and decoy receptors. Science 277 (5327), 818–21. [DOI] [PubMed] [Google Scholar]
- 5.Pobezinskaya YL and Liu Z (2012) The role of TRADD in death receptor signaling. Cell Cycle 11 (5), 871–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morlon A et al. (2005) TAB2, TRAF6 and TAK1 are involved in NF-kappaB activation induced by the TNF-receptor, Edar and its adaptator Edaradd. Hum Mol Genet 14 (23), 3751–7. [DOI] [PubMed] [Google Scholar]
- 7.Micheau O and Tschopp J (2003) Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 114 (2), 181–90. [DOI] [PubMed] [Google Scholar]
- 8.Peter ME and Krammer PH (2003) The CD95(APO-1/Fas) DISC and beyond. Cell Death Differ 10 (1), 26–35. [DOI] [PubMed] [Google Scholar]
- 9.Li H et al. (1998) Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell 94 (4), 491–501. [DOI] [PubMed] [Google Scholar]
- 10.Srinivasula SM et al. (2001) A conserved XIAP-interaction motif in caspase-9 and Smac/DIABLO regulates caspase activity and apoptosis. Nature 410 (6824), 112–6. [DOI] [PubMed] [Google Scholar]
- 11.Li P et al. (1997) Cytochrome c and dATP-dependent formation of Apaf-1/Caspase-9 complex initiates an apoptotic protease cascade. Cell 91, 479–489. [DOI] [PubMed] [Google Scholar]
- 12.Lenardo MJ (1991) lnterleukin-2 programs mouse αβ T lymphocytes for apoptosis. Nature 353 (6347), 858–861. [DOI] [PubMed] [Google Scholar]
- 13.Critchfield JM et al. (1994) T cell deletion in high antigen dose therapy of autoimmune encephalomyelitis. Science 263 (5150), 1139–43. [DOI] [PubMed] [Google Scholar]
- 14.Dhein J et al. (1995) Autocrine T-cell suicide mediated by APO-1/(Fas/CD95). Nature 373 (6513), 438–41. [DOI] [PubMed] [Google Scholar]
- 15.Ju ST et al. (1995) Fas(CD95)/FasL interactions required for programmed cell death after T-cell activation. Nature 373 (6513), 444–8. [DOI] [PubMed] [Google Scholar]
- 16.Karin M and Greten FR (2005) NF-kappaB: linking inflammation and immunity to cancer development and progression. Nat Rev Immunol 5 (10), 749–59. [DOI] [PubMed] [Google Scholar]
- 17.Li X and Stark GR (2002) NFkappaB-dependent signaling pathways. Exp Hematol 30 (4), 285–96. [DOI] [PubMed] [Google Scholar]
- 18.Oeckinghaus A et al. (2011) Crosstalk in NF-kappaB signaling pathways. Nat Immunol 12 (8), 695–708. [DOI] [PubMed] [Google Scholar]
- 19.Ashany D et al. (1999) Dendritic cells are resistant to apoptosis through the Fas (CD95/APO-1) pathway. J Immunol 163 (10), 5303–11. [PubMed] [Google Scholar]
- 20.Guo Z et al. (2003) Fas ligation induces IL-1beta-dependent maturation and IL-1beta-independent survival of dendritic cells: different roles of ERK and NF-kappaB signaling pathways. Blood 102 (13), 4441–7. [DOI] [PubMed] [Google Scholar]
- 21.Hohlbaum AM et al. (2001) Fas ligand engagement of resident peritoneal macrophages in vivo induces apoptosis and the production of neutrophil chemotactic factors. J Immunol 167 (11), 6217–24. [DOI] [PubMed] [Google Scholar]
- 22.Miwa K et al. (1998) Caspase 1-independent IL-1beta release and inflammation induced by the apoptosis inducer Fas ligand. Nat Med 4 (11), 1287–92. [DOI] [PubMed] [Google Scholar]
- 23.Bossaller L et al. (2012) Cutting Edge: FAS (CD95) mediates noncanonical IL-1beta and IL-18 maturation via caspase-8 in an RIP3-independent manner. J Immunol 189 (12), 5508–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gurung P et al. (2014) FADD and caspase-8 mediate priming and activation of the canonical and noncanonical Nlrp3 inflammasomes. J Immunol 192 (4), 1835–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Uchiyama R et al. (2013) Fas-mediated inflammatory response in Listeria monocytogenes infection. J Immunol 190 (8), 4245–54. [DOI] [PubMed] [Google Scholar]
- 26.Fukui M et al. (2003) Pathogen-associated molecular patterns sensitize macrophages to Fas ligand-induced apoptosis and IL-1 beta release. J Immunol 171 (4), 1868–74. [DOI] [PubMed] [Google Scholar]
- 27.Gregory MS et al. (2005) A novel treatment for ocular tumors using membrane FasL vesicles to activate innate immunity and terminate immune privilege. Invest Ophthalmol Vis Sci 46 (7), 2495–502. [DOI] [PubMed] [Google Scholar]
- 28.Gregory MS et al. (2002) Membrane Fas ligand activates innate immunity and terminates ocular immune privilege. J Immunol 169 (5), 2727–35. [DOI] [PubMed] [Google Scholar]
- 29.George JF et al. (1998) An essential role for Fas ligand in transplantation tolerance induced by donor bone marrow. Nat Med 4 (3), 333–5. [DOI] [PubMed] [Google Scholar]
- 30.Askenasy EM et al. (2011) Engineering of bone marrow cells with fas-ligand protein-enhances donor-specific tolerance to solid organs. Transplant Proc 43 (9), 3545–8. [DOI] [PubMed] [Google Scholar]
- 31.Kang SM et al. (1997) Immune response and myoblasts that express Fas ligand. Science 278 (5341), 1322–4. [DOI] [PubMed] [Google Scholar]
- 32.Restifo NP (2000) Not so Fas: Re-evaluating the mechanisms of immune privilege and tumor escape. Nat Med 6 (5), 493–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen JJ et al. (1998) Regulation of the proinflammatory effects of fas ligand (CD95L). Science 282 (5394), 1714–7. [DOI] [PubMed] [Google Scholar]
- 34.Corsini NS et al. (2009) The death receptor CD95 activates adult neural stem cells for working memory formation and brain repair. Cell Stem Cell 5 (2), 178–90. [DOI] [PubMed] [Google Scholar]
- 35.Demjen D et al. (2004) Neutralization of CD95 ligand promotes regeneration and functional recovery after spinal cord injury. Nat Med 10 (4), 389–95. [DOI] [PubMed] [Google Scholar]
- 36.Cullen SP et al. (2013) Fas/CD95-induced chemokines can serve as “find-me” signals for apoptotic cells. Mol Cell 49 (6), 1034–48. [DOI] [PubMed] [Google Scholar]
- 37.Park DR et al. (2003) Fas (CD95) induces proinflammatory cytokine responses by human monocytes and monocyte-derived macrophages. J Immunol 170 (12), 6209–16. [DOI] [PubMed] [Google Scholar]
- 38.Umemura M et al. (2004) Involvement of IL-17 in Fas ligand-induced inflammation. Int Immunol 16 (8), 1099–108. [DOI] [PubMed] [Google Scholar]
- 39.Kataoka T et al. (2000) The caspase-8 inhibitor FLIP promotes activation of NF-kappaB and Erk signaling pathways. Curr Biol 10 (11), 640–8. [DOI] [PubMed] [Google Scholar]
- 40.Henry CM and Martin SJ (2017) Caspase-8 Acts in a Non-enzymatic Role as a Scaffold for Assembly of a Pro-inflammatory “FADDosome” Complex upon TRAIL Stimulation. Mol Cell 65 (4), 715–729 e5. [DOI] [PubMed] [Google Scholar]
- 41.Varfolomeev E et al. (2005) Molecular determinants of kinase pathway activation by Apo2 ligand/tumor necrosis factor-related apoptosis-inducing ligand. J Biol Chem 280 (49), 40599–608. [DOI] [PubMed] [Google Scholar]
- 42.O’ Reilly LA et al. (2009) Membrane-bound Fas ligand only is essential for Fas-induced apoptosis. Nature 461 (7264), 659–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fisher GH et al. (1995) Dominant interfering Fas gene mutations impair apoptosis in a human autoimmune lymphoproliferative syndrome. Cell 81 (6), 935–46. [DOI] [PubMed] [Google Scholar]
- 44.Watanabe-Fukunaga R et al. (1992) Lymphoproliferation disorder in mice explained by defects in Fas antigen that mediates apoptosis. Nature 356 (6367), 314–7. [DOI] [PubMed] [Google Scholar]
- 45.Brunner T et al. (1995) Cell-autonomous Fas (CD95)/Fas-ligand interaction mediates activation-induced apoptosis in T-cell hybridomas. Nature 373 (6513), 441–4. [DOI] [PubMed] [Google Scholar]
- 46.Siegel RM et al. (2000) The multifaceted role of Fas signaling in immune cell homeostasis and autoimmunity. Nat Immunol 1 (6), 469–74. [DOI] [PubMed] [Google Scholar]
- 47.Alderson MR et al. (1993) Fas transduces activation signals in normal human T lymphocytes. J Exp Med 178 (6), 2231–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kreuz S et al. (2004) NFkappaB activation by Fas is mediated through FADD, caspase-8, and RIP and is inhibited by FLIP. J Cell Biol 166 (3), 369–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kennedy NJ et al. (1999) Caspase activation is required for T cell proliferation. J Exp Med 190 (12), 1891–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Su H et al. (2005) Requirement for caspase-8 in NF-kappaB activation by antigen receptor. Science 307 (5714), 1465–8. [DOI] [PubMed] [Google Scholar]
- 51.Salmena L and Hakem R (2005) Caspase-8 deficiency in T cells leads to a lethal lymphoinfiltrative immune disorder. J Exp Med 202 (6), 727–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chun HJ et al. (2002) Pleiotropic defects in lymphocyte activation caused by caspase-8 mutations lead to human immunodeficiency. Nature 419 (6905), 395–9. [DOI] [PubMed] [Google Scholar]
- 53.Kaiser WJ et al. (2011) RIP3 mediates the embryonic lethality of caspase-8-deficient mice. Nature 471 (7338), 368–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Oberst A et al. (2011) Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature 471 (7338), 363–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Peter ME et al. (1997) Resistance of cultured peripheral T cells towards activation-induced cell death involves a lack of recruitment of FLICE (MACH/caspase 8) to the CD95 death-inducing signaling complex. European Journal of Immunology 27 (5), 1207–12. [DOI] [PubMed] [Google Scholar]
- 56.Ramaswamy M et al. (2011) Specific elimination of effector memory CD4+ T cells due to enhanced Fas signaling complex formation and association with lipid raft microdomains. Cell Death Differ 18 (4), 712–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Klebanoff CA et al. (2016) Memory T cell-driven differentiation of naive cells impairs adoptive immunotherapy. J Clin Invest 126 (1), 318–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Feig C et al. (2007) Palmitoylation of CD95 facilitates formation of SDS-stable receptor aggregates that initiate apoptosis signaling. EMBO J 26 (1), 221–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chakrabandhu K et al. (2007) Palmitoylation is required for efficient Fas cell death signaling. EMBO J 26 (1), 209–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cruz AC et al. (2016) Fas/CD95 prevents autoimmunity independently of lipid raft localization and efficient apoptosis induction. Nat Commun 7, 13895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Stranges PB et al. (2007) Elimination of antigen-presenting cells and autoreactive T cells by fas contributes to prevention of autoimmunity. Immunity 26 (5), 629–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hao Z et al. (2008) Fas receptor expression in germinal-center B cells is essential for T and B lymphocyte homeostasis. Immunity 29 (4), 615–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Martin-Villalba A et al. (2013) CD95 in cancer: tool or target? Trends Mol Med 19 (6), 329–35. [DOI] [PubMed] [Google Scholar]
- 64.Barnhart BC et al. (2004) CD95 ligand induces motility and invasiveness of apoptosis-resistant tumor cells. Embo J 23 (15), 3175–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chen L et al. (2010) CD95 promotes tumour growth. Nature 465 (7297), 492–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lee JK et al. (2003) Lack of FasL-mediated killing leads to in vivo tumor promotion in mouse Lewis lung cancer. Apoptosis 8 (2), 151–60. [DOI] [PubMed] [Google Scholar]
- 67.Kleber S et al. (2008) Yes and PI3K bind CD95 to signal invasion of glioblastoma. Cancer Cell 13 (3), 235–48. [DOI] [PubMed] [Google Scholar]
- 68.Wick W et al. (2014) A phase II, randomized, study of weekly APG101+reirradiation versus reirradiation in progressive glioblastoma. Clin Cancer Res 20 (24), 6304–13. [DOI] [PubMed] [Google Scholar]
- 69.Neelapu SS et al. (2017) Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N Engl J Med 377 (26), 2531–2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Maude SL et al. (2018) Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N Engl J Med 378 (5), 439–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Klebanoff CA et al. (2016) Prospects for gene-engineered T cell immunotherapy for solid cancers. Nat Med 22 (1), 26–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Louis CU et al. (2011) Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood 118 (23), 6050–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tran E et al. (2014) Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science 344 (6184), 641–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tran E et al. (2016) T-Cell Transfer Therapy Targeting Mutant KRAS in Cancer. N Engl J Med 375 (23), 2255–2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Robbins PF et al. (2015) A pilot trial using lymphocytes genetically engineered with an NY-ESO-1-reactive T-cell receptor: long-term follow-up and correlates with response. Clin Cancer Res 21 (5), 1019–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Goff SL et al. (2016) Randomized, Prospective Evaluation Comparing Intensity of Lymphodepletion Before Adoptive Transfer of Tumor-Infiltrating Lymphocytes for Patients With Metastatic Melanoma. J Clin Oncol 34 (20), 2389–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Brown CE et al. (2016) Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy. N Engl J Med 375 (26), 2561–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chandran SS et al. (2017) Treatment of metastatic uveal melanoma with adoptive transfer of tumour-infiltrating lymphocytes: a single-centre, two-stage, single-arm, phase 2 study. Lancet Oncol 18 (6), 792–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lu YC et al. (2017) Treatment of Patients With Metastatic Cancer Using a Major Histocompatibility Complex Class II-Restricted T-Cell Receptor Targeting the Cancer Germline Antigen MAGE-A3. J Clin Oncol, JCO2017745463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ahmed N et al. (2017) HER2-Specific Chimeric Antigen Receptor-Modified Virus-Specific T Cells for Progressive Glioblastoma: A Phase 1 Dose-Escalation Trial. JAMA Oncol 3 (8), 1094–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Stevanovic S et al. (2017) Landscape of immunogenic tumor antigens in successful immunotherapy of virally induced epithelial cancer. Science 356 (6334), 200–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Crompton JG et al. (2016) Lineage relationship of CD8(+) T cell subsets is revealed by progressive changes in the epigenetic landscape. Cell Mol Immunol 13 (4), 502–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Henning AN et al. (2018) Silencing stemness in T cell differentiation. Science 359 (6372), 163–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gattinoni L et al. (2011) A human memory T cell subset with stem cell-like properties. Nat Med 17 (10), 1290–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Klebanoff CA et al. (2017) Inhibition of AKT signaling uncouples T cell differentiation from expansion for receptor-engineered adoptive immunotherapy. JCI Insight 2 (23). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sukumar M et al. (2013) Inhibiting glycolytic metabolism enhances CD8+ T cell memory and antitumor function. J Clin Invest 123 (10), 4479–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sukumar M et al. (2016) Mitochondrial Membrane Potential Identifies Cells with Enhanced Stemness for Cellular Therapy. Cell Metab 23 (1), 63–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bowers JS et al. (2017) PI3Kdelta Inhibition Enhances the Antitumor Fitness of Adoptively Transferred CD8(+) T Cells. Front Immunol 8, 1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Klebanoff CA et al. (2012) Sorting through subsets: which T-cell populations mediate highly effective adoptive immunotherapy? J Immunother 35 (9), 651–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gattinoni L et al. (2012) Paths to stemness: building the ultimate antitumour T cell. Nat Rev Cancer 12 (10), 671–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Klebanoff CA et al. (2005) Central memory self/tumor-reactive CD8+ T cells confer superior antitumor immunity compared with effector memory T cells. Proc Natl Acad Sci U S A 102 (27), 9571–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gattinoni L et al. (2009) Wnt signaling arrests effector T cell differentiation and generates CD8+ memory stem cells. Nat Med 15 (7), 808–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Klebanoff CA et al. (2011) Determinants of successful CD8+ T-cell adoptive immunotherapy for large established tumors in mice. Clin Cancer Res 17 (16), 5343–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Barrett DM et al. (2014) Relation of clinical culture method to T-cell memory status and efficacy in xenograft models of adoptive immunotherapy. Cytotherapy 16 (5), 619–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sommermeyer D et al. (2016) Chimeric antigen receptor-modified T cells derived from defined CD8+ and CD4+ subsets confer superior antitumor reactivity in vivo. Leukemia 30 (2), 492–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hurton LV et al. (2016) Tethered IL-15 augments antitumor activity and promotes a stem-cell memory subset in tumor-specific T cells. Proc Natl Acad Sci U S A 113 (48), E7788–E7797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Rosenberg SA et al. (2011) Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res 17 (13), 4550–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Xu Y et al. (2014) Closely related T-memory stem cells correlate with in vivo expansion of CAR.CD19-T cells and are preserved by IL-7 and IL-15. Blood 123 (24), 3750–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kochenderfer JN et al. (2017) Lymphoma Remissions Caused by Anti-CD19 Chimeric Antigen Receptor T Cells Are Associated With High Serum Interleukin-15 Levels. J Clin Oncol, JCO2016713024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Turtle CJ et al. (2016) CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J Clin Invest 126 (6), 2123–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Pauken KE et al. (2016) Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science 354 (6316), 1160–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zhou J et al. (2018) A kinetic investigation of interacting, stimulated T cells identifies conditions for rapid functional enhancement, minimal phenotype differentiation, and improved adoptive cell transfer tumor eradication. PLoS One 13 (1), e0191634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Schmueck-Henneresse M et al. (2017) Comprehensive Approach for Identifying the T Cell Subset Origin of CD3 and CD28 Antibody-Activated Chimeric Antigen Receptor-Modified T Cells. J Immunol 199 (1), 348–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Russ BE et al. (2014) Distinct epigenetic signatures delineate transcriptional programs during virus-specific CD8(+) T cell differentiation. Immunity 41 (5), 853–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Chen L and Flies DB (2013) Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol 13 (4), 227–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Riou C et al. (2007) Convergence of TCR and cytokine signaling leads to FOXO3a phosphorylation and drives the survival of CD4+ central memory T cells. J Exp Med 204 (1), 79–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Feig C et al. (2007) Palmitoylation of CD95 facilitates formation of SDS-stable receptor aggregates that initiate apoptosis signaling. EMBO J 26 (1), 221–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Terakura S et al. (2012) Generation of CD19-chimeric antigen receptor modified CD8+ T cells derived from virus-specific central memory T cells. Blood 119 (1), 72–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Stemberger C et al. (2012) Novel serial positive enrichment technology enables clinical multiparameter cell sorting. PLoS One 7 (4), e35798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Crompton JG et al. (2015) Akt inhibition enhances expansion of potent tumor-specific lymphocytes with memory cell characteristics. Cancer Res 75 (2), 296–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Majchrzak K et al. (2017) beta-catenin and PI3Kdelta inhibition expands precursor Th17 cells with heightened stemness and antitumor activity. JCI Insight 2 (8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ramaswamy M et al. (2011) Harnessing programmed cell death as a therapeutic strategy in rheumatic diseases. Nat Rev Rheumatol 7 (3), 152–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Gajewski TF (2007) On the TRAIL toward death receptor-based cancer therapeutics. J Clin Oncol 25 (11), 1305–7. [DOI] [PubMed] [Google Scholar]
- 114.Thorburn A et al. (2008) TRAIL receptor-targeted therapeutics: resistance mechanisms and strategies to avoid them. Drug Resist Updat 11 (1–2), 17–24. [DOI] [PMC free article] [PubMed] [Google Scholar]