

Abstract
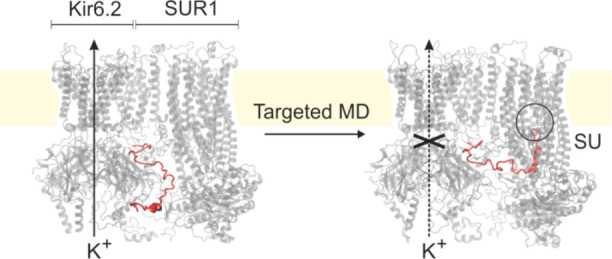
Inward rectifying potassium ion channels (KATP), sensitive to the ATP/ADP concentration ratio, play an important, control role in pancreatic β cells. The channels close upon the increase of this ratio, which, in turn, triggers insulin release to blood. Numerous mutations in KATP lead to severe and widespread medical conditions such as diabetes. The KATP system consists of a pore made of four Kir6.2 subunits and four accompanying large SUR1 proteins belonging to the ABCC transporters group. How SUR1 affects KATP function is not yet known; therefore, we created simplified models of the Kir6.2 tetramer based on recently determined cryo-EM KATP structures. Using all-atom molecular dynamics (MD) with the CHARMM36 force field, targeted MD, and molecular docking, we revealed functionally important rearrangements in the Kir6.2 pore, induced by the presence of the SUR1 protein. The cytoplasmic domain of Kir6.2 (CTD) is brought closer to the membrane due to interactions with SUR1. Each Kir6.2 subunit has a conserved, functionally important, disordered N-terminal tail. Using molecular docking, we found that the Kir6.2 tail easily docks to the sulfonylurea drug binding region located in the adjacent SUR1 protein. We reveal, for the first time, dynamical behavior of the Kir6.2/SUR1 system, confirming a physiological role of the Kir6.2 disordered tail, and we indicate structural determinants of KATP-dependent insulin release from pancreatic β cells.
I. Introduction
ATP-sensitive inwardly rectifying potassium channels (KATPs) are transmembrane proteins present in the pancreas, cardiac myocytes, skeletal muscles, and neurons.1 The great interest in understanding their function stems, i.e., from their critical role in regulation of insulin secretion from pancreatic β cells.2 In response to a change in the ratio of ATP to ADP concentrations, the channels close and stop the potassium ions outflow, which results in a rapid depolarization of the cell membrane. Upon such a signal, voltage-gated calcium channels open, and the intracellular calcium level rises in β cells. More calcium triggers insulin secretion into the blood. A change of ATP/ADP concentrations is the physiological response to the increase of the blood glucose level in healthy individuals. However, some mutations in the KATP complex may affect its function and lead to neonatal diabetes (ND)2,3 or congenial hyperinsulinism (HI).4 To date, more than 200 mutations in genes encoding the KATP channel (KCNJ11 and ABCC8 for Kir6.2 and SUR1 components, respectively) have been reported. Spatial localizations of these mutations give useful hints on KATP architecture and function.5 The KATP channels are natural targets in type 2 diabetes treatment. Drugs from the sulfonylurea (SU) group, as well as other secretogogues, are known to exert an inhibitory function on these channels.6 Despite its fundamental physiological role, the molecular mechanism of the closing and opening of KATP channels is not known yet.
A number of KATP channels structures has been revealed in 2017–2019 by three independent groups using a cryo-EM technique.7−10 These structural data have opened the door to gaining a better insight into the mechanism of KATP action.11 Cryo-EM confirmed the earlier notion12 that a KATP channel is an octamer (Figure 1a,b,d) and consists of two types of protein subunits: four Kir6.2 (inward rectifier potassium channel) and four SUR1 (sulfonylurea receptor).13 All eight protein subunits are required to make a fully functional channel.
Figure 1.

Structural details of the KATP models used in this work. (a) Ribbon representation of two Kir6.2 in one SUR1 system. The disordered region of KN tail is placed outside the SUR1 cavity (model KS) and is shown as a thick black ribbon. (b) Domain architecture of the system. Black bold lines denote regions where no clear density was observed in the cryo-EM outward open structure. (c) The sequence of the KN tail region, with three N-terminal peptides used in the tail model building indicated. (d) Top view of the KATP octamer (outward open). The shaded part represents domains which were not included in our modeling. The colored part is included in model K (four Kir6.2 proteins) and model KS (four Kir6.2 and one SUR1 protein). (e) Possible positions of the disordered KN tail region (red) in model KS and (f) N tail-constrained variants of model KS named models KNt. A color representation of domains remains the same throughout the paper.
Unfortunately, the structural picture is not yet complete. When we initiated this project, several important regions of the KATP channel remained unresolved in the available cryo-EM structures.7−10 In particular, for the N-terminus of Kir6.2, the 31 amino acid long fragment (KN tail) was not localized. A lot of experimental data indicate that this tail is a functionally important part of KATP. The possible involvement of the Kir6.2 N-terminus in channel gating modulated by SUR1 was first reported in 1999. Babenko et al.14 and Koster et al.15 noticed that the truncation of the N-terminus of Kir6.2 increases the probability of the channel opening in ligand-free solution. Riemann et al.16 have shown that such an effect manifests only in the presence of SUR1. The KN tail critically affects binding of antidiabetic drugs. The shortening of the KN tail by as few as five amino acids reduces the inhibitory effects of SUs.17,18 How SUR1 affects Kir6.2 is not clear. Lack of knowledge on the whereabouts of the KN tail seriously hampers our understanding of the KATP gating.
Recent (2018) reports from Wu et al.6,8 and Martin et al.18 suggest a possible position of the KN tail in the complex. The authors hypothesize that this unit, despite being a disordered polypetide part of the Kir6.2, interacts with SUR1 and has a critical role in the regulation of KATP conductance. Difficulties with experimental determination of the KN tail position are inherent to KATP, since it is a dynamical, signaling part of the complex. Disordered regions of proteins, for example, N- or C-termini, often have regulatory roles and currently are subjects of vigorous research.19 Should such a disordered tail play a control role in the Kir6.2/SUR1 system, it would advance our understanding of an important class of KATP ion channels.
Given these controversies regarding the position of the KN tail, and the lack of any dynamical studies of KATP models, here we have used computational modeling methods to gain a better insight into interactions between Kir6.2 and SUR1, and the role of the KN tail in the physiology of KATP. We address three questions: (1) What is the effect of association of the Kir6.2 pore with SUR1 modules on a pore’s geometry and dynamics? (2) How does the localization of the disordered KN tail affect the structure of KATP? (3) What are the local changes in the KATP pore geometry and dynamics induced by remote interactions of the KN tail with SUR1? Answering these questions brings a better understanding of the allosteric interactions and heterogeneous architecture of KATP.
As a starting point, we used the inhibitory mechanism of KATP action proposed by Wu et al.8 The following steps are distinguished in this working model: (1) The Kir6.2 pore is closed in the ATP bounded state whereas the SUR1 subunits remain in the inward facing conformation. The KN tail wedges into a large central cavity formed by the transmembrane domains of SUR1, and this insertion maintains the SUR1/ABC subunit in the open state. At the same time, restraining the KN tail prevents the CTD domain of Kir6.2 from rotation (critical for the pore opening), which results in the KATP inhibition. No potassium ions are transported. (2) The situation changes when the intracellular ADP level increases and more MgADP is produced. MgADP binds to NBDs of SUR1 (see Figure 1a), which results in a conformational change of SUR1. (3) The central cavity of SUR1 can no longer accommodate the KN tail, and this release, by not yet known signal transduction, activates the KATP channel. Potassium ions leak out of the β cell. (4) Introducing sulfonylureas to the central cavity of SUR1 inhibits the conformational change induced by MgADP binding and increases KN tail binding affinity. Thus, SU drugs promote insulin release.
In this work, we focus exclusively on step 1; therefore, we do not include any ligands in our modeling. First, we describe how the presence of SUR1 affects Kir6.2 dynamics and changes the channel pore (Section IV.1. and KATP gating (Section IV.2)
Next, we analyze the role of the N-terminal part of Kir6.2 in the KATP channel activity (Section IV.3). Using computational docking, we predict energetically favorable binding modes of N-terminal-derived peptides and propose the positioning of the Kir 6.2 N-terminus within the SU receptor cavity (Section IV.3.1). Finally, using a combination of steered and targeted MD, we drive the whole system to the conformation with the KN tail docked into SUR1, and we investigate the behavior of the systems using all-atom MD (Section IV.3.2).
To the best of our knowledge, this is the first MD study of the Kir6.2/SUR1 interface and the role of the disordered KN tail fragment in the nanomechanics of KATP channels. Structural data collected from the simulations support our hypothesis that the disordered tail is a major factor in controlling the state of KATP and thus affecting insulin release from pancreatic β cells. Our data are more general and of interest to many researchers, since KATPs are involved in the regulation of neuronal excitability,20 cardiac ischemia and stress adaptation,21 vascular smooth muscle tone,22 skeletal muscle fatigue,23 and hormonal secretion.24
II. Methods
II.1. Initial Structures and Systems Preparation
The fully solvated KATP complex model has nearly 1 000 000 atoms and is not practical for studies focused on the Kir6.2/SUR1 interface. Therefore, two basic model systems were prepared: model K (i) and model KS (ii) (see Figure 1).
-
(i)
Model K is limited to the four main pore-forming units of Kir6.2 based on the human KATP channel structure (Protein Data Bank ID 6C3P, outward open SUR1 conformation).7 Model K contains only amino acids which were already present in this structure (residues 32–353 of four Kir6.2 chains).
-
(ii)
Model KS is larger, since it is model K with one SUR1 subunit added (Figure 1a,e). We wanted to investigate the human SUR1 protein in the inward open conformation, but no such data were available. Therefore, the inward open structure of SUR1 was prepared using the targeted MD (TargMD) method, by application of external forces to the available human outward open channel. A template of the inward open conformation was necessary. We used the Cricetus cricetus channel (PDB ID 6BAA)10 as the required target to drive NBD domains of the SUR1 ABC transporter from the outward to the inward open conformation. Then, we added one such TargMD prepared inward open human SUR1 subunit to the model K and constructed the model KS. Comparisons of these two systems dynamics allowed us to study the structural impacts of SUR1 on the KATP gating. To study the role of the KN tail, variants of model KS, described in detail in Supporting Information Table S1, were prepared as well.
During construction of our models Kir6.2 and SUR1 proteins were examined for missing side chains and loops using the Schrodinger software.25 The structure of both missing terminal parts of Kir6.2 subunits in model KS were predicted using the QUARK server.26,27 Four missing regions in SUR1 structure were noticed (Figure 1b, thick black lines). Two of them interact directly with the Kir6.2 protein: the intercellular loop (ICL1) and L0 loop (Figure 1a). Structures of these loops, as well as the structures of linkers joining the NBD1 domain with TMD1 and TMD2, were predicted using the I-TASSER server.28−3026,27The initial model K and model KS structures were equilibrated during 50 ns MD simulation with implicit solvent. We used standard parameters (solvent dielectric constant 78.5, ion concentration 0.15 M). The original parts of the protein, derived from the cryo-EM experiments, were constrained during the equilibration stage.
Models K and KS were embedded into an explicit palmitoyloleoyl-phosphatidylcholine (POPC) bilayer generated by the VMD membrane module.31 The entire protein–membrane systems were solvated with the TIP3P water and 150 mM NaCl. The protonation states were assigned with the Epic algorithm (pH 7.0) in Maestro.25 Periodic boundary conditions were used in MD. The model K simulation box was 110 × 110 × 190 Å3 and consisted of approximately 150 000 atoms. For model KS, the size of a simulation box was 190 × 110 × 190 Å3 and involved ∼380 000 atoms. The prepared membrane embedded systems underwent 100 ns equilibration runs. In Section III.4, we describe details of N tail-constrained variants of model KS, named for brevity models KNt (Figure 1f, SI Table S1).
II.2. Molecular Dynamics Protocols
All MD simulations were performed with the NAMD 2.13 code.32 Bonds between hydrogens and oxygen atoms were held rigid only for water molecules (SHAKE algorithms33). The 1 fs time step was used. Long-range electrostatic forces were calculated using the Particle Mesh Ewald method (188 × 120 × 196 grid points).34 The simulations were performed with a constant temperature of 310 K maintained using Langevin dynamics and a constant pressure of 1 atm (Langevin piston). The barostat oscillation and damping time scale were set to 200 and 50 fs, respectively. The CHARMM36 force field was used in all the MD simulations.35 The VMD code was used for molecular visualization diagrams.31
II.3. Peptide Docking as a Tool for Guiding the Tail
The exact position of the KN tail is not yet known. To find the key residues which warrant the tail binding, we used automated ligand docking to model KS. We have identified the individual energetic contributions of all residues from the glibenclimide binding SUR1 cavity (further called SU region) to binding energies of different variants of KN tail pentapeptides. To select such peptides, the first 20 amino acids from the amino terminus of Kir6.2 (chain A) were considered. On the basis of the KN tail sequence (Figure 1c), we constructed 21 individual peptides having a length of five aa each.
The Glide tool (Schrodinger Inc.) was used to prepare grid maps and flexible peptide ligands for docking.25 The grid space for the docking simulation was limited to the region of a putative binding site of the tail suggested in works by Martin18 and Wu.6,8 The searched region covered the two transmembrane domains and the TMD0 domain of SUR1. Experiments were performed using a molecular structure with SUR1 protein in the inward open conformation. For each KN tail peptide, 5000 poses were generated for 10 starting conformations. The 10 poses with the lowest docking score are shown in Tables S2 and S3 in SI. Three of them were chosen as the anchor positions for the KN tail in the TargMD simulations. In that way, three alternative variants of model KS, named model KNt26, model KNt37, and model KNt48, were further investigated. Additionally, we performed a set of simulations in which the target position of the KN tail was based on the N-terminal fragment from the cryo-EM structure published by Martin et al. (courtesy of Professor Shyng)18 (model KNtM). Details are described in SI.
II.4. MD Trajectories Analysis
Prior to further analysis, the MD trajectories were aligned by the transmembrane domains of Kir6.2 subunits (residues 70–170) which were not in direct contact with SUR1.
To quantitatively monitor conformational changes of the KATP channel during simulations, carefully selected geometrical parameters were calculated using Python scripts and the MDAnalysis toolkit.36 The same toolkit was used to assess the number of hydrogen bonds.37 Couplings among parts of molecular systems were analyzed using the dynamic cross-correlation (DCC) matrices.38,39 In both model K and model KS, cross-correlation matrices were obtained by averaging data for one selected Kir6.2 subunit from three simulation runs.
III. Results and Discussion
The KATP channel is the octameric complex, and the role of SUR1 in regulation of potassium ion transport through its Kir pore is of great interest. We present and discuss the results of the MD simulations, focusing on
-
(i)
general effects of SUR1 on Kir6.2 dynamics (IV.1),
-
(ii)
modulation of the KATP gating by SUR1 (IV.2), and
-
(iii)
a functional role of conserved, disordered KN tail in KATP structure control (IV.3).
III.1. How the Presence of SUR1 Affects Dynamics of Kir6.2 Subunits?
The presence of the TMD0 domain of SUR1 is indispensable for the activity of Kir6.2 pore. SUR1 proteins were therefore called “gatekeepers”.40 As a reference for further comparisons, we run three 100 ns long unbiased simulations of an isolated pore, formed solely by four Kir6.2 subunits, embedded in a membrane and an appropriate water box (model K). All subunits exhibited similar dynamics, after 30 ns the RMSD stabilizes at the level of around 4 Å with respect to the original structure (see SI Table S2).
Root-mean-square fluctuations (RMSF) of residues from individual Kir6.2 modules are presented in Figure 2 a.
Figure 2.

RMSF of a single Kir6.2 chain. (a) Three 100 ns simulations of model K (each consists of four Kir6.2 subunits, so 12 data sets are averaged). Three 100 ns simulations of model KS (gray scale) compared to the averaged RMSF of model K (cyan) (b) for Kir6.2 chain having no direct contact with SUR1 and (c) for the Kir6.2 subunit with a direct contact with SUR1. Red triangles and yellow stars denote residues participating in ATP and PIP2 binding, respectively. Black dots refer to residues directly interacting with SUR1. Dashed rectangles denote regions in which SUR1 affects fluctuations on Kir6.2 amino acids.
One can see that TM helical regions are quite stable, and only the Turret loop (100–110) and the extracellular loop (ECL) located next to the selectivity filter (SF) (140–150) exhibit increased flexibility. The cytoplasmic CTD region (170–350) is more prone to deformations, with high mobility in 180–200, 220–230, and 252–280 areas. These areas contain flexible loops joining parts of CTD β sheets. Two such linker regions are associated with intradomain interactions (a salt bridge between R192 and E229 from adjacent Kir6.2 subunits) contributing to the stability of CTD. Elevated values of RMSF at 350 region (Figure 2) are natural, since this part of our models represents the 30 AA long C-term which is perhaps disordered, and therefore absent in the cryo-EM structures.
The model K is extended to model KS by having one extra SUR1 module added (see Figure 1d, model KS). We assessed the possible effects of the SUR1 presence on Kir6.2 dynamics by examining three 100 ns MD simulations of model KS. The RMS data indicate that the equilibrium was achieved after 30 ns of the simulation time. The RMSF data for model KS are presented in Figure 2b,c. In panel b, we show data for three Kir6.2 modules localized opposite to SUR1 and having no direct contact with this protein and in panel c for three Kir6.2 subdomains in contact with SUR1. On the basis of PDB 6BAA (inward open) and 6C3P (outward open) structures, we identified contact regions between Kir6.2 and SUR1. The interface residues are marked by black circles in Figure 2c. Detailed data are presented in Figure S3 (SI). We monitored dynamics of these interfaces during SUR1 association with Kir6.2 and enforced binding of the KN tail into the SU pocket (vide infra).
The main piece of information shown in Figure 2 is that Kir6.2 residues from the membrane embedded part (green bars in Figure 2a), including the interface region, are rather insensitive to the presence of the SUR1 protein. Differences in their dynamics (Figure 2b,c), measured by fluctuations, are localized in the loop regions and are within statistical errors.
Interestingly, the only two exceptions showing the clear sensitivity to the SUR1 presence are (i) the area near the 48–50 region which corresponds to the ATP binding site and (ii) the selectivity filter region 130–134. Thus, we conclude that SUR1 is perhaps mechanically coupled to the Kir6.2 ATP binding site and has a stabilizing role for this region. The presence of SUR1 increases also fluctuations in the SF region of the adjacent Kir6.2 subunit. MD simulations indicate a small stabilizing effect of SUR1 on the PIP2 binding region, located in the vicinity of R54, K67, R176, R177, R206, R301 (see Figure S5).
Correlations of residues in model K and model KS help to pinpoint the possible remote effects of Kir6.2–SUR1 interactions on the KATP pore dynamics. Cross-correlation matrices calculated for Cα atoms of Kir6.2 residues in models K and KS are presented in Figure 3a,b. The difference in cross-correlations between these two models is presented in Figure 3c.
Figure 3.

Cross-correlation matrices calculated for Cα atoms of Kir6.2 residues in (a) model K and (b) model KS. (c) The difference in cross-correlations between these two models.
The presence of SUR1 (model KS) clearly affects the motion of the N-ter part already resolved in cryo-EM structures. Due to interactions with the SUR1 protein, a much longer region of N-ter (47 amino acids in model KS vs 14 amino acids in model KS) is correlated with the CTD domain motions. A pattern of internal correlations/anticorrelations in the N-ter (the first 30 AA, Figure 3a,b) also changes upon SUR1 binding. Initially, there are relatively strong internal correlations within the large CTD fragment, which are attenuated by the SUR1 presence. We noticed also that SUR1 enhances interdomain correlations between CTDs located in the adjacent Kir6.2 subunits.
The TM helix M1 has a direct contact with the TMD0 domain of SUR1 (Figure 1a). We observed that M1/M2 motions change upon SUR1 attachment, and they become more correlated if SUR1 is present. Indeed, detailed analysis of M1–M2 distances showed that these helices are in closer contact in model KS than in model K (see Section IV.2.2). Also, correlations of the N-ter with M1 substantially increase when SUR1 interacts with Kir6.2. The M1/SF regions, being correlated with the CTD domain in model K, upon SUR1 binding, become anticorrelated (see Figure 3 b,c). We studied also cross-correlation matrices in the regions attributed to gating. The G-loop gate (295–296), the HBC gate (164–168), and the SF (130–134) regions do not exhibit interesting features in correlation matrices. We conclude that motions of these classical gating regions are not substantially affected by SUR1, at least on a short, 100 ns time scale.
Thus, based on the cross-correlation matrices analysis, we infer that the presence of SUR1 affects mainly cytoplasmic regions of the Kir6.2 moiety. In particular, the N-ter is affected. The dynamics of the direct interface between Kir6.2 and TMD0 of SUR1 is only slightly modified in the M1 region by the association between these two subunits. No clear signal transduction between SUR1 and Kir6.2 in the transmembrane region, especially related to helix–helix interaction, was found in our trajectories. This observation is consistent with the fact that only a fraction of mutations affecting insulin release were found in the Kir/SUR1 hydrophobic interface. The mutations W91R and P45L in Kir6.2 and SUR1, respectively, prevent the correct formation of the whole KATP system.41,42
III.2. How Does Association with SUR1 Affect KATP Gating?
There are numerous experimental observations that SUR1 affects KATP gating. At least two mechanisms may be involved: (1) direct interactions with Kir6.2 regions considered to be gates, and (2) modulation of ATP/ADP/PIP2 ligand binding affinity. Here, we focus on computational analysis of group 1 factors since we work with apo models of KATP. Calculation of the average water flux through the gating regions for all models indicates that the presence of only one SUR1 subunit (model KS and models KNt) significantly reduces permeation of water molecules through the pore (Figure 4 d). On the basis of this observation, we infer that potassium ion conductance will be affected as well.
Figure 4.

KATP channel gating. (a) Water occupancy along the pore during an example simulation run; every blue dot represents a single water molecule in the pore cavity. Right: average water occupancy per 1 Å slice along the pore axis for model K (cyan) and model KS (black). (b) Pair of Kir6.2 subunits forming the gate region in two crystal structures (inward open 6BAA and outward open 6C3P). Pore residues directly interacting with SUR1 are in red, and residues responsible for gating are shown in yellow. Volumes representing the diameter of the pore were calculated using HOLE: wide areas of the pore (pore radius >2.3 Å) are blue, water accessible parts (1.15 Å > pore radius < 2.30 Å) are green, and parts that are inaccessible to water (pore radius <1.15 Å) are red. (c) Comparison of the HOLE profiles of the pore radius of inward open 6BAA (red) and outward open 6C3P (green).43 (d) Average water flux per 1 ns of simulation for four gating regions calculated for all models: model K (cyan), model KS (black), model KNt26 (green), model KNt37 (yellow), model KNt48 (blue), and model KNtM (magenta).
The detailed mechanism in which the KATP pore alternates between the conducting state and nonconducting one has not been determined yet. Nevertheless, the Kir6.2 channels share several characteristic structural features controlling the flux of K+ ions along their electrochemical gradient (see Figure 4 b,c):
-
(a)
G-loop gate (CTD domain, close to the inner membrane),
-
(b)
helix bundle crossing gate (HBC), and
-
(c)
selectivity filter (close to the outer membrane).
Next, we discuss how SUR1 modulates regions a–c using our MD results.
III.2.1. G-Loop Gate
G-loop is a region at the apex of the cytoplasmic domain (residues 295–296) (see Figures 1a and 4b). Crystallographic and functional studies of other members of the Kir channels family suggest the gating role of this region.44−47 A mutation within the loop region I296L causes a significant decrease in ATP inhibition and gives rise to DEND syndrome (neonatal diabetes in association with developmental delay, epilepsy, or muscle weakness).48 All available cryo-EM structures of Kir6.2 determine the closed conformation of the channel, with the G-loop diameter ranging from 10.7 to 12.8 Å for the outward open 6C3P structure9 and 5YKG (“relaxed” state of Kir6.2),8 respectively. Computational studies of the Kir3.2 channel gating performed by Bernsteiner at al.47 indicated that the gate diameter, calculated as the distance between the centers of mass of the G-loop gate-forming residues G318 and M319 from the opposite Kir subunits, varies from ∼3 to 11 Å during the simulations, with the narrowest diameter allowing K+ ion permeation to be 5.9 Å. They observe also that the G-loop is remarkably narrower than the HBC gate during MD simulations. Considered here, the Kir6.2 channels have methionine replaced with isoleucine in the G-loop region. The diameter of the gate in our simulations varies from 5.9 to 17 Å. The average distance between opposite subunits oscillates around 10 Å for a narrower pair and around 14 Å for a most distant pair. Considering a 4-fold symmetry of the channel, despite its closed conformation, both pairs exhibit the G-loop gate configuration allowing the passage of hydrated K+ ions. Neither the presence of SUR1 nor a change in the position of the KN tail affects the dynamics of this gate (see SI Figure S4). This finding supports a hypothesis that this region might be important for KATP functioning, but the gate itself does not produce any significant steric obstacles for ion permeation, even in the closed channel conformation. Notably, mutations in this regions affect the proper physiology of β cells.49
III.2.2. HBC Gate and Changes in the Dynamical Properties of the Pore
In many K+ channels, the helix bundle crossing is assumed to be a gate which physically opens and closes in response to a variety of stimuli.50 The HBC region contains hydrophobic, pore facing residues F168 and L164, which form the narrowest part of the channel (see Figure 4a–c). The diameter of the gate (calculated as the minimal distance between pore occluding F168 residues) varies from 3.11 to 7.15 Å in MD simulations. It results in the average of 4.4 Å for all of our models, except one simulation run for model K, in which just one pair of the residues stays further apart (∼13 Å). A comparison of these diameters with those present in cryo-EM structures (from 5.36 to 7.1 Å) confirms that all our MD models remain in the closed state.
HBC is localized in the vicinity of the interface between Kir6.2 and SUR1. Therefore, we analyzed how SUR1 changes the conformation and dynamics of the surroundings of this gate. It is worth noting that several gain-of-function mutations in the HBC area were reported. They lead to neonatal diabetes (L170R/N/T and V64L)51,52 or to the DEND syndrome (C166Y/F and I167L).53−55 This supports our pursuit to look for the Kir6.2/SUR1 communication in this particular region.
In model KS, we observe a different pattern of H-bonds in comparison with model K. In model KS, the newly formed hydrogen bonds join L170 (M2), T71 (M1), and D65 (Slide helix) providing the direct connection with the intercellular loop of SUR1 (IL1) (see Figure 5b,d).The distance between Cα atoms of residues F168 and D65 (Figure 5 c, lower panel) is lower in model KS with respect to model K.
Figure 5.

Influence of SUR1 on the HBC gate. (a) Distortion of 2-fold symmetry of Kir6.2 upon one SUR1 binding: initial structure (left) and final structure (right). Cα atoms of residues forming the HBC gate (F168 and L164) are shown as black dots. (b) Hydrogen bonds formed in the presence of SUR1. (c) The time evolution of the distances between the Cα atom of F168 and the axis of the pore (upper panel) and between Cα atoms of F168 and D65 from the slide helix (lower panel). (d) The probability of hydrogen bond formation. As in Figure 2, data related to model K is shown in cyan, that for model KS is in black, and the gray color denotes the chain of Kir6.2 in model KS which does not interact directly with SUR1.
The presence of one SUR1 protein distorts the 2-fold symmetry of the channel in the HBC region by pulling two (out of four) opposite M2 chains of Kir6.2 toward SUR1 (see Figure 5a). As a result, the distance between the Cα atom of the F168 residue and the axis of the pore slightly increases in model KS (Figure 5c, upper panel), whereas the pairwise distance between Cα atoms remains nearly the same in all models. One can expect that adding three more SUR1 proteins to the simplified model KS (as in the available cryo-EM structures) will cause a widening of a pore with a magnitude around 1 Å, which might facilitate opening of the pore. This is in agreement with the increased P0 probability of the Kir6.2-SUR1 channels compared to those made by Kir6.2 alone.56
III.2.3. How Does the Association with SUR1 Affect Kir6.2 Selectivity Filter?
A selectivity filter (SF) is a common feature in ion channels.57 In KATP, it is composed of TIGFG (residues 130–134) and is located in the outer membrane part of the KATP pore (Figure 1a). The proper functioning of SF is critical to channel function. Each Kir6.2 subunit has two transmembrane helices M1 and M2 separated by a pore-loop that contains a characteristic GFG motif (G132–F133–G134 here) and an important E126–R136 salt bridge which stabilizes the SF region. The SF is linked directly to the pore-helix (S116–V129) (see Figure 6c). McCoy proposed several interactions within SF which might be crucial for its stability.57 We checked how the presence of SUR1 affects these interactions.
Figure 6.

Histograms showing the differences in the SF region dynamics between model K (cyan) and model KS (black): (a) the pore-helix rotation, (b) the distances between Cα atoms of the opposite SF residues, and (c) cartoon diagrams depicting interactions within the selectivity filters and surrounding. Selectivity filter residue carbon atoms are yellow, and calculated bonds are shown as dashed black lines. (d) The dihedral angle ψ of F133, (e) the distance of the hydrogen bond forming residues E126 and I131, and (f) the distance between the salt bridge forming residues E126 and R136.
From the aggregated MD data, we constructed histograms of parameters used to monitor an impact of the association between SUR1 and Kir6.2 on the SF structure. In Figure 6a, we show that in the absence of SUR1 (model K) the inclination of the pore-helix relative to the pore axis (θ, Figure 6c) varies with a magnitude of 7°. Binding one SUR1 protein to the Kir6.2 tetramer (model KS) restrains such movement to a much narrower distribution, and it shifts the mean value back to those measured in the experimental structures (θ = 47.4° and θ = 48° for inward open and outward open structure, respectively). The SF is linked to the pore-helix, and its structure is also affected by the presence of SUR1. Distances between opposing Cα atoms of SF forming residues are shown in Figure 6b. While distances calculated for model KS show a single narrow distribution, those calculated for model K show much higher variability.50Additionally, we calculated histograms of distances between two critical pairs of residues in the SF region (Figure 6e,f) and dihedral ψ angles of residue F133 (Figure 6d). The distribution of distances between E126–I132 is narrower in model KS than that calculated for model K. This means that SUR1 stabilizes the geometry of the SF region. The fact that carbonyl-oxygen flips of F133 are not observed in model KS (Figure 6d) also corroborates this hypothesis. The stabilization of the SF region is further supported by the data shown in Figure 6f. The E126–R136 salt bridge is crucial for proper functioning of SF. It was present practically all the time during model KS simulations, while in the model K case a wide distribution of E126–R136 distances indicates only a transient character of this link. This salt bridge is physiologically important since mutations of R136 lead to serious dysfunction of KATP.42
Proks et al.58 distinguished slow and fast gating of KATP. The authors linked conformational changes in SF to the fast gating. Our MD data confirm that remote interactions with SUR1 do affect the SF region. However, the perturbation we studied here is quite large (on/off type). What the effects are of subtle conformational changes in SUR1 itself, for example, upon KN tail or drug binding to the SU region, on fast gating is still an open question. Perhaps SF may be controlled indirectly by changes in CTD as well, as suggested by Clarke et al.59 These hypotheses require more extensive MD simulations.
III.2.4. Association of Kir6.2 Complex with SUR1 Affects Position and Orientation of CTD Domain
The cytoplasmic domain of Kir6.2 (CTD, Figure 1a) and the SF are considered to be major factors controlling Kir6.2 gating.59 All components of the KATP channel (ATP binding site, PIP2 binding site, KN tail orientation, a contact with SUR1 governed by open/close orientation of SUR1) affect the CTD domain. Additionally, the relative position of CTD with respect to the transmembrane part of the channel is different in “activated” and “inactivated” states of KATP.
Numerous mutations changing the KATP function were found in this region.5 Those mutations affect both binding sites of ligands and the proper intersubunit communication. For example, the disruption of intersubunit salt bridges (R192–E229, R301–E292, and R314–E229) at the interface of CTDs induces a loss of the channel activity.60,61
To check what structural transformations of CTD are induced solely by the contacts between SUR1 and Kir6.2, we compared the dynamics of CTD domains in model K and model KS. We have monitored the position along the z-axis (Figure 7c) and the orientation (angle β, Figure 7a,b) of the CTD domain with respect to the axis of the KATP channel, as well as the stability of salt bridges R192–E229′, and R301–E292′ (Figure 7d,e).
Figure 7.

Dynamics of the CTD domain. (a) Ribbon diagram of Kir6.2 tetramer showing interdomain interactions of CTD. (b) An angle β between CTD and the pore axis. (c) Position of the center of mass of CTD along the pore axis for model K (cyan) and model KS. Gray color states for the Kir6.2 chain without direct contact with SUR1 in model KS. (d, e) The closest distance between the residues forming salt bridges.
The close proximity of a large SUR1 ABC transporter in model KS affects the orientation of CTD substantially. The center of mass of the CTD domain upon contact with SUR1 is shifted upward (closer to the membrane) by 4 Å with respect to the coordinates of the opposite Kir6.2 module separated from SUR1. This shift is also noticeable (2 Å) with respect to the positions of CTD domains in model K (Figure 7c). At the same time, a rotation (from 35° to 50°) of the whole CTD domain induced by interactions with SUR1 is observed (Figure 7b). Thus, the presence of SUR1 in the open conformation clearly brings the whole Kir6.2 structure closer to the activated state. Additionally, we observe that only in the presence of SUR1 are two important “cavities” serving as the biding sites for ATP and PIP2 properly formed (see SI).
III.3. How Does the Localization of a Disordered KN Tail of Kir6.2 Affect KATP Function?
During evolution, protein sequences change in time due to random mutations and deletions. This alters their physicochemical properties and structural characteristics. Parts of proteins that are maintained by the natural selection often have an important biological function which is therefore protected.
The presence of a long and disordered N-terminal part is a common feature of Kir6 channels, and its activity might warrant proper Kir6.2-SUR1 coupling and molecular signal transduction.6,8,18 In known organisms having functional KATP channels (chordates), both the sequence and the length of the KN tail are highly conserved. In comparison, the level of conservation of the C-terminus in those channels is very low.
A localization of a disordered KN tail in the known cryo-EM structures of KATP is a matter of debate.6,8,18,62 Importantly, there is no open conformation of the SUR1 part of human KATP channels determined yet. In order to determine possible binding modes of the KN tail to the SU region in SUR1, we generated, using SMD and TargMD protocols, a new, structural homology based, structure of the open form of human SUR1 (see Methods section and SI). The SU region is important for binding antidiabetic drugs that keep the KATP channel closed.6,18,63
III.3.1. Docking Studies Reveal Possible KN Tail Binding Modes to the SU Binding Region
The KN tail is disordered and has no defined secondary structure, but it apparently participates in KATP closing and opening. Wu et al.8 hypothesize that binding of the KN tail to open the SUR1 protein transiently locks the KATP channel. Only very recently has a putative position of the KN tail been proposed on the basis of cryo-EM data.18 We asked a question regarding whether there is any “preferable” way for the binding of the KN tail into the SU binding region. We docked all tail-derived consecutive pentapeptides to our open form of human SUR1 (results are shown in SI Table 2).
Only three pentapeptides, representing the first part of the KN tail, were successfully docked into the SU pocket. All the other 18 peptides docked in remote places of SUR1. The docking scores of pentapeptides immersed in the SU site were similar (KNt37, −7.8 kcal/mol; KNt48, −7.0 kcal/mol; KNt26, −6.5 kcal/mol). Only those three structures were considered to be promising starting points for further MD simulations. The interactions of the KN tail end with the SUR1 SU pocket are shown in Figure 8a–g.
Figure 8.

Docking of pentapeptides to the SUR1 cavity. (a) General view of the SUR1 structure with the KN tail peptide shown in black and the approximate binding position of GBM63 shown as a pink volume. (b–d) KNt26, KNt37, and KNt48 pentapetide binding position in a top view, and (e–g) side view, with the main residues forming the binding spaces shown in a cyan (TMD1) and orange (TMD2) stick representation.
We monitored a number of hydrogen bonds between the KN tail and Kir (and SUR1) protein moieties during enforced docking of the tail. We found that the diffusion of the tail on the surface does not change that number much; it oscillates around 6. The number of H-bonds between the tail and SUR1 increases from 1 to 5 during this process.
Our data indicate that all three configurations of the tail ending are plausible. However, the best (in terms of energy) docking pose was observed for the KNt37 model. This indicates that the tail penetrates the SU pocket in the open form of SUR1. Obviously, the tail blocks a fast return of SUR1 to the closed form and regulates KATP conductivity.
III.3.2. How Does the Tail Localization Affect Kir6.2 and SUR1 Structure/Dynamic?
One may expect that the exact embedding of the KN tail in the SU pocket modifies Kir6.2 dynamics. We monitored RMSF of Kir6.2 residues during simulations. We did not notice any substantial changes in the Kir6.2 RMSF patterns (SI, Figure S1ab). More interesting effects were observed in the SUR1 RMSF data (Figure 9).
Figure 9.

Impact of the KN tail embedding mode into the SU region on the SUR1 dynamics. RMSF of SUR1 are calculated for five models: KNt26 (green), KNt37 (yellow), KNt48 (blue), and KNtM (magenta) and compared to KS (black) shown in each panel.
Two main observations from SUR1 RMSF data are the following: (1) In the SU region (residues no. ∼500 and ∼1100), higher fluctuations are noted, induced by the KN tail presence. (2) The NBD2 domain is much more delocalized by a rigid motion during MD than the NBD1 unit. That means that NBD2 domain dynamics is indeed affected by the presence of an NKt tail docked in the SU region. More data on KN mechanics and interactions with SUR1 residues may be found in the SI.
The general effect of KN tail docking on SUR1 is presented in Figure 10a–c. The presence of the tail, as expected, stabilized SUR1 in the open form. The size of the SU region in the open SUR1 is bigger that in the closed one (Figure 10c). The size of the SU region allows for multiple binding modes of the KN tail; we found that at least three modes (KNt26, KNt37, KNt48, and KNtM) are possible. Notably, the distances between NBD domains depend on the KN tail positions (Figure 11b) ranging from ∼34 Å for model KS to above 50 Å in model KNt26. Values of NDB separations measured for cryo-EM structures vary from 28.6 Å, for the structure of SUR1 in the outward open form, to 41.3–44.1 Å for SUR1 in the inward open form.9,10
Figure 10.

Structural changes in SUR1 upon KN tail binding. (a) General view of SUR1, (b) bottom view of NBD domains, (c) bottom view of TMD domains showing the SU/tail binding cavity for model KNt26 (left), model KS (middle), and preopen state of model KS (right).
Figure 11.

Disordered KN tail embedding mode (models KNt) into the SU region in SUR1 affects dynamics of KATP: (a) slide helix rotation, (b) distance between NBD1 and NBD2, and (c) distance between interdomain salt bridge forming residues D58 and R206′. Colors denote KNt models; data for model KS are shown in black.
Rotation of the slide helix (AA 55–65) is affected by the docking of the KN tail to the SU region. This is one of the most important findings of this study. The slide helix amino acids participate in formation of the PIP2 binding site and the ATP binding site. Moreover, this helix interacts directly with the HBC gate in the Kir6.2 pore (Figure 7b,c). The MD data presented in Figure 11a show that the slide helix orientation is sensitive to the presence of SUR1 and the specific orientation of the KN tail. Once the tail is docked into the SU pocket, the slide helix rotates by some 10°.
We also found that potentially important interdomain CTD-CTD′ salt bridge D58–R206′ is strongly affected by the KN tail localization (Figure11c). In the absence of the localized tail, the salt bridge is sometimes broken, but when the tail is captured in the SU pocket, as in model KNt37 and model KNtM, the presence of the bridge is much more probable. Thus, we infer that that interdomain D58–R206′ salt bridge is an important component of the allosteric regulation of KATP. In the real KATP systems, a chain of such salt bridges couples together all CTD domains. Notably, the slide helix mutations heavily affect the KATP functioning.3,5
IV. Conclusions
The recent discovery of relatively high resolution cryo-EM structures of KATP channels7−10,18 provided an excellent basis for computer modeling of possible allosteric regulations involved in the insulin release pathway and other physiological processes. We used the targeted MD approach to construct simplified models of Kir6.2 coupled with an open form of the human SUR1 protein. We focused on the postulated physiological role of the well-conserved, structurally disordered KN tail of the Kir6.2 subunit possibly interacting with the adjacent SUR1. Presumably this tail “sneaks in” into the sulfonylurea drug binding region of SUR1. Using molecular docking, we determined preferred conformations of all (21) pentapeptides derived from the KN tail in that SU region. Modeling indicates that only 3 pentapetides (KNt26, KNt37, KNt48) effectively come into stable interactions with SUR1.
We systematically compared the dynamics of the Kir6.2 pore model having 4 bare subunits (model K) with a bigger system (model KS) having just one full SUR1 unit in the inward open conformation attached to model K. The MD results reveal a number of conformational changes induced in the generic model K by the interactions with SUR1. Major conformational effects observed already on the 100 ns time scale include the following: (1) The HBC gate of Kir6.2 has closer distances between the M1 and M2 helices due to the interaction with SUR1. This effect may modulate the K+ ion transfer rate. (2) SUR1 limits rotations in the pore-helix which in turn provides stabilization of SF. (3) The SUR1 presence favors the activated KATP state versus the inactivated one by bringing the TMD domain closer to the cell membrane. On the basis of the present modeling, we may conclude that the disordered KN tail is a major factor in controlling KATP function. It keeps SUR1 in the open form and is able to limit rotation of the CTD domain. This underlines the role of the conserved, disordered protein tail in the molecular pathway controlling insulin release from pancreatic β cells.
Observations from dynamical modeling of our potassium channel models provide, perhaps for the first time, the basis and a starting point for further exploration and explanation of the structural mechanisms and allosteric regulation of the KATP systems. The local interactions, revealed in the present MD analysis, may be experimentally tested. The simulations of more extended KATP models, i.e., having ATP/MgADP/SU ligands, are under way in our laboratory.
Acknowledgments
The authors acknowledge funding from the National Science Centre, Poland (Grant 2016/23/B/ST4/01770). The computational results used in this paper were obtained with the support of the Interdisciplinary Centre for Mathematical and Computational Modelling ICM, University of Warsaw, under Grant GA76-10, the TASK computational centre in Gdansk, and the facilities of the Interdisciplinary Centre for Modern Technologies, NCU, Poland.
Glossary
Abbreviations
- KATP
ATP-sensitive potassium channel
- KN tail
Kir6.2 N-terminus
- MD
molecular dynamics
- TargMD
targeted molecular dynamics
- SMD
steered molecular dynamics
- ND
neonatal diabetes
- HI
congenital hyperinsulinism of infancy
- SU
sulfonylurea
- GBM
glibenclamide
- HBC
helix bundle crossing
- SF
selectivity filter
- CTD
Kir6.2 cytoplasmic domain
- TMD
Kir6.2 transmembrane domain
- NBD
SUR1 nucleotide binding domain
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jpcb.0c02720.
Additional data as figures and tables along with SMD/TargMD protocol details (PDF)
Author Contributions
W.N. and K.W.-S. designed the research. K.W.-S. performed simulations. W.N. and K.W.-S. analyzed data and wrote the manuscript. Both authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Hibino H.; Inanobe A.; Furutani K.; Murakami S.; Findlay I.; Kurachi Y. Inwardly rectifying potassium channels: their structure, function, and physiological roles. Physiol. Rev. 2010, 90 (1), 291–366. 10.1152/physrev.00021.2009. [DOI] [PubMed] [Google Scholar]
- Ashcroft F. M. ATP-sensitive potassium channelopathies: focus on insulin secretion. J. Clin. Invest. 2005, 115 (8), 2047–58. 10.1172/JCI25495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashcroft F. M.; Puljung M. C.; Vedovato N. Neonatal Diabetes and the KATP Channel: From Mutation to Therapy. Trends Endocrinol. Metab. 2017, 28 (5), 377–387. 10.1016/j.tem.2017.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunne M. J.; Cosgrove K. E.; Shepherd R. M.; Aynsley-Green A.; Lindley K. J. Hyperinsulinism in infancy: from basic science to clinical disease. Physiol. Rev. 2004, 84 (1), 239–75. 10.1152/physrev.00022.2003. [DOI] [PubMed] [Google Scholar]
- Walczewska-Szewc K.; Nowak W. Spacial models of malfunctioned protein complexes help to elucidate signal transduction critical for insulin release. BioSystems 2019, 177, 48–55. 10.1016/j.biosystems.2018.11.001. [DOI] [PubMed] [Google Scholar]
- Wu J. X.; Ding D.; Wang M.; Chen L. Structural Insights into the Inhibitory Mechanism of Insulin Secretagogues on the Pancreatic ATP-Sensitive Potassium Channel. Biochemistry 2020, 59 (1), 18–25. 10.1021/acs.biochem.9b00692. [DOI] [PubMed] [Google Scholar]
- Lee K. P. K.; Chen J.; MacKinnon R. Molecular structure of human KATP in complex with ATP and ADP. eLife 2017, 6, e32481. 10.7554/eLife.32481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J. X.; Ding D.; Wang M.; Kang Y.; Zeng X.; Chen L. Ligand binding and conformational changes of SUR1 subunit in pancreatic ATP-sensitive potassium channels. Protein Cell 2018, 9 (6), 553–567. 10.1007/s13238-018-0530-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N.; Wu J.-X.; Ding D.; Cheng J.; Gao N.; Chen L. Structure of a pancreatic ATP-sensitive potassium channel. Cell 2017, 168 (1–2), 101–110. 10.1016/j.cell.2016.12.028. [DOI] [PubMed] [Google Scholar]
- Martin G. M.; Yoshioka C.; Rex E. A.; Fay J. F.; Xie Q.; Whorton M. R.; Chen J. Z.; Shyng S.-L. Cryo-EM structure of the ATP-sensitive potassium channel illuminates mechanisms of assembly and gating. eLife 2017, 6, e24149. 10.7554/eLife.24149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puljung M. C. Cryo-electron microscopy structures and progress toward a dynamic understanding of KATP channels. J. Gen. Physiol. 2018, 150 (5), 653–669. 10.1085/jgp.201711978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tung R. T.; Kurachi Y. On the mechanism of nucleotide diphosphate activation of the ATP-sensitive K+ channel in ventricular cell of guinea-pig. J. Physiol. 1991, 437, 239–56. 10.1113/jphysiol.1991.sp018593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikhailov M. V.; Campbell J. D.; de Wet H.; Shimomura K.; Zadek B.; Collins R. F.; Sansom M. S. P.; Ford R. C.; Ashcroft F. M. 3-D structural and functional characterization of the purified KATP channel complex Kir6.2-SUR1. EMBO J. 2005, 24 (23), 4166–4175. 10.1038/sj.emboj.7600877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babenko A. P.; Gonzalez G.; Bryan J. The N-Terminus of KIR6. 2 Limits Spontaneous Bursting and Modulates the ATP-Inhibition of KATPChannels. Biochem. Biophys. Res. Commun. 1999, 255 (2), 231–238. 10.1006/bbrc.1999.0172. [DOI] [PubMed] [Google Scholar]
- Koster J.; Marshall B.; Ensor N. e. a.; Corbett J.; Nichols C. Targeted overactivity of β cell KATP channels induces profound neonatal diabetes. Cell 2000, 100 (6), 645–654. 10.1016/S0092-8674(00)80701-1. [DOI] [PubMed] [Google Scholar]
- Reimann F.; Tucker S. J.; Proks P.; Ashcroft F. M. Involvement of the N-terminus of Kir6. 2 in coupling to the sulphonylurea receptor. J. Physiol. 1999, 518 (2), 325–336. 10.1111/j.1469-7793.1999.0325p.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devaraneni P. K.; Martin G. M.; Olson E. M.; Zhou Q.; Shyng S.-L. Structurally distinct ligands rescue biogenesis defects of the KATP channel complex via a converging mechanism. J. Biol. Chem. 2015, 290, 7980. 10.1074/jbc.M114.634576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin G. M.; Sung M. W.; Yang Z.; Innes L. M.; Kandasamy B.; David L. L.; Yoshioka C.; Shyng S.-L. Mechanism of pharmacochaperoning in a mammalian KATP channel revealed by cryo-EM. eLife 2019, 8, e46417. 10.7554/eLife.46417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uversky V. N., Intrinsically Disordered Proteins and Their “Mysterious” (Meta)Physics. Front. Phys. 2019, 7 ( (10), ). 10.3389/fphy.2019.00010 [DOI] [Google Scholar]
- Hernández-Sánchez C.; Basile A. S.; Fedorova I.; Arima H.; Stannard B.; Fernandez A. M.; Ito Y.; LeRoith D. Mice transgenically overexpressing sulfonylurea receptor 1 in forebrain resist seizure induction and excitotoxic neuron death. Proc. Natl. Acad. Sci. U. S. A. 2001, 98 (6), 3549–54. 10.1073/pnas.051012898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zingman L. V.; Hodgson D. M.; Bast P. H.; Kane G. C.; Perez-Terzic C.; Gumina R. J.; Pucar D.; Bienengraeber M.; Dzeja P. P.; Miki T.; Seino S.; Alekseev A. E.; Terzic A. Kir6.2 is required for adaptation to stress. Proc. Natl. Acad. Sci. U. S. A. 2002, 99 (20), 13278–83. 10.1073/pnas.212315199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miki T.; Suzuki M.; Shibasaki T.; Uemura H.; Sato T.; Yamaguchi K.; Koseki H.; Iwanaga T.; Nakaya H.; Seino S. Mouse model of Prinzmetal angina by disruption of the inward rectifier Kir6.1. Nat. Med. 2002, 8 (5), 466–72. 10.1038/nm0502-466. [DOI] [PubMed] [Google Scholar]
- Gong B.; Miki T.; Seino S.; Renaud J. M. A K(ATP) channel deficiency affects resting tension, not contractile force, during fatigue in skeletal muscle. Am. J. Physiol. Cell physiol 2000, 279 (5), C1351–8. 10.1152/ajpcell.2000.279.5.C1351. [DOI] [PubMed] [Google Scholar]
- MacDonald P. E.; De Marinis Y. Z.; Ramracheya R.; Salehi A.; Ma X.; Johnson P. R.; Cox R.; Eliasson L.; Rorsman P. A K ATP channel-dependent pathway within alpha cells regulates glucagon release from both rodent and human islets of Langerhans. PLoS Biol. 2007, 5 (6), e143. 10.1371/journal.pbio.0050143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrödinger LLC : New York, NY, 2018.
- Xu D.; Zhang Y. Ab initio protein structure assembly using continuous structure fragments and optimized knowledge-based force field. Proteins: Struct., Funct., Genet. 2012, 80 (7), 1715–1735. 10.1002/prot.24065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu D.; Zhang Y. Toward optimal fragment generations for ab initio protein structure assembly. Proteins: Struct., Funct., Genet. 2013, 81 (2), 229–239. 10.1002/prot.24179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy A.; Kucukural A.; Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat. Protoc. 2010, 5 (4), 725–38. 10.1038/nprot.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J.; Yan R.; Roy A.; Xu D.; Poisson J.; Zhang Y. The I-TASSER Suite: protein structure and function prediction. Nat. Methods 2015, 12 (1), 7–8. 10.1038/nmeth.3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y. I-TASSER server for protein 3D structure prediction. BMC Bioinf. 2008, 9, 40. 10.1186/1471-2105-9-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. VMD: Visual molecular dynamics. J. Mol. Graphics 1996, 14 (1), 33–38. 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Phillips J. C.; Braun R.; Wang W.; Gumbart J.; Tajkhorshid E.; Villa E.; Chipot C.; Skeel R. D.; Kalé L.; Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26 (16), 1781–1802. 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryckaert J.-P.; Ciccotti G.; Berendsen H. J. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23 (3), 327–341. 10.1016/0021-9991(77)90098-5. [DOI] [Google Scholar]
- Darden T.; York D.; Pedersen L. Particle mesh Ewald: An N· log (N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98 (12), 10089–10092. 10.1063/1.464397. [DOI] [Google Scholar]
- Huang J.; Rauscher S.; Nawrocki G.; Ran T.; Feig M.; de Groot B. L.; Grubmüller H.; MacKerell A. D. CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat. Methods 2017, 14 (1), 71–73. 10.1038/nmeth.4067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaud-Agrawal N.; Denning E. J.; Woolf T. B.; Beckstein O. MDAnalysis: A toolkit for the analysis of molecular dynamics simulations. J. Comput. Chem. 2011, 32 (10), 2319–2327. 10.1002/jcc.21787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregoret L. M.; Rader S. D.; Fletterick R. J.; Cohen F. E. Hydrogen bonds involving sulfur atoms in proteins. Proteins: Struct., Funct., Genet. 1991, 9 (2), 99–107. 10.1002/prot.340090204. [DOI] [PubMed] [Google Scholar]
- McCammon J. Protein dynamics. Rep. Prog. Phys. 1984, 47 (1), 1. 10.1088/0034-4885/47/1/001. [DOI] [Google Scholar]
- Kasahara K.; Fukuda I.; Nakamura H. A novel approach of dynamic cross correlation analysis on molecular dynamics simulations and its application to Ets1 dimer-DNA complex. PLoS One 2014, 9 (11), e112419–e112419. 10.1371/journal.pone.0112419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babenko A. P.; Bryan J. Sur domains that associate with and gate KATP pores define a novel gatekeeper. J. Biol. Chem. 2003, 278 (43), 41577–80. 10.1074/jbc.C300363200. [DOI] [PubMed] [Google Scholar]
- Crane A.; Aguilar-Bryan L. Assembly, maturation, and turnover of K(ATP) channel subunits. J. Biol. Chem. 2004, 279 (10), 9080–90. 10.1074/jbc.M311079200. [DOI] [PubMed] [Google Scholar]
- Lang V.; Light P. E. The molecular mechanisms and pharmacotherapy of ATP-sensitive potassium channel gene mutations underlying neonatal diabetes. Pharmacogenomics Pers. Med. 2010, 3, 145–161. 10.2147/PGPM.S6969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smart O. S.; Neduvelil J. G.; Wang X.; Wallace B. A.; Sansom M. S. HOLE: a program for the analysis of the pore dimensions of ion channel structural models. J. Mol. Graphics 1996, 14 (6), 354–60. 10.1016/S0263-7855(97)00009-X. [DOI] [PubMed] [Google Scholar]
- Pegan S.; Arrabit C.; Zhou W.; Kwiatkowski W.; Collins A.; Slesinger P. A.; Choe S. Cytoplasmic domain structures of Kir2.1 and Kir3.1 show sites for modulating gating and rectification. Nat. Neurosci. 2005, 8 (3), 279–87. 10.1038/nn1411. [DOI] [PubMed] [Google Scholar]
- Whorton M. R.; MacKinnon R. X-ray structure of the mammalian GIRK2-beta gamma G-protein complex. Nature 2013, 498 (7453), 190–7. 10.1038/nature12241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen S. B.; Tao X.; MacKinnon R. Structural basis of PIP2 activation of the classical inward rectifier K+ channel Kir2.2. Nature 2011, 477 (7365), 495–498. 10.1038/nature10370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernsteiner H.; Zangerl-Plessl E. M.; Chen X.; Stary-Weinzinger A. Conduction through a narrow inward-rectifier K(+) channel pore. J. Gen. Physiol. 2019, 151 (10), 1231–1246. 10.1085/jgp.201912359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proks P.; Girard C.; Haider S.; Gloyn A. L.; Hattersley A. T.; Sansom M. S.; Ashcroft F. M. A gating mutation at the internal mouth of the Kir6.2 pore is associated with DEND syndrome. EMBO Rep. 2005, 6 (5), 470–5. 10.1038/sj.embor.7400393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushman J. D.; Zhou Q.; Shyng S.-L. A Kir6.2 pore mutation causes inactivation of ATP-sensitive potassium channels by disrupting PIP2-dependent gating. PLoS One 2013, 8 (5), e63733–e63733. 10.1371/journal.pone.0063733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopec W.; Rothberg B. S.; de Groot B. L. Molecular mechanism of a potassium channel gating through activation gate-selectivity filter coupling. Nat. Commun. 2019, 10 (1), 5366. 10.1038/s41467-019-13227-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massa O.; Iafusco D.; D’Amato E.; Gloyn A. L.; Hattersley A. T.; Pasquino B.; Tonini G.; Dammacco F.; Zanette G.; Meschi F.; et al. KCNJ11 activating mutations in Italian patients with permanent neonatal diabetes. Hum. Mutat. 2005, 25 (1), 22–27. 10.1002/humu.20124. [DOI] [PubMed] [Google Scholar]
- Männikkö R.; Jefferies C.; Flanagan S. E.; Hattersley A.; Ellard S.; Ashcroft F. M. Interaction between mutations in the slide helix of Kir6.2 associated with neonatal diabetes and neurological symptoms. Hum. Mol. Genet. 2010, 19 (6), 963–972. 10.1093/hmg/ddp554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Della Manna T.; Battistim C.; Radonsky V.; Savoldelli R. D.; Damiani D.; Kok F.; Pearson E. R.; Ellard S.; Hattersley A. T.; Reis A. F. Glibenclamide unresponsiveness in a Brazilian child with permanent neonatal diabetes mellitus and DEND syndrome due to a C166Y mutation in KCNJ11 (Kir6. 2) gene. Arq. Bras. Endocrinol. Metabol. 2008, 52 (8), 1350–1355. 10.1590/S0004-27302008000800024. [DOI] [PubMed] [Google Scholar]
- Gloyn A. L.; Pearson E. R.; Antcliff J. F.; Proks P.; Bruining G. J.; Slingerland A. S.; Howard N.; Srinivasan S.; Silva J. M.; Molnes J. Activating mutations in the gene encoding the ATP-sensitive potassium-channel subunit Kir6. 2 and permanent neonatal diabetes. New Eng. J. Med. 2004, 350 (18), 1838–1849. 10.1056/NEJMoa032922. [DOI] [PubMed] [Google Scholar]
- Shimomura K.; Horster F.; de Wet H.; Flanagan S. E.; Ellard S.; Hattersley A. T.; Wolf N. I.; Ashcroft F.; Ebinger F. A novel mutation causing DEND syndrome: a treatable channelopathy of pancreas and brain. Neurology 2007, 69 (13), 1342–9. 10.1212/01.wnl.0000268488.51776.53. [DOI] [PubMed] [Google Scholar]
- Nichols C. G. KATP channels as molecular sensors of cellular metabolism. Nature 2006, 440 (7083), 470–6. 10.1038/nature04711. [DOI] [PubMed] [Google Scholar]
- McCoy J. G.; Nimigean C. M. Structural correlates of selectivity and inactivation in potassium channels. Biochim. Biophys. Acta, Biomembr. 2012, 1818 (2), 272–85. 10.1016/j.bbamem.2011.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proks P.; Capener C. E.; Jones P.; Ashcroft F. M. Mutations within the P-loop of Kir6.2 modulate the intraburst kinetics of the ATP-sensitive potassium channel. J. Gen. Physiol. 2001, 118 (4), 341–353. 10.1085/jgp.118.4.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke O. B.; Caputo A. T.; Hill A. P.; Vandenberg J. I.; Smith B. J.; Gulbis J. M. Domain Reorientation and Rotation of an Intracellular Assembly Regulate Conduction in Kir Potassium Channels. Cell 2010, 141 (6), 1018–1029. 10.1016/j.cell.2010.05.003. [DOI] [PubMed] [Google Scholar]
- Borschel W. F.; Wang S.; Lee S.; Nichols C. G. Control of Kir channel gating by cytoplasmic domain interface interactions. J. Gen. Physiol. 2017, 149 (5), 561–576. 10.1085/jgp.201611719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y. W.; Bushman J. D.; Yan F. F.; Haidar S.; MacMullen C.; Ganguly A.; Stanley C. A.; Shyng S. L. Destabilization of ATP-sensitive potassium channel activity by novel KCNJ11 mutations identified in congenital hyperinsulinism. J. Biol. Chem. 2008, 283 (14), 9146–56. 10.1074/jbc.M708798200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding D.; Wang M.; Wu J. X.; Kang Y.; Chen L. The Structural Basis for the Binding of Repaglinide to the Pancreatic KATP Channel. Cell Rep. 2019, 27 (6), 1848–1857. 10.1016/j.celrep.2019.04.050. [DOI] [PubMed] [Google Scholar]
- Martin G. M.; Kandasamy B.; DiMaio F.; Yoshioka C.; Shyng S. L. Anti-diabetic drug binding site in a mammalian KATP channel revealed by Cryo-EM. eLife 2017, 6, e31054. 10.7554/eLife.31054. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.