

DSCAM regulates neuronal delamination from the ventricular surface by suppressing RapGEF2/Rap1 and N-cadherin.
Abstract
For normal neurogenesis and circuit formation, delamination of differentiating neurons from the proliferative zone must be precisely controlled; however, the regulatory mechanisms underlying cell attachment are poorly understood. Here, we show that Down syndrome cell adhesion molecule (DSCAM) controls neuronal delamination by local suppression of the RapGEF2–Rap1–N-cadherin cascade at the apical endfeet in the dorsal midbrain. Dscam transcripts were expressed in differentiating neurons, and DSCAM protein accumulated at the distal part of the apical endfeet. Cre-loxP–based neuronal labeling revealed that Dscam knockdown impaired endfeet detachment from ventricles. DSCAM associated with RapGEF2 to inactivate Rap1, whose activity is required for membrane localization of N-cadherin. Correspondingly, Dscam knockdown increased N-cadherin localization and ventricular attachment area at the endfeet. Furthermore, excessive endfeet attachment by Dscam knockdown was restored by co-knockdown of RapGEF2 or N-cadherin. Our findings shed light on the molecular mechanism that regulates a critical step in early neuronal development.
INTRODUCTION
During brain development, most nascent neurons detach their apical endfeet from the ventricular surface and initiate migration toward their final positions from their site of production (1). The impairment of neuronal migration has been implicated in human brain disorders, such as periventricular nodular heterotopia and autism spectrum disorder (2). Thus, normal neuronal migration is fundamental to the architectural formation and functional wiring of the nervous system.
Progenitor cells are located in the ventricular zone (VZ). The ventricular surface is joined by intercellular adhesions known as adherence junctions (AJs). In the dorsal midbrain, cell division giving rise to progenitors and/or neurons occurs only on the ventricular surface (3, 4). Therefore, nascent neurons need to detach their apical endfeet from the cadherin-based AJ belt (5–8), which is initiated by the down-regulation of key components of AJs, classical cadherin family between neighboring cells at the apical surface (7), followed by cytoskeletal disassembly for morphological changes (5, 9). Elevated expression of transcription factors such as FoxP (8) and Scratch1/2 (6) in nascent neurons has been reported to repress transcription of the cadherin family, promoting apical process detachment from the proliferative zone in the spinal cord (8) and developing cortex (6). These findings have clarified the importance of transcriptional repression of the cadherin family in delaminating cells (7). However, overexpression of Scratch1/2, as well as their upstream molecule Neurogenin2 (Ngn2, also called Neurog2), reduced E-cadherin, but not N-cadherin, in the developing cortex (6). N-cadherin is required for multiple developmental steps immediately following (or concurrently with) the delamination process (10–12). These observations suggest that mechanisms other than transcriptional suppression of N-cadherin account for the delamination or detachment of newborn neurons from the ventricular surface (6). It may be possible that the N-cadherin protein is localized or activated/inactivated posttranscriptionally in certain compartments of intracellular regions. However, the underlying mechanism of posttranscriptional down-regulation of N-cadherin at the apical endfeet remains unknown.
Down syndrome cell adhesion molecule (DSCAM) is a homophilic cell adhesion molecule in the immunoglobulin superfamily that is essential for multiple aspects of neuronal wiring in flies and vertebrates (13). Some of the reported functions of DSCAM, such as synaptic targeting, are consistent with homophilic cell adhesion. In the chick retina, Dscam and related adhesion molecules define distinct sublaminae and form synaptic contact based on the homophilic interactions in the synaptic inner plexiform layer (14). In contrast, other reported functions, such as self-avoidance or tiling, indicate a notable role for DSCAM in balancing cell attachment and separation (15). In mice retina, there is excessive adhesion and fasciculation between DSCAM-depleted soma and processes (15). These reports suggest that DSCAM functions in the separation/cancellation of allogeneic cell adhesion. Consistently, it has been reported that DSCAM masks the functions of some cell adhesion molecules, such as the cadherin family, contributing to self-avoidance and tiling (16). However, the underlying molecular mechanism of the antagonistic effect of DSCAM on cadherins remains unknown.
Here, we investigated the role of DSCAM proteins in neuronal development in the midbrain and discovered a key role for DSCAM in neuronal delamination of nascent neurons. Specifically, we found that DSCAM suppressed the RapGEF2/Rap1 pathway in nascent neurons attached to the ventricular surface, and this subsequently suppressed N-cadherin, which leads to proper delamination of neurons. This study demonstrates the molecular mechanism by which DSCAM and its signaling molecules suppress cell adhesion molecules in a cell-autonomous manner, and it provides insight into various biological events that cell adhesion molecules may control.
RESULTS
DSCAM modulates midbrain apoptosis
DSCAM ablation induces severe midbrain hypertrophy (fig. S1A) (17). To understand the functional roles of DSCAM in the dorsal midbrain, we examined the midbrain constitution in mice carrying a spontaneous mutation in Dscam (Dscamdel17/del17) with vGluT2-GFP or GAD-GFP alleles, in which excitatory or inhibitory neurons were labeled with green fluorescent protein (GFP). In Dscamdel17/del17 mice, prominent cell clustering of GFP-labeled excitatory and inhibitory neurons was observed in the superior colliculus (SC) and inferior colliculus (IC) of the dorsal midbrain on postnatal days (P) 30 (fig. S1, B and C). Cell clusters were observed as early as embryonic day (E) 18.5 (fig. S1D). In contrast, midbrain hypertrophy became prominent after birth. Next, we performed immunohistochemical analysis using an anti–cleaved caspase-3 antibody, which is a marker of apoptotic cells. At P0, when apoptosis peaks during dorsal midbrain development, cleaved caspase-3–positive cell numbers were significantly reduced in the mutant midbrain (fig. S1, E and F), consistent with reports in DSCAMdel17/del17 retina (18). These results suggest that DSCAM suppresses cell death in the developing dorsal midbrain, and loss of DSCAM function reduces apoptosis, leading to hypertrophy.
DSCAM is expressed at early neurogenesis in the dorsal midbrain
We investigated the temporal expression profiles of DSCAM in the midbrain during development by Western blot analysis (Fig. 1A). Mouse midbrain extracts were prepared from E11.5—the early stage of neurogenesis (4)—to the adult stage. DSCAM expression was observed at all stages examined, with particularly strong expression from E13.5 to P30. On E14.5, Dscam transcripts were detected in the intermediate zone (IZ), which included soma of immature neurons, but transcripts were barely observed in the marginal zone (MZ), a layer with fibrous structures, in the dorsal midbrain (Fig. 1B). Notably, we observed some cells in the VZ expressing DSCAM mRNA slightly (arrows in Fig. 1B).
Fig. 1. Expression and localization of DSCAM in the dorsal midbrain during early neurogenesis.
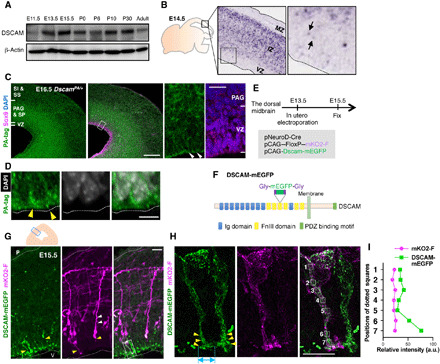
(A) Temporal expression profiles of DSCAM protein in the midbrain from E11.5 to adult stage (P90). (B) In situ hybridization analysis of a mouse embryo (GenePaint.org). Black arrows indicate weak signals in the VZ. (C) Immunohistochemical analyses using anti-PA tag and Sox9 antibodies. Scale bar, 200 μm. Areas surrounded by white dotted lines are shown at higher magnification on the right. White arrowheads indicate accumulation of DSCAM-PA signals. Scale bar, 20 μm. (D) DSCAM-PA accumulated at the apical ventricular surface (yellow arrowheads) just beneath DAPI-positive nuclei. Scale bar, 5 μm. (E) Experimental design. (F) Diagram of the DSCAM-mEGFP protein domain structure. (G) DSCAM-mEGFP and mKO2-F expression in the dorsal midbrain. Upper schematic depicts coronal section of dorsal midbrain. Small blue box indicates area showing fluorescence image. White and yellow arrowheads indicate basal and apical processes, respectively. Higher magnification in (H) represents area surrounded by dotted box in (G). P, pia; V, ventricle. Scale bar, 20 μm. (H) Enlarged image of mKO2-F–positive neurons abutting apical endfeet into the ventricular surface. Yellow arrowheads indicate the accumulation of DSCAM-mEGFP. Light blue double-headed arrow indicates size of widely spread palm-like structures at the endfeet tip. Numbered areas in the merged image correspond to the fluorescence intensity measurement position in (I). Scale bar, 10 μm. (I) Relative intensity of mKO2-F and DSCAM-mEGFP. a.u., arbitrary units.
Next, we investigated DSCAM protein localization using several anti-DSCAM antibodies. Upon comparison of immunostained samples between control and Dscam knockout (KO) mice (Dscamflox/flox; CreCAG), we observed high background staining in the developing midbrain, indicating that these antibodies did not work in the dorsal midbrain, probably due to unknown antigens similar to DSCAM (fig. S2, A to C). To investigate the localization of DSCAM in vivo, we generated a knock-in (KI) mouse allele using the CRISPR-Cas9 method to produce DSCAM-PA fusion protein, in which tandem PA tags were integrated into the intracellular region (DscamPA/+; fig. S3, A to D). DSCAM expression levels in whole brain lysates from wild-type and DscamPA/PA mice were similar (fig. S4A); the DSCAM-PA protein was primarily distributed on the membrane along with untagged DSCAM in COS-7 cells (fig. S5, A and C). The expression of DSCAM-PA protein was confirmed in DscamPA/+ embryos by Western blot analysis (fig. S3D), which was comparable to the distribution of Dscam transcripts (Fig. 1B), suggesting that these signals correctly reflect the distribution of endogenous wild-type DSCAM.
In the E16.5 dorsal midbrain, strong signals for DSCAM-PA were prominent in the intermediate area [comprising the periaqueductal gray (PAG) and strata profundum (SP)]. Prominent signals were also observed in the VZ and outer area [comprising the strata intermedium (SI) and superficiale (SS)] (Fig. 1, C and D, and fig. S3F) (4). Furthermore, DSCAM-PA signals prominently accumulated in the VZ (Figs. 1D and 4D and figs. S3F and S7, A and B). A dense array of granular DSCAM-PA labeling was observed beneath 4P,6-diamidino-2-phenylindole (DAPI)–labeled nuclei facing the ventricular surface (Figs. 1D and 4D).
Fig. 4. DSCAM associates with RapGEF2 and MAGI1c/β-catenin.
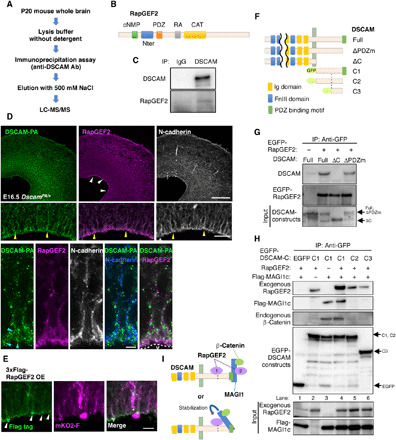
(A) Procedure for searching for DSCAM-binding molecules. (B) Diagram of the RapGEF2 protein domain structure. (C) Coimmunoprecipitation and Western blot assays using E15.5 brain and indicated antibodies. (D) Immunohistochemistry of RapGEF2 and N-cadherin in E16.5 DscamPA/+ midbrains. (Top) RapGEF2 localization was prominent at the ventricular surface (white arrowheads). Scale bar, 200 μm. (Middle) Higher-magnification image of the area enclosed by a dotted rectangle in the top-right panel. Yellow arrowheads indicate radially fibrous signals. V, ventricle. Scale bar, 10 μm. (Bottom) High-resolution images of thin sections. Light blue arrowheads indicate DSCAM accumulation. (E) Specific accumulation of 3xFlag-RapGEF2 was observed at the most distal part of the endfeet (white arrowheads) at E18.5. The outline of this experiment is described in Fig. 3A. Scale bar, 20 μm. (F) Diagram of the domain structure of DSCAM deletion constructs. (G) Coimmunoprecipitation assay revealed the association of EGFP-RapGEF2 with DSCAM cytoplasmic domain. The input (bottom column, 5%) and immunoprecipitants (upper two columns) were analyzed by Western blotting using anti-DSCAM and anti-GFP antibodies. (H) Coimmunoprecipitation assay revealed association of DSCAM with RapGEF2/MAGI1/β-catenin ternary complex. Experimental conditions were the same as in (G). The input (bottom two columns) and immunoprecipitants (upper four columns) were analyzed by Western blotting with anti-RapGEF2, anti-Flag, anti–β-catenin, and anti-GFP antibodies. The experiment was repeated at least three times. (I) Model of the complex formation of DSCAM, RapGEF2, MAGI1, and β-catenin.
During immunohistochemical analysis of DscamPA/+ animals, it was difficult to distinguish DSCAM-PA signals on one nascent neuron from those derived from other cells. To investigate the detailed subcellular localization of DSCAM, we electroporated pCAG-Dscam-mEGFP with Cre-loxP–based neuronal labeling to the ventricular surface of the dorsal midbrain at E13.5 and fixed embryos at E15.5 (Fig. 1, E and F). We validated DSCAM-mEGFP in terms of its dimerization potential with nontagged DSCAM, its binding ability to RapGEF2 (a DSCAM interacting protein, see below), and its subcellular localization upon transfection into cultured cells, all of which were the same for untagged DSCAM (fig. S5, A, B, and D). To label nascent neurons, we selected the promotor of NeuroD, an early marker for newborn excitatory neurons in the embryonic dorsal midbrain (fig. S6A) (3), and performed in utero co-electroporation with pNeuroD-Cre and pCAG-loxP-polyA-loxP (FloxP)–mKO2-F (Fig. 1E and fig. S6B) (19). While membrane-localizing monomer KusabiraOrange2 (mKO2-F) labeling was barely observed in Ki67 (mitotic cell marker)–positive cells, many neuron-shaped cells with long processes were labeled with mKO2-F (magenta signals in Fig. 1G and fig. S5C). Some mKO2-F–expressing neurons harbored apical processes abutting the ventricular surface (yellow arrowheads in Fig. 1G) with wide palm-like structures (light blue double-headed arrow in Fig. 1H), suggesting that this labeling system can specifically visualize nascent neurons even before endfeet detachment from the ventricular surface. DSCAM-mEGFP expression was low; however, an anti-GFP antibody enabled visualization of its localization (Fig. 1, G and H). mKO2-F–positive neurons separated from the ventricular plane had apical and/or basal processes, with accumulation of DSCAM-mEGFP in the apical processes (Fig. 1G). In mKO2-F–positive neurons abutting the apical endfeet into the ventricular surface, DSCAM-mEGFP tended to accumulate at the endfeet (Fig. 1G). Higher-magnification images revealed that elongated or circular DSCAM-mEGFP signals were localized at the soma and apical endfeet of mKO2-F–positive newborn neurons, concentrated at the terminals of apical endfeet (Fig. 1, H and I). These findings suggest that DSCAM is involved in developmental events at the endfeet of newborn neurons.
Dscam knockdown impairs newborn neuron delamination and neuronal migration
We next examined the function of DSCAM at the endfeet in knockdown (KD) experiments using short-hairpin RNA interference (shRNAi) for Dscam (sh-Dscam) (14). shRNA vectors were introduced with Cre-loxP–based neuronal labeling plasmids into the E13.5 dorsal midbrain wall through in utero electroporation, followed by fixation at E15.5 (Fig. 2A). pCAG-H2B-EGFP was co-electroporated to visualize the nuclei of transfected cells (Fig. 2B). In control (sh-scramble)–introduced midbrain, most mKO2-F–positive neurons were delaminated from the ventricular surface and migrated toward the pia. Only a few neurons attached to the ventricular surface via their short endfeet (Fig. 2, B and C). Many sh-Dscam–introduced neurons retained significantly longer endfeet attached to the ventricular surface and remained apically attached (Fig. 2, B and C). In control, only 5% of neurons in mKO2-F–labeled neurons attached their apical endfeet to the ventricular surface. However, more than 20% of Dscam KD neurons abnormally retained these attachments (Fig. 2D). This excess in endfeet due to Dscam KD was significantly rescued by the shRNA-resistant Dscam expression vector (Kruskal-Wallis test with Dunn’s multiple comparisons test, Dscam KD, P = 0.0056; Fig. 2D), confirming that DSCAM is involved in endfeet detachment of newly formed neurons in the midbrain.
Fig. 2. Dscam KD prevents delamination of newborn neurons.
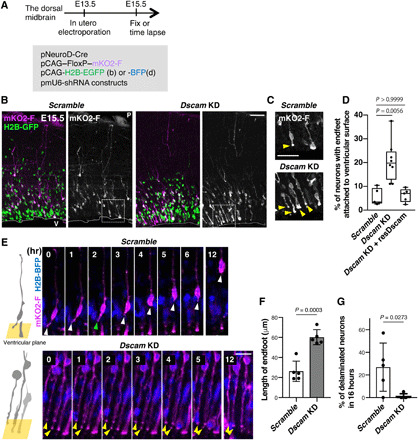
(A to F) Embryonic dorsal midbrains were electroporated in utero at E13.5 with Cre-loxP–based neuron labeling. pCAG-H2B-EGFP (B) or BFP (D) plasmid was introduced to monitor nuclear position. At E15.5, midbrain coronal slices were analyzed. (A) Experimental design. (B) E15.5 dorsal midbrain coronal sections expressing mKO2-F and H2B-EGFP in electroporated cells with control (scramble) or Dscam KD. Higher magnification in (C) represents areas surrounded by dotted boxes. P, pia; V, ventricle. Scale bar, 50 μm. (C) Excessive abnormal apical processes (yellow arrowheads) observed in Dscam KD condition. Scale bar, 20 μm. (D) Percentage of neurons (mKO2-F–positive cells) with endfeet attached to the ventricular surface. Box plots show median (horizontal line), quartiles (box), and range (whiskers) from five to eight brains; Kruskal-Wallis test with Dunn’s multiple comparisons test. (E) Endfeet live imaging in slice culture. Left-side cartoons represent the angle of the ventricular surface in each movie. Each frame comprises z-stacked images. Control mKO2-F–positive neurons had apical endfeet (white arrowhead) first, which were then retracted (green arrowhead). In Dscam KD neurons, most mKO2-F–positive neurons bore long endfeet (yellow arrowheads). The ventricle is toward the bottom. Scale bar, 30 μm. (F) Length of mKO2-F–positive endfeet (Scramble, n = 5; Dscam-KD, n = 5). (G) Ratio of mKO2-F–positive delaminating neurons in 16 hours (Scramble, n = 5; Dscam KD, n = 5). Data are presented as the mean ± SEM; unpaired two-tailed t test.
To monitor the dynamic behavior of nascent neurons, a time-lapse analysis was performed using similar electroporation conditions (Fig. 2, A and E to G). Here, we focused on mKO2-F–positive neurons, the endfeet of which were attached to the ventricular surface at the beginning of time-lapse imaging. At 16 hours, numerous control neurons displayed detachments of their endfeet from the ventricular surface, and their soma had moved radially without forming long apical processes (Fig. 2, E and G, and movie S1). However, Dscam KD neurons tended to have significantly longer apical endfeet that maintained their attachment to the ventricular surface than those of controls (P = 0.0003; Fig. 2, E and F, and movie S2). At 16 hours, almost 30% of control neurons were delaminated, whereas Dscam KD neurons were rarely detached from the ventricular surface (Fig. 2, E and G). These findings suggest that DSCAM is involved in endfeet detachment and delamination of newborn neurons.
To further examine the function of DSCAM in neuronal migration after neuronal delamination, midbrains were electroporated at E13.5 with pCAG-EGFP and KD vectors, followed by fixation at E18.5 (Fig. 3A). We stained the sections with an anti–phospho–JNK (c-Jun N-terminal kinase) antibody to demarcate the superficial SC (sSC)/intermediate SC (iSC), deeper SC (dSC), and PAG layers in the dorsal midbrain at E18.5 (Fig. 3B) (3). As reported previously, control-electroporated neurons were widely distributed throughout the dorsal midbrain (Fig. 3, C and D) (3, 4). Conversely, Dscam KD resulted in impaired radial migration in the dorsal midbrain. Although excessive endfeet in Dscam KD neurons disappeared at E18.5, the lower layer (PAG)–localizing neuron number was significantly increased, while the upper layer (sSC/iSC, dSC)–localizing number decreased in the KD experiment (Fig. 3, C and D). Furthermore, we often observed abnormal Dscam KD cell clusters in the PAG layer (P < 0.0001; yellow arrowheads in Fig. 3, C and E). Impaired migration and abnormal cluster formation were rescued by the shRNA-resistant expression construct (resDscam; P = 0.0006; Fig. 3, C and E). These observations suggest that DSCAM is involved in radial neuronal migration and avoidance of inappropriate cell-cell clustering in the developing midbrain.
Fig. 3. Dscam KD prevents neuronal migration and isolated behavior.
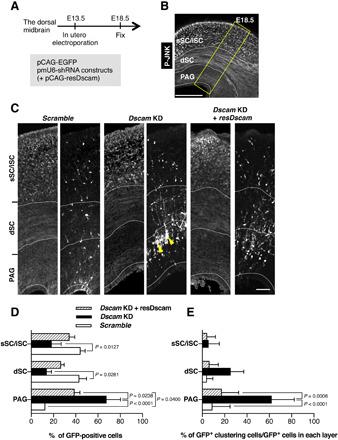
(A to E) Dorsal midbrains were electroporated in utero at E13.5 with plasmids encoding pCAG-EGFP and indicated shRNA constructs and pCAG vector encoding shRNA-resistant expression construct (resDscam). At E18.5, midbrain coronal slices were analyzed. (A) Experimental design for in utero electroporation and analyses. (B) Schematic of coronal section of the dorsal midbrain at E18.5. Slices were stained with anti–phospho-JNK (P-JNK) antibody, a marker that demarcates the sSC/iSC, dSC, and PAG layers in the dorsal midbrain. Yellow box indicates the area shown in (C). Scale bar, 300 μm. (C) E18.5 dorsal midbrain coronal sections expressing EGFP in the electroporated cells with shRNA vectors and pCAG vector coding resDscam. Yellow arrowheads indicate impaired neuronal migration and abnormal cell clustering. Scale bar, 50 μm. (D) Distribution of EGFP-positive cells in three layers demarcated by anti–phospho-JNK antibody (Scramble, n = 4; Dscam KD, n = 4; Dscam KD and resDscam, n = 4). (E) Ratios of abnormal cell cluster formation in each layer (Scramble, n = 4; Dscam KD, n = 4; Dscam KD and resDscam, n = 4). Data are presented as the mean ± SEM; two-way analysis of variance (ANOVA) and Tukey’s multiple comparison test.
DSCAM binds to RapGEF2 and down-regulates its activity
Next, we investigated how DSCAM regulates endfeet delamination. We searched for previously unidentified DSCAM-binding molecules by immunoprecipitation followed by mass spectrometry and identified candidate proteins (Fig. 4A and table S1). Among them, RapGEF2 (also known as PDZ-GEF1/RA-GEF1), a Rap1-specific guanine nucleotide exchange factor (GEF) (20, 21), was identified as a DSCAM-interacting protein (Fig. 4B). RapGEF2 contains the cyclic nucleotide-binding (cNMP), Ras GEFs N-terminal (Nter), PDZ, Ras-associating (RA), and Ras GEFs catalytic (CAT) domains (Fig. 4B). During neocortical neurogenesis, Rap1 regulates plasma membrane localization of N-cadherin (10, 11, 22), and RapGEF2 acts on multipolar-bipolar transitions during neuronal migration via the Rap1/N-cadherin pathway (23). These reports raised the possibility that the DSCAM-RapGEF2 interaction may alter RapGEF2 and Rap1 activities, which somehow affect N-cadherin localization and neuronal migration/delamination.
First, we confirmed that RapGEF2 coimmunoprecipitated with DSCAM from embryonic brain lysates (Fig. 4C and fig. S4B). Immunostaining using DscamPA/+ mice revealed that endogenous RapGEF2 was distributed throughout the dorsal midbrain and accumulated with DSCAM-PA and N-cadherin at the ventricular surface (white arrowheads in Fig. 4D). In contrast, endogenous RapGEF6 was not prominent at the ventricular surface (fig. S7B). The DSCAM-PA and RapGEF2 signals colocalized with the N-cadherin signals (yellow arrowheads in Fig. 4D). High-resolution images of thin sections of the dorsal midbrain revealed that RapGEF2 signals were concentrated near the ventricular surface and overlapped with those of DSCAM-PA and N-cadherin (light blue arrowheads in Fig. 4D).
Consistent with these observations, electroporated 3xFlag-RapGEF2 accumulated at the most distal part of the endfeet (Fig. 4E). Given the accumulation of exogenous DSCAM-mEGFP and endogenous DSCAM-PA at the ventricular surface (Figs. 1, G and H, and 4D), these observations suggest that those two molecules, DSCAM, and RapGEF2 were colocalized at the apex of the endfeet with N-cadherin (5, 6). Next, we investigated the binding region of DSCAM to RapGEF2 by a coimmunoprecipitation assay using deletion constructs and COS-7 cells. EGFP-RapGEF2 bound to full-length DSCAM and mutants lacking the PDZ binding motif (ΔPDZm) but not to mutants lacking the cytoplasmic domain (ΔC; Fig. 4, F and G), indicating that the cytoplasmic region of DSCAM, excluding the PDZ binding motif, binds to RapGEF2.
RapGEF2 associates with MAGI1/2, a multi-PDZ domain scaffolding protein at cell-cell junctions (24), while MAGIs associate with β-catenin (25). Depletion of MAGI1 suppressed cell-cell contact-induced Rap1 activation and cadherin-mediated cell adhesion (26). As DSCAM interacts with MAGIs via its PDZ binding motif (27) and both MAGI1/2 and β-catenin are expressed in the E14.5 dorsal midbrain (fig. S8), we investigated whether DSCAM associates with the RapGEF2/MAGI1/β-catenin ternary complex. Immunoprecipitation followed by Western blotting revealed that the DSCAM-cytoplasmic domain (C1) can associate with exogenous RapGEF2 and Flag-MAGI1c, respectively (Fig. 4H, lanes 2 and 3). Endogenous β-catenin coimmunoprecipitated with DSCAM-C1 only when Flag-MAGI1c was transfected (Fig. 4H, lanes 3 and 4), suggesting that β-catenin interacts with DSCAM via MAGI1c.
RapGEF2 binding to DSCAM-C1 increased when Flag-MAGI1c was transfected simultaneously (Fig. 4H, lane 4). However, if the PDZ binding motif of DSCAM was deleted (DSCAM-C2), its association with RapGEF2 decreased, and Flag-MAGI1c and β-catenin were barely detectable in the immunoprecipitants (Fig. 4H, lane 5). These findings suggest that the PDZ binding motif of DSCAM associates with MAGI1c and that this interaction increases DSCAM and RapGEF2 association. Deletion of the N-half of C1 (DSCAM-C3) decreased RapGEF2 binding to DSCAM (Fig. 4H, lane 6). These results propose two modes of RapGEF2 association with DSCAM: (i) direct binding to the cytoplasmic region including the N-half or (ii) indirect binding via MAGI1 (with β-catenin) through the PDZ binding motif of DSCAM (Fig. 4I). Alternatively, MAGI1c and/or β-catenin association with DSCAM may stabilize the direct interaction of RapGEF2 with DSCAM (Fig. 4I).
DSCAM negatively regulates RapGEF2 and Rap1 pathways
We then investigated the DSCAM-binding region of RapGEF2 using a series of RapGEF2 deletion constructs and immunoprecipitation assays (Fig. 5A). DSCAM associated with the RapGEF2 N-half region (N-half), cNMP, and Nter domains (Fig. 5B). It is known that the cNMP domain in RapGEF2 does not interact with cyclic adenosine monophosphate (cAMP) but serves as a negative regulatory domain (21). Therefore, we hypothesized that DSCAM binding to the cNMP domain may regulate the ability of RapGEF2 to activate Rap1. Thus, we evaluated Rap1 activity by a pull-down assay using the Rap1-binding domain of Ral1 (RBD domain; Fig. 5, C and D). As expected, active Rap1 was increased by the expression of EGFP-RapGEF2 compared with control EGFP (P = 0.0007). This RapGEF2-induced Rap1 activation was significantly decreased by coexpression of the Flag-DSCAM-C1 fragment (P = 0.0475), suggesting that DSCAM association with RapGEF2 suppresses the ability of RapGEF2 to activate Rap1. Next, we examined the effect of MAGI1c on Rap1 activity. The effect of the DSCAM-C1–mediated suppression of Rap1 activity in the presence of RapGEF2 was significantly enhanced upon co-introduction of MAGI1c (P = 0.0440; fig. S9, A and B). Because MAGI1c enhances the interaction between the cytoplasmic domain of DSCAM and RapGEF2 (Fig. 4H), MAGI1 may stabilize the DSCAM/RapGEF2 interaction and enhance the inhibitory effect of DSCAM on RapGEF2 activity.
Fig. 5. DSCAM associates with RapGEF2 and prevents Rap1 activation.
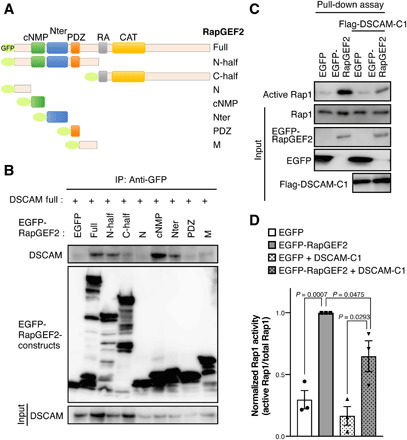
(A) Diagram of the domain structure of RapGEF2 deletion constructs. (B) Coimmunoprecipitation assay revealed that DSCAM associates with the RapGEF2 cNMP and Nter domains. Lysates of COS-7 cells expressing RapGEF2 constructs described in (A) and DSCAM were incubated with an anti-GFP antibody. The input (bottom column, 5%) and immunoprecipitants (upper two columns) were analyzed by Western blotting using anti-DSCAM and anti-GFP antibodies. The experiment was repeated at least three times. (C) Pull-down assay to evaluate active Rap1 using COS-7 cells expressing control EGFP or EGFP-RapGEF2 with or without Flag-Dscam cytoplasmic region (Flag-DSCAM-C1; described in Fig. 4F). The input (bottom four columns, 5%) and immunoprecipitants (upper column) were analyzed by Western blotting using anti-Rap1, anti-GFP, and anti-Flag antibodies. (D) Quantification of fold change of active Rap1 levels. Normalized active Rap1 activity was calculated by active Rap1/total Rap1. Data are presented as the mean ± SEM from three independent experiments; unpaired two-tailed t test.
DSCAM controls neuronal delamination via RapGEF2 and N-cadherin
As mentioned above, endfeet retraction is initiated by the down-regulation of N-cadherin/AJ (5, 9) and Rap1 regulates the plasma membrane localization of N-cadherin in cortical neurons (10, 11, 22). Therefore, we considered the possibility that excessive endfeet formation induced by Dscam KD was caused by overactivation of RapGEF2/Rap1 and extra stabilization of N-cadherin. To assess this, we investigated the following two parameters in Dscam KD neurons concurrently, the size of the apex of the apical endfeet and the fluorescence level of N-cadherin at the apical junction, in the en face view of the dorsal midbrain visualized by Cre-loxP–based neuron labeling (Fig. 6A). We collected data from mKO2-F–expressing cells, but not from mKO2-F and H2B-GFP (a nuclear marker) double-positive cells, to exclude somal AJ data. The en face view observation revealed that the apex size of the endfeet in Dscam KD neurons was significantly larger than in controls (P = 0.0236; Fig. 6, B and C), suggesting a key role of DSCAM in the reduction of the apex area, which is maintained by AJ and cytoskeletal networks. Intensities of immunolabeled N-cadherin at the apex of the endfeet were not significantly different between control and Dscam KD neurons when observing the whole population (P = 0.3749, Mann-Whitney test; Fig. 6D). However, when the apex areas were categorized into small and large halves, the intensity of immunolabeled N-cadherin was significantly higher in the small half of the apex areas in the Dscam KD condition (P = 0.0345; Fig. 6E). Furthermore, we constructed a scatter plot for N-cadherin intensity and the apex area. The covalence of N-cadherin intensity and the apex area was statistically evaluated via analysis of covariance (ANCOVA), which revealed significant differences between the control and Dscam KD (P = 0.0154; Fig. 6F). These results suggest that DSCAM plays a critical role in the local disassembly of N-cadherin from cell-cell contact in the process of reducing the apex area and subsequent neuronal delamination.
Fig. 6. DSCAM regulates neuronal delamination via RapGEF2 and N-cadherin down-regulation.
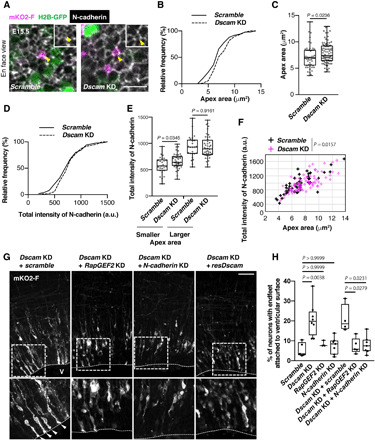
(A) En face view of E15.5 dorsal midbrain that was electroporated in utero at E13.5. The outline of this experiment is described in Fig. 2A. Yellow arrowheads indicate collected apex areas. Scale bar, 10 μm. (B) Cumulative distribution and (C) quantification of the size of the endfeet apex area. (B and C) Scramble, n = 50 cells from three animals; Dscam KD, n = 88 cells from three animals. Box plots show median (horizontal line), quartiles (box), and range (whiskers); Mann-Whitney test. (D) Cumulative distribution of normalized fluorescence intensity of N-cadherin in apex of endfeet. This was calculated by the ratio of the average pixel intensity within the apical circumference of one transfected cell versus the mean of average pixel intensity of four to seven of its close nontransfected neighbors. (E) Quantification of normalized fluorescence intensity of N-cadherin level ratio. A smaller population was defined as the size of the apex area of endfeet being smaller than the median size, and the other half was defined as larger [smaller, n = 25 cells; Dscam KD, n = 44 cells, box plots show median (horizontal line), quartiles (box), and range (whiskers), Mann-Whitney test]. (F) Scatter plot of N-cadherin intensity and apex area for control and Dscam KD brains. ANCOVA test. (G) Coronal sections of E15.5 dorsal midbrain expressing mKO2-F that were coelectroporated with indicated vectors. White arrowheads indicate excess endfeet formation. Higher magnification in bottom panels represents areas surrounded by the dotted box. V, ventricle. Scale bar, 50 μm. (H) Analysis of the number of mKO2-F–positive neurons bearing endfeet. Box plots show median (horizontal line), quartiles (box), and range (whiskers) from five to eight brains; Kruskal-Wallis test with Dunn’s multiple comparisons test.
Last, to test whether RapGEF2 and N-cadherin mediate excessive endfeet formation in Dscam KD neurons, we electroporated shRNA of RapGEF2 or N-cadherin with that of Dscam. RapGEF2 or N-cadherin KD significantly rescued the excessive endfeet induced by Dscam KD (Kruskal-Wallis test with Dunn’s multiple comparisons test; Dscam and RapGEF2 co-KD, P = 0.0279; Dscam and N-cadherin co-KD, P = 0.0231; Fig. 6, G and H), suggesting that RapGEF2 and N-cadherin function downstream of DSCAM in the delaminating neurons. On the basis of these observations, we proposed a working model (fig. S10) in which DSCAM is critical for local N-cadherin reduction in the delamination process via inactivation of the RapGEF2/Rap1 signaling pathway, leading to apical endfeet retraction and neuronal delamination.
DISCUSSION
Despite the important linkage of cell adhesion to neuronal delamination, its underlying post-transcriptional mechanisms are largely unexplored. Here, we showed that DSCAM was locally accumulated at the distal part of endfeet. KD of Dscam caused excess abnormal endfeet in nascent neurons and prevented neuronal delamination. DSCAM was shown to associate with and down-regulate RapGEF2/Rap1, and co-KD of RapGEF2 or N-cadherin rescued the abnormal endfeet formation induced by Dscam KD. These results suggest that DSCAM plays a critical role in delamination by preventing the RapGEF2–Rap1–N-cadherin signaling cascade in newborn neurons.
In this study, we found that DSCAM plays a critical role in neuronal delamination, canceling the cell attachment with surrounding cells at the ventricular surface. The apical endfeet of differentiating cells securely prohibits inappropriate differentiation of surrounding progenitors, thus acting as the central sites of active Notch signaling (28). Therefore, the cancellation of cell attachment may occur between neuronal endfeet and their surrounding cells, probably the progenitor cells or radial glia. However, it remains unclear whether the function of DSCAM in neuronal delamination is mediated by trans-DSCAM homophilic interaction between newborn neurons and radial glia. If radial glia express DSCAM, trans-homodimer DSCAM interaction between newborn neurons and radial glia might be formed to suppress N-cadherin at the site. However, this appears unlikely because DSCAM expression was found to be notably low in the VZ and radial glia in our study. Because we do not have appropriate antibodies against DSCAM that can be used for immunohistochemical analysis in the midbrain, we cannot exclude the possibility of trans-DSCAM interaction between newborn neurons and radial glia. Alternatively, because DSCAM interacts with other ligands such as Netrin (29), it is possible that DSCAM on newborn neurons interacts with an unknown molecule(s) expressed on surrounding cells (e.g., radial glia) to suppress N-cadherin at the site.
While Drosophila dscam1 gene produces extraordinary DSCAM alternative splicing variants (30, 31), mammalian Dscam gene does not generate as many variants. However, previous studies have suggested that Drosophila and mammalian DSCAM have similar functions in neural development and share common intracellular pathways. This protein regulates the axon guidance (31), dendrite arborization (32), and synaptic contact (14, 33), which are mediated by trans-homophilic binding. Conversely, DSCAM plays other roles in self-avoidance of dendrites (18) and tilling of allogeneic cells, in which DSCAM functions via homophilic repulsion. In this study, we observed that Dscam KD neurons formed abnormal clusters (Fig. 3, C to E) during neuronal migration. This might also be caused by loss of homo-repulsion between cells, although we do not have any direct evidence. DSCAM-mediated self-avoidance and tiling are thought to require the separation/cancellation of allogeneic cell adhesion (16); however, the underlying molecular mechanisms remain unclear. A previous study reported that DSCAM masks the function of the cadherin superfamily, cadherin-3/6 and γ-protocadherins (16). The authors showed that cadherin-3 was abnormally accumulated on Dscam-deficient cortical neurons, which was not observed on wild-type neurons. However, abnormal accumulation was prevented by trans-homodimer DSCAM interactions, suggesting that DSCAM can cancel the functions of cadherin proteins. Although the DSCAM cytoplasmic region has been reported to bind intracellular molecules, such as PSD95 and MAGI1 (14), and Importin 5 (34), the intracellular molecular machinery required to cancel the adhesive ability of cadherins remains unclear. Therefore, we hypothesize that DSCAM-mediated self-avoidance and tiling are also elicited by RapGEF/Rap family proteins, although we do not have any direct evidence. Further analyses are required to elucidate the function of homodimer DSCAM in suppressing allogeneic cell adhesion.
As cells of the nervous system originate from the neuroepithelium during early development, delamination of neurons has been discussed in models of epithelial-mesenchymal transformation (EMT) (6, 35). EMT is a process in which cells lose epithelial characteristics, such as cell-cell junctions and apico-basal polarity, and acquire mesenchymal polarity capable of high mobility. Therefore, it is plausible that the molecular machinery mediating cell polarity is conserved in both epithelial and neuronal cells, underscoring EMT (35). Rap1 activity is critical for apico-basal polarity of epithelial cells via its regulation of AJ and polar complexes, such as N-cadherin and Par3/Cdc42, on the apical surface of radial glia (36). In Rap1a and Rap1b double KO (dKO) mice, the localization of apical radial glia in the ventricular plane was lost, glial fibers were mislocalized outside the ventricular plane, and localization of polar complexes was abnormal. Moreover, in the RapGEF2/6-dKO cortex (and to a lesser degree in RapGEF2-conditional KO cortex), the dense array of apical surface AJs was almost nonexistent such that a substantial population of cells was apparently exfoliated from the surface into the ventricle (37). Because RapGEF2, rather than RapGEF6, prominently accumulated at the ventricular surface of the dorsal midbrain, single KD of RapGEF2 might adequately induce the disruption of apical AJs in the dorsal midbrain. The KO mice used in these previous studies (36, 37) are consistent with our current model, in which RapGEF2/Rap1 inactivation leads to withdrawal of apical endfeet from the AJ belt and delamination of nascent neurons. Regarding the interaction between MAGI1 and RapGEF2, previous studies have reported that the C-terminal PDZ-binding motif of RapGEF2 interacts with MAGI1 (24), and the PDZ1 domain of MAGI1 (BAP1) interacts with RapGEF2 (21). To further elucidate the molecular mechanisms underlying delamination, verification of the involvement of polar regulatory molecules is necessary.
The critical functions of Rap1 and its downstream molecule N-cadherin in neuronal migration have repeatedly been reported. During the early stage of neocortical development, Rap1 and N-cadherin in the neuroepithelium maintain the AJ belt to regulate neurogenesis through Notch signaling (28). Upon commitment to a neuronal cell, N-cadherin is down-regulated when neurons leave the VZ (7). Subsequently, Rap1 activity that regulates N-cadherin localization is required for multipolar-bipolar transition (11, 23) in glial-guided radial migration (12, 36) and glial-independent somal translocation (10). To complete these various multistep functions, Rap1 activity and AJ stability appear to be dynamically and locally regulated by sensing directional and environmental cues. Although the transcriptional repression of adhesion molecules (e.g., by Scratch and Foxp) (6, 8) is required for the departure of cells from the VZ, these transcriptional effects are considered to affect the whole cell body and may be insufficient for the recruitment of N-cadherin for subsequent steps. Nascent neurons may migrate radially through the efficient reuse of remaining adhesion molecules for subsequent migration steps, e.g., the attachment of radial glia. Therefore, as post-transcriptional machinery, we propose that DSCAM down-regulates N-cadherin function via suppression of RapGEF2/Rap1.
In Dscam KD cells, the detachment of apical endfeet was transiently suppressed but not completely inhibited. These results suggest that mechanisms other than DSCAM-mediated suppression of the AJ may exist to complete the delamination or detachment of nascent neurons, e.g., transcriptional suppression of cadherin family, interdependent actomyosin contractility, and centrosome and ciliary membrane dissociation (9). It was recently reported that Lzts1, a molecule associated with microtubule components, is involved in neuronal and outer radial glia delamination from the AJ through cytoskeletal rearrangements (38). The transcription factor Insm1 induces basal progenitor delamination by repressing the apical AJ belt–specific protein Plekha (39). These reports suggest that delamination is mediated by multiple cascades to regulate cell adhesion and cytoskeletal architecture. As DSCAM is expressed in differentiated neurons only before migration, the machinery for neuronal delamination may be strictly defined by the neuronal fate commitment system.
Recently, evidence has been accumulating regarding mutations in genes associated with neuronal migration in human brain disorders (2). A de novo mutation in the Dscam coding region has been identified in individuals with autism spectral disorder (40). Our findings on the molecular functions associated with neuronal migration raise the possibility of identifying drug targets on the basis of pathology of periventricular heterotopia and autism. Further investigations on the functions of disorder-related Dscam mutations may shed light on the physiological and pathological significance of DSCAM and its associating molecules.
MATERIALS AND METHODS
Animals
All animal experiments in this study were approved by the Animal Care and Use Committee of the National Institute of Neuroscience, Japan. Pregnant ICR mice purchased from Japan SLC Inc. (Shizuoka, Japan) were used for in utero electroporation. E1 was defined as 12 hours after detection of vaginal plugs. Dscamdel17/+ allele (B6.CBy-Dscamdel17/RwbJ) was obtained from The Jackson Laboratory (18). Dscamflox/+; CreCAG allele was obtained from K. Yamakawa [RIKEN Center for Brain Science (CBS), Saitama, Japan] (41). Because the C57BL/6 background Dscamdel17/ del17 and Dscamflox/flox; CreCAG mice die neonatally as previously noted (18, 41), the experiments described were conducted on a mixed but predominantly BALB/cByJ background. Littermates of experimental mutants were used as controls. GAD67+/GFP allele has been described previously (3). vGlut2+/GFP allele (Tg[Slc17a6-EGFP]FY115Gsat) was obtained from The Jackson Laboratory. In total, 200 pregnant female mice were used in this study. Two or three pregnant mice were housed in one cage and maintained on a 12-hour light/dark cycle with free access to food and water.
DNA constructs
Complementary DNAs (cDNAs) encoding the full-length mouse Dscam were generated using a cDNA library prepared from mouse embryonic brains. Polymerase chain reaction (PCR) was performed using PrimeSTAR Max (R045A, Takara Bio) and the following primers: 5′-TCTCATCATTTTGGCAAAGAATTCGGCATGTGGATACTGGCTCTCTCCT-3′ and 5′-GGGGGGCGGAATTTACGTAGCGGCCGCTTATACCAAGGTGTAAGATTTTG-3′. The obtained product was subcloned into the pBS vector using the In-Fusion HD Cloning Kit (Clontech Laboratories, Mountain View, CA). The cDNA encoding human RapGEF2 was obtained as previously described (20). Flag-tagged RapGEF2 was amplified using the following primers: 5′-AACTCGAGATGGACTACAAAGACCACGATGGCGACTACAAAGACCACGATATCGACTACAAAGACGACGATGACAAGATGAAACCACTAGCAATCCC-3′ and 5′-.
AAGCGGCCGCTCAAACAGCAGAAACTTGTT-3′; the obtained PCR products were subcloned into pCAG (#89684; Addgene, Watertown, MA). The cDNA encoding mouse MAGI1c was obtained from Addgene (#107814). The NeuroD promotor was amplified using ICR mice tail genome by PrimeSTAR Max and the following primers: 5′-AAAGTCGACGAGCTCGGAGGACACTTG-3′ and 5′-TTTGAATTCCTCGTGTCCCGG-3′ (section S1). All sequences were confirmed by DNA sequencing as described below. pCAG–loxP–polyA–loxP–mKO2-F (pCAG–FloxP–mKO2-F) was provided by A. Shitamukai (RIKEN CBS). To generate Dscam-mEGFP, the PCR product of monomer EGFP (mEGFP) with 10 glycine repeat linkers (Gly) was inserted in frame into the Dscam full-length coding sequence at the Mro I restriction enzyme site [in fibronectin type III domain (FnIII)], which is a usable and unique enzyme site within Dscam, using the In-Fusion HD Cloning Kit (Fig. 1F). To generate Dscam-PA, PCR was performed to amplify the Dscam N-terminal [5745 base pairs (bp)] and C-terminal fragments (369 bp) using PrimeSTAR Max and the following primers: 5′-TTTTGGCAAAGAATTCATGTGGATACTGGCTCTCT-3′ and 5′-TGGCATGGCAACGCCTGCGCCCCGGTTTAACAA-3′ for the Dscam-N fragment and 5′-ggcgttgccatgccaggcgccgaagatgatgtggtgggcgttgccatgccaggtgccgaagatgatgtggtgCCAGGCACCAGCAGGGACCT-3′ and 5′-CTGAGGAGTGCGGCCGCTTATACCAAGGTGTAAGATTTT-3′ for the Dscam-C fragment (the sequence of PA tag is underlined). The amplified products were subcloned into the pCAG vector using the In-Fusion HD Cloning Kit. GFP was obtained from Clontech Laboratories, and blue fluorescent protein (BFP) was obtained from Evrogen (#FP176, pTagBFP-H2B; Moscow, Russia), and H2B-BFP was subcloned into the pCAG vector. Dscam and RapGEF2 fragments, DSCAM-Full (1 to 2013 amino acids), DSCAM-ΔPDZm (1 to 2007 amino acids), DSCAM-ΔC (1 to 1615 amino acids), DSCAM-C1 (1616 to 2013 amino acids), DSCAM-C2 (1616 to 2007 amino acids), DSCAM-C3 (1792 to 2007 amino acids), RapGEF2-Full (1 to 1499 amino acids), RapGEF2–N-half (1 to 605 amino acids), RapGEF2–C-half (606 to 1499 amino acids), RapGEF2-N (1 to 134 amino acids), RapGEF2-cNMP (135 to 235 amino acids), RapGEF2-Nter (267 to 380 amino acids), RapGEF2-PDZ (385 to 455 amino acids), and RapGEF2-M (456 to 605 amino acids) were amplified by PCR and subcloned into pCAG, pCAG-Flag, pEGFP-C2, or pEGFP-C3 vectors. All fragments were confirmed by DNA sequencing. We constructed shRNA-expressing vectors using the previously verified targeting sequences sh-Dscam-1 (5′-AAAGAGTTTAGCTGAAATGCT-3′) (42), sh-Dscam-2 (5′-GTACATGGACTTGCTGACGTCT-3′) (14), sh-Dscam-3 (5′-GTGGGAGAGGAAGTGATAT-3′) (29), sh-RapGEF2 (5′-GTCATTAACCAGGAAACAT-3′) (23), sh-N-cadherin (5′-GACTGGATTTCCTGAAGAT-3′) (7), or a control Scramble (5′-AATGCATCTCTGCAAGAGGTA-3′) (42). These targeting sequences and their complementary sequences were inserted into the mU6pro vector. We used the data of sh-Dscam-2 for each figure (Figs. 2, B to F, 3, C to E, and 6, A to G). The shRNA-resident Dscam expression construct against sh-Dscam-2 (resDscam) was generated by introducing three silent mutations into the cDNA sequence by PCR-mediated site-directed mutagenesis using PrimeSTAR Max and the following primers: 5′-TCAGTAAATCGATGTACCTCACAGTGAAA-3′ and 5′-ACATCGATTTACTGACGTCTGCGCCCACA-3′ (the silenced mutations are underlined).
In utero electroporation
Pregnant E13.5 mice were anaesthetized by intraperitoneal administration of dexmedetomidine (0.3 mg/kg; Zenoaq, Tokyo, Japan), midazolam (2 mg/kg; Astellas Pharma, Tokyo, Japan), and butorphanol tartrate (2.5 mg/kg; Meiji Seika Pharma, Tokyo, Japan) on a heating pad. The uterine horns were exposed, and approximately 1 μl of plasmids mixed with Fast Green (Sigma-Aldrich, St. Louis, MO) in TE buffer was manually injected into the developing aqueduct using a pulled glass micropipette (G-1.0; Narishige, Tokyo, Japan). Concentrations were as follows: pNeuroD-Cre (0.25 μg/μl), pCAG–FloxP–mKO2-F (0.25 μg/μl), pCAG-Dscam-mEGFP (2 μg/μl), pCAG-H2B-EGFP (BFP) (0.25 μg/μl), pmU6-shRNA constructs (1 μg/μl), pCAG-EGFP (0.25 μg/μl), pCAG-resDscam (2 μg/μl), and pCAG-3xFlag-RapGEF2 (2 μg/μl). Electric pulses (33 V, four pulses; 30 ms on, 970 ms off) were delivered across the heads of the embryos targeting the dorsal-medial part of the midbrain with forceps-type electrodes (CUY650P5; Nepa Gene, Ichikawa City) connected to an electroporator (CUY21E, Nepa Gene). After electroporation, uteri were placed back in the abdominal cavity, allowing embryos to continue developing. All surgical procedures were completed within 30 min, after which the mice recovered on a heating pad for 30 min. After 2 or 5 days, embryos were collected and subjected to immunohistochemical analysis.
Antibodies and immunohistochemistry
Embryos were dissected in ice-cold phosphate-buffered saline (PBS) and fixed in 4% paraformaldehyde for 2 hours at 4°C. Fixed embryos and dissected tissues were cryoprotected by overnight immersion in 30% sucrose in PBS, embedded in optimum cutting temperature (Tissue-Tek O.C.T. compound; Sakura Finetek Japan, Tokyo), and cryosectioned at 20 or 40 μm for fluorescence observation of neuronal morphology (CM3050 S; Leica, Wetzlar, Germany). Sections of rostro-dorsal midbrain were used for experiments. The sections were soaked in PBS for 5 min, preincubated in blocking buffer containing 0.03% Triton X-100 and 10% normal donkey serum (Millipore) at room temperature (RT) for 30 min, and subsequently immunolabeled using the following primary antibodies in blocking buffer at 4°C overnight. The following primary antibodies were used: rat anti-Ki67 (1:50; 14-5698; eBioscience, San Diego, CA), rat anti-GFP (1:100; RQ1; gift from A. Imura, BRI, Kobe, Japan), rat anti-PA tag (1:1000; GVAMPGAEDDVV; NZ-1; 012-25863; Fujifilm, Tokyo, Japan), goat anti-Sox9 (1:500; AF3075; R&D Systems, Minneapolis, MN), rabbit anti–active-JNK (1:200; V793A; Promega, Madison, WI), rabbit anti-RapGEF2 and RapGEF6 (1:50; gift from T. Kataoka, Kobe University, Kobe, Japan) (37), mouse anti–N-cadherin (1:500; 610921; BD Biosciences, Franklin Lakes, NJ), rabbit anti–cleaved caspase-3 (1:100; Asp175; 5A1E; 9664S; Cell Signaling Technology, Danvers, MA), goat anti-NeuroD (1:100; N-19; sc-1084; Santa Cruz Biotechnology, Dallas, TX), and mouse anti-HuC/D (1:500; 16A11; A-21271; Invitrogen, Carlsbad, CA). In fig. S2, the following three Dscam antibodies were used: rabbit anti-Dscam (1:100 to 1:5000; HPA019324; Atlas Antibodies, Stockholm, Sweden), rabbit anti-Dscam (1:100 to 1:5000; N-16, sc-79437; Santa Cruz Biotechnology), and goat anti-Dscam (1:100 to 1:5000; AF3666; R&D Systems). Specimens were subsequently rinsed with PBS and incubated with secondary antibodies conjugated with Alexa Fluor 488, Alexa Fluor 568, Alexa Fluor 594, or Alexa Fluor 647 (1:1000; Abcam, Cambridge, UK) and DAPI (5 μg/ml; Invitrogen) in blocking buffer in PBS at RT for 2 hours. Fluorescence imaging was performed using a Zeiss LSM 780 confocal microscope system (Carl Zeiss) and ZEN 2009 software (Carl Zeiss AG, Oberkochen, Germany). Quantification of fluorescence intensity and endfeet length of the immunolabeled cells was performed using the “Measure” and “Plot Profile” functions of ImageJ. The size of the apex of endfeet was traced using N-cadherin staining. The normalized fluorescence intensity of N-cadherin in the endfeet apex was calculated by the ratio of the average pixel intensity within the apical circumference of one transfected cell versus the mean of average pixel intensity of 4 to 7 of its close nontransfected neighbors. A “smaller” or “larger” population in Fig. 6E was defined as the size of the apex area of endfeet lesser or greater than the median size.
Brain lysate preparation and Western blot analyses
For the investigation of temporal expression profiles of DSCAM in the midbrain (Fig. 1A), the whole midbrain (E11.5 and E13.5) or dorsal midbrain (from E15.5 to adult) was isolated in PBS and homogenized in homogenization buffer [20 mM tris, 1 mM EDTA, 1 mM dithiothreitol, 150 mM NaCl, 100 mM sucrose, and protease inhibitor cocktail (cOmplete; Roche, Basel, Switzerland); pH 7.4 at 4°C]. Samples were clarified by centrifugation at 20,000g for 10 min at 4°C. Resultant pellets were pipetted with lysis buffer (10 mM tris, 1 mM EDTA, 150 mM NaCl, 1% NP-40, and protease inhibitor cocktail; pH 7.4 at 4°C), sonicated for 15 s on ice, and boiled in SDS sample buffer for 5 min at 95°C. Collected samples were subjected to SDS–polyacrylamide gel electrophoresis (PAGE) and Western blotting using anti-DSCAM (1:1000; Atlas Antibodies) or β-actin (1:1000; sc-47778; Santa Cruz Biotechnology) antibodies. To confirm DSCAM-PA expression using E15.5 control or Dscam PA/+ mice (fig. S3D), caudal brain lysate containing the midbrain, cerebellum, and medulla was adjusted in the same way as the Fig. 1A samples. Collected samples were subjected to SDS-PAGE and Western blotting using the anti-PA (1:1000; NZ-1, Fujifilm) or DSCAM (1:1000; Atlas Antibodies) antibodies.
Immunoprecipitation assay and LC-MS/MS analysis
To identify DSCAM-interacting proteins (Fig. 4A), P20 mice’s whole brains were extracted in lysis buffer [20 mM tris, 150 mM NaCl, 1 mM EDTA, and protease inhibitor cocktail (cOmplete); pH 7.4 at 4°C] and clarified by centrifugation at 12,000g for 5 min at 4°C. Collected supernatant was incubated with protein A Sepharose (Amersham Pharmacia Biotech, Little Chalfont, UK) and 10 μg of control rabbit immunoglobulin G (IgG), rotated for 30 min at 4°C, and clarified by centrifugation at 12,000g for 1 min at 4°C. Resultant supernatant was incubated with fresh protein A Sepharose and 10 μg of rabbit anti-DSCAM antibody (Santa Cruz Biotechnology) or control rabbit IgG, rotated for 2 hours at 4°C, and then clarified by centrifugation at 12,000g for 1 min at 4°C. After three washes in PBS, immunocomplexes were eluted by the addition of PBS containing 500 mM NaCl. Liquid chromatography–tandem mass spectrometry (LC-MS/MS) analysis was performed as previously described (43). However, we cannot exclude the possibility that some proteins in this list are false positive.
Coimmunoprecipitation assay using embryonic brain lysates
To confirm the endogenous DSCAM-RapGEF2 interaction (Fig. 4C), E15.5 mouse whole brains were extracted in lysis buffer [20 mM tris, 150 mM NaCl, 1 mM EDTA, 0.1% NP-40, and protease inhibitor cocktail (cOmplete); pH 7.4 at 4°C] and clarified by centrifugation at 20,000g for 10 min at 4°C. The soluble supernatants were incubated with 2 μg of rabbit IgG or rabbit anti-DSCAM (Atlas Antibodies) for 2 hours at 4°C. The immunocomplexes were then precipitated with protein G Sepharose 4B (Amersham) for 1 hour at 4°C. After three washes with lysis buffer, the obtained eluates were subjected to SDS-PAGE and immunoblotting using the DSCAM (1:1000; DS2-176; Millipore) or PDZ-GEF1 (1:1000; RapGEF2; Bethyl Laboratories) antibodies.
Cell culture and coimmunoprecipitation analysis
COS-7 cells were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum. Transfection was performed using Lipofectamine LTX (Invitrogen) according to the manufacturer’s instructions. Twenty-four hours after transfection, cells were scraped in lysis buffer [20 mM tris, 150 mM NaCl, 1 mM EDTA, 0.1% NP-40, and protease inhibitor cocktail (Roche); pH 7.4 at 4°C], sonicated for 15 s on ice, and then clarified by centrifugation at 20,000g for 10 min at 4°C. The soluble supernatants were incubated with 1 μg of rabbit IgG or rabbit anti-GFP (598; MBL, Woburn, MA) for 2 hours at 4°C. The immunocomplexes were then precipitated with protein A Sepharose 4B for 1 hour at 4°C. After three washes with lysis buffer, the obtained eluates were subjected to SDS-PAGE and immunoblotting with the following antibodies: anti-DSCAM (1:1000; Atlas Antibodies), anti-GFP (1:1000; 1E4; MBL), anti–PDZ-GEF1 (1:1000; RapGEF2; Bethyl Laboratories), anti-Flag (1:1000; M2; Sigma-Aldrich), and anti–β-catenin (1:1000; 610153; BD Biosciences).
Rap1 pull-down assay
Rap1 pull-down assays were performed using the Active GTPase Pull-Down and Detection Kit (16120; Thermo Fisher Scientific, Waltham, MA) according to the manufacturer’s instructions. Briefly, the active guanosine triphosphate (GTP)–bound form of Rap1 was pulled down from the cell lysates using 20 μg of glutathione S-transferase (GST)–RalGDS–RBD proteins beads. Bound GTP-Rap1 proteins were detected by immunoblotting using attached anti-Rap1 antibody (Thermo Fisher Scientific) in 5% BSA (bovine serum albumin)/TBST (tris-buffered saline with Tween 20) buffer. Exogenous protein expression was confirmed by anti-GFP (1:1000; 1E4; MBL) or anti-Flag (1:1000; M2; Sigma-Aldrich) antibodies.
Rigorous selection/optimization of the tag-insertion site in DSCAM
Because DSCAM has functionally critical regions at the N terminus (signal peptide) and C terminus (PDZ binding motif), terminal fusion of EGFP or PA-tag to these domains can disrupt the molecular functions. To narrow the adequate tag-insertion site, amino acid sequences for mouse Dscam were compared with those for DscamL1 or DSCAM from other species using the web-based protein structure prediction tool Jpred 4 (www.compbio.dundee.ac.uk/jpred/) to find candidate target regions with low stringencies. The selected region was analyzed using the web-based CRISPR design tool CRISPOR (http://crispor.org) to choose the guide RNA sequence with minimized off-target effects. First, either EGFP or tandem PA-tag was inserted into the extracellular region close to the mEGFP insertion site of pCAG-Dscam-mEGFP, which did not disturb molecular function (e.g., membrane localization and association with RapGEF2/MAGI1c). In these KI mice, expression levels of EGFP/PA-tag fused DSCAM and endogenous DSCAM were severely decreased because of unknown reasons. Second, tandem PA-tag was inserted into the cytoplasmic region to obtain the DscamPA KI mouse line (fig. S3), which was confirmed to show no changes in total DSCAM expression levels. Mice homozygous for Dscam-PA were viable and fertile, exhibiting normal development with regard to body size and weight, and did not present any abnormality throughout the study period. This carefully evaluated line was used accordingly in this study.
Generation of DscamPA KI allele by CRISPR-Cas9–mediated genome editing
Two parts of CRISPR guide RNA, Dscam-crRNA (CRISPR RNA) (5′-TTGTTAAACCGGGGCGCACCGTTTTAGAGCTATGCTGTTTTG-3′) and tracrRNA (trans-acting crRNA) (5′-AAACAGCAUAGCAAGUUAAAAUAAGGCUAGUCCGUUAUCAACUUGAAAAAGUGGCACCGAGUCGGUGCU-3′), were chemically synthesized and purified by reversed-phase column purification (FASMAC). Recombinant Cas9 protein (EnGen Cas9 NLS) was purchased from New England Biolabs (NEB; Ipswich, MA). As described below, cleavage activities of the guide RNAs with Cas9 protein were evaluated in vitro before electroporation. The single-stranded DNA (ssDNA) donor containing tandem PA-tag sequence flanked by 44-bp homology arms on both sides was subsequently designed. The left (5′-TGCCTCCATACCTACGAATGGACTTCTTGTTAAACCGGGGCGCA-3′) and right (5′-CCAGGCACCAGCAGGGACCTGAGTTTAGGACAAGCGTGCTTGGA-3′) homology arms were connected to tandem PA-tag sequences containing the Nar I recognition site (5′-GGCGTTGCCATGCCAGGCGCCGAAGATGATGTGGTGGGCGTTGCCATGCCAGGTGCCGAAGATGATGTGGTG-3′) for easier genotyping. The ssDNA was chemically synthesized by Eurofins Genomics (Luxembourg). The guide RNAs, Cas9 proteins, and donor ssDNA were electroporated into B6C3F1 mouse zygotes following the standard protocol. Using PCR with three sets of primers and sequence analyses, 5 of 34 (14.7%) pups were confirmed to carry the correct PA-tag KI allele. A pair of primers, forward (5′-TTCATGTCTTGGGTGGGCTC-3′) and reverse (5′-TTTGCGCTGTCTGTGGTTTC-3′), was used to genotype the progenies.
In vitro digestion assay
The genomic region (754 bp) containing the Dscam-crRNA target sequences was PCR-amplified using PrimeSTAR Max and a pair of primers, forward (5′-TTCATGTCTTGGGTGGGCTC-3′) and reverse (5′-TTTGCGCTGTCTGTGGTTTC-3′). Cas9 protein (50 ng/μl), Dscam-crRNA (15.8 ng/μl), and tracrRNA (25.8 ng/μl) were incubated with the Dscam target PCR products in Cas9 Nuclease Reaction Buffer (NEB) for 60 min at 37°C. Reactions were stopped with 10× DNA loading buffer containing 40% glycerol, 2% SDS, and 180 mM EDTA, after which the samples were analyzed by electrophoresis using 1.5% agarose gels.
Midbrain slice cultures
Two days after in utero electroporation, E15.5 midbrains were embedded in 3% low-gelling temperature agarose (A9045-25G; Sigma-Aldrich) in Hanks’ balanced salt solution (HBSS), after which 250-μm coronal slices were obtained with a vibratome (VTI1000S; Leica). E15.5 midbrain slices were placed on a Millicell-CM (PICM0RG50; Millipore, Burlington, MA), mounted in 70% collagen gel (KP-2100; Nitta Gelatin, Fayetteville, NC), and soaked in culture medium containing Neurobasal medium minus phenol red (12348017; Thermo Fisher Scientific), 1× B27 supplement (17504044; Invitrogen), and 1 mM l-glutamine in a glass-bottom dish (3910-035; Iwaki, Tokyo, Japan). The slices in Millicell-CM dishes were kept at 37°C in a CO2 incubator (Olympus, Tokyo, Japan) equipped with a motorized inverted research microscope with focus-drift compensation IX81 (Olympus) and set at the heat stage. Neurons were observed under 5% CO2 and 60% O2. Time-lapse recordings were performed by epifluorescence microscopy with a 20× long operation distance objective lens (LMPLFLN-BD 20×, 0.40 numerical aperture; Olympus). Image acquisition was performed with Dell computers using an FV10-ASW (Olympus) control camera. The time interval of time-lapse recording was 10 min for all movies. The medium intensity projection was prepared from 10 to 15 Z-stack images at each time point.
Statistical analysis
Statistical analyses were performed using GraphPad Prism 7.0 (GraphPad Software Inc., La Jolla, CA) or using RStudio (RStudio, Boston, MA). All data are expressed as the mean ± SEM unless otherwise stated. Binary continuous variables were compared using an unpaired two-tailed t test, while binary continuous variables with different numbers were compared with Mann-Whitney test. Multiple continuous variables were compared using two-way analysis of variance (ANOVA) and Tukey’s multiple comparison test. A P value of <0.05 was considered statistically significant. No statistical methods were used to predetermine the sample size, but sample sizes were similar to those described in previous related studies (7, 8, 10–12, 43). No randomization of samples was performed. No blinding was conducted. The number of samples or cells examined in each analysis is indicated in the figure legends.
Supplementary Material
Acknowledgments
We thank A. Shitamukai (RIKEN CDB) for pCAG-FloxP-mKO2-F. We also thank T. Kawauchi (FBRI) for fruitful discussion. Funding: This work was supported by Grants-in-Aid for Challenging Exploratory Research (grant number 15K14337 to M.H.), KAKENHI (grant numbers 16H06528, 18H02538, and 19H03536 to M.H.; 17K07126 to S.T.; JP17K07071 and 17J40150 to N.A.), and Innovative Areas (grant numbers 15H01304 and 16H06528 to M.H.) from MEXT, as well as AMED (grant number 20dm0107085h0005), Naito Foundation, an Intramural Research Grant (grant numbers 27-7 and 28-4 to M.H.), and the Japan Epilepsy Research Foundation (S.T.). Author contributions: N.A., M.H., and S.T. supervised the project. N.A. designed the study and analyzed the data. N.A., M.O., S.M., K.-i.D., A.T., H.U., K.H., K.S., S.E., T.N., Y.U.I., T.I., Y.Y., K.Y., and K.K. performed the experiments. N.A. and M.H. wrote the manuscript. N.A., K.-i.D., S.T., and M.H. discussed the experimental results and commented on the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors. The DscamPA KI mouse line and used research materials can be provided by M.H., as well as pending scientific review and a completed material transfer agreement. Requests for these materials should be submitted to M.H.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/36/eaba1693/DC1
REFERENCES AND NOTES
- 1.Hatta K., Takeichi M., Expression of N-cadherin adhesion molecules associated with early morphogenetic events in chick development. Nature 320, 447–449 (1986). [DOI] [PubMed] [Google Scholar]
- 2.D’Gama A. M., Walsh C. A., Somatic mosaicism and neurodevelopmental disease. Nat. Neurosci. 21, 1504–1514 (2018). [DOI] [PubMed] [Google Scholar]
- 3.Arimura N., Dewa K.-I., Okada M., Yanagawa Y., Taya S.-I., Hoshino M., Comprehensive and cell-type-based characterization of the dorsal midbrain during development. Genes Cells 24, 41–59 (2019). [DOI] [PubMed] [Google Scholar]
- 4.Edwards M. A., Caviness V. S. Jr., Schneider G. E., Development of cell and fiber lamination in the mouse superior colliculus. J. Comp. Neurol. 248, 395–409 (1986). [DOI] [PubMed] [Google Scholar]
- 5.Das R. M., Storey K. G., Apical abscission alters cell polarity and dismantles the primary cilium during neurogenesis. Science 343, 200–204 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Itoh Y., Moriyama Y., Hasegawa T., Endo T. A., Toyoda T., Gotoh Y., Scratch regulates neuronal migration onset via an epithelial-mesenchymal transition-like mechanism. Nat. Neurosci. 16, 416–425 (2013). [DOI] [PubMed] [Google Scholar]
- 7.Zhang J., Woodhead G. J., Swaminathan S. K., Noles S. R., McQuinn E. R., Pisarek A. J., Stocker A. M., Mutch C. A., Funatsu N., Chenn A., Cortical neural precursors inhibit their own differentiation via N-cadherin maintenance of β-catenin signaling. Dev. Cell 18, 472–479 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rousso D. L., Pearson C. A., Gaber Z. B., Miquelajauregui A., Li S., Portera-Cailliau C., Morrisey E. E., Novitch B. G., Foxp-mediated suppression of N-cadherin regulates neuroepithelial character and progenitor maintenance in the CNS. Neuron 74, 314–330 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kasioulis I., Das R. M., Storey K. G., Inter-dependent apical microtubule and actin dynamics orchestrate centrosome retention and neuronal delamination. eLife 6, e26215 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Franco S. J., Martinez-Garay I., Gil-Sanz C., Harkins-Perry S. R., Müller U., Reelin regulates cadherin function via Dab1/Rap1 to control neuronal migration and lamination in the neocortex. Neuron 69, 482–497 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jossin Y., Cooper J. A., Reelin, Rap1 and N-cadherin orient the migration of multipolar neurons in the developing neocortex. Nat. Neurosci. 14, 697–703 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kawauchi T., Sekine K., Shikanai M., Chihama K., Tomita K., Kubo K.-i., Nakajima K., Nabeshima Y.-I., Hoshino M., Rab GTPases-dependent endocytic pathways regulate neuronal migration and maturation through N-cadherin trafficking. Neuron 67, 588–602 (2010). [DOI] [PubMed] [Google Scholar]
- 13.Hattori D., Millard S. S., Wojtowicz W. M., Zipursky S. L., Dscam-mediated cell recognition regulates neural circuit formation. Annu. Rev. Cell Dev. Biol. 24, 597–620 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yamagata M., Sanes J. R., Dscam and Sidekick proteins direct lamina-specific synaptic connections in vertebrate retina. Nature 451, 465–469 (2008). [DOI] [PubMed] [Google Scholar]
- 15.Garrett A. M., Tadenev A. L., Burgess R. W., DSCAMs: Restoring balance to developmental forces. Front. Mol. Neurosci. 5, 86 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Garrett A. M., Khalil A., Walton D. O., Burgess R. W., DSCAM promotes self-avoidance in the developing mouse retina by masking the functions of cadherin superfamily members. Proc. Natl. Acad. Sci. U.S.A. 115, E10216–E10224 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maynard K. R., Stein E., DSCAM contributes to dendrite arborization and spine formation in the developing cerebral cortex. J. Neurosci. 32, 16637–16650 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fuerst P. G., Koizumi A., Masland R. H., Burgess R. W., Neurite arborization and mosaic spacing in the mouse retina require DSCAM. Nature 451, 470–474 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shitamukai A., Konno D., Matsuzaki F., Oblique radial glial divisions in the developing mouse neocortex induce self-renewing progenitors outside the germinal zone that resemble primate outer subventricular zone progenitors. J. Neurosci. 31, 3683–3695 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kawajiri A., Itoh N., Fukata M., Nakagawa M., Yamaga M., Iwamatsu A., Kaibuchi K., Identification of a novel β-catenin-interacting protein. Biochem. Biophys. Res. Commun. 273, 712–717 (2000). [DOI] [PubMed] [Google Scholar]
- 21.de Rooij J., Boenink N. M., van Triest M., Cool R. H., Wittinghofer A., Bos J. L., PDZ-GEF1, a guanine nucleotide exchange factor specific for Rap1 and Rap2. J. Biol. Chem. 274, 38125–38130 (1999). [DOI] [PubMed] [Google Scholar]
- 22.Gil-Sanz C., Franco S. J., Martinez-Garay I., Espinosa A., Harkins-Perry S., Müller U., Cajal-Retzius cells instruct neuronal migration by coincidence signaling between secreted and contact-dependent guidance cues. Neuron 79, 461–477 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ye T., Ip J. P., Fu A. K. Y., Ip N. Y., Cdk5-mediated phosphorylation of RapGEF2 controls neuronal migration in the developing cerebral cortex. Nat. Commun. 5, 4826 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mino A., Ohtsuka T., Inoue E., Takai Y., Membrane-associated guanylate kinase with inverted orientation (MAGI)-1/brain angiogenesis inhibitor 1-associated protein (BAP1) as a scaffolding molecule for Rap small G protein GDP/GTP exchange protein at tight junctions. Genes Cells 5, 1009–1016 (2000). [DOI] [PubMed] [Google Scholar]
- 25.Dobrosotskaya I. Y., James G. L., MAGI-1 interacts with β-catenin and is associated with cell-cell adhesion structures. Biochem. Biophys. Res. Commun. 270, 903–909 (2000). [DOI] [PubMed] [Google Scholar]
- 26.Sakurai A., Fukuhara S., Yamagishi A., Sako K., Kamioka Y., Masuda M., Nakaoka Y., Mochizuki N., MAGI-1 is required for Rap1 activation upon cell-cell contact and for enhancement of vascular endothelial cadherin-mediated cell adhesion. Mol. Biol. Cell 17, 966–976 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yamagata M., Sanes J. R., Synaptic localization and function of Sidekick recognition molecules require MAGI scaffolding proteins. J. Neurosci. 30, 3579–3588 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hatakeyama J., Wakamatsu Y., Nagafuchi A., Kageyama R., Shigemoto R., Shimamura K., Cadherin-based adhesions in the apical endfoot are required for active Notch signaling to control neurogenesis in vertebrates. Development 141, 1671–1682 (2014). [DOI] [PubMed] [Google Scholar]
- 29.Ly A., Nikolaev A., Suresh G., Zheng Y., Tessier-Lavigne M., Stein E., DSCAM is a netrin receptor that collaborates with DCC in mediating turning responses to netrin-1. Cell 133, 1241–1254 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen B. E., Kondo M., Garnier A., Watson F. L., Püettmann-Holgado R., Lamar D. R., Schmucker D., The molecular diversity of Dscam is functionally required for neuronal wiring specificity in Drosophila. Cell 125, 607–620 (2006). [DOI] [PubMed] [Google Scholar]
- 31.Schmucker D., Clemens J. C., Shu H., Worby C. A., Xiao J., Muda M., Dixon J. E., Zipursky S. L., Drosophila Dscam is an axon guidance receptor exhibiting extraordinary molecular diversity. Cell 101, 671–684 (2000). [DOI] [PubMed] [Google Scholar]
- 32.Hughes M. E., Bortnick R., Tsubouchi A., Bäumer P., Kondo M., Uemura T., Schmucker D., Homophilic Dscam interactions control complex dendrite morphogenesis. Neuron 54, 417–427 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kamiyama D., McGorty R., Kamiyama R., Kim M. D., Chiba A., Huang B., Specification of dendritogenesis site in Drosophila aCC motoneuron by membrane enrichment of Pak1 through Dscam1. Dev. Cell 35, 93–106 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sachse S. M., Lievens S., Ribeiro L. F., Dascenco D., Masschaele D., Horré K., Misbaer A., Vanderroost N., De Smet A. S., Salta E., Erfurth M.-L., Kise Y., Nebel S., Van Delm W., Plaisance S., Tavernier J., De Strooper B., De Wit J., Schmucker D., Nuclear import of the DSCAM-cytoplasmic domain drives signaling capable of inhibiting synapse formation. EMBO J. 38, e99669 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Famulski J. K., Solecki D. J., New spin on an old transition: Epithelial parallels in neuronal adhesion control. Trends Neurosci. 36, 163–173 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shah B., Lutter D., Tsytsyura Y., Glyvuk N., Sakakibara A., Klingauf J., Püschel A. W., Rap1 GTPases are master regulators of neural cell polarity in the developing neocortex. Cereb. Cortex 27, 1253–1269 (2017). [DOI] [PubMed] [Google Scholar]
- 37.Maeta K., Edamatsu H., Nishihara K., Ikutomo J., Bilasy S. E., Kataoka T., Crucial role of Rapgef2 and Rapgef6, a family of guanine nucleotide exchange factors for Rap1 small GTPase, in formation of apical surface adherens junctions and neural progenitor development in the mouse cerebral cortex. eNeuro 3, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kawaue T., Shitamukai A., Nagasaka A., Tsunekawa Y., Shinoda T., Saito K., Terada R., Bilgic M., Miyata T., Matsuzaki F., Kawaguchi A., Lzts1 controls both neuronal delamination and outer radial glial-like cell generation during mammalian cerebral development. Nat. Commun. 10, 2780 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tavano S., Taverna E., Kalebic N., Haffner C., Namba T., Dahl A., Wilsch-Bräuninger M., Paridaen J. T. M. L., Huttner W. B., Insm1 induces neural progenitor delamination in developing neocortex via downregulation of the adherens junction belt-specific protein Plekha7. Neuron 97, 1299–1314.e8 (2018). [DOI] [PubMed] [Google Scholar]
- 40.Iossifov I., O’Roak B. J., Sanders S. J., Ronemus M., Krumm N., Levy D., Stessman H. A., Witherspoon K. T., Vives L., Patterson K. E., Smith J. D., Paeper B., Nickerson D. A., Dea J., Dong S., Gonzalez L. E., Mandell J. D., Mane S. M., Murtha M. T., Sullivan C. A., Walker M. F., Waqar Z., Wei L., Willsey A. J., Yamrom B., Lee Y.-h., Grabowska E., Dalkic E., Wang Z., Marks S., Andrews P., Leotta A., Kendall J., Hakker I., Rosenbaum J., Ma B., Rodgers L., Troge J., Narzisi G., Yoon S., Schatz M. C., Ye K., McCombie W. R., Shendure J., Eichler E. E., State M. W., Wigler M., The contribution of de novo coding mutations to autism spectrum disorder. Nature 515, 216–221 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Amano K., Fujii M., Arata S., Tojima T., Ogawa M., Morita N., Shimohata A., Furuichi T., Itohara S., Kamiguchi H., Korenberg J. R., Arata A., Yamakawa K., DSCAM deficiency causes loss of pre-inspiratory neuron synchroneity and perinatal death. J. Neurosci. 29, 2984–2996 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Purohit A. A., Li W., Qu C., Dwyer T., Shao Q., Guan K.-L., Liu G., Down syndrome cell adhesion molecule (DSCAM) associates with uncoordinated-5C (UNC5C) in netrin-1-mediated growth cone collapse. J. Biol. Chem. 287, 27126–27138 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Itoh N., Nakayama M., Nishimura T., Fujisue S., Nishioka T., Watanabe T., Kaibuchi K., Identification of focal adhesion kinase (FAK) and phosphatidylinositol 3-kinase (PI3-kinase) as Par3 partners by proteomic analysis. Cytoskeleton 67, 297–308 (2010). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/36/eaba1693/DC1