

Abstract
Therapeutic proteins are an indispensable class of drugs and often therapeutics of last resort. They are sensitive to oxidation, which is of critical concern, because it can affect drug safety and efficacy. Protein oxidation, with methionine and tryptophan as the most susceptible moieties, is mainly monitored by HPLC–MS techniques. However, since several oxidation products display the same mass difference, their identification by MS is often ambiguous. Therefore, an alternative analytical method able to unambiguously identify and, ideally, also quantify oxidation species in proteins is highly desired. Here, we present an NMR-based approach to monitor oxidation in full-length proteins under denaturing conditions, as demonstrated on two biotherapeutic monoclonal antibodies (mAbs). We show that methionine sulfoxide, methionine sulfone, N-formylkynurenine, kynurenine, oxindolylalanine, hydroxypyrroloindole, and 5-hydroxytryptophan result in characteristic chemical shift correlations suited for their identification and quantification. We identified the five most abundant oxidation products in forced degradation studies of two full-length therapeutic mAbs and can also unambiguously distinguish oxindolylalanine from 5-hydroxytryptophan, which are undistinguishable by MS due to the same mass shift. Quantification of the abundant methionine sulfoxide by NMR and MS gave highly comparable values. These results underline the suitability of NMR spectroscopy for the identification and quantification of critical quality attributes of biotherapeutics.
Oxidation of amino acid side chains in biotherapeutics is a common alteration that can occur during production due to suboptimal processing and purification protocols as well as during prolonged storage and can lead to conformational changes or instability of proteins. Thus, the efficacy, half-life, and safety of a drug may be significantly affected.1 The most observed oxidation product in proteins and biotherapeutics is methionine sulfoxide (Met(O)).2−4 However, oxidation of tryptophan was also shown to have an impact on the activity, structure, and stability of biopharmaceuticals.5−8 Since oxidation products are generally considered as critical quality attributes in biotherapeutics, tight monitoring during drug development is generally performed. Liquid chromatography together with MS are the preferred techniques to analyze post-translational modifications (PTMs). However, due to identical mass changes for structurally different oxidation products, other methods are needed for their unambiguous identification and for cross-validation of currently applied analytical methods. For this purpose, NMR spectroscopy is ideally suited, because it is the most common technique to elucidate the structure of molecules in solution.
Several recent publications have demonstrated the applicability of 2D NMR spectroscopy to detect PTMs in proteins without the necessity of isotope labeling.9−11 This approach relies on 2D 1H–13C HSQC spectra of denatured proteins containing 13C at natural abundance, which provides unique chemical shift pairs for the unambiguous identification of certain PTMs, e.g., succinimide,9N-glycans,10−12 and methylated and glycated amino acids.13 In a very recent work, we have successfully applied this method even to intact mAbs for the detection and quantification of pyroglutamate.14 The results agreed very well with HPLC–MS so that both methods are delivering orthogonal results and can potentially be used for cross-validation. However, protein oxidation was not investigated so far by NMR spectroscopy, in particular not in denatured full-length proteins. This could be due to the large variety of oxidation products.15−18 Denatured proteins with oxidized residues are expected to show characteristic cross-peaks in addition to those of the random-coil chemical shifts of the 20 natural amino acids.19,20 For an unambiguous identification of any oxidation product, their random-coil chemical shifts have to be assigned, typically by using model peptides containing the PTM of interest as a reference, as it was done, for example, for phosphorylated amino acids,13,21 succinimide,9 and pyroglutamate.14
Methionine residues are most susceptible to oxidation, leading mainly to methionine sulfoxide (Figure 1), as MS studies of a variety of biotherapeutics show.2−4 For example, two methionine residues in the Fc/2 region of the heavy chain of rituximab (Met256, Met432) are significantly prone to H2O2-mediated oxidation.4,22 Less frequent but important for certain proteins is the oxidation of tryptophan residues,15,16 especially for mAbs.8,23−27 Tryptophan may convert into different oxidation products, as suggested by MS studies.6,8,23−26,28 Several oxidation mechanisms were described for tryptophan and indole-containing substances, which depend also on the oxidation conditions.30 In any case, N-formylkynurenine (NFK), kynurenine (Kyn), oxindolylalanine (Oia), and hydroxytryptophans like 5-hydroxytryptophan (5HTP) have been described as the main oxidation products,15,16,18,31−33 which are depicted in Figure 1. The chemical structures of most oxidation products in proteins are based on MS data, which is often ambiguous, because several products show identical masses (e.g., Oia and 5HTP, both characterized by a mass shift of +16 Da). Only in very few cases an unambiguous identification of a modification at a Trp residue in a protein was achieved, e.g., the detection of Kyn by X-ray crystallography in the bacterial copper binding protein MopE.34 A comprehensive analysis of oxidation products by NMR spectroscopy has been lacking so far. Only for free 4-, 5-, and 7-HTP chemical shift assignments in D2O were reported35 but unrelated to peptides or proteins.
Figure 1.

Possible Met and Trp oxidation products occurring in proteins. Chiral centers are indicated by an asterisk (a red asterisk represents the chiral centers generated upon the transformation of the natural residues). In the case of HPI, two stereoisomers are expected, which correspond to the cis or trans configurations of the OH group with respect to the α-carbonyl within the pyrrole ring, as previously defined by Ronsein et al.29 In the case of N-formylkynurenine (NFK), two species can form, due to the cis and trans isomerization of the N-aryl-formamide moiety (indicated by ‡).
Here, we present the random-coil chemical shifts of Met(O), methionine sulfone (Met(O2)), NFK, Kyn, Oia, 5HTP, and hydroxypyrroloindole (HPI) under denaturing conditions using small synthetic peptides as well as a protocol that allows the analysis of full-length proteins without the necessity of additional sample treatment, like digestion. The NMR fingerprints of these PTMs enable the unambiguous identification and quantification of methionine and tryptophan oxidation products in proteins using 2D NMR spectra as demonstrated by the two protein therapeutics rituximab and adalimumab.
Experimental Section
Peptides
Reference peptides were synthesized by solid phase peptide synthesis using Fmoc chemistry and analyzed by HPLC and MALDI-TOF-MS. H–Oia–OH, which was prepared from H–Trp–OH as described previously,36 H–Kyn–OH, and H–5HTP–OH were Fmoc-protected as reported before37 and coupled using a protocol described previously.33 For experimental details, see the Supporting Information.
Forced Oxidation of Met and Trp Residues in Peptides and Proteins
To oxidze Met and Trp in reference peptides or in proteins, we used H2O2 at concentrations ranging from 0.35 to 1% using various incubation times at RT as described in detail in the Supporting Information.
NMR Spectroscopy
A 600 MHz Bruker Avance III HD spectrometer equipped with a 1H/13C/15N/31P quadruple-resonance probe was used to record the spectra at 298 K in 5 mm NMR tubes (ARMAR, Type 5TA) with a sample volume of 500 μL. To assign the reference peptide signals in the spectrum, the following experiments were recorded: 1H–13C HSQC, 1H–13C HMBC (hmbcgpndqf), 1H–1H TOCSY, 1H–1H COSY (cosygpppqf), 1H–1H ROESY, 1H–15N HSQC, 1H–13C HQQC,38 and 1H–13C H–CO.9 For data processing and analysis, Topspin 3.5/3.6 (Bruker) and Sparky 3.114 (T. D. Goddard and D. G. Kneller, SPARKY 3, University of California, San Francisco, USA) were used, respectively. For chemical shift referencing, approximately 0.5 mM 2,2-dimethyl-2-silapentane-5-sulfonic acid (DSS) (ARMAR Chemicals) was added to the sample after the 2D NMR measurements, and a 1D 1H spectrum was recorded. The chemical shifts of carbon and nitrogen were referenced using the IUPAC-IUB recommended chemical shift referencing ratios Ξ of 0.251449530 (carbon) and 0.10132918 (nitrogen).39 The chemical shift assignments are made publicly available at the BioMagResBank40 under the accession codes 28123 (Met(O)–Gly), 28126 (Met(O)–Ala), 28127 (Met(O–Pro), 28124 (5-HTP), 28125(Kyn), 28128 (Met(O2)), 28129 (NFK), and 28130 (Oia).
Quantification of Methionine Oxidation in NMR Spectra
Quantification of induced Met(O) was performed for the NMR samples of rituximab and adalimumab, which were treated with 0.35% H2O2 for 30 min at RT, lyophilized, and then denatured and reduced as described previously. Four approaches (a–d) were tested and compared. Approach a: in the measured 1H–13C HSQC spectra, the Hγ–Cγ cross-peak of Met(O) was integrated (normalized to the number of methionine residues present in the mAbs; adalimumab: 10, rituximab: 12) and compared to the integrated and normalized random-coil chemical shift peaks of Cδ−Hδ of Arg (number of Arg in adalimumab: 42, in rituximab: 28), Cε–Hε of Lys (number of Lys in adalimumab: 88, in rituximab: 98), or Cβ–Hβ of Leu (number of Leu in adalimumab: 104, in rituximab: 90) (Table S1). The amino acid sequences for rituximab and adalimumab were obtained from the Web site https://www.drugbank.ca (access date 9/23/2019). Approach b: the Hε–Cε cross-peak of Met(O) was compared with the sum of the integrals of the Hε–Cε cross-peaks of nonoxidized Met and Met(O). Approach c: the integral of the Hε–Cε cross-peak of Met(O) was compared to CH3 cross-peaks of other amino acids like Ala, Ile, and Leu. Approach d: the Hε–Cε cross-peak of CH3 Met was compared to CH3 cross-peaks of other amino acids like Ala, Ile, and Leu to obtain the amount of nonoxidized Met, from which the amount of Met(O) was then extrapolated. Approaches b and d are based on the assumption that the only oxidation product of Met is Met(O). More details and formulas regarding quantification are provided in the Supporting Information.
Quantification of Met(O) by MS
Experimental details for quantification by MS can be found in the Supporting Information. Briefly, both mAbs were trypsinized followed by peptide analysis by HPLC–ESI–Quadrupole-Orbitrap MS. Identification of modified sites was based on peptide fragmentation, and relative quantification of oxidation was based on peak areas on extracted ion current chromatograms derived from full-scan data.
Results
Random-Coil Chemical Shifts of Synthetic Oxidation Products in Peptides
To determine the NMR signature of the previously described oxidation products of methionine and tryptophan as shown in Figure 1, we synthesized short peptides containing Met(O), Kyn, Oia, and 5HTP (Table S2). 2D NMR 1H–13C and 1H–1H correlations were used to obtain the 1H and 13C random-coil chemical shifts of these moieties as part of a peptide chain (Tables S3 and S4). For comparison with chemical shift data of denatured proteins, all spectra of the model peptides were recorded under denaturing conditions (7 M urea) at pH values of either 2.3 or 7.4. The acidic pH was chosen, because it aids complete denaturation of some proteins; moreover, random-coil chemical shifts at pH 2.3 had been reported previously.19 Physiological pH (7.4) might be required for moieties that are unstable at acidic pH. To achieve maximal sensitivity and avoid disturbances of the solvent resonances, deuterated urea and D2O were used. The chemical shifts of all investigated oxidation variants did not change significantly at the two different pH values suggesting that the presented method is also viable at pH values between 2.3 and 7.4.
The random-coil chemical shifts of Met(O) showed significantly different correlations compared to nonoxidized Met (Figure 2a). In particular, the cross-peaks of Cγ–Hγ and Cε–Hε originating of the nuclei directly adjacent to the oxidized sulfur moved to significantly different regions. Surprisingly, the chirality of the sulfur41 did not lead to peak doubling except for a small separation of the Cα–Hα correlation (Figure 2a). Comparing these shifts with the random-coil chemical shifts of all 20 natural amino acids shows that the Hγ–Cγ cross-peaks of Met(O) are unique, well-isolated, and therefore ideally suited to unambiguously detect Met(O) in proteins (Figure 2b). The Cε–Hε cross-peak may partly overlap with Cβ–Hβ shifts of Asn; however, to resolve this degeneracy, either spectra are recorded with higher resolution in the 13C dimension (Figures 2c and S1) or a 1H triple-quantum filtered HQQC experiment can be performed, which is selective for CH3 groups but equally sensitive compared to an HSQC38 (Figure S2). The HQQC experiment is suitable for an unambiguous identification of Met(O) by its unique Cε–Hε cross-peak, without the need of high resolution in 13C and thus resulting in shorter measurement time. However, its application for quantitative purposes is not recommended, since pulse imperfections and variations in scalar couplings have a much larger impact and will influence the peak intensities.
Figure 2.

1H–13C HSQC spectra of the reference peptides for the detection of Met(O), lysozyme, and H2O2-treated rituximab. (a) Overlay of the reference spectra of peptides Ac–Gly–Gly–Met(O)–Gly–Gly–NH2 (red) and Ac–Gly–Gly–Met–Gly–Gly–NH2 (blue) under denaturing conditions (7 M urea, pH 2.3). (b) Overlay of the spectra of the reference peptide Ac–Gly–Gly–Met(O)–Gly–Gly–NH2 (red) and denatured lysozyme (blue), to identify unique chemical shifts of Met(O) that differ from the random-coil chemical shift correlations of the 20 natural amino acids. (c) 1H–13C HSQC spectrum of treated (0.35% H2O2 for 30 min at RT) rituximab (512 × 512 complex points, 104 scans, a recycle delay of 3 s, total measurement time of 2 days and 14 h) under denaturing conditions (7 M urea-d4 in D2O) at pH 2.3.
As control experiments, we analyzed if neighboring amino acids influence these characteristic chemical shift correlations (Table S5). In particular, a Pro residue at the i + 1 position, which is known to have the highest impact on neighboring random-coil chemical shifts,20,42 showed only insignificant differences of the positions of the Cγ–Hγ and Cε–Hε cross-peaks (Tables S3 and S5). An Ala residue at the i + 1 position did not change the Met(O) chemical shifts (Table S5). This suggests that the characteristic random-coil chemical shifts for Met(O) are not impaired by the surrounding peptide sequence.
In principle, Met(O) can be further oxidized to methionine sulfone Met(O2), but this reaction is very slow. In order to obtain NMR data of Met(O2), the model peptide Ac–Gly–Gly–Met–Gly–Gly–NH2 was treated extensively with H2O2 to detect possible side products apart of Met(O). Indeed, a second spin system was detected (Figure S3 and Table S6), which was identified as Met(O2), in agreement with previously reported 1H and 13C chemical shifts.43
The 1H–13C random-coil chemical shift correlations of Kyn, Oia, and 5HTP showed all at least one characteristic cross-peak in the aromatic region (Figure 3). A peculiarity of Oia is that it contains a second chiral center at Cγ in addition to the chirality of Cα, and the resulting diastereomers give rise to two sets of very similar signals, e.g., at Hβ2 and Hβ3 (Table S4). In addition, keto–enol tautomerism leads to an exchange of Hγ with deuterium of the solvent so that the Hγ signal is unobservable in spectra recorded in D2O.
Figure 3.
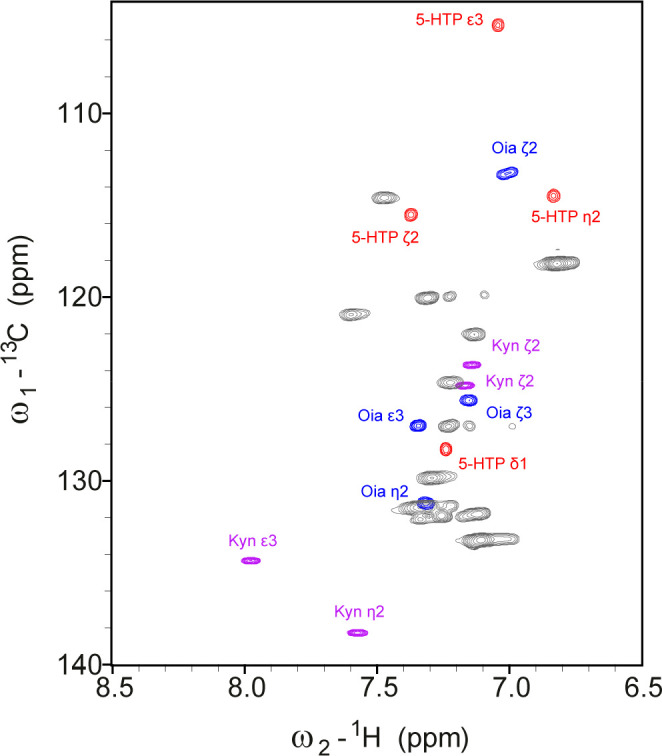
Overlay of the aromatic region of 1H–13C HSQC spectra of the three reference peptides containing 5-HTP (red), Kyn (purple), or Oia (blue) under denaturing conditions (7 M urea-d4 in D2O, pH 2.3), compared with random-coil chemical shift correlations of the natural aromatic amino acids of denatured lysozyme (gray). Several signals of the oxidation products are unique and suitable for an unambiguous identification of the different oxidation products of Trp.
NMR Characterization of Oxidation Products of Trp in a Model Peptide
Although a large variety of Trp oxidation products were described previously (Figure 1), it is unclear which variants are predominant, short-lived, or low populated. Moreover, the reactivity of the indole moiety in large, folded biomolecules strongly depends on its environment and solvent exposure as well as on the strength of the oxidant, which makes it difficult to predict the type of Trp oxidation. For example, partial conversion of the solvent-accessible Trp32 (but not of Trp35) from the light chain of an mAb to Kyn and Oia or 5HTP was observed upon prolonged storage or tert-butylhydroperoxide treatment.6 For another mAb (MEDI-493), it was shown that only the solvent-exposed Trp105 in the heavy chain was susceptible to oxidation: however, upon UV irradiation, the observed degradation products were Oia or 5HTP (both +16 Da) and NKF or DiOia (both +32 Da), whereas upon treatment with ozone, the major product was NFK (with Kyn, 5HTP, or Oia being only minor products).8
To investigate which Trp oxidation products are observable by NMR spectroscopy, we treated the peptide Ac–Gly–Gly–Trp–Gly–Gly–NH2 extensively with H2O2 and analyzed the resulting mixture. The choice of H2O2 as the oxidant was made because of its frequent use to prepare oxidatively stressed protein samples for analytical purposes.3,4,6,44 A 1H–13C fingerprint spectrum is shown in Figure 4. In addition to the expected signals of Kyn and Oia, we could identify four additional spin systems. Two of those could be assigned to NFK. These two signal sets are exchanging during the NMR experiment, as illustrated by exchange signals between their Hζ2 resonances in a 2D ROESY as well as between Hδ1 resonances (Figure S4). The duplication of signal sets was caused by a cis/trans isomerization of the N-aryl-formamide moiety (Figure 4). Since only one cross-peak between Hδ1 and Cε2 was detected in the 1H–13C HMBC spectrum (Figure S4d), the most populated species must be the one in which Hδ1 and Cε2 are in trans, for which a higher value of 3JHδ1Cε2 is expected compared to the cis-Hδ1/Cε2 configuration. This is further supported by the 1H and 13C chemical shifts of the formyl group, which are more downfield for the cis form, according to previous NMR studies of N-formyl-group-containing compounds.45,46 The downfield chemical shift of Hζ2 at 8.14 ppm can be explained in the case of cis-NFK by the proximity to the formyl oxygen. The other two similar sets of signals matched previously reported chemical shifts of the two diasteriomers of hydroxypyrroloindole (HPI), which the authors named trans- and cis-HPI (Figure 4).29 However, the formation of HPI, which involves a bond between the backbone nitrogen and the former Cδ1 of Trp, was so far only reported as an oxidation product of the free amino acid tryptophan. Now, we show that this tricyclic product can be formed also when the tryptophan is incorporated into a peptide backbone. Interestingly, we observed that the intensities of the NFK signals decreased over time, while those of Kyn increased. A spectrum recorded immediately after the oxidation treatment contained mainly NFK and barely Kyn, whereas a sample measured after several days displayed predominantly Kyn signals. This indicates a conversion of NFK into Kyn over time (Figure S5). To our surprise, we did not observe any signals matching to 5HTP nor even vaguely to other HTP species.35 However, we cannot rule out that 5HTP is formed under different oxidation conditions.
Figure 4.

1H–13C HSQC spectrum of the aromatic region of the Trp-containing peptide Ac–Gly–Gly–Trp–Gly–Gly–NH2 under denaturing conditions (7 M urea-d4 in D2O) after treatment with 1% H2O2 for 5 h, dialysis, and lyophilization. Four different oxidation products of Trp could be detected and are shown on the right. For HPI, Oia, and NFK, two sets of shifts can be observed due to different stereoisomers.
Identification of Oxidation Products in Biotherapeutics Using 2D NMR
In order to detect Met(O) in full-length proteins, we chose the two mAbs rituximab and adalimumab as model systems. For positive controls, we introduced methionine oxidation by treating the proteins with H2O2. All protein samples were buffer exchanged, lyophilized, and subsequently denatured by using 7 M urea-d4 and tris(2-carboxyethyl)phosphine hydrochloride (TCEP) as a reducing agent. These conditions resulted in a completely denatured state even for intact mAbs both at acidic and neutral conditions. Therefore, no digestion and fragment separation was necessary using our approach. The H2O2-treated proteins (30 min at room temperature, 0.35% H2O2), investigated at the two pH conditions (2.3 and 7.4), revealed characteristic Cγ–Hγ and Cε–Hε cross-peaks indicative for Met(O) (Figures 2c and S6). Signals indicating the presence of Met(O2) and Trp oxidation products were not observed after these conditions were applied. However, by performing extended oxidation under denaturing conditions (1% H2O2 for 25 h in the formulation buffer, measured in 7 M urea and 11 mM TCEP), it was possible to detect Met(O2) (Figures S7 and S8) and the Trp oxidation products Oia, and Kyn. In particular, Cε3–Hε3 and Cζ3–Hζ3 cross-peaks of Oia and weak signals of Cε2–Hε2 and Cη2–Hη2 of Kyn were detected in a 2D 1H–13C HSQC spectrum (Figure S9a) of rituximab. Interstingly, in a sample treated for more than five days, Kyn but not Oia was observed (Figure S9c,d). Weak NFK signals were also detected, which, however, dissapeared over time.
Quantification of Met(O) in (H2O2-Treated) mAbs
NMR spectroscopy is, in principle, a quantitative method as long as sufficiently long recycle delays ensure the restoration of equilibrium magnetization and as long as signals do not overlap. For the quantification of the most abundant oxidation product Met(O) using 2D 1H–13C HSQC spectra of rituximab and adalimumab treated with H2O2, we evaluated several approaches that were based on the following considerations: (i) The well-isolated CH3 group of nonoxidized Met is sharp and well-suited for quantification, whereas the CH3 group of Met(O) is located close to other random-coil correlations, mainly of Asn. Therefore, due to the difficulty of integrating overlapping signals, the Cε–Hε integral of Met(O) might have a larger error. However, very high resolution in the 13C dimension results in an isolated signal and thus overcomes this challenge and provides an excellent handle for quantification (Figure S10). (ii) The signal of CH2 at Cγ of Met(O) is well-isolated, in contrast to the corresponding signal of nonoxidized Met. However, it is quite weak, which results in large integration errors. (iii) Suitable reference signals for cross-peak integration were chosen by taking into account the different multiplicities and slightly different 1JCH scalar couplings so that integrals of CH2 were only compared to other CH2 signals, and integrals of CH3 were only compared to other CH3 signals. Therefore, the resulting approaches to obtain the fraction of Met(O) were as follows (a–d): (a) the integral of Cγ–Hγ of Met(O) was compared to those of Lys Cε–Hε, Arg Cδ−Hδ, and Leu Cβ–Hβ signals; (b) the integral of CH3 of Met(O) (Cε–Hε cross-peak) was divided by the sum of the integrals of Met(O) and Met methyl signals (the latter have the advantage of being more intense than the methylene signals); (c) alternatively, the integral of CH3 of Met(O) was compared to each of the methyl groups of Ile, Ala, and Leu; (d) in addition, we tested an indirect approach for the quantification of Met(O), based on the typically intense CH3 signal of nonoxidized Met Cε–Hε, which was compared to the methyl groups of Ile, Ala, and Leu under the assumption that methionine sulfoxide is the only modification of Met occurring in the protein. Further details and applied equations for calculating the Met(O) fractions and their associated errors are given in the Supporting Information.
The results of all approaches applied to rituximab and adalimumab treated with H2O2 are summarized in Table 1. For both mAbs, all approaches gave comparable values. However, the quantification based on CH2 groups showed larger errors than the other direct approaches based on CH3 groups. This can be explained by the weaker Met(O) CH2 signals and the associated lower accuracy of their integrals. Neither was Met(O2) detected nor was the sum of the integrals of Met and Met(O) smaller than expected, indicating Met(O) was the only PTM of Met. However, an important parameter for a reliable quantification is a sufficiently long recycle delay. In our case, the recycle delay of 3 s was sufficient, whereas 2 s led to a reduction of the Hε–Cε signal of nonoxidized Met, which resulted in a smaller total integral of all methionine species and, consequently, in overestimation of Met(O) using quantification methods (b) and (d).
Table 1. Quantification of Induced Met(O) in Rituximab and Adalimumab Treated with 0.35% H2O2 for 30 min at RT.
| cross-peaks used for quantificationa | rituximab Met(O) (%) | adalimumab Met(O) (%) |
|---|---|---|
| a: Met(O) Cγ/Hγ compared to | ||
| Arg Cδ/Hδ | 14.3 ± 1.9c | 15.3 ± 1.0c |
| Leu Cβ/Hβ | 18.7 ± 4.6c | 17.3 ± 1.9c |
| Lys Cε/Hε | 16.2 ± 2.3c | 14.7 ± 1.6c |
| b:b Met(O) Cε/Hε compared to | ||
| Met Cε/Hε | 18.3 ± 1.0c | 19.1 ± 1.2c |
| c: Met(O) Cε/Hε compared to | ||
| Ala Cβ/Hβ | 18.3 ± 3.8c | 19.3 ± 0.5c |
| Ile Cδ1/Hδ1 | 18.6 ± 1.3c | 19.6 ± 0.7c |
| Ile Cγ2/Hγ2 | 17.8 ± 1.8c | 19.7 ± 1.2c |
| Leu Cδ1/Hδ1 | 18.1 ± 0.9c | 18.7 ± 0.5c |
| Leu Cδ2/Hδ2 | 18.2 ± 1.5c | 18.7 ± 0.8c |
| d:b Met Cε/Hε compared to | ||
| Ala Cβ/Hβ2 | 18.4 ± 3.8c | 18.3 ± 4.7c |
| Ile Cδ1/Hδ1 | 16.9 ± 3.6c | 17.4 ± 5.5c |
| Ile Cγ2/Hγ2 | 20.8 ± 5.8c | 16.8 ± 7.4c |
| Leu Cδ1/Hδ1 | 19.1 ± 2.1c | 21.0 ± 4.6c |
| Leu Cδ2/Hδ2 | 18.7.4 ± 4.8c | 21.1 ± 5.6c |
| MS | 17.9 ± 1.3 | 21.3 ± 1.2 |
1H–13C HSQC spectra used for quantification shown in Figure S6.
Based on the assumption that Met(O) is the only oxidation product of Met.
Error determined using eqs 3, 5, and 7 (see Supplementary Methods).
The quantification of oxidation of the same samples was also performed with MS (Tables 1 and S7). The relative quantification of total methionine oxidation showed that on average 17.9% of all methionine residues in rituximab and 21.3% of all methionine residues in adalimumab were oxidized, which is in the same range as the values obtained by NMR. The relative oxidation rates of every single methionine residue are provided in Table S7. As previously reported, the methionine residues Met256 and Met432 in the Fc domain of the mAbs were most prone to oxidation22 and, indeed, showed the highest percentage of oxidation. Figure S11 depicts one example of the identification and relative quantification by MS: oxidized and nonoxidized peptide species were identified, based on the fragment ion spectra (Figure S11b,c) and subsequently relatively quantified based on peak areas of the full-scan mass spectra (Figure S11a). It should be noted that consistent quantification data were only obtained when the sample was kept under argon atmosphere during the sample preparation steps in order to exclude oxidation by atmospheric oxygen.
Discussion
The presented random-coil chemical shift assignment of the most important oxidation products of Met and Trp (Figure 1) enables a straightforward and unambiguous identification of these modifications in proteins under denaturing conditions. Due to standardized conditions, the random-coil chemical shifts are generally applicable, independent of the NMR instrument and without the necessity of reference peptides. Owing to the flexibility of the protein chain in the unfolded state, our approach results in narrow line widths even for large proteins. With this method, there is per se no size limit, and large proteins like full-length mAbs can be analyzed, as long as it is possible to denature them. This has been demonstrated here by using rituximab and adalimumab as model systems. In contrast to MS studies that, in general, require the digestion of mAbs for the detection of Met(O),4,47,48 we can use intact proteins. Middle-down or bottom-up approaches have the disadvantage of laborious sample preparation, which can also lead to artifacts due to the formation of additional degradation products. With regard to oxidation, the atmospheric oxygen can significantly increase Met(O) formation during long incubation times. As an orthogonal and complementary approach to MS, we propose to use a straightforward NMR-based protocol for the investigation of the oxidation of small proteins, like lysozyme, as well as of large ones, like mAbs, at different pH values. This is especially suited for forced degradation studies generating modifications identical to those that are spontaneously formed during the lifetime of a drug but typically with much higher amounts (>10%). Our method can unambiguously assess the identity of the oxidation products, which is very important in the case of chemical modifications characterized by the same mass difference (e.g., Oia and HTP). In this way, ambiguous assignment or even overestimation of side products with identical masses can be avoided. Since our NMR protocol relies on the random-coil chemical shift fingerprints of modified residues, it provides the chemical identity but not the position of the modification. Therefore, additional MS measurements would be necessary, which, again, underlines the complementarity of the NMR and MS approaches for the complete characterization of protein degradation products. Besides the application to large proteins, like mAbs, also small-to-medium-size therapeutic peptides are ideally suited for our NMR-based approach. These peptides may be particularly susceptible to spontaneous chemical changes during production and storage, due to the lack of a well-defined three-dimensional structure. Because of the small molecular weight of peptides and higher achievable concentrations, it seems even feasible to use NMR for product quality control.
Although the amount of protein required, the detection limit, and the relatively long measurement time might represent some limitations to the NMR approach, it should be considered that biotherapeutics are usually produced in large scale, and thus, the quantities required for NMR are accessible. The required NMR measurement time will depend on the sensitivity of the spectrometer, obviously being dramatically shortened with higher fields. Similarly, the detection limit reflects the type of equipment utilized for the NMR measurements: for example, we have recently shown that the lowest detectable amount of pyroglutamate (pGlu) in mAbs was about 55 μM by using a 600 MHz spectrometer with a cryogenic probe (TCI) and 2 days measurement time.14 Therefore, we assume that, also in the case of Met and Trp oxidation products, the limit of detection will be in the two-digit micromolar range. However, the use of higher field magnets in combination with cryogenic probes will allow reaching even lower limits of detections, as shown by Peng et al., who could detect glycosylation in mAb fragments down to about 10% by using an 850 MHz spectrometer with a cryogenic probe with a measurement time of 11 h.10
To quantify Met(O) by NMR spectroscopy, we propose that the most reliable approach is the one based on the integrals of the methyl group of Met(O) and of other isolated methyl groups of the protein (approach (c)). This requires very high resolution in the 13C dimension to prevent overlap of the Met(O) methyl signal with nearby random-coil signals. Depending on the reference methyl groups, the obtained Met(O) amount varied by approximately 2%, which is within the error estimated for each value. Alternatively, the integrals of the well-isolated Cγ/Hγ2 + Hγ3 methylene group signals of Met(O) and of other isolated methylene signals of the protein can be used (approach a) even at lower resolution, but the error of the Met(O) fraction will be higher (approximately 4%) due to the weaker intensity of the methylene group of Met(O). The other two approaches ((b) and (d)), which are based on the assumption that Met(O) is the only oxidation product of Met and thus that the sum of the integrals of Met and Met(O) represent all methionine species, are less recommended, because they are sensitive to the presence of any other oxidation products of methionine. Another factor to consider is that a sufficiently long recycle delay is crucial for the experiment. In case of the quantification of Met(O), a recycle delay of 3 s was required to obtain reliable quantification results.
Conclusion
Here, we provide for the first time the complete NMR characterization of the seven most relevant oxidation products potentially occurring in proteins under denaturing conditions (detailed summary in Figure S12). These chemical shift assignments revealed for each of the species unique cross-peaks in 2D fingerprint spectra, allowing an unambiguous identification of these chemical moieties in any protein under denaturing conditions. The applicability of this method in a biopharmaceutical context is demonstrated in a forced degradation study of two biotherapeutic mAbs through the identification of the oxidation products, but its use in quality control of peptide pharmaceuticals is feasible as well. The characteristic chemical shift fingerprints enable an unambiguous identification even for cases, which are undistinguishable by MS. The fact that we could identify the most common oxidized species in such large proteins like rituximab and adalimumab (treated) shows how powerful these chemical shift assignments are in combination with 1H–13C spectra under denaturing conditions.
Acknowledgments
We acknowledge Novartis GmbH for kindly providing expired batches of rituximab (MabThera, Roche) and adalimumab (Humira, AbbVie) and Dr. Urs Lohrig from Novartis GmbH as well as Dr. Frank Steiner and Dr. Kai Scheffler from Thermo Fisher Scientific for comments on the manuscript as well as scientific discussions. We thank Dr. Peter Schmieder (FMP Berlin) and Dr. Tammo Dierks (CIC bioGUNE Bilbao) for providing HQQC pulse sequences and fruitful discussions. We gratefully acknowledge the Biomolecular NMR Spectroscopy Platform at ETH Zürich for access to Bruker spectrometers with cryogenic probes. The financial support by the Austrian Federal Ministry of Science, Research, and Economy and by a Start-up Grant of the State of Salzburg is gratefully acknowledged.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.analchem.0c00965.
2D 1H–13C HSQC spectra, chemical shift assignment tables, HPLC and MS characterization data, Met(O) quantification data, and supplementary methods (PDF)
Author Contributions
M.S., C.C., and A.H. designed the experiments. V.S. synthesized the reference peptides and noncommercially available building blocks. A.H. and M.S. performed the NMR experiments and assigned the NMR resonances. C.R. performed and interpreted the MS experiments. C.G.H. supervised the MS analysis. M.S., C.C., and A.H. wrote the manuscript. All authors reviewed and approved the manuscript.
The authors declare the following competing financial interest(s): The salary of A.H. is fully funded and C.G.H.'s salary is partly funded by the Christian Doppler Laboratory for Biosimilar Characterization, which is partly supported by Novartis GmbH and Thermo Fisher Scientific. The authors declare no other competing financial interest.
Supplementary Material
References
- Torosantucci R.; Schöneich C.; Jiskoot W. Oxidation of therapeutic proteins and peptides: structural and biological consequences. Pharm. Res. 2014, 31 (3), 541–553. 10.1007/s11095-013-1199-9. [DOI] [PubMed] [Google Scholar]
- Davies M. J. The oxidative environment and protein damage. Biochim. Biophys. Acta, Proteins Proteomics 2005, 1703 (2), 93–109. 10.1016/j.bbapap.2004.08.007. [DOI] [PubMed] [Google Scholar]
- Forstenlehner I. C.; Holzmann J.; Toll H.; Huber C. G. Site-specific characterization and absolute quantification of pegfilgrastim oxidation by top-down high-performance liquid chromatography-mass spectrometry. Anal. Chem. 2015, 87 (18), 9336–9343. 10.1021/acs.analchem.5b02029. [DOI] [PubMed] [Google Scholar]
- Regl C.; Wohlschlager T.; Holzmann J.; Huber C. G. A Generic HPLC Method for Absolute Quantification of Oxidation in Monoclonal Antibodies and Fc-Fusion Proteins Using UV and MS Detection. Anal. Chem. 2017, 89 (16), 8391–8398. 10.1021/acs.analchem.7b01755. [DOI] [PubMed] [Google Scholar]
- Gao X.; Ji J. A.; Veeravalli K.; Wang Y. J.; Zhang T.; Mcgreevy W.; Zheng K.; Kelley R. F.; Laird M. W.; Liu J.; Cromwell M. Effect of individual Fc methionine oxidation on FcRn binding: Met252 oxidation impairs FcRn binding more profoundly than Met428 oxidation. J. Pharm. Sci. 2015, 104 (2), 368–377. 10.1002/jps.24136. [DOI] [PubMed] [Google Scholar]
- Hensel M.; Steurer R.; Fichtl J.; Elger C.; Wedekind F.; Petzold A.; Schlothauer T.; Molhoj M.; Reusch D.; Bulau P. Identification of potential sites for tryptophan oxidation in recombinant antibodies using tert-butylhydroperoxide and quantitative LC-MS. PLoS One 2011, 6 (3), e17708. 10.1371/journal.pone.0017708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D.; Ren D.; Huang H.; Dankberg J.; Rosenfeld R.; Cocco M. J.; Li L.; Brems D. N.; Remmele R. L. Structure and stability changes of human IgG1 Fc as a consequence of methionine oxidation. Biochemistry 2008, 47 (18), 5088–5100. 10.1021/bi702238b. [DOI] [PubMed] [Google Scholar]
- Wei Z.; Feng J.; Lin H.-Y.; Mullapudi S.; Bishop E.; Tous G. I.; Casas-Finet J.; Hakki F.; Strouse R.; Schenerman M. A. Identification of a single tryptophan residue as critical for binding activity in a humanized monoclonal antibody against respiratory syncytial virus. Anal. Chem. 2007, 79 (7), 2797–2805. 10.1021/ac062311j. [DOI] [PubMed] [Google Scholar]
- Grassi L.; Regl C.; Wildner S.; Gadermaier G.; Huber C. G.; Cabrele C.; Schubert M. Complete NMR assignment of succinimide, its detection and quantification in peptides and intact proteins. Anal. Chem. 2017, 89 (22), 11962–11970. 10.1021/acs.analchem.7b01645. [DOI] [PubMed] [Google Scholar]
- Peng J.; Patil S. M.; Keire D. A.; Chen K. Chemical Structure and Composition of Major Glycans Covalently Linked to Therapeutic Monoclonal Antibodies by Middle-Down Nuclear Magnetic Resonance. Anal. Chem. 2018, 90 (18), 11016–11024. 10.1021/acs.analchem.8b02637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert M.; Walczak M. J.; Aebi M.; Wider G. Posttranslational modifications of intact proteins detected by NMR spectroscopy. Angew. Chem., Int. Ed. 2015, 54 (24), 7096–7100. 10.1002/anie.201502093. [DOI] [PubMed] [Google Scholar]
- Unione L.; Lenza M. P.; Ardá A.; Urquiza P.; Laín A.; Falcón-Pérez J. M.; Jiménez-Barbero J.; Millet O. Glycoprofile Analysis of an Intact Glycoprotein As Inferred by NMR Spectroscopy. ACS Cent. Sci. 2019, 5 (9), 1554–1561. 10.1021/acscentsci.9b00540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conibear A. C.; Rosengren K. J.; Becker C. F. W.; Kaehlig H. J. Biomol. NMR 2019, 73 (10–11), 587–599. 10.1007/s10858-019-00270-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinterholzer A.; Stanojlovic V.; Cabrele C.; Schubert M. Unambiguous Identification of Pyroglutamate in Full-Length Biopharmaceutical Monoclonal Antibodies by NMR Spectroscopy. Anal. Chem. 2019, 91 (22), 14299–14305. 10.1021/acs.analchem.9b02513. [DOI] [PubMed] [Google Scholar]
- Ehrenshaft M.; Deterding L. J.; Mason R. P. Tripping up Trp: Modification of protein tryptophan residues by reactive oxygen species, modes of detection, and biological consequences. Free Radical Biol. Med. 2015, 89, 220–228. 10.1016/j.freeradbiomed.2015.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finley E. L.; Dillon J.; Crouch R. K.; Schey K. L. Identification of tryptophan oxidation products in bovine alpha-Crystallin. Protein Sci. 1998, 7 (11), 2391–2397. 10.1002/pro.5560071116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grassi L.; Cabrele C. Susceptibility of protein therapeutics to spontaneous chemical modifications by oxidation, cyclization, and elimination reactions. Amino Acids 2019, 51, 1409–1431. 10.1007/s00726-019-02787-2. [DOI] [PubMed] [Google Scholar]
- Nakagawa M.; Watanabe H.; Kodato S.; Okajima H.; Hino T.; Flippen J. L.; Witkop B. A valid model for the mechanism of oxidation of tryptophan to formylkynurenine-25 years later. Proc. Natl. Acad. Sci. U. S. A. 1977, 74 (11), 4730–4733. 10.1073/pnas.74.11.4730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarzinger S.; Kroon G. J.A.; Foss T. R.; Wright P. E.; Dyson H. J. Random coil chemical shifts in acidic 8 M urea: implementation of random coil shift data in NMRView. J. Biomol. NMR 2000, 18 (1), 43–48. 10.1023/A:1008386816521. [DOI] [PubMed] [Google Scholar]
- Wishart D. S.; Bigam C. G.; Holm A.; Hodges R. S.; Sykes B. D. 1H, 13C and 15N random coil NMR chemical shifts of the common amino acids. I. Investigations of nearest-neighbor effects. J. Biomol. NMR 1995, 5 (1), 67–81. 10.1007/BF00227471. [DOI] [PubMed] [Google Scholar]
- Bienkiewicz E. A.; Lumb K. J. Random-coil chemical shifts of phosphorylated amino acids. J. Biomol. NMR 1999, 15, 203–206. 10.1023/A:1008375029746. [DOI] [PubMed] [Google Scholar]
- Gaza-Bulseco G.; Faldu S.; Hurkmans K.; Chumsae C.; Liu H. Effect of methionine oxidation of a recombinant monoclonal antibody on the binding affinity to protein A and protein G. J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2008, 870 (1), 55–62. 10.1016/j.jchromb.2008.05.045. [DOI] [PubMed] [Google Scholar]
- Pavon J. A.; Xiao L.; Li X.; Zhao J.; Aldredge D.; Dank E.; Fridman A.; Liu Y.-H. Selective Tryptophan Oxidation of Monoclonal Antibodies: Oxidative Stress and Modeling Prediction. Anal. Chem. 2019, 91 (3), 2192–2200. 10.1021/acs.analchem.8b04768. [DOI] [PubMed] [Google Scholar]
- Folzer E.; Diepold K.; Bomans K.; Finkler C.; Schmidt R.; Bulau P.; Huwyler J.; Mahler H.-C.; Koulov A. V. Selective Oxidation of Methionine and Tryptophan Residues in a Therapeutic IgG1Molecule. J. Pharm. Sci. 2015, 104 (9), 2824–2831. 10.1002/jps.24509. [DOI] [PubMed] [Google Scholar]
- Li Y.; Polozova A.; Gruia F.; Feng J. Characterization of the degradation products of a color-changed monoclonal antibody: tryptophan-derived chromophores. Anal. Chem. 2014, 86 (14), 6850–6857. 10.1021/ac404218t. [DOI] [PubMed] [Google Scholar]
- Wong C.; Strachan-Mills C.; Burman S. Facile method of quantification for oxidized tryptophan degradants of monoclonal antibody by mixed mode ultra performance liquid chromatography. J. Chromatogr. A 2012, 1270, 153–161. 10.1016/j.chroma.2012.10.064. [DOI] [PubMed] [Google Scholar]
- Sreedhara A.; Lau K.; Li C.; Hosken B.; Macchi F.; Zhan D.; Shen A.; Steinmann D.; Schöneich C.; Lentz Y. Role of surface exposed tryptophan as substrate generators for the antibody catalyzed water oxidation pathway. Mol. Pharmaceutics 2013, 10 (1), 278–288. 10.1021/mp300418r. [DOI] [PubMed] [Google Scholar]
- Domingues M. R. M.; Domingues P.; Reis A.; Fonseca C.; Amado F. M. L.; Ferrer-Correia A. J. V. Identification of oxidation products and free radicals of tryptophan by mass spectrometry. J. Am. Soc. Mass Spectrom. 2003, 14 (4), 406–416. 10.1016/S1044-0305(03)00127-2. [DOI] [PubMed] [Google Scholar]
- Ronsein G. E.; Oliveira M. C. B.; Miyamoto S.; Medeiros M. H. G.; Di Mascio P. Tryptophan oxidation by singlet molecular oxygen O2(1Deltag). Chem. Res. Toxicol. 2008, 21 (6), 1271–1283. 10.1021/tx800026g. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Foote C. S.; Khan S. I. Reactions of N-acylated indoles with singlet oxygen. J. Org. Chem. 1993, 58 (1), 47–51. 10.1021/jo00053a014. [DOI] [Google Scholar]
- Maskos Z.; Rush J. D.; Koppenol W. H. The hydroxylation of tryptophan. Arch. Biochem. Biophys. 1992, 296 (2), 514–520. 10.1016/0003-9861(92)90605-V. [DOI] [PubMed] [Google Scholar]
- Simat; Steinhart J. Agric. Food Chem. 1998, 46 (2), 490–498. 10.1021/jf970818c. [DOI] [PubMed] [Google Scholar]
- Todorovski T.; Fedorova M.; Hennig L.; Hoffmann R. Synthesis of peptides containing 5-hydroxytryptophan, oxindolylalanine, N-formylkynurenine and kynurenine. J. Pept. Sci. 2011, 17 (4), 256–262. 10.1002/psc.1322. [DOI] [PubMed] [Google Scholar]
- Helland R.; Fjellbirkeland A.; Karlsen O. A.; Ve T.; Lillehaug J. R.; Jensen H. B. An oxidized tryptophan facilitates copper binding in Methylococcus capsulatus-secreted protein MopE. J. Biol. Chem. 2008, 283 (20), 13897–13904. 10.1074/jbc.M800340200. [DOI] [PubMed] [Google Scholar]
- van den Berg E. M. M.; Jansen F. J. H. M.; de Goede A. T. J. W.; Baldew A. U.; Lugtenburg J. Chemo-enzymatic synthesis and characterization of L-tryptophans selectively 13C-enriched or hydroxylated in the six-membered ring using transformed Escherichia coli cells. Recl. Trav. Chim. Pays-Bas 1990, 109 (4), 287–297. 10.1002/recl.19901090405. [DOI] [Google Scholar]
- Labroo R. B.; Cohen L. A. Preparative separation of the diastereoisomers of dioxindolyl-L-alanine and assignment of stereochemistry at C-3. J. Org. Chem. 1990, 55 (16), 4901–4904. 10.1021/jo00303a027. [DOI] [Google Scholar]
- Raillard S. P.; Mann A. D.; Baer T. A. An efficient procedure for the preparation of Fmoc-amino acids. Org. Prep. Proced. Int. 1998, 30 (2), 183–186. 10.1080/00304949809355277. [DOI] [Google Scholar]
- Kessler H.; Schmieder P.; Köck M.; Reggelin M. Selection of methyl resonances in proton-detected heteronuclear shift correlation, the hqqc experiment. J. Magn. Reson. 1991, 91 (2), 375–379. 10.1016/0022-2364(91)90199-4. [DOI] [Google Scholar]
- Markley J. L.; Bax A.; Arata Y.; Hilbers C. W.; Kaptein R.; Sykes B. D.; Wright P. E.; Wüthrich K. Recommendations for the presentation of NMR structures of proteins and nucleic acids. J. Biomol. NMR 1998, 12, 1–23. 10.1023/A:1008290618449. [DOI] [PubMed] [Google Scholar]
- Ulrich E. L.; Akutsu H.; Doreleijers J. F.; Harano Y.; Ioannidis Y. E.; Lin J.; Livny M.; Mading S.; Maziuk D.; Miller Z.; Nakatani E.; Schulte C. F.; Tolmie D. E.; Kent Wenger R.; Yao H.; Markley J. L. BioMagResBank. Nucleic Acids Res. 2007, 36, D402–8. 10.1093/nar/gkm957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee B. C.; Gladyshev V. N. The biological significance of methionine sulfoxide stereochemistry. Free Radical Biol. Med. 2011, 50 (2), 221–227. 10.1016/j.freeradbiomed.2010.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarzinger S.; Kroon G. J. A.; Foss T. R.; Chung J.; Wright P. E.; Dyson H. J. Sequence-Dependent Correction of Random Coil NMR Chemical Shifts. J. Am. Chem. Soc. 2001, 123 (13), 2970–2978. 10.1021/ja003760i. [DOI] [PubMed] [Google Scholar]
- Takizawa Y.; Fukushima H.; Usui T.; Yoshihara N.; Takizawa J. Isolation of Oxidation Products of Methionine Formed under the Condition of Active Oxygen Method. J. Jpn. Oil. Chem. Soc. 1992, 41 (5), 428–430. 10.5650/jos1956.41.428. [DOI] [Google Scholar]
- Holzmann J.; Hausberger A.; Rupprechter A.; Toll H. Top-down MS for rapid methionine oxidation site assignment in filgrastim. Anal. Bioanal. Chem. 2013, 405 (21), 6667–6674. 10.1007/s00216-013-7138-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X.; Zhang W.; Carmichael I.; Serianni A. S. Amide cis-trans isomerization in aqueous solutions of methyl N-formyl-D-glucosaminides and methyl N-acetyl-D-glucosaminides: chemical equilibria and exchange kinetics. J. Am. Chem. Soc. 2010, 132 (13), 4641–4652. 10.1021/ja9086787. [DOI] [PubMed] [Google Scholar]
- Quintanilla-Licea R.; Colunga-Valladares J.; Caballero-Quintero A.; Rodríguez-Padilla C.; Tamez-Guerra R.; Gómez-Flores R.; Waksman N. NMR Detection of Isomers Arising from Restricted Rotation of the C-N Amide Bond of N-Formyl-o-toluidine and N,N′-bis-Formyl-o-tolidine. Molecules 2002, 7 (8), 662–673. 10.3390/70800662. [DOI] [Google Scholar]
- Huang L.-J.; Chiang C.-W.; Lee Y.-W.; Wang T.-F.; Fong C.-C.; Chen S.-H. Characterization and comparability of stress-induced oxidation and deamidation on vulnerable sites of etanercept products. J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2016, 1032, 189–197. 10.1016/j.jchromb.2016.05.007. [DOI] [PubMed] [Google Scholar]
- Mo J.; Yan Q.; So C. K.; Soden T.; Lewis M. J.; Hu P. Understanding the Impact of Methionine Oxidation on the Biological Functions of IgG1 Antibodies Using Hydrogen/Deuterium Exchange Mass Spectrometry. Anal. Chem. 2016, 88 (19), 9495–9502. 10.1021/acs.analchem.6b01958. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.