

Conspectus
Complexes of lanthanide(III) ions are being actively studied because of their unique ground and excited state properties and the associated optical and magnetic behavior. In particular, they are used as emissive probes in optical spectroscopy and microscopy and as contrast agents in magnetic resonance imaging (MRI). However, the design of new complexes with specific optical and magnetic properties requires a thorough understanding of the correlation between molecular structure and electric and magnetic susceptibilities, as well as their anisotropies. The traditional Judd–Ofelt–Mason theory has failed to offer useful guidelines for systematic design of emissive lanthanide optical probes. Similarly, Bleaney’s theory of magnetic anisotropy and its modifications fail to provide accurate detail that permits new paramagnetic shift reagents to be designed rather than discovered.
A key determinant of optical and magnetic behavior in f-element compounds is the ligand field, often considered as an electrostatic field at the lanthanide created by the ligands. The resulting energy level splitting is a sensitive function of several factors: the nature and polarizability of the whole ligand and its donor atoms; the geometric details of the coordination polyhedron; the presence and extent of solvent interactions; specific hydrogen bonding effects on donor atoms and the degree of supramolecular order in the system. The relative importance of these factors can vary widely for different lanthanide ions and ligands. For nuclear magnetic properties, it is both the ligand field splitting and the magnetic susceptibility tensor, notably its anisotropy, that determine paramagnetic shifts and nuclear relaxation enhancement.
We review the factors that control the ligand field in lanthanide complexes and link these to aspects of their utility in magnetic resonance and optical emission spectroscopy and imaging. We examine recent progress in this area particularly in the theory of paramagnetic chemical shift and relaxation enhancement, where some long-neglected effects of zero-field splitting, magnetic susceptibility anisotropy, and spatial distribution of lanthanide tags have been accommodated in an elegant way.
Key References
Suturina E. A.; Mason K.; Geraldes C. F.; Kuprov I.; Parker D.. Beyond Bleaney’s Theory: Experimental and Theoretical Analysis of Periodic Trends in Lanthanide-Induced Chemical Shift. Angew. Chem., Int. Ed. 2017, 56, 12215–12218.1The orientation of the main component of the magnetic susceptibility tensor differs significantly for lanthanide complexes of a common ligand; thus, one of the key assumptions in Bleaney’s theory is incorrect.
Vonci M.; Mason K.; Suturina E. A.; Frawley A. T.; Worswick S. G.; Kuprov I.; Parker D.; McInnes E. J.; Chilton N. F.. Rationalization of Anomalous Pseudocontact Shifts and Their Solvent Dependence in a Series of C3-Symmetric Lanthanide Complexes. J. Am. Chem. Soc. 2017, 139, 14166–14172.2The sign and magnitude of the pseudocontact chemical shift, determined by the anisotropy of the magnetic susceptibility tensor, can be extremely sensitive to minimal structural changes, such as differential complex solvation.
Harnden A. C.; Suturina E. A.; Batsanov A. S.; Senanayake P. K.; Fox M. A.; Mason K.; Vonci M.; McInnes E. J.; Chilton N. F.; Parker D.. Unravelling the Complexities of Pseudocontact Shift Analysis in Lanthanide Coordination Complexes of Differing Symmetry. Angew. Chem. 2019, 131, 10396–10400.3A switch in the sign of the dominant ligand field parameter and large changes in the sense, amplitude, and orientation of the main component of the magnetic susceptibility tensor may occur simultaneously and hence hide smaller NMR pseudocontact shift changes.
Suturina E. A.; Mason K.; Geraldes C. F.; Chilton N. F.; Parker D.; Kuprov I.. Lanthanide-induced relaxation anisotropy. Phys. Chem. Chem. Phys. 2018, 20, 17676–17686. Detailed variable field proton relaxation rate analyses for isostructural series of lanthanide complexes reveal an angular dependence in both the dipolar and Curie mechanisms, demonstrated both experimentally and theoretically in a revised approach.
Electronic Structure Introduction
The unique electronic structure of trivalent 4f ions determines the distinctive properties of their coordination complexes. The electrostatic shielding of the electrons in 4f orbitals by fully occupied 5s and 5p orbitals makes the effects from surrounding ligands and other molecules far smaller than the interelectron repulsion and spin–orbit coupling (Figure 1). Due to these order-of-magnitude differences, electronic transitions in lanthanide(III) complexes are often independent of the ligand environment, and the ligand field splitting can be considered on the basis of the ground-state total angular momentum, J.
Figure 1.

Schematic representation of electronic states for Eu(III) (4f6): six electrons occupy seven degenerate 4f orbitals giving a 7F ground term in the Russell–Saunders coupling scheme (spectroscopic notation, 2S+1LJ), with total spin S = 3 and total orbital angular momentum L = 3. Spin–orbit coupling splits this term into seven J multiplets separated by about 103 cm–1. Each J state is (2J + 1)-fold degenerate for the free-ion; this degeneracy is partially removed upon loss of spherical symmetry. The separation of mJ states due to the ligand field is around 102 cm–1 but can be much larger.
The energy of mJ sublevels can be calculated using the crystal field theory that neglects mixing of f-orbitals with the orbitals of the ligands. For a given J multiplet, the model Hamiltonian has the form given in eq 1:
| 1 |
where Bqk are ligand field parameters, Ok are Stevens operators, and θk are operator equivalent coefficients (Table 1), defined for each term and multiplet in a given configuration.5,6 The Bqk parameters are defined in a particular reference frame; in symmetric molecules, the z-axis is usually aligned with the principal axes of the symmetry group, in which case the number of nonzero parameters is reduced.7,8 In the absence of symmetry, the expansion in eq 1 has 27 independent parameters. However, given sufficiently high symmetry or enough spectroscopic data, all nonzero ligand field parameters may be determined by luminescence spectroscopy.9,10 The principal parameter of interest to the NMR community is B0, due to the prevalence of Bleaney’s theory.11 As an example, for Eu(III), it may be extracted directly from the 5D0 to 7F1 transition (Figure 2).12−14 The Bqk parameters can be estimated from experimental data but are nowadays commonly obtained from multireference ab initio electronic structure methods, such as complete active space self-consistent field (CASSCF) calculations.15
Table 1. Equivalence Coefficients for the Low-Energy Terms of Late Ln(III) Ions6.
| Ln(III) | term | θ2 | θ4 | θ6 |
|---|---|---|---|---|
| Eu | 7F0 | 0 | 0 | 0 |
| 7F1 | –1/5 | 0 | 0 | |
| Tb | 7F6 | –1/99 | 2/(11 × 1485) | –1/(13 × 33 × 2079) |
| Dy | 6H15/2 | –2/(9 × 35) | –8/(11 × 45 × 273) | 4/(112×132×33×7) |
| Ho | 5I8 | –1/(30 × 15) | –1/(11 × 10 × 273) | –5/(112×132×33×7) |
| Er | 4I15/2 | 4/(45 × 35) | 2/(11 × 15 × 273) | 8/(112×132×33×7) |
| Tm | 3H6 | 1/99 | 8/(3 × 11 × 1485) | –5/(13 × 33 × 2079) |
| Yb | 2F7/2 | 2/63 | –2/(77 × 15) | 4/(13 × 33 × 63) |
Figure 2.

Europium emission spectra (295 K, MeOH, λexc 270 nm of [EuL8b] (lower)
and [EuL9]+ (upper) highlighting different splittings
of the ΔJ = 1 manifold for 5D0 → 7F1; in the spherical operator
formalism 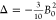 ,
, 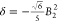 .15,16
.15,16
Because the emissive state 5D0 is nondegenerate, the splitting of the transition must arise from the ligand field splitting of the 7F1 multiplet (excluding vibrational effects). Since J = 1, the series in eq 1 terminates at k = 2, and when the complex has symmetry higher than C2, only B02 is nonzero and the spectrum exhibits two bands corresponding to the degenerate mJ = ±1 pair and the mJ = 0 singlet, whose separation is ∝B0. In lower symmetry, the degeneracy of the mJ = ±1 states is lifted and B±22 is nonzero. Therefore, the 5D0 → 7F1 band can be modeled with band-specific B0 and B22, which may differ slightly from the parameters determined by fitting all observable bands.14,16 The splittings are given as Δ = 3θ2B0 and δ = 2θ2B22, and θ2 = −1/5 (Table 1), where ligand field parameters are defined for Stevens operators, and the renormalization for more commonly used spherical tensors is given in the Figure 2 caption.13 The sign of Δ is positive if the mJ = 0 component of 7F1 is lower in energy than the barycenter of mJ = ±1 components, giving a singlet transition at higher energy than the doublet. Comparing the aza-phosphinate complexes [EuL8b] and [EuL9]+ (Figure 2), there is a change in the sign of B0, which is positive for the latter. The sign of these crystal field parameters is tightly linked to the local symmetry at the Eu(III) ion.12,14,17,18 Even though, Bqk parameters determined for Eu(III) complexes can be very similar to isostructural complexes of other lanthanide ions, Bq depends on the radial part of the f-electron wave function, which changes with nuclear charge, and small changes in bond lengths and angles may also affect the angular part of Bqk unexpectedly.
When the ligand field splitting is comparable to the splitting between spin–orbit multiplets, J is no longer a good quantum number, and the coupling scheme breaks down, for example, for Sm(III),19 leading to the phenomenon of “J mixing”, commonly invoked to explain unusual oscillator strengths and odd transitions in polarized emission spectra.20−22 Despite this, many other spectral phenomena defy explanation, and “J mixing” is often cited as a “catch-all”, highlighting limitations in current understanding.12,14,23
Lanthanide magnetic moments,24 which are often assumed to be independent of coordination environment25,26 also routinely show reductions in room-temperature susceptibility values compared to the free-ion due to the ligand field effect; a notable 11% reduction was found for Ho(III).27 Apart from the reduction of the average magnetic susceptibility, the ligand field also induces magnetic anisotropy that is the origin of paramagnetic NMR shifts and dramatically alters nuclear spin relaxation.
Overview of Factors Determining Ligand Field Splitting
The spectral behavior of several series of macrocyclic lanthanide(III) complexes [LnL1–9] has been studied, owing to their interest as emissive probes in optical spectroscopy and microscopy28−30 or contrast agents in magnetic resonance imaging.13,17,18
Design of complexes with desired optical and magnetic properties requires an understanding of correlations between molecular structure and the electromagnetic susceptibility tensors.31 These correlations are often assumed to follow simple models. However, Judd–Ofelt–Mason theory fails to offer guidelines for the design of emissive lanthanide optical probes,32−34 and similarly Bleaney’s theory of magnetic anisotropy11,35,36 has been widely used for NMR spectral fitting but provides no guidance for paramagnetic shift reagent design.37,38
Examination of the emission spectral properties of the [LnL1–9] series permits a dissection of the key factors determining the most important contribution to the ligand field. The size and sign of B02 varies widely across these series of complexes (Chart 1 and Table 2).
Chart 1.

Table 2. Values of Second Order Crystal Field Terms for Eu(III) Complexesa.
| complex | B02, cm–1 | B22, cm–1 |
|---|---|---|
| [EuLla] | <−200d | 0 |
| [EuL2a] | <+200d | 0 |
| [EuL3]3+ | +230d | 0 |
| [EuL4(H2O)]3+ | –470b | 0 |
| [EuL5a]− | –700 | 0 |
| [EuL5b]− | –650 | 0 |
| [EuL6] | –550 | –145 |
| [EuL7] | –455 | –120 |
| [EuL8a] | –660 | –122 |
| [EuL8b] | –650 | –80 |
| [EuL9]+ | +735c | –220c |
From emission spectra at 295 K in H2O. Ligand field parameter values quoted in the spherical tensor formalism.
With different axial donors, values changed dramatically, for example, MeCN (−630), DMF (−340), DMSO (−150), and HMPA (−85), and with fluoride replacing the coordinated water molecule, B02 has a positive sign.
In methanol, values were +920 and −153 cm–1.
Data recorded in methanol, not water, where values are smaller; the value for [EuL2a] represents an upper limit, owing to the lack of spectral resolution.
Variation of Complex Constitution and Symmetry
-
(i)
In the series of C3-symmetric complexes, [EuL1–3], the triacetate, triphosphinate, and triamide ligands gave values for B02, [EuL2] = [EuL1] < [Eu.L3]3+. The sign is negative for [EuL1] but positive for the other two in methanol.39 The polarizability of the oxygen donor atoms can be hypothesized to determine the multipolar ion–oxygen interaction energy.
-
(ii)
In the series of square antiprismatic complexes, [LnL4(S)]3+, the axial donor, S, can be permuted.40−43 When S is MeCN, B02 = −630 cm–1 and replacement of MeCN by a more polarizable oxygen donor is energetically favorable in the sequence: H2O < DMF < DMSO < HMPA (B0, −470 < −340 < −150 < −85 cm–1), correlating with the dipole moment change.44 When the axial donor is replaced by fluoride, B02 inverts sign causing a large change in magnetic susceptibility anisotropy, as the order of the mJ sublevels switches.31 The importance of the “axial component” of the ligand field was highlighted by Di Bari,45 examining spectral behavior of [YbL5]− complexes. Another example of switching sign in B0 for Yb(III) complexes combined NMR, EPR, and computational studies to track changes in the anisotropy of the magnetic susceptibility tensor.46
-
(iii)
For [EuL1a–e] (Chart 1), B02 changes as the para substituent in the pyridine ring varies. A linear correlation between the Hammett parameter, σp, and B0 (R2 = 0.97, in acetonitrile), is consistent with the strongly dipolar nature of the Ln–Npy interaction.46 The variation of overall ligand polarizability and its directionality, involving the electrostatic interaction between induced dipoles on the ligand and the quadrupole moment on the Ln3+ ion, is important in determining the “allowedness” of f–f electronic transitions.47,48 Thus, it is the overall ligand molecular polarizability that is important in determining the ligand field.
-
(iv)
Other examples of switches in the sign of B02 can be identified when complex constitution and local symmetry vary. The difference between the emission spectra of [EuL8b] and [EuL9]+ (Figure 2) is consistent with a change in sign, as symmetry changes from C1 to C2.3,14 Other cases have been reported, including systems involving reversible coordination of a polarizable N atom, which following protonation is replaced by water.49−51
Polyhedral Distortion
In point-charge ligand field theory, the geometric position and charge of each atom determine contributions to the ligand field potential. An axial anionic donor gives a positive contribution to B02, which becomes negative if it is in an equatorial position (switching at the “magic angle” θ ≈ 54.7° or 125.3°),52−55 leading to sensitivity of the ligand field potential to polyhedral distortion. The tricapped trigonal prismatic geometry is particularly sensitive, as noted by Binnemans, if all nine ligands are equivalent and the two sets of axial donors have polar angles 45° and 135°, leading to exact cancellation of all contributions and hence B0 = 0.56
The situation with [LnL1–3] is different. The first coordination sphere has three sets of donors: nitrogen atoms from the macrocycle (Nax) lie in axial positions (polar angle θ ≈ 142°); pyridyl N atoms in equatorial positions (Neq, θ ≈ 90°); carboxylate oxygens in axial sites (θ ≈ 50°).2 In [LnL1a], the two sets of N donors (Nax, Neq) give contributions to B02 of similar magnitude but opposite sign and cancel out; this is because the opposite of a ligand in an axial position is a ring of donors in the equatorial plane, and here the 3-fold equatorial disposition of Neq balances the Nax contribution. However, the oxygen donors lie close to the magic angle, and thus the ligand field is almost entirely ascribed to the oxygen atoms, resulting in an exquisite sensitivity of ligand field and magnetic anisotropy to very small variations in their angular position (Figure 3).2 Emission studies with [EuL1a] showed a pronounced dependence of B0 on solvent, suggesting that hydrogen bonding interactions with the oxygen donors could alter their effective polar angle θ. Indeed, the X-ray structure of [YbL1b] shows hydrogen bonding of water to the coordinated carboxylate oxygen, demonstrating this “tugging” on the donor oxygen. For [YbL1b], [YbL1e], and [EuL1e], both carbonyl and carboxylate oxygen atoms served as hydrogen bond acceptors to the water hydrogen atom.46
Figure 3.
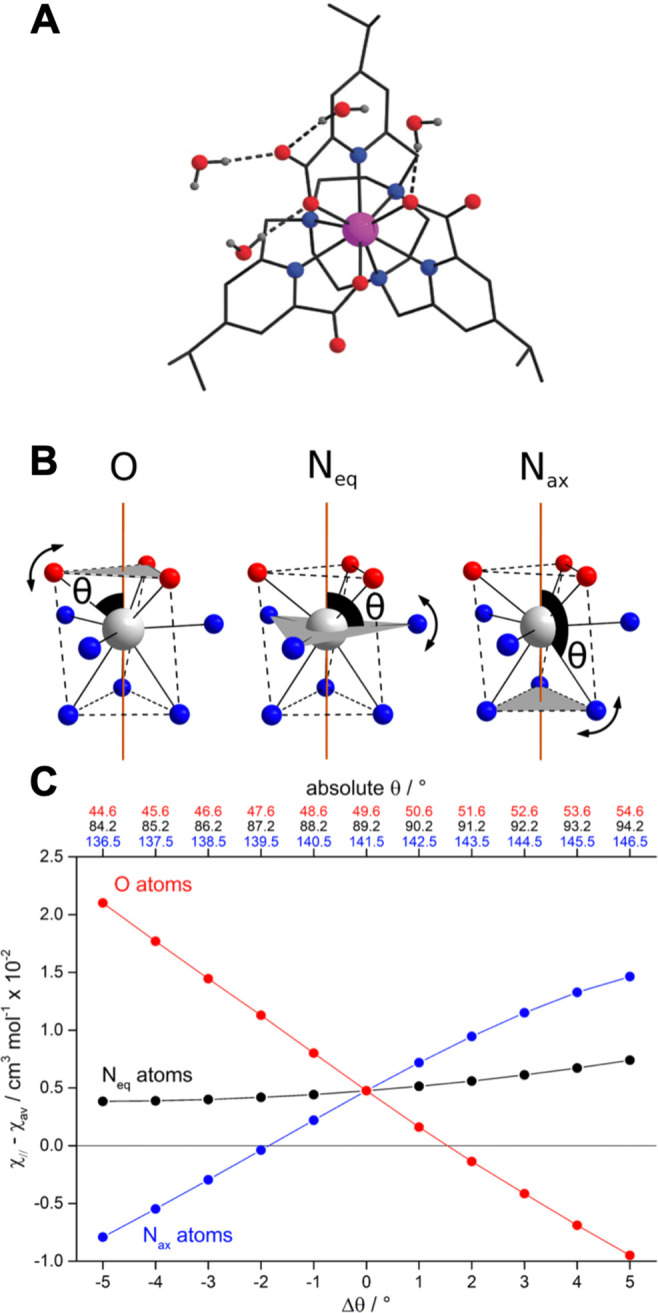
(A) X-ray crystal structure of [YbL1b], showing hydrogen bonding of water, tugging at the ligand oxygen atoms. (B) Schematic representation of the change in the polar angles θ for the oxygen, Neq (py) and Nax (ring) donor atoms in [DyL1a]. (C) Calculated room temperature magnetic susceptibility anisotropy arising from distortion, where Δθ is the deviation from the lowest energy structure.2
In 9-coordinate lanthanide complexes based on 12-N4 (e.g., DOTA18), the most common geometries are a monocapped square antiprism (SAP) and a twisted version (TSAP). The twist angle between upper and lower planes of four donor atoms found in X-ray analyses vary around 40° and 25°, respectively. Values of B02 for [EuL10–15(OH2)] (Chart 2, Table 3) show larger parameters in the SAP series.40−44,57 These variations relate to polyhedral distortion but may also be ascribed to changes in the axial water distances that are systematically longer (<0.3 Å) in the TSAP series due to increased steric demand. Such behavior is consistent with the concept of nonintegral metal ion hydration states, reducing in value between unity (Eu) and zero (Yb), through certain TSAP series, as the bond length to the water oxygen increases.40−43,58
Chart 2.

Table 3. Values of B02 (Spherical Tensor Formalism) Determined by Emission Analysis for [EuL10–15(OH2)].
|
B02, cm–1 |
||
|---|---|---|
| complex | SAP isomer | TSAP isomer |
| [EuL10] | –630 | –400 |
| (RRRR)-[EuL11]5– | –760 | –425 |
| (RRRS)-[EuL12]5– | –780 | –445 |
| (SSSS)-[EuL13]5– | –700 | –410 |
| [EuL14]3+ | –475 | –205 |
| [EuL15]3+ | –450 | –185 |
Supramolecular Effects: Solvation and the Degree of Aggregation
The nature of the solvent and the state of complex aggregation are supramolecular effects. For [EuL1b] and [EuL1a] where no solvent is bound, emission spectra change significantly with solvent, highlighted in the ΔJ = 1 manifold (Figure 4).46 The variation can be attributed to differing time-averaged orientations of solvent dipoles, perturbing the Ln–O and Ln–Npy dipolar and quadrupolar interactions, consistent with solvent multipolar effects.47,48,59−61 DOSY NMR studies of the diamagnetic analogue [YL1b] revealed clear evidence for aggregation that was greatest in chloroform and was positively correlated with the ligand field splitting.62 With [YL1a], in water only the monomer was evident, whereas in CD3CO2D and CF3CO2D, the aggregation state was 4 to 5.
Figure 4.

(left) ΔJ = 1 manifold for [EuL1a] in the stated solvents revealing the sign change of B02 in CF3CO2H. (right) Related emission spectra for [EuL1b] in the given solvents (298 K, λexc = 268 nm).46,62
In summary, ligand field splitting of lanthanide complexes is a sensitive function of several factors: the nature and polarizability of the ligand and its donors; the type and degree of polyhedral distortion; the presence and extent of solvent dipolar interactions; hydrogen bonding effects and the degree of supramolecular order. Each factor may be non-negligible in defining the ligand field, and their relative importance varies for different lanthanides.
Pseudocontact Shift and Bleaney’s Theory of Magnetic Anisotropy
When a lanthanide is treated as a point with second-rank magnetic susceptibility and infinitely fast magnetic relaxation, an additional isotropic shielding experienced by nearby nuclei is given by63
| 2 |
where θ, ϕ, and r are nuclear coordinates in the eigenframe of the magnetic susceptibility tensor. The eigenvalues of the traceless susceptibility tensor are labeled to satisfy the relation |χx| < |χy| < |χz|, with axiality χax = 3χz/2 and rhombicity χrh = (χx – χy)/2. Below we also use terms of χav = Tr(χ)/3 and χ∥ = χz + χav.
Bleaney’s theory of magnetic anisotropy11,35,36,64 shows that for a well-isolated J multiplet in the high temperature approximation, the anisotropy of the susceptibility tensor depends only on the second rank B02 and B2 ligand field parameters:
| 3 |
where CJ is Bleaney’s constant, defined for each lanthanide(III) ion (CJ = −158 (Tb), −181 (Dy), −71.2 (Ho), +58.8 (Er), +95.3 (Tm), and +39.2 (Yb)), and μB is the Bohr magneton.
Approximations and Their Limits
Assuming that the ligand field parameters do not vary between lanthanide ions, eq 3 suggests that χax/χrh remains constant within the series and PCS only varies due to the change in the value of CJ. However, if the overall ligand field splitting is greater than kT (Figure 5),28,63,65 the Bleaney formula is no longer valid, and χax/χrh and the eigenframe of the susceptibility tensor will depend on temperature.66 It is evident from low temperature measurements of [LnL10](H2O)]− that the principal axis changes direction by up to 90° from Tb to Yb.67−69
Figure 5.
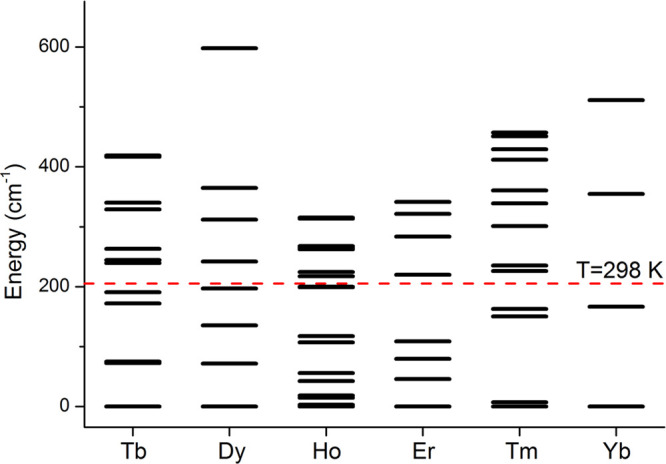
Energy splitting of the ground terms of [LnL8a] due to the ligand field, computed with CASSCF-SO in MOLCAS 8.0.1
Wave function calculations accounting for orbital degeneracy and correlation among the 4f electrons, as well as spin–orbit coupling (CASSCF-SO method), are used to determine ligand field splittings in lanthanide complexes.70,71 Such calculations (e.g., for [LnL8a], Figure 5) clearly show that in all cases the splitting is larger than kT. Thus, if eqs 2 and 3 are used to determine B02 and B2 from PCS data, the parameters may appear to be very different for each lanthanide simply because Bleaney’s approximations do not hold.72
Equation 2 also assumes a point magnetic source at the nuclear position of the lanthanide ion; a revised approach has recently emerged where the distribution of 4f electron density can be accounted for.73 There are two distinct reasons for such a distribution to occur: (i) spin delocalization and (ii) fast tag mobility. Disregarding the nature of the distribution, the mathematical formulation is the same. The effect of spin delocalization across ligands can be easily accounted for by ab initio calculation of the dipolar hyperfine tensors, but the tag mobility is often ignored despite the possible ∼30% deviation from a point model for nuclei close to the tag.74
Contact Contribution to Paramagnetic Shift
In most of the cases considered in this review, the proton contact shift is negligible compared to the PCS, and the point-dipole approximation in eq 2 is valid. The contact shift is proportional to the isotropic hyperfine coupling (itself related to spin density at the nucleus) and the isotropic magnetic moment of the lanthanide ion. Accounting for admixture of excited states with different J to the ground term, the isotropic magnetic moment can be corrected,75 and the ratio of contact contribution to the PCS can be estimated for different lanthanides provided that all other parameters in the series stay the same.76 Such estimations suggest that in the Tb–Yb series the most pronounced effect of a contact contribution is expected for Ho/Er.
NMR Shift Behavior of Systems with Large Magnetic Anisotropy
Detailed analyses of PCS data have been undertaken for isostructural complexes, with known solution speciation.28,38,77−81 A semiautomated combinatorial assignment procedure using PCS data, XRD structure and NMR relaxation rates to limit the combinatorial space (in Spinach82) was deployed for [LnL8a], enabling assignment of almost every proton resonance.1 Subsequently, the traceless part of the magnetic susceptibility tensor was obtained by fitting eq 2 to experimental data, giving excellent agreement (R2 > 0.99).
The experimentally determined susceptibility tensor can be displayed as a PCS field (Figure 6), revealing significant variations in the amplitude, shape, and orientation for the [LnL8a] series. Bleaney’s theory predicts that only the amplitude and sign should vary. However, the tensors change from almost fully rhombic (Dy and Tb; PCS field resembles dxz orbital) to near axial (Tm, PCS field resembles dz2 orbital). Critically the tilt angle β of the main anisotropy axis, relative to the molecular pseudosymmetry axis, varies significantly between complexes: Tb 8°; Dy 20°; Ho 22°; Er 8°; Tm 6°; Yb 23°.
Figure 6.
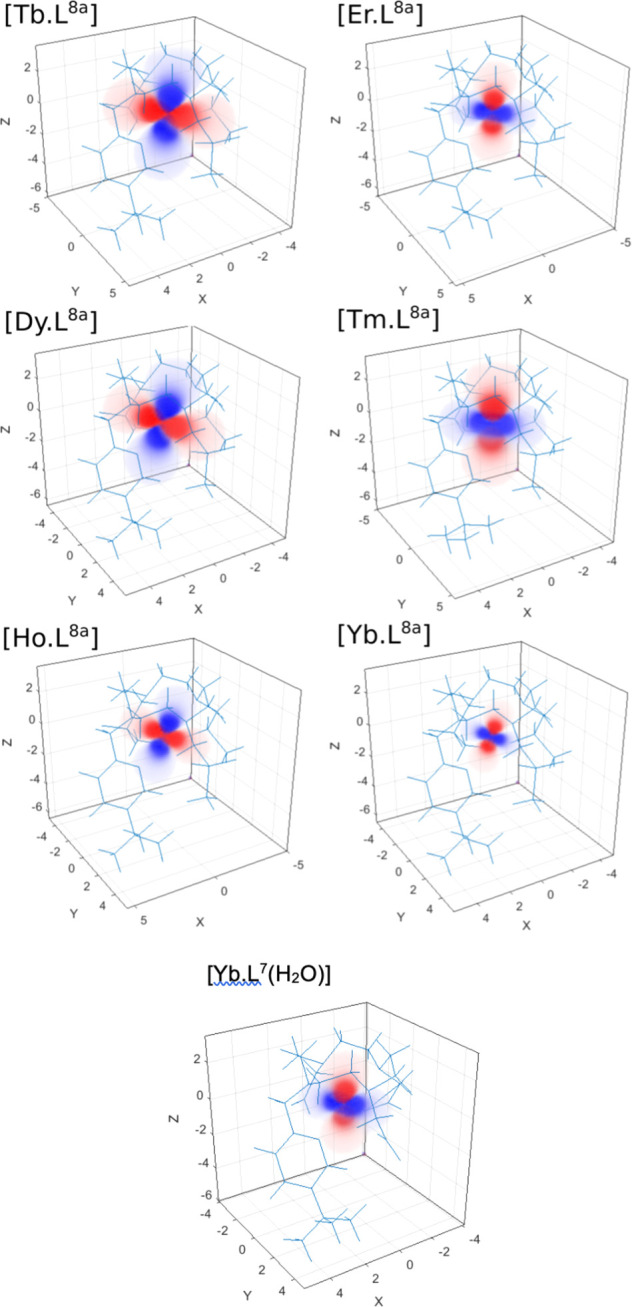
Pseudocontact shift fields for [LnL8a], reconstructed using Spinach81 with the “best-fit” magnetic susceptibility tensor. Positive PCS, red; negative, blue. Note changes in the orientation, size, and tilt of fields between [LnL8a] complexes1 and how the change in coordination in [YbL7(H2O)] vs [YbL8a] affects the PCS field.83
To illustrate the sensitivity of magnetic susceptibility anisotropy to structural change, consider PCS shifts for [YbL8b] and [YbL9]+ (Chart 3). The tBu NMR chemical shifts vary markedly across the series but appear in the same order, notwithstanding the B02 sign inversion (Figure 7 and Figure 2);3 Bleaney’s theory predicts the shift sense should be inverted.
Chart 3.

Figure 7.

(top) Schematic representation of tBu NMR shifts: [LnL8b] (upper), [LnL9]Cl (lower) (CD3OD, 11.7 T, 295 K) (yellow, Tm; green, Er; magenta, Yb; black, Ho; red, Dy; blue, Tb). (bottom) Pseudocontact shift fields for [YbL8b] (left) and [YbL9]+ (right). Positive PCS, red (+200 ppm); negative, blue (−200 ppm). Twist angles of each TSAP complex were 26.4° and 18.5°, that is, greater distortion in the cationic complex.3
The explanation lies in the magnetic susceptibility tensors, expressed in their very different PCS shift fields. While the second-order magnetic anisotropy changes sign, the negative PCS lobe is still oriented in the “equatorial plane”, because along with the change in sign of B02, there is a 90° rotation in orientation of the principal magnetic axis. Thus, the combined effect of the change in sign and orientation of the ligand field were shown to give rise to similar PCS fields for the tBu protons, explaining the “hidden” changes in PCS behavior.3
NMR Shift Behavior of Systems with Small Ligand Field Splittings
Complexes [LnL1–3] adopt tricapped trigonal prismatic structures and possess small ligand field splittings close to kT. Yet, their PCS values do not conform to Bleaney’s theory.31,37,38 Both the sign and magnitude of their ligand field parameters are sensitive to local polarity changes and polyhedral distortion. They are particularly sensitive to perturbation of the polar angle of oxygen donor atoms, θ, defining the angle subtended by the Ln–O vector compared to the C3 axis. As θ lies close to the “magic” angle, small variations cause major changes in magnetic susceptibility anisotropy.2,46
For [YbL1b], DFT was used to determine a pseudosolution structure with imposed C3 symmetry and CASSCF-SO calculations gave the anisotropy of the susceptibility tensor (squares, Figure 8). The experimental values of χ∥ – χav were determined assuming a fixed structural model based on experimental PCS, referenced to the diamagnetic Y(III) complex. A comparison was then made with the CASSCF-SO-calculated susceptibility anisotropy, to determine the “spectroscopic” average value of θ in solution. In [YbL1b], the diastereotopic methyl groups of the isopropyl substituent serve as a local probe of magnetic anisotropy. The PCS fields in acetone, water, and methanol highlight the sensitivity to solvent. The PCS field changes sign as the magnetic susceptibility anisotropy switches from “easy axis” to “easy plane” in D2O.46 Similar solvent dependences were found for [LnL1a] (Dy, Er, Eu).2
Figure 8.

(left) Schematic representation of PCS (295 K, 4.7 T) for pyridyl H3, H5, and iPr resonances of [YbL1b] and variation in the susceptibility anisotropy with θ: D2O (blue); CD3OD (green); CD3CN (purple), DMSO-d6 (red), acetone-d6 (orange); diastereotopic methyl resonances are isochronous in D2O. (right) PCS fields for [YbL1b], (using Spinach82): positive PCS, red; negative, blue.46
The sensitivity of magnetic anisotropy in these lanthanide complexes with small ligand field splittings was shown to have a major impact on solid-state EPR behavior.84 The magnetic and spectroscopic properties depend upon a number of factors that cannot be disentangled: a distribution of structural parameters generates a range of B02 values; an electronic structure sensitive to thermal changes of the ligand structure; thermally accessible EPR-active excited states; disordered solvation influencing the local ligand field. Each effect is present across the [LnL1–3] series, making interpretation of EPR spectra very difficult for systems with small magnetic anisotropy.84
NMR Shift Behavior of Mobile Lanthanide Tags on Proteins
Complexes with large magnetic anisotropies are often used as tags in protein NMR to provide structural constraints,85,86 where large magnetic anisotropy is preferred so that PCS is measurable even at distances of 40 Å. The tag is often attached by a flexible linker, but mobility results in big deviations from the point-dipole approximation, at <15 Å.
A generalization of McConnell’s expression, eq 4, was derived for lanthanide tag mobility in protein NMR:73,74,87
| 4 |
where ∇⃗ is the gradient operator, ρ(r) is the probability distribution of the spatial position of the lanthanide tag, and subscript t indicates the traceless part of the magnetic susceptibility tensor. The susceptibility is assumed to be the same in every point of the probability density.74 The latter assumption may be lifted, but the corresponding equation is considerably harder to solve. The partial differential eq 4 can be solved using three-dimensional Fourier transforms:74
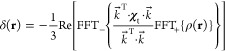 |
5 |
where FFT+ refers to the forward fast-Fourier transform and FFT– the inverse. If the probability density is defined on a grid, numerical solution of eq 5 gives the PCS values. The solution of the inverse problem is possible; one can extract probability density from PCS data. Numerical solvers for both direct and inverse problems are available.82 The resulting lanthanide probability densities from PCS are in agreement with Double Electron–Electron Resonance (DEER) spectroscopy, and PCS fits are significantly improved near the tag (Figure 9).87
Figure 9.
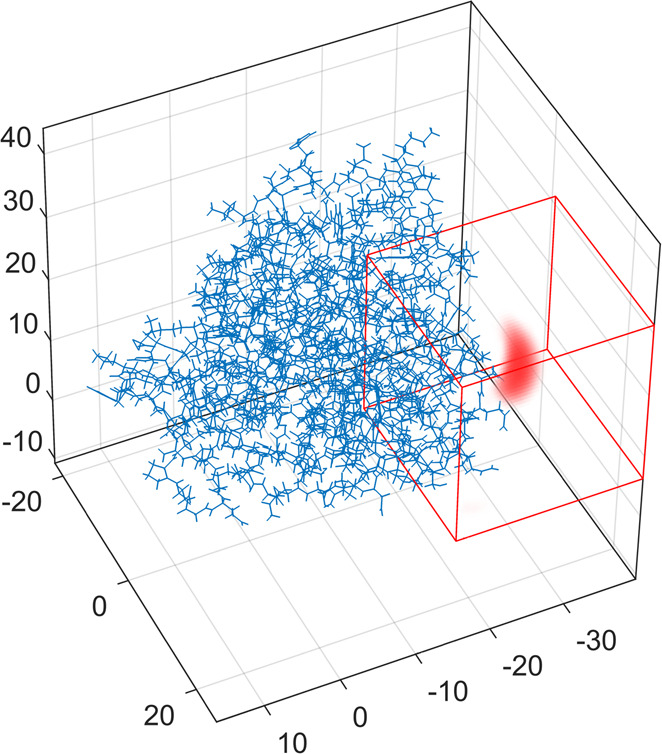
Tm3+ ion distribution (red) in a DOTA-M8 tagged S50C mutant of human carbonic anhydrase II (blue), extracted from PCS data. The red cube indicates the volume where the probability density can vary during fitting. The source code is available in Spinach;82 axes in Å.
Lanthanide Relaxation and Its Anisotropy
Common MRI contrast agents contain magnetically isotropic Gd3+ ions. Their long electron relaxation times mean that PCS is absent, and the effect is only to accelerate nuclear relaxation.88 Likewise, relaxation enhancement experiments in NMR often use magnetically isotropic Mn2+ or Gd3+ complexes to maximize the volume affected by the metal and minimize PCS.89 Following nuclear relaxation enhancement models designed for these ions, it has often been assumed that the enhancements show a simple 1/r6 dependence on the electron–nuclear distance, without angular terms in the molecular frame of reference.
Nuclear relaxation enhancement by unpaired f electrons of lanthanide complexes has two principal components. One (“dipolar relaxation”) comes from stochastic modulation of the electron–nuclear dipolar interaction and the other (“Curie relaxation”) from rotational modulation of extra nuclear shielding caused by the presence of the unpaired electron. The angular dependence in non-Gd lanthanides4 was first acknowledged for Curie relaxation.90 The reasons are twofold. First, magnetic susceptibility tensor anisotropy can be as large as the isotropic part, contradicting the assumption made by Gueron when he derived Curie relaxation theory.91 Second, zero field splitting can be much stronger than the electron Zeeman interaction, the opposite limit from the classical Solomon–Bloembergen–Morgan theory of lanthanide-induced dipolar relaxation.92
Experimental proof came from relaxation rate measurements in complexes where all nuclei in the ligand cages could be unambiguously assigned, and atomic coordinate estimates were available from DFT calculations28,37,38,93−95 (Chart 1 and Figure 10).
Figure 10.

Longitudinal relaxation rates in [LnL8a] complexes as functions of Ln–H distance (r–6, D2O, 295 K, 1 T), for 12-N4 ring protons (blue circles), ligand arms (red triangles), and pyridine protons (black squares). In the Yb set, axial and equatorial protons are indicated. A pure r–6 dependence is a straight line.4
It is obvious from Figure 10 that nuclear relaxation enhancements at low magnetic field (1 T) do not depend simply on the distance to the lanthanide ion. The relaxation rates also appear to depend on the sign of the magnetic anisotropy: in complexes with easy-plane anisotropy (Tb, Dy, Ho), ligand arm protons relax faster than macrocyclic ring protons; the opposite is true for the complexes with easy-axis anisotropy (Er, Tm, Yb).
Encouraged by these findings, we updated the dipolar relaxation theory4 to include the direction of the Ln–H vector in the molecular frame:
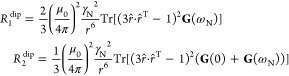 |
6 |
| 7 |
where the spectral power density G(ω) is no longer a scalar but a tensor accommodating stochastic dynamics of the electron spin as well as molecular accommodating stochastic dynamics of the electron spin as well as molecular rotation, and r̂ is the unit vector pointing in the same direction as r⃗; further details may be found in the paper cited above.
Similar observations were made at higher field 9.4 T for [LnL8a] complexes (Ln = Tb–Yb), where the Curie contribution dominates. For Curie relaxation, it turned out to be essential to account for the antisymmetric component in the total nuclear shielding tensor
| 8 |
that includes diamagnetic shielding tensor σ0 and paramagnetic shielding tensor, which is proportional to the dipolar matrix D and magnetic susceptibility tensor χ. This is necessary because the antisymmetric part is significant here; the product of two symmetric matrices is only symmetric when they commute. With the relevant extra terms in place, the Curie relaxation rates become
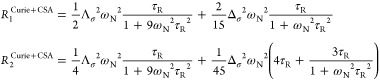 |
9 |
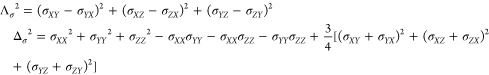 |
10 |
here, Λσ2 is the first and Δσ2 the second rank invariant of the chemical shielding tensor. These equations have been incorporated into Spinach;82 for [LnL8a], the modifications yielded a much better agreement with experiment (Figure 11).4
Figure 11.

Experimental longitudinal relaxation rates (black dots) for the ligand cage nuclei of [LnL8a] in D2O at 295 K and 1 T. The calculated rates are shown as bars and color-coded by the mechanism.4
In summary, the presence of magnetic anisotropy required a fundamental update of the relevant nuclear relaxation theories. These updates revealed strong molecular-frame angular dependencies in paramagnetic relaxation enhancements. In systems with large magnetic anisotropy and at short electron–nuclear distances, the classical Solomon–Bloembergen–Morgan and Gueron expressions should not be used.
Summary and Conclusions
The ligand field for a lanthanide complex varies with the nature of the ligand, metal ion, and its environment. The size and sign of ligand field parameters are difficult to determine experimentally, but information can be gained using optical spectroscopy with Eu(III) complexes.9 They are sensitive to several factors including the nature and polarizability of the overall ligand and donor atoms, the type and degree of geometric distortion, the extent of solvent dipolar interactions, and specific hydrogen bonding effects and the degree of supramolecular order.
Bleaney’s magnetic anisotropy theory provided guidance in rationalizing NMR PCS data. However, its crude approximations and limitations are apparent. In explaining the nature and magnitude of PCS data, both the ligand field splitting and the type, size, and orientation of the principal component of the magnetic susceptibility tensor are key. The latter can be determined by careful magneto-structural correlations27,31,2,46 assessed by VT magnetic susceptibility measurements, low temperature EPR studies, and modern computational methods.
Considerable caution is needed using PCS data for structural refinement. Such methods are used in biomolecular analyses but may fail when the lanthanide ion is permuted. Delving more deeply, the ordering, nature, and relative Boltzmann population of the mJ sublevels for a given lanthanide ion complex is key to understanding the overall magnetic susceptibility and its directional dependences.
The nuclear relaxation induced by lanthanide ions can be anisotropic in the molecular frame, and accounting for this anisotropy can drastically improve agreement between experiment and theoretical models. Analyses based only on distance variations are a crude approximation for both dipolar and Curie relaxation mechanisms. Biomolecular structural refinement using lanthanide spin tags must account for this anisotropy or risk significant errors; any work using simple 1/r6 models for lanthanide labeled systems should be considered with appropriate caution.
Acknowledgments
We thank EPSRC for grant support (EP/N007034/1, EP/N006909/1, EP/N006895/1) and talented colleagues in Manchester, Southampton, Bath, and Durham whose names appear in the reference list for their conscientiousness, resilience, and excellence.
Biographies
David Parker was born in Leadgate, England, on 30th July 1956. Educated in the state sector, he read Chemistry at Oxford University (1974–1978) and remained there to study with John Brown for a DPhil, which he gained in 1980. Following a NATO Fellowship in Strasbourg with Jean-Marie Lehn, he returned to the NE of England in early 1982 as a Lecturer in Chemistry at Durham University. He gained several national and international awards and prizes for work that embraces several aspects of contemporary chemistry, including the design of sensors, targeted imaging probes, and therapeutic agents, as well as major contributions to chiral analysis and mechanistic studies. He is a Professor of Chemistry at Durham University.
Elizaveta Suturina studied physics at Novosibirsk State University where she obtained her undergraduate degree and DPhil in chemical physics working in the group of Nina Gritsan. She then moved to work in Max-Planck Institute with Frank Neese and Mihail Atanasov on ab initio modeling of molecular magnetic properties and then joined Ilya Kuprov’s group to work on NMR theory and modelling of paramagnetic systems. In 2018, she was awarded the Prize fellowship from the University of Bath where she is currently establishing her research group.
Ilya Kuprov is a magnetic resonance specialist and the leading author of Spinach, an advanced spin dynamics simulation package. He did his undergraduate degrees at Novosibirsk State University before moving to Oxford for a DPhil. He was a Lecturer at Durham University and is presently an Associate Professor at Southampton. He is the Secretary of the Electron Spin Resonance Group of the Royal Society of Chemistry and an Associate Editor at Science Advances. He has published about a hundred papers, principally on the theoretical and computational aspects of magnetic resonance spectroscopy and imaging.
Nicholas Chilton obtained his B.Sc. Adv. (Hons.) in chemistry in the group of Prof. Keith Murray at Monash University, Australia, before moving to the UK in 2013 to undertake his Ph.D. with Profs. Richard Winpenny and Eric McInnes at The University of Manchester. In 2016, he obtained a Ramsay Memorial Research Fellowship and started his own research group at The University of Manchester, followed by a Presidential Fellowship in 2018 and a Royal Society University Research Fellowship in 2019. He is currently a Senior Lecturer at The University of Manchester and holds a number of prizes and awards.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
References
- Suturina E. A.; Mason K.; Geraldes C. F.; Kuprov I.; Parker D. Beyond Bleaney’s Theory: Experimental and Theoretical Analysis of Periodic Trends in Lanthanide-Induced Chemical Shift. Angew. Chem., Int. Ed. 2017, 56, 12215–12218. 10.1002/anie.201706931. [DOI] [PubMed] [Google Scholar]
- Vonci M.; Mason K.; Suturina E. A.; Frawley A. T.; Worswick S. G.; Kuprov I.; Parker D.; McInnes E. J.; Chilton N. F. Rationalization of Anomalous Pseudocontact Shifts and Their Solvent Dependence in a Series of C3-Symmetric Lanthanide Complexes. J. Am. Chem. Soc. 2017, 139, 14166–14172. 10.1021/jacs.7b07094. [DOI] [PubMed] [Google Scholar]
- Harnden A. C.; Suturina E. A.; Batsanov A. S.; Senanayake P. K.; Fox M. A.; Mason K.; Vonci M.; McInnes E. J.; Chilton N. F.; Parker D. Unravelling the Complexities of Pseudocontact Shift Analysis in Lanthanide Coordination Complexes of Differing Symmetry. Angew. Chem. 2019, 131, 10396–10400. 10.1002/ange.201906031. [DOI] [PubMed] [Google Scholar]
- Suturina E. A.; Mason K.; Geraldes C. F.; Chilton N. F.; Parker D.; Kuprov I. Lanthanide-induced relaxation anisotropy. Phys. Chem. Chem. Phys. 2018, 20, 17676–17686. 10.1039/C8CP01332B. [DOI] [PubMed] [Google Scholar]
- Stevens K. Matrix elements and operator equivalents connected with the magnetic properties of rare earth ions. Proc. Phys. Soc., London, Sect. A 1952, 65, 209. 10.1088/0370-1298/65/3/308. [DOI] [Google Scholar]
- Stevens K. W. H. Matrix Elements and Operator Equivalents Connected with the Magnetic Properties of Rare Earth Ions. Proc. Phys. Soc., London, Sect. A 1952, 65, 209–215. 10.1088/0370-1298/65/3/308. [DOI] [Google Scholar]
- Mulak J.; Gajek Z.. The Effective Crystal Field Potential; Elsevier, 2000. [Google Scholar]
- Eyring L.; Gschneidner K. A.; Lander G. H.. Handbook on the Physics and Chemistry of Rare Earths; Elsevier, 2002; Vol. 32. [Google Scholar]
- Wybourne B. G.; Smentek L.. Optical Spectroscopy of Lanthanides: Magnetic and Hyperfine Interactions; CRC Press, 2007. [Google Scholar]
- Bünzli J.-C. G.; Eliseeva S. V.. Basics of Lanthanide Photophysics. In Lanthanide Luminescence: Photophysical, Analytical and Biological Aspects; Hänninen P., Härmä H., Eds.; Springer Berlin Heidelberg: Berlin, Heidelberg, 2011; pp 1–45. [Google Scholar]
- Bleaney B. Nuclear magnetic resonance shifts in solution due to lanthanide ions. J. Magn. Reson. (1969-1992) 1972, 8, 91–100. 10.1016/0022-2364(72)90027-3. [DOI] [Google Scholar]
- Binnemans K.; Görller-Walrand C. A simple model for crystal field splittings of the 7F1 and 5D1 energy levels of Eu3+. Chem. Phys. Lett. 1995, 245, 75–78. 10.1016/0009-2614(95)00984-C. [DOI] [Google Scholar]
- Finney K. L. N.; Harnden A. C.; Rogers N. J.; Senanayake P. K.; Blamire A. M.; O’Hogain D.; Parker D. Simultaneous triple imaging with two PARASHIFT probes: encoding anatomical, pH and temperature information using magnetic resonance shift imaging. Chem. - Eur. J. 2017, 23, 7976–7989. 10.1002/chem.201700447. [DOI] [PubMed] [Google Scholar]
- Binnemans K. Interpretation of europium(III) spectra. Coord. Chem. Rev. 2015, 295, 1–45. 10.1016/j.ccr.2015.02.015. [DOI] [Google Scholar]
- Ungur L.; Chibotaru L. F. Ab Initio Crystal Field for Lanthanides. Chem. - Eur. J. 2017, 23, 3708–3718. 10.1002/chem.201605102. [DOI] [PubMed] [Google Scholar]
- Görller-Walrand C.; Binnemans K. Chapter 155 Rationalization of crystal-field parametrization. In Handbook on the Physics and Chemistry of Rare Earths; Elsevier 1996, 23, 121–283. 10.1016/S0168-1273(96)23006-5. [DOI] [Google Scholar]
- Senanayake P. K.; Rogers N. J.; Finney K. L. N.; Harvey P.; Funk A. M.; Wilson J. I.; O’Hogain D.; Maxwell R.; Parker D.; Blamire A. M. A new paramagnetically shifted imaging probe for MRI. Magn. Reson. Med. 2017, 77, 1307–1317. 10.1002/mrm.26185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harnden A. C.; Parker D.; Rogers N. J. Employing paramagnetic shift for responsive MRI probes. Coord. Chem. Rev. 2019, 383, 30–42. 10.1016/j.ccr.2018.12.012. [DOI] [Google Scholar]
- Liu J.; Reta D.; Cleghorn J. A.; Yeoh Y. X.; Ortu F.; Goodwin C. A.; Chilton N. F.; Mills D. P. Light Lanthanide Metallocenium Cations Exhibiting Weak Equatorial Anion Interactions. Chem. - Eur. J. 2019, 25, 7749–7758. 10.1002/chem.201901167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma C.-G.; Brik M.; Kiisk V.; Kangur T.; Sildos I. Spectroscopic and crystal-field analysis of energy levels of Eu3+ in SnO2 in comparison with ZrO2 and TiO2. J. Alloys Compd. 2011, 509, 3441–3451. 10.1016/j.jallcom.2010.12.071. [DOI] [Google Scholar]
- Auzel F.; Malta O. A scalar crystal field strength parameter for rare-earth ions: meaning and usefulness. J. Phys. (Paris) 1983, 44, 201–206. 10.1051/jphys:01983004402020100. [DOI] [Google Scholar]
- Malta O.; Antic-Fidancev E.; Lemaitre-Blaise M.; Milicic-Tang A.; Taibi M. The crystal field strength parameter and the maximum splitting of the 7F1 manifold of the Eu3+ ion in oxides. J. Alloys Compd. 1995, 228, 41–44. 10.1016/0925-8388(95)01645-7. [DOI] [Google Scholar]
- Gendron F.; Moore B. II; Cador O.; Pointillart F.; Autschbach J.; Le Guennic B. Ab Initio Study of Circular Dichroism and Circularly Polarized Luminescence of Spin-Allowed and Spin-Forbidden Transitions: From Organic Ketones to Lanthanide Complexes. J. Chem. Theory Comput. 2019, 15, 4140–4155. 10.1021/acs.jctc.9b00286. [DOI] [PubMed] [Google Scholar]
- Souza A.; Dos Santos M. C. The J-mixing effect in Ln3+ ions crystal field levels. Chem. Phys. Lett. 2012, 521, 138–141. 10.1016/j.cplett.2011.10.060. [DOI] [Google Scholar]
- Jensen J.; Mackintosh A. R.. Rare Earth Magnetism; Clarendon Press Oxford, 1991. [Google Scholar]
- Kurzen H.; Bovigny L.; Bulloni C.; Daul C. Electronic structure and magnetic properties of lanthanide 3+ cations. Chem. Phys. Lett. 2013, 574, 129–132. 10.1016/j.cplett.2013.04.070. [DOI] [Google Scholar]
- Hölsä J.; Lastusaari M.; Niittykoski J.; Puche R. S. Interplay between crystal structure and magnetic susceptibility of tetragonal ROBr. Phys. Chem. Chem. Phys. 2002, 4, 3091–3097. 10.1039/b110745n. [DOI] [Google Scholar]
- Butler S. J.; Delbianco M.; Lamarque L.; McMahon B. K.; Neil E. R.; Pal R.; Parker D.; Walton J. W.; Zwier J. M. EuroTracker® dyes: design, synthesis, structure and photophysical properties of very bright europium complexes and their use in bioassays and cellular optical imaging. Dalton Transactions 2015, 44, 4791–4803. 10.1039/C4DT02785J. [DOI] [PubMed] [Google Scholar]
- Delbianco M.; Sadovnikova V.; Bourrier E.; Mathis G.; Lamarque L.; Zwier J. M.; Parker D. Bright, Highly Water-Soluble Triazacyclononane Europium Complexes To Detect Ligand Binding with Time-Resolved FRET Microscopy. Angew. Chem., Int. Ed. 2014, 53, 10718–10722. 10.1002/anie.201406632. [DOI] [PubMed] [Google Scholar]
- Shuvaev S.; Starck M.; Parker D. Responsive, Water-Soluble Europium (III) Luminescent Probes. Chem. - Eur. J. 2017, 23, 9974–9989. 10.1002/chem.201700567. [DOI] [PubMed] [Google Scholar]
- Blackburn O. A.; Edkins R. M.; Faulkner S.; Kenwright A. M.; Parker D.; Rogers N. J.; Shuvaev S. Electromagnetic susceptibility anisotropy and its importance for paramagnetic NMR and optical spectroscopy in lanthanide coordination chemistry. Dalton Transactions 2016, 45, 6782–6800. 10.1039/C6DT00227G. [DOI] [PubMed] [Google Scholar]
- Ofelt G. Intensities of crystal spectra of rare-earth ions. J. Chem. Phys. 1962, 37, 511–520. 10.1063/1.1701366. [DOI] [Google Scholar]
- Judd B. R. Optical absorption intensities of rare-earth ions. Phys. Rev. 1962, 127, 750. 10.1103/PhysRev.127.750. [DOI] [PubMed] [Google Scholar]
- Jørgensen C. K.; Judd B. Hypersensitive pseudoquadrupole transitions in lanthanides. Mol. Phys. 1964, 8, 281–290. 10.1080/00268976400100321. [DOI] [Google Scholar]
- Piguet C.; Geraldes C. F. Paramagnetic NMR lanthanide induced shifts for extracting solution structures. Handbook on the Physics and Chemistry of Rare Earths 2003, 33, 353–463. 10.1016/S0168-1273(02)33005-8. [DOI] [Google Scholar]
- Bertini I.; Luchinat C.; Parigi G. Magnetic susceptibility in paramagnetic NMR. Prog. Nucl. Magn. Reson. Spectrosc. 2002, 40, 249. 10.1016/S0079-6565(02)00002-X. [DOI] [Google Scholar]
- Funk A. M.; Finney K.-L. N.; Harvey P.; Kenwright A. M.; Neil E. R.; Rogers N. J.; Senanayake P. K.; Parker D. Critical analysis of the limitations of Bleaney’s theory of magnetic anisotropy in paramagnetic lanthanide coordination complexes. Chemical Science 2015, 6, 1655–1662. 10.1039/C4SC03429E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro G.; Regueiro-Figueroa M.; Esteban-Gómez D.; Pérez-Lourido P.; Platas-Iglesias C.; Valencia L. Magnetic anisotropies in rhombic lanthanide (III) complexes do not conform to Bleaney’s theory. Inorg. Chem. 2016, 55, 3490–3497. 10.1021/acs.inorgchem.5b02918. [DOI] [PubMed] [Google Scholar]
- Suturina E. A.; Mason K.; Botta M.; Carniato F.; Kuprov I.; Chilton N. F.; McInnes E. J.; Vonci M.; Parker D. Periodic trends and hidden dynamics of magnetic properties in three series of triazacyclononane lanthanide complexes. Dalton Transactions 2019, 48, 8400–8409. 10.1039/C9DT01069F. [DOI] [PubMed] [Google Scholar]
- Beeby A.; Clarkson I. M.; Dickins R. S.; Faulkner S.; Parker D.; Royle L.; de Sousa A. S.; Williams J. A. G.; Woods M. Non-radiative deactivation of the excited states of europium, terbium and ytterbium complexes by proximate energy-matched OH, NH and CH oscillators: an improved luminescence method for establishing solution hydration states. J. Chem. Soc., Perkin Trans. 2 1999, 2, 493–504. 10.1039/a808692c. [DOI] [Google Scholar]
- Aime S.; Botta M.; Parker D.; Williams J. G. Extent of hydration of octadentate lanthanide complexes incorporating phosphinate donors: Solution relaxometry and luminescence studies. J. Chem. Soc., Dalton Trans. 1996, 17–23. 10.1039/dt9960000017. [DOI] [Google Scholar]
- Woods M.; Aime S.; Botta M.; Howard J. A.; Moloney J. M.; Navet M.; Parker D.; Port M.; Rousseaux O. Correlation of water exchange rate with isomeric composition in diastereoisomeric gadolinium complexes of tetra (carboxyethyl) dota and related macrocyclic ligands. J. Am. Chem. Soc. 2000, 122, 9781–9792. 10.1021/ja994492v. [DOI] [Google Scholar]
- Kotek J.; Rudovský J.; Hermann P.; Lukeš I. Three in One: TSA, TSA ‘, and SA Units in One Crystal Structure of a Yttrium (III) Complex with a Monophosphinated H4dota Analogue. Inorg. Chem. 2006, 45, 3097–3102. 10.1021/ic060006a. [DOI] [PubMed] [Google Scholar]
- Dickins R. S.; Parker D.; Bruce J. I.; Tozer D. J. Correlation of optical and NMR spectral information with coordination variation for axially symmetric macrocyclic Eu (III) and Yb (III) complexes: axial donor polarisability determines ligand field and cation donor preference. Dalton Transactions 2003, 1264–1271. 10.1039/b211939k. [DOI] [Google Scholar]
- Bari L. D.; Pintacuda G.; Salvadori P.; Dickins R. S.; Parker D. Effect of axial ligation on the magnetic and electronic properties of lanthanide complexes of octadentate ligands. J. Am. Chem. Soc. 2000, 122, 9257–9264. 10.1021/ja0012568. [DOI] [Google Scholar]
- Mason K.; Harnden A. C.; Patrick C. W.; Poh A. W.; Batsanov A. S.; Suturina E. A.; Vonci M.; McInnes E. J.; Chilton N. F.; Parker D. Exquisite sensitivity of the ligand field to solvation and donor polarisability in coordinatively saturated lanthanide complexes. Chem. Commun. 2018, 54, 8486–8489. 10.1039/C8CC04995E. [DOI] [PubMed] [Google Scholar]
- Richardson F. S. On the calculation of electric dipole strengths of 4f→ 4f transitions in lanthanide complexes. Chem. Phys. Lett. 1982, 86, 47–50. 10.1016/0009-2614(82)83114-X. [DOI] [Google Scholar]
- Kuroda R.; Mason S. F.; Rosini C. Crystal structure and single-crystal spectra of Gd (Eu) Al 3 (BO 3) 4. Anisotropic ligand polarization contributions to the f–f transition probabilities in Eu III. J. Chem. Soc., Faraday Trans. 2 1981, 77, 2125–2140. 10.1039/F29817702125. [DOI] [Google Scholar]
- Lowe M. P.; Parker D.; Reany O.; Aime S.; Botta M.; Castellano G.; Gianolio E.; Pagliarin R. pH-dependent modulation of relaxivity and luminescence in macrocyclic gadolinium and europium complexes based on reversible intramolecular sulfonamide ligation. J. Am. Chem. Soc. 2001, 123, 7601–7609. 10.1021/ja0103647. [DOI] [PubMed] [Google Scholar]
- Krchová T.; Herynek V.; Gálisová A.; Blahut J.; Hermann P.; Kotek J. Eu (III) Complex with DO3A-amino-phosphonate Ligand as a Concentration-Independent pH-Responsive Contrast Agent for Magnetic Resonance Spectroscopy (MRS). Inorg. Chem. 2017, 56, 2078–2091. 10.1021/acs.inorgchem.6b02749. [DOI] [PubMed] [Google Scholar]
- Shuvaev S.; Suturina E. A.; Mason K.; Parker D. Chiral probes for α-1-AGP reporting by species-specific induced circularly polarised luminescence. Chemical Science 2018, 9, 2996–3003. 10.1039/C8SC00482J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinehart J. D.; Long J. R. Exploiting single-ion anisotropy in the design of f-element single-molecule magnets. Chemical Science 2011, 2, 2078–2085. 10.1039/c1sc00513h. [DOI] [Google Scholar]
- Sievers J. Asphericity of 4f-shells in their Hund’s rule ground states. Z. Phys. B: Condens. Matter Quanta 1982, 45, 289–296. 10.1007/BF01321865. [DOI] [Google Scholar]
- Chilton N. F.; Collison D.; McInnes E. J.; Winpenny R. E.; Soncini A. An electrostatic model for the determination of magnetic anisotropy in dysprosium complexes. Nat. Commun. 2013, 4, 2551. 10.1038/ncomms3551. [DOI] [PubMed] [Google Scholar]
- Sorace L.; Benelli C.; Gatteschi D. Lanthanides in molecular magnetism: old tools in a new field. Chem. Soc. Rev. 2011, 40, 3092–3104. 10.1039/c0cs00185f. [DOI] [PubMed] [Google Scholar]
- Mironov V. S.; Galyametdinov Y. G.; Ceulemans A.; Görller-Walrand C.; Binnemans K. Room-temperature magnetic anisotropy of lanthanide complexes: A model study for various coordination polyhedra. J. Chem. Phys. 2002, 116, 4673–4685. 10.1063/1.1450543. [DOI] [Google Scholar]
- Dai L.; Zhang J.; Chen Y.; Mackenzie L. E.; Pal R.; Law G.-L. Synthesis of Water-Soluble Chiral DOTA Lanthanide Complexes with Predominantly Twisted Square Antiprism Isomers and Circularly Polarized Luminescence. Inorg. Chem. 2019, 58, 12506–12510. 10.1021/acs.inorgchem.9b01799. [DOI] [PubMed] [Google Scholar]
- Woods M.; Payne K. M.; Valente E. J.; Kucera B. E.; Young V. G. Jr Crystal structures of DOTMA chelates from Ce3+ to Yb3+: evidence for a continuum of metal ion hydration states. Chem. - Eur. J. 2019, 25, 9997–10005. 10.1002/chem.201902068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason S. F.; Peacock R. D.; Stewart B. Ligand-polarization contributions to the intensity of hypersensitive trivalent lanthanide transitions. Mol. Phys. 1975, 30, 1829–1841. 10.1080/00268977500103321. [DOI] [Google Scholar]
- Mason S. F.The ligand polarization model for the spectra of metal complexes: The dynamic coupling transition probabilities. In Electrons and Transitions; Springer, 1980; pp 43–81. [Google Scholar]
- Reid M. F.; Richardson F. S. Anisotropic ligand polarizability contributions to lanthanide 4f→ 4f intensity parameters. Chem. Phys. Lett. 1983, 95, 501–506. 10.1016/0009-2614(83)80341-8. [DOI] [Google Scholar]
- Poh A. W.; Aguilar J. A.; Kenwright A. M.; Mason K.; Parker D. Aggregation of Rare Earth Coordination Complexes in Solution Studied by Paramagnetic and DOSY NMR. Chem. - Eur. J. 2018, 24, 16170–16175. 10.1002/chem.201803766. [DOI] [PubMed] [Google Scholar]
- McConnell H. M. Theory of nuclear magnetic shielding in molecules. I. Long-range dipolar shielding of protons. J. Chem. Phys. 1957, 27, 226–229. 10.1063/1.1743676. [DOI] [Google Scholar]
- Peters J.; Huskens J.; Raber D. Lanthanide induced shifts and relaxation rate enhancements. Prog. Nucl. Magn. Reson. Spectrosc. 1996, 28, 283–350. 10.1016/0079-6565(95)01026-2. [DOI] [Google Scholar]
- Mironov V. S.; Galyametdinov Y. G.; Ceulemans A.; Görller-Walrand C.; Binnemans K. Influence of crystal-field perturbations on the room-temperature magnetic anisotropy of lanthanide complexes. Chem. Phys. Lett. 2001, 345, 132–140. 10.1016/S0009-2614(01)00842-9. [DOI] [Google Scholar]
- McGarvey B. R. Temperature dependence of the pseudocontact shift in lanthanide shift reagents. J. Magn. Reson. (1969-1992) 1979, 33, 445–455. 10.1016/0022-2364(79)90261-0. [DOI] [Google Scholar]
- Boulon M. E.; Cucinotta G.; Luzon J.; Degl’Innocenti C.; Perfetti M.; Bernot K.; Calvez G.; Caneschi A.; Sessoli R. Magnetic anisotropy and spin-parity effect along the series of lanthanide complexes with DOTA. Angew. Chem., Int. Ed. 2013, 52, 350–354. 10.1002/anie.201205938. [DOI] [PubMed] [Google Scholar]
- Cucinotta G.; Perfetti M.; Luzon J.; Etienne M.; Car P. E.; Caneschi A.; Calvez G.; Bernot K.; Sessoli R. Magnetic anisotropy in a dysprosium/DOTA single-molecule magnet: beyond simple magneto-structural correlations. Angew. Chem., Int. Ed. 2012, 51, 1606–1610. 10.1002/anie.201107453. [DOI] [PubMed] [Google Scholar]
- Briganti M.; Garcia G. F.; Jung J.; Sessoli R.; Le Guennic B.; Totti F. Covalency and magnetic anisotropy in lanthanide single molecule magnets: the DyDOTA archetype. Chemical Science 2019, 10, 7233–7245. 10.1039/C9SC01743G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung J.; Islam M. A.; Pecoraro V. L.; Mallah T.; Berthon C.; Bolvin H. Derivation of lanthanide series crystal field parameters from first principles. Chem. - Eur. J. 2019, 25, 15112–15122. 10.1002/chem.201903141. [DOI] [PubMed] [Google Scholar]
- Ungur L.; Chibotaru L. F. Ab initio crystal field for lanthanides. Chem. - Eur. J. 2017, 23, 3708–3718. 10.1002/chem.201605102. [DOI] [PubMed] [Google Scholar]
- Golding R.; Pyykkö P. On the theory of pseudocontact NMR shifts due to lanthanide complexes. Mol. Phys. 1973, 26, 1389–1396. 10.1080/00268977300102561. [DOI] [Google Scholar]
- Charnock G.; Kuprov I. A partial differential equation for pseudocontact shift. Phys. Chem. Chem. Phys. 2014, 16, 20184–20189. 10.1039/C4CP03106G. [DOI] [PubMed] [Google Scholar]
- Suturina E. A.; Kuprov I. Pseudocontact shifts from mobile spin labels. Phys. Chem. Chem. Phys. 2016, 18, 26412–26422. 10.1039/C6CP05437D. [DOI] [PubMed] [Google Scholar]
- Golding R.; Halton M. A theoretical study of the 14N and 17O N.M.R. shifts in lanthanide complexes. Aust. J. Chem. 1972, 25, 2577–2581. 10.1071/CH9722577. [DOI] [Google Scholar]
- Reilley C. N.; Good B. W.; Allendoerfer R. D. Separation of contact and dipolar lanthanide induced nuclear magnetic resonance shifts: evaluation and application of some structure independent methods. Anal. Chem. 1976, 48, 1446–1458. 10.1021/ac50005a010. [DOI] [Google Scholar]
- Kreidt E.; Bischof C.; Platas-Iglesias C.; Seitz M. Magnetic Anisotropy in Functionalized Bipyridyl Cryptates. Inorg. Chem. 2016, 55, 5549–5557. 10.1021/acs.inorgchem.6b00591. [DOI] [PubMed] [Google Scholar]
- Esteban-Gómez D.; Büldt L. A.; Pérez-Lourido P.; Valencia L.; Seitz M.; Platas-Iglesias C. Understanding the Optical and Magnetic Properties of Ytterbium (III) Complexes. Inorg. Chem. 2019, 58, 3732–3743. 10.1021/acs.inorgchem.8b03354. [DOI] [PubMed] [Google Scholar]
- Parigi G.; Benda L.; Ravera E.; Romanelli M.; Luchinat C. Pseudocontact shifts and paramagnetic susceptibility in semiempirical and quantum chemistry theories. J. Chem. Phys. 2019, 150, 144101. 10.1063/1.5037428. [DOI] [PubMed] [Google Scholar]
- Benda L.; Mareš J.; Ravera E.; Parigi G.; Luchinat C.; Kaupp M.; Vaara J. Pseudo-Contact NMR Shifts over the Paramagnetic Metalloprotein CoMMP-12 from First Principles. Angew. Chem., Int. Ed. 2016, 55, 14713–14717. 10.1002/anie.201608829. [DOI] [PubMed] [Google Scholar]
- Benda L.; Mareš J.; Ravera E.; Parigi G.; Luchinat C.; Kaupp M.; Vaara J. Pseudo-Contact NMR Shifts over the Paramagnetic Metalloprotein CoMMP-12 from First Principles. Angew. Chem. 2016, 128, 14933–14937. 10.1002/ange.201608829. [DOI] [PubMed] [Google Scholar]
- Hogben H.; Krzystyniak M.; Charnock G.; Hore P.; Kuprov I. Spinach–a software library for simulation of spin dynamics in large spin systems. J. Magn. Reson. 2011, 208, 179–194. 10.1016/j.jmr.2010.11.008. [DOI] [PubMed] [Google Scholar]
- Mason K.; Rogers N. J.; Suturina E. A.; Kuprov I.; Aguilar J. A.; Batsanov A. S.; Yufit D. S.; Parker D. PARASHIFT probes: solution NMR and X-ray structural studies of macrocyclic ytterbium and yttrium complexes. Inorg. Chem. 2017, 56, 4028–4038. 10.1021/acs.inorgchem.6b02291. [DOI] [PubMed] [Google Scholar]
- Vonci M.; Mason K.; Neil E. R.; Yufit D. S.; McInnes E. J.; Parker D.; Chilton N. F. Sensitivity of Magnetic Anisotropy in the Solid State for Lanthanide Complexes with Small Crystal Field Splitting. Inorg. Chem. 2019, 58, 5733–5745. 10.1021/acs.inorgchem.9b00060. [DOI] [PubMed] [Google Scholar]
- Bertini I.; Luchinat C.; Parigi G.; Pierattelli R. NMR spectroscopy of paramagnetic metalloproteins. ChemBioChem 2005, 6, 1536–1549. 10.1002/cbic.200500124. [DOI] [PubMed] [Google Scholar]
- Otting G. Protein NMR using paramagnetic ions. Annu. Rev. Biophys. 2010, 39, 387–405. 10.1146/annurev.biophys.093008.131321. [DOI] [PubMed] [Google Scholar]
- Suturina E. A.; Häussinger D.; Zimmermann K.; Garbuio L.; Yulikov M.; Jeschke G.; Kuprov I. Model-free extraction of spin label position distributions from pseudocontact shift data. Chemical science 2017, 8, 2751–2757. 10.1039/C6SC03736D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caravan P. Protein-targeted gadolinium-based magnetic resonance imaging (MRI) contrast agents: design and mechanism of action. Acc. Chem. Res. 2009, 42, 851–862. 10.1021/ar800220p. [DOI] [PubMed] [Google Scholar]
- Clore G. M.; Iwahara J. Theory, practice, and applications of paramagnetic relaxation enhancement for the characterization of transient low-population states of biological macromolecules and their complexes. Chem. Rev. 2009, 109, 4108–4139. 10.1021/cr900033p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vega A. J.; Fiat D. Nuclear relaxation processes of paramagnetic complexes the slow-motion case. Mol. Phys. 1976, 31, 347–355. 10.1080/00268977600100261. [DOI] [Google Scholar]
- Guéron M. Nuclear relaxation in macromolecules by paramagnetic ions: a novel mechanism. J. Magn. Reson. (1969-1992) 1975, 19, 58–66. 10.1016/0022-2364(75)90029-3. [DOI] [Google Scholar]
- Bloembergen N. Proton relaxation times in paramagnetic solutions. J. Chem. Phys. 1957, 27, 572–573. 10.1063/1.1743771. [DOI] [Google Scholar]
- Funk A. M.; Harvey P.; Finney K.-L. N.; Fox M. A.; Kenwright A. M.; Rogers N. J.; Senanayake P. K.; Parker D. Challenging lanthanide relaxation theory: erbium and thulium complexes that show NMR relaxation rates faster than dysprosium and terbium analogues. Phys. Chem. Chem. Phys. 2015, 17, 16507–16511. 10.1039/C5CP02210J. [DOI] [PubMed] [Google Scholar]
- Rogers N. J.; Finney K.-L. N.; Senanayake P. K.; Parker D. Another challenge to paramagnetic relaxation theory: a study of paramagnetic proton NMR relaxation in closely related series of pyridine-derivatised dysprosium complexes. Phys. Chem. Chem. Phys. 2016, 18, 4370–4375. 10.1039/C5CP06755C. [DOI] [PubMed] [Google Scholar]
- Funk A. M.; Fries P. H.; Harvey P.; Kenwright A. M.; Parker D. Experimental measurement and theoretical assessment of fast lanthanide electronic relaxation in solution with four series of isostructural complexes. J. Phys. Chem. A 2013, 117, 905–917. 10.1021/jp311273x. [DOI] [PubMed] [Google Scholar]