

Abstract
Recent years have witnessed an explosion of interest in understanding the role of conformational dynamics both in the evolution of new enzymatic activities from existing enzymes and in facilitating the emergence of enzymatic activity de novo on scaffolds that were previously non-catalytic. There are also an increasing number of examples in the literature of targeted engineering of conformational dynamics being successfully used to alter enzyme selectivity and activity. Despite the obvious importance of conformational dynamics to both enzyme function and evolvability, many (although not all) computational design approaches still focus either on pure sequence-based approaches or on using structures with limited flexibility to guide the design. However, there exist a wide variety of computational approaches that can be (re)purposed to introduce conformational dynamics as a key consideration in the design process. Coupled with laboratory evolution and more conventional existing sequence- and structure-based approaches, these techniques provide powerful tools for greatly expanding the protein engineering toolkit. This Perspective provides an overview of evolutionary studies that have dissected the role of conformational dynamics in facilitating the emergence of novel enzymes, as well as advances in computational approaches that allow one to target conformational dynamics as part of enzyme design. Harnessing conformational dynamics in engineering studies is a powerful paradigm with which to engineer the next generation of designer biocatalysts.
Introduction
Enzymes are conformationally dynamic, and there has been significant debate in the literature about the extent to which their flexibility corresponds to their catalytic activity.1−6 However, in recent years, the focus has shifted toward trying to understand the extent to which conformational dynamics contributes to enzyme evolvability, and the acquisition of new enzyme functions.7−13 This so-called “New View” of enzyme catalysis7 describes proteins as existing on an energy landscape with multiple local minima, corresponding to discrete conformations with different energy levels. These different conformations can potentially bind different substrates and facilitate different chemistry, allowing for enzyme promiscuity (the ability to catalyze multiple, distinct chemical reactions).7,8 One would expect the landscape for the wild-type enzyme to be dominated by one of these minima, which binds the native substrate and corresponds to the native activity of the enzyme. However, mutations introduced over the course of an evolutionary trajectory can shift the equilibrium between conformational states, such that previously minor conformations increase in population, leading to the emergence of new activities (Figure 1).
Figure 1.

Schematic illustration of the relationship between ligand binding, conformational dynamics, and protein evolution.7,8 The major conformation adopted by the enzyme is responsible for its native activity, however the existence of minor conformations (that may or may not also be able to interconvert directly) can give rise to promiscuous activities for non-natural substrates. Mutations accumulated over the course of evolutionary trajectories (through either natural or directed evolution) with the appropriate selective pressure(s) can ultimately lead to population shifts, with these minor conformations now becoming the major conformers and the promiscuous activity becoming the new “native” activity. For further discussion, see e.g. refs (7, 8, and 31).
It is by now well established that enzyme promiscuity plays an important role in enzyme evolvability.7,8,10,12,14−16 This has generated great interest in trying to use Nature’s tricks to harness promiscuity in enzyme design.16−19 But what about conformational dynamics? Many enzymes harbor decorating loops on their scaffolds (this is very common for example in the case of TIM barrel proteins20,21) that theoretically could be manipulated to alter activity. Or, activity could be altered by the controlled introduction of mutations to fine-tune enzymes’ conformational ensembles (as has been observed in a number of directed evolution studies22−27). This can even be coupled with the incorporation of non-canonical amino acids, thus expanding the genetic code and either facilitating completely novel chemistry in existing active sites or allowing for the emergence of catalytic activity on previously non-catalytic scaffolds.28−30
While harnessing conformational diversity has tremendous potential in enzyme engineering, several technical challenges remain that make laboratory engineering of conformational dynamics far from routine.31,32 However, recent advances in computational approaches targeting conformational dynamics may allow for further progress in this area, opening up a highly powerful new avenue for targeted enzyme design. This is particularly important, as many computational enzyme design studies still focus on either exclusively sequence-based approaches, or, where structure-based approaches are used, they are limited in scope as they frequently focus on static structures and disregard dynamical properties of the system (e.g., refs (23, 33−42)).
This Perspective will discuss the role of conformational dynamics in enzyme evolution, as well as providing examples where manipulation of conformational dynamics has been successfully harnessed as a tool for enzyme engineering. We will particularly discuss recent work from both our own as well as the Tawfik laboratories, while also showcasing important contributions to the field from other research teams. We will also discuss recent developments and advances in computational methodologies that incorporate conformational dynamics as part of the design process, providing a promising avenue for the generation of novel enzymes with tailored catalytic and dynamical profiles.
Repeating Structural Motifs Facilitate the Emergence of Novel Proteins
There has been substantial interest in understanding the evolution of enzymatic activity on existing enzymatic scaffolds, either through specialization toward one of a set of generalist functionalities, or through emergence of completely new activities in existing active sites (see e.g. refs (16, 43−49)). This focus on existing enzymes makes sense considering that at least 87% of all existing enzyme functions have been estimated to have either evolved from another pre-existing catalytic function, or evolved through specialization of a generalist enzyme.50 What then about the remaining enzymes—what is their origin? Clearly, these enzymes must have, in some format, evolved on scaffolds that were previously non-catalytic. In addition, even if one assumes that all modern enzymes have evolved from pre-existing enzymes, still, the first enzymes must have somehow evolved at some point in our evolutionary history. This is likely to have occurred at a very early stage in the evolution of life on Earth, as it has been estimated that a wide array of enzymes already existed in the last universal common ancestor (LUCA).51,52 So how then are non-enzymatic scaffolds repurposed for enzymatic function, and how do new proteins emerge to provide these scaffolds in the first place?
In this context, Tawfik and co-workers have explored the evolutionary constraints and driving forces that underlie the emergence of β-propeller proteins.53 Specifically, by combining ancestral reconstruction with biochemical and structural analysis, the authors were able to trace the emergence of functional 5-bladed lectin β-propeller via tandem duplication from short (<50 amino acids) motifs that are present in known genomes. This is in perfect agreement with Dayhoff’s hypothesis that the first folded functional protein domains arose through the fusion, duplication, and diversification of short polypeptide sequences,54 as discussed in detail in ref (55). In a related study, Voet and co-workers were recently able to exploit the modular structure of WD40 proteins to design a symmetrical 8-bladed β-propeller protein through pure computational design.56
Following from this, Tawfik and co-workers have traced an “ancient fingerprint” in Rossman-fold enzymes, comprised of an interaction between a carboxylate (Asp or Glu) sitting at the tip of the second β-strand of these enzymes and bound ribose (ribose- β2-Asp/Glu).57 This interaction appears to both have unique geometrical features and also be exclusively present in Rossman-fold enzymes that bind cofactors. In addition, the authors demonstrated that ribose–carboxylate interactions found in other protein folds are both rare and topologically different from those observed in the Rossman-fold enzymes. This led to the suggestion that the presence of this fingerprint indicates the divergence of Rossman-fold enzyme from a common pre-LUCA ancestor that possessed the same binding motif.57 In subsequent analysis, Grishin and co-workers58 defined a minimal Rossman-like structure motif (RLM) involved in ligand binding, comprised of a “doubly-wound” α/β/α sandwich structure, and used this as a baseline for analysis of RLM domains in the Protein Data Bank (PDB).59 This structural analysis was coupled with evolutionary analysis, using the Evolutionary Classification of Protein Structure Domains (ECOD) database,60,61 and indicated that the RLM binding motif likely arose several times during the evolution of these proteins; it was likely used already by the LUCA.
How then do these motifs get translated into function? Phosphate binding proteins are an excellent model system to address this question, as phosphate binding is ubiquitous in biology, and phosphate esters are the building blocks of life itself, being involved in essentially all cellular processes.62 We recently performed detailed sequence and structural analysis of all phosphate binding motifs in the PDB, combined with evolutionary analysis using ECOD.63 Curiously, across 4 billion years of evolutionary time, the dominant mode of phosphate binding appears to be mediated through side-chain interactions, with no involvement at all from the protein backbone. However, in the earliest proteins (particularly αβα sandwich enzyme domains), the dominant binding mode (to any of mono-, di-, or triphosphate binding) involves interactions with the N-terminus of an α-helix, primarily through interactions with the backbones and/or side chains of the Prebiotic amino acids Gly, Ser, and Thr64−66 (Figure 2). This provides a putative snapshot of the first phosphate binding interactions in proteins and a baseline for further engineering of binding or catalytic activity. As an example of this, Tawfik and co-workers have used phylogenetic analysis to identify the ancestral “sequence logo” of the Walker-A P-loop element, which is absolutely critical for facilitating binding and phosphoryl transfer in modern P-loop NTPases.67 They then used computational design to incorporate this sequence logo into de novo designed scaffolds, obtaining soluble and stable proteins with an expanded binding repertoire. In addition to polynucleotides and both RNA and single-stranded DNA, they were also able to bind adenosine triphosphate (ATP) without the involvement of metal cofactors. In addition, phosphate binding was apparently facilitated by complex cooperative conformational changes that were likely only feasible due to the structural plasticity of these designed proteins.67 This highlights the engineering potential of transferring minimal motifs capable of conferring binding ability to conformationally diverse scaffolds to generate new functionality.
Figure 2.
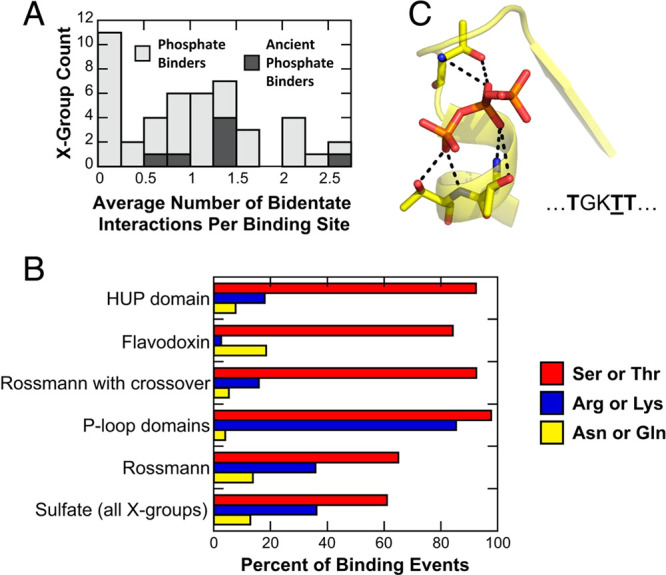
Observed prevalence of bidentate interactions in phosphate binding (where “phosphate” in this case refers broadly to mono-, di-, and triphosphates), based on combined analysis of structural data in the Protein Data Bank59 and evolutionary information in the ECOD database.60,61 X-groups provide the broadest level of classification in ECOD, corresponding to discrete events of evolutionary emergence with no detectable sequence homology or fold identity. Shown here are (A) the frequency of bidentate phosphate binding interactions across all X-groups, including also ancient phosphate binders, and (B) the amino acids involved in forming these bidentate interactions across X-groups and in specific protein folds. Here, it can be seen that Thr and Ser (both prebiotic amino acids) are essential for the formation of bidentate interactions in the N-helix binding mode at the tip N-terminus of an α-helix, an illustrative example of which is shown in the case of the binding of a triphosphate in panel (C). For more details, see ref (63). Reproduced with permission from ref (63). Copyright 2020 National Academy of Sciences.
Conformational Dynamics and the Emergence of Novel Enzymes
It is clear that the modular nature of protein structure can act as a driving force to facilitate the evolution of novel scaffolds with novel functionalities, and the modular structure of proteins has been frequently used also in protein engineering studies to control protein structure and function.68−74 But it is one thing to simply assemble a stable, folded scaffold, and another to confer enzymatic activity to that scaffold. One of the easiest ways to confer enzymatic activity to a non-catalytic scaffold is simply by repurposing existing functionality, in particular a binding site, as ligand binding is an important first step toward efficient catalysis. There exist several examples in the literature of the emergence of novel enzymatic activity on previously non-catalytic scaffolds,25,75−80 for example through the functionalization of binding sites. The evolutionary trajectories that can lead to the emergence of enzymatic activity can be characterized by ancestral sequence reconstruction,47,81 alongside any combination of structural, biochemical, and computational characterization, and it appears that conformational dynamics can play an important role in the emergence of enzymatic activity. Here, we showcase two systems where conformational dynamics appears to play a role in the transition from a solute binding protein to an enzyme.
The first of these is the enzyme cyclohexadienyl dehydratase (CDT), which catalyzes the cofactor-independent Grob-type fragmentation of prephenate and l-arogenate respectively to yield phenylpyruvate and l-phenylalanine.82 Sequence and structural analysis of this enzyme has suggested that CDT has evolved from solute-binding proteins.75,79 Jackson and co-workers have recently harnessed the power of ancestral sequence reconstruction47,81 coupled with biochemical characterization in order to explore the physiochemical parameters that allowed for the evolutionary transition of CDT from a solute-binding protein to an enzyme.79 The key feature leading to the emergence of CDT activity appeared to be the incorporation of a desolvated general base into the ancestral active site, conferring catalytic activity to this scaffold. Directed evolution indicated the presence of multiple independent mutational pathways leading to higher catalytic activity once the key catalytic residues were introduced, as well as separate mutational pathways from the historic mutational pathway observed in the ancestral proteins, suggesting that the enhancement of CDT activity on this scaffold occurred non-deterministically. Other mutations reshaped the active site and introduced hydrogen-bonding networks that improved enzyme–substrate complementarity as well as placement of the reacting fragments in the active site. Finally, remote mutations refined the conformational ensemble of the enzyme, by dampening the sampling of catalytically non-productive conformations.79 More recent experimental work performing double electron–electron resonance (DEER)83 on putative evolutionary intermediates along the trajectory toward a modern catalytically efficient CDT has further illustrated the role of remote mutations in reducing the sampling of catalytically non-productive conformations of the enzyme.
In parallel work, we have studied the evolution of chalcone isomerases (CHI) from solute binding proteins.25,84 Chalcone isomerases catalyze the enantioselective intramolecular Michael addition of chalconaringenin, to yield the plant flavonoid (2S)-naringenin, making it a key enzyme in plant flavonoid biosynthesis.85 Ancestral sequence inference suggests that both modern CHIs and a related group of CHI-like proteins (CHILs) that lack enzymatic activity86,87 have evolved from fatty acid binding proteins (FAPs, which are enzymes that are important for plant fatty acid biosynthesis88) via a common ancestor lacking isomerase activity.25,88 By combining ancestral sequence reconstruction,47,81 X-ray crystallography, NMR, and simulations, we were able to identify four founder mutations that each, individually, are able to confer chalcone isomerase activity.25
One important factor in examining the effect of these founder mutations is whether the effect of these mutations is additive or not. In epistasis, the effect of the mutations is not additive (i.e., the order in which the mutations are introduced becomes important). As discussed in ref (25), epistasis is significant from an evolutionary point of view, because where present, epistasis will limit the number of accessible evolutionary pathways, as mutations need to be introduced in a specific sequence in order to reach the desired effect. There is evidence in the literature for epistasis, including sign epistasis (where new mutations can change the effect of previous mutations from beneficial to deleterious, or vice versa), playing an important role in protein evolution.89−96 Curiously, however, a laboratory reconstructed mutational trajectory of CHI showed only weak functional epistasis between key founder mutations, with multiple subsequent trajectories that each could confer isomerase activity. This suggests that the order in which these founder mutations are introduced is not important, which is indicative of a smooth evolutionary landscape underlying the emergence of CHI activity. This suggests that the gain of enzymatic activity is relatively facile despite the evolutionary origin of this enzyme from a non-catalytic ancestor.
Our combined analysis also indicated a combined role for reshaping of the active site by mutations toward a productive substrate-binding mode, as well as repositioning of a key catalytic arginine inherited from the ancestral FAPs (Figure 3) as major driving forces for the emergence of isomerase activity.25 We later demonstrated that the side chain of this arginine acts as a combined Brønsted and Lewis acid in bifunctional substrate activation during the Michael addition catalyzed by CHI.84 Such bifunctional activation is also observed when employing the guanidine- and urea-based chemical reagents that are frequently used for asymmetric organocatalysis.97,98 This highlights the potential application of the CHI scaffold in the design of biocatalysts for guanidine-based asymmetric catalysis. A critical observation here, however, is the fact that even the inactive CHI ancestor possessed all key catalytic residues in the correct position in the active site,25 demonstrating that simply having the correct catalytic residues in the correct position is not alone sufficient for catalysis to actually occur.
Figure 3.

Changes in the conformational ensemble of the catalytic arginine, R34, during the evolution of chalcone isomerases (CHIs) from binders to catalysts. (A) Structural alignment of crystal structures of different CHIs, showing how widely the conformation of R34 varies. This is in agreement with (B) NMR steady-state heteronuclear NOE values for the catalytic arginine, and the corresponding (C) HSQC signals of the arginine side chain. These indicate both the changes in the mobility of this residue during the evolution of CHI from ancCC to ancR1 to ancR7, as well as the corresponding changes in the electrostatic environment of this side chain. Finally, this is corroborated by long-time-scale molecular dynamics simulations, where the corresponding χ1 and χ3 dihedral angles of R34 during simulation of the different variants once again show the changes in the conformational space of this residue. Reproduced with permission from ref (25). Copyright 2018 Springer Nature.
Conformational Dynamics Modulates the Activity of Extant Enzymes
Functionally important conformational dynamics in enzymes can manifest themselves in different ways, and on different time scales, spanning several orders of magnitude. Such conformational dynamics can range from simple side chain fluctuations through to larger-scale conformational changes such as loop dynamics, domain movements, or large allosteric motions.99 From a catalytic perspective, these may or may not be ligand-gated, in that substrate binding energy can be used to drive an otherwise catalytically unfavorable conformational change.100,101 The relevance of conformational dynamics and enzyme evolution has been reviewed in great detail elsewhere,7,8,10−13,32 and therefore we will only touch briefly upon selected relevant systems in this section. We note that in this section, we focus in particular on evolutionary fine-tuning of enzyme loop dynamics, as this can be targeted for protein engineering;102 however, clearly, other forms of conformational dynamics can also be evolutionarily important.
One of the classical examples of an important enzyme for understanding the role of conformational dynamics in enzyme function and evolution has been dihydrofolate reductase (DHFR).3,4,6,9,103−106 DHFR uses NADPH as a cofactor to catalyze the reduction of dihydrofolate (DHF), through a two-step mechanism (Figure 4). In E. coli DHFR (EcDHFR), the catalytic mechanism is aided by the movement of multiple loops close to the binding pocket, including the catalytically important “Met20 loop”. This is a highly flexible loop that acts as a lid to hold the cofactor tightly in the binding pocket. It can occupy three distinct conformations: open, closed, and occluded (Figure 4).104 Upon cofactor binding, it undergoes a conformational transition from an open to a closed conformation, thus placing the reacting fragments in a catalytically competent conformation and increasing the probability of productive binding.107 In between the first and second mechanistic steps, another conformational change occurs from the closed to the occluded state, where the cofactor binding pocket is obstructed by the Met20 loop, thus forcing the nicotinamide ring out of its bound position and facilitating the rate-limiting product release step.108,109 The conformational dynamics of DHFR’s Met20 loop has been probed by using NMR, with loop rearrangements occurring on the millisecond time scale having been demonstrated to be responsible for the required changes in the active-site configuration throughout the catalytic cycle.108
Figure 4.

(A) Overlay of three X-ray crystal structures of E. coli DHFR with the Met20 loop crystallized in different conformations. Shown here are structures of DHFR with the Met20 loop in the closed (red, PDB: 1RX2), occluded (magenta, PDB: 1RX4), and open (cyan, PDB: 1RE7) conformations, based on data provided in conjunction with ref (107). (B) Schematic overview of the hydride transfer reaction catalyzed by DHFR, in which dihydrofolate is reduced to tetrahydrofolate, and NADPH is oxidized to NADP+.
DHFR has historically been an important model system for probing the role of conformational dynamics in enzyme catalysis.4,6,103,104,106 More recently, there has also been increasing interest in understanding the role of conformational dynamics in DHFR evolution.9,105,110−113 In particular, despite high structural similarity, human DHFR (hDHFR) exhibits very different conformational movements throughout the catalytic cycle compared to EcDHFR.110 That is, the loop analogous to the Met20 loop in EcDHFR remains in a closed position throughout the catalytic cycle of hDHFR. In addition, millisecond time scale fluctuations facilitate flux through the catalytic cycle in EcDHFR.114−117 Such millisecond fluctuations are not observed in hDHFR which instead exhibits pervasive fluctuations on the microsecond time scale, including in regions which border the binding pocket, suggesting that these fluctuations may be productive for product release.110 Other studies have explored how DHFR dynamics has changed over the course of evolution, focusing in particular on whether the conformational fluctuations of the wild-type enzyme are conserved, or whether they are dampened or amplified during evolution (see e.g. refs (105 and 112)), as well as exploring the coupling of fast dynamics to the reaction coordinate.111
Another example of systems where evolution appears to have focused on fine-tuning loop dynamics are TIM barrel proteins. The TIM barrel is highly evolvable20,118,119 and one of the most common protein folds observed in the PDB.20,59,120 The name giving enzyme, triosephosphate isomerase, possesses several decorating loops that are active within the catalytic cycle.100,121,122 Of these, loop 6, undergoes a large ligand-gated conformational change upon substrate binding, moving up to 7 Å from the open to closed position, thus creating a catalytic cage that sequesters the active site from solvent.100 Despite the persistent image of this loop as a classical example of a two-state rigid-body motion,123−128 simulation studies have shown that this loop is highly flexible and can take on multiple conformations, thus yielding multiple different potential trajectories that can lead from the inactive open conformation of the enzyme to the catalytically competent closed conformation of the enzyme (Figure 5).129
Figure 5.

Superimposition of Markov state models (MSMs)130,131 of (A) unliganded triosephosphate isomerase (TIM) and (B) TIM in complex with substrate dihydroxyacetone phosphate (DHAP), onto the corresponding free energy surfaces (T = 300 K) obtained from performing principal component analysis (PCA) on conventional MD simulations of each system. The free energy surfaces are defined in terms of the first two principal components, PC1 and PC2. The area of the nodes representing each of the metastable states in panels (A) and (B), and the thickness of the arrows connecting them, correspond to the populations of each node and the transition probabilities between them, respectively (note that areas and thicknesses do not scale linearly with transition probabilities). Shown here also are (C) overlays of representative structures from each of the metastable states sampled in these simulations, with the crystallographic “open” and “closed” conformations of the loop shown in red and blue, respectively, and the loop conformation at each state shown in yellow. Note that the metastable state 2 is virtually identical for simulations of both the liganded and unliganded forms of the enzyme. For details, see ref (129). Reprinted with permission from ref (129). Copyright 2018 American Chemical Society.
Another family of enzymes where loop dynamics appears to be evolutionarily important are protein tyrosine phosphatases (PTPs).132 PTPs catalyze the dephosphorylation of phospho-tyrosine residues through a two-step “ping-pong” mechanism, in an active site composed of three highly conserved loops (Figure 6).133 Of these, the “P-loop” is responsible for coordinating the reacting phosphate group and providing a nucleophilic cysteine to dephosphorylate the phospho-tyrosine residue in the first step. Furthermore, this reaction is promoted by the closure of a highly flexible “WPD-loop”, which contains an active-site aspartic acid that acts as a general acid to stabilize the leaving group. In the second step, the thiol-phosphate group is subjected to nucleophilic attack by an active-site water molecule, which is again promoted by the aspartic acid on the WPD-loop, in this case acting as a general base, deprotonating the active-site water to enhance its nucleophilicity. Finally, vital to the second step is the coordination of a glutamine on the “Q-loop” to the nucleophilic water molecule. An NMR study on two different PTPs demonstrated the rate of WPD-loop closure to be highly correlated with the rate of the first chemical step.132 Given that PTPs are responsible for regulating many cellular signaling processes (meaning their catalytic rates will have been subjected to strict evolutionary pressure), and that throughout nature the rate of PTP catalysis can vary by several orders of magnitude,140 this data may suggest that evolution has fine-tuned individual PTP loop dynamics to regulate their catalytic rates. Further, numerous PTPs have known allosteric sites (Figure 6),134,135,141,142 and a recent combined bioinformatics and biomolecular simulation study has identified evolutionarily conserved allosteric communication within PTPs, suggesting that PTPs have been subjected to both local and distal mutagenesis in order to regulate the conformational dynamics of its active-site loops.143
Figure 6.

(A, B) Aligned crystal structures of PTP1B with the WPD-loop in its closed and open conformations, respectively (PDB: 6B90,134 this structure contains both conformations of the loop). Panel (A) depicts the overall structure of PTP1B, highlighting the three major loops which make up the active site (WPD-loop: cyan, P-loop: green, and Q-loop: purple) indicated. The two known allosteric drug binding sites on PTP1B are labeled and depicted with a representative drug bound to each site (BB site, PDB ID: 1T49,135 and K197 site, PDB ID: 6B95(134)). Dark green spheres are residues not located within the active site, but where single-point substitutions have been shown to alter PTP1B’s kcat or Km by |>50%| (data collated from refs (134, 136−139)). (B) A close-up of the PTP1B active site, with a model substrate p-nitrophenyl phosphate (pNPP) bound. The backbone nitrogen atoms and the arginine side chain on the P-loop that are harnessed to coordinate the phosphate group are also shown. (C) Conserved two-step reaction mechanism utilized by PTPs.133
Fructose-1,6-bisphosphate (FBP) is another enzyme with an active site primarily composed of loops, with these loops used to catalyze a two-step reaction in which FBP first acts as an aldolase before undergoing a large-scale conformational change in order to act as a phosphatase in its second catalytic step.144,145 This dual aldolase/phosphatase activity likely emerged in FBP to prevent degradation of the reaction intermediates if they were released back into the high temperature environment that FBP is natively found in. In many other cases in which an unstable intermediate is formed, modular catalytic systems are directly connected to one another, allowing for a cascade of chemical reactions to occur before releasing the reactant back into the environment (see e.g. refs (146 and 147)). The solution adopted by FBP in which the active site is able to (re)organize itself in order to allow for a different form of catalysis is striking, and it could be argued that this approach is notably more accessible due to the large amount of conformational plasticity available to the active site. That is, an active site composed primarily of loops (as opposed to more defined secondary structure) is likely to have a wider range of accessible conformational substates from which dual aldolase and phosphatase activity could emerge. While FBP represents a remarkable instance of evolutionary ingenuity, the competing interests associated with using one active site to engineer multiple different reactivities is likely to be particularly challenging. Indeed, several identified single point mutations that enhanced aldolase activity came at the cost of reduced phosphatase activity and vice versa.144
We note that there exist many other systems where conformational dynamics appears to be evolutionarily important, including organophosphate hydrolases,148−150 β-lactamases,151−155 tryptophan synthase,156Pseudomonas aeruginosa arylsulfatase,157 thioredoxins,158 cold-adapted enzymes,159,160 and guanylate kinase161 as just some examples. For economy of space we have not discussed these systems in detail here, but instead refer readers to the cited references for more details on each of these systems.
Enzyme Engineering by Fine-Tuning Protein Conformational Dynamics
It is becoming clear that fine-tuning of conformational dynamics plays a crucial role in enzyme evolution. Being able to enhance enzyme activity through manipulating conformational dynamics requires either being able to increase the population of catalytically productive conformations and/or being able to dampen the population of catalytically unproductive conformations of an enzyme. This is challenging, but not impossible, to achieve in silico or in the laboratory. There are a number of examples, both where the conformational ensemble has been serendipitously optimized through directed evolution, and where the conformational ensemble has been successfully targeted for enhancing an enzyme’s activity, indicating that there is great potential for doing this more systematically. In particular, it appears that maintaining conformational dynamics similar to that of the native enzyme is not critical for the engineering of functional proteins,162 suggesting that there is significant scope for the manipulation of conformational dynamics while at the same time maintaining catalytic activity. We discuss here some examples of the engineering of enzyme conformational dynamics (for detailed reviews see e.g. refs (12, 31, and 102)), with a particular focus on the engineering of enzyme specificity and activity.
Retro-aldolases (RAs) are among the most complex computationally designed enzymes to date.163−165 They catalyze the amine-assisted cleavage of a methodol substrate through a multi-step mechanism involving an enzyme-bound Schiff base intermediate. In 2012, Baker and co-workers performed an expansive study introducing a catalytic motif likely to be capable of Kemp elimination onto a variety of scaffolds, including TIM barrel and Jelly Roll folds.164 The resulting de novo designs only exhibited modest catalytic activity, but were enhanced substantially through directed evolution (from initial kcat/KM values of <1 M–1 s–1 for all designed variants with subsequent improvements between 7-fold and 88-fold). Following from this, Hilvert and co-workers used directed evolution to increase the catalytic efficiency of a de novo RA, and were able to successfully reach catalytic efficiencies comparable to those of natural enzymes (Figure 7).165,166 During the evolutionary pathway, the binding site underwent a complete remodeling event, with the catalytic lysine being abandoned in favor of another lysine in the binding pocket. In addition, mutations were observed both in the binding site and at distal positions. With the introduction of distal mutations, both significant changes to loop conformations and more extensive conformational changes were observed.
Figure 7.

Overview of directed evolution experiments of the in silico designed retro-aldolase RA95, showing the increase in activity toward that of natural enzymes. Shown here are (A) the catalytic efficiencies of various evolved RA95 variants toward (R)-methodol, (B) the mutations introduced in each variant, and (C) the corresponding position of the different mutations on the structure, based on kinetic and structural data presented in refs (165 and 166). Reproduced with permission from ref (10). Copyright 2018 the authors, published by the Royal Society. All rights reserved.
Biochemical analysis indicated a shift in rate-limiting step from C–C bond scission to product release for the evolved variants, with a catalytic tetrad that emerges in the later rounds of evolution playing an important role in facilitating the tremendous rate acceleration (>9000 fold increase in kcat/KM for the most evolved variants) observed in these enzymes.167 In addition, computational modeling indicated that the conformational space sampled by the highly efficient enzyme contains a high percentage of catalytically competent conformations, in contrast to variants from earlier rounds of evolution which sample only small populations of catalytically competent substates.23 Finally, further computational modeling identified fast time scale motions that were present only in the most catalytically efficient evolved variant of the de novo RA.168 The change in conformational ensemble during directed evolution thus occurs as an unintentional but essential consequence of the mutations introduced during laboratory evolution.
Optimization of conformational dynamics has played an unforeseen but important role in several other successful de novo enzyme design studies.10−12,31,169 In a series of studies, Baker and co-workers first generated a de novo Kemp eliminase (KE07) catalyzing proton elimination from 5-nitrobenzisoxazole with modest catalytic activity, which was then further optimized by directed evolution to increase kcat/KM by 200-fold.170 This led to a further study to improve KE07 through (1) optimizing the electrostatic environment of the active site by removal of a catalytically unfavorable “quenching” interaction between an active-site lysine and the catalytic base as well as fine-tuning the pKa of the catalytic base, and (2) stabilizing the active site in a conformation optimal for catalysis.171 There have subsequently been several experimental and computational studies of KE07,171−175 which have provided significant insight into catalysis by the original design and the evolved variants. However, accounting for the effect of mutations that emerge in later rounds of evolution has been challenging. In this context, we have performed detailed crystallographic and computational analysis of the evolutionary trajectory of KE07,24 where we showed that across the trajectory, the instability of the original designed active site leads to the emergence of two additional active-site configurations, involving significant active-site reorganization (Figure 8). The most efficient of these is then gradually stabilized by evolutionary conformational selection. Our computational analysis indicates that the new active-site configurations are not only catalytically active, they are, in fact, catalytically preferred over the original design. In particular, our work demonstrated that substitution of residues remote from the active site appeared to play an important role in allowing for the emergence of these new active-site configurations, and thus in controlling and shaping the active site for efficient catalysis.24
Figure 8.

Directed evolution (DE) of Kemp eliminase KE07 involves binding pocket restructuring and conformational diversification of a key catalytic residue. (A) Binding pocket restructuring from the first round of directed evolution (R1, gray) to the fourth round of directed evolution (R4, cyan). (B) Conformational plasticity in Trp50 introduces three distinct conformations, conformation A (green) is present prior to DE, conformation B (magenta) is present in intermediate DE steps, and conformation C (cyan) is present in later DE steps. Representative structures for Michaelis complexes (i–iii) and transition states (iv–vi) obtained from simulations of the reaction mechanism are shown. (C) Kinetic isotope effects (KIEs) on (i) activation energies and (ii) pre-exponential factors. Conformational mixing of different Trp50 conformations leads to anomalous KIEs in R5-R6, while the values at the end points of the evolutionary trajectory remain similar. Adapted from ref (24). Copyright 2018 Springer Nature. Published under a CC-BY license (http://creativecommons.org/licenses/by/4.0/).
Following this, in 2013, Hilvert and co-workers were able to obtain a de novo Kemp eliminase (KE), HG3, which was further optimized by directed evolution, with the most efficient evolved variant after 17 rounds of evolution (HG3.17) being able to cleave 5-nitrobenzisoxazole with kcat = 700 ± 60 s–1 and kcat/KM = 230 000 ± 20 000 M–1 s–1.176 Structural analysis suggested three potential origins for this tremendous enhancement of catalytic activity: (1) improved shape complementarity of the evolved active site toward the substrate, which includes the elimination of a non-productive substrate binding mode, (2) improved alignment of the catalytic base, and (3) the introduction of a new catalytic group contributing to the stabilization of negative charge developed during the reaction.176 More recently, Chica and co-workers used room-temperature crystallography to study changes in the conformational ensemble of the HG3 series of Kemp eliminases during directed evolution.27 They observed a number of key changes across the evolutionary trajectory, specifically, rigidification of key catalytic residues, improved active-site preorganization, and enlargement of the entrance to the active site, which in turn facilitates substrate entry and product release. They then created a construct, HG4, which contained the minimal subset of mutations observed in the HG3 series, all of which are in or close to the active site, in order to establish the conformational changes necessary to enhance the activity of HG3. The designed variant (HG4, kcat/KM = 120 000 M–1 s–1) is >700-fold more effective than HG3 itself (kcat/KM = 160 M–1 s–1), but not as efficient as HG3.17 (kcat/KM = 230 000 M–1 s–1), since only a minimal subset of mutations was introduced.27 Significantly, these key changes in the conformational ensemble could be predicted using computational design, indicating again the importance of including conformational flexibility as part of the design procedure.
In another example of using conformational flexibility to design efficient Kemp eliminases, we harnessed the conformational flexibility of Precambrian β-lactamases, identified through ancestral inference,47,81 and used these enzymes as a scaffold to insert a de novo active site capable of Kemp elimination.177 This was achieved through a single hydrophobic-to-ionizable substitution of a tryptophan to an aspartic acid side chain (due to both shape congruity with the substrate for Kemp elimination, as well as introduction of a general base to the active site). Our most proficient Kemp eliminase, an ancestral eliminase at the GNCA node, showed catalytic parameters of kcat ≈ 10 s–1 and kcat/KM ≈ 5 × 103 M–1 s–1, only 2 orders of magnitude below that of HG3.17.176 Curiously, while our design strategy was highly effective in the ancestral lactamases, it was unsuccessful in modern lactamases. Combined structural and computational analysis suggested that this was due to the increased rigidity of the evolved active sites, which could not adapt to bind the substrate and catalyze Kemp elimination with optimal electrostatic preorganization. Subsequently, we performed computationally focused ultra-low-throughput screening of variants of our most efficient lactamase predicted by FuncLib,40 and were able to further enhance our most proficient lactamase from our earlier study177 to kcat ≈ 102 s–1 and kcat/KM ≈ 2 × 104 M–1 s–1,49 bringing it to the range of the catalytic activities of naturally occurring enzymes.178 We note that the catalytic base (D229) introduced into the de novo active site lies on the end of a flexible loop. Therefore, subsequent studies could potentially target the flexibility of this loop, in order to optimize its placement in the active site.102
As more is discovered about the connection between loop dynamics and enzyme catalysis, directed evolution of loops and conformational dynamics is being harnessed to produce more efficient enzymes. For example, Kim and co-workers performed concerted insertion and deletion of dynamic loops using SIAFE (Simultaneous Incorporation and Adjustment of Functional Elements) and directed evolution, in order to successfully confer β-lactamase activity onto a glyoxalase II αβ/βα hydrolase scaffold.179 More recently, Zhu and co-workers focused on mutations in the active-site decorating loops of PpADI (Pseudomonas plecoglossicida arginine deaminase).180 Through targeted mutations, they determined that loop flexibility appears to be a critical basis for efficient substrate affinity, not only by reducing the amount the loop blocks access to the active site, but that synergy between the motions of the two decorating loops plays a role in determining the binding efficiency. As another example, Fraser and co-workers performed directed evolution on a catalytically impaired variant of cyclophilin A (CypA), and were able to partially restore the catalytic activity of the enzyme through the introduction of two second-shell mutations that “rescued” activity through modulation of conformational dynamics.181 For several more examples, we refer the readers to refs (26, 181, and 182). Taken together, these successful examples of modulating enzyme activity through targeting conformational dynamics, either deliberately or serendipitously, indicate their importance and further highlight the vast opportunity still present in the field.
Computational Approaches to Engineer Conformational Dynamics
Experimental approaches for the laboratory evolution of functional enzyme conformational dynamics has been discussed in detail in refs (31 and 183). A wide array of techniques exist that can be used to probe conformational dynamics on a variety of time scales, including NMR,184 single-molecule FRET,185 fluorescence anisotropy,186 time-resolved187 or multi-temperature188 X-ray crystallography, and mass spectrometry.189 As these techniques have also been reviewed in detail elsewhere, we refer the reader to e.g. refs (106, 184, 187, and 190) for further discussion of relevant techniques and the contributions they have made to our understanding of the role of conformational dynamics in enzyme function (not just enzyme evolvability). In parallel, molecular simulation has also played an important part in dissecting the physico-chemical parameters that lead to the emergence of new enzyme functions.10,11,191 Simulation is also playing an increasingly important role in enzyme design, combining both sequence- and structural-based approaches, including approaches that take into account conformational dynamics as part of the design process,23,33−42 with increasing contributions from machine-learning approaches.192−195 Clearly, both conventional and even enhanced molecular dynamics-based approaches are far too computationally expensive for the extensive screening necessary for efficient design of conformational dynamics, and are more suited to characterization of a select number of variants from a pool of different designs. However, coupling structural bioinformatics/loop engineering with experimental design strategies has tremendous potential for the targeted engineering of enzyme–substrate selectivity and catalytic activity. In this section we will present some relevant techniques that are likely to play an important role in protein engineering efforts in the coming years.
The information obtained from studying the conservation and co-evolution patterns of residues in a protein/enzyme family has been used to great benefit in homology modeling,196 protein–protein docking,197,198 and protein/enzyme engineering.38,40,199 In enzyme engineering, these methods can be used to massively reduce the sequence search space, under the principle that deleterious mutations will largely not be preserved by natural selection.169 PROSS38 combines the above-described phylogenetic analysis with Rosetta design calculations and has been successfully used to improve the stability and/or expression of several proteins.38,200,201 Building on the successes of PROSS, FuncLib40 (Figure 9) was designed specifically for enzyme engineering, with the aim to generate large increases in activity with a minimal set of mutations. Further, FuncLib can be performed with or without a model of the substrate or transition state, and a repertoire of enzymes with different actives, specificities, and enantioselectivities can be obtained.40 While, strictly, techniques such as PROSS38 and FuncLib40 focus on optimizing stability rather than conformational dynamics, they hold great potential as tools that can move a significant part of in vitro screening approaches in silico, and techniques such as these will likely become the “go-to” starting point in future enzyme engineering studies; therefore we have included these techniques in this section. In addition, while these methods do not directly target conformational dynamics (they focus on optimizing stability), they do so indirectly by preferentially optimizing one conformation of the enzyme over all others. In doing so, they induce a population shift toward the desired state, thus reducing unproductive “floppiness”.
Figure 9.

Overview of the FuncLib workflow,40 using the steps involved in generating a repertoire of phosphotriesterase enzymes starting from a bacterial phosphotriesterase as an illustrative example. (A) First, the active-site positions to be modified for design are selected, and, at each position, the sequence space is constrained through a combination of evolutionarily-conservation analysis (PCCM) and mutational-scanning calculations (based on calculated folding–free energy differences, ΔΔG). Following from this, (B) the resulting multipoint mutants are exhaustively enumerated with the aid of Rosetta atomistic design calculations. This allows for (C) ranking of the resulting constructs by energy, followed by (D) sequence clustering to obtain a final repertoire of diverse designs, ranked by energy, which can be subjected to experimental testing. For further details, see ref (40). Reproduced with permission from ref (40). Copyright 2018 Elsevier.
Molecular dynamics (MD) simulations have been used extensively to provide insight into the conformational dynamics of enzymes and its relationship with catalysis.23,24,129,156,202−206 MD simulations can also be coupled with QM/MM calculations, to explicitly link conformational dynamics to chemistry.207,208 The insights gained from MD simulations can be directly applied toward the (semi-)rational design of variants with altered conformational dynamics.182,203,209 Dodani et al. utilized extensive MD simulations to identify a single residue that was responsible for controlling the conformational dynamics of the F/G loop of a nitrating cytochrome P450 TxtE, with point variants ultimately able to switch substrate regioselectivity.203 Extensive MD simulations can be used to construct MSMs, which can provide thermodynamic and kinetic characterization of conformational substates.130,131 While MSMs are information rich, they often require at least many μs of aggregate sampling in order to be produced. Unbiased enhanced sampling techniques such as accelerated or Gaussian accelerated MD (aMD or GaMD),210,211 scaled MD,212 and temperature or Hamiltonian replica exchange (TREX or HREX)213,214 offer a means to much more efficiently sample available conformational space. For example, a 500 ns long aMD simulation of bovine pancreatic trypsin inhibitor was able to sample equivalent phase space as compared to a 1 ms long conventional MD simulation.210 The identification of rarely sampled conformational states from MD simulations of a WT enzyme could be used as the starting point for computational design efforts. For example, HREX-MD simulations of a promiscuous P450 enzyme identified numerous conformational states available to the WT enzyme’s active site that would ultimately lead to different products.206 Semi-rational design using information from the HREX-MD simulations and MMPBSA calculations was then used to generate distal variants with altered preferences for the available conformational states, ultimately leading to different product distributions for the enzyme variants.206
In cases such as the above where one wishes to stabilize a specific conformational state(s) over others, enhanced sampling techniques that bias along user specified reaction coordinate(s) may be beneficial for low-to-medium throughput screening of variants, with methods such as metadynamics,215 steered MD,216 umbrella sampling (US),217 and adaptive biasing force218 all falling into this category. Michielssens et al. performed US MD simulations to screen 15 distal variants that tune the binding selectivity of ubiquitin through altering the relative populations of the two major ubiquitin binding-site conformations, ultimately taking forward six variants for experimental validation.219 As another example, MD simulations can be used to identify correlated motions and allosteric networks in enzymes, providing a means to identify the impact of distal mutations on enzyme catalysis.12,220−222 Numerous methodologies based on analysis of correlated motions allow one to probe allostery, including: WISP,223 CNA224 and CARDS.225 The “shortest path map” (SPM) method23 allows one to identify the key residues distributed throughout the entire enzyme that play a significant role in regulating the overall conformational dynamics. The potential of this approach toward enzyme engineering was demonstrated by evaluating several intermediates along a multi-step evolutionary trajectory of a retro-aldolase enzyme, in which distal residues mutated throughout the directed evolution trajectory were repeatedly found on or very close to the SPM.23 SPM could thus be applied to guide further design efforts, by targeting a specific set of residues, allowing for a more exhaustive search at these positions. Another potentially valuable tool is the “dynamic flexibility index”, which can be used to calculate the contribution of each residue to the enzyme’s functionally important dynamics.226
Machine learning (ML) is finding increasing applicability in the field of biomolecular simulation and more specifically enzyme engineering.195,227 ML has been shown to improve the efficiency of directed evolution experiments,193 as well as predict allosteric mutations that increase the activity of beta-lactamases toward antibiotics.228 In addition, databases such as ProtMiscuity may provide valuable insight into selecting an optimal starting enzyme for further optimization.229 Furthermore, there are many enzyme design approaches that focus more directly on the active site, such as CASCO,37 CADEE,230 multi-state design approaches,231 the “inside/out” approach from Rosetta232 and also Rosetta-based de novo design approaches as in ref (170). While these approaches do not specifically focus on targeting conformational dynamics (similarly to PROSS38 and FuncLib40), they provide nevertheless a powerful complementary tool to drive the engineering of designer enzymes with tailored physico-chemical properties.
Conclusions and Future Perspectives
Almost two decades since James and Tawfik presented their “New View” of enzyme catalysis,7 it is becoming increasingly clear that conformational dynamics are critical to enzyme evolvability.7−13 This manifests itself in all contexts: from the emergence of novel enzymes, through to the natural evolution of existing enzymes, and even to the fine-tuning of dynamical properties of designed enzymes during laboratory evolution, whether incidentally or intentionally. As has been discussed elsewhere,31 and as we show in this Perspective, the role of conformational dynamics in evolution is two-fold: on the one hand, an expanded repertoire of conformational states being available to an enzyme allows for a greater diversity of catalytically competent conformations, that can facilitate the emergence of new activities (Figure 1).7,8 However, with this also comes an expanded repertoire of catalytically non-productive conformations, and once an initial activity has been established, the subsequent focus of evolution appears to be dampening of catalytically non-productive conformations.31
Here, it is possible to learn from the tricks Nature uses in natural evolution for enzyme design. Engineering of conformational dynamics has already been effectively applied to, for instance, improve binding219,233,234 or stability.200,201,234,235 Clearly, this suggests that conformational dynamics is therefore also a feature that can be manipulated in protein engineering, to generate new designer enzymes with targeted substrate specificities or improved catalytic activity, and there are a number of such success stories in the literature.11,31,49 Computational approaches have played a big role in protein engineering, in particular in the context of designing de novo enzymes.164,170,236 As an illustration, the topic of de novo enzyme design has been recently reviewed extensively by Korendovcyh and DeGrado,237 who describe three key stages of de novo design: (1) manual protein design (based on work from the 1970s and 1980s), (2) computational design guided by fundamental physico-chemical principles (from the mid 1980s to the early 2000s), and (3) fragment-based and bioinformatically informed computational design (starting in the early 2000s). Only the first of these three stages (manual protein design) is arguably non-computational. Historically, however, the computational approaches harnessed for protein design either have been purely sequence based or have focused mainly on design based on static structures,238 with the major enhancements in activity coming from subsequent laboratory evolution.164−166,171,176 This is changing, as greater awareness of the importance of conformational dynamics, as well as the role of remote mutations in modulating activity,239−245 means that both conformational dynamics and mutations of outer shell residues are starting to be incorporated into design approaches.11,31,49
There exist already a large number of computational approaches that can be used to incorporate dynamical properties into the design process, for example those presented in refs (40, 206, and 219). However, their use in computational design is at present far from routine, in part due to the not insubstantial computational cost involved. However, approaches such as PROSS38 and FuncLib40 enable large-scale in silico screening of potential enzyme variants, allowing for the design of novel enzymes. Further, coupling structural bioinformatic approaches with machine learning could be used to help predict enzyme variants that are optimized for a given physico-chemical property. Analysis using structural bioinformatics approaches will further help guide the design process, and it is not inconceivable that computational protein design will become a pipeline of multiple different approaches with varying levels of complexity, a portion of which will be focused on targeting conformational dynamics.
One of the biggest challenges that we currently face in incorporating conformational dynamics in either computational or laboratory engineering of protein function is simply that not enough is known about the precise way in which the dynamical properties of a given system affect its activity, and alterations to dynamical properties of an enzyme can just as easily be catalytically detrimental as beneficial. For example, it would be tremendously useful if one could define a list of requirements that should be satisfied in order to determine that conformational dynamics is important for evolution for a given case study. However, the problem is that creating such a list would be non-trivial, because the role of conformational dynamics can be important in different ways for different systems. To take just a few of the examples discussed in this work, in the case of chalcone isomerases (CHI),25 the role of conformational dynamics is easy to assign, as all the key catalytic residues are already in place in the non-catalytic ancestor, and evolution appears to be primarily fine-tuning both side-chain conformational dynamics (through optimizing the position of the catalytic arginine, Figure 3) and substrate positioning (through elimination of non-productive substrate binding conformations in the evolved enzyme). In the case of the β-lactamases we have repurposed as Kemp eliminases,49,177 once again, scaffold flexibility appears to play an important role both in the process of specialization from a generalist to a specialist β-lactamase,151 and for whether the Precambrian vs modern enzymes are capable of accommodating our de novo active site for catalyzing Kemp elimination.49,177 In the case of the designed Kemp eliminase, KE07, we observed that the introduction of remote mutations facilitates the stabilization of completely new active-site conformations through evolutionary conformational selection.24 However, the question remains of whether the changes in conformational dynamics drive the changes in function, or the selection pressure on the changes in function drives the changes in conformational dynamics.
Following from this, and as pointed out by a reviewer, it is unclear whether it will be necessary to “dial-in” or “dial-out” conformational dynamics for a given system, as one needs to balance sampling catalytically competent (productive) conformations with dampening the sampling of catalytically non-productive conformations. This can potentially be achieved in targeted way through engineering, provided that the behavior of the system is sufficiently well understood. Ultimately, however, this will be a system-specific balancing act, driven by the intrinsic physico-chemical properties of a given system, and will therefore need to be determined on a case-by-case basis. However, clearly, considering dynamical properties in the design process is critical, as simply having the catalytic residues in the correct place is not always enough to impart efficient catalytic activity.25 Shifting this paradigm is essential for overcoming one of the next big barriers on the path to designing green biocatalysts for a sustainable future.
Acknowledgments
This work was supported by the Knut and Alice Wallenberg Foundation (Wallenberg Academy Fellowship and Wallenberg Scholar grant to S.C.L.K., grants 2018.0140 and 2019.0431), the Human Frontier Science Program (grant RGP0041/2017), and the Carl Tryggers Foundation for Scientific Research (postdoctoral fellowship to R.M.C., grant CTS 19:172). We thank Dr. Sarel Fleishman for permission to reuse Figure 9, and Profs. Alvan Hengge and Jose Manuel Sanchez-Ruiz, as well as Dr. Irmeli Barkefors, for helpful discussions and comments on the manuscript.
The authors declare no competing financial interest.
References
- McGeagh J. D.; Ranaghan K. E.; Mulholland A. J. Protein Dynamics and Enzyme Catalysis: Insights from Simulations. Biochim. Biophys. Acta, Proteins Proteomics 2011, 1814, 1077–1092. 10.1016/j.bbapap.2010.12.002. [DOI] [PubMed] [Google Scholar]
- Doshi U.; Hamelberg D. The Dilemma of Conformational Dynamics in Enzyme Catalysis: Perspectives from Theory and Experiment. Adv. Exp. Med. Biol. 2014, 805, 221–243. 10.1007/978-3-319-02970-2_10. [DOI] [PubMed] [Google Scholar]
- Kohen A. Role of Dynamics in Enzyme Catalysis: Substantial versus Semantic Controversies. Acc. Chem. Res. 2015, 48, 466–473. 10.1021/ar500322s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanoian P.; Liu C. T.; Hammes-Schiffer S.; Benkovic S. J. Perspectives on Electrostatics and Conformational Motions in Enzyme Catalysis. Acc. Chem. Res. 2015, 48, 482–489. 10.1021/ar500390e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klinman J. P. Dynamically Achieved Active Site Precision in Enzyme Catalysis. Acc. Chem. Res. 2015, 48, 449–456. 10.1021/ar5003347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warshel A.; Bora R. P. Perspective: Defining and Quantifying the Role of Dynamics in Enzyme Catalysis. J. Chem. Phys. 2016, 144, 180901. 10.1063/1.4947037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James L. C.; Tawfik D. S. Conformational Diversity and Protein Evolution - A 60-year-old Hypothesis Revisited. Trends Biochem. Sci. 2003, 28, 361–368. 10.1016/S0968-0004(03)00135-X. [DOI] [PubMed] [Google Scholar]
- Tokuriki N.; Tawfik D. S. Protein Dynamism and Evolvability. Science 2009, 324, 203–207. 10.1126/science.1169375. [DOI] [PubMed] [Google Scholar]
- Klinman J. P.; Kohen A. Evolutionary Aspects of Enzyme Dynamics. J. Biol. Chem. 2014, 289, 30205–30212. 10.1074/jbc.R114.565515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrović D.; Risso V. A.; Kamerlin S. C. L.; Sanchez-Ruiz J. M. Conformational Dynamics and Enzyme Evolution. J. R. Soc., Interface 2018, 15, 20180330. 10.1098/rsif.2018.0330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson K. E.; Lindorff-Larsen K. Structural Heterogeneity and Dynamics in Protein Evolution and Design. Curr. Opin. Struct. Biol. 2018, 48, 157–163. 10.1016/j.sbi.2018.01.010. [DOI] [PubMed] [Google Scholar]
- Maria-Solano M. A.; Serrano-Hervás E.; Romero-Rivera A.; Iglesias-Fernández J.; Osuna S. Role of Conformational Dynamics in the Evolution of Novel Enzyme Functions. Chem. Commun. 2018, 54, 6622–6634. 10.1039/C8CC02426J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campitelli P.; Modi T.; Kumar S.; Ozkan S. B. The Role of Conformational Dynamics and Allostery in Modulating Protein Evolution. Annu. Rev. Biophys. 2020, 49, 267–288. 10.1146/annurev-biophys-052118-115517. [DOI] [PubMed] [Google Scholar]
- Jensen R. A. Enzyme Recruitment in Evolution of New Function. Annu. Rev. Microbiol. 1976, 30, 409–425. 10.1146/annurev.mi.30.100176.002205. [DOI] [PubMed] [Google Scholar]
- O’Brien P. J.; Herschlag D. Catalytic Promiscuity and the Evolution of New Enzymatic Activities. Chem. Biol. 1999, 6, R91–R105. 10.1016/S1074-5521(99)80033-7. [DOI] [PubMed] [Google Scholar]
- Khersonsky O.; Tawfik D. S. Enzyme Promiscuity: A Mechanistic and Evolutionary Perspective. Annu. Rev. Biochem. 2010, 79, 471–505. 10.1146/annurev-biochem-030409-143718. [DOI] [PubMed] [Google Scholar]
- Bornscheuer U. T.; Kazlauskas R. J. Catalytic Promiscuity in Biocatalysis: Using Old Enzymes to Form New Bonds and Follow New Pathways. Angew. Chem., Int. Ed. 2004, 43, 6032–6040. 10.1002/anie.200460416. [DOI] [PubMed] [Google Scholar]
- Zeymer C.; Hilvert D. Directed Evolution of Protein Catalysts. Annu. Rev. Biochem. 2018, 87, 131–157. 10.1146/annurev-biochem-062917-012034. [DOI] [PubMed] [Google Scholar]
- Leveson-Gower R. B.; Mayer C.; Roelfes G. The Importance of Catalytic Promiscuity for Enzyme Design and Evolution. Nat. Rev. Chem. 2019, 3, 687–705. 10.1038/s41570-019-0143-x. [DOI] [Google Scholar]
- Wierenga R. K. The TIM-Barrel Fold: A Versatile Framework for Efficient Enzymes. FEBS Lett. 2001, 492, 193–198. 10.1016/S0014-5793(01)02236-0. [DOI] [PubMed] [Google Scholar]
- Varma Kadumuri R.; Vadrevu R. Diversity in αβ and βα Loop Connections in TIM Barrel Proteins: Implications for Stability and Design of the Fold. Interdiscip. Sci.: Comput. Life Sci. 2018, 10, 805–812. 10.1007/s12539-017-0250-7. [DOI] [PubMed] [Google Scholar]
- Campbell E.; Kaltenbach M.; Correy G. J.; Carr P. D.; Porebski B. T.; Livingstone E. K.; Afriat-Jurnou L.; Buckle A. M.; Weik M.; Hollfelder F.; Tokuriki N.; Jackson C. J. The Role of Protein Dynamics in the Evolution of New Enzyme Function. Nat. Chem. Biol. 2016, 12, 944–950. 10.1038/nchembio.2175. [DOI] [PubMed] [Google Scholar]
- Romero-Rivera A.; Garcia-Borràs M.; Osuna S. Role of Conformational Dynamics in the Evolution of Retro-Aldolase Activity. ACS Catal. 2017, 7, 8524–8532. 10.1021/acscatal.7b02954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong N. S.; Petrović D.; Lee R.; Gryn’ova G.; Purg M.; Saunders J.; Bauer P.; Carr P. D.; Lin C. Y.; Mabbitt P. D.; Zhang W.; Altamore T.; Easton C.; Coote M. L.; Kamerlin S. C. L.; Jackson C. J. The Evolution of Multiple Active Site Configurations in a Designed Enzyme. Nat. Commun. 2018, 9, 3900. 10.1038/s41467-018-06305-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaltenbach M.; Burke J. R.; Dindo M.; Pabis A.; Munsberg F. S.; Rabin A.; Kamerlin S. C. L.; Noel J. P.; Tawfik D. S. Evolution of Chalcone Isomerase from a Noncatalytic Ancestor. Nat. Chem. Biol. 2018, 14, 548–555. 10.1038/s41589-018-0042-3. [DOI] [PubMed] [Google Scholar]
- Buller A. R.; van Roye P.; Cahn J. K. B.; Scheele R. A.; Herger M.; Arnold F. H. Directed Evolution Mimics Allosteric Activation by Stepwise Tuning of the Conformational Ensemble. J. Am. Chem. Soc. 2018, 140, 7256–7266. 10.1021/jacs.8b03490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broom A.; Rakotoharisoa R. V.; Thompson M. C.; Zarifi N.; Nguyen E.; Mukhametzhanov N.; Liu L.; Fraser J. S.; Chica R. A. Evolution of an Enzyme Conformational Ensemble Guides Design of an Efficient Biocatalyst. bioRxiv 2020, 999235 10.1101/2020.03.19.999235. [DOI] [Google Scholar]
- Drienovská I.; Mayer C.; Dulson C.; Roelfes G. A Designer Enzyme for Hydrazone and Oxime Formation Featuring an Unnatural Catalytic Aniline Residue. Nat. Chem. 2018, 10, 946–952. 10.1038/s41557-018-0082-z. [DOI] [PubMed] [Google Scholar]
- Yang B.; Wang N.; Schnier P. D.; Zheng F.; Zhu H.; Polizzi N. F.; Ittuveetil A.; Saikam V.; DeGrado W. F.; Wang Q.; Wang P. G.; Wang L. Genetically Introducing Biochemically Reactive Amino Acids Dehydroalanine and Dehydrobutyrine in Proteins. J. Am. Chem. Soc. 2019, 141, 7698–7703. 10.1021/jacs.9b02611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drienovská I.; Roelfes G. Expanding the Enzyme Universe with Genetically Encoded Unnatural Amino Acids. Nat. Catal. 2020, 3, 193–202. 10.1038/s41929-019-0410-8. [DOI] [Google Scholar]
- Campbell E. C.; Correy G. J.; Mabbitt P. D.; Buckle A. M.; Tokuriki N.; Jackson C. J. Laboratory Evolution of Protein Conformational Dynamics. Curr. Opin. Struct. Biol. 2018, 50, 49–57. 10.1016/j.sbi.2017.09.005. [DOI] [PubMed] [Google Scholar]
- Gardner J. M.; Biler M.; Risso V. A.; Sanchez-Ruiz J. M.; Kamerlin S. C. L. Manipulating Conformational to Repurpose Ancient Proteins for Catalytic Functions. ACS Catal. 2020, 10, 4863–4870. 10.1021/acscatal.0c00722. [DOI] [Google Scholar]
- Pavelka A.; Chovancova E.; Damborsky J. HotSpot Wizard: A Web Server for Identification of Hot Spots in Protein Engineering. Nucleic Acids Res. 2009, 37, W376–W383. 10.1093/nar/gkp410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C.-Y.; Georgiev I.; Anderson A. C.; Donald B. R. Computational Structure-Based Redesign of Enzyme Activity. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 3764–3769. 10.1073/pnas.0900266106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiss G.; Röthlisberger D.; Baker D.; Houk K. N. Evolution and Ranking of Enzyme Designs. Protein Sci. 2010, 19, 1760–1773. 10.1002/pro.462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindert S.; Meiler J.; McCammon J. A. Iterative Molecular Dynamics—Rosetta Protein Structure Refinement Protocol to Improve Model Quality. J. Chem. Theory Comput. 2013, 9, 3843–3847. 10.1021/ct400260c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wijma H. J.; Floor R. J.; Bjelic S.; Marrink S. J.; Baker D.; Janssen D. B. Enantioselective Enzymes by Computational Design and In Silico Screening. Angew. Chem., Int. Ed. 2015, 54, 3726–3730. 10.1002/anie.201411415. [DOI] [PubMed] [Google Scholar]
- Goldenzweig A.; Goldsmith M.; Hill S. E.; Gertman O.; Laurino P.; Ashani Y.; Dym O.; Unger T.; Albeck S.; Prilusky J.; Lieberman R. L.; Aharoni A.; Silman I.; Sussman J. L.; Tawfik D. S.; Fleishman S. J. Automated Structure- and Sequence-Based Design of Proteins for High Bacterial Expression and Stability. Mol. Cell 2016, 63, 337–346. 10.1016/j.molcel.2016.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Childers M. C.; Daggett V. Insight from Molecular Dynamics Simulations for Computational Protein Design. Mol. Syst. Des. Eng. 2017, 2, 9–33. 10.1039/C6ME00083E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khersonsky O.; Lipsh R.; Avizemer Z.; Ashani Y.; Goldsmith M.; Leader H.; Dym O.; Rogotner S.; Trudeau D. L.; Prilusky J.; Amengual-Rigo P.; Guallar V.; Tawfik D. S.; Fleishman S. J. Automated Design of Efficient and Functionally Diverse Enzyme Repertoires. Mol. Cell 2018, 72, 178–186. 10.1016/j.molcel.2018.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Currin A.; Kwok J.; Sadler J. C.; Bell E. L.; Swainston N.; Ababi M.; Day P.; Turner N. J.; Kell D. B. GeneORator: An Effective Strategy for Navigating Protein Sequence Space More Efficiently through Boolean OR-Type DNA Libraries. ACS Synth. Biol. 2019, 8, 1371–1378. 10.1021/acssynbio.9b00063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St-Jacques A. D.; Eyahpaise M.-E. C.; Chica R. A. Computational Design of Multisubstrate Enzyme Specificity. ACS Catal. 2019, 9, 5480–5485. 10.1021/acscatal.9b01464. [DOI] [Google Scholar]
- Pandya C.; Farelli J. D.; Dunaway-Mariano D.; Allen K. N. Enzyme Promiscuity: Engine of Evolutionary Innovation. J. Biol. Chem. 2014, 289, 30229–30236. 10.1074/jbc.R114.572990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baier F.; Copp J. N.; Tokuriki N. Evolution of Enzyme Superfamilies: Comprehensive Exploration of Sequence-Function Relationships. Biochemistry 2016, 55, 6375–6388. 10.1021/acs.biochem.6b00723. [DOI] [PubMed] [Google Scholar]
- Copley S. D. Shining a Light on Enzyme Promiscuity. Curr. Opin. Struct. Biol. 2017, 47, 167–175. 10.1016/j.sbi.2017.11.001. [DOI] [PubMed] [Google Scholar]
- Tyzack J. D.; Furnham N.; Sillitoe I.; Orengo C. M.; Thornton J. M. Understanding Enzyme Evolution from a Computational Perspective. Curr. Opin. Struct. Biol. 2017, 47, 131–139. 10.1016/j.sbi.2017.08.003. [DOI] [PubMed] [Google Scholar]
- Siddiq M. A.; Hochberg G. K.; Thornton J. M. Evolution of Protein Specificity: Insights From Ancestral Protein Reconstruction. Curr. Opin. Struct. Biol. 2017, 47, 113–122. 10.1016/j.sbi.2017.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidi D.; Longo L. M.; Jabłońska J.; Milo R.; Tawfik D. S. A Bird’s-Eye View of Enzyme Evolution: Chemical, Physicochemical, and Physiological Considerations. Chem. Rev. 2018, 118, 8786–8797. 10.1021/acs.chemrev.8b00039. [DOI] [PubMed] [Google Scholar]
- Risso V. A.; Romero-Rivera A.; Gutierrez-Rus L. I.; Ortega-Muñoz M.; Santoyo-Gonzalez F.; Gavira J. A.; Sanchez-Ruiz J. M.; Kamerlin S. C. L. Enhancing a De Novo Enzyme Activity by Computationally-Focused, Ultra-Low-Throughput Sequence Screening. Chem. Sci. 2020, 10.1039/D0SC01935F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furnham N.; Dawson N. L.; Rahman S. A.; Thornton J. M.; Orengo C. A. Large-Scale Analysis Exploring Evolution of Catalytic Machineries and Mechanisms in Enzyme Superfamilies. J. Mol. Biol. 2016, 428, 253–267. 10.1016/j.jmb.2015.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouzounis C. A.; Kunin V.; Darzentas N.; Goldovsky L. A Minimal Estimate for the Gene Content of the Last Universal Common Ancestor - Exobiology From a Terrestrial Perspective. Res. Microbiol. 2006, 157, 57–68. 10.1016/j.resmic.2005.06.015. [DOI] [PubMed] [Google Scholar]
- Weiss M. C.; Sousa F. L.; Mrnjavac N.; Neukirchen S.; Roettger M.; Nelson-Sathi S.; Martin W. F. The Physiology and Habitat of the Last Universal Common Ancestor. Nat. Microbiol. 2016, 1, 16116. 10.1038/nmicrobiol.2016.116. [DOI] [PubMed] [Google Scholar]
- Smock R. G.; Yadid I.; Dym O.; Clarke J.; Tawfik D. S. De Novo Evolutionary Emergence of a Symmetrical Protein is Shaped by Folding Constraints. Cell 2016, 164, 476–486. 10.1016/j.cell.2015.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eck R. V.; Dayhoff M. O. Evolution of the Structure of Ferredoxin Based on Living Relics of Primitive Amino Acid Sequences. Science 1966, 152, 363–366. 10.1126/science.152.3720.363. [DOI] [PubMed] [Google Scholar]
- Romero Romero M. L.; Rabin A.; Tawfik D. S. Functional Proteins from Short Peptides: Dayhoff’s Hypothesis Turns 50. Angew. Chem., Int. Ed. 2016, 55, 15966–15971. 10.1002/anie.201609977. [DOI] [PubMed] [Google Scholar]
- Noguchi H.; Addy C.; Simoncini D.; Wouters S.; Mylemans B.; Van Meervelt L.; Schiex T.; Zhang K. Y. J.; Tame J. R. H.; Voet A. R. D. Computational Design of Symmetrical Eight-Bladed β-Propeller PRoteins. IUCrJ 2019, 6, 46–55. 10.1107/S205225251801480X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurino P.; Tóth-Petróczy Á.; Meana-Pañeda R.; Lin W.; Truhlar D. G.; Tawfik D. S. An Ancient Fingerprint Indicates the Common Ancestry of Rossmann-Fold Enzymes Utilizing Different Ribose-Based Cofactors. PLoS Biol. 2016, 14, e1002396. 10.1371/journal.pbio.1002396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medvedev K. E.; Kinch L. N.; Schaeffer R. D.; Grishin N. V. Functional Analysis of Rossman-like Domains Reveals Convergent Evolution of Topology and Reaction Pathways. PLoS Comput. Biol. 2019, 15, e1007569. 10.1371/journal.pcbi.1007569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman H. M.; Westbrook J.; Feng Z.; Gilliland G.; Bhat T. N.; Weissig H.; Sindyalov I. N.; Bourne P. E. The Protein Data Bank. Nucelic Acids Res. 2000, 28, 235–242. 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng H.; Schaeffer R. D.; Liao Y.; Kinch L. N.; Pei J.; Shi S.; Kim B.-H.; Grishin N. V. ECOD: An Evolutionary Classification of Protein Domains. PLoS Comput. Biol. 2014, 10, e1003926. 10.1371/journal.pcbi.1003926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng H.; Liao Y.; Schaeffer R. D.; Grishin N. V. Manual Classification Strategies in the ECOD Database. Proteins: Struct., Funct., Genet. 2015, 83, 1238–1251. 10.1002/prot.24818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westheimer F. H. Why Nature Chose Phosphates. Science 1987, 235, 1173–1178. 10.1126/science.2434996. [DOI] [PubMed] [Google Scholar]
- Longo L. M.; Petrović D.; Kamerlin S. C. L.; Tawfik D. S. Short and Simple Sequences Favored the Emergence of N-helix Phospho-Ligand Binding Sites in the First Enzymes. Proc. Natl. Acad. Sci. U. S. A. 2020, 117, 5310–5318. 10.1073/pnas.1911742117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longo L. M.; Blaber M. Protein Design at the Interface of the Pre-Biotic and Biotic Worlds. Arch. Biochem. Biophys. 2012, 526, 16–21. 10.1016/j.abb.2012.06.009. [DOI] [PubMed] [Google Scholar]
- Longo L. M.; Blaber M. Prebiotic Protein Design Supports a Halophile Origin of Foldable Proteins. Front. Microbiol. 2014, 4, 418. 10.3389/fmicb.2013.00418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trifonov E. N. Consensus Temporal Order of Amino Acids and Evolution of the Triplet Code. Gene 2000, 261, 139–151. 10.1016/S0378-1119(00)00476-5. [DOI] [PubMed] [Google Scholar]
- Romero Romero M. L.; Yang F.; Lin Y.-R.; Toth-Petroczy A.; Berezovsky I. N.; Goncearenco A.; Yang W.; Wellner A.; Kumar-Deshmukh F. K.; Sharon M.; Baker D.; Varani G.; Tawfik D. S. Simple Yet Functional Phosphate-Loop Proteins. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, E11943–E11950. 10.1073/pnas.1812400115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaccai N. R.; Chi B.; Thomson A. R.; Boyle A. L.; Bartlett G. J.; Bruning M.; Linden N.; Sessions R. B.; Booth P. J.; Brady R. L.; Woolfson D. N. A De Novo Peptide Hexamer with a Mutable Channel. Nat. Chem. Biol. 2011, 7, 935–941. 10.1038/nchembio.692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khersonsky O.; Fleishman S. J. Why Reinvent the Wheel? Building New Proteins Based on Ready-Made Parts. Protein Sci. 2016, 25, 1179–1187. 10.1002/pro.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voet A. R. D.; Simoncini D.; Tame J. R. H.; Zhang K. Y. J. Evolution-Inspired Computational Design of Symmetric Proteins. Methods Mol. Biol. 2017, 1529, 309–322. 10.1007/978-1-4939-6637-0_16. [DOI] [PubMed] [Google Scholar]
- Parmeggiani F.; Huang P.-S. Designing Repeat Proteins: A Molecular Approach to Protein Design. Curr. Opin. Struct. Biol. 2017, 45, 116–123. 10.1016/j.sbi.2017.02.001. [DOI] [PubMed] [Google Scholar]
- Dou J.; Vorobieva A. A.; Sheffler W.; Doyle L. A.; Park H.; Bick M. J.; Mao B.; Foight G. W.; Lee M. Y.; Gagnon L. A.; Carter L.; Sankaran B.; Ovchinnikov S.; Marcos E.; Huang P.-S.; Vaughan J. C.; Stoddard B. L.; Baker D. De Novo Design of a Fluorescence-Activating β-Barrel. Nature 2018, 561, 485–491. 10.1038/s41586-018-0509-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J.; Selvaraj B.; McShan A. C; Boyken S. E; Wei K. Y; Oberdorfer G.; DeGrado W.; Sgourakis N. G; Cuneo M. J; Myles D. A.; Baker D. De Novo Design of a Homo-Trimeric Amantadine-Binding Protein. eLife 2019, 8, e47839. 10.7554/eLife.47839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombardi A.; Pirro F.; Maglio O.; Chino M.; DeGrado W. F. De Novo Design of Four-Helix Bundle Metalloproteins: One Scaffold, Diverse Reactivities. Acc. Chem. Res. 2019, 52, 1148–1159. 10.1021/acs.accounts.8b00674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam R.; Saier M. H. Jr. A Bacterial Periplasmic Receptor Homologue With Catalytic Activity: Cyclohexadienyl Dehydratase of Pseudomonas Aeruginosa is Homologous to Receptors Specific for Polar Amino Acids. Res. Microbiol. 1993, 144, 165–169. 10.1016/0923-2508(93)90041-Y. [DOI] [PubMed] [Google Scholar]
- Taga M. E.; Larsen N. A.; Howard-Jones A. R.; Walsh C. T.; Walker G. C. BluB Cannibalizes Flavin to Form the Lower Ligand of Vitamin B12. Nature 2007, 446, 449–453. 10.1038/nature05611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuhara K.; Yonehara H.; Hattori T.; Kobayashi K.; Kirimura K. Enzymatic Characterization and Gene Identification of Aconitate Isomerase, an Enzyme Involved in Assimilation of Trans-Aconitic Acid, from Pseudomonas sp. WU-0701. FEBS J. 2015, 282, 4257–4267. 10.1111/febs.13494. [DOI] [PubMed] [Google Scholar]
- Ortmayer M.; Lafite P.; Menon B. R. K.; Tralau T.; Fisher K.; Denkhaus L.; Scrutton N. S.; Rigby S. E. J.; Munro A. W.; Hay S.; Leys D. An Oxidative N-Demethylase Reveals PAS Transition from Ubiquitous Sensor to Enzyme. Nature 2016, 539, 593–597. 10.1038/nature20159. [DOI] [PubMed] [Google Scholar]
- Clifton B. E.; Kaczmarski J. A.; Carr P. D.; Gerth M. L.; Tokuriki N.; Jackson C. J. Evolution of Cyclohexadienyl Dehydratase from an Ancestral Solute-Binding Protein. Nat. Chem. Biol. 2018, 14, 542–547. 10.1038/s41589-018-0043-2. [DOI] [PubMed] [Google Scholar]
- Kaczmarski J. A.; Mahawaththa M. C.; Feintuch A.; Clifton B. E.; Adams L. A.; Goldfarb D.; Otting G.; Jackson C. J. Altered Conformational Sampling Along an Evolutionary Trajectory Changes the Catalytic Activity of an Enzyme. bioRxiv 2020, 932491 10.1101/2020.02.03.932491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochberg G. K. A.; Thornton J. W. Reconstructing Ancient Proteins to Understand the Causes of Structure and Function. Annu. Rev. Biophys. 2017, 46, 247–269. 10.1146/annurev-biophys-070816-033631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao G. S.; Xia T. H.; Fischer R. S.; Jensen R. A. Cyclohexadienyl Dehydratase from Pseudomonas Aeruginosa. Molecular Cloning of the Gene and Characterization of the Gene Product. J. Biol. Chem. 1992, 267, 2487–2493. [PubMed] [Google Scholar]
- Jeschke G. DEER Distance Measurements on Proteins. Annu. Rev. Phys. Chem. 2012, 63, 419–446. 10.1146/annurev-physchem-032511-143716. [DOI] [PubMed] [Google Scholar]
- Burke J. R.; La Clair J. L.; Philippe R. N.; Pabis A.; Corbella M.; Jez J. M.; Cortina G. A.; Kaltenbach M.; Bowman M. E.; Louie G.; Woods K. B.; Nelson A. T.; Tawfik D. S.; Kamerlin S. C. L.; Noel J. P. Bifunctional Substrate Activation via an Arginine Residue Drives Catalysis in Chalcone Isomerases. ACS Catal. 2019, 9, 8388–8396. 10.1021/acscatal.9b01926. [DOI] [Google Scholar]
- Koes R. E.; Quattrocchio F.; Mol J. N. M. The Flavonoid Biosynthetic Pathway in Plants: Function and Evolution. BioEssays 1994, 16, 123–132. 10.1002/bies.950160209. [DOI] [Google Scholar]
- Morita Y.; Takagi K.; Fukuchi-Mizutani M.; Ishiguro K.; Tanaka Y.; Nitasaka E.; Nakayama M.; Saito N.; Kagami T.; Hoshino A.; Iida S. A Chalcone Isomerase-Like Protein Enhances Flavonoid Production and Flower Pigmentation. Plant J. 2014, 78, 294–304. 10.1111/tpj.12469. [DOI] [PubMed] [Google Scholar]
- Jiang W.; Yin Q.; Wu R.; Zheng G.; Liu J.; Dixon R. A.; Pang Y. Role of a Chalcone Isomerase-Like Protein in Flavonoid Biosynthesis in Arabidopsis Thaliana. J. Exp. Bot. 2015, 66, 7165–7179. 10.1093/jxb/erv413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngaki M. N.; Louie G. V.; Philippe R. N.; Manning G.; Pojer F.; Bowman M. E.; Li L.; Larsen E.; Wurtele E. S.; Noel J. P. Evolution of the Chalcone Isomerase Fold from Fatty Acid-Binding to Stereospecific Enzyme. Nature 2012, 485, 530–533. 10.1038/nature11009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitlock M C; Phillips P C; Moore F B; Tonsor S J Multiple Fitness Peaks and Epistasis. Annu. Rev. Ecol. Syst. 1995, 26, 601–629. 10.1146/annurev.es.26.110195.003125. [DOI] [Google Scholar]
- Poelwijk F. J.; Kiviet D. J.; Weinreich D. M.; Tans S. J. Empirical Fitness Landscapes Reveal Accessible Evolutionary Paths. Nature 2007, 445, 383–386. 10.1038/nature05451. [DOI] [PubMed] [Google Scholar]
- de Visser J. A.; Cooper T. F.; Elena S. F. The Causes of Epistasis. Proc. R. Soc. London, Ser. B 2011, 278, 3617–3624. 10.1098/rspb.2011.1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokuriki N.; Jackson C. J.; Afriat-Jurnou L.; Wyganowski K. T.; Tang R.; Tawfik D. S. Diminishing Returns and Tradeoffs Constrain the Laboratory Optimization of an Enzyme. Nat. Commun. 2012, 3, 1257. 10.1038/ncomms2246. [DOI] [PubMed] [Google Scholar]
- Breen M. S.; Kemena C.; Vlasov P. K.; Notredame C.; Kondrashov F. A. Epistasis as the Primary Factor in Molecular Evolution. Nature 2012, 490, 535–538. 10.1038/nature11510. [DOI] [PubMed] [Google Scholar]
- Harms M. J.; Thornton J. W. Evoluationary Biochemistry: Revealing the Historical and Physical Causes of Protein Properties. Nat. Rev. Genet. 2013, 14, 559–571. 10.1038/nrg3540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCandlish D. M.; Rajon E.; Shah P.; Ding Y.; Plotkin J. B. The Role of Epistasis in Protein Evolution. Nature 2013, 497, E1–2. 10.1038/nature12219. [DOI] [PubMed] [Google Scholar]
- Kaltenbach M.; Tokuriki N. Dynamics and Constraints of Enzyme Evolution. J. Exp. Zool., Part B 2014, 322, 468–487. 10.1002/jez.b.22562. [DOI] [PubMed] [Google Scholar]
- Taylor M. S.; Jacobsen E. N. Asymmetric Catalysis by Chiral Hydrogen-Bond Donors. Angew. Chem., Int. Ed. 2006, 45, 1520–1543. 10.1002/anie.200503132. [DOI] [PubMed] [Google Scholar]
- Bernardi L.; Fochi M.; Franchini M. C.; Ricci A. Bioinspired Organocatalyic Asymmetric Reactions. Org. Biomol. Chem. 2012, 10, 2911–2922. 10.1039/c2ob07037e. [DOI] [PubMed] [Google Scholar]
- Orellana L. Large-Scale Conformational Changes and Protein Function: Breaking the In Silico Barrier. Front. Mol. Biosci. 2019, 6, 117. 10.3389/fmolb.2019.00117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard J. P.; Amyes T. L.; Goryanova B.; Zhai X. Enzyme Architecture: On the Importance of Being in a Protein Cage. Curr. Opin. Chem. Biol. 2014, 21, 1–10. 10.1016/j.cbpa.2014.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreira C.; Calixto A. R.; Richard J. P.; Kamerlin S. C. L. The Role of Ligand-Gated Conformational Changes in Enzyme Catalysis. Biochem. Soc. Trans. 2019, 47, 1449–1460. 10.1042/BST20190298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nestl B. M.; Hauer B. Engineering of Flexible Loops in Enzymes. ACS Catal. 2014, 4, 3201–3211. 10.1021/cs500325p. [DOI] [Google Scholar]
- Rajagopalan P. T. R.; Benkovic S. J. Preorganization and Protein Dynamics in Enzyme Catalysis. Chem. Rec. 2002, 2, 24–36. 10.1002/tcr.10009. [DOI] [PubMed] [Google Scholar]
- Schnell J. R.; Dyson H. J.; Wright P. E. Structure, Dynamics, and Catalytic Function of Dihydrofolate Reductase. Annu. Rev. Biophys. Biomol. Struct. 2004, 33, 119–140. 10.1146/annurev.biophys.33.110502.133613. [DOI] [PubMed] [Google Scholar]
- Francis K.; Stojković V.; Kohen A. Preservation of Protein Dynamics in Dihydrofolate Reductase Evolution. J. Biol. Chem. 2013, 288, 35961–35968. 10.1074/jbc.M113.507632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhabha G.; Biel J. T.; Fraser J. S. Keep on Moving: Discovering and Perturbing the Conformational Dynamics of Enzymes. Acc. Chem. Res. 2015, 48, 423–430. 10.1021/ar5003158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawaya M. R.; Kraut J. Loop and Subdomain Movements in the Mechanism of Escherichia Coli Dihydrofolate Reductase: Crystallographic Evidence. Biochemistry 1997, 36, 586–603. 10.1021/bi962337c. [DOI] [PubMed] [Google Scholar]
- Boehr D. D.; McElheny D.; Dyson H. J.; Wright P. E. The Dynamic Energy Landscape of Dihydrofolate Reductase Catalysis. Science 2006, 313, 1638–1642. 10.1126/science.1130258. [DOI] [PubMed] [Google Scholar]
- Cao H.; Gao M.; Zhou H.; Skolnick J. The Crystal Structure of a Tetrahydrofolate-Bound Dihydrofolate Reductase Reveals the Origin of Slow Product Release. Commun. Biol. 2018, 1, 226. 10.1038/s42003-018-0236-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhabha G.; Ekiert D. C.; Jennewein M.; Zmasek C. M.; Tuttle L. M.; Kroon G.; Dyson H. J.; Godzik A.; Wilson I. A.; Wright P. E. Divergent Evolution of Protein Conformational Dynamics in Dihydrofolate Reductase. Nat. Struct. Mol. Biol. 2013, 20, 1243–1249. 10.1038/nsmb.2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masterson J. E.; Schwartz S. D. Evolution Alters the Enzymatic Reaction Coordinate of Dihydrofolate Reductase. J. Phys. Chem. B 2015, 119, 989–996. 10.1021/jp506373q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz-Pernía J. J.; Behiry E.; Luk L. Y. P.; Loveridge E. J.; Tuñón I.; Moliner V.; Allemann R. K. Minimization of Dynamic Effects in the Evolution of Dihydrofolate Reductase. Chem. Sci. 2016, 7, 3248–3255. 10.1039/C5SC04209G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mhashal A. R.; Pshetitsky Y.; Cheatum C. M.; Kohen A.; Major D. T. Evolutionary Effects on Bound Substrate pKa in Dihydrofolate Reductase. J. Am. Chem. Soc. 2018, 140, 16650–16660. 10.1021/jacs.8b09089. [DOI] [PubMed] [Google Scholar]
- Bhabha G.; Lee J.; Ekiert D. C.; Gam J.; Wilson I. A.; Dyson H. J.; Benkovic S. J.; Wright P. E. A Dynamic Knockout Reveals That Conformational Fluctuations Influence the Chemical Step of Enzyme Catalysis. Science 2011, 332, 234–238. 10.1126/science.1198542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller G. P.; Wahnon D. C.; Benkovic S. J. Interloop Contacts Modulate Ligand Cycling During Catalysis by Escherichia Coli Dihydrofolate Reductase. Biochemistry 2001, 40, 867–875. 10.1021/bi001608n. [DOI] [PubMed] [Google Scholar]
- Boehr D. D.; McElheny D.; Dyson H. J.; Wright P. E. Millisecond Timescale Fluctuations in Dihydrofolate Reductase are Exquisitely Sensitive to the Bound Ligands. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 1373–1378. 10.1073/pnas.0914163107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller G. P.; Benkovic S. J. Strength of an Interloop Hydrogen Bond Determines the Kinetic Pathway in Catalysis by Escherichia Coli Dihydrofolate Reductase. Biochemistry 1998, 37, 6336–6342. 10.1021/bi973065w. [DOI] [PubMed] [Google Scholar]
- Nagano N.; Orengo C. A.; Thornton J. M. One Fold with Many Functions: The Evolutaionary Relationships Between TIM Barrel Families Based on their Sequences, Structures and Functions. J. Mol. Biol. 2002, 321, 741–765. 10.1016/S0022-2836(02)00649-6. [DOI] [PubMed] [Google Scholar]
- Sterner R.; Höcker B. Catalytic Versatility, Stability, and Evolution of the (βα)8-Barrel Enzyme Fold. Chem. Rev. 2005, 105, 4038–4055. 10.1021/cr030191z. [DOI] [PubMed] [Google Scholar]
- Jansen R.; Gerstein M. Analysis of the Yeast Transcriptome with Structural and Functional Categories: Characterizing Highly Expressed Proteins. Nucleic Acids Res. 2000, 28, 1481–1488. 10.1093/nar/28.6.1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malabanan M. M.; Amyes T. L.; Richard J. P. A Role for Flexible Loops in Enzyme Catalysis. Curr. Opin. Struct. Biol. 2010, 20, 702–710. 10.1016/j.sbi.2010.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkarni Y. S.; Liao Q.; Byléhn F.; Amyes T. L.; Richard J. P.; Kamerlin S. C. L. The Role of Ligand-Driven Conformational Changes in Enzyme Catalysis: Modeling the Reactivity of the Catalytic Cage of Triosephosphate Isomerase. J. Am. Chem. Soc. 2018, 140, 3854–3857. 10.1021/jacs.8b00251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown F. K.; Kollman P. A. Molecular Dynamics Simulations of “Loop Closing” in the Enzyme Triose Phosphate Isomerase. J. Mol. Biol. 1987, 198, 533–546. 10.1016/0022-2836(87)90298-1. [DOI] [PubMed] [Google Scholar]
- Joseph D.; Petsko G.; Karplus M. Anatomy of a Conformational Change: Hinged “Lid” Motion of the Triosephosphate Isomerase Loop. Science 1990, 249, 1425–1428. 10.1126/science.2402636. [DOI] [PubMed] [Google Scholar]
- Williams J. C.; McDermott A. E. Dynamics of the Flexible Loop of Triose-Phosphate Isomerase: The Loop Motion is Not Ligand Gated. Biochemistry 1995, 34, 8309–8319. 10.1021/bi00026a012. [DOI] [PubMed] [Google Scholar]
- Rozovsky S.; Jogl G.; Tong L.; McDermott A. E. Solution-State NMR Investigations of Triosephosphate Isomerase Active Site Loop Motion: Ligand Release in Relation to Active Site Loop Dynamics. J. Mol. Biol. 2001, 310, 271–280. 10.1006/jmbi.2001.4673. [DOI] [PubMed] [Google Scholar]
- Kempf J. G.; Jung J.-Y.; Ragain C.; Sampson N. S.; Loria J. P. Dynamic Requirements for a Functional Protein Hinge. J. Mol. Biol. 2007, 368, 131–149. 10.1016/j.jmb.2007.01.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wierenga R. K.; Kapetaniou E. G.; Venkatesan R. Triosephosphate Isomerase: A Highly Evolved Biocatalyst. Cell. Mol. Life Sci. 2010, 67, 3961–3982. 10.1007/s00018-010-0473-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Q.; Kulkarni Y.; Sengupta U.; Petrović D.; Mulholland A. J.; van der Kamp M. W.; Strodel B.; Kamerlin S. C. L. Loop Motion in Triosephosphate Isomerase Is Not a Simple Open and Shut Case. J. Am. Chem. Soc. 2018, 140, 15889–15903. 10.1021/jacs.8b09378. [DOI] [PubMed] [Google Scholar]
- Chodera J. D.; Noé F. Markov State Models of Biomolecular Conformational Dynamics. Curr. Opin. Struct. Biol. 2014, 25, 135–144. 10.1016/j.sbi.2014.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla D.; Hernández C. X.; Weber J. K.; Pande V. S. Markov State Models Provide Insights into Dynamic Modulation of Protein Function. Acc. Chem. Res. 2015, 48, 414–422. 10.1021/ar5002999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittier S. K.; Hengge A. C.; Loria J. P. Conformational Motions Regulate Phosphoryl Transfer in Related Protein Tyrosine Phosphatases. Science 2013, 341, 899–903. 10.1126/science.1241735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandao T. A. S.; Hengge A. C.; Johnson S. J. Insights into the Reaction of Protein Tyrosine Phosphatase 1B. Crystal Structures for Transition-State Analogs of Both Catalytic Steps. J. Biol. Chem. 2010, 285, 15874–15883. 10.1074/jbc.M109.066951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keedy D. A.; Hill Z. B.; Biel J. T.; Kang E.; Rettenmaier T. J.; Brandão-Neto T. S.; Pearce N. M.; von Delft F.; Wells J. A.; Fraser J. S. An Expanded Allosteric Network in PTP1B by Multitemperature Crystallography, Fragment Screening, and Covalent Tethering. eLife 2018, 7, e36307. 10.7554/eLife.36307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiesmann C.; Barr K. J.; Kung J.; Zhu J.; Erlanson D. A.; Shen W.; Fahr B. J.; Zhong M.; Taylor L.; Randal M.; McDowell R. S.; Hansen S. K. Allosteric Inhibition of Protein Tyrosine Phosphatase 1B. Nat. Struct. Mol. Biol. 2004, 11, 730–737. 10.1038/nsmb803. [DOI] [PubMed] [Google Scholar]
- Peters G. H.; Iversen L. F.; Branner S.; Andersen H. S.; Mortensen S. B.; Hvilsted Olsen O.; Møller K. B.; Hundahl Møller N. P. Residue 259 Is a Key Determinant of Substrate Specificity of Protein-tyrosine Phosphatases 1B and α. J. Biol. Chem. 2000, 275, 18201–18209. 10.1074/jbc.M910273199. [DOI] [PubMed] [Google Scholar]
- Gunawardana J.; Chan F. C.; Telenius A.; Woolcock B.; Kridel R.; Tan K. L.; Ben-Neriah S.; Mottok A.; Lim R. S.; Boyle M.; Rogic S.; Rimsza L. M.; Guiter C.; Leroy K.; Gaulard P.; Haioun C.; Marra M. A.; Savage K. J.; Connors J. M.; Shah S. P.; Gascoyne R. D.; Steidl C. Recurrent Somatic Mutations of PTPN1 in Primary Mediastinal B Cell Lymphoma and Hodgkin Lymphoma. Nat. Genet. 2014, 46, 329–335. 10.1038/ng.2900. [DOI] [PubMed] [Google Scholar]
- Choy M. S.; Li Y.; Machado L. E. S. F.; Kunze M. B. A.; Connors C. R.; Wei X.; Lindorff-Larsen K.; Page R.; Peti W. Conformational Rigidity and Protein Dynamics at Distinct Timescales Regulate PTP1B Activity and Allostery. Mol. Cell 2017, 65, 644–658. 10.1016/j.molcel.2017.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui D. S.; Beaumont V.; Ginther P. S.; Lipchock J. M.; Loria J. P. Leveraging Reciprocity to Identify and Characterize Unknown Allosteric Sites in Protein Tyrosine Phosphatases. J. Mol. Biol. 2017, 429, 2360–2372. 10.1016/j.jmb.2017.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moise G.; Morales Y.; Beaumont V.; Caradonna T.; Loria J. P.; Johnson S. J.; Hengge A. C. A YopH PTP1B Chimera Shows the Importance of the WPD-Loop Sequence to the Activity, Structure, and Dynamics of Protein Tyrosine Phosphatases. Biochemistry 2018, 57, 5315–5326. 10.1021/acs.biochem.8b00663. [DOI] [PubMed] [Google Scholar]
- Marsh-Armstrong B.; Fajnzylber J. M.; Korntner S.; Plaman B. A.; Bishop A. C. The Allosteric Site on SHP2’s Protein Tyrosine Phosphatase Domain is Targetable With Druglike Small Molecules. ACS Omega 2018, 3, 15763–15770. 10.1021/acsomega.8b02200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pádua R. A. P.; Sun Y.; Marko I.; Pitsawong W.; Stiller J. B.; Otten R.; Kern D. Mechanism of Activating Mutaions and Allosteric Drug Inhibition of the Phosphatase SHP2. Nat. Commun. 2018, 9, 4507. 10.1038/s41467-018-06814-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hjortness M. K.; Riccardi L.; Hongdusit A.; Zwart P. H.; Sankaran B.; de Vivo M.; Fox J. M. Evolutionarily Conserved Alloseric Communication in Protein Tyrosine Phosphatases. Biochemistry 2018, 57, 6443–6451. 10.1021/acs.biochem.8b00656. [DOI] [PubMed] [Google Scholar]
- Du J.; Say R. F.; Lü W.; Fuchs G.; Einsle O. Active-Site Remodelling in the Bifunctional Fructose-1,6-Bisphosphate Aldolase/Phosphatase. Nature 2011, 478, 534–537. 10.1038/nature10458. [DOI] [PubMed] [Google Scholar]
- Fushinobu S.; Nishimasu H.; Hattori D.; Song H.-J.; Wakagi T. Structural Basis for the Bifunctionality of Fructose-1,6-Bisphosphate Aldolase/Phosphatase. Nature 2011, 478, 538–541. 10.1038/nature10457. [DOI] [PubMed] [Google Scholar]
- Miles E. W.; Rhee S.; Davies D. R. The Molecular Basis of Substrate Channeling. J. Biol. Chem. 1999, 274, 12193–12196. 10.1074/jbc.274.18.12193. [DOI] [PubMed] [Google Scholar]
- Wheeldon I.; Minteer S. D.; Banta S.; Barton S. C.; Atanassov P.; Sigman M. Substrate Channeling as an Approach to Cascade Reactions. Nat. Chem. 2016, 8, 299–309. 10.1038/nchem.2459. [DOI] [PubMed] [Google Scholar]
- Hiblot J.; Gotthard G.; Elias M.; Chabriere E. Differential Active Site Loop Conformations Mediate Promiscuous Activities in the Lactonase SsoPox. PLoS One 2013, 8, e75272. 10.1371/journal.pone.0075272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaha-Nelson D.; Krüger D. M.; Szeler K.; Ben-David M.; Kamerlin S. C. L. Active Site Hydrophobicity and the Convergent Evolution of Paraoxonase Activity in Structurally Divergent Enzymes: The Case of Serum Paraoxonase 1. J. Am. Chem. Soc. 2017, 139, 1155–1167. 10.1021/jacs.6b10801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purg M.; Elias M.; Kamerlin S. C. L. Similar Active Sites and Mechanisms Do Not Lead to Cross-Promiscuity in Organophosphate Hydrolysis: Implications for Biotherapeutic Engineering. J. Am. Chem. Soc. 2017, 139, 17533–17546. 10.1021/jacs.7b09384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou T.; Risso V. A.; Gavira J. A.; Sanchez-Ruiz J. M.; Ozkan S. B. Evolution of Conformational Dynamics Determines the Conversion of a Promiscuous Generalist into a Specialist Enzyme. Mol. Biol. Evol. 2015, 32, 132–143. 10.1093/molbev/msu281. [DOI] [PubMed] [Google Scholar]
- Hart K. M.; Ho C. M. W.; Dutta S.; Gross M. L.; Bowman G. R. Modelling Proteins’ Hidden Conformations to Predict Antibiotic Resistance. Nat. Commun. 2016, 7, 12965. 10.1038/ncomms12965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortina G. A.; Kasson P. M. Excess Positional Mutual Information Predicts both Local and Allosteric Mutations Affecting Beta Lactamase Drug Resistance. Bioinformatics 2016, 32, 3420–3427. 10.1093/bioinformatics/btw492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortina G. A.; Hays J. M.; Kasson P. M. Conformational Intermediate that Controls KPC-2 Catalysis and Beta Lactam Drug Resistance. ACS Catal. 2018, 8, 2741–2747. 10.1021/acscatal.7b03832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baier F.; Hong N.; Yang G.; Pabis A.; Miton C. M.; Barrozo A.; Carr P. D.; Kamerlin S. C. L.; Jackson C. J.; Tokuriki N. Cryptic Genetic Variation Shapes the Adaptive Evolutionary Potential of Enzymes. eLife 2019, 8, e40789. 10.7554/eLife.40789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maria-Solano M. A.; Iglesias-Fernández J.; Osuna S. Deciphering the Allosterically Driven Conformational Ensemble in Tryptophan Synthase Evolution. J. Am. Chem. Soc. 2019, 141, 13049–13056. 10.1021/jacs.9b03646. [DOI] [PubMed] [Google Scholar]
- Miton C. M.; Jonas S.; Fischer R. S.; Duarte F.; Mohamed M. F.; van Loo B.; Kintses B.; Kamerlin S. C. L.; Tokuriki N.; Hyvönen M.; Hollfelder F. Evolutionary Repurposing of a Sulfatase: A New Michaelis Complex Leads to Efficient Transition State Charge Offset. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, E7293–E7302. 10.1073/pnas.1607817115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modi T.; Huihui J.; Ghosh K.; Ozkan S. B. Ancient Thioredoxins Evolved to Modern-Day Stability—Function Requiremen by Altering Native State Ensemble. Philos. Trans. R. Soc., B 2018, 373, 20170184. 10.1098/rstb.2017.0184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaksen G. V.; Åqvist J.; Brandsdal B. O. Protein Surface Softness is the Origin of Enzyme Cold-Adaptation of Trypsin. PLoS Comput. Biol. 2014, 10, e1003813. 10.1371/journal.pcbi.1003813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saavedra H. G.; Wrabl J. O.; Anderson J. A.; Li J.-K.; Hilser V. J. Dynamic Allosery can Drive Cold Adaptation in Enzymes. Nature 2018, 558, 324–328. 10.1038/s41586-018-0183-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitney D. S.; Volkman B. F.; Prehoda K. E. Evolution of a Protein Interaction Domain Family by Tuning Conformational Flexibility. J. Am. Chem. Soc. 2016, 138, 15150–15156. 10.1021/jacs.6b05954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gobeil S. M. C.; Clouthier C. M.; Park J.; Gagné D.; Berghuis A. M.; Doucet N.; Pelletier J. N. Maintenance of Native-Like Protein Dynamics May Not Be Required for Engineering Functional Proteins. Chem. Biol. 2014, 21, 1330–1340. 10.1016/j.chembiol.2014.07.016. [DOI] [PubMed] [Google Scholar]
- Jiang L.; Althoff E. A.; Clemente F. R.; Doyle L.; Röthlisberger D.; Zanghellini A.; Gallaher J. L.; Betker J. L.; Tanaka F.; Barbas C. F. III; Hilvert D.; Houk K. N.; Stoddard B. L.; Baker D. De Novo Computational Design of Retro-Aldol Enzymes. Science 2008, 319, 1387–1391. 10.1126/science.1152692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Althoff E. A.; Wang L.; Jiang L.; Giger L.; Lassila J. K.; Wang Z.; Smith M.; Hari S.; Kast P.; Herschlag D.; Hilvert D.; Baker D. Robust Design and Optimization of Retroaldol Enzymes. Protein Sci. 2012, 21, 717–726. 10.1002/pro.2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giger L.; Caner S.; Obexer R.; Kast P.; Baker D.; Ban N.; Hilvert D. Evolution of a Designed Retro-Aldolase Leads to Complete Active Site Remodeling. Nat. Chem. Biol. 2013, 9, 494–498. 10.1038/nchembio.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obexer R.; Godina A.; Garrabou X.; Mittl P. R. E.; Baker D.; Griffiths A. D.; Hilvert D. Emergence of a Catalytic Tetrad During Evolution of a Highly Active Artificial Aldolase. Nat. Chem. 2017, 9, 50–56. 10.1038/nchem.2596. [DOI] [PubMed] [Google Scholar]
- Zeymer C.; Zschoche R.; Hilvert D. Optimization of Enzyme Mechanism along the Evolutionary Trajectory of a Computationally Designed (Retro-)Aldolase. J. Am. Chem. Soc. 2017, 139, 12541–12549. 10.1021/jacs.7b05796. [DOI] [PubMed] [Google Scholar]
- Schafer J. W.; Zoi I.; Antoniou D.; Schwartz S. D. Optimization of the Turnover in Artificial Enzymes via Directed Evolution Results in the Coupling of Protein Dynamics to Chemistry. J. Am. Chem. Soc. 2019, 141, 10431–10439. 10.1021/jacs.9b04515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trudeau D. L.; Tawfik D. S. Protein Engineers Turned Evolutionists—the Quest for the Optimal Starting Point. Curr. Opin. Biotechnol. 2019, 60, 46–52. 10.1016/j.copbio.2018.12.002. [DOI] [PubMed] [Google Scholar]
- Röthlisberger D.; Khersonsky O.; Wollacott A. M.; Jiang L.; DeChancie J.; Betker J.; Gallaher J. L.; Althoff E. A.; Zanghellini A.; Dym O.; Albeck S.; Houk K. N.; Tawfik D. S.; Baker D. Kemp Elimination Catalysts by Computational Enzyme Design. Nature 2008, 453 (7192), 190–195. 10.1038/nature06879. [DOI] [PubMed] [Google Scholar]
- Khersonsky O.; Röthlisberger D.; Dym O.; Albeck S.; Jackson C. J.; Baker D.; Tawfik D. S. Evolutionary Optimization of Computationally Designed Enzymes: Kemp Eliminases of the KE07 Series. J. Mol. Biol. 2010, 396, 1025–1042. 10.1016/j.jmb.2009.12.031. [DOI] [PubMed] [Google Scholar]
- Labas A.; Szabo E.; Mones L.; Fuxreiter M. Optimization of Reorganization Energy Drives Evolution of the Designed Kemp Eliminase KE07. Biochim. Biophys. Acta, Proteins Proteomics 2013, 1834, 908–917. 10.1016/j.bbapap.2013.01.005. [DOI] [PubMed] [Google Scholar]
- Bhowmick A.; Sharma S. C.; Honma H.; Head-Gordon T. The Role of Side Chain Entropy and Mutual Information for Improving the De Novo Design of Kemp Eliminases KE07 and KE70. Phys. Chem. Chem. Phys. 2016, 18, 19386–19396. 10.1039/C6CP03622H. [DOI] [PubMed] [Google Scholar]
- Jindal G.; Ramachandran B.; Bora R. P.; Warshel A. Exploring the Development of Ground-State Destabilization and Transition State Stabilization in Two Directed Evolution Paths of Kemp Eliminases. ACS Catal. 2017, 7, 3301–3305. 10.1021/acscatal.7b00171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhowmick A.; Sharma S. C.; Head-Gordon T. The Importance of the Scaffold for de Novo Enzymes: A Case Study with Kemp Eliminase. J. Am. Chem. Soc. 2017, 139, 5793–5800. 10.1021/jacs.6b12265. [DOI] [PubMed] [Google Scholar]
- Blomberg R.; Kries H.; Pinkas D. M.; Mittl P. R. E.; Grütter M. G.; Privett H. K.; Mayo S. L.; Hilvert D. Precision is Essential for Efficient Catalysis in an Evolved Kemp Eliminase. Nature 2013, 503, 418–421. 10.1038/nature12623. [DOI] [PubMed] [Google Scholar]
- Risso V. A.; Martinez-Rodriguez S.; Candel A. M.; Krüger D. M.; Pantoja-Uceda D.; Ortega-Muñoz M.; Santoyo-Gonzalez F.; Gaucher E. A.; Kamerlin S. C. L.; Bruix M.; Gavira J. A.; Sanchez-Ruiz J. M. De Novo Active Sites for Resurrected Precambrian Enzymes. Nat. Commun. 2017, 8, 16113. 10.1038/ncomms16113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar-Even A.; Noor E.; Savir Y.; Liebermeister W.; Davidi D.; Tawfik D. S.; Milo R. The Moderately Efficient Enzyme: Evolutionary and Physicochemical Trends Shaping Enzyme Parameters. Biochemistry 2011, 50, 4402–4410. 10.1021/bi2002289. [DOI] [PubMed] [Google Scholar]
- Park H. S.; Nam S. H.; Lee J. K.; Yoon C. N.; Mannervik B.; Benkovic S. J.; Kim H. S. Design and Evolution of New Catalytic Activity with an Existing Protein Scaffold. Science 2006, 311, 535–538. 10.1126/science.1118953. [DOI] [PubMed] [Google Scholar]
- Cheng F.; Yang J.; Bocola M.; Schwaneberg U.; Zhu L. Loop Engineering Reveals the Importance of Active-Site-Decorating Loops and Gating Residue in Substrate Affinity Modulation of Arginine Deaminase (an Anti-Tumor Enzyme). Biochem. Biophys. Res. Commun. 2018, 499, 233–238. 10.1016/j.bbrc.2018.03.134. [DOI] [PubMed] [Google Scholar]
- Otten R.; Liu L.; Kenner L. R.; Clarkson M. W.; Mavor D.; Tawfik D. S.; Kern D.; Fraser J. S. Rescue of Conformational Dynamics in Enzyme Catalysis by Directed Evolution. Nat. Commun. 2018, 9, 1314. 10.1038/s41467-018-03562-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B.; Qu G.; Li J.-K.; Fan W.; Ma J.-A.; Xu Y.; Nie Y.; Sun Z. Conformational Dynamics-Guided Loop Engineering of an Alcohol Dehydrogenase: Capture, Turnover and Enantioselective Transformation of Difficult-to-Reduce Ketones. Adv. Synth. Catal. 2019, 361, 3182–3190. 10.1002/adsc.201900249. [DOI] [Google Scholar]
- Boehr D. D.; D’Amico R. N.; O’Rourke K. F. Engineered Control of Enzyme Structural Dynamics and Function. Protein Sci. 2018, 27, 825–838. 10.1002/pro.3379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer A. G. III Enzyme Dynamics from NMR Spectroscopy. Acc. Chem. Res. 2015, 48, 457–465. 10.1021/ar500340a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazal H.; Haran G. Single-Molecular FRET Methods to Study the Dynamics of Proteins at Work. Curr. Opin. Biomed. Eng. 2019, 12, 8–17. 10.1016/j.cobme.2019.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yengo C. M.; Berger C. L. Fluorescence Anisotropy and Resonance Energy Transfer: Powerful Tools for Measuring Real Time Protein Dynamics in a Physiological Environment. Curr. Opin. Pharmacol. 2010, 10, 731–737. 10.1016/j.coph.2010.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J. G.; Kim T. W.; Kim J.; Ihee H. Protein Structural Dynamics Revealed by Time-Resolved X-ray Solution Scattering. Acc. Chem. Res. 2015, 48, 2200–2208. 10.1021/acs.accounts.5b00198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keedy D. A.; Kenner L. R.; Warkentin M.; Woldeyes R. A.; Hopkins J. B.; Thompson M. C.; Brewster A. S.; van Benschoten A. H.; Baxter E. L.; Uervirojnangkoorn U.; McPhillips S.; Song J.; Alonso-Mori R.; Holton J. M.; Weis W. I.; Brunger A. T.; Soltis S. M.; Lemke H.; Gonzalez A.; Sauter N. K.; Cohen A. E.; van den Bedem H.; Thorne R. E.; Fraser J. S. Mapping the Conformational Landscape of a Dynamic Enzyme by Multitemperature and XFEL Crystallography. eLife 2015, 4, e07574. 10.7554/eLife.07574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konermann L.; Pan J.; Liu Y.-H. Hydrogen Exchange Mass Spectrometry for Studying Protein Structure and Dynamics. Chem. Soc. Rev. 2011, 40, 1224–1234. 10.1039/C0CS00113A. [DOI] [PubMed] [Google Scholar]
- Henzler-Wildman K.; Kern D. Dynamic Personalities of Proteins. Nature 2007, 450, 964–972. 10.1038/nature06522. [DOI] [PubMed] [Google Scholar]
- Pabis A.; Risso V. A.; Sanchez-Ruiz J. M.; Kamerlin S. C. L. Cooperativity and Flexibility in Enzyme Evolution. Curr. Opin. Struct. Biol. 2018, 48, 83–92. 10.1016/j.sbi.2017.10.020. [DOI] [PubMed] [Google Scholar]
- Cadet F.; Fontaine N.; Li G.; Sanchis J.; Ng Fuk Chong M.; Pandjaitan R.; Vetrivel I.; Offmann B.; Reetz M. T. A Machine Learning Approach for Reliable Prediction of Amino Acid Interactions and its Application in the Directed Evolution of Enantioselective Enzymes. Sci. Rep. 2018, 8, 16757. 10.1038/s41598-018-35033-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z.; Kan S. B. J.; Lewis R. D.; Wittmann B. J.; Arnold F. H. Machine Learning-Assisted Directed Protein Evolution with Combinatorial Libraries. Proc. Natl. Acad. Sci. U. S. A. 2019, 116, 8852–8858. 10.1073/pnas.1901979116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G.; Dong Y.; Reetz M. T. Can Machine Learning Revolutionize Directed Evolution of Selective Enzymes?. Adv. Synth. Catal. 2019, 361, 2377–2386. 10.1002/adsc.201900149. [DOI] [Google Scholar]
- Mazurenko S.; Prokop Z.; Damborsky J. Machine Learning in Enzyme Engineering. ACS Catal. 2020, 10, 1210–1223. 10.1021/acscatal.9b04321. [DOI] [Google Scholar]
- Kamisetty H.; Ovchinnikov S.; Baker D. Assessing the Utility of Coevolution-Based Residue-Residue Contact Predictions in a Sequence- and Structure-Rich Era. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 15674–15679. 10.1073/pnas.1314045110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ovchinnikov S.; Kamisetty H.; Baker D. Robust and Accurate Prediction of Residue–Residue Interactions Across Protein Interfaces Using Evolutionary Information. eLife 2014, 3, e02030. 10.7554/eLife.02030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ovchinnikov S.; Kinch L.; Park H.; Liao Y.; Pei J.; Kim D. E.; Kamisetty H.; Grishin N. V.; Baker D. Large-Scale Determination of Previously Unsolved Protein Structures Using Evolutionary Information. eLife 2015, 4, e09248. 10.7554/eLife.09248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M. D.; Warner E. A.; Morrissey C. E.; Fick C. W.; Wu T. S.; Ornelas M. Y.; Ochoa G. V.; Zhang B. S.; Rathbun C. M.; Porterfield W. B.; Prescher J. A.; Leconte A. M. Statistical Coupling Analysis-Guided Library Design for the Discovery of Mutant Luciferases. Biochemistry 2018, 57, 663–671. 10.1021/acs.biochem.7b01014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campeotto I.; Goldenzweig A.; Davey J.; Barfod L.; Marshall J. M.; Silk S. E.; Wright K. E.; Draper S. J.; Higgins M. K.; Fleishman S. J. One-Step Design of a Stable Variant of the Malaria Invasion Protein RH5 for Use as a Vaccine Immunogen. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, 998–1002. 10.1073/pnas.1616903114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malladi S. K.; Schreiber D.; Pramanick I.; Sridevi M. A.; Goldenzweig A.; Dutta S.; Fleishman S. J.; Varadarajan R. One-Step Sequence and Strucure-Guided Optimization of HIV-1 Envelope GP140. Curr. Res. Struct. Biol. 2020, 2, 45–55. 10.1016/j.crstbi.2020.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiménez-Osés G.; Osuna S.; Gao X.; Sawaya M. R.; Gilson L.; Collier S. J.; Huisman G. W.; Yeates T. O.; Tang Y.; Houk K. N. The Role of Distant Mutations and Allosteric Regulation on LovD Active Site Dynamics. Nat. Chem. Biol. 2014, 10, 431–436. 10.1038/nchembio.1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodani S. C.; Kiss G.; Cahn J. K. B.; Su Y.; Pande V. S.; Arnold F. H. Discovery of a Regioselectivity Switch in Nitrating P450s Guided by Molecular Dynamics Simulations and Markos Models. Nat. Chem. 2016, 8, 419–425. 10.1038/nchem.2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrović D.; Frank D.; Kamerlin S. C. L.; Hoffmann K.; Strodel B. Shuffling Active Site Substate Populations Affects Catalytic Activity: The Case of Glucose Oxidase. ACS Catal. 2017, 7, 6188–6197. 10.1021/acscatal.7b01575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sultan M. M.; Kiss G.; Pande V. S. Towards Simple Kinetic Models of Functional Dynamics in a Kinase Subfamily. Nat. Chem. 2018, 10, 903–909. 10.1038/s41557-018-0077-9. [DOI] [PubMed] [Google Scholar]
- Petrović D.; Bokel A.; Allan M.; Urlacher V. B.; Strodel B. Simulation-Guided Design of Cytochrome P450 for Chemo- and Regioselective Macrocyclic Oxidation. J. Chem. Inf. Model. 2018, 58, 848–858. 10.1021/acs.jcim.8b00043. [DOI] [PubMed] [Google Scholar]
- Senn H. M.; Thiel W. QM/MM Methods for Biomolecular Systems. Angew. Chem., Int. Ed. 2009, 48, 1198–1229. 10.1002/anie.200802019. [DOI] [PubMed] [Google Scholar]
- van der Kamp M. W.; Mulholland A. J. Combined Quantum Mechanics/Molecular Mechanics (QM/MM) Methods in Computational Enzymology. Biochemistry 2013, 52, 2708–2728. 10.1021/bi400215w. [DOI] [PubMed] [Google Scholar]
- de Luca F.; Benvenuti M.; Carboni F.; Pozzi C.; Rossolini G. M.; Mangani S.; Docquier J.-D. Evolution to Carbapenem-Hydrolyzing Activity in Noncarbapenemase Class D β-Lactamase OXA-10 by Rational Protein Design. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 18424–18429. 10.1073/pnas.1110530108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce L. C. T.; Salomon-Ferrer R.; de Oliveira C. A. F.; McCammon J. A.; Walker R. C. Routine Access to Millisecond Time Scale Events with Accelerated Molecular Dynamics. J. Chem. Theory Comput. 2012, 8, 2997–3002. 10.1021/ct300284c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao Y.; Feher V. A.; McCammon J. A. Gaussian Accelerated Molecular Dynamics: Unconstrained Enhanced Sampling and Free Energy Calculation. J. Chem. Theory Comput. 2015, 11, 3584–3595. 10.1021/acs.jctc.5b00436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsujishita H.; Moriguchi I.; Hirono S. Potential-Scaled Molecular Dynamics and Potential Annealing: Effective Conformational Search Techniques for Biomolecules. J. Phys. Chem. 1993, 97, 4416–4420. 10.1021/j100119a028. [DOI] [Google Scholar]
- Sugita Y.; Okamoto Y. Replica-Exchange Molecular Dynamics Method for Protein Folding. Chem. Phys. Lett. 1999, 314, 141–151. 10.1016/S0009-2614(99)01123-9. [DOI] [Google Scholar]
- Wang L.; Friesner R. A.; Berne B. J. Replica Exchange With Solute Scaling: A More Efficient Version of Replica Exchange With Solute Tempering (REST2). J. Phys. Chem. B 2011, 115, 9431–9438. 10.1021/jp204407d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laio A.; Parrinello M. Escaping Free-Energy Minima. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 12562–12566. 10.1073/pnas.202427399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H.; Schulten K. Steered Molecular Dynamics Simulations of Force-Induced Protein Domain Unfolding. Proteins: Struct., Funct., Genet. 1999, 35, 453–463. 10.1002/(SICI)1097-0134(19990601)35:4<453::AID-PROT9>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- Torrie G. M.; Valleau J. P. Nonphysical Sampling Distributions in Monte Carlo Free-energy Estimation: Umbrella Sampling. J. Comput. Phys. 1977, 23, 187–199. 10.1016/0021-9991(77)90121-8. [DOI] [Google Scholar]
- Comer J.; Gumbart J. C.; Hénin J.; Lelièvre T.; Pohorille A.; Chipot C. The Adaptive Biasing Force Method: Everything You Always Wanted To Know but Were Afraid To Ask. J. Phys. Chem. B 2015, 119, 1129–1151. 10.1021/jp506633n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michielssens S.; Henning Peters J.; Ban D.; Pratihar S.; Seeliger D.; Sharma M.; Giller K.; Sabo T. M.; Becker S.; Lee D.; Griesinger C.; de Groot B. L. A Designed Conformational Shift to Control Protein Binding Specificity. Angew. Chem., Int. Ed. 2014, 53, 10367–10371. 10.1002/anie.201403102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowerman S.; Wereszczynski J. Detecting Allosteric Networks Using Molecular Dynamics Simulation. Methods Enzymol. 2016, 578, 429–447. 10.1016/bs.mie.2016.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrović D.; Kamerlin S. C. L. Molecular Modeling of Conformational Dynamics and its Role in Enzyme Evolution. Curr. Opin. Struct. Biol. 2018, 52, 50–57. 10.1016/j.sbi.2018.08.004. [DOI] [PubMed] [Google Scholar]
- Liang Z.; Verkhivker G. M.; Hu G. Integration of Network Models and Evoluationary Analysis into High-Throughput Modeling of Protein Dynamics and Allosteric Regulation: Theory, Tools and Applications. Briefings Bioinf. 2020, 21, 815–835. 10.1093/bib/bbz029. [DOI] [PubMed] [Google Scholar]
- van Wart A. T.; Durrant J.; Votapka L.; Amaro R. E. Weighted Implementation of Suboptimal Paths (WISP): An Optimized Algorithm and Tool for Dynamical Network Analysis. J. Chem. Theory Comput. 2014, 10, 511–517. 10.1021/ct4008603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sethi A.; Eargle J.; Black A. A.; Luthey-Schulten Z. Dynamical Networks in tRNA:Protein Complexes. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 6620–6625. 10.1073/pnas.0810961106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh S.; Bowman G. R. Quantifying Allosteric Communication via Both Concerted Structural Changes and Conformational Disorder with CARDS. J. Chem. Theory Comput. 2017, 13, 1509–1517. 10.1021/acs.jctc.6b01181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerek Z. N.; Kumar S.; Ozkan S. B. Structural Dynamics Flexibility Informs Function and Evolution at a Proteome Scale. Evol. Appl. 2013, 6, 423–433. 10.1111/eva.12052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Ribeiro J. M. L.; Tiwary P. Machine Learning Approaches for Analyzing and Enhancing Molecular Dynamics Simulations. Curr. Opin. Struct. Biol. 2020, 61, 139–145. 10.1016/j.sbi.2019.12.016. [DOI] [PubMed] [Google Scholar]
- Latallo M. J.; Cortina G. A.; Faham S.; Nakamoto R. K.; Kasson P. M. Predicting Allosteric Mutants that Increase Activity of a Major Antibiotic Resistance Enzyme. Chem. Sci. 2017, 8, 6484–6492. 10.1039/C7SC02676E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velez Rueda A. J.; Palopoli N.; Zacarías M.; Sommese L. M.; Parisi G. ProtMiscuity: A Database of Promiscuous Proteins. Database 2019, 2019, baz103. 10.1093/database/baz103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amrein B. A.; Steffen-Munsberg F.; Szeler I.; Purg M.; Kulkarni Y.; Kamerlin S. C. L. CADEE: Computer-Aided Directed Evolution of Enzymes. IUCrJ 2017, 4, 50–64. 10.1107/S2052252516018017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davey J. A.; Chica R. A. Multistate Approaches in Computational Protein Design. Protein Sci. 2012, 21, 1241–1252. 10.1002/pro.2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiss G.; Çelebi-Ölçüm N.; Moretti R.; Baker D.; Houk K. N. Computational Enzyme Design. Angew. Chem., Int. Ed. 2013, 52, 5700–5725. 10.1002/anie.201204077. [DOI] [PubMed] [Google Scholar]
- Koenig P.; Lee C. V.; Walters B. T.; Janakiraman V.; Stinson J.; Patapoff T. W.; Fuh G. Mutational Landscape of Antibody Variable Domains Reveals a Switch Modulating the Interdomain Conformational Dynamics and Antigen Binding. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, E486–E495. 10.1073/pnas.1613231114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warszawski S.; Borenstein Katz A.; Lipsh R.; Khmelnitsky L.; Ben Nissan G.; Javitt G.; Dym O.; Unger T.; Knop O.; Albeck S.; Diskin R.; Fass D.; Sharon M.; Fleishman S. J. Optimizing Antibody Affinity and Stability by the Automated Design of the Variable Light-Heavy Chain Interfaces. PLoS Comput. Biol. 2019, 15, e1007207. 10.1371/journal.pcbi.1007207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aalbers F. S.; Fürst M. J. L. J.; Rovida S.; Trajkovic M.; Gómez Castellanos J. R.; Bartsch S.; Vogel A.; Mattevi A.; Fraaije M. W. Approaching Boiling Point Stability of an Alcohol Dehydrogenase Through Computationally Guided Engineering. eLife 2020, 9, e54639. 10.7554/eLife.54639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel J. B.; Zanghellini A.; Lovick H. M.; Kiss G.; Lambert A. R.; St.Clair J. L.; Gallaher J. L.; Hilvert D.; Gelb M. H.; Stoddard B. L.; Houk K. N.; Michael F. E.; Baker D. Computational Design of an Enzyme Catalyst for a Stereoselective Bimolecular Diles-Alder Reaction. Science 2010, 329, 309–313. 10.1126/science.1190239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korendovych I. V.; DeGrado W. F. De Novo Protein Design, A Restrospective. Q. Rev. Biophys. 2020, 53, e3. 10.1017/S0033583519000131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kries H.; Blomberg R.; Hilvert D. De Novo Enzymes by Computational Design. Curr. Opin. Chem. Biol. 2013, 17, 221–228. 10.1016/j.cbpa.2013.02.012. [DOI] [PubMed] [Google Scholar]
- Tomatis P. E.; Rasia R. M.; Segovia L.; Vila A. J. Mimicking Natural Evolution in Metallo-β-Lactamases Through Second-Shell Ligand Mutations. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 13761–13766. 10.1073/pnas.0503495102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morley K. L.; Kazlauskas R. J. Improving Enzyme Properties: When Are Closer Mutations Better?. Trends Biotechnol. 2005, 23, 231–237. 10.1016/j.tibtech.2005.03.005. [DOI] [PubMed] [Google Scholar]
- El Omari K.; Liekens S.; Bird L. E.; Balzarini J.; Stammers D. K. Mutations Distal to the Substrate Site Can Affect Varicella Zoster Virus Thymidine Kinase Activity: Implications for Drug Design. Mol. Pharmacol. 2006, 69, 1891–1896. 10.1124/mol.106.023002. [DOI] [PubMed] [Google Scholar]
- Lee J.; Goodey N. M. Catalytic Contributions from Remote Regions of Enzyme Structure. Chem. Rev. 2011, 111, 7595–7624. 10.1021/cr100042n. [DOI] [PubMed] [Google Scholar]
- Yang G.; Hong N.; Baier F.; Jackson C. J.; Tokuriki N. Conformational Tinkering Drives Evolution of a Promiscuous Activity through Indirect Mutational Effects. Biochemistry 2016, 55, 4583–4593. 10.1021/acs.biochem.6b00561. [DOI] [PubMed] [Google Scholar]
- Wrenbeck E. E.; Azouz L. R.; Whitehead T. A. Single-Mutation Fitness Landscapes for an Enzyme on Multiple Substrates Reveal Specificity is Globally Encoded. Nat. Commun. 2017, 8, 15695. 10.1038/ncomms15695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold F. H. Directed Evolution: Bringing New Chemistry to Life. Angew. Chem., Int. Ed. 2018, 57, 4143–4148. 10.1002/anie.201708408. [DOI] [PMC free article] [PubMed] [Google Scholar]