

Abstract
Lidocaine is one of the most common local anesthetics (LA) used in clinical practice and it is neurotoxic. Recent studies suggested that LA, including lidocaine, could exert protective effect over neurotoxicity by promoting autophagy. However, the underlying mechanism was not sufficiently elucidated. This study aimed to explore the mechanism behind. Human neuroblastoma cell line SH-SY5Y was used throughout the whole study. The effect of lidocaine on viability, toxicity of SH-SY5Y cells were analyzed by MTT and lactate dehydrogenase (LDH) assays, respectively. The relative expression of miR-145 was assessed by quantitative reverse transcription-polymerase chain reaction. The impact which lidocaine brought on PI3K/AKT/mTOR pathway and autophagy-related proteins were examined by the western blot assay. LC3B was assessed by immunofluorescence staining. The interaction between miR-145 and AKT3 was conducted by the dual-luciferase reporting assay. Lidocaine inhibited viability of SH-SY5Y cells in a time and dose dependent manner and enhanced the release of LDH in SH-SY5Y cells. Furthermore, the expression of miR-145 and autophagy were enhanced by lidocaine. Transfection with miR-145 inhibitor inhibited the release of LDH and autophagy. miR-145 targeted AKT3 to inhibit PI3K/AKT/mTOR pathway. Finally, lidocaine inactivated PI3K/AKT/mTOR pathways via upregulation of miR-145, and it subsequently promoted autophagy of SH-SY5Y cells. However, silence of miR-145 could reverse the promotion of the autophagy of SH-SY5Y cells. Our results showed that lidocaine promoted autophagy of nerve cells via regulating miR-145 expression and further inactivation of PI3K/AKT/mTOR signaling pathway.
Keywords: lidocaine, neurotoxicity, autophagy, miR-145
Introduction
Local anesthetics (LA) are universally used medicines in clinical settings. When interventional therapies are performed, they are normally used as a substitute for pain management, which makes patients suffer less during surgeries. Despite the immense benefits of LA, contemporary studies suggested that LA are toxic to various tissues [1,2] and may involve in perioperative nerve injury [3]. Studies also indicated that LA induced direct nerve damage could already occur at clinical concentration levels [4,5]. Lidocaine, first synthesized in 1943, is one of them, which is the most widely used LA in clinical practice [6]. However, studies showed that it might be a potential neuroprotectant, which is to protect neurons from injury or degeneration [7, 8].
Autophagy is a pivotal process for degradation of cellular components in a lysosome-dependent manner [9]. Under certain pathological conditions, autophagy activation could be beneficial or harmful to cells, depending on cell type and stimulation [10,11]. Recent study suggests that autophagy is a common protective response to neurotoxicity caused by different LA. It has an inhibitory effect on neurotoxicity; however, the regulatory mechanism is still not sufficiently elucidated [12]. Some studies indicated that PI3K/AKT/mTOR signaling pathway negatively regulated autophagy [13,14].
Micro RNAs (miRNAs) are highly conservative non-coding small RNA molecules, which involve in a variety of cellular processes by interfering with messenger RNA degradation and protein expression [15]. Several studies demonstrated that miRNAs play a critical role in the major cascades of autophagy [16–20]. A study showed that miR-145 promoted autophagy by inhibiting PI3K/AKT/mTOR signaling pathway [21]. Furthermore, another research indicated that lidocaine inhibited proliferation, migration and invasion of gastric cancer cells by upregulating miR-145 [22].
Taken together, we hypothesized that lidocaine promoted neuronal autophagy and protected nerve cells by upregulating miR-145 and inhibiting PI3K/AKT/mTOR pathway.
Materials and Methods
Cell culture
Human neuroblastoma cells SH-SY5Y were obtained from ATCC and were cultivated in Dulbecco’s modified Eagles medium (Gibco, Carlsbad, CA, USA), which contained 10% fetal bovine serum. SH-SY5Y cells were kept in the environment at a temperature of 37°C and 5% CO2.
MTT assay
Lidocaine was purchased from Sigma Chemical Company (St Louis, MO). Cell viability was measured by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) method. Briefly, cells were treated with lidocaine (0, 0.1, 0.5, 1, 2, 4 and 8 mM) for 24 h or 48 h, respectively. Cells grown in 96-well plates were treated with MTT (Sigma, M2128), in a final concentration of 500 μg/mL, at 37°C for 4 h. The formed crystals were solubilized in 100 μL of dimethyl sulfoxide and the color was read photometrically at 570 nm on Synergy HT plate reader (Bio-Tek Inc., Winooski, VT, USA). Assays were performed on six parallel wells. At least five independent viability assays were carried out. The percentage of survived cells over controls was calculated.
LDH assay
The CytoTox96 nonradioactive assay kit (Cell Biolabs inc., San Diego, CA, USA) was used in the LDH assay. SH-SY5Y cells were seeded in 96-well plates, exposed to different dose of lidocaine and cultured for 48 h. Then the plates were centrifuged (at 430 × g) for 5 min, and the cells was moved to another new 96-well plates (50 μL/well). They were kept under a dark environment for 30 min after adding LDH substrate to each well (50 μL/well). To terminate the reaction, 1 N hydrochloric acid (HCl) (25 μL/well) was added to each well. The absorbance was assessed at 490 nm. Control experiments were performed with 0.1% (w/v) Triton X-100 set as 100% cytotoxicity. All experiments were carried out in triplicate.
Western blot assay
Western blotting was carried out as described before [18, 19]. Cell lysates were then prepared using lysis buffer (50 mM Tris,150 mM NaCl,0.5% sodium deoxycholate, pH 7.4, 0.5 mM ethylenediaminetetraacetic acid, 0.1% sodium dodecyl sulfate [SDS] and 1% NP-40). After centrifugation at 7900 × g for 10 min, the supernatants were subjected to protein assay. An equivalent amount of protein (30 μg) was separated by SDS-polyacrylamide gel and transferred to Immobilon-P membrane (Millipore Corp., Bedford, MA, USA). After blocking with 5% fat-free milk, the membrane was incubated with the corresponding primary antibodies (anti-LC3I, anti-LC3II, anti-Beclin 1, anti-p62, anti-p-mTOR, anti-mTOR, anti-p-AKT, anti-AKT, anti-p-PI3K, anti-PI3K and anti-GAPDH) overnight at 4°C and then incubated with the appropriate secondary antibody (HRP-conjugated). The blots against GAPDH were used as a loading control. Signals were detected using the ECL kit and quantified by scanning densitometry.
Immunofluorescence Staining
Cells seeded in 96-well plates were fixed with 4% paraformaldehyde solution, permeabilized with 0.5% Triton X-100 for 10 min, and incubated for 30 min at room temperature in a blocking buffer containing 5% bovine serum albumin. The cells were then incubated with LC3B antibody (1:50 dilution) overnight at 4°C. Ultimately, we incubated the cells with a fluorescently labeled secondary antibody (Alexa Fluor 488, 1:400, Gongsi) for 1 h at room temperature in the dark, and then for 10 min with 0.1% DAPI. We observed and recorded LC3B positive staining using an inverted fluorescence microscope.
Cell transfection
miR-145 inhibitor and negative control were transfected into SH-SY5Y cells and incubated for 24 h. This method was used to decrease the expression of miR-145 in SH-SY5Y cells. Then they were diluted into a density of 2 × 105 cells/well, seeded in new plates and incubated until 70–80% confluent cells were reached. miR-145 inhibitor (5′-AGGGAUUCCUGGGAAAACUGGAC-3′) and negative control (5′-CAGUACUUUUGUGUAGUACAA-3′) were supplied by GenePharma Co. (Shanghai, China). For transfection, Lipofectamine 3000 reagen was used (Invitrogen, Carlsbad, CA, USA).
Dual-luciferase reporting assay
The 3′-UTR of the wild-type AKT3 and a variant containing mutations in the putative binding site were inserted downstream of the firefly luciferase reporter into the psiCHECK-2 vector (Promega, Madison, WI, USA). Constructed luciferase reporter plasmids (wild-type or mutant AKT3 plasmids) were co-transfected with miR-145 inhibitor or miR-NC into cells using Lipofectamine 2000. After 48 h, the luciferase activity was determined using Dual-Luciferase Reporter Assay System according to the manufacturer’s protocol (Promega, Madisoon, WI, USA).
qRT-PCR analysis
Quantitative reverse transcription-polymerase chain reaction (qRT-PCR) assay was used to evaluate miR-145 expression. TRIzol reagent (Life Technologies Corporation, Carlsbad, CA, USA) was used to extract total cellular RNA. Reverse transcription was achieved by the Taqman MicroRNA Reverse Transcription Kit (Thermo Fisher scientific, Waltham, MA, US). Moreover, to amplify complementary DNA (cDNA), Taqman Universal Master Mix II was used. The TaqMan MicroRNA Assay used new target-specific stem-loop primers during cDNA synthesis to generate a template for real-time PCR, and then to determine the expression of miR-145 (Applied Biosystems, Foster City, CA, USA). miR-145 was normalized to U6 small nuclear RNA.
Statistical analysis
All results are presented as means ± standard deviation (SD). Data analysis was performed using Graphpad Prism version 6.0 software (Graph Pad Software, San Diego California, USA). The Student t-test and one-way analysis of variance were used according to the data characteristics. The statistical significance level was set at P < 0.05.
Results
Lidocaine inhibited growth and induced death of SH-SY5Y cells
The SH-SY5Y cell viability was assessed when cells were treated with lidocaine. In MTT assay, cell viability was inhibited after cells were incubated with different concentrations of lidocaine (0, 0.1, 0.5, 1, 2, 4 and 8 mM). The inhibition of cells was in a time and dose dependent manner (Fig. 1). As show in Fig. 1, we determined the induction concentrations of lidocaine were: low concentration 0.5 mM, medium concentration 2 mM and high concentration 8 mM. The toxicity of lidocaine was also detected by the LDH assay. The rate of cell death increased with the dosage increment (Fig. 2A). However, when miR-145 was silenced, the LDH leakage level decreased compared with the lidocaine alone group (Fig. 4B).
Figure 1.
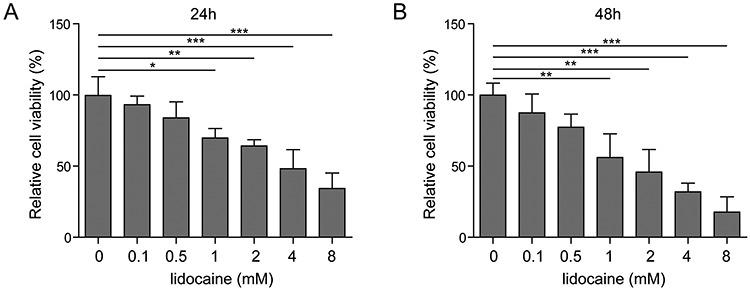
The growth of SH-SY5Y cells was inhibited by lidocaine. Effects of different concentrations of lidocaine on the viability of SH-SY5Y cells treated with 24 h (A) and 48 h (B).
Figure 2.
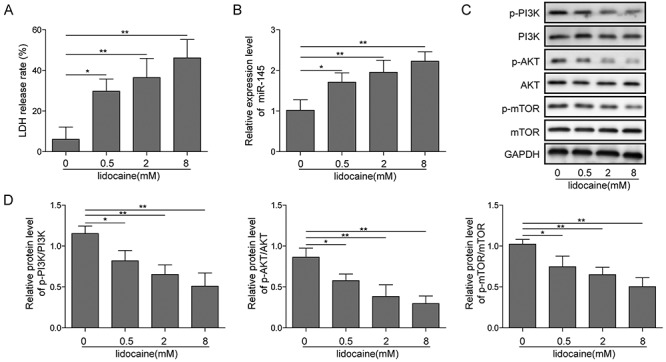
Lidocaine upregulated miR-145 and inhibited PI3K/AKT/mTOR pathway. (A) LDH release level on different concentrations of lidocaine. (B) Effect of different concentrations of lidocaine on the expression of miR-145 tested by qRT-PCR. (C) PI3K/AKT/mTOR pathway-associated protein expression in SH-SY5Y cells detected by the western blot assay.
Figure 4.
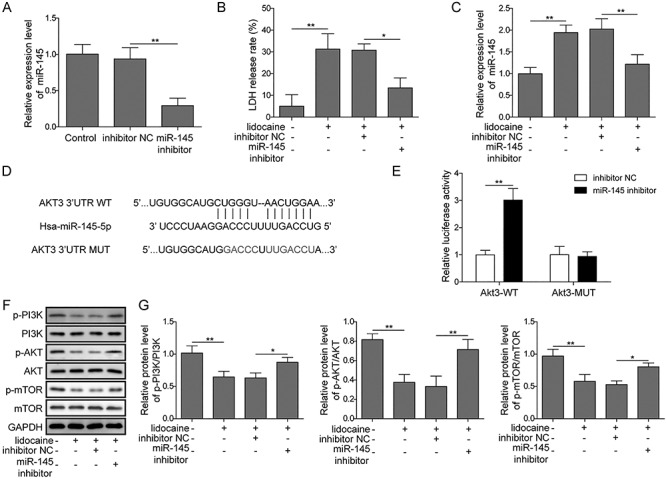
Inhibition of miR-145 expression relieved the inhibition of lidocaine on the PI3K/AKT/mTOR pathway. (A) miR-145 expression after transfection of miR-145 inhibitor measured by qRT-PCR. (B) Effect of lidocaine on the release of LDH in cells after inhibition of miR-145. (C) Effect of lidocaine on the expression of miR-145 in cells after inhibition of miR-145.(D,E) miR-145 and its putative binding sequence in the 3′-UTR of AKT3, Luciferase reporter assay showed the luciferase activity in miR-145 inhibitor and AKT3 wild-type/mutant co-transfected SH-SY5Y cells. (F, G) Effect of lidocaine on the expression of PI3K/AKT/mTOR pathway-related proteins after inhibiting miR-145.
Lidocaine upregulated miR-145 and inhibited PI3K/AKT/mTOR pathway
Increasing evidence suggested that miR-145 was involved in the process induced by lidocaine [22, 23]. To reveal the behind mechanism of lidocaine in SH-SY5Y cells, the relative expression of miR-145 was detected. Results from qRT-PCR showed that the relative expression of miR-145 was significantly increased when lidocaine was added, and it was in a dose dependent manner (Fig. 3). It indicated that miR-145 might participate in the process of lidocaine-induced activity on SH-SY5Y cells. The signal pathway involved in the lidocaine-induced event was also investigated. PI3K/AKT/mTOR pathway was found to be related with the effect of lidocaine and miR-145. Western blot assay showed that lidocaine significantly decreased the phosphorylation levels of PI3K, AKT and mTOR when the dose of lidocaine increased (Fig. 2C and D). Interestingly, when miR-145 was silenced, the PI3K/AKT/mTOR pathway was activated. Phosphorylation levels of PI3K, AKT and mTOR increased (Fig. 4F and G). These data indicated that lidocaine inhibited activation of PI3K/AKT/mTOR pathway possibly by upregulating miR-145. To find out the potential target of miR-145 in SH-SY5Y cells, the predicted targeting 3′-UTR region of human AKT3 was mutated and cloned into the psiCHECK-2 vector (Fig. 4D, labeled as AKT3 3′-UTR mutation). In parallel, the corresponding wild-type 3′-UTR region of AKT3 was constructed as well. Co-transfecting the wild-type AKT3 3′-UTR plasmid and miR-145 inhibitors into SH-SY5Y cells significantly increased the luciferase activity, which was not affected by AKT3 mutation (Fig. 4E). These results implied that AKT3 might be one direct target of miR-145 in SH-SY5Y cells.
Figure 3.
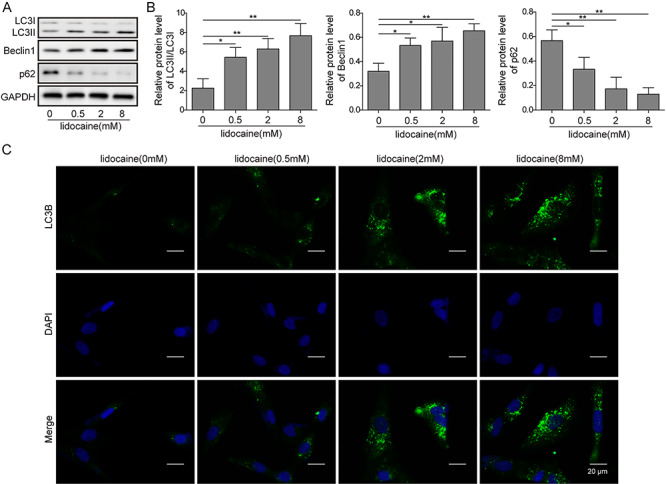
Lidocaine promoted autophagy in SH-SY5Y cells. (A,B) Autophagy-related protein expression detected by the western blot assay. (C) Effect of different concentrations of lidocaine on the expression of LC3B tested by immunofluorescence staining.
Lidocaine promoted autophagy in SH-SY5Y cells
Previous study suggested that mTOR served as a negative regulator of autophagy activation [24]. Moreover, a study also suggested that PI3K/AKT/mTOR signaling was closely associated with autophagy [25]. According to the results, we hypothesized that lidocaine might lead to the activation of autophagy in SH-SY5Y cells. Western blotting revealed that lidocaine enhanced the LC3II and Beclin1 upregulation and p62 and LC3I downregulation as the dose increased (Fig. 3A and B). When the miR-145 was silenced, the expression of LC3II and Beclin1 was downregulated, whereas the expression of p62 and LC3I was upregulated comparing to the cells without silence of miR-145 (Fig. 5A and B). Immunofluorescence staining assay showed that autophagy-related protein LC3B was upregulated when the dose of lidocaine increased (Fig. 3C). However, when miR-145 was silenced, the expression of LC3B was decreased as compared to the inhibitor-NC cells (Fig. 5C).
Figure 5.
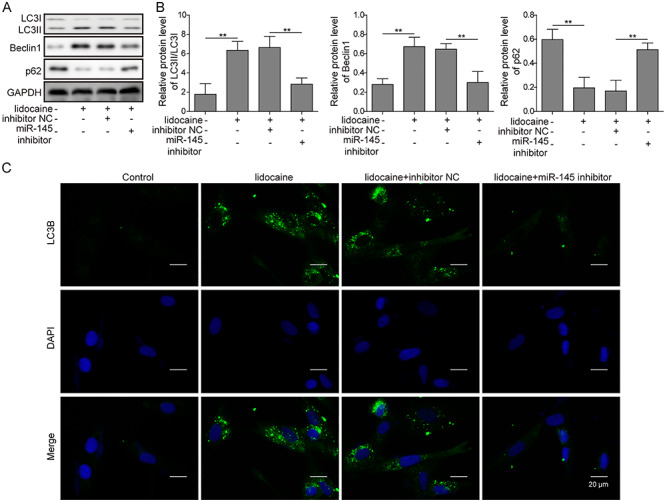
Inhibition of miR-145 expression relieved the promotion of lidocaine on autophagy in SH-SY5Y cells. (A,B) Autophagy-related protein expression after inhibiting miR-145 detected by the western blot assay. (C) LC3B expression after inhibiting miR-145 tested by immunofluorescence staining.
Discussion
The most remarkable finding of this study is that autophagy was promoted by lidocaine in 5H-SY5Y cells. MiR-145 was upregulated when treated with lidocaine. Moreover, PI3K/AKT/mTOR pathway might involve in this process, and negatively regulated autophagy activation.
MiR-145 is located on chromosome 5 (5q32–33) [26], a well-known fragile location in the human genome [27]. Downregulation of miR-145 has been observed in multiple types of cancers, indicating that it may function as a tumor suppressor. Moreover, a recent published article demonstrated that lidocaine inhibited growth, invasion and migration of gastric carcinoma cells by upregulation of miR-145 [22]. However, the role that miR-145 could play in lidocaine-induced neuronal toxicity is still unknown. Our study, first ever, showed that lidocaine could up-regulate miR-145 in nerve cells.
Another study suggested that upregulation of miRNA-145 could inhibit the PI3K/AKT/mTOR signaling pathway and enhance autophagy in HK-2 cells [21]. In our case, the results showed that the expression miR-145 was significantly increased when 5H-SY5Y cells were exposed to lidocaine, and subsequently, the PI3K/AKT/mTOR signaling pathway was suppressed. However, when the miR-145 was silenced, the pathway was activated again. Similar to aforementioned study, our study also showed that the autophagy was enhanced when miR-145 was upregulated.
The autophagy is related to the formation and clearance of autophagosomes [10,28]. The formation of these structures is tremendously related to the cytosolic form of LC3 (LC3I), which is coupled to phosphatidylethanolamine and converted to LC3-phosphatidylethanolamine conjugate (LC3II) [29]. LC3II is located in the autophagosomal membrane. After the fusion of autophagosome and lysosome, autophagosomal ingredients such as LC3II are hydrolyzed. Therefore, the LC3 protein and the formation of LC3II is the major indicator of autophagy [30]. In our study, lidocaine resulted in an increase and decrease in the protein level of LC3II and LC3I, respectively, which correspondingly reached to the highest and the lowest expression in SH-SY5Y cells after treatment with 8 mM lidocaine. Similar results were obtained using fluorescence methods, which showed that the expression of LC3B increased with the increasing concentration of lidocaine. In addition to LC3, autophagy is regulated by a wide range of regulatory factors, such as Beclin-1, an important initiator of autophagy. Beclin-1 plays a key role in the recruitment of autophagic proteins to pre-autophagosomal structures, and also plays a critical role in the formation of core complexes, including Beclin-1, vacuolar protein sorting (Vps) 34 and Vps15 [31]. In the present study, it was found that lidocaine upregulated the protein expression of Beclin-1, which confirmed the importance of elevated Beclin-1 levels during lidocaine-induced autophagy [12]. Furthermore, p62 (also known as SQSTM1) is also a vital player for autophagosome formation and is selectively degraded in an autophagy-dependent mechanism [32, 33]. In our study, it showed that p62 levels decreased when treated with lidocaine, which the results were in accordance with the previous study from Xiong et al as well [12]. Our study also demonstrated that autophagy inhibition was achieved by transfecting cells with microR-145 small interfering RNA; knockdown in SH-SY5Y cells reduced autophagosome formation-related proteins.
In conclusion, lidocaine promoted autophagy by upregulation of miR-145, and miR-145 inhibited PI3K/AKT/mTOR signaling pathway through targeting AKT3. Manipulation of autophagy by upregulating miR145, might be an effective approach for preventing lidocaine-induced nerve damage.
Funding
None.
Conflict of Interest
None declared.
Reference
- 1. Brun A. Effect of procaine, carbocain and xylocaine on cutaneous muscle in rabbits and mice. Acta Anaesthesiol Scand 1959;3:59–73. [DOI] [PubMed] [Google Scholar]
- 2. Nouette-Gaulain K, Capdevila X, Rossignol R. Local anesthetic “in-situ” toxicity during peripheral nerve blocks: update on mechanisms and prevention. Curr Opin Anaesthesiol 2012;25:589–95. [DOI] [PubMed] [Google Scholar]
- 3. Hogan QH. Pathophysiology of peripheral nerve injury during regional anesthesia. Reg Anesth Pain Med 2008;33:435–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Selander D, Brattsand R, Lundborg G et al. Local anesthetics: importance of mode of application, concentration and adrenaline for the appearance of nerve lesions: an experimental study of axonal degeneration and barrier damage after intrafascicular injection or topical application of bupivacaine (marcah®). Acta Anaesthesiol Scand 1979;23:127–36. [DOI] [PubMed] [Google Scholar]
- 5. Lirk P, Haller I, Myers RR et al. Mitigation of direct neurotoxic effects of lidocaine and amitriptyline by inhibition of p38 mitogen-activated protein kinase in vitro and in vivo. Anesthesiology 2006;104:1266–73. [DOI] [PubMed] [Google Scholar]
- 6. Gordh T. Lidocaine: the origin of a modern local anesthetic. 1949. Anesthesiology 2010;113:1433–7. [DOI] [PubMed] [Google Scholar]
- 7. Wang D, Wu X, Li J et al. The effect of lidocaine on early postoperative cognitive dysfunction after coronary artery bypass surgery. Anesth Analg 2002;95:1134–41. [DOI] [PubMed] [Google Scholar]
- 8. Klinger RY, Cooter M, Berger M et al. Effect of intravenous lidocaine on the transcerebral inflammatory response during cardiac surgery: a randomized-controlled trial. Can J Anaesth 2016;63:1223–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Czaja MJ, Ding W-X, Donohue TM et al. Functions of autophagy in normal and diseased liver. Autophagy 2013;9:1131–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chen G, Ke Z, Xu M et al. Autophagy is a protective response to ethanol neurotoxicity. Autophagy 2012;8:1577–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wang WJ, Wang Y, Chen HZ et al. Orphan nuclear receptor TR3 acts in autophagic cell death via mitochondrial signaling pathway. Nat Chem Biol 2014;10:133–40. [DOI] [PubMed] [Google Scholar]
- 12. Xiong J, Kong Q, Dai L et al. Autophagy activated by tuberin/mTOR/p70S6K suppression is a protective mechanism against local anaesthetics neurotoxicity. J Cell Mol Med 2017;21:579–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Heras-Sandoval D, Pérez-Rojas JM, Hernández-Damiá J et al. The role of PI3K/AKT/mTOR pathway in the modulation of autophagy and the clearance of protein aggregates in neurodegeneration. Cell Signal 2014;26:2694–701. [DOI] [PubMed] [Google Scholar]
- 14. Wang S, Zhu M, Wang Q et al. Alpha-fetoprotein inhibits autophagy to promote malignant behaviour in hepatocellular carcinoma cells by activating PI3K/AKT/mTOR signalling. Cell Death Dis 2018;9:1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Krol J, Loedige I, Filipowicz W. The widespread regulation of microRNA biogenesis, function and decay. Nat Rev Genet 2010;11:597–610. [DOI] [PubMed] [Google Scholar]
- 16. Wu H, Wang F, Hu S et al. MiR-20a and miR-106b negatively regulate autophagy induced by leucine deprivation via suppression of ULK1 expression in C2C12 myoblasts. Cell Signal 2012;24:2179–86. [DOI] [PubMed] [Google Scholar]
- 17. Ju J-A, Huang C-T, Lan S-H et al. Characterization of a colorectal cancer migration and autophagy-related microRNA miR-338-5p and its target gene PIK3C3. Biomarkers Genomic Med 2013;5:74–8. [Google Scholar]
- 18. Zhu H, Wu H, Liu X et al. Regulation of autophagy by a beclin 1-targeted microRNA, miR-30a, in cancer cells. Autophagy 2009;5:816–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Korkmaz G, Le Sage C, Tekirdag KA et al. miR-376b controls starvation and mTOR inhibition-related autophagy by targeting ATG4C and BECN1. Autophagy 2012;8:165–76. [DOI] [PubMed] [Google Scholar]
- 20. Menghini R, Casagrande V, Marino A et al. MiR-216a: a link between endothelial dysfunction and autophagy. Cell Death Dis 2014;5:e1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Xiang J, Jiang T, Zhang W et al. Human umbilical cord-derived mesenchymal stem cells enhanced HK-2 cell autophagy through MicroRNA-145 by inhibiting the PI3K/AKT/mTOR signaling pathway. Exp Cell Res 2019;378:198–205. [DOI] [PubMed] [Google Scholar]
- 22. Sui H, Lou A, Li Z, Yang J. Lidocaine inhibits growth, migration and invasion of gastric carcinoma cells by up-regulation of microR-145. BMC Cancer 2019;19:233. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 23. Rancan L, Simón C, Marchal-Duval E et al. Lidocaine administration controls MicroRNAs alterations observed after lung ischemia-reperfusion injury. Anesth Analg 2016;123:1437–47. [DOI] [PubMed] [Google Scholar]
- 24. Saiki S, Sasazawa Y, Imamichi Y et al. Caffeine induces apoptosis by enhancement of autophagy via PI3K/Akt/mTOR/p70S6K inhibition. Autophagy 2011;7:176–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yang J, Pi C, Wang G. Inhibition of PI3K/Akt/mTOR pathway by apigenin induces apoptosis and autophagy in hepatocellular carcinoma cells. Biomed Pharmacother 2018;103:699–707. [DOI] [PubMed] [Google Scholar]
- 26. Le Beau MM, Lemons RS, Espinosa R et al. Interleukin-4 and interleukin-5 map to human chromosome 5 in a region encoding growth factors and receptors and are deleted in myeloid leukemias with a del(5q). Blood 1989;73:647–50. [PubMed] [Google Scholar]
- 27. Zhou J, Hu SE, Tan SH et al. Andrographolide sensitizes cisplatin-induced apoptosis via suppression of autophagosome-lysosome fusion in human cancer cells. Autophagy 2012;8:338–49. [DOI] [PubMed] [Google Scholar]
- 28. Yin Z, Pascual C, Klionsky DJ. Autophagy: machinery and regulation. Microb Cell 2016;3:588–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kang R, Zeh HJ, Lotze MT et al. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ 2011;18:571–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Roger S, Rollin J, Barascu A et al. Voltage-gated sodium channels potentiate the invasive capacities of human non-small-cell lung cancer cell lines. Int J Biochem Cell Biol 2007;39:774–86. [DOI] [PubMed] [Google Scholar]
- 31. Gao R, Shen Y, Cai J et al. Expression of voltage-gated sodium channel alpha subunit in human ovarian cancer. Oncol Rep 2010;23:1293–9. [DOI] [PubMed] [Google Scholar]
- 32. Haidar M, Asselbergh B, Adriaenssens E et al. Neuropathy-causing mutations in HSPB1 impair autophagy by disturbing the formation of SQSTM1/p62 bodies. Autophagy 2019;15:1051–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Young MM, Takahashi Y, Khan O et al. Autophagosomal membrane serves as platform for intracellular death-inducing signaling complex (iDISC)-mediated caspase-8 activation and apoptosis. J Biol Chem 2012;15:12455–68. [DOI] [PMC free article] [PubMed] [Google Scholar]