

ABSTRACT
Background
The mechanistic target of rapamycin complex 1 (mTORC1) is a nutrient-sensing pathway and a key regulator of amino acid and glucose metabolism. Dysregulation of the mTOR pathways is implicated in the pathogenesis of metabolic syndrome, obesity, type 2 diabetes, and pancreatic cancer.
Objectives
We investigated the impact of inhibition of mTORC1/mTORC2 and synergism with metformin on pancreatic tumor growth and metabolomics.
Methods
Cell lines derived from pancreatic tumors of the KPC (KrasG12D/+; p53R172H/+; Pdx1-Cre) transgenic mice model were implanted into the pancreas of C57BL/6 albino mice (n = 10/group). Two weeks later, the mice were injected intraperitoneally with daily doses of 1) Torin 2 (mTORC1/mTORC2 inhibitor) at a high concentration (TH), 2) Torin 2 at a low concentration (TL), 3) metformin at a low concentration (ML), 4) a combination of Torin 2 and metformin at low concentrations (TLML), or 5) DMSO vehicle (control) for 12 d. Tissues and blood samples were collected for targeted xenometabolomics analysis, drug concentration, and cell signaling.
Results
Metabolomic analysis of the control and treated plasma samples showed differential metabolite profiles. Phenylalanine was significantly elevated in the TLML group compared with the control (+426%, P = 0.0004), whereas uracil was significantly lower (–38%, P = 0.009). The combination treatment reduced tumor growth in the orthotopic mouse model. TLML significantly decreased pancreatic tumor volume (498 ± 104 mm3; 37%; P < 0.0004) compared with control (1326 ± 134 mm3; 100%), ML (853 ± 67 mm3; 64%), TL (745 ± 167 mm3; 54%), and TH (665 ± 182 mm3; 50%) (ANOVA and post hoc tests). TLML significantly decreased tumor weights (0.66 ± 0.08 g; 52%) compared with the control (1.28 ± 0.19 g; 100%) (P < 0.002).
Conclusions
The combination of mTOR dual inhibition by Torin 2 and metformin is associated with an altered metabolomic profile and a significant reduction in pancreatic tumor burden compared with single-agent therapy, and it is better tolerated.
Keywords: metabolomics, phenylalanine, KPC mouse model, pancreatic cancer, glycolysis, tricarboxylic acid cycle, xenometabolomics, antidiabetic drug, metformin
We investigated a nutrient-sensing pathway, mTORC1/mTORC2, inhibition, and synergism with metformin on pancreatic tumor metabolism. Combined treatment showed a differential metabolomics profile and a reduction in pancreatic tumor size and weight.
Introduction
The mechanistic target of rapamycin (mTOR) is a protein kinase that tightly controls nutrient metabolism and cell proliferation. mTOR is a highly conserved, 289-kDa serine/threonine kinase and a master regulator of cell growth and energy metabolism (1–3). mTOR integrates inputs from nutrients, energy levels, and growth signals and coordinates anabolic cell growth, nutrient metabolism, and inhibits catabolic autophagy (4). As such, mTOR regulates protein, lipid, and nucleotide synthesis, and it inhibits catabolism in normal cell and cancer metabolism.
Pancreatic cancer is the third leading cause of cancer-related death in the United States. This outcome is attributable to its late diagnosis and lack of response to current therapies. Furthermore, recent studies have linked both type 2 diabetes and new-onset diabetes to pancreatic cancer as either risk factors or symptoms. Alarmingly, the 5-y survival rate of pancreatic cancer patients was a dismal 8.5% in the United States in 2019, with an incidence (56,770 cases/y) nearly equal to the mortality (45,750 cases/y) (5). Thus, there is an urgent need to develop new strategies for diagnosis and treatment. Rapamycin, a prototype allosteric mTOR inhibitor, acutely inhibits mTOR complex 1 (mTORC1). Chronic administration of rapamycin may inhibit mTORC2 in some cell types (2). However, ATP-competitive mTOR kinase inhibitors block both mTORC1 and mTORC2 complexes. Although the mTOR inhibitors have pharmacological applications, the fundamental value of mTOR chemical knockdown is gaining insight into the mechanistic regulation of nutrient anabolism and catabolism. We hacked the mTOR pathways with drugs to determine their functionality, monitor the inner working of the mTORC1/mTORC2 signaling network, and advance the disease therapeutics. Several lines of evidence have revealed that the mTOR network regulates energy and nutrient sensing, cell growth and proliferation, and cancer metabolism via both transcriptional mechanisms and posttranslational modifications (1, 6–10).
mTOR is a druggable protein that is dysregulated in a multitude of chronic diseases, including type 2 diabetes and pancreatic cancer, and therefore is a promising target for diabetes and pancreatic cancer interventions. mTOR has also been identified as a driver of stem cell growth and pancreatic progenitor cell differentiation (11–13).
In addition, the antidiabetic drug metformin inhibits mTORC1 indirectly by activating AMP-activated protein kinase (AMPK) (14) and has emerged as a potential therapeutic target for the treatment of several types of cancer, including pancreatic ductal adenocarcinoma (PDAC) (15–18). Thus, mTOR is an attractive actionable gene due to its central role in cellular metabolism, pancreatic progenitor cell growth, and tumor metabolism. By nucleating 2 functionally distinct complexes, namely mTORC1 and mTORC2, mTOR plays a central role in pancreatic progenitor cell growth and proliferation and tumor metabolism.
The tumor microenvironment is not a static entity but, rather, functions as a sophisticated network communicating between tumor cells, stroma, immune cells, soluble cytokines, proteases, and components of the extracellular matrix. These complex interactions are critical drivers of tumor growth advantage, differentiation of multipotent stem cells, cancer metabolism and progression, and acquired treatment resistance (19). Mounting evidence has linked glycolytic metabolic disturbances to adverse pancreatic cancer prognosis. The tumor microenvironment can influence the tumor immune-editing process (20) and tumor heterogeneity (21) and thus may lead to therapeutic resistance (22). In this context, PDAC is considered one of the most stroma-rich tumors. Therefore, factors that influence the tumor microenvironment are attractive targets for pancreatic cancer early detection, control, and intervention (23, 24). Moreover, it has been reported that the tumor microenvironment mediates mTOR-induced tumor resistance to therapy (22).
In this study, we surgically implanted pancreatic cancer cells derived from pancreatic ductal adenocarcinoma of genetically engineered triple-mutant [KrasG12D/+, p53R172H/+, Pdx1-Cre (KPC)] transgenic mice in the pancreas of C57BL/6 albino mice. This orthotopic mouse model provides a physiologically relevant model that emulates human PDAC in the milieu of an intact tumor microenvironment in vivo. This model allowed us to, directly and indirectly, investigate the role of mTOR signaling and the impact of mTOR inhibition on cellular transformation during the progression from premalignancy to PDAC. In addition, because single-agent regimens do not work in pancreatic cancer (23), this study addressed the possible synergistic effects of mTORC1/mTORC2 inhibition by Torin 2 and interactions with metformin. We hypothesized that a combination of an mTOR inhibitor and pharmacological AMPK activator might act synergistically on different targets to overcome cancer cell heterogeneity, thereby reducing drug resistance, which is a significant obstacle in pancreatic cancer therapy. Thus, in this study, we evaluated the synergistic effects of mTOR inhibition and metformin activity on pancreatic cancer tumor size and volume and the associated metabolomics changes.
Methods
Reagents
Torin 2 (9-(6-aminopyridine-3-yl)-1-(3-trifluromethyl)-phenyl)benzos[h] [1,6]naphthyridin-2 (1H) (catalog no. 4248) was obtained from Tocris Bioscience (R & D Systems). Metformin hydrochloride (N,N-dimethyllimidodicarbonimidic diamide hydrochloride) (Tocris; catalog no. 2864) and rapamycin were obtained from Cell Signaling (catalog no. 9904). Other chemicals were obtained from either Sigma or Fisher Scientific. Immobilon-P polyvinylidene difluoride membrane (0.45 µm) and the reagents for enhanced chemiluminescence (ECL) were obtained from Millipore (Immobilon Western chemiluminescent horseradish peroxidase), as we previously reported (25). HPLC-grade methanol, acetonitrile, ammonium acetate, acetic acid, propylene glycol, and PBS (1×) were obtained from Fisher Scientific, as previously described (26). Isoflurane was obtained from Halocarbon Product Corporation.
Antibodies
Antibodies against the following proteins were purchased from Cell Signaling: total mTOR (catalog no. 2983), serine P-2481 mTOR (catalog no. 2976), S6 (catalog no. 2217), serine 235/236 phospho-S6 ribosomal protein (catalog no. 2211), total Akt (catalog no. 4691), serine P-473 Akt (catalog no. 4060), as we previously reported (25). The actin antibody was obtained from Sigma (catalog no. A2103). Sheep anti-rabbit secondary antibodies were obtained from GE Health Care Bioscience.
Orthotopic mouse model
We used an orthotopic mouse model of pancreatic cancer, which allowed for tumor growth at the natural organ site of the primary tumor and within an intact tumor microenvironment. Pancreatic tumors were harvested after 25 wk under sterile conditions and cultured in DMEM supplemented with 20% FBS, penicillin–streptomycin, and antimycotics. The tumors were cut into small pieces, washed in PBS, and placed in 10-cm dishes in DMEM. Once cells reached 80% confluence, tumor pieces were removed, and cells were trypsinized and seeded at very low density to minimize fibroblast contamination. KPC cell lines were confirmed with sequencing following PCR amplification. The KPC cell lines were characterized and their tumorigenic potential was tested after 40 passages, as previously described (27, 28).
We surgically implanted pancreatic cancer cell lines derived from pancreatic ductal adenocarcinoma of triple-mutant (KPC) genetically engineered mice in the gastric lobe of the pancreas of C57BL/6 albino mice. The abdomen of the mice was opened by a 1-cm incision in the upper medial abdomen. The transplanted KPC adenocarcinoma cell lines (5 × 103 cells) in 30 μL PBS were injected in the gastric lobe of the pancreas. The abdomen was closed by using a 2-layer suture with 5–0 Vicryl absorbable suture material and Ethilon sutures. Tumor growth was monitored by palpitation by calipers. After 2 wk, the pancreatic tumors were palpable. At that time, the albino mice were injected intraperitoneally daily with DMSO, Torin 2 (mTORC1/mTORC2 inhibitor) at a low concentration (5 mg/kg) or a high concentration (25 mg/kg), metformin (10 mg/kg), or a combination of Torin 2 (5 mg/kg) and metformin (10 mg/kg). Mice were sacrificed, and pancreatic tumors were dissected, measured, and weighed. Tissues were harvested, and blood samples were collected to conduct the metabolomics, drug tissue distribution, and cell signaling studies. The Institutional Animal Care and Use Committee (IACUC) at the University of Nebraska Medical Center approved the protocol for the orthotopic mouse model (no. 15-101-12) and the animal protocol for breeding colonies (no. 13-011-04EP). The procedures followed the preapproval protocols, and the number of animals used and euthanized conformed to the numbers approved by IACUC. For this study, the power calculations were as follows: the sample size of n = 50 mice (10/group × 5 groups) achieves 80% power to detect a moderate effect size of 0.3 using 1-way ANOVA with a significance level of 0.05. In addition, 10% extra animals were added to account for surgical error and inevitable losses (n = 10).
Pancreatic tissue lysis and immunoblotting
Pancreatic tissues were weighed, snap-frozen in liquid nitrogen, and stored at –80°C until the time of analysis. The details of the immunoblot analysis have been reported previously (25, 29, 30). Briefly, the tissues were washed twice with ice-cold PBS (pH 7.4) and collected in ice-cold lysis Buffer A containing 10 mM KPO4, 1 mM EDTA, 5 mM EGTA, 10 mM MgCl2, 50 mM β-glycerophosphate, 1 mM sodium orthovanadate (Na3VO4), 5 µg/mL pepstatin A, 10 µg/mL leupeptin, and 40 µg/mL PMSF. Cells were lysed in the presence of the detergent Nonidet P-40. Cell lysates were centrifuged at 13,200 × g for 5 min at 4°C, and the supernatants were collected. Protein concentration was measured with a Bradford assay to normalize the concentration for immunoblotting, as we described previously (25, 29, 30). Briefly, samples were heated at 95°C for 5 min and electrophoresed in SDS-PAGE gels to resolve protein bands according to the molecular weight. The protein bands were transferred to polyvinylidene difluoride membranes in Tobin buffer (24 mM Tris, 192 mM glycine, 10% methanol, and 0.02% SDS). Western blotting was performed by blocking the membranes in Tris-buffered saline with Tween 20 [TBST; 40 mM Tris HCl (pH 7.5), 0.9% NaCl, and 0.1% Tween 20] containing 3% nonfat milk. The membranes were then incubated in TBST with 2% BSA containing either the primary or the secondary horseradish peroxidase-conjugated antibodies. The blots were developed via ECL.
Targeted metabolomics analysis using LC-MS/MS
Metabolites from frozen serum were extracted using 1.5 mL chilled methanol:chloroform:water at a ratio of 8:1:1, and 13C-labeled glycolysis and tricarboxylic acid (TCA) cycle standards were utilized. Following extraction, the samples were sonicated on ice (20% duty cycle and 20% maximum power for 20 s). Briefly, polar metabolites were extracted and analyzed using LC-MS/MS, as described previously (31). The selected reaction monitoring LC-MS/MS method was employed using positive/negative ion polarity switching on a Xevo Tandem Quadrupole-TQ-S mass spectrometer (32). The peak chromatographic areas were integrated using MassLynx 4.1 software (Water) and normalized to the corresponding tissue protein concentration. The resultant peaks were identified and quantified using MetaboAnalyst 3 (http://www.metaboanalyst.ca) (33).
Quantification and distribution of Torin 2 and metformin in plasma and tissues
Mouse tissues were snap-frozen in liquid nitrogen and stored at –80°C until the time of analysis. Plasma samples were separated from RBCs via centrifugation at 15,000 × g for 5 min at 4°C within 1 h of sample collection and stored at –80°C until analysis. Torin 2 and metformin quantification was performed using LC-MS/MS. For plasma samples, 50 µL of plasma was mixed with 1 mL ice-cold acetonitrile and spiked with 10 µL of internal standard (IS; maraviroc at a final concentration of 25 ng/mL for Torin 2 and indinavir at a final concentration of 50 ng/mL for metformin). Samples were then vortexed for 3 min and centrifuged at 17,000 × g for 10 min at 4°C. Supernatants were dried, reconstituted in 100 µL 50% methanol in H2O, and injected for LC-MS/MS analysis. For tissue samples, ∼100 mg of tissues was homogenized in 4 volumes of 90% methanol in H2O. The homogenates (100 µL) were mixed with 90 µL methanol containing IS and 10 µL of 50% methanol in H2O containing no IS. Samples were then vortexed for 3 min and centrifuged at 17,000 × g for 10 min. Then, 100 µL of supernatants was collected and mixed with 50 µL H2O before LC-MS/MS analysis. Plasma and tissue standards were prepared at a final concentration of 0.5–1000 ng/mL.
For LC-MS/MS analysis, 10 µL of the samples prepared above was injected onto an LC-MS/MS Waters Acquity UPLC system coupled to an Applied Biosystems 4000 Q TRAP quadrupole linear ion trap hybrid mass spectrometer (Applied Biosystems, MDS Sciex). (26, 34). Chromatographic separation was performed using a phenyl column for Torin 2 and HILIC column for metformin (2.1 × 100 mm, 1.7 µm; Waters). Mobile phase A consisted of 7.5 mM ammonium bicarbonate (pH 7), and mobile phase B was methanol. The flow rate was 0.25 mL/min. For Torin 2 separation, the initial mobile phase composition was 60% B for the first 4.75 min, increased to 95% B over 0.1 min, and then held constant for 1.05 min. Mobile phase B was then reset to 60% over 0.1 min, and the column was equilibrated for 1 min before the next injection. For metformin separation, the initial mobile phase composition was 50% B for the first 4.25 min, increased to 95% B over 0.25 min, and then held constant for 1 min. Mobile phase B was then reset to 50% over 0.25 min, and the column was equilibrated for 1 min before the next injection. The mass spectrometer was operated in the positive ion mode using multiple reaction monitoring. The following transitions were monitored: m/z 433.2 → 373.2 for Torin 2, m/z 130 → 71.1 for metformin, m/z 514.2 → 280 for the IS maraviroc, and m/z 614.4 → 421.2 for the IS indinavir (26, 34).
Data processing and statistical analysis
Metabolites were identified based on retention time, which is based on the solubility of the analyte in the stationary phase and m/z spectrum match to injections of known standards. The data are expressed as the mean ± SE. ANOVA was performed using SPSS software (IBM) to determine whether the treatment groups differed with respect to a given outcome compared with the control group. If the overall ANOVA was significant, then a secondary post hoc statistical test was employed to identify the pairs of groups that differed. Statistical significance was assigned as P < 0.05 if an overall statistical significance was detected between groups.
Results
mTORC1/mTORC2 inhibitor (Torin 2) and metformin exhibit a combinatorial effect on pancreatic tumor growth
After 12 d of daily metformin and Torin 2 injections, Torin 2 at low and high concentrations led to a significant decrease in pancreatic tumor weight and volume, and a combination of the drug with metformin augmented these effects (Figure 1, Table 1). However, a high dose of Torin 2 was associated with lower survival compared with the control, a low dose of Torin 2, and the combination of Torin 2 and metformin. Only 3 of 10 mice/group tolerated the high dose of Torin 2 compared with 8 mice on the TLML treatment, 6 mice on metformin, 5 mice on TL, and the control group (18).
FIGURE 1.
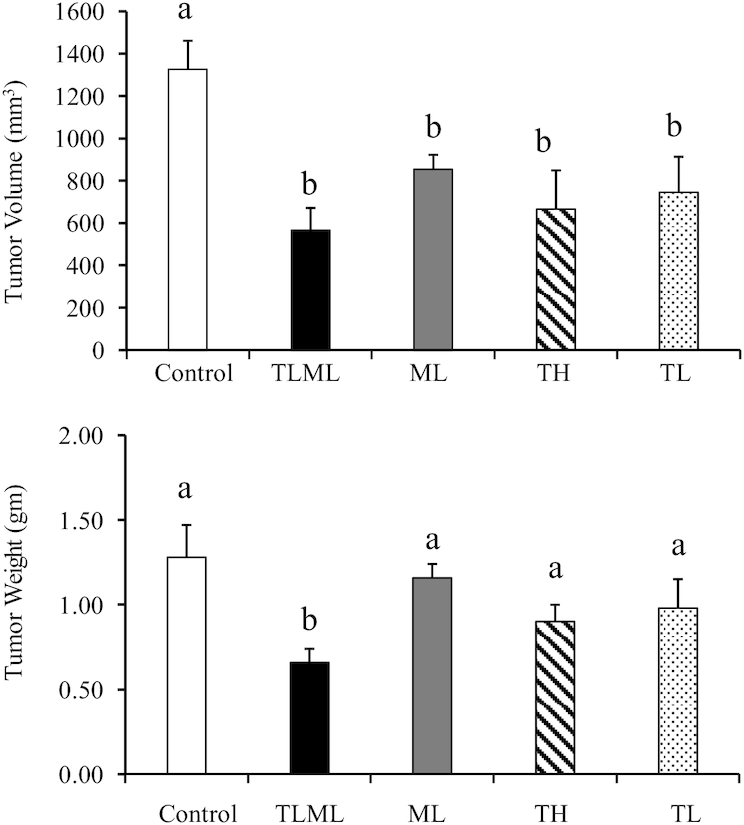
mTORC1/mTORC2 inhibitor (Torin 2) and metformin exhibit a combinatorial effect on pancreatic cancer tumor size and volume in the orthotopic mouse model. C57BL/6 albino mice were implanted with KPC pancreatic cancer cells and injected intraperitoneally daily with Torin 2 at a high dose, Torin 2 at a low dose, metformin at a low dose, both Torin 2 and metformin at low doses, or DMSO control for 12 d. Mice were killed, and the pancreas was collected, weighed, and measured. The values are means ± SDs (n = 3–7). Means with different letters were significantly different: a > b (P < 0.02). ANOVA was performed using SPSS software (IBM) to determine whether the treatment groups differed with respect to a given outcome compared with the control group. A post hoc test was employed to identify the pairs of groups that differed. Statistical significance was assigned as P < 0.05 if an overall statistical difference was detected between groups. ML, metformin at low dose; mTORC, mechanistic target of rapamycin complex; TH, Torin 2 at high dose; TL, Torin 2 at low dose; TLML, Torin 2 and metformin at low doses.
TABLE 1.
mTORC1/mTORC2 inhibitor (Torin 2) and metformin exhibit a combinatorial effect on pancreatic cancer tumor size and volume in the orthotopic mouse model1
| Survival (n = x/10) | |||||
|---|---|---|---|---|---|
| Control (n = 5/10) | TLML (n = 8/10) | ML (n = 6/10) | TH (n = 3/10) | TL (n = 5/10) | |
| Tumor volume, mm3 | 1326.50 ± 134.4 | 498.05 ± 104.8 | 853.97 ± 67.8 | 665.62 ± 182.6 | 745.40 ± 167.5 |
| ANOVA | P < 0.001 | ||||
| Post hoc test | — | P < 0.0004 | P < 0.004 | P < 0.01 | P < 0.01 |
| Tumor weight, g | 1.28 ± 0.19 | 0.66 ± 0.08 | 1.16 ± 0.08 | 0.90 ± 0.10 | 0.98 ± 0.17 |
| ANOVA | P < 0.01 | ||||
| Post hoc test | — | P < 0.002 | NS | P = 0.08 | NS |
The C57/BL6 mice were intrapancreatically implanted with KPC cells and developed palpable pancreatic cancer. The mice were then injected intraperitoneally with Torin 2 (mTORC1/mTORC2 inhibitor) high concentration, Torin 2 low concentration, metformin low concentration, combination of Torin 2 and metformin low concentrations, or DMSO vehicle (control). The mice were injected with daily doses for 12 d, after which they were killed and tumor weight and volumes were recorded. ML, metformin at low dose; mTORC, mechanistic target of rapamycin complex; NS, not significant; TH, Torin 2 at high dose; TL, Torin 2 at low dose; TLML, Torin 2 and metformin at low doses.
The TLML combination significantly decreased tumor volume (498 ± 104 mm3; 37%; P < 0.0004) compared with the control group (1326 ± 134; 100%), ML (853 ± 67; 64%), TL (745 ± 167; 54%) and TH (665 ± 182; 50%) (ANOVA and post hoc protected t tests). However, a high concentration of Torin 2 was toxic to the animals (only 3 of 10 mice survived). In addition, TLML significantly decreased (0.66 ± 0.08 g; 52%) tumor weights compared with the control (1.28 ± 0.19; 100%) (P < 0.002). Torin 2 synergistically acted with metformin and led to a significant decrease in pancreatic cancer tumor size and volume (Figure 1, Table 1) compared with either agent alone. All treatment groups were significantly different from the DMSO control.
mTORC1/mTORC2 inhibition modulates cell signaling
Torin 2 (TH and TL) and the combination of Torin 2 and metformin (TLML) inhibited mTORC1 signaling based on analysis of the phosphorylation of the mTORC1 downstream target ribosomal protein S6. In contrast, metformin (ML) alone did not affect the phosphorylation state (Figure 2). In addition, Torin 2 inhibited mTORC2, as shown by the level of Akt phosphorylation at serine 473, which is selectively phosphorylated and activated by mTORC2 (Figure 2). mTORC2 also activates other members of the AGC kinase family, such as glucocorticoid inducible kinase 1 and protein kinase C. We also quantified the concentrations of Torin 2 in blood and tissue byLC-MS/MS.
FIGURE 2.
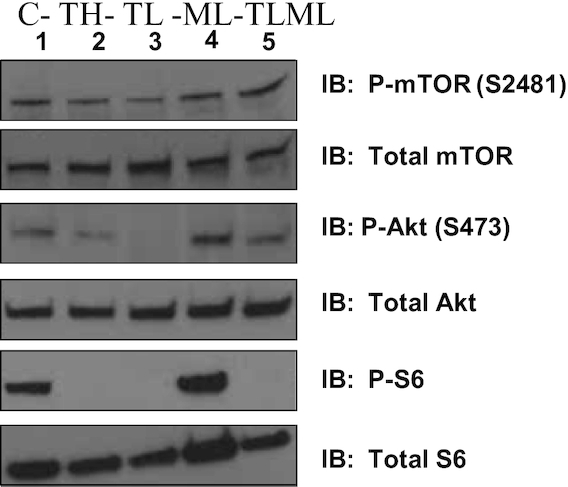
mTORC1/mTORC2 inhibition decreases the phosphorylation of downstream targets. C57BL/6 albino mice were implanted with KPC pancreatic cancer cells and injected intraperitoneally daily with Torin 2 at a high dose, Torin 2 at a low dose, metformin at a low dose, both Torin 2 and metformin at low doses (TLML), or DMSO control for 12 d. Mice were killed, and the pancreas was collected, flash-frozen in liquid nitrogen, and stored at –80°C until analysis. Whole-cell lysates were harvested and immunoblotted with total mTOR, P-mTOR Ser2481, total Akt, P-Akt-Ser473, total S6, P-S6, or β-actin antibody. C, control; ML, metformin at low dose; mTORC, mechanistic target of rapamycin complex; TH, Torin 2 at high dose; TL, Torin 2 at low dose; TLML, Torin 2 and metformin at low doses.
Dual mTORC1/mTORC2 inhibition decreases concentrations of glycolysis and TCA cycle intermediates
A heat map of the metabolites was constructed based on the relative metabolite analysis (Figure 3). Metabolomics analysis of the control and treated plasma samples (Torin 2 and metformin) showed significantly different metabolite profiles compared with the control group. The concentrations of both blood glucose and lactate decreased significantly upon treatment with Torin 2 and combined treatment with Torin 2 and metformin (Figure 4).
FIGURE 3.

The heat map and PLS-DA analysis show that the control and the mTORC1/mTORC2-treated mice have distinctive metabolomic profiles. An orthotopic pancreatic cancer mouse model was generated from KPC cells. The mice were injected intraperitoneally daily with Torin 2, metformin, both, or DMSO control for 12 d. Plasma was collected, and targeted metabolomics was performed using LC-MS/MS. Comparison of the control and TLML metabolite distinction between the 2 groups. (A) Comparison of the control and treated orthotopic KPC tumor metabolomes via principal component analysis. (B) Heat map analysis showing differences in the TCA cycle and glycolysis metabolites between the control group and the TLML group. mTORC, mechanistic target of rapamycin complex; PLS-DA, partial least square-discriminant analysis; TCA, tricarboxylic acid; TLML, Torin 2 and metformin at low doses.
FIGURE 4.

Dual mTORC1/mTORC2 inhibition decreases concentrations of glycolysis and TCA cycle intermediates: glycolysis and TCA intermediate concentrations in an orthotopic pancreatic cancer mouse model after Torin 2 and metformin treatment. C57BL/6 albino mice were implanted with KPC pancreatic cancer cells and injected intraperitoneally daily with Torin 2 at a high dose, Torin 2 at a low dose, metformin at a low dose, both Torin 2 and metformin at low doses, or DMSO control for 12 d. Mice were killed, plasma was collected, and targeted metabolomics was performed using LC-MS/MS. (A) Glucose, (B) 3-phosphoglycerate, (C) galactose, (D) α-ketoglutarate, (E) lactate;, and (F) citrate. ANOVA was performed using SPSS software (IBM) to determine whether the treatment groups differed with respect to a given outcome compared with the control group. If the overall ANOVA was significant, then a post hoc test was employed to identify the pairs of groups that differed. Statistical significance was assigned as P < 0.05 if an overall statistical difference was detected between groups. The values are means ± SEs (n = 3–7). Means with different letters were significantly different: a < b (P < 0.05). ML, metformin at low dose; mTORC, mechanistic target of rapamycin complex; TCA, tricarboxylic acid; TH, Torin 2 at high dose; TL, Torin 2 at low dose; TLML, Torin 2 and metformin at low doses.
The metabolite concentrations of some glycolysis intermediates, such as 3-phosphoglycerate and the TCA cycle intermediates citrate and α-ketoglutarate, were decreased after treatment with Torin 2 at a high concentration (TH), Torin 2 at a low concentration (TL), metformin (ML), and combined Torin 2 low/metformin low (TLML) compared with the DMSO control group, as shown in Figure 4. Thus, the dual mTOR inhibitor may synergize with metformin to decrease the electron donors NAD+ and FAD in the TCA cycle.
Dual mTORC1/mTORC2 inhibition and amino acid metabolism
We leveraged the use of the MetaboAnalyst platform and machine learning to link our experimental data to publicly available genomic and metabolic databases such as the global KEGG and BioCyc metabolic networks. This approach allowed us to transform the raw LC/MS data to a comprehensive and integrative global metabolomic analysis. As such, we were able to identify the metabolic pathways altered by the combinational inhibition of mTOR by ATP competitive catalytic inhibitor and metformin. These affected pathways included amino acids, nucleotides, and glucose metabolism (Figure 5A).
FIGURE 5.

(A) Combinatorial dual mTORC1/mTORC2 inhibition and metformin increases uracil, 5-thymidylates, and phenylalanine concentrations and decreases amounts of glucose and galactose in the circulation. C57BL/6 mice implanted with KPC pancreatic cancer cells were injected intraperitoneally with Torin 2 at a low dose, Torin 2 at a high dose, metformin at a low dose, Torin 2 and metformin at low doses, or control for 12 d. Mice were killed, plasma samples were collected, and targeted metabolomics was performed using LC-MS/MS. The values are means ± SEs (n = 3–7). Means with different letters were significantly different: a < b (P < 0.02). The data show that phenylalanine, lysine, glucose (hexose), and nucleotide (pyrimidine) metabolic pathways were significantly impacted by treatment compared with the control. (B) Dual mTORC1/mTORC2 inhibition increases phenylalanine concentrations in the circulation. Amino acids impacted by Torin 2 and metformin treatment in the orthotopic pancreatic cancer mouse model. C57BL/6 albino mice were implanted with KPC pancreatic cancer cells and injected intraperitoneally daily with Torin 2 at a low dose, Torin 2 at a high dose, metformin at a low dose, Torin 2 and metformin at low doses, or control for 12 d. Mice were killed, plasma samples were collected, and targeted metabolomics was performed using LC-MS/MS. The values are means ± SEs (n = 3–7). Means with different letters were significantly different: a < b (P < 0.02). ANOVA was performed using SPSS software (IBM) to determine whether the treatment groups differed with respect to a given outcome compared with the control group. A post hoc test was employed to identify the pairs of groups that differed. The data show that phenylalanine was increased in all treatment groups (TLML, ML, TL, and TH), whereas tryptophan, lysine, and threonine concentrations were significantly increased by Torin 2 (TL and TH) and asparagine was increased by ML compared with the control group. ML, metformin at low dose; mTORC, mechanistic target of rapamycin complex; TH, Torin 2 at high dose; TL, Torin 2 at low dose; TLML, Torin 2 and metformin at low doses.
Our data from the orthotopic pancreatic cancer mouse model also showed that treatment with Torin 2 (TL and TH) or with a combination of metformin and Torin 2 (TLML) significantly increased phenylalanine concentrations (4-fold) in the plasma of mice orthotopically implanted with pancreatic tumor cells derived from KPC mice (Figure 5). However, tyrosine, which is derived from phenylalanine, was not elevated; on the contrary, its concentration showed a decreasing trend that did not reach statistical significance (P = 0.07). This finding suggests that phenylalanine may be metabolized to its byproducts phenylpyruvate and phenylacetate or that it enters the TCA cycle via acetoacetate or fumarate. The finding that phenylalanine is elevated when both mTORC1 and mTORC2 are inhibited is novel in the current study; perturbation of nucleotide metabolism is a new finding as well. In addition, TL and TH increased the threonine concentration by 2-fold (P = 0.0002), tryptophan by 1.6-fold (P = 0.01), and lysine by 0.25-fold (P = 0.04). However, the TLML combination had no effect on these amino acids, whereas ML alone increased the amino acid asparagine.
The mTORC1/mTORC2 inhibitor and metformin combination facilitates tissue availability of the drugs
Torin 2 and metformin concentrations were quantified via LC-MS/MS in several tissues, including muscle, spleen, kidney, liver, lung, and plasma (Figure 6).
FIGURE 6.
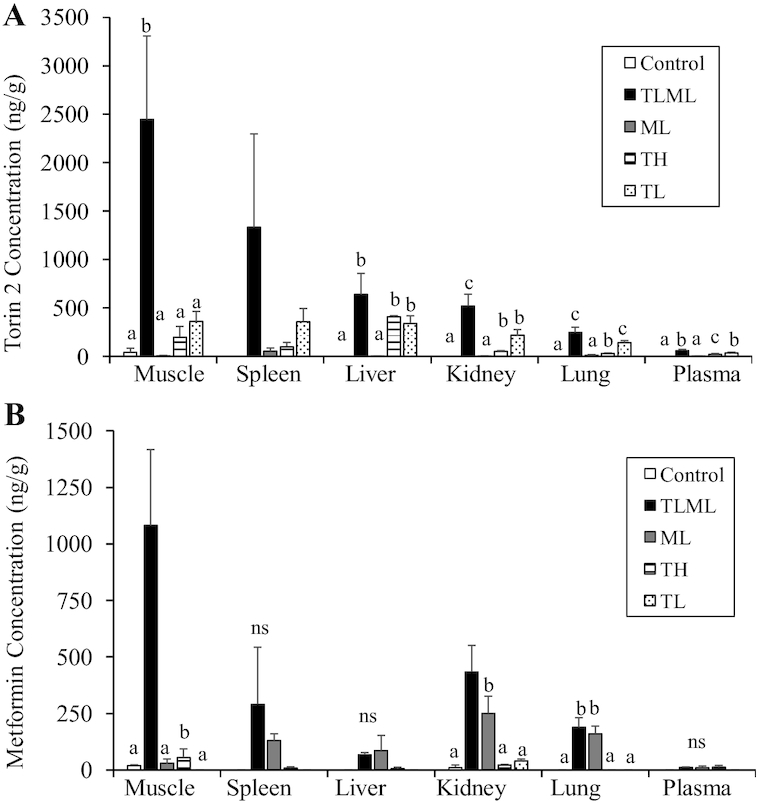
The mTORC1/mTORC2 inhibitor and metformin combination facilitates tissue availability of the drugs: Tissue distribution of Torin 2 (A) and metformin (B) in an orthotopic mouse model injected with mTORC1/2 inhibitor or metformin. KPC cells were implanted into the pancreas in C57/BL6 mice. Once tumors became palpable, the mice were injected intraperitoneally daily for 12 d with Torin 2 (mTORC1 + mTORC2 inhibitor) at a high concentration (TH); Torin 2 at a low concentration (TL), metformin at a low concentration (ML), a combination of Torin 2 and metformin at low concentrations (TLML), or DMSO vehicle (control). At the end of the 12 d, the mice were sacrificed, and tissues were harvested and flash-frozen. Torin 2 and metformin concentrations were analyzed and quantified using LC-MS/MS. The data are reported as the average ± SD. One animal in the low-dose Torin 2 treatment group had a very high concentration of Torin 2 (∼4-fold higher) in all tissues and was considered an outlier and was not included in the statistical analysis. Drug concentrations were measured using LC-MS/MS. ANOVA test was performed using SPSS software (IBM) to determine the overall differences between groups. A post hoc test was employed to identify the pairs of groups that differed. Statistical significance was assigned as P < 0.05 if an overall statistical difference was detected between groups. Means with different letters were significantly different. ML, metformin at low dose; mTORC, mechanistic target of rapamycin complex; ns, nonsignificant results; TH, Torin 2 at high dose; TL, Torin 2 at low dose; TLML, Torin 2 and metformin at low doses.
As shown in Figure 6, our data indicate that Torin 2 and metformin were differentially distributed in different organs. The muscles retained the highest concentrations of both drugs. The accumulation of Torin 2 in the muscle after treatment with a low concentration (TL) was 360 ± 104 ng/g tissue and that with Torin 2 administration at a high concentration (TH) was 197 ± 112 ng/g, whereas the combination of Torin 2-low and metformin-low (TLML) treatment significantly elevated Torin 2 accumulation (2447 ± 806 ng/g; P = 0.001). Because the tissue concentrations of both drugs in the combination TLML group are much higher than ML or TL alone, these data suggest a synergism between Torin 2 and metformin that is more than an additive effect. The data also indicate that a low concentration of Torin 2 is more effective than a high concentration, possibly due to the toxicity of the higher dose.
Similarly, when metformin was measured in the muscle, low metformin administration led to an accumulation of metformin of 29.9 ± 18.6 ng/g, and the combination of Torin 2 and metformin led to an accumulation of 1081 ± 335 ng/g. Taken together, these findings suggest a synergistic effect rather than an additive effect of the Torin 2 and metformin combination. A similar pattern was observed in the spleen. However, the difference did not reach the level of significance due to high variability in the amount of Torin 2 accumulation in the spleen (TLML: 1333.5 ± 936 ng/g).
Discussion
The mTOR pathway is critical for nutrient metabolism. It integrates nutrition status, energy level, and environmental cues to coordinate cellular metabolism and physiological homeostasis. We targeted the mTOR pathway by pharmacological inhibitors to determine the functionality of the mTORC1 and mTORC2 complexes in the disease state. Drugs that inhibit mTOR catalytic functions serve as tools to investigate the molecular mechanism of the protein functions and the metabolic outcome.
As an anabolic kinase, mTORC1 controls energy metabolism and metabolic integration while suppressing the catabolic autophagy. mTORC1 is activated by amino acids, glucose, and fat as a source of energy, and in turn increases protein and nucleotide synthesis, glycolysis, fatty acid synthesis, and esterification. mTORC1 also inhibits the catabolic lysosomal degradation of intracellular components and lysosomal biogenesis, known as autophagy. mTORC2 has been shown to inhibit insulin receptor substrate 1 ubiquitination and thus stabilize insulin receptor substrate and insulin signaling. Rictor, the mTORC2 exclusive partner, was shown to contribute to glucose and lipid homeostasis, as evidenced by conditional tissue-specific deletion of rictor in mouse models (35–37). Also, Sestrin 2 has been shown to regulate glucose metabolism via the mTORC2/Akt pathway (37, 38).
Intriguingly, the mTOR-mediated amino acid sensing in normal cells is also utilized by cancer cells to drive metabolic reprogramming. It is therefore critical to identify the metabolite readout of mTOR inhibition to provide insight into mTOR regulation of protein synthesis and metabolic integration.
mTORC1 regulates amino acid metabolism via several amino acid sensors. For example, leucine is sensed via Sestrins, GATOR2, and CASTOR sensors (39). Leucine recruits mTORC1 to the lysosomal surface for activation (40). CASTOR also senses arginine via GTP-RagA and GDP-RagC heterodimerization (41, 42). Furthermore, arginine is sensed via SLC38A9 lysosomal transporter, which mediates the efflux of arginine from the lysosome (43). Glutamine signaling is relayed via the Arf-1 rag-independent mechanism and drives the glutaminolysis pathway (44). Methionine is sensed via SAMTOR (S-adenosylmethionine sensor upstream of mTORC1), which is a GATOR1/KICSTOR-interacting protein. As such, S-adenosylmethoinine binds to SAMTOR and interrupts SAMTOR–GATOR1 interaction, which is a negative regulator of mTORC1. The knowledge gap regarding amino acid metabolism stems from 2 possibilities. First, it is not known whether the previously mentioned sensors are specific to 1 amino acid or can cross-talk and detect related amino acids. It is known that some amino acids are more potent in mTORC1 activation than others, so it is possible that these amino acid sensors are specific to individual amino acids. Second, it is not known whether amino acid sensing is tissue-specific, whether all amino acids are sensed by mTORC1, or whether there are mTORC1-independent pathways. Here, we report for the first time that phenylalanine (but not tyrosine) concentrations are significantly elevated when mTORC1 and mTORC2 are inhibited in the pancreas, suggesting that concentrations of phenylalanine and its metabolic channeling may be regulated by mTOR. This novel observation warrants further research to understand the basic physiology and biology and to identify the scientific bases for disease management.
Pancreatic cancer is the third leading cause of cancer death in the United States and has a high mortality rate despite current therapies. Thus, there is an urgent public health need to develop new strategies to combat this devastating disease. In this regard, the protein kinase mTOR is an attractive candidate due to its fundamental role in metabolism and its deregulation in pancreatic diseases, including diabetes and pancreatic cancer. We investigated the impact of inhibition of mTOR complexes by Torin 2, an ATP-competitive inhibitor that inhibits both mTORC1 and mTORC2 complexes. We also evaluated possible synergistic effects of Torin 2 and the antidiabetic drug metformin on targeted metabolomics and pancreatic tumor size and volume. We tested the hypothesis that inhibitors of mTOR complexes will synergize with AMPK activators to augment tumor responsiveness to treatments.
In this study, we orthotopically implanted pancreatic cancer cells derived from triple-mutant KPC mice. The KPC genetically engineered mouse is a model of PDAC, which undergoes spontaneous pancreatic cancer transformation. This unique KPC model activates a Kras knock-in allele (KrasG12D) and inactivates the tumor suppressor p53R172H/+, to conditionally target the pancreas via PDX1, which is required for pancreatic islet β-cell development, using the Cre–LoxP system. This Kras single point mutation is sufficient to initiate ductal premalignant pancreatic intraepithelial neoplasia (PanIN) transformation (24). The subsequent loss of p53 [Lox–Stop–Lox (LSL)–p53R172H/+] aids Kras in promoting tumor invasion in a time- and tissue-specific manner (45). The presence of the Cre–recombinase allows for the deletion of the LSL transcription termination sequence and thereby enables the expression of the oncogenic protein Kras. Thus, this KPC mouse model faithfully mimics pancreatic cancer in humans in terms of progression from intraepithelial neoplasia (premalignant) to malignant adenocarcinoma. These mice develop PanIN at 10 wk, progress to PanIN grade II and III by 25 wk, and advance to PDAC at 50 wk (46). Thus, the KPC transgenic mouse genetic model recapitulates the progression of PanIN to pancreatic cancer in humans.
Given that mTORC1/mTORC2 inhibitors were injected daily for 12 d, the significant changes in tumor weight and volume cannot be entirely explained by transient posttranslational mTOR kinase-mediated phosphorylation of downstream targets (Figure 2). Thus, these findings suggest additional long-term mTOR functions via the transcriptional and epigenetic machinery. Indeed, a possible role of mTOR in transcriptional regulation has been proposed (8, 47). Rohde and Cardenas (48) reported that in yeast, TOR regulates gene expression through acetylation in response to nutrients.
Taken together, our data obtained in an orthotopic pancreatic cancer mouse model indicate that mTOR inhibition has a favorable impact on pancreatic cancer outcome, evidenced by decreased pancreatic tumor weight (62% reduction in TLML compared with control) and tumor volume (49% reduction in TLML compared with control) (Figure 1) and the associated changes in glucose and energy metabolism (Figure 4). In humans, researchers conducted an untargeted metabolomics analysis in serum from patients and found 9 metabolites that might differentiate between PDAC and control (49, 50), including ceramide and phospholipids. Other investigators employed an NMR approach to identify a metabolite signature of pancreatic cancer (51). They reported elevated concentrations of triglycerides, leucine, isoleucine, and creatinine in patients with pancreatic cancer compared with the control group. However, these studies did not address the mechanistic underpinning of these observations. We reasoned that our results of changes in targeted metabolomics that were altered by mTOR inhibition could explain such findings. mTOR signaling could be an essential contributor to the development of pancreatic cancer. Thus, changes in mTOR signaling could serve as an early diagnostic biomarker and a therapeutic target.
Although insulin resistance, hyperinsulinemia, increased BMI, and type 2 diabetes are risk factors for pancreatic cancer and are linked to aberrant PI3K-Akt-mTOR signaling, whether they are causal targets of intervention remains unclear because it is difficult to distinguish cause and effect in observational studies. A recent Mendelian randomization study using genetic variants as an unconfounded proxy for exposure failed to show a causal association between type 2 diabetes and the development of pancreatic cancer (52)—a finding that refutes the results of observational studies (53). Instead, pancreatic cancer may lead to the development of type 2 diabetes, generating a reverse causality.
Metformin is a biguanide drug that is considered the first-line of treatment in type 2 diabetes, and it has been employed as an antineoplastic agent (54, 55). Metformin functions via several mechanisms. First, it inhibits complex 1 of the respiratory chain in the mitochondria, leading to uncoupling of oxidative phosphorylation. Second, it increases reactive oxygen species concentrations and reduces mitochondrial transmembrane potential, thereby hampering the self-renewal capacity of cancer stem cells (56, 57). Third, in diabetes-associated cancer, metformin decreases hyperlipidemia and hyperinsulinemia (58). Epidemiological studies have shown that treatment with metformin is associated with decreased cancer risk and/or improved survival. As a member of the biguanide family, metformin activates AMPK and decreases gluconeogenesis (59, 60). Metformin, which also indirectly inhibits mTOR via activation of AMPK as well as AMPK-independent mechanisms, has emerged as a potential therapeutic agent for the treatment of pancreatic cancer and diabetes-associated cancer. Intriguingly, studies conducted in xenograft models revealed that metformin decreases pancreatic cancer growth in a dose-dependent manner (61, 62). The impact of metformin on mTORC1 inhibition indirectly via AMPK activation is notably different from that of the rapamycin binding domain or the catalytic active-sites mTOR inhibitors (62). This observation suggests that metformin may augment the effects of mTOR inhibition by an additional mechanism. In our study, we found that metformin reduced tumor volume to a lesser extent than Torin 2 and that a combination of metformin and Torin 2 at low doses led to a synergistic reduction in tumor weight and volume (Figure 1).
The metabolomics data revealed that some glycolysis and TCA cycle intermediates were decreased with administration of Torin 2 at a high concentration (TH), Torin 2 at a low concentration (TL), and combined Torin 2 low/metformin low (TLML) treatment compared with the DMSO control group, as shown in Figure 4. These findings suggest decreased ATP production via the mitochondrial TCA cycle. We also noted that the lactate concentration was reduced in all treatment groups, indicating decreased ATP production via aerobic glycolysis (Warburg effect). Our results also showed that Torin 2, together with metformin, significantly altered phenylalanine, lysine, glucose (hexose), and nucleotide (pyrimidine) metabolism (Figure 5). It is possible that this metabolomic profile can be utilized for early detection, diagnosis, monitoring, and targeted therapy. As such, metabolomic profiling, which provides a snapshot of metabolic alterations, can be utilized as a metabolic laboratory test to improve clinical outcomes.
Our data suggest a synergistic interaction between the highly conserved mTOR pathway and the well-known antidiabetes drug metformin in pancreatic cancer. We found that muscles isolated from the orthotopic pancreatic cancer model mice in the combined TLML group accumulated Torin 2 and metformin (6-fold for Torin 2 and 36-fold for metformin) compared with accumulated concentrations in the TL or ML mice (Figure 6). Similarly, Wang and colleagues (63) reported that metformin acted synergistically with everolimus (a rapamycin analog) in inhibition of breast cancer growth and abrogation of S6 phosphorylation. In agreement with our data, other investigators reported that rapamycin synergized with metformin to inhibit pancreatic cancer growth in SW1990 pancreatic cancer cell lines and a xenograft mouse model of pancreatic cancer (16). Intriguingly, metformin, which activates the AMPK pathway, indirectly inhibits mTOR signaling and has been investigated as an antitumor agent (64).
In conclusion, our data indicate that mTORC1/mTORC2 inhibition by Torin 2 reduced glycolytic intermediate and TCA metabolite pools and increased phenylalanine concentrations in mice. mTOR inhibition by ATP competitive catalytic inhibitors also altered amino acid and nucleotide metabolism. This dual mTOR inhibitor may synergize with metformin to decrease the electron donors NAD+ and FAD in the TCA cycle, which may lead to reduced energy production. The combination of dual mTOR inhibition by Torin 2 and metformin treatment was associated with a significant reduction in pancreatic tumor size and weight, altered concentrations of glycolysis and TCA cycle intermediates compared with single-agent treatment, and was better tolerated in mice. Metabolomics profiling can be employed for risk assessment, early detection, monitoring, as well as personalized targeted therapy to improve clinical outcomes. The findings suggest that a combination of drugs may act synergistically to provide better tumor reduction and might be a better treatment approach than single-agent therapies.
ACKNOWLEDGEMENTS
We thank the Structural Biology Initiative of the Graduate Center/Advanced Science Research Center at City University of New York for providing laboratory research space for this work. The authors’ responsibilities were as follows—GS: designed the experiments, wrote the first draft of the manuscript, and participated in data collection, analysis, and interpretation; SKS: performed surgical implantation of the KPC cells in the pancreas of albino mice and helped with the mouse injections; GS, SKS, and AE: conducted the mouse drug injections, harvested the mouse tissues, and collected the mouse blood samples; VG: conducted the metabolomics analysis; SMS: helped with cell signaling and animal studies; NG: conducted drug distribution studies; GS, YA, and PKS: conceptualized the studies, interpreted the results, and revised the manuscript; and all authors: read and approved the final manuscript.
Notes
Funding provided by institutional grants from the Cancer Prevention and Control Pilot Grant, University of Nebraska Medical Center, COPH, to GS (principal investigator) and YA and PKS (co-investigators); City University of New York, Professional Staff Congress (PSC)-CUNY Research Award grant 61335-00-49 to GS; and City University of New York Advanced Science Research Center (CUNY ASRC) Seed Award grant 95649-00-01 to GS; . The research was also supported by the National Cancer Institute of the National Institutes of Health under award numbers R01CA163649 and R01CA216853 to PKS. The funders had no role in designing, implementing, analyzing, or interpreting the results.
Author disclosures: The authors report no conflicts of interest.
Abbreviations: AMPK, AMP-activated protein kinase; ECL, enhanced chemiluminescence; IACUC, Institutional Animal Care and Use Committee; IS, internal standard; KPC, KrasG12D/+, p53R172H/+, Pdx1-Cre; LSL, Lox–Stop–Lox; ML, metformin low concentration; mTOR, mechanistic target of rapamycin; mTORC, mechanistic target of rapamycin complex; PanIN, pancreatic intraepithelial neoplasia; PDAC, pancreatic ductal adenocarcinoma; TBST, Tris-buffered saline with Tween 20; TCA, tricarboxylic acid cycle; TH, Torin 2 high concentration; TL, Torin 2 low concentration; TLML, Torin low, metformin low.
Contributor Information
Ghada A Soliman, Email: ghada.soliman@sph.cuny.edu, Department of Environmental, Occupational, and Geospatial Health Sciences, CUNY Graduate School of Public Health and Health Policy, City University of New York, New York, NY, USA.
Surendra K Shukla, The Eppley Institute for Research in Cancer and Allied Diseases, University of Nebraska Medical Center, Omaha, NE, USA.
Asserewou Etekpo, Nebraska Department of Health, Lincoln, NE, USA.
Venugopal Gunda, The Eppley Institute for Research in Cancer and Allied Diseases, University of Nebraska Medical Center, Omaha, NE, USA.
Sharalyn M Steenson, Department of Health Promotion, College of Public Health, University of Nebraska Medical Center, Omaha, NE, USA.
Nagsen Gautam, Department of Pharmaceutical Sciences, College of Pharmacy, University of Nebraska Medical Center, Omaha, NE, USA.
Yazen Alnouti, Department of Pharmaceutical Sciences, College of Pharmacy, University of Nebraska Medical Center, Omaha, NE, USA.
Pankaj K Singh, The Eppley Institute for Research in Cancer and Allied Diseases, University of Nebraska Medical Center, Omaha, NE, USA.
References
- 1. Lien EC, Lyssiotis CA, Cantley LC. Metabolic reprogramming by the PI3K-Akt-mTOR pathway in cancer. Recent Results Cancer Res. 2016;207:39–72. [DOI] [PubMed] [Google Scholar]
- 2. Sabatini DM. Twenty-five years of mTOR: Uncovering the link from nutrients to growth. Proc Natl Acad Sci USA. 2017;114(45):11818–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sarbassov DD, Ali SM, Sabatini DM. Growing roles for the mTOR pathway. Curr Opin Cell Biol. 2005;17(6):596–603. [DOI] [PubMed] [Google Scholar]
- 4. Ben-Sahra I, Manning BD. mTORC1 signaling and the metabolic control of cell growth. Curr Opin Cell Biol. 2017;45:72–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. National Cancer Institute Cancer stat facts: pancreatic cancer. [Internet] 2019; [cited 2019 Sep 18]. Available from: https://seer.cancer.gov/statfacts/html/pancreas.html. [Google Scholar]
- 6. Martinez-Outschoorn UE, Peiris-Pages M, Pestell RG, Sotgia F, Lisanti MP. Cancer metabolism: a therapeutic perspective. Nat Rev Clin Oncol. 2017;14(1):11–31. [DOI] [PubMed] [Google Scholar]
- 7. Lamb R, Harrison H, Smith DL, Townsend PA, Jackson T, Ozsvari B, Martinez-Outschoorn UE, Pestell RG, Howell A, Lisanti MP et al. Targeting tumor-initiating cells: eliminating anabolic cancer stem cells with inhibitors of protein synthesis or by mimicking caloric restriction. Oncotarget. 2015;6(7):4585–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Vadla R, Haldar D. Mammalian target of rapamycin complex 2 (mTORC2) controls glycolytic gene expression by regulating histone H3 lysine 56 acetylation. Cell Cycle. 2018;17(1):110–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Masui K, Tanaka K, Akhavan D, Babic I, Gini B, Matsutani T, Iwanami A, Liu F, Villa GR, Gu Y et al. mTOR complex 2 controls glycolytic metabolism in glioblastoma through FoxO acetylation and upregulation of c-Myc. Cell Metab. 2013;18(5):726–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hall MN. mTOR: what does it do? Transplant Proc. 2008;40(10 Suppl):S5–8. [DOI] [PubMed] [Google Scholar]
- 11. Hobbs RM, Seandel M, Falciatori I, Rafii S, Pandolfi PP. Plzf regulates germline progenitor self-renewal by opposing mTORC1. Cell. 2010;142(3):468–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Elghazi L, Blandino-Rosano M, Alejandro E, Cras-Meneur C, Bernal-Mizrachi E. Role of nutrients and mTOR signaling in the regulation of pancreatic progenitors development. Mol Metab. 2017;6(6):560–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nardella C, Carracedo A, Alimonti A, Hobbs RM, Clohessy JG, Chen Z, Egia A, Fornari A, Fiorentino M, Loda M et al. Differential requirement of mTOR in postmitotic tissues and tumorigenesis. Sci Signal. 2009;2(55):ra2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ben Sahra I, Regazzetti C, Robert G, Laurent K, Le Marchand-Brustel Y, Auberger P, Tanti JF, Giorgetti-Peraldi S, Bost F. Metformin, independent of AMPK, induces mTOR inhibition and cell-cycle arrest through REDD1. Cancer Res. 2011;71(13):4366–72. [DOI] [PubMed] [Google Scholar]
- 15. Mallik R, Chowdhury TA. Metformin in cancer. Diabetes Res Clin Pract. 2018;143:409–19. [DOI] [PubMed] [Google Scholar]
- 16. Zhang JW, Zhao F, Sun Q. Metformin synergizes with rapamycin to inhibit the growth of pancreatic cancer in vitro and in vivo. Oncol Lett. 2018;15(2):1811–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang Y, Xu W, Yan Z, Zhao W, Mi J, Li J, Yan H. Metformin induces autophagy and G0/G1 phase cell cycle arrest in myeloma by targeting the AMPK/mTORC1 and mTORC2 pathways. J Exp Clin Cancer Res. 2018;37(1):63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Soliman GA, Shukla SK, Etekpo E, Gunda V, Gautam N, Alnouti Y, Singh PK. The impact of mTOR nutrient-sensing metabolic pathway and AMPK activator metformin on the growth of pancreatic cancer cells lines in C57/BL6 mice. American Society for Nutrition 2018 Annual Conference Proceedings, Abstract 511,2018;P10–112. [Google Scholar]
- 19. Haqq J, Howells LM, Garcea G, Metcalfe MS, Steward WP, Dennison AR. Pancreatic stellate cells and pancreas cancer: current perspectives and future strategies. Eur J Cancer. 2014;50(15):2570–82. [DOI] [PubMed] [Google Scholar]
- 20. Mittal D, Gubin MM, Schreiber RD, Smyth MJ. New insights into cancer immunoediting and its three component phases—elimination, equilibrium and escape. Curr Opin Immunol. 2014;27:16–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lou E, Subramanian S, Steer CJ. Pancreatic cancer: modulation of KRAS, microRNAs, and intercellular communication in the setting of tumor heterogeneity. Pancreas. 2013;42(8):1218–26. [DOI] [PubMed] [Google Scholar]
- 22. Weekes CD, Song D, Arcaroli J, Wilson LA, Rubio-Viqueira B, Cusatis G, Garrett-Mayer E, Messersmith WA, Winn RA, Hidalgo M. Stromal cell-derived factor 1α mediates resistance to mTOR-directed therapy in pancreatic cancer. Neoplasia. 2012;14(8):690–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Neesse A, Krug S, Gress TM, Tuveson DA, Michl P. Emerging concepts in pancreatic cancer medicine: targeting the tumor stroma. Onco Targets Ther. 2013;7:33–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Feig C, Gopinathan A, Neesse A, Chan DS, Cook N, Tuveson DA. The pancreas cancer microenvironment. Clin Cancer Res. 2012;18(16):4266–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Soliman GA, Steenson SM, Etekpo AH. Effects of metformin and a mammalian target of rapamycin (mTOR) ATP-competitive inhibitor on targeted metabolomics in pancreatic cancer cell line. Mol Biol. 2016;6(3):183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gautam N, Roy U, Balkundi S, Puligujja P, Guo D, Smith N, Liu XM, Lamberty B, Morsey B, Fox HS et al. Preclinical pharmacokinetics and tissue distribution of long-acting nanoformulated antiretroviral therapy. Antimicrob Agents Chemother. 2013;57(7):3110–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Shukla SK, Dasgupta A, Mulder SE, Singh PK. Molecular and physiological evaluation of pancreatic cancer-induced cachexia. Methods Mol Biol. 2019;1882:321–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Vernucci E, Abrego J, Gunda V, Shukla SK, Dasgupta A, Rai V, Chaika N, Buettner K, Illies A, Yu F et al. Metabolic alterations in pancreatic cancer progression. Cancers. 2019;12(1):2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Soliman GA, Acosta-Jaquez HA, Dunlop EA, Ekim B, Maj NE, Tee AR, Fingar DC. mTOR Ser-2481 autophosphorylation monitors mTORC-specific catalytic activity and clarifies rapamycin mechanism of action. J Biol Chem. 2010;285(11):7866–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Soliman GA, Acosta-Jaquez HA, Fingar DC. mTORC1 inhibition via rapamycin promotes triacylglycerol lipolysis and release of free fatty acids in 3T3-L1 adipocytes. Lipids. 2010;45(12):1089–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shukla SK, Purohit V, Mehla K, Gunda V, Chaika NV, Vernucci E, King RJ, Abrego J, Goode GD, Dasgupta A et al. MUC1 and HIF-1α signaling crosstalk induces anabolic glucose metabolism to impart gemcitabine resistance to pancreatic cancer. Cancer Cell. 2017;32(3):392. [DOI] [PubMed] [Google Scholar]
- 32. Gunda V, Yu F, Singh PK. Validation of metabolic alterations in microscale cell culture lysates using hydrophilic interaction liquid chromatography (HILIC)–tandem mass spectrometry-based metabolomics. PLoS One. 2016;11(4):e0154416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Xia J, Wishart DS. Using MetaboAnalyst 3.0 for comprehensive metabolomics data analysis. Curr Protoc Bioinformatics. 2016;55:14.10.191. [DOI] [PubMed] [Google Scholar]
- 34. Huang J, Gautam N, Bathena SP, Roy U, McMillan J, Gendelman HE, Alnouti Y. UPLC-MS/MS quantification of nanoformulated ritonavir, indinavir, atazanavir, and efavirenz in mouse serum and tissues. J Chromatogr B. 2011;879(23):2332–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kumar A, Harris TE, Keller SR, Choi KM, Magnuson MA, Lawrence JC Jr. Muscle-specific deletion of rictor impairs insulin-stimulated glucose transport and enhances basal glycogen synthase activity. Mol Cell Biol. 2008;28(1):61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kumar A, Lawrence JC Jr, Jung DY, Ko HJ, Keller SR, Kim JK, Magnuson MA, Harris TE. Fat cell-specific ablation of rictor in mice impairs insulin-regulated fat cell and whole-body glucose and lipid metabolism. Diabetes. 2010;59(6):1397–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hagiwara A, Cornu M, Cybulski N, Polak P, Betz C, Trapani F, Terracciano L, Heim MH, Ruegg MA, Hall MN. Hepatic mTORC2 activates glycolysis and lipogenesis through Akt, glucokinase, and SREBP1c. Cell Metab. 2012;15(5):725–38. [DOI] [PubMed] [Google Scholar]
- 38. Tao R, Xiong X, Liangpunsakul S, Dong XC. Sestrin 3 protein enhances hepatic insulin sensitivity by direct activation of the mTORC2-Akt signaling. Diabetes. 2015;64(4):1211–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chantranupong L, Wolfson RL, Orozco JM, Saxton RA, Scaria SM, Bar-Peled L, Spooner E, Isasa M, Gygi SP, Sabatini DM. The Sestrins interact with GATOR2 to negatively regulate the amino-acid-sensing pathway upstream of mTORC1. Cell Rep. 2014;9(1):1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Settembre C, Zoncu R, Medina DL, Vetrini F, Erdin S, Erdin S, Huynh T, Ferron M, Karsenty G, Vellard MC et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. 2012;31(5):1095–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sha Y, Rao L, Settembre C, Ballabio A, Eissa NT. STUB1 regulates TFEB-induced autophagy-lysosome pathway. EMBO J. 2017;36(17):2544–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chantranupong L, Scaria SM, Saxton RA, Gygi MP, Shen K, Wyant GA, Wang T, Harper JW, Gygi SP, Sabatini DM. The CASTOR proteins are arginine sensors for the mTORC1 pathway. Cell. 2016;165(1):153–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lei HT, Ma J, Sanchez Martinez S, Gonen T. Crystal structure of arginine-bound lysosomal transporter SLC38A9 in the cytosol-open state. Nat Struct Mol Biol. 2018;25(6):522–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Scalise M, Galluccio M, Pochini L, Cosco J, Trotta M, Rebsamen M, Superti-Furga G, Indiveri C. Insights into the transport side of the human SLC38A9 transceptor. Biochim Biophys Acta Biomembr. 2019;1861(9):1558–67. [DOI] [PubMed] [Google Scholar]
- 45. Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, Rustgi AK, Chang S, Tuveson DA. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005;7(5):469–83. [DOI] [PubMed] [Google Scholar]
- 46. Rachagani S, Torres MP, Kumar S, Haridas D, Baine M, Macha MA, Kaur S, Ponnusamy MP, Dey P, Seshacharyulu P et al. Mucin (Muc) expression during pancreatic cancer progression in spontaneous mouse model: potential implications for diagnosis and therapy. J Hematol Oncol. 2012;5:68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chen H, Xiong T, Qu Y, Zhao F, Ferriero D, Mu D. mTOR activates hypoxia-inducible factor-1α and inhibits neuronal apoptosis in the developing rat brain during the early phase after hypoxia–ischemia. Neurosci Lett. 2012;507(2):118–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rohde JR, Cardenas ME. The Tor pathway regulates gene expression by linking nutrient sensing to histone acetylation. Mol Cell Biol. 2003;23(2):629–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Macias RIR, Munoz-Bellvis L, Sanchez-Martin A, Arretxe E, Martinez-Arranz I, Lapitz A, Gutierrez ML, La Casta A, Alonso C, Gonzalez LM et al. A novel serum metabolomic profile for the differential diagnosis of distal cholangiocarcinoma and pancreatic ductal adenocarcinoma. Cancers (Basel). 2020;12(6):1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Mayerle J, Kalthoff H, Reszka R, Kamlage B, Peter E, Schniewind B, Gonzalez Maldonado S, Pilarsky C, Heidecke CD, Schatz P et al. Metabolic biomarker signature to differentiate pancreatic ductal adenocarcinoma from chronic pancreatitis. Gut. 2018;67(1):128–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. OuYang D, Xu J, Huang H, Chen Z. Metabolomic profiling of serum from human pancreatic cancer patients using 1H NMR spectroscopy and principal component analysis. Appl Biochem Biotechnol. 2011;165(1):148–54. [DOI] [PubMed] [Google Scholar]
- 52. Carreras-Torres R, Johansson M, Gaborieau V, Haycock PC, Wade KH, Relton CL, Martin RM, Davey Smith G, Brennan P. The role of obesity, type 2 diabetes, and metabolic factors in pancreatic cancer: a Mendelian randomization study. J Natl Cancer Inst. 2017;109(9):djx012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Makhoul I, Yacoub A, Siegel E. Type 2 diabetes mellitus is associated with increased risk of pancreatic cancer: a Veteran Administration registry study. SAGE Open Med. 2016;4:2050312116682257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Pollak MN. Investigating metformin for cancer prevention and treatment: the end of the beginning. Cancer Discov. 2012;2(9):778–90. [DOI] [PubMed] [Google Scholar]
- 55. Pollak M. Metformin and pancreatic cancer: a clue requiring investigation. Clin Cancer Res. 2012;18(10):2723–5. [DOI] [PubMed] [Google Scholar]
- 56. Algire C, Moiseeva O, Deschenes-Simard X, Amrein L, Petruccelli L, Birman E, Viollet B, Ferbeyre G, Pollak MN. Metformin reduces endogenous reactive oxygen species and associated DNA damage. Cancer Prev Res. 2012;5(4):536–43. [DOI] [PubMed] [Google Scholar]
- 57. Lonardo E, Cioffi M, Sancho P, Sanchez-Ripoll Y, Trabulo SM, Dorado J, Balic A, Hidalgo M, Heeschen C. Metformin targets the metabolic Achilles’ heel of human pancreatic cancer stem cells. PLoS One. 2013;8(10):e76518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. He H, Ke R, Lin H, Ying Y, Liu D, Luo Z. Metformin, an old drug, brings a new era to cancer therapy. Cancer J. 2015;21(2):70–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ben-Sahra I, Howell JJ, Asara JM, Manning BD. Stimulation of de novo pyrimidine synthesis by growth signaling through mTOR and S6K1. Science. 2013;339(6125):1323–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Howell JJ, Hellberg K, Turner M, Talbott G, Kolar MJ, Ross DS, Hoxhaj G, Saghatelian A, Shaw RJ, Manning BD. Metformin inhibits hepatic mTORC1 signaling via dose-dependent mechanisms involving AMPK and the TSC complex. Cell Metab. 2017;25(2):463–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kisfalvi K, Moro A, Sinnett-Smith J, Eibl G, Rozengurt E. Metformin inhibits the growth of human pancreatic cancer xenografts. Pancreas. 2013;42(5):781–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Soares HP, Ni Y, Kisfalvi K, Sinnett-Smith J, Rozengurt E. Different patterns of Akt and ERK feedback activation in response to rapamycin, active-site mTOR inhibitors and metformin in pancreatic cancer cells. PLoS One. 2013;8(2):e57289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Wang Y, Wei J, Li L, Fan C, Sun Y. Combined use of metformin and everolimus is synergistic in the treatment of breast cancer cells. Oncol Res. 2015;22(4):193–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Goodwin PJ, Stambolic V, Lemieux J, Chen BE, Parulekar WR, Gelmon KA, Hershman DL, Hobday TJ, Ligibel JA, Mayer IA et al. Evaluation of metformin in early breast cancer: a modification of the traditional paradigm for clinical testing of anti-cancer agents. Breast Cancer Res Treat. 2011;126(1):215–20. [DOI] [PubMed] [Google Scholar]