

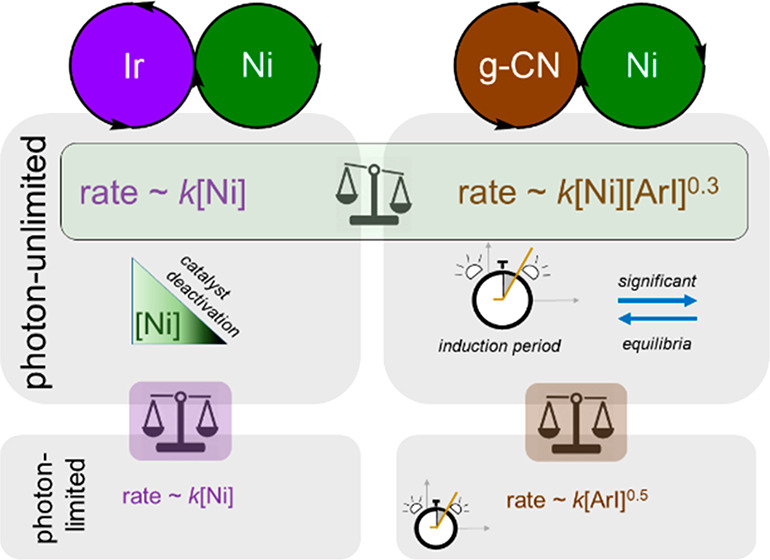
Abstract
Dual photocatalysis and nickel catalysis can effect cross-coupling under mild conditions, but little is known about the in situ kinetics of this class of reactions. We report a comprehensive kinetic examination of a model carboxylate O-arylation, comparing a state-of-the-art homogeneous photocatalyst (Ir(ppy)3) with a competitive heterogeneous photocatalyst (graphitic carbon nitride). Experimental conditions were adjusted such that the nickel catalytic cycle is saturated with excited photocatalyst. This approach was designed to remove the role of the photocatalyst, by which only the intrinsic behaviors of the nickel catalytic cycles are observed. The two reactions did not display identical kinetics. Ir(ppy)3 deactivates the nickel catalytic cycle and creates more dehalogenated side product. Kinetic data for the reaction using Ir(ppy)3 supports a turnover-limiting reductive elimination. Graphitic carbon nitride gave higher selectivity, even at high photocatalyst-to-nickel ratios. The heterogeneous reaction also showed a rate dependence on aryl halide, indicating that oxidative addition plays a role in rate determination. The results argue against the current mechanistic hypothesis, which states that the photocatalyst is only involved to trigger reductive elimination.
Introduction
Palladium catalysis is key to many conceptual and practical advances in modern organic chemistry, but the scarcity of palladium bodes poorly for long-term use. As such, transferring well-defined reactivities of palladium to nickel, an isoelectronic and more abundant metal, has been an explicit goal of catalysis for more than 50 years. Low-valent nickel species are capable of undergoing oxidative additions readily, but the subsequent reductive eliminations (REs), particularly for Ni(II) complexes, are thought to be comparatively difficult.1 Nickel couplings are hence believed to go through higher-valent intermediates,2,3 accessed by stoichiometric redox modulators, such that the RE is more favorable.4,5
The combination of photocatalysis with nickel catalysis has provided an elegant pathway by which nickel can turn over without adding excessive reductants or harsh conditions.6,7 Iridium polypyridyl complexes were found to act as photocatalysts (PCs) to effect C–N, C–O, C–S, and C–C couplings using nickel(II) precatalysts.8−11 Subsequent reports showed that the homogeneous iridium photocatalysts can be replaced with heterogeneous graphitic carbon nitride (g-CN) semiconductors, rendering the overall transformations noble-metal-free.12−16
The current mechanistic proposal for a relatively well-studied carboxylate O-arylation (Figure 1A) in this family of reactions is depicted in Figure 1B.17 Numerous common nickel(II) precatalysts are viable, which are thought to undergo two single-electron transfers to a bipyridyl Ni(0) 4. From this reduced species, oxidative addition to form 5 is facile. The catalyst resting state 6 is unable to undergo the necessary reductive elimination. To close the cycle, 6 receives an energy transfer from the excited state of the photocatalyst, which enables C–O bond formation.18,19 Stern–Volmer quenching studies,17 transient absorption spectroscopy,18 and computational studies19 have been used to support quenching of Ir(ppy)3 by a potential nickel(II) intermediate.
Figure 1.
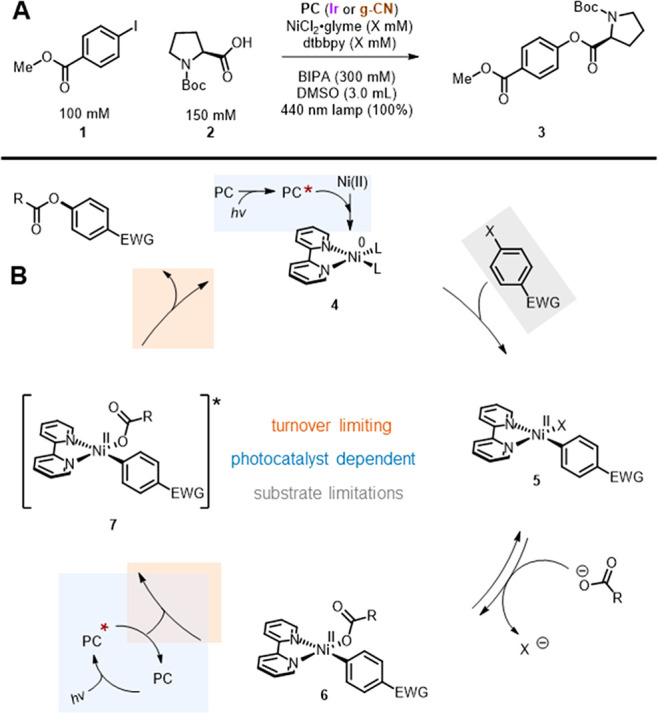
(A) The two dual photocatalyzed and nickel-catalyzed reactions studied in this work. Standard conditions are shown. Deviations from these conditions in any subsequent figure will be noted. (B) Mechanistic hypothesis presented in 2017.17
Broadly, the substrate scopes for both homogeneous (Ir(ppy)3) and heterogeneous (g-CN) variations of carbon–heteroatom couplings contain only electron-poor aryl halides and electron-neutral or rich coupling partners (HNR2, HOR, HSR, etc.). To our knowledge, these limitations have not been explicitly discussed in a mechanistic context. The Ir(ppy)3-catalyzed homogeneous carboxylate O-arylation accommodates only electron-withdrawing aryl bromides, and electron-withdrawing carboxylic acids are absent. Such a combination could be explained by arguing that an SNAr-type reductive elimination is required.
However, tension between the existing mechanism and the resulting substrate scopes surfaced during our development of the heterogeneous congener using graphitic carbon nitride photocatalysts.12 During a cursory kinetic study, methyl 4-iodobenzoate 1 (96%, 4 h) coupled considerably faster than methyl 4-bromobenzoate (88%, 48 h) with N-Boc-proline 2. Once the nickel catalytic cycle is at the resting state 6, the differences between an aryl iodide and bromide are irrelevant, as only the aryl ring remains on-cycle. This discrepancy in rate must therefore arise from the oxidative addition or ligand exchange step. As even aryl chlorides show facile proclivities toward oxidative addition with low-valent nickel in non-photochemical reactions,20 and the homogeneous analogue converted aryl bromides efficiently, we began to question whether the currently accepted mechanism is operational with the heterogeneous congener. Given that we found the same electronic substituent limitations with g-CN as for Ir(ppy)3, albeit with iodides instead of bromides, we further wondered if there were mechanistic commonalities among the two O-arylations.
Electron deficiency of an aryl halide can accelerate oxidative additions, as the aryl halide is itself formally reduced. In this vein, aryl halide limitations in this class of reactions could be explained by scission of the aryl halide bond that pushes at the energetic boundaries of photocatalytic reductions. Reduction potentials of common aryl halides range from −1.0 to −3.5 V vs SCE,21,22 with electron-donating groups and higher-row halides ranking as more difficult. The iridium polypyridyl complex Ir(ppy)3 used in the homogeneous O-arylation has a reduction potential of −1.73 V, and substrate limitations could be a marker of the Ir(III)*/Ir(IV) potential within the aforementioned range.23 Such logic has been invoked for the thioetherification of aryl iodides24 as well as dehalogenations25,26 with this exact photocatalyst. Stern–Volmer and transient absorption studies showed that aryl iodides could in fact quench the excited triplet state of Ir(ppy)3,24 forming an aryl radical.
With these considerations in mind, we set out to answer three basic questions: (1) Do the heterogeneous and homogeneous reactions proceed by the same mechanism? If so, (2) do they both conform to the extant hypothesis, in which reductive elimination is turnover-limiting, and (3) are the turnover-limiting steps and the steps which require a photocatalyst the same?
Research Plan and Hypotheses
Numerous mechanistic studies have been undertaken for photocatalytic reactions in general.27 Inquiries on dual nickel-catalyzed and photocatalyzed carbon–heteroatom cross-couplings have largely focused on the character of the interaction between photocatalyst and metal catalyst.19,28 However, no in-depth kinetics studies of any of these reactions have been disclosed.
Kinetics studies of catalytic systems that observe native reaction conditions throughout their entire course can afford a wealth of qualitative and quantitative information usually inaccessible from studies of a narrower scope.29,30 However, “native” reaction conditions are typically based on the methodology studies where the primary aim is the optimization of yield. Studies of dual photocatalytic and nickel catalytic reactions under such conditions may give data obfuscated by rate-limiting processes derived from interaction between the PC and metal catalyst. Kinetically, this may paint an incomplete picture of the reaction, but these data could arguably be useful to synthetic chemists considering using such a method. We therefore sought to observe the homogeneous and heterogeneous dual photo- and nickel-catalyzed O-arylations in two regimes each. The first would be closer to the standard conditions obtained during methodology development (“photon-limited”), geared toward understanding overall reaction behavior. The second would be in a regime in which the metal catalyst is saturated with excited photocatalyst species (“photon-unlimited”). Under such conditions, photons are essentially treated as a reagent in high excess. Data obtained in this regime allow us to both evaluate the intrinsic kinetics of the nickel cycle, as well as enable a more rigorous comparison between data sets of different photocatalysts.
As no in situ studies exist to date for any dual nickel-catalyzed and photocatalyzed cross-couplings, comparison of the photon-limited and photon-unlimited regimes with each other for the two reactions would be of general interest. We anticipated that we would see differences between the photon-limited and photon-unlimited regimes, potentially in the magnitude of the reagents’ coefficients in the rate laws.
Most importantly, the intrinsic, photon-unlimited kinetics of both reactions should be compared with each other and to the extant mechanistic hypothesis. The kinetics in photon-unlimited regimes of the homogeneous and heterogeneous reactions should be identical since the PC should drop out of the rate law. The extant hypothesis for the dual photo- and nickel-catalyzed carboxylate O-arylation, congruent with the accrued understanding of nickel catalysis, is that the PC enables a turnover-limiting reductive elimination.17 Thus, we predicted that the photon-unlimited regime for both reactions would give rate ∼k[Ni].
Results
Heterogeneous (Graphitic Carbon Nitride) PC
Photon-Limited (3.33 mg/mL g-CN, 5 mM Ni·L, 50% Lamp Power)
First, the kinetics of the carboxylate O-arylation of aryl iodides using the graphitic carbon nitride CN-OA-m31 as a photocatalyst in photon-limited conditions were analyzed. These photocatalytic reactions can be tracked by attaching a custom vial to an in situ infrared probe.12 Using a stronger LED lamp for increased photon flux and improved reproducibility, we ensured that the infrared technique tracked reaction progress by verification with an orthogonal method (Figure S2).
From a cursory, qualitative glance, these reactions at the native conditions display no unusual behavior, except for exhibiting a short induction period. A series of experiments were conducted to assess which reagents were responsible for this induction (Figure S5). Delaying the injection of NiCl2·glyme and its bipyridyl ligand or aryl iodide 1 resulted in immediate productive catalysis. Delayed injection of carboxylic acid 2 retained the induction period. Conspicuously, delaying addition of the base N-tert-butylisopropylamine (BIPA) resulted in a reaction profile slightly suggestive of catalyst activation,32 indicating that the secondary amine plays a role more significant than simply that of a Brønsted base.
Varying the nickel concentration from 5 to 20 mM unsurprisingly resulted in no rate change (Figure S6). At 5 mM Ni, no catalyst deactivation or product inhibition was observed (Figure S8), which is expected at such a high metal catalyst concentration. Surprisingly, modulation in [1] resulted in a fractionally positive order (Figure S9) for the aryl iodide. Carboxylic acid 2 showed a zero-order dependence (Figure S10). Reagent orders obtained in this regime should be considered with some measure of caution as the interactions between PC and Ni are ill-defined. However, we were curious if the reagent order of the aryl iodide would persist in the photon-unlimited regime, as it would be a significant clue to help resolve the questions posed above.
Photon-Unlimited (3.33 mg/mL g-CN, 0.2 mM Ni·L, 100% Lamp Power)
To switch from the photon-limited to the photon-unlimited regime, we needed to increase the ratio of photons and/or PC to nickel. Although numerous photocatalytic reactions with metal centers have been tracked with in situ apparatuses,33 to our knowledge only one study has described finding a region in which the metal’s access to excitation is carefully controlled to be unimpeded.34 In the work of Lehnherr et al., light directly excited a metal catalyst, with no photocatalytic intermediary. To find a region in which their metal catalyst was not limited by throughput of photons, initial rate was plotted against catalyst concentration, and at lower concentrations a roughly linear correlation is apparent. To make a comparable plot, we retained a high PC loading and maximum lamp power (Figure 2A).
Figure 2.
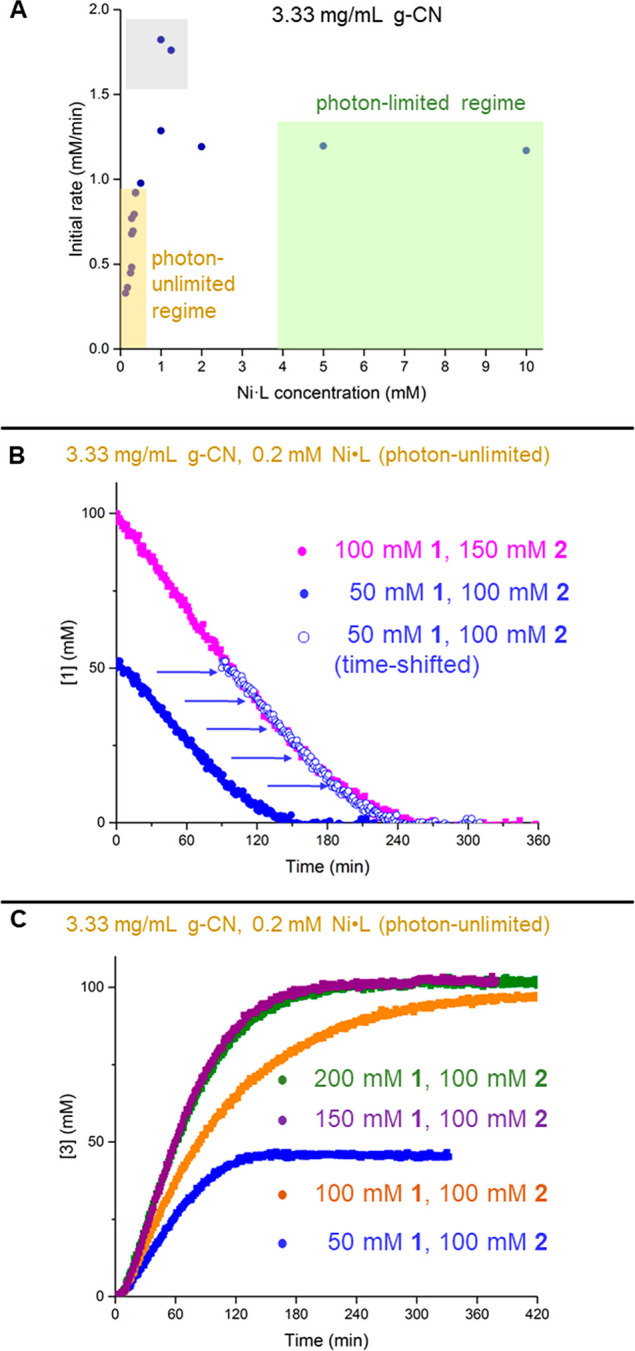
Key experiments of heterogeneous congener. (A) Overview of where the photon-limited and unlimited regimes could be identified. (B) Time-shifted same excess experiments. Overlay indicates lack of catalyst deactivation or product inhibition. (C) Positive-order dependence of aryl iodide observed.
Moving from high to low nickel concentrations along this plot surprisingly did not provide an identical shape to that of the literature precedent. Specifically, we observed an unexpected “peak” where catalysis has a much higher rate, around 1 mM. Although further studies are required to account for this behavior, it is possible that, above a certain threshold, the metal catalyst engages in some combination of comproportionation35,36 and/or higher-order nickel species thought to be catalytically relevant.37,38
The area at very low concentration of nickel would likely provide a regime in which the nickel catalytic cycle was not limited by access to an excited photocatalytic species. To confirm this was the case, at low concentrations we used variable time normalization analysis (VTNA)39 to find a first-order dependence on nickel between 0.15–0.25 mM (Figure S11). Kinetic studies in this area are expected to produce data that reflect the intrinsic characteristics of the nickel cycle.
To probe the robustness of the nickel catalyst, two “same-excess” experiments were conducted29,40 under photon-unlimited conditions and the less concentrated was time-shifted on the x-axis (Figure 2B). Overlay here indicates that the product does not inhibit catalysis, nor does meaningful or disproportionate catalyst deactivation occur.
Experiments to determine the reagent orders of the substrates revealed that aryl iodide again exhibited a positive order dependence (Figure 2C). Due to the induction period, first attempts at VTNA overlay were found to be problematic (Figure S12). Elegant mathematical solutions to this common issue have been disclosed,41 but the most rigorous would require spectroscopic determination of the active catalyst concentration. Repeating the different excess experiments with delayed catalyst injections (Figure S13), which bypass the induction period as described above, cleanly leads to a reagent order of 0.3 under 150 mM [1].
Examination of the dependence on reagents’ concentrations for 2, the base, and the photocatalyst provided a much less straightforward picture. All three reagents exhibit positive order dependence at lower concentrations and inhibitory effects at higher concentrations (Figures S14–S16).
Homogeneous (Ir(ppy)3) PC
We began the homogeneous studies by attempting to determine a proper photocatalyst loading that would enable photon-limited conditions. Initial experiments with 0.1 mM Ni·L and the standard (1 mM) photocatalyst concentration17 gave unsatisfactory yields. We observed dehalogenation accompanying productive catalysis (Table 1). Formation of the phenol side product 9 appears to be independent of an unfavorable PC-to-Ni ratio.
Table 1. Screening to Determine Photocatalyst Loading for Photon-Limited Regimes of the Homogeneous Reactiona.

| mM PC | 3 | 1 | 8 | 9 | Mass balance |
|---|---|---|---|---|---|
| 1 | 62 | 0 | 18 | 6 | 86 |
| 0.5 | 83 | 0 | 10 | 4 | 97 |
| 0.25 | 82 | 0 | 8 | 3 | 94 |
| 0.125 | 90 | 0 | 5 | 5 | 100 |
Final concentrations of product and side products were determined by 1H NMR using 1,3,5-trimethoxybenzene as an internal standard.
High concentrations of Ir(ppy)3 accelerate the consumption of 1 but not significantly the rate of product formation (Figure 3A). This finding is in striking contrast to the heterogeneous analogue, where slower product formation is a function of slower consumption of 1 at higher PC loading (Figure S16). Experiments with 0.125 mM PC loading (Figure 3A, blue) appear to be limited by the relay of energy from PC to Ni (Figure S17), and are the basis for the photon-limited studies.
Figure 3.
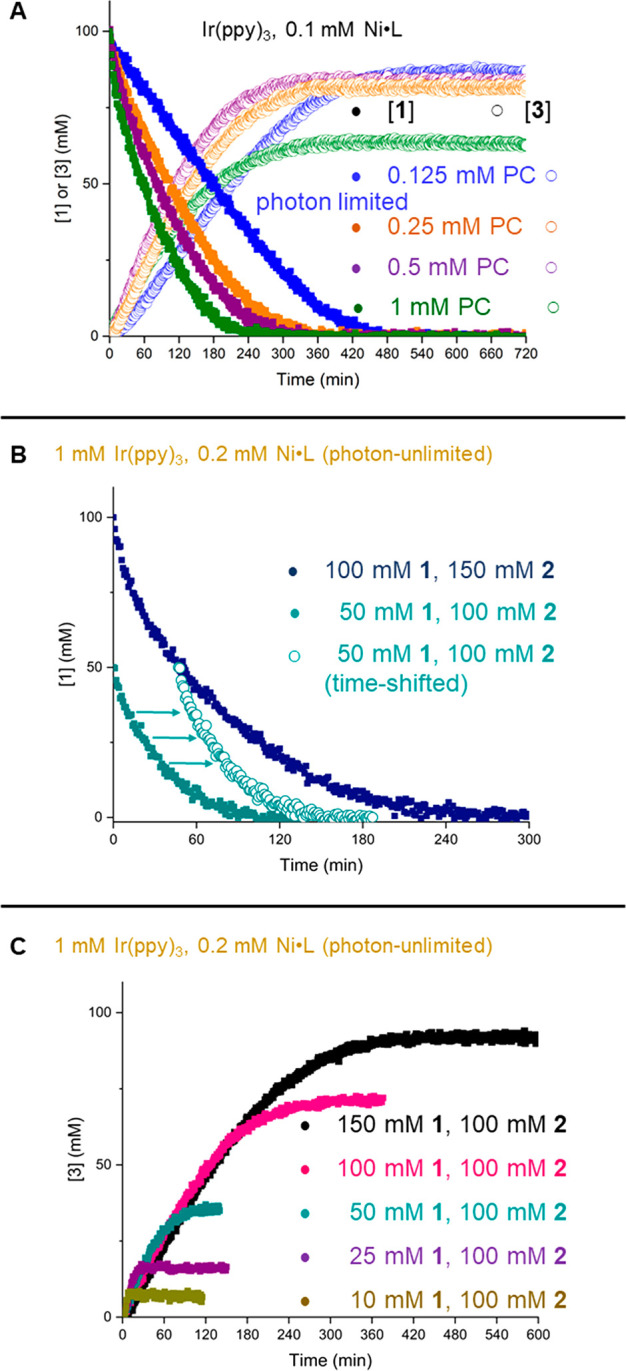
Key experiments of homogeneous congener. (A) Identification of photon-limited regime by reaction fingerprint; reactions progress of blue curve appears to be limited by interaction between PC and Ni. (B) Same excess experiment shows catalyst deactivation intrinsic to iridium-catalyzed O-arylations. (C) Rate is independent of [1].
Photon-Limited (0.125 mM Ir(ppy)3, 0.1 mM Ni·L, 100% Lamp Power)
The first noteworthy qualitative observation of iridium-catalyzed O-arylations is the lack of induction period. Catalysis begins immediately in a positive-order fashion. Increasing concentration of nickel leads to increased rate, which was determined to be a first-order dependence by VTNA (Figure S18). A same-excess experiment shows no deactivation of the nickel throughout the course of the reaction (Figure S19). In contrast to the heterogeneous congener, no rate dependence was found for 1 or 2 (Figure S20). Interestingly, increasing concentration of base increases the rate (Figure S21). Again, some caution is advised in mechanistic interpretation from these data, as these conditions are designed to maximize yield. However, the lack of rate dependence on aryl iodide could be a significant finding if it persisted in photon-unlimited conditions.
Photon-Unlimited (1 mM Ir(ppy)3, 0.2 mM Ni·L, 100% Lamp Power)
In order to draw meaningful comparisons between the two photon-unlimited data sets, we sought to use the same [Ni]t=0 as was used for the heterogeneous system. As such, finding the photon-unlimited regime for the homogeneous reaction occurred through a reverse process from the heterogeneous case. A short series of experiments was conducted in which the nickel concentration was kept constant and the photocatalyst concentration was raised, similar to Figure 3A (Figure S22). VTNA analysis showed first-order nickel dependence in the concentration range around 0.2 mM Ni at 1 mM Ir(ppy)3 (Figure S24). Corroboration that this approach gives a photon-unlimited regime was found through data obtained in a similar regime derived from an initial-rate plot (Figures S27–S31).
A major difference between the heterogeneous and homogeneous reaction was apparent from the same-excess experiment, where no overlay was observed (Figure 3B). Should the product inhibit catalysis, an effect in the heterogeneous reaction would have been observed, so we can attribute this behavior to catalyst deactivation.
Experiments varying the amounts of substrate and coupling partner were conducted to obtain the reagent orders of our reactants. In contrast to the heterogeneous reaction, we found the rate to be essentially independent of the concentration of 1 (Figure 3C). The dependence on [2] was not found to be significant (Figure S25). Last, we modified the amount of base while keeping all other reagents constant. Similar to the observation from the photon-limited data set, this change was the sole factor that accelerates the homogeneous reaction (Figure S26).
Discussion
The most conspicuous finding of this study was the rejection of the hypothesis that the kinetics of both systems would give identical rate laws. In other words, as particular care was taken such that the nickel cycle was saturated with excited photocatalyst species for both systems, the intrinsic kinetics of a nickel cross-coupling cycle should have been identical between the two had the mechanisms been the same.
The chief quantitative finding of this study is the fractional dependence of aryl iodide 1 in the heterogeneous rate law. To some degree in the heterogeneous congener, oxidative addition is turnover-limiting. We propose that this is a result of direct photocatalytic activation of the substrate (Figure 4B). The fractional order arises as this activation occurs off of the nickel cycle. Nickel is first-order as it intercepts the radical species on the nickel catalytic cycle.
Figure 4.
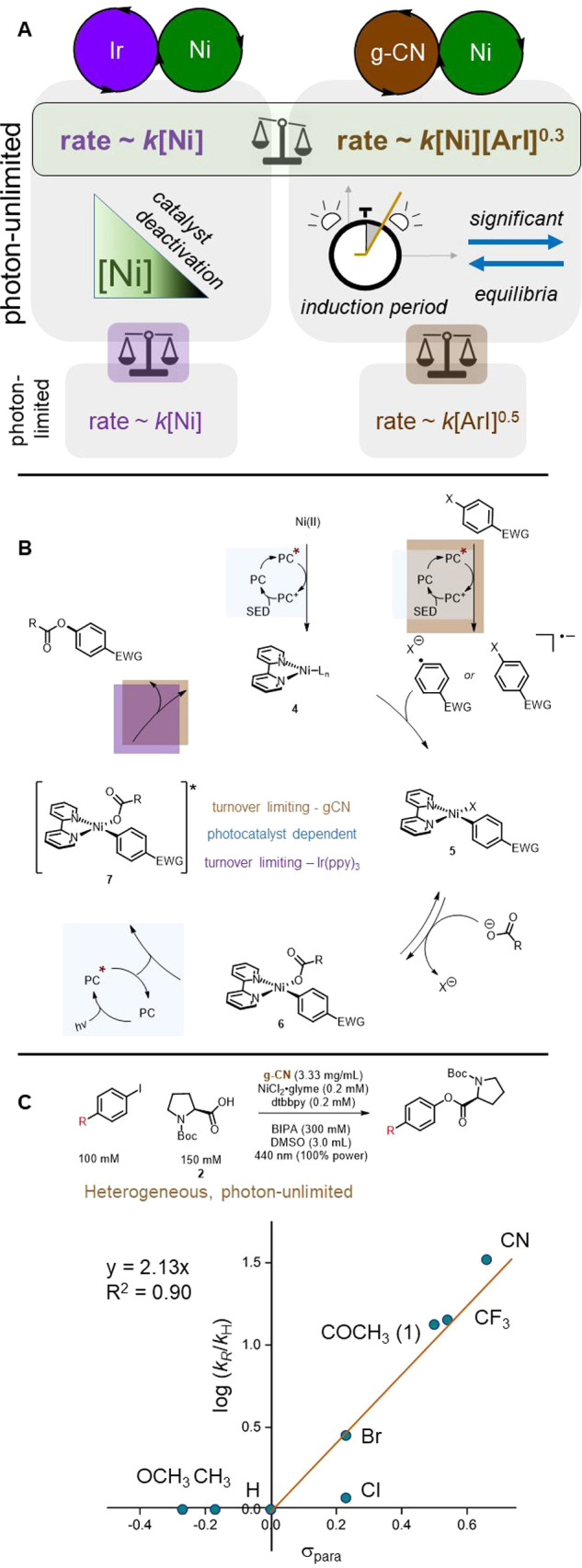
(A) Key findings of this study. (B) Updated mechanistic proposal. Secondary amine base is proposed to close the photoredox cycle by serving as single electron donor (SED). (C) Hammett plot for heterogeneous congener supports that oxidative addition is to some degree turnover-limiting.
This process may have been overlooked because it is not necessarily turnover-limiting. The reduction potential of Ir(III)*/Ir(IV), −1.73 V, is sufficient to donate an electron to the aryl halide 1;25,42,43 this step is photocatalyst-dependent but fast for aryl iodides (Figure 4B). In contrast, the analogous excited state of the graphitic carbon nitride used in this study is less reducing (up to −1.65 V vs SCE). The capacity for electron transfer is worse, and therefore even in conditions that can be considered photon-unlimited, formation of oxidative addition intermediate 5 is still limited by the kinetically relevant halide fragmentation.
We reject the hypothesis that photocatalysts are required only as a late-cycle trigger for RE. We contend that the photocatalyst is required for at least one additional point on the catalytic cycle, namely for single-electron transfer to the aryl halide. Substrate limitations for dual photo- and nickel-catalyzed carbon–heteroatom cross-couplings derive from this elementary step. The most obvious example is the far superior conversion of aryl bromides in this homogeneous O-arylation compared to the heterogeneous congener. Such a framework also harmonizes the mechanisms by which these transformations occur with the existing body of photocatalytic dehalogenations and radical-based transformations.25,26
To augment our understanding of the heterogeneous reaction, we created a Hammett plot examining rate dependencies on aryl iodides with substituents of varying electronic character (Figure 4C). Qualitatively, this shape is also seen for the oxidative addition of tris(triphenylphosphine)nickel(0) to aryl chlorides.20 Our ρ value (2.1) is far below that of this more traditional oxidative addition (8.8), thought to proceed through an SNAr-type mechanism. This could be explained by differences in ligand systems and halides, but also could point to a lowered dependence on the electronic substituent due to a non-concerted oxidative addition.
With a rate law proportional only to the nickel concentration, data for the homogeneous reaction could reasonably fit three turnover-limiting steps: a unimolecular rearrangement, ligand dissociation, or reductive elimination. We have observed nothing to support or reject the first two. Given that the incumbent hypothesis is that RE is turnover-limiting, our data support the existing proposals. As we had originally predicted, our kinetic data here likely reflects the limitations inherent to nickel catalysis.
Whether or not 6 requires a photocatalyst to trigger RE likely depends on the oxidation state of this intermediate. It has been clearly demonstrated that a Ni(II) resting state cannot undergo RE without photocatalytic excitation.17−19 However, extensive literature precedent points to the viability of multiple nickel oxidation states in cross-coupling reactions across very different conditions,35,44,45 and the oxidation state of the on-cycle intermediate for these photochemical reactions has yet to be convincingly resolved. Should this intermediate require activation via energy/electron transfer in the homogeneous reactions, it would not be turnover-limiting, given the photon-unlimited environment.
Of note is the fact that the only way to accelerate the homogeneous reaction was by addition of more secondary amine base, which is already in large excess. We offer three potential explanations. First, and most likely, the secondary amine donates an electron to [Ir(ppy)3]+ to regenerate the photocatalytic cycle. We can assume that the concentration of 2 requires equimolar amounts of base for deprotonation, and extra amine beyond that point fulfills various roles including weak ligand and electron source. Second, the base is potentially involved in the organization of the nickel catalyst to some degree, evinced by the “reaction fingerprint” of the heterogeneous reaction when the base is injected after a delay (Figure S5). As such, it is possible that its presence does not outright accelerate the reaction as much as it is as guard against decelerating or deactivating processes. Alternatively, or additionally, weak coordination of the base to the square planar pre-RE complex could create a more encumbered five-coordinate complex that is more prone to elimination,46,47 or is better tuned to absorb an energy transfer.28
Qualitatively, two main observations stand out for iridium-catalyzed carboxylate O-arylations. First, this system inherently tends to be less selective. In the photon-unlimited regime, at reactions with reasonable concentrations of substrates, yields were frequently in the 70–80% range, often accompanied by significant dehalogenation of the aryl iodide. We believe that this results from speeding up photocatalytic activation of the aryl iodide beyond the capacity of the nickel species to trap the radical on-cycle.
Second, the homogeneous reactions have an intrinsic propensity for catalyst deactivation. Kinetics for the photon-limited and photon-unlimited regimes were essentially the same outside of the fact that catalyst deactivation was not observed in the photon-limited regime. We contend that dual iridium- and nickel-catalyzed processes operate at inflated nickel concentrations to compensate for this inherent deactivation.
Qualitatively, the induction period in the heterogeneous congener notably differs from the homogeneous reaction. We observe small amounts of consumption of aryl iodide during the delay before product begins to form. If reduction of the Ni(II) precatalyst to a lower-valent species is requisite for productive catalysis, it is possible that this reduction is parallel to or intertwined with dehalogenation.
An unexpected revelation of the heterogeneous reaction is seen in the studies modulating the photocatalyst loading (Figure S16). In contrast to the homogeneous reaction, in which additional PC accelerates consumption of 1 indiscriminately, excess g-CN loading shows slower catalysis but with reasonable preservation of selectivity. We again attribute this to the weaker reduction potential of graphitic carbon nitrides, and is a potential upside to the more limited scope. Such a finding portends well for scalability as well as for sensitive or late-stage functionalization. Careful optimization of the ratios of photocatalyst, nickel, and substrate is less likely to be necessary with graphitic carbon nitrides.
Conclusion
Efforts to swap photocatalysts in dual photocatalytic and metal catalytic systems may not preserve the inherent reactivity of the metal catalytic cycle. Homogeneous and heterogeneous esterifications differ primarily in that the heterogeneous congener showed a rate dependence on the aryl halide substrate. We ascribe this to the nickel trapping an off-cycle radical anion generated by the photocatalyst and the aryl halide. In this sense, the requirements for electron-withdrawing substrates may be ascribed to an energetic barrier that is independent of the nickel catalytic cycle. Overcoming such limitations likely must occur through photocatalyst development and tuning to maximize the reduction potential of the excited PC. Alternatively, methods to front-load the energetic relay from external source to photocatalyst to substrate show great promise.48 However, as our PC and nickel screenings to find different regimes show, using a PC that is too reducing for the chosen substrate and/or metal catalyst can lead to over-reduction and catalyst deactivation. Countering this requires careful modulation of PC and metal catalyst concentrations. Hence, for the Ir(ppy)3-catalyzed O-arylation shown here, a photon-limited approach is desirable. Optimization of the graphitic carbon nitride conditions did not require such considerations. At least partially due to the reduction potential of the PC*, nickel catalyst concentration could be lowered up to 50 times from what was used in methodological development without observing a significant amount of nickel catalyst deactivation. Additionally, peak catalytic activity was found at a much lower concentration of nickel than used in method development. The choice of photocatalyst may, among other things, be determined by the electronic character of the substrate, as less dehalogenation was observed in the g-CN case. We believe that photocatalysts with weaker reduction potentials, such as graphitic carbon nitrides, are less likely to participate in a Ni(0)/Ni(II) cycle.
Acknowledgments
We gratefully acknowledge the Max-Planck Society for generous financial support. A.M. and B.P. acknowledge the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany’s Excellence Strategy–EXC 2008/1 (UniSysCat)–390540038 for financial support. B.P. acknowledges financial support by a Liebig Fellowship from the German Chemical Industry Fund (Fonds der Chemischen Industrie, FCI). We also gratefully thank Dr. Matt Mower (Janssen Pharmaceutical), Prof. Keary Mark Engle (TSRI), and anonymous reviewers for helpful discussions. A version of this research was previously posted to ChemRxiv: DOI: 10.26434/chemrxiv.11973141.v1
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.0c02848.
Experimental procedures and data (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Tasker S. Z.; Standley E. A.; Jamison T. F. Recent advances in homogeneous nickel catalysis. Nature 2014, 509, 299–309. 10.1038/nature13274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz J. W.; Fuchigami K.; Zheng B.; Rath N. P.; Mirica L. M. Isolated Organometallic Nickel(III) and Nickel(IV) Complexes Relevant to Carbon-Carbon Bond Formation Reactions. J. Am. Chem. Soc. 2016, 138, 12928–12934. 10.1021/jacs.6b06862. [DOI] [PubMed] [Google Scholar]
- Jones G. D.; Martin J. L.; McFarland C.; Allen O. R.; Hall R. E.; Haley A. D.; Brandon R. J.; Konovalova T.; Desrochers P. J.; Pulay P.; Vicic D. A. Ligand Redox Effects in the Synthesis, Electronic Structure, and Reactivity of an Alkyl-Alkyl Cross-Coupling Catalyst. J. Am. Chem. Soc. 2006, 128, 13175–13183. 10.1021/ja063334i. [DOI] [PubMed] [Google Scholar]
- Colon I.; Kelsey D. R. Coupling of aryl chlorides by nickel and reducing metals. J. Org. Chem. 1986, 51, 2627–2637. 10.1021/jo00364a002. [DOI] [Google Scholar]
- Meucci E. A.; Ariafard A.; Canty A. J.; Kampf J. W.; Sanford M. S. Aryl-Fluoride Bond-Forming Reductive Elimination from Nickel(IV) Centers. J. Am. Chem. Soc. 2019, 141, 13261–13267. 10.1021/jacs.9b06896. [DOI] [PubMed] [Google Scholar]
- Skubi K. L.; Blum T. R.; Yoon T. P. Dual Catalysis Strategies in Photochemical Synthesis. Chem. Rev. 2016, 116, 10035–10074. 10.1021/acs.chemrev.6b00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavedon C.; Seeberger P. H.; Pieber B. Photochemical Strategies for Carbon-Heteroatom Bond Formation. Eur. J. Org. Chem. 2020, 2020, 1379–1392. 10.1002/ejoc.201901173. [DOI] [Google Scholar]
- Twilton J.; Le C.; Zhang P.; Shaw M. H.; Evans R. W.; MacMillan D. W. C. The Merger of Transition Metal and Photocatalysis. Nat. Rev. Chem. 2017, 1, 0052. 10.1038/s41570-017-0052. [DOI] [Google Scholar]
- Milligan J. A.; Phelan J. P.; Badir S. O.; Molander G. A. Alkyl Carbon-Carbon Bond Formation by Nickel/Photoredox Cross-Coupling. Angew. Chem., Int. Ed. 2019, 58, 6152–6163. 10.1002/anie.201809431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oderinde M. S.; Frenette M.; Robbins D. W.; Aquila B.; Johannes J. W. Photoredox Mediated Nickel Catalyzed Cross-Coupling of Thiols With Aryl and Heteroaryl Iodides via Thiyl Radicals. J. Am. Chem. Soc. 2016, 138, 1760–1763. 10.1021/jacs.5b11244. [DOI] [PubMed] [Google Scholar]
- Oderinde M. S.; Jones N. H.; Juneau A.; Frenette M.; Aquila B.; Tentarelli S.; Robbins D. W.; Johannes J. W. Highly Chemoselective Iridium Photoredox and Nickel Catalysis for the Cross-Coupling of Primary Aryl Amines with Aryl Halides. Angew. Chem., Int. Ed. 2016, 55, 13219–13223. 10.1002/anie.201604429. [DOI] [PubMed] [Google Scholar]
- Pieber B.; Malik J. A.; Cavedon C.; Gisbertz S.; Savateev A.; Cruz D.; Heil T.; Zhang G.; Seeberger P. H. Semi-heterogeneous Dual Nickel/Photocatalysis using Carbon Nitrides: Esterification of Carboxylic Acids with Aryl Halides. Angew. Chem., Int. Ed. 2019, 58, 9575–9580. 10.1002/anie.201902785. [DOI] [PubMed] [Google Scholar]
- Cavedon C.; Madani A.; Seeberger P. H.; Pieber B. Semiheterogeneous Dual Nickel/Photocatalytic (Thio)etherification Using Carbon Nitrides. Org. Lett. 2019, 21, 5331–5334. 10.1021/acs.orglett.9b01957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh I.; Khamrai J.; Savateev A.; Shlapakov N.; Antonietti M.; König B. Organic Semiconductor Photocatalyst Can Bifunctionalize Arenes and Heteroarenes. Science 2019, 365, 360–366. 10.1126/science.aaw3254. [DOI] [PubMed] [Google Scholar]
- Khamrai J.; Ghosh I.; Savateev A.; Antonietti M.; König B. Photo-Ni-Dual-Catalytic C(sp2)-C(sp3) Cross-Coupling Reactions with Mesoporous Graphitic Carbon Nitride as a Heterogeneous Organic Semiconductor Photocatalyst. ACS Catal. 2020, 10, 3526–3532. 10.1021/acscatal.9b05598. [DOI] [Google Scholar]
- Gisbertz S.; Reischauer S.; Pieber B. Overcoming Limitations in Dual Photoredox/Nickel catalyzed C-N Cross-Couplings due to Catalyst Deactivation. ChemRxiv 2019, 10298735. 10.26434/chemrxiv.10298735.v1. [DOI] [Google Scholar]
- Welin E. R.; Le C.; Arias-Rotondo D. M.; McCusker J. K.; MacMillan D. W. C. Photosensitized, energy transfer-mediated organometallic catalysis through electronically excited nickel(II). Science 2017, 355, 380–385. 10.1126/science.aal2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian L.; Till N. A.; Kudisch B.; MacMillan D. W. C.; Scholes G. D. Transient Absorption Spectroscopy Offers Mechanistic Insights for an Iridium/Nickel-Catalyzed C-O Coupling. J. Am. Chem. Soc. 2020, 142, 4555–4559. 10.1021/jacs.9b12835. [DOI] [PubMed] [Google Scholar]
- Ma P.; Wang S.; Chen H. Reactivity of Transition-Metal Complexes in Excited States: C-O Bond Coupling Reductive Elimination of a Ni(II) Complex Is Elicited by the Metal-to-Ligand Charge Transfer State. ACS Catal. 2020, 10, 1–6. 10.1021/acscatal.9b03827. [DOI] [Google Scholar]
- Foà M.; Cassar L. Oxidative addition of aryl halides to tris(triphenylphosphine)nickel(0). J. Chem. Soc., Dalton Trans. 1975, 2572–2576. 10.1039/dt9750002572. [DOI] [Google Scholar]
- Roth H. G.; Romero N. A.; Nicewicz D. A. Experimental and Calculated Electrochemical Potentials of Common Organic Molecules for Applications to Single-Electron Redox Chemistry. Synlett 2016, 27, 714–723. 10.1055/s-0035-1561297. [DOI] [Google Scholar]
- All redox potentials in this article are given in reference to the saturated calomel electrode (SCE).
- These values may not serve as a hard limit and are often found to be surpassed in reaction conditions. See the following for a seminal discussion, and refs (24) and (25) for examples:; King K. A.; Spellane P. J.; Watts R. J. Excited-State Properties of a Triply Ortho-metalated Iridium(III) Complex. J. Am. Chem. Soc. 1985, 107, 1431–1432. 10.1021/ja00291a064. [DOI] [Google Scholar]
- Czyz M. L.; Weragoda G. K.; Monaghan R.; Connell T. U.; Brzozowski M.; Scully A. D.; Burton J.; Lupton D. W.; Polyzos A. A visible-light photocatalytic thiolation of aryl, heteroaryl and vinyl iodides. Org. Biomol. Chem. 2018, 16, 1543–1551. 10.1039/C8OB00238J. [DOI] [PubMed] [Google Scholar]
- Nguyen J. D.; D’Amato E. M.; Narayanam J. M. R.; Stephenson C. R. J. Engaging unactivated alkyl, alkenyl and aryl iodides in visible-light-mediated free radical reactions. Nat. Chem. 2012, 4, 854–859. 10.1038/nchem.1452. [DOI] [PubMed] [Google Scholar]
- Discekici E. H.; Treat N. J.; Poelma S. O.; Mattson K. M.; Hudson Z. M.; Luo Y.; Hawker C. J.; de Alaniz J. R. A highly reducing metal-free photoredox catalyst: design and application in radical dehalogenations. Chem. Commun. 2015, 51, 11705–11708. 10.1039/C5CC04677G. [DOI] [PubMed] [Google Scholar]
- Buzzetti L.; Crisenza G. E. M.; Melchiorre P. Mechanistic Studies in Photocatalysis. Angew. Chem., Int. Ed. 2019, 58, 3730–3747. 10.1002/anie.201809984. [DOI] [PubMed] [Google Scholar]
- Kudisch M.; Lim C.-H.; Thordarson P.; Miyake G. M. Energy Transfer to Ni-Amine Complexes in Dual Catalytic, Light-Driven C-N Cross-Coupling Reactions. J. Am. Chem. Soc. 2019, 141, 19479–19486. 10.1021/jacs.9b11049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackmond D. G. Kinetic Profiling of Catalytic Organic Reactions as a Mechanistic Tool. J. Am. Chem. Soc. 2015, 137, 10852–10866. 10.1021/jacs.5b05841. [DOI] [PubMed] [Google Scholar]
- As an example, ex situ mechanistic studies with the same techniques are able to support the two competing hypotheses in the Introduction, with no real way to resolve the discrepancy.
- Zhang G.; Li G.; Lan Z.-A.; Lin L.; Savateev A.; Heil T.; Zafeiratos S.; Wang X.; Antonietti M. Optimizing Optical Absorption, Exciton Dissociation, and Charge Transfer of a Polymeric Carbon Nitride with Ultrahigh Solar Hydrogen Production Activity. Angew. Chem., Int. Ed. 2017, 56, 13445–13449. 10.1002/anie.201706870. [DOI] [PubMed] [Google Scholar]
- Mower M. P.; Blackmond D. G. Mechanistic Rationalization of Unusual Sigmoidal Kinetic Profiles in the Machetti-De Sarlo Cycloaddition Reaction. J. Am. Chem. Soc. 2015, 137, 2386–2391. 10.1021/ja512753v. [DOI] [PubMed] [Google Scholar]
- Ji Y.; DiRocco D. A.; Kind J.; Thiele C. M.; Gschwind R. M.; Reibarkh M. LED-Illuminated NMR Spectroscopy: A Practical Tool for Mechanistic Studies of Photochemical Reactions. ChemPhotoChem. 2019, 3, 984–992. 10.1002/cptc.201900109. [DOI] [Google Scholar]
- Lehnherr D.; Ji Y.; Neel A. J.; Cohen R. D.; Brunskill A. P. J.; Yang J.; Reibarkh M. Discovery of a Photoinduced Dark Catalytic Cycle Using in Situ LED-NMR Spectroscopy. J. Am. Chem. Soc. 2018, 140, 13843–13853. 10.1021/jacs.8b08596. [DOI] [PubMed] [Google Scholar]
- Sun R.; Qin Y.; Ruccolo S.; Schnedermann C.; Costentin C.; Nocera D. G. Elucidation of a Redox-Mediated Reaction Cycle for Nickel-Catalyzed Cross Coupling. J. Am. Chem. Soc. 2019, 141, 89–93. 10.1021/jacs.8b11262. [DOI] [PubMed] [Google Scholar]
- Sun R.; Qin Y.; Nocera D. G. General Paradigm in Photoredox Nickel-Catalyzed Cross-Coupling Allows for Light-Free Access to Reactivity. Angew. Chem., Int. Ed. 2020, 59, 9527–9533. 10.1002/anie.201916398. [DOI] [PubMed] [Google Scholar]
- Somerville R. J.; Hale L. V. A.; Gómez-Bengoa E.; Burés J.; Martin R. Intermediacy of Ni-Ni Species in sp2 C-O Bond Cleavage of Aryl Esters: Relevance in Catalytic C-Si Bond Formation. J. Am. Chem. Soc. 2018, 140, 8771–8780. 10.1021/jacs.8b04479. [DOI] [PubMed] [Google Scholar]
- Mohadjer Beromi M.; Brudvig G. W.; Hazari N.; Lant H. M. C.; Mercado B. Q. Synthesis and Reactivity of Paramagnetic Nickel Polypyridyl Complexes Relevant to C(sp2)-C(sp3)Coupling Reactions. Angew. Chem., Int. Ed. 2019, 58, 6094–6098. 10.1002/anie.201901866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen C. D. T.; Burés J. Visual kinetic analysis. Chem. Sci. 2019, 10, 348–353. 10.1039/C8SC04698K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackmond D. G. Reaction Progress Kinetic Analysis: A Powerful Methodology for Mechanistic Studies of Complex Catalytic Reactions. Angew. Chem., Int. Ed. 2005, 44, 4302–4320. 10.1002/anie.200462544. [DOI] [PubMed] [Google Scholar]
- Martínez-Carrión A.; Howlett M. G.; Alamillo-Ferrer C.; Clayton A. D.; Bourne R. A.; Codina A.; Vidal-Ferran A.; Adams R. W.; Burés J. Kinetic Treatments for Catalyst Activation and Deactivation Processes based on Variable Time Normalization Analysis. Angew. Chem., Int. Ed. 2019, 58, 10189–10193. 10.1002/anie.201903878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh I.; Marzo L.; Das A.; Shaikh R.; König B. Visible Light Mediated Photoredox Catalytic Arylation Reactions. Acc. Chem. Res. 2016, 49, 1566–1577. 10.1021/acs.accounts.6b00229. [DOI] [PubMed] [Google Scholar]
- Cheng Y.; Gu X.; Li P. Visible-Light Photoredox in Homolytic Aromatic Substitution: Direct Arylation of Arenes with Aryl Halides. Org. Lett. 2013, 15, 2664–2667. 10.1021/ol400946k. [DOI] [PubMed] [Google Scholar]
- Manzoor A.; Wienefeld P.; Baird M. C.; Budzelaar P. H. M. Catalysis of Cross-Coupling and Homocoupling Reactions of Aryl Halides Utilizing Ni(0), Ni(I), and Ni(II) Precursors; Ni(0) Compounds as the Probable Catalytic Species but Ni(I) Compounds as Intermediates and Products. Organometallics 2017, 36, 3508–3519. 10.1021/acs.organomet.7b00446. [DOI] [Google Scholar]
- Kawamata Y.; Vantourout J. C.; Hickey D. P.; Bai P.; Chen L.; Hou Q.; Qiao W.; Barman K.; Edwards M. A.; Garrido-Castro A. F.; deGruyter J. N.; Nakamura H.; Knouse K.; Qin C.; Clay K. J.; Bao D.; Li C.; Starr J. T.; Garcia-Irizarry C.; Sach N.; White H. S.; Neurock M.; Minteer S. D.; Baran P. S. Electrochemically Driven, Ni-Catalyzed Aryl Amination: Scope, Mechanism, and Applications. J. Am. Chem. Soc. 2019, 141, 6392–6402. 10.1021/jacs.9b01886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson J. B.; Rovis T. More than Bystanders: The Effect of Olefins on Transition-Metal-Catalyzed Cross-Coupling Reactions. Angew. Chem., Int. Ed. 2008, 47, 840–871. 10.1002/anie.200700278. [DOI] [PubMed] [Google Scholar]
- Ujaque G.; Maseras F.; Eisenstein O.; Liable-Sands L.; Rheingold A. L.; Yao W.; Crabtree R. H. Breaking an Electronically Preferred Symmetry by Steric Effects in a Series of [Ir(biph)X(QR3)2] Compounds (X = Cl or I, Q = P or As). New J. Chem. 1998, 22, 1493–1498. 10.1039/a804840a. [DOI] [Google Scholar]
- Cowper N. G. W.; Chernowsky C. P.; Williams O. P.; Wickens Z. K. Potent Reductants via Electron-Primed Photoredox Catalysis: Unlocking Aryl Chlorides for Radical Coupling. J. Am. Chem. Soc. 2020, 142, 2093–2099. 10.1021/jacs.9b12328. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.