

Abstract
To account for the charge transfer and covalent character in bonding between P and Bi centers, the electronic structures of [P(C6H4-o-CH2SCH3)3BiCln](3–n)+ (n = 0–3) model species have been investigated computationally. On the basis of this survey a synthetic target compound with a dative P→Bi bond has been selected. Consecutively, the highly reactive bismuth cage [P(C6H4-o-CH2SCH3)3Bi]3+ has been accessed experimentally and characterized. Importantly, our experiments (single-crystal X-ray diffraction and solid-state NMR spectroscopy) and computations (NBO and AIM analysis) reveal that the P···Bi bonding in this trication can be described as a dative bond. Here we have shown that our accordion-like molecular framework allows for tuning of the interaction between P and Bi centers.
Short abstract
The bonding situation in species with the general formula of [P(C6H4-o-CH2SCH3)3BiCln](3−n)+ (n = 0−3) has been investigated computationally, and on this basis a selected example has been accessed synthetically. The structural and solid-state NMR studies reveal that the weak secondary pnictogen interaction in P(C6H4-o-CH2SCH3)3BiCl3 can be tuned into dative bonding by enhancing the charge transfer from the phosphorus to the bismuth center, which is accompanied by a change in the spin−spin coupling mechanism between the 31P and 209Bi nuclei.
1. Introduction
The hundred years old concept of coordinative covalent or dative bonding has evolved from a fundamental physical theory1,2 to cornerstones of undergraduate chemical education. In contrast to electron-sharing covalent bonding, dative bonding arises between two closed-shell systems, an electron pair donor (Lewis base) and an electron pair acceptor (Lewis acid), and significant electron density is transferred from the donor to the acceptor (charge transfer). More recently, the concept of σ-hole interactions introduced just a decade ago3,4 has gained increasing attention5−7 (note that some examples such as hydrogen bonds and halogen bonds have been known for much longer5). σ-Hole interactions, similarly to dative bonds, also arise between two closed-shell entities, but they are regarded as noncovalent in nature. A relatively new congener of noncovalent interactions is the pnictogen bond (PnB),8−10 which (in analogy with the IUPAC definition of a halogen bond11) can be defined as an attractive interaction between the electron-deficient region of a pnictogen (group 15 element) called a pnictogen bond donor and a Lewis base (pnictogen bond acceptor, acting as an electron pair donor).12 In the past few years, the potential of PnB in structural assembly, supramolecular architecture, anion sensing, (organo)catalysis, and molecular recognition has also been highlighted.7,13−21 On the basis of thorough computational studies,6,9,22−41 pnictogen bonding is chiefly electrostatic in nature (attraction between the oppositely charged regions around the two centers). Moreover, charge transfer effects (donation from the lone pair of the Lewis base into the σ*-antibonding orbitals at the pnictogen center) may also contribute, though to a much lesser extent. This also means that the charge transfer and thus the covalent character in pnictogen bonding are rather low. Importantly, the strength of a pnictogen interaction can be comparable to that of a hydrogen bond. However, the experimental observation is still challenging, especially with spectroscopic methods such as NMR spectroscopy.9,15,42−45
Even though the concepts of both dative bonds and σ-hole interactions are well-known, to our knowledge the relationship and connectivity between them has not yet been explored experimentally. In the present study we aim to investigate the bonding situation in P/Bi complexes. Due to its highest polarizability among the group 15 elements, bismuth has the strongest ability to form complexes with conventional dative bonds. On the other hand, phosphorus stands out because of its excellent NMR properties (broad chemical shift range, 100% natural abundance) and can be employed as a sensor to detect delicate structural changes.
Several prototype donor–acceptor complexes are known in the literature exhibiting P–Bi distances described as dative bonds such as the peri-substituted acenaphthyl derivative A (2.7696(8) Å46 and other recently reported congeners of this family47) and the monocation B shown in Figure 1 (2.6883(14) and 2.6750(13) Å).48 Similarly, a dative bond was reported for a phosphane coordinated diphenylbismuthenium cation (see C 2.6672(19) Å).49 In contrast, significantly longer P–Bi distances of 2.968(3) and 2.937(3) Å were observed for the related complex D, in which two phosphane ligands coordinate to the bismuthenium cation. These bonds were described rather as an electrostatic, induced dipole–ion attraction than a conventional dative bond.50 The Bi···P distance can be stretched further as in the geminally substituted tris(acenaphthyl) bismuthine E (3.218(3)–3.279(4) Å), in which only weak interactions between the P and Bi centers were found.51 Recently we have reported a series of phosphane–trihalobismuth complexes (PS3BiX3, X = Cl, Br, I), featuring even longer P···Bi distances (in the range of 3.365(1)–3.792(9) Å). The interactions between the P and Bi centers in these PS3BiX3 compounds exhibit a remarkable strength of 7.1–8.8 kcal mol–1.52
Figure 1.
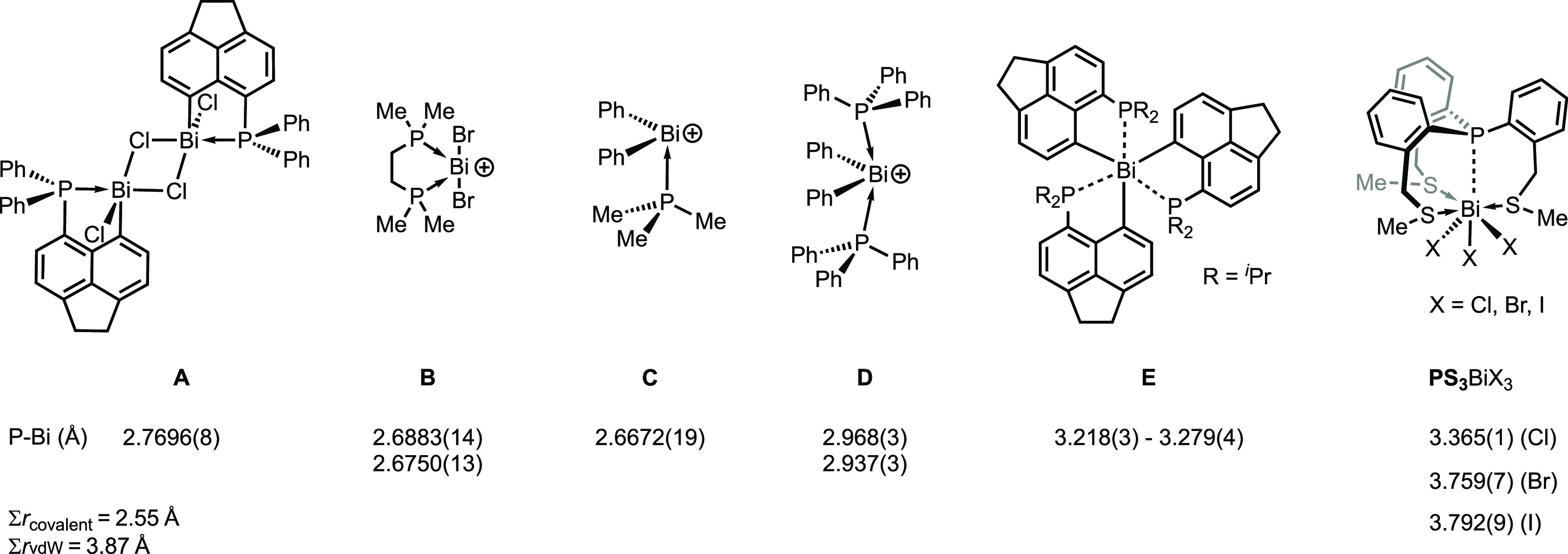
Selected examples of P–Bi distances determined by X-ray crystallography. For B, C and D the counteranions are not shown. ∑rcovalent and ∑rvdW denote the sum of covalent53 and van der Waals54 radii, respectively, for the P Bi couple.
The goal of the present study is to gain more insight into the bonding situation in bismuth(III) complexes with the PS3 ligand. Although other multidentate ligand systems are known in the literature,55−64 due to their accordion-like structural flexibility the molecular skeleton of type PS3BiX3 seems to be suitable for experimental investigations. First, we aim to predict computationally how the pnictogen interaction between the bridgehead atoms P and Bi observed in the neutral PS3BiX3 compounds can be shifted into a dative (covalent) regime, and subsequently, we provide experimental evidence based on X-ray crystallography and NMR studies.
2. Results and Discussion
2.1. Computational Considerations
To search for possible candidates in which the interaction between the P and Bi can be described with dative bonding, first we investigated a series of species related to PS3BiX3. As the P–Bi atom distance in PS3BiX3 depends on the halide substituent on the bismuth and decreases in the order I > Br > Cl,52 an obvious choice could be the complex of P(C6H4-o-CH2SCH3)3 (PS3) with bismuth trifluoride; however, this compound could not be synthetically realized due to the extreme low solubility of BiF3.52 Alternatively, the halides could be changed for weakly coordinating anions. In our computational survey we studied a series of model species arising by sequential abstraction of chlorides from the compound PS3BiCl3. Thus, the monocation [PS3BiCl2]+, the dication [PS3BiCl]2+, the trication [PS3Bi]3+, and the complex of the trication with three acetonitrile (ACN) molecules, [PS3Bi(ACN)3]3+ were investigated alongside the neutral compound PS3BiCl3. In order to gain insight into the bonding situation between the P and Bi atoms we performed dispersion-corrected DFT calculations (for details and the results at the B3LYP-D3/cc-pVDZ(-PP) level, see the Supporting Information). Herein, we present the results obtained at the ωB97XD/cc-pVDZ(-PP) level, which was successfully employed for similar systems.52 Furthermore, the solvent effects were simulated with the polarized continuum model (PCM), employing acetonitrile as solvent. On the optimized structures bond valences have been obtained according to the method described by Brown,65 employing the data set reported by Brese and O’Keeffe.66 Furthermore, NBO (natural bonding orbital) computations67 delivering the Wiberg bond indices as well as atoms in molecules (AIM) analyses68 have been carried out and the results are collected in Table 1. The NPA (natural population analysis) charges show the same tendencies as the AIM charges and, therefore, they can be found in Table S5 in the Supporting Information.
Table 1. Calculated Bi···P Atomic Distances (d, Å) in [PS3BiCln]3–n (n = 0–3) and [PS3Bi(ACN)3]3+, bond valences (s, valence units), Wiberg bond indices (WBIs), AIM Charges (q, electron) and Net Charge Donation from the Ligand to the [BiCln]3-n moiety (Δq Calculated As the Sum of Partial AIM Charges in the Ligand Fragment), Properties at the Bond Critical Point of Electron Density (ρbcp, e/Å3), Laplacian of the Electron Density (∇2ρbcp, e/Å5), Total Electronic Energy Density (H, au), and Ratio of Potential and Kinetic Energy Density (|V|/G)a.

The gas-phase data without PCM for PS3BiCl3 are taken from ref (52). All data were obtained at the ωB97XD/cc-pVDZ(-PP) level of theory. The values obtained with the solvent model PCM are shown in italics.
The P···Bi atom distance decreases systematically upon abstraction of the chlorides in the row of PS3BiCl3 → [PS3Bi]3+, both in the gas-phase calculations and with the PCM solvent model. If these two data sets (without and with solvent model) are compared, the largest difference (0.154 Å) is observed for the neutral PS3BiCl3, while the deviation is significantly smaller for the other species. The PCM model results in somewhat shorter P···Bi distances in the case of the neutral PS3BiCl3 and the monocationic [PS3BiCl2]+, whereas it results in slightly longer distances for the di- and trications. Altogether, the differences between the results obtained with or without the solvent model are much smaller than those resulting from the change in the substituents/charge. Therefore, the general tendencies among the different species are not affected, and in the following we only discuss the parameters computed using the solvent model.
Importantly, the shortest P···Bi distance is found in the trication [PS3Bi]3+ (2.743 Å), being clearly in the range of dative bonds reported for compounds such as A, B, and C (see Figure 1). As the direct coordination of solvent molecules to the bismuth center may have an effect on the interaction between the P and Bi centers, we have also computed the model species [PS3Bi(ACN)3]3+. As expected, the competition between the donor atoms around the Bi center leads to an elongation of the P···Bi distance (2.849 Å in [PS3Bi(ACN)3]3+). This bond, however, is significantly shorter than that in the dication (2.923 Å in [PS3BiCl]2+), and thus the effect of three coordinating solvent molecules is inferior to that of one chloride anion.
The strengthening of the interaction between the P and Bi centers in the sequence of PS3BiCl3, [PS3BiCl2]+, [PS3BiCl]2+, [PS3Bi(ACN)3]3+, and [PS3Bi]3+ is also reflected in the increasing bond valence values (s) and Wiberg bond indices (WBI). These two nicely correlating parameter sets indicate a tendentious rise in covalent character on going from the neutral to the tricationic species. Surprisingly, the AIM charge at the bismuth center shows a decreasing trend upon abstraction of the chlorides, which is moderate if the solvent model is employed and is more pronounced in the gas-phase calculations. This descending charge can be explained by substantial charge transfer from the PS3 ligand toward the bismuth center reaching Δq = 1.875 e in the gas phase (Δq = 1.663 e with the PCM solvent model) for [PS3Bi]3+. The intensification of the charge transfer is also nicely visible on the AIM charges at the phosphorus centers, which gradually grow from the neutral PS3BiCl3 (q(P) = 1.695 e in the gas phase, 1.693 e with PCM) to the tricationic [PS3Bi]3+ (q(P) = 1.924 e in the gas phase, 1.867 e with PCM).
The results of the NBO investigations and partial charges are further bolstered by an atoms in molecules (AIM) analysis68 of the electron density, which located bond critical points (bcp) between the P and Bi nuclei in each of the complexes. The trend of the electron density at the bond critical points (ρbcp) again indicates gradual strengthening of the interaction between the P and Bi atoms from PS3BiCl3 to [PS3Bi]3+. The Laplacian of the electron density at the P···Bi bcp (∇2ρbcp) is positive for each of these complexes, which suggests closed-shell interactions (dative, ionic, or σ-hole interaction). The electron density and its Laplacian at the bcp of the Bi–P bond in the tricationic [PS3Bi]3+ (0.424 e/Å3 and 0.697 e/Å5, respectively) are similar to those reported for the acenaphthyl derivative A in Figure 1 (0.42 e/Å3 and 0.6 e/Å5, respectively46), which was described with a polar covalent Bi–P interaction. While the Cl–Bi–Cl arrangement in A is close to linear (174.32(2)°), a second rotamer exhibiting a pyramidalized Bi environment and an elongated Bi–P bond (3.005 Å) could also be optimized. The Bi–P interaction in the latter rotamer was described as rather ionic (electrostatic), and its characteristics (ρbcp = 0.24 e/Å3 and ∇2ρbcp = 1.0 e/Å5) are similar to those of the monocationic [PS3BiCl2]+ (ρbcp = 0.252 e/Å3 and ∇2ρbcp = 1.183 e/Å5).
The neutral PS3BiCl3 and the tricationic [PS3Bi]3+ present the two extremes of noncovalent and dative bonding, respectively; however, there is clearly a continuum between them. The gradual conversion of the electrostatic nature of the interaction into covalency is visible in a smooth change of all the bonding descriptors shown in Table 1. The question may arise where the borderline is between weak interaction and dative bonding. Necessarily, this separation is arbitrary and here we refer to a simple tool based on the relative amount of kinetic and potential energy at the bcp (Cremer–Kraka criteria).69,70 At the bcp of a covalent bond the potential energy density V (always negative) is more substantial than the kinetic energy density G (always positive), resulting in a total electronic energy density H = V + G < 0 or alternatively |V|/G > 1. These characteristics show that the P···Bi interaction in the trichloride PS3BiCl3 is chiefly electrostatic, whereas the cationic complexes [PS3BiCln](3–n)+ (n = 0–2) all exhibit H < 0 and |V|/G > 1. Thus, in all these cationic species the P···Bi bond has a highly covalent character and can be classified as a dative bond (P→Bi).
On the basis of the results of the above analysis we propose a simple model based on the competing effects of the donor atoms to understand the different P···Bi bonding situations in the complexes outlined in Table 1. To discuss the changes in the coordination sphere of the Bi center, we employ the bond valences obtained from the geometries optimized with the PCM solvent model (see Table 1 and Table S7 in the Supporting Information). The rather strongly donating chloride ligands and the sulfur donors in the coordination sphere around the bismuth center in PS3BiCl3 hamper the phosphorus from developing significant charge transfer toward the bismuth. Therefore, a weak, mainly electrostatic pnictogen bond can only be formed, which results in a large P···Bi distance. The valence of the bismuth center in PS3BiCl3 is 2.793 vu (valence units), calculated as the sum of bond valences (∑s). The abstraction of one chloride ligand formally frees up a coordination site by 0.663 vu (taken as the least strongly bound chloride among the three; see Table S7), which initiates reorganization to reach a new equilibrium structure. Thus, the remaining donors can coordinate more strongly to the bismuth, which is reflected in the larger P···Bi and S···Bi bond valences (for details see Table S7 in the Supporting Information) and the valence of the bismuth center is almost re-established (∑s = 2.700 vu in [PS3BiCl2]+). Similar phenomena occur for the abstraction of the second and third chloride anions. However, the “buffering effect” of the remaining donors to re-establish the valence of the bismuth center decreases somewhat, which can clearly be seen in the slight gradual dropping of the ∑s values in the direction PS3BiCl3 to [PS3Bi]3+ (see Table 1), indicating slight undercoordination. Nevertheless, the intensification of the charge transfer (due to the sequential abstraction of chloride ions) results in a gradual strengthening of the P···Bi and S···Bi interactions in the same direction.
On the basis of the computations above, all three cationic species seem to be suitable for experimental detection of a dative interaction between the P and Bi centers. However, to target the strongest possible P→Bi interaction, we chose the trication [PS3Bi]3+ with weakly coordinating anions as the best candidate for our experimental study.
2.3. Experimental Validation
In order to access the aimed tricationic coordination compound [PS3Bi]3+, we decided to employ trifluoromethanesulfonate (triflate) as a counteranion for two reasons: the triflate anion is reasonably weakly coordinating,71 and bismuth triflate is an easily accessible metal salt, which is also widely used as a catalyst in various organic syntheses.72,73
To form the target complex, the ligand P(C6H4-o-CH2SCH3)3 (PS3)52 and 1 equiv of commercially available bismuth trifluoromethanesulfonate Bi[OTf]3(H2O)n (n ≈ 14 according to a TGA measurement) were reacted in acetonitrile at room temperature. In the 31P{1H} NMR spectrum of this reaction mixture only a singlet at −22.0 ppm was observed, which appears in the 31P NMR spectrum as a doublet with 1JPH = 535.0 Hz, indicating protonation at the phosphorus center. This phosphorus-containing product was unambiguously identified by 31P, 13C, and 1H NMR spectroscopy and a single-crystal X-ray diffraction study as the phosphonium salt [HP(C6H4-o-CH2SCH3)3]+[OTf]− (see Figure S7 in the Supporting Information). Note that bismuth salts in general are prone to hydrolysis in the presence of water and in this case the hydrolysis of bismuth triflate resulted in the formation of triflic acid, which as a strong acid can protonate the phosphane ligand PS3.
According to the literature it is practically impossible to obtain strictly anhydrous bismuth triflate.74,75 To decrease the possibility of hydrolysis, we dried Bi[OTf]3 for 10 days at 160 °C under a dynamic vacuum,76 which resulted in a solid with the formula of Bi[OTf]3(H2O)n (n ≈ 2.8, on the basis of elemental analysis). We repeated the reaction described above employing Bi[OTf]3(H2O)n (n ≈ 2.8) in toluene instead of acetonitrile, which resulted in the formation of a yellow precipitate. On the basis of its solid-state 31P CP-MAS NMR spectrum this material is a mixture of products. However, recrystallization from acetonitrile delivered yellow crystals isolated in a low yield (15%). These crystals were also suitable for single-crystal X-ray diffraction analysis (see below), showing that their composition is [PS3Bi]2{Bi6O4(OH)4[OTf]12}(H2O)(CH3CN)6, which was also confirmed by elemental analysis of the isolated product. [PS3Bi]2{Bi6O4(OH)4[OTf]12}(H2O)(CH3CN)6 is in the following abbreviated as [PS3Bi]2[BOT], in which the “bismuth oxo triflate” cluster hexaanion [BOT]6– stands for {Bi6O4(OH)4[OTf]12}6– (Figure 2). This compound is only stable in the solid state and was further characterized by CP-MAS 31P, 1H, 13C, and 19F NMR spectroscopy (for details see section 2.5 and Figure 4). The low yield is attributed to the observation that [PS3Bi]2[BOT] decomposes in solution. Indeed, when crystals of [PS3Bi]2[BOT] are dissolved in dry acetonitrile, the 31P NMR spectrum of the solution (see Figure S2) shows two resonances: a very small singlet resonance at δ +67.6 ppm is observed (likely indicating the [PS3Bi]3+ species) while the larger doublet resonance at δ −22.0 ppm with the coupling constant 1JPH = 535.0 Hz again indicates the phosphonium salt [HPS3]+[OTf]− as a decomposition product.
Figure 2.

Schematic depiction of [PS3Bi]2[BOT].
Figure 4.
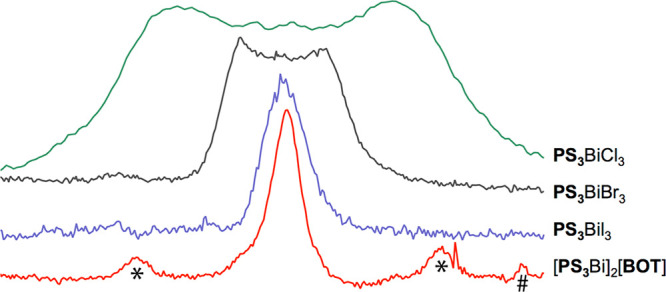
Room-temperature solid-state CP-MAS 31P NMR spectra of the adduct [PS3Bi]2[BOT], PS3BiI3, PS3BiBr3, and PS3BiCl3 (at spinning rates of 8, 6, 6, and 10 kHz, respectively). For an easier comparison of the band widths, the resonances of [PS3Bi]2[BOT] and PS3BiX3 (X = I, Br, Cl)52 have been centered at the same position. For all samples the line widths of the bands have been proven to be independent from the applied spinning rates. The symbols * and # indicate spinning side bands and [HPS3]+[OTf]− impurity, respectively.
2.4. Structural Studies
We suggest describing the compound [PS3Bi]2[BOT] as an assembly of two tricationic [PS3Bi]3+ units and a central {Bi6O4(OH)4[OTf]12}6– cluster anion (Figure 3), on the basis of the analogy of the latter to known cluster anions such as [Bi6O4(OH)4(F3CCO2)12]6–.77 Alternatively, [PS3Bi]2[BOT] could also be considered as {[PS3Bi][OTf]3}{Bi6O4(OH)4[OTf]6}{[PS3Bi][OTf]3} with two neutral capping {[PS3Bi][OTf]3} moieties and a central neutral {Bi6O4(OH)4[OTf]6} cluster.
Figure 3.

(a) Plot of the adduct [PS3Bi]2[BOT] (Bi, purple; S, yellow; P, orange; F, green; O, red; C, gray). Hydrogen atoms and solvent molecules have been omitted for clarity. (b) Side view of the section marked with asterisk in (a) showing the central core (CF3 groups are not shown). (c) ORTEP representation of the C3-symmetric [PS3Bi]3+ moiety (thermal ellipsoids are drawn at 50% probability). Hydrogen atoms, solvent molecules, and the [BOT]6– cluster have been omitted for clarity. Selected atomic distances (Å): Bi–P 2.800(3), Bi–S 2.749(9), Bi–O[triflate] 2.760(1).
The so far unknown {Bi6O4(OH)4[OTf]12}6– ([BOT]6–) cluster hexaanion formally consists of a cationic [Bi6(μ3-O)4(μ3-OH)4]6+ core surrounded by 12 triflate anions. In the core, which is a common motif in hydrolysis products of bismuth salts,77−84 six bismuth(III) centers occupy the corners of an octahedron, whose faces are capped by four oxo and four hydroxo ligands in a μ3 fashion (the hydroxy protons could not be located crystallographically). The 12 edges of the octahedron are each capped by a triflate anion in a μ2-O fashion, six of which form the ring arrangement shown in Figure 3b. The additional (2 times 3) triflate anions bridge the central core with one of the two tricationic [PS3Bi]3+ moieties on the two sides of the structure (Figure 3a).
The bismuth centers of these [PS3Bi]3+ fragments are coordinated by three triflate anions as well as the three sulfur and the phosphorus centers of ligand PS3; the C3 symmetry around the bismuth centers suggests stereochemical inactivity of their lone pairs. The Bi–S atom distances of 2.749(9) Å reflect a dative bond between the sulfur donor and a Bi(III) cation (for example 2.6873(3)–3.013(2) Å in Bi(III) chalcogenone complexes).85 The Bi–O[triflate] distances (2.760(1) Å) are between the sum of the covalent and van der Waals radii of the respective elements (2.1453 and 3.59 Å,54 respectively) and are in the range of those found in reported bismuth triflato compounds (2.386(11)–3.010(3) Å).48,86
Most importantly, the P–Bi distance is 2.800(3) Å, which is only slightly longer than those in compounds A, B, and C in Figure 1 (from 2.6672(19) to 2.7696(8) Å); however, it is significantly shorter than the “pnictogen-bonded” P–Bi distances in PS3BiX3 (3.365(1)–3.792(9) Å). The corresponding bond valences have also been calculated65,66 (see Table S8 in the Supporting Information) and show that the P–Bi bond in [PS3Bi]2[BOT] (s = 0.632 vu) is remarkably stronger than those in PS3BiX3 (s = 0.137, 0.047, and 0.043 vu for X = Cl, Br, I, respectively). The sum of the bond valences at the bismuth center indicates that the Bi in [PS3Bi]2[BOT] (∑s = 2.874 vu) is undercoordinated in comparison to the halide analogues PS3BiX3 (∑s = 3.233, 3.217, and 3.191 vu for X = Cl, Br, I, respectively), which is in accord with the observations discussed for the gas-phase structures (vide supra).
We also compared the solid-state structure of [PS3Bi]2[BOT] to the gas-phase structure of the tricationic adduct [PS3Bi]3+ obtained by DFT computations (vide supra). The structure corresponding to the energy minimum (in the gas phase) is not symmetrical and shows a stereochemically active lone pair at the bismuth center. Furthermore, a structure with a constrained C3 symmetry was also obtained (as a second-order saddle point) lying only 3.7 kcal/mol higher in energy. This indicates that the coordination geometry around the bismuth is very flexible and, thus, the difference between the gas-phase and solid-state structures is most likely caused by crystal-packing effects. Nevertheless, the computed P–Bi distances in the gas phase (2.697 Å for the minimum and 2.715 Å for the C3-symmetric structure) are similar but somewhat shorter than that in the solid-state structure (2.800(3) Å). We attribute this difference to weak coordination of the triflate anions in the solid state, transferring some electron density to the bismuth center and thus elongating the P–Bi atom distance.
2.5. Solid-State NMR Investigations
As 31P NMR spectroscopy is a useful tool in studying bonding situations, we investigated specifically the compound [PS3Bi]2[BOT]. The chemical shifts obtained in both solid-state CP-MAS and solution 31P NMR spectra (+56.0 and +67.6 ppm, respectively) are similar to that of B, in which the phosphorus coordinates to a bismuth center (Figure 1, δ(31P) 50.8 ppm),48 confirming a remarkable change in the chemical environment around the phosphorus nucleus in comparison to the free ligand PS3 (δ(31P) −38.0 ppm52). This substantial coordination chemical shift change (Δδ ≈ 100 ppm) is attributable to the significant P→Bi charge transfer, nicely agreeing with the results of the computations (the partial charge difference between the trication [PS3Bi]3+ and the ligand PS3: Δq(P) = 0.205 e).
In the CP MAS 31P NMR spectra of compounds PS3BiX3 scalar indirect spin–spin coupling between the 209Bi and 31P nuclei was observed (typically as an unresolved broad band), which is consistent with the overlap of the P and Bi lone pairs, resulting in a Mallory-type through-space coupling mechanism.87−89 Furthermore, the strength of the pnictogen interaction in these compounds was found to correlate with the width of the band arising from this coupling, which increases in the order PS3BiI3 < PS3BiBr3 < PS3BiCl3.52 Knowing that the coupling mechanism in all of these PS3BiX3 compounds is the same, and assuming that it is the same in [PS3Bi]2[BOT] as well, we would expect that intensifying the interaction between P and Bi would lead to an increase in the coupling constant and as a consequence to a broadening of the band. Thus, the band of [PS3Bi]2[BOT] would be significantly broader than that of PS3BiCl3. Surprisingly, the CP MAS 31P NMR spectrum of [PS3Bi]2[BOT] (see Figure 4) exhibits a band which is narrower than that in the case of PS3BiI3. The remarkable half-width of 1.6 kHz for [PS3Bi]2[BOT] again indicates an unresolved, indirect spin–spin coupling between the 31P and the quadrupolar 209Bi nuclei. However, since the width of [PS3Bi]2[BOT] does not fit into the expected tendency, the coupling mechanism should be starkly different in the compounds PS3BiX3 and [PS3Bi]2[BOT]. To verify this assumption, we have simulated the spin–spin coupling constant in the gas phase at the PBE1/TZ2P level with a scalar ZORA approximation (see the Supporting Information). The J(209Bi–31P) value calculated for the gas-phase optimized structure of [PS3Bi]3+ is −773 Hz, the absolute value of which is indeed smaller than that calculated for PS3BiI3 at the same level (908 Hz52), agreeing with the experimental findings for the band widths. A striking difference is, however, found in the sign of the computed coupling constant, which was determined to be positive for the compounds PS3BiX3 (X = Cl, Br, I), while it was negative for [PS3Bi]3+. The former is characteristic for a Mallory type through-space coupling mechanism resulting from the overlap of the Bi and P lone pairs,52 while we attribute the latter to a real through-bond coupling in line with the presence of a dative P→Bi interaction.
3. Summary and Conclusions
In summary, it is demonstrated that a weak secondary pnictogen interaction previously reported for PS3BiX3 can be tuned into a dative bond employing the same molecular framework. On the basis of our DFT computations on the structures of [PS3BiCln](3–n)+ (n = 0–3) and [PS3Bi(ACN)3]3+ model species, the increase in covalency and thus the shift toward dative bonding can be achieved by intensifying the charge transfer from the P to the Bi center already in the case of the monocation [PS3BiCl2]+. Experimentally this was realized by employing weakly coordinating triflate anions and a new coordination compound exhibiting a dative P→Bi bond was synthesized and characterized, which shows marked contrast to pnictogen-bonded complexes PS3BiX3 described previously. Furthermore, our investigations reveal a connection between the nature of the P···Bi interaction and the spin–spin coupling mechanism between the two nuclei involved, offering the possibility to distinguish between these two types of interactions. Our results also show that ligand P(C6H4-o-CH2SCH3)3 is “flexidentate”, which means it can adopt different coordination modes and in these accordion-like complexes the distance between the bridgehead atoms may deviate by nearly 1 Å. Therefore, this system could also be of interest for the development of new catalytic systems: for example, with transition metals.
Acknowledgments
The authors thank Dr. P. W. Dyer, Dr. D. Yufit, Dr. A. Batsanov, and Dr. G. Müller for their help and acknowledge the support of a European Union COFUND/Durham Junior Research Fellowship under EU grant agreement number 609412, the ETH Zurich, a BME-Nanotechnology FIKP grant of EMMI (BME FIKP-NAT), NKFIH (PD 116329), a Janos Bolyai Research Scholarship, a UNKP-19-4-BME-422 grant, Varga József Alapítvány, and Pro Progressio Alapítvány.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.inorgchem.0c00734.
Description of experimental procedures, characterization of compounds, X-ray crystallographic studies, and computational details (PDF)
Accession Codes
CCDC 1941566 and 1941695 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
The authors declare no competing financial interest.
Dedication
Dedicated to Prof. Magdolna Hargittai on the occasion of her 75th birthday.
Supplementary Material
References
- Lewis G. N. The atom and the molecule. J. Am. Chem. Soc. 1916, 38 (4), 762–785. 10.1021/ja02261a002. [DOI] [Google Scholar]
- Nandi A.; Kozuch S. History and Future of Dative Bonds. Chem. - Eur. J. 2020, 26 (4), 759–772. 10.1002/chem.201903736. [DOI] [PubMed] [Google Scholar]
- Clark T.; Hennemann M.; Murray J. S.; Politzer P. Halogen bonding: the σ-hole. J. Mol. Model. 2007, 13 (2), 291–296. 10.1007/s00894-006-0130-2. [DOI] [PubMed] [Google Scholar]
- Politzer P.; Murray J. S.; Concha M. C. Sigma-hole bonding between like atoms; a fallacy of atomic charges. J. Mol. Model. 2008, 14 (8), 659–665. 10.1007/s00894-008-0280-5. [DOI] [PubMed] [Google Scholar]
- Politzer P.; Murray J. S.; Clark T. Halogen bonding and other σ-hole interactions: a perspective. Phys. Chem. Chem. Phys. 2013, 15 (27), 11178–11189. 10.1039/c3cp00054k. [DOI] [PubMed] [Google Scholar]
- Kolář M. H.; Hobza P. Computer Modeling of Halogen Bonds and Other σ-Hole Interactions. Chem. Rev. 2016, 116 (9), 5155–5187. 10.1021/acs.chemrev.5b00560. [DOI] [PubMed] [Google Scholar]
- Lim J. Y. C.; Beer P. D. Sigma-Hole Interactions in Anion Recognition. Chem. 2018, 4 (4), 731–783. 10.1016/j.chempr.2018.02.022. [DOI] [Google Scholar]
- Moilanen J.; Ganesamoorthy C.; Balakrishna M. S.; Tuononen H. M. Weak Interactions between Trivalent Pnictogen Centers: Computational Analysis of Bonding in Dimers X3E···EX3 (E = Pnictogen, X = Halogen). Inorg. Chem. 2009, 48 (14), 6740–6747. 10.1021/ic900635f. [DOI] [PubMed] [Google Scholar]
- Zahn S.; Frank R.; Hey-Hawkins E.; Kirchner B. Pnicogen Bonds: A New Molecular Linker?. Chem. - Eur. J. 2011, 17 (22), 6034–6038. 10.1002/chem.201002146. [DOI] [PubMed] [Google Scholar]
- Scheiner S. The Pnicogen Bond: Its Relation to Hydrogen, Halogen, and Other Noncovalent Bonds. Acc. Chem. Res. 2013, 46 (2), 280–288. 10.1021/ar3001316. [DOI] [PubMed] [Google Scholar]
- Desiraju G. R.; Ho P. S.; Kloo L.; Legon A. C.; Marquardt R.; Metrangolo P.; Politzer P.; Resnati G.; Rissanen K. Definition of the halogen bond (IUPAC Recommendations 2013). Pure Appl. Chem. 2013, 85 (8), 1711–1713. 10.1351/PAC-REC-12-05-10. [DOI] [Google Scholar]
- Scheiner S.Noncovalent forces; Springer International: Cham, Switzerland, 2015; online resource. [Google Scholar]
- Moaven S.; Yu J.; Vega M.; Unruh D. K.; Cozzolino A. F. Self-assembled reversed bilayers directed by pnictogen bonding to form vesicles in solution. Chem. Commun. 2018, 54 (64), 8849–8852. 10.1039/C8CC05187A. [DOI] [PubMed] [Google Scholar]
- Schmauck J.; Breugst M. The potential of pnicogen bonding for catalysis – a computational study. Org. Biomol. Chem. 2017, 15 (38), 8037–8045. 10.1039/C7OB01599B. [DOI] [PubMed] [Google Scholar]
- Benz S.; Poblador-Bahamonde A. I.; Low-Ders N.; Matile S. Catalysis with Pnictogen, Chalcogen, and Halogen Bonds. Angew. Chem., Int. Ed. 2018, 57 (19), 5408–5412. 10.1002/anie.201801452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Q. J.; Li Q. Z. Enhancing effect of metal coordination interaction on pnicogen bonding. J. Mol. Model. 2016, 22 (3), 1–7. 10.1007/s00894-016-2929-9. [DOI] [PubMed] [Google Scholar]
- Mahmudov K. T.; Gurbanov A. V.; Guseinov F. I.; da Silva M. F. C. G. Noncovalent interactions in metal complex catalysis. Coord. Chem. Rev. 2019, 387, 32–46. 10.1016/j.ccr.2019.02.011. [DOI] [Google Scholar]
- Moaven S.; Andrews M. C.; Polaske T. J.; Karl B. M.; Unruh D. K.; Bosch E.; Bowling N. P.; Cozzolino A. F. Triple-Pnictogen Bonding as a Tool for Supramolecular Assembly. Inorg. Chem. 2019, 58 (23), 16227–16235. 10.1021/acs.inorgchem.9b02761. [DOI] [PubMed] [Google Scholar]
- Scilabra P.; Terraneo G.; Daolio A.; Baggioli A.; Famulari A.; Leroy C.; Bryce D. L.; Resnati G. 4,4′-Dipyridyl Dioxide·SbF3 Cocrystal: Pnictogen Bond Prevails over Halogen and Hydrogen Bonds in Driving Self-Assembly. Cryst. Growth Des. 2020, 20 (2), 916–922. 10.1021/acs.cgd.9b01306. [DOI] [Google Scholar]
- Lu L.; Lu Y.; Zhu Z.; Liu H. Pnictogen, chalcogen, and halogen bonds in catalytic systems: theoretical study and detailed comparison. J. Mol. Model. 2020, 26 (1), 16. 10.1007/s00894-019-4275-1. [DOI] [PubMed] [Google Scholar]
- Yang M. X.; Hirai M.; Gabbai F. P. Phosphonium-stibonium and bis-stibonium cations as pnictogen-bonding catalysts for the transfer hydrogenation of quinolines. Dalton Trans. 2019, 48 (20), 6685–6689. 10.1039/C9DT01357A. [DOI] [PubMed] [Google Scholar]
- Legon A. C. Tetrel, pnictogen and chalcogen bonds identified in the gas phase before they had names: a systematic look at non-covalent interactions. Phys. Chem. Chem. Phys. 2017, 19 (23), 14884–14896. 10.1039/C7CP02518A. [DOI] [PubMed] [Google Scholar]
- Klinkhammer K. W.; Pyykko P. Ab Initio Interpretation of the Closed-Shell Intermolecular E···E Attraction in Dipnicogen (H2E-EH2)2 and Dichalcogen (HE-EH)2 Hydride Model Dimers. Inorg. Chem. 1995, 34 (16), 4134–4138. 10.1021/ic00120a018. [DOI] [Google Scholar]
- Oliveira V.; Kraka E. Systematic Coupled Cluster Study of Noncovalent Interactions Involving Halogens, Chalcogens, and Pnicogens. J. Phys. Chem. A 2017, 121 (49), 9544–9556. 10.1021/acs.jpca.7b10196. [DOI] [PubMed] [Google Scholar]
- Setiawan D.; Kraka E.; Cremer D. Strength of the Pnicogen Bond in Complexes Involving Group Va Elements N, P, and As. J. Phys. Chem. A 2015, 119 (9), 1642–1656. 10.1021/jp508270g. [DOI] [PubMed] [Google Scholar]
- Setiawan D.; Kraka E.; Cremer D. Description of pnicogen bonding with the help of vibrational spectroscopy-The missing link between theory and experiment. Chem. Phys. Lett. 2014, 614, 136–142. 10.1016/j.cplett.2014.09.030. [DOI] [Google Scholar]
- Marin-Luna M.; Alkorta I.; Elguero J. A computational study on [(PH2X)(2)](·+) homodimers involving intermolecular two-center three-electron bonds. Struct. Chem. 2016, 27 (3), 753–762. 10.1007/s11224-015-0617-5. [DOI] [Google Scholar]
- Roohi H.; Tondro T. Exploring the pnicogen bond non-covalent interactions in 4-XPhNH2:PFnH3-n complexes (n = 1–3, X = H, F, CN, CHO, NH2, CH3, NO2 and OCH3). J. Fluorine Chem. 2017, 202, 19–33. 10.1016/j.jfluchem.2017.08.009. [DOI] [Google Scholar]
- Trubenstein H. J.; Moaven S.; Vega M.; Unruh D. K.; Cozzolino A. F. Pnictogen bonding with alkoxide cages: which pnictogen is best?. New J. Chem. 2019, 43 (36), 14305–14312. 10.1039/C9NJ03648B. [DOI] [Google Scholar]
- Radha A.; Kumar S.; Sharma D.; Jassal A. K.; Zareba J. K.; Franconetti A.; Frontera A.; Sood P.; Pandey S. K. Indirect influence of alkyl substituent on sigma-hole interactions: The case study of antimony(III) diphenyldithiophosphates with covalent Sb-S and non-covalent Sb center dot center dot center dot S pnictogen bonds. Polyhedron 2019, 173, 114126. 10.1016/j.poly.2019.114126. [DOI] [Google Scholar]
- Tondro T.; Roohi H. Substituent effects on the halogen and pnictogen bonds characteristics in ternary complexes 4-YPhNH2 ···PH2F ··· ClX (Y = H, F, CN, CHO, NH2, CH3, NO2 and OCH3, and X = F, OH, CN, NC, FCC and NO2): A theoretical study. J. Chem. Sci. 2020, 132 (1), 1–21. 10.1007/s12039-019-1715-5. [DOI] [Google Scholar]
- Shukla R.; Chopra D. Pnicogen bonds” or “chalcogen bonds”: exploiting the effect of substitution on the formation of P···Se noncovalent bonds. Phys. Chem. Chem. Phys. 2016, 18 (20), 13820–13829. 10.1039/C6CP01703G. [DOI] [PubMed] [Google Scholar]
- Wysokiński R.; Zierkiewicz W.; Michalczyk M.; Scheiner S. How Many Pnicogen Bonds can be Formed to a Central Atom Simultaneously?. J. Phys. Chem. A 2020, 124, 2046. 10.1021/acs.jpca.0c00257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zierkiewicz W.; Michalczyk M.; Wysokinski R.; Scheiner S. On the ability of pnicogen atoms to engage in both σ and π-hole complexes. Heterodimers of ZF(2)C(6)H(5) (Z = P, As, Sb, Bi) and NH3. J. Mol. Model. 2019, 25 (6), 1–13. 10.1007/s00894-019-4031-6. [DOI] [PubMed] [Google Scholar]
- Esrafili M. D.; Vakili M.; Solimannejad M. Cooperative effects in pnicogen bonding: (PH2F)2–7 and (PH2Cl)2–7 clusters. Chem. Phys. Lett. 2014, 609, 37–41. 10.1016/j.cplett.2014.06.050. [DOI] [Google Scholar]
- Adhikari U.; Scheiner S. Comparison of P ··· D (D = P,N) with other noncovalent bonds in molecular aggregates. J. Chem. Phys. 2011, 135 (18), 184306. 10.1063/1.3660355. [DOI] [PubMed] [Google Scholar]
- Scheiner S. Comparison of halide receptors based on H, halogen, chalcogen, pnicogen, and tetrel bonds. Faraday Discuss. 2017, 203, 213–226. 10.1039/C7FD00043J. [DOI] [PubMed] [Google Scholar]
- Scheiner S.; Michalczyk M.; Zierkiewicz W. Structures of clusters surrounding ions stabilized by hydrogen, halogen, chalcogen, and pnicogen bonds. Chem. Phys. 2019, 524, 55–62. 10.1016/j.chemphys.2019.05.005. [DOI] [Google Scholar]
- Scheiner S.; Michalczyk M.; Wysokinski R.; Zierkiewicz W. Structures and energetics of clusters surrounding diatomic anions stabilized by hydrogen, halogen, and other noncovalent bonds. Chem. Phys. 2020, 530, 110590. 10.1016/j.chemphys.2019.110590. [DOI] [Google Scholar]
- Scheiner S. Effects of Substituents upon the P···N Noncovalent Interaction: The Limits of Its Strength. J. Phys. Chem. A 2011, 115 (41), 11202–11209. 10.1021/jp203964b. [DOI] [PubMed] [Google Scholar]
- Buzsáki D.; Kelemen Z.; Nyulászi L. Stretching the P–C Bond. Variations on Carbenes and Phosphanes. J. Phys. Chem. A 2020, 124, 2660. 10.1021/acs.jpca.0c00641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi P. R.; Ramanathan N.; Sundararajan K.; Sankaran K. Phosphorous bonding in PCl3:H2O adducts: A matrix isolation infrared and ab initio computational studies. J. Mol. Spectrosc. 2017, 331, 44–52. 10.1016/j.jms.2016.11.005. [DOI] [Google Scholar]
- Zong J.; Mague J. T.; Kraml C. M.; Pascal R. A. A Congested in,in-Diphosphine. Org. Lett. 2013, 15 (9), 2179–2181. 10.1021/ol400728m. [DOI] [PubMed] [Google Scholar]
- Joshi P. R.; Ramanathan N.; Sundararajan K.; Sankaran K. Evidence for Phosphorus Bonding in Phosphorus Trichloride-Methanol Adduct: A Matrix Isolation Infrared and ab Initio Computational Study. J. Phys. Chem. A 2015, 119 (14), 3440–3451. 10.1021/jp511156d. [DOI] [PubMed] [Google Scholar]
- Xu Y.; Szell P. M. J.; Kumar V.; Bryce D. L. Solid-state NMR spectroscopy for the analysis of element-based non-covalent interactions. Coord. Chem. Rev. 2020, 411, 213237. 10.1016/j.ccr.2020.213237. [DOI] [Google Scholar]
- Hupf E.; Lork E.; Mebs S.; Checinska L.; Beckmann J. Probing Donor-Acceptor Interactions in pen-Substituted Diphenylphosphinoacenaphthyl-Element Dichlorides of Group 13 and 15 Elements. Organometallics 2014, 33 (24), 7247–7259. 10.1021/om501036c. [DOI] [Google Scholar]
- Nejman P. S.; Curzon T. E.; Bühl M.; McKay D.; Woollins J. D.; Ashbrook S. E.; Cordes D. B.; Slawin A. M. Z.; Kilian P. Phosphorus–Bismuth Peri-Substituted Acenaphthenes: A Synthetic, Structural, and Computational Study. Inorg. Chem. 2020, 59, 5616. 10.1021/acs.inorgchem.0c00317. [DOI] [PubMed] [Google Scholar]
- Chitnis S. S.; Burford N.; Decken A.; Ferguson M. J. Coordination Complexes of Bismuth Triflates with Tetrahydrofuran and Diphosphine Ligands. Inorg. Chem. 2013, 52 (12), 7242–7248. 10.1021/ic400875a. [DOI] [PubMed] [Google Scholar]
- Wielandt J. W.; Petrie S.; Kilah N. L.; Willis A. C.; Dewhurst R. D.; Belaj F.; Orthaber A.; Stranger R.; Wild S. B. Self-Assembly of Square-Planar Halide Complexes of Trimethylphosphine-Stabilized Diphenyl-Arsenium, -Stibenium, and -Bismuthenium Hexafluorophosphates. Aust. J. Chem. 2016, 69 (5), 524–532. 10.1071/CH15701. [DOI] [Google Scholar]
- Kilah N. L.; Petrie S.; Stranger R.; Wielandt J. W.; Willis A. C.; Wild S. B. Triphenylphosphine-Stabilized Diphenyl-Arsenium, -Stibenium, and -Bismuthenium Salts. Organometallics 2007, 26 (25), 6106–6113. 10.1021/om700512h. [DOI] [Google Scholar]
- Chalmers B. A.; Meigh C. B. E.; Nejman P. S.; Bühl M.; Lébl T.; Woollins J. D.; Slawin A. M. Z.; Kilian P. Geminally Substituted Tris(acenaphthyl) and Bis(acenaphthyl) Arsines, Stibines, and Bismuthine: A Structural and Nuclear Magnetic Resonance Investigation. Inorg. Chem. 2016, 55 (14), 7117–7125. 10.1021/acs.inorgchem.6b01079. [DOI] [PubMed] [Google Scholar]
- Mokrai R.; Barrett J.; Apperley D. C.; Batsanov A. S.; Benkő Z.; Heift D. Weak Pnictogen Bond with Bismuth: Experimental Evidence Based on Bi-P Through-Space Coupling. Chem. - Eur. J. 2019, 25 (16), 4017–4024. 10.1002/chem.201900266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordero B.; Gomez V.; Platero-Prats A. E.; Reves M.; Echeverria J.; Cremades E.; Barragan F.; Alvarez S. Covalent radii revisited. Dalton Trans. 2008, 21, 2832–2838. 10.1039/b801115j. [DOI] [PubMed] [Google Scholar]
- Mantina M.; Chamberlin A. C.; Valero R.; Cramer C. J.; Truhlar D. G. Consistent van der Waals Radii for the Whole Main Group. J. Phys. Chem. A 2009, 113 (19), 5806–5812. 10.1021/jp8111556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clardy J. C.; Milbrath D. S.; Springer J. P.; Verkade J. G. Characterization and molecular structure of 1-hydro-2,8,9-trioxa-1-phospha-5-aza-tricyclo[3.3.3.0]undecane tetrafluoroborate. Pentacoordination of phosphorus. J. Am. Chem. Soc. 1976, 98 (2), 623–624. 10.1021/ja00418a058. [DOI] [Google Scholar]
- Verkade J. G. Main-Group Atranes - Chemical and Structural Features. Coord. Chem. Rev. 1994, 137, 233–295. 10.1016/0010-8545(94)03007-D. [DOI] [Google Scholar]
- Nyulaszi L.; Veszpremi T.; DSa B. A.; Verkade J. Photoelectron spectra and structures of proazaphosphatranes. Inorg. Chem. 1996, 35 (21), 6102–6107. 10.1021/ic960532+. [DOI] [Google Scholar]
- Liu X.; Bai Y.; Verkade J. G. Synthesis and structural features of new sterically hindered azaphosphatrane systems: ZP(RNCH2CH2)(3)N. J. Organomet. Chem. 1999, 582 (1), 16–24. 10.1016/S0022-328X(98)01180-2. [DOI] [Google Scholar]
- Suter R.; Swidan A. a.; Macdonald C. L. B.; Burford N. Oxidation of a germanium(II) dication to access cationic germanium(IV) fluorides. Chem. Commun. 2018, 54 (33), 4140–4143. 10.1039/C8CC01799A. [DOI] [PubMed] [Google Scholar]
- Swidan A.; Suter R.; Macdonald C. L. B.; Burford N. Tris(benzoimidazol)amine (L) complexes of pnictogen(III) and pnictogen(V) cations and assessment of the [LP]3+/[LPF2]3+ redox couple. Chem. Sci. 2018, 9 (26), 5837–5841. 10.1039/C8SC01682H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green J. P.; Wells J. A. L.; Orthaber A. Heavier pnictogens – treasures for optical electronic and reactivity tuning. Dalton Trans. 2019, 48 (14), 4460–4466. 10.1039/C9DT00574A. [DOI] [PubMed] [Google Scholar]
- Weigand J. J.; Feldmann K.-O.; Echterhoff A. K. C.; Ehlers A. W.; Lammertsma K. Preparation of Ligand-Stabilized [P4O4]2+ by Controlled Hydrolysis of a Janus Head Type Diphosphorus Trication. Angew. Chem., Int. Ed. 2010, 49 (35), 6178–6181. 10.1002/anie.201001363. [DOI] [PubMed] [Google Scholar]
- Šimon P.; de Proft F.; Jambor R.; Růžička A.; Dostál L. Monomeric Organoantimony(I) and Organobismuth(I) Compounds Stabilized by an NCN Chelating Ligand: Syntheses and Structures. Angew. Chem., Int. Ed. 2010, 49 (32), 5468–5471. 10.1002/anie.201002209. [DOI] [PubMed] [Google Scholar]
- Kremláček V.; Hyvl J.; Yoshida W. Y.; Růžička A.; Rheingold A. L.; Turek J.; Hughes R. P.; Dostál L.; Cain M. F. Heterocycles Derived from Generating Monovalent Pnictogens within NCN Pincers and Bidentate NC Chelates: Hypervalency versus Bell-Clappers versus Static Aromatics. Organometallics 2018, 37 (15), 2481–2490. 10.1021/acs.organomet.8b00290. [DOI] [Google Scholar]
- Brown I. D. VALENCE: A program for calculating bond valences. J. Appl. Crystallogr. 1996, 29, 479–480. 10.1107/S002188989600163X. [DOI] [Google Scholar]
- Brese N. E.; O’Keeffe M. Bond-Valence Parameters for Solids. Acta Crystallogr., Sect. B: Struct. Sci. 1991, 47, 192–197. 10.1107/S0108768190011041. [DOI] [Google Scholar]
- Glendening E. D.; Reed A. E.; Carpenter J. E.; Bohmann J. A.; Morales C. M.; Weinhold F.. NBO 5.0.; Theoretical Chemistry Institute, University of Wisconsin: Madison, WI, 2001.
- Bader R. F. W. A quantum theory of molecular structure and its applications. Chem. Rev. 1991, 91 (5), 893–928. 10.1021/cr00005a013. [DOI] [Google Scholar]
- Cremer D.; Kraka E. Chemical-Bonds without Bonding Electron-Density - Does the Difference Electron-Density Analysis Suffice for a Description of the Chemical-Bond. Angew. Chem., Int. Ed. Engl. 1984, 23 (8), 627–628. 10.1002/anie.198406271. [DOI] [Google Scholar]
- Cremer D.; Kraka E. A Description of the Chemical-Bond in Terms of Local Properties of Electron-Density and Energy. Croat. Chem. Acta 1984, 57 (6), 1259–1281. [Google Scholar]
- Riddlestone I. M.; Kraft A.; Schaefer J.; Krossing I. Taming the Cationic Beast: Novel Developments in the Synthesis and Application of Weakly Coordinating Anions. Angew. Chem., Int. Ed. 2018, 57 (43), 13982–14024. 10.1002/anie.201710782. [DOI] [PubMed] [Google Scholar]
- Ollevier T. New trends in bismuth-catalyzed synthetic transformations. Org. Biomol. Chem. 2013, 11 (17), 2740–2755. 10.1039/c3ob26537d. [DOI] [PubMed] [Google Scholar]
- Ollevier T. Bismuth-Mediated Organic Reactions. Top. Curr. Chem. 2012, 311, 1–277. 10.1007/978-3-642-27239-4. [DOI] [PubMed] [Google Scholar]
- Labrouillere M.; Le Roux C.; Gaspard H.; Laporterie A.; Dubac J.; Desmurs J. R. An efficient method for the preparation of bismuth(III) trifluoromethanesulfonate. Tetrahedron Lett. 1999, 40 (2), 285–286. 10.1016/S0040-4039(98)02397-1. [DOI] [Google Scholar]
- Gaspard-Iloughmane H.; Le Roux C. Bismuth(III) triflate in organic synthesis. Eur. J. Org. Chem. 2004, 2004 (12), 2517–2532. 10.1002/ejoc.200300754. [DOI] [Google Scholar]
- Chitnis S. S.; Vos K. A.; Burford N.; McDonald R.; Ferguson M. J. Distinction between coordination and phosphine ligand oxidation: interactions of di- and triphosphines with Pn(3+) (Pn = P, As, Sb, Bi). Chem. Commun. 2016, 52 (4), 685–688. 10.1039/C5CC08086J. [DOI] [PubMed] [Google Scholar]
- Loera Fernandez I. I.; Donaldson S. L.; Schipper D. E.; Andleeb S.; Whitmire K. H. Anionic Bismuth-Oxido Carboxylate Clusters with Transition Metal Countercations. Inorg. Chem. 2016, 55 (21), 11560–11569. 10.1021/acs.inorgchem.6b02092. [DOI] [PubMed] [Google Scholar]
- Dehnen S.; Corrigan J. F.. Clusters - contemporary insight in structure and bonding; Springer International: Cham, Switzerland, 2017; p 379. [Google Scholar]
- Senevirathna D. C.; Blair V. L.; Werrett M. V.; Andrews P. C. Polynuclear Bismuth Oxido Sulfonato Clusters, Polymers, and Ion Pairs from Bi2O3 under Mild Conditions. Inorg. Chem. 2016, 55 (21), 11426–11433. 10.1021/acs.inorgchem.6b01937. [DOI] [PubMed] [Google Scholar]
- Senevirathna D. C.; Werrett M. V.; Blair V. L.; Mehring M.; Andrews P. C. 2D and 3D Coordination Networks of Polynuclear Bismuth Oxido/Hydroxido Sulfonato Clusters from Low Temperature Solid-State Metathesis Reactions. Chem. - Eur. J. 2018, 24 (26), 6722–6726. 10.1002/chem.201705981. [DOI] [PubMed] [Google Scholar]
- Rogow D. L.; Fei H. H.; Brennan D. P.; Ikehata M.; Zavalij P. Y.; Oliver A. G.; Oliver S. R. J. Hydrothermal Synthesis of Two Cationic Bismuthate Clusters: An Alkylenedisulfonate Bridged Hexamer, [Bi6O4(OH)(4)(H2O)(2)][(CH2)(2)(SO3)(2)](3) and a Rare Nonamer Templated by Triflate, [Bi9O8(OH)(6)][CF3SO3](5). Inorg. Chem. 2010, 49 (12), 5619–5624. 10.1021/ic1004402. [DOI] [PubMed] [Google Scholar]
- Pye C. C.; Gunasekara C. M.; Rudolph W. W. An ab initio investigation of bismuth hydration. Can. J. Chem. 2007, 85 (11), 945–950. 10.1139/v07-108. [DOI] [Google Scholar]
- Sundvall B. Crystal and Molecular-Structure of Tetraoxotetrahydroxobismuth(III) Nitrate Monohydrate, Bi6O4(HO)4(NO3)6.H2O. Acta Chem. Scand. A 1979, 33 (3), 219–224. 10.3891/acta.chem.scand.33a-0219. [DOI] [Google Scholar]
- Sattler D.; Schlesinger M.; Mehring M.; Schalley C. A. Mass Spectrometry and Gas-Phase Chemistry of Bismuth-Oxido Clusters. ChemPlusChem 2013, 78 (9), 1005–1014. 10.1002/cplu.201300122. [DOI] [PubMed] [Google Scholar]
- Srinivas K.; Sathyanarayana A.; Babu C. N.; Prabusankar G. Bismuth(III)dichalcogenones as highly active catalysts in multiple C-C bond formation reactions. Dalton Trans. 2016, 45 (12), 5196–5209. 10.1039/C5DT04738B. [DOI] [PubMed] [Google Scholar]
- Mazieres S.; Le Roux C.; Peyronneau M.; Gornitzka H.; Roques N. Structural characterization of bismuth(III) and antimony(III) chlorotriflates: Key intermediates in catalytic Friedel-Crafts transformations. Eur. J. Inorg. Chem. 2004, 2004 (14), 2823–2826. 10.1002/ejic.200400281. [DOI] [Google Scholar]
- Mallory F. B.; Luzik E. D.; Mallory C. W.; Carroll P. J. Nuclear spin-spin coupling via nonbonded interactions. 7. Effects of molecular structure on nitrogen-fluorine coupling. J. Org. Chem. 1992, 57 (1), 366–370. 10.1021/jo00027a063. [DOI] [Google Scholar]
- Manatt S. L.; Cooper M. A.; Mallory C. W.; Mallory F. B. Evidence for a Steric Effect on Directly Bonded Carbon-Fluorine and Carbon-Proton Nuclear Magnetic-Resonance Couplings. J. Am. Chem. Soc. 1973, 95 (3), 975–977. 10.1021/ja00784a086. [DOI] [Google Scholar]
- Hierso J.-C.; Fihri A.; Ivanov V. V.; Hanquet B.; Pirio N.; Donnadieu B.; Rebière B.; Amardeil R.; Meunier P. “Through-Space” Nuclear Spin–Spin JPP Coupling in Tetraphosphine Ferrocenyl Derivatives: A 31P NMR and X-ray Structure Correlation Study for Coordination Complexes. J. Am. Chem. Soc. 2004, 126 (35), 11077–11087. 10.1021/ja048907a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.