

Abstract
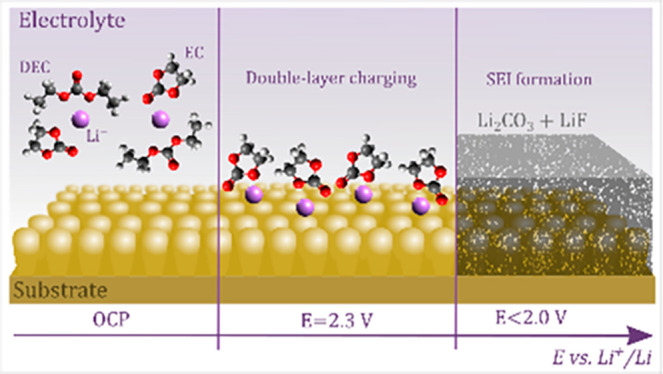
The solid electrolyte interphase (SEI) is the most critical yet least understood component to guarantee stable and safe operation of a Li-ion cell. Herein, the early stages of SEI formation in a typical LiPF6 and organic carbonate-based Li-ion electrolyte are explored by operando surface-enhanced Raman spectroscopy, on-line electrochemical mass spectrometry, and electrochemical quartz crystal microbalance. The electric double layer is directly observed to charge as Li+ solvated by ethylene carbonate (EC) progressively accumulates at the negatively charged electrode surface. Further negative polarization triggers SEI formation, as evidenced by H2 evolution and electrode mass deposition. Electrolyte impurities, HF and H2O, are reduced early and contribute in a multistep (electro)chemical process to an inorganic SEI layer rich in LiF and Li2CO3. This study is a model example of how a combination of highly surface-sensitive operando characterization techniques offers a step forward to understand interfacial phenomena in Li-ion batteries.
Li-ion batteries (LIBs) are today transforming both the transportation sector and energy infrastructure of our societies toward the use of electric vehicles and renewable electricity. Among the various aspects of the LIB technology, the solid electrolyte interphase (SEI) is one of the most critical yet least understood components. The SEI is known to be a protective layer that forms in situ on the negative electrode primarily in the first cycle of the Li-ion cell and thereafter stabilizes its operation until the end of life. However, despite more than two decades of intense research activity, the formation mechanism, composition, and morphology of the SEI remain debated and not completely understood.1−3
Numerous advanced micro/spectroscopic characterization techniques have been developed and applied to study the SEI.4 The technological advancement in the field has recently been immense and provided us with a multitude of insights into the nature and operating mechanism of these electrode layers. However, because of the nanometer dimensions and multicomponent composite structure of the SEI, no characterization approach is straightforward, and much work remains. Particularly, the complex multistep (electro)chemical formation process of the SEI raises several major experimental challenges, and real-time in situ- or operando-based methodologies are imperative.5 Fundamental understanding of factors underlining the formation and stability of the SEI promises to provide tailored SEI design and optimization principles and therefore more stable and safer Li-ion batteries for the future.
Raman spectroscopy has traditionally been employed to study bulk electrodes6,7 rather than interfacial processes because of the limited spatial resolution (∼1–10 μm with a modern microscope) and the weak Raman signal (only 1 in 107 photons is inelastically scattered). In order to overcome these drawbacks, surface-enhanced Raman spectroscopy (SERS) has been extensively developed and applied in the past decades.8 The SERS effect typically relies on the ability of nanostructured metal surfaces (mainly Ag, Au, and Cu) to enhance signal intensity from near-surface molecules (up to 108 times), although the exact physical mechanism behind the enhancement is still under debate.8,9 SERS has in recent years been applied to investigate battery electrochemistry in various configurations, such as “classical” SERS,10,11 tip-enhanced Raman spectroscopy (TERS),12,13 and shell-isolated nanoparticle-enhanced Raman spectroscopy (SHINERS).14,15 The “classical” SERS configuration was employed by the Hardwick group to study the mechanism of oxygen reduction in nonaqueous electrolytes using roughened Au electrodes.10 Nanda et al. employed TERS to study both the chemical composition and topography of the SEI on Si as a function of cycle number.12 Yang et al. employed “classical” (or nanogap) SERS to study electrolyte solvation structures at the electrode–electrolyte interface.16,17 In particular, the Li+ solvation in the common Li-ion solvents ethylene carbonate (EC) and diethyl carbonate (DEC) at different ionic and solvent fractions was investigated, and a deviating Li+ solvation structure at the solid–liquid interface compared with the bulk electrolyte was found. However, the same authors did not study those effects under an applied potential.
Herein we explore the initial stages of double-layer charging and SEI formation in a typical carbonate–LiPF6-based battery electrolyte on a model Au SERS substrate. Interpretation of the results is supported by both operando gas analysis and the measurement of interphase mass deposition on a model Au electrode substrate using an electrochemical quartz crystal microbalance (EQCM). The aim of this simplified model system is to disentangle often competing electrochemical reactions governing electrode interphase formation and to provide fundamental understanding that can be extrapolated to practical systems.
Figure 1a shows the nonresonant (normal) and surface-enhanced Raman spectra of LP40 (1 M LiPF6 in 1:1 EC/DEC) as recorded in the operando Raman cell (see section S1 in the Supporting Information (SI)). The spectra display essentially the same characteristic peaks stemming from the EC, DEC, and PF6– electrolyte components (marked by dashed lines; cf. SI section S2 for details).16,18−20 The EC vibrational bands are assigned to ring breathing (oO–C–O), C–C stretching (νC–C), and CH2 twisting (τCH2) and stretching (νCH2) modes.16,18−20 DEC is identified by the O–C–O bending (δO–C–O), symmetric stretching (νO–C–O), and CH3 bond rocking and bending (δCH3) bands, while the PF6– anion is here represented by the symmetric stretching of the P–F bond.21 Compared with the nonenhanced approach, the SERS spectrum displays several additional bands, primarily in the 1200–1600 cm–1 range, which we relate to vibrational modes either associated with the electrolyte–substrate interaction (e.g., EC/DEC adsorption on Au) or more likely to electrolyte impurities (e.g., glycols, etc.) attracted to the Au surface. Nanda et al. have for instance shown that carboxylate RCOOLi-type compounds result from electrolyte degradation and appear in the 1500–1600 cm–1 range.12 In any case, minor concentrations of electrolyte impurities are generally known to interfere with the spectroscopic signal (and electrochemistry) in comparable studies in the field (e.g., electrocatalysis).8,22,23 Most importantly, the two peaks of EC solvated to Li+ (at 730 and 903 cm–1) labeled EC+Li+, are more intense relative to the nonsolvated EC peaks (at 717 and 894 cm–1) when the spectrum is resonance-enhanced. Yang et al. recently made a similar observation, though for a different SERS substrate, but related the increase in the EC+Li+ peak to the geometric confinement effect of the electrolyte in the hotspot nanogap of their SERS substrate. However, turning to operando SERS shown in Figure 1b, we clearly observe a potential-dependent growth of the EC+Li+ peaks during negative polarization of the Au, which can be explained by the charging of the electric double layer and accumulation of Li+ and solvation of EC at the SERS substrate surface. The intensities of the respective peaks can be extracted by fitting, and Figure 2a shows that the EC+Li+ peaks clearly increase in intensity, whereas the EC peaks remain constant until ∼2.3 V. On the basis of the EC+Li+ to EC peak area ratio, a Li+ concentration at the Au surface can be estimated (according to the relationship derived by Yang et al.16) and was found to increase from the expected 1 M at the OCP up to 1.5 M at 2.3 V vs Li+/Li (see SI section S3 for details). However, all of the peaks associated with the electrolyte decrease in intensity thereafter because of SEI formation. The gradual coverage of the Au surface with degradation products also affects the EC+Li+/EC peak ratio, which returns to its initial value corresponding to 1 M as the LP40 electrolyte loses contact with the charged electrode surface.
Figure 1.

(a) Nonenhanced normal Raman spectrum and SERS spectra (at the open-circuit potential (OCP), 3.0 V, and the end of sweep, 0.5 V) of the LP40 electrolyte. (b) Operando SERS performed on nanostructured gold electrodes in LP40 electrolyte during potentiostatic steps (100 mV, 3.0 to 0.5 V).
Figure 2.
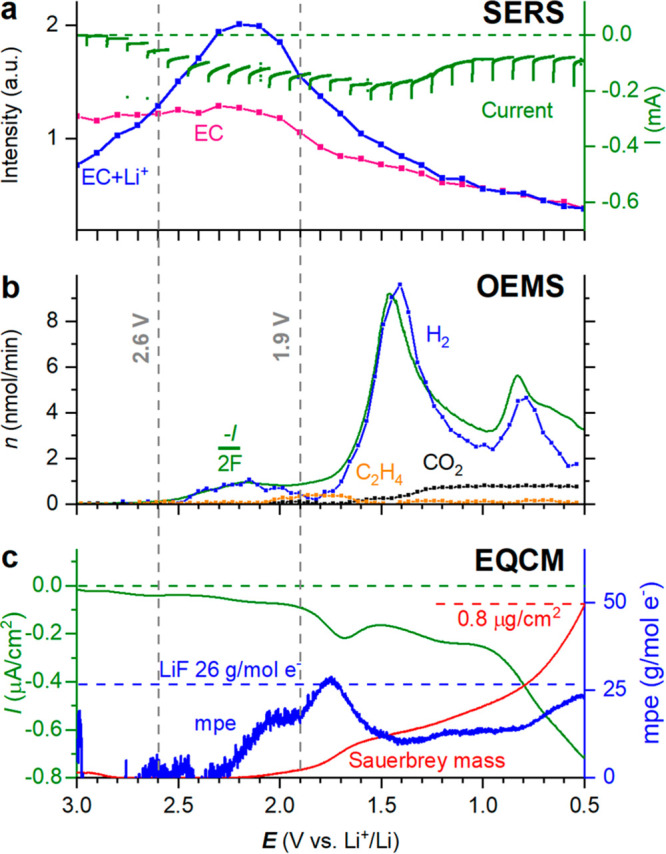
(a) Fitted intensities of the Raman peaks at 894 and 904 cm–1 and the current recorded during the potentiostatic steps. (b) Faradaic current and H2 and CO2 evolution rates during the reductive sweep. (c) Current density, Sauerbrey mass, and mass change per mole of electrons transferred (mpe) of the Au-EQCM sensor as a function of applied potential.
In order to strengthen our conclusions, complementary operando surface characterization was performed. On-line electrochemical mass spectrometry (OEMS) and EQCM results are presented in Figure 2b,c, respectively. The SEI formation process is electrochemically initiated, as evidenced by a reduction current concurrent with both an increase in gas evolution and electrode mass deposition. The onset of H2 evolution at ∼2.6 V is consistent with the reduction of hydrogen fluoride impurities in the electrolyte as reported by Strmcnik et al.24 according to
| 1 |
However, both the Raman signal intensity loss and EQCM mass deposition onset occur later at about 2.3 V. The lag in deposition could be explained by partial LiF solubility in the electrolyte.25 The H2 formed at the first evolution maximum (>1.9 V) was integrated (Figure S4) and corresponded to ∼16 ppm HF in the electrolyte (according to reaction 1), which was also confirmed by measurement of the F– concentration separately.24 LiF is also Raman-inactive, which explains the loss of the electrolyte bands without the appearance of any new major species. However, LiF formation is not the only process, as several other minor and intermediate Raman bands reminiscent of organic fragments in the 1000–1050, 1230–1430, and 1500–1650 cm–1 regions appear and vanish during the reduction sweep (Figure 1b). Moreover, the calculated electrode mass change per electron transferred, known as mass per electron (mpe), from EQCM clearly deviates from the 26 g/mol of e– as expected for LiF (Figure 2c), thus indicating that the reduction current is consumed by other processes. Indeed, a band at 1085 cm–1 characteristic of Li2CO3 appears below ∼1.9 V (Figure 1). Li2CO3 has been claimed26,27 to form in a multistep process starting with reduction of water impurities in the electrolyte:
| 2 |
The hydroxide ions can in turn trigger a chemical ring-opening reaction of EC:28
 |
3 |
The products of reactions 2 and 3, OH– and CO2, are known to react to form the intermediate hydrogen carbonate anion:
| 4 |
A final electrochemical step then results in lithium carbonate:
| 5 |
Although nearly all of the current is associated with H2 evolution according to OEMS (Figure 2b), reactions 1, 2, and 5 all fulfill the criterion of 2e–/H2, and no discrimination can be made between them on that basis. However, the spectroscopic observation of Li2CO3 and the concomitant onset of CO2 evolution suggest that water reduction (reaction 2) sets in at 1.8 V. The H2 associated with that process was integrated and corresponded to about 120 ppm H2O in the electrolyte (see SI section S3), which is possible considering the difficulty of completely drying several components in the OEMS cell. However, the band at 1085 cm–1 associated with Li2CO3 remains weak, and no other bands appear, from which it may be concluded that the electrode–electrolyte interphase as formed herein is dominated by LiF and Li2CO3. No evidence of lithium oxide, peroxide, or hydroxide formation is found (reference Raman spectra of LiOH, Li2O2, Li2CO3, and LiF can be found in SI section S2). These results suggest that the loss of SERS signal is due to expulsion of the electrolyte from the Au surface by LiF. With the assumption that the interphase layer consists predominantly of LiF with a density of 2.64 g/cm3, the measured total interphase mass of 0.8 μg/cm2 equals the formation of a uniform ∼3 nm thick LiF layer, which would significantly reduce the surface enhancement of any electrolyte bands. Below 1 V, a third process slightly deviating from 2e–/H2 kicks in, but the SERS substrate is already largely passivated, as neither C2H4 nor any other volatile species apart from H2 were observed around 1 V. A very minor evolution of C2H4 is electrochemically triggered around 2 V, but the mechanism will not be further investigated here.
In summary, operando SERS complemented by OEMS and EQCM is shown to provide unique insights into several critical processes governing the performance of electrochemical interphases in Li-ion battery electrolytes. Upon negative polarization of the model substrate, the double layer is directly observed to charge up to the point where the SEI formation overtakes the spectral response. The electrochemistry is dominated by the reduction of HF and H2O electrolyte impurities, as evidenced by H2 evolution and the deposition of the Raman-inactive LiF. Li2CO3 forms at ∼1.8 V in a multistep (electro)chemical process based on H2O reduction and EC decomposition. The presented operando methodology provides powerful means to explore the intricate dynamics of Li+ solvation/coordination and electrolyte/impurity side reactions and promises to reveal the formation and operation of the SEI layer itself.
Acknowledgments
E.F. and E.J.B. acknowledge the Swiss National Science Foundation (SNSF) under the “Ambizione Energy” funding scheme (Grant 160540). E.J.B., N.M., R.L., V.N., and P.G.K. acknowledge the Knut and Alice Wallenberg (KAW) Foundation (Grant 2017.0204). Base funding from StandUp for Energy is acknowledged.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jpclett.0c01089.
Experimental details, Raman spectra and assignments of the electrolyte and individual components, OEMS data for H2 evolution, and surface Li+ concentration calculations (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Peled E.; Menkin S. Review—SEI: Past, Present and Future. J. Electrochem. Soc. 2017, 164 (7), A1703–A1719. 10.1149/2.1441707jes. [DOI] [Google Scholar]
- Xu K. Electrolytes and Interphases in Li-Ion Batteries and Beyond. Chem. Rev. 2014, 114, 11503–11618. 10.1021/cr500003w. [DOI] [PubMed] [Google Scholar]
- Winter M. The Solid Electrolyte Interphase - The Most Important and the Least Understood Solid Electrolyte in Rechargeable Li Batteries. Z. Phys. Chem. 2009, 223, 1395–1406. 10.1524/zpch.2009.6086. [DOI] [Google Scholar]
- Hui J.; Gossage Z. T.; Sarbapalli D.; Hernández-Burgos K.; Rodríguez-López J. Advanced Electrochemical Analysis for Energy Storage Interfaces. Anal. Chem. 2019, 91, 60–83. 10.1021/acs.analchem.8b05115. [DOI] [PubMed] [Google Scholar]
- Edström K.; Herstedt M.; Abraham D. P. A New Look at the Solid Electrolyte Interphase on Graphite Anodes in Li-Ion Batteries. J. Power Sources 2006, 153 (2), 380–384. 10.1016/j.jpowsour.2005.05.062. [DOI] [Google Scholar]
- Julien C. M.; Mauger A. In Situ Raman Analyses of Electrode Materials for Li-Ion Batteries. AIMS Mater. Sci. 2018, 5 (4), 650–698. 10.3934/matersci.2018.4.650. [DOI] [Google Scholar]
- Flores E.; Novák P.; Berg E. J. In Situ and Operando Raman Spectroscopy of Layered Transition Metal Oxides for Li-Ion Battery Cathodes. Front. Energy Res. 2018, 6, 82. 10.3389/fenrg.2018.00082. [DOI] [Google Scholar]
- Sharma B.; Frontiera R. R.; Henry A.; Ringe E.; Van Duyne R. P. SERS: Materials, Applications, and the Future Surface Enhanced Raman Spectroscopy (SERS) Is a Powerful Vibrational. Mater. Today 2012, 15 (1–2), 16–25. 10.1016/S1369-7021(12)70017-2. [DOI] [Google Scholar]
- Fleischmann M.; Hendra P. J.; McQuillan A. J. Raman Spectra of Pyridine Adsorbed at a Silver Electrode. Chem. Phys. Lett. 1974, 26 (2), 163–166. 10.1016/0009-2614(74)85388-1. [DOI] [Google Scholar]
- Radjenovic P. M.; Hardwick L. J. Time-Resolved SERS Study of the Oxygen Reduction Reaction in Ionic Liquid Electrolytes for Non-Aqueous Lithium–Oxygen Cells. Faraday Discuss. 2018, 206 (0), 379–392. 10.1039/C7FD00170C. [DOI] [PubMed] [Google Scholar]
- Matsuo Y.; Kostecki R.; McLarnon F. Surface Layer Formation on Thin-Film LiMn[Sub 2]O[Sub 4] Electrodes at Elevated Temperatures. J. Electrochem. Soc. 2001, 148 (7), A687. 10.1149/1.1373658. [DOI] [Google Scholar]
- Nanda J.; Yang G.; Hou T.; Voylov D. N.; Li X.; Ruther R. E.; Naguib M.; Persson K.; Veith G. M.; Sokolov A. P. Unraveling the Nanoscale Heterogeneity of Solid Electrolyte Interphase Using Tip- Enhanced Raman Spectroscopy Unraveling the Nanoscale Heterogeneity of Solid Electrolyte Interphase Using Tip-Enhanced Raman Spectroscopy. Joule 2019, 3 (8), 2001–2019. 10.1016/j.joule.2019.05.026. [DOI] [Google Scholar]
- Touzalin T.; Joiret S.; Maisonhaute E.; Lucas I. T. Capturing Electrochemical Transformations by Tip-Enhanced Raman Spectroscopy. Curr. Opin. Electrochem. 2017, 6 (1), 46–52. 10.1016/j.coelec.2017.10.016. [DOI] [Google Scholar]
- Galloway T. A.; Cabo-Fernandez L.; Aldous I. M.; Braga F.; Hardwick L. J. Shell Isolated Nanoparticles for Enhanced Raman Spectroscopy Studies in Lithium-Oxygen Cells. Faraday Discuss. 2017, 205, 469–490. 10.1039/C7FD00151G. [DOI] [PubMed] [Google Scholar]
- Hy S.; Felix; Chen Y.-H.; Liu J.; Rick J.; Hwang B.-J. In Situ Surface Enhanced Raman Spectroscopic Studies of Solid Electrolyte Interphase Formation in Lithium Ion Battery Electrodes. J. Power Sources 2014, 256, 324–328. 10.1016/j.jpowsour.2014.01.092. [DOI] [Google Scholar]
- Yang G.; Sacci R. L.; Ivanov I. N.; Ruther R. E.; Hays K. A.; Zhang Y.; Cao P.-F.; Veith G. M.; Dudney N. J.; Saito T.; et al. Probing Electrolyte Solvents at Solid/Liquid Interface Using Gap-Mode Surface-Enhanced Raman Spectroscopy. J. Electrochem. Soc. 2019, 166 (2), A178–A187. 10.1149/2.0391902jes. [DOI] [Google Scholar]
- Yang G.; Ivanov I. N.; Ruther R. E.; Sacci R. L.; Subjakova V.; Hallinan D. T.; Nanda J. Electrolyte Solvation Structure at Solid–Liquid Interface Probed by Nanogap Surface-Enhanced Raman Spectroscopy. ACS Nano 2018, 12, 10159–10170. 10.1021/acsnano.8b05038. [DOI] [PubMed] [Google Scholar]
- Fortunato B.; Mirone P.; Fini G. Infrared and Raman Spectra and Vibrational Assignment of Ethylene Carbonate. Spectrochim. Acta Part A Mol. Spectrosc. 1971, 27 (9), 1917–1927. 10.1016/0584-8539(71)80245-3. [DOI] [Google Scholar]
- Klassen B.; Aroca R.; Nazri M.; Nazri G. A. Raman Spectra and Transport Properties of Lithium Perchlorate in Ethylene Carbonate Based Binary Solvent Systems for Lithium Batteries. J. Phys. Chem. B 1998, 102 (24), 4795–4801. 10.1021/jp973099d. [DOI] [Google Scholar]
- Allen J. L.; Borodin O.; Seo D. M.; Henderson W. A. Combined Quantum Chemical/Raman Spectroscopic Analyses of Li+ Cation Solvation: Cyclic Carbonate Solvents - Ethylene Carbonate and Propylene Carbonate. J. Power Sources 2014, 267, 821–830. 10.1016/j.jpowsour.2014.05.107. [DOI] [Google Scholar]
- Aroca R.; Nazri M.; Nazri G. A.; Camargo A. J.; Trsic M. Vibrational Spectra and Ion-Pair Properties of Lithium Hexafluorophosphate in Ethylene Carbonate Based Mixed-Solvent Systems for Lithium Batteries. J. Solution Chem. 2000, 29 (10), 1047–1060. 10.1023/A:1005151220893. [DOI] [Google Scholar]
- Garcia-Araez N.; Rodriguez P.; Bakker H. J.; Koper M. T. M. Effect of the Surface Structure of Gold Electrodes on the Coadsorption of Water and Anions. J. Phys. Chem. C 2012, 116 (7), 4786–4792. 10.1021/jp211782v. [DOI] [Google Scholar]
- Cabo-Fernandez L.; Neale A. R.; Braga F.; Sazanovich I. V.; Kostecki R.; Hardwick L. J. Kerr Gated Raman Spectroscopy of LiPF6 Salt and LiPF6-Based Organic Carbonate Electrolyte for Li-Ion Batteries. Phys. Chem. Chem. Phys. 2019, 21 (43), 23833–23842. 10.1039/C9CP02430A. [DOI] [PubMed] [Google Scholar]
- Strmcnik D.; Castelli I. E.; Connell J. G.; Haering D.; Zorko M.; Martins P.; Lopes P. P.; Genorio B.; Østergaard T.; Gasteiger H. A.; et al. Electrocatalytic Transformation of HF Impurity to H 2 and LiF in Lithium-Ion Batteries. Nat. Catal. 2018, 1, 255–262. 10.1038/s41929-018-0047-z. [DOI] [Google Scholar]
- Jones J.; Anouti M.; Caillon-Caravanier M.; Willmann P.; Lemordant D. Thermodynamic of LiF Dissolution in Alkylcarbonates and Some of Their Mixtures with Water. Fluid Phase Equilib. 2009, 285 (1), 62–68. 10.1016/j.fluid.2009.07.020. [DOI] [Google Scholar]
- Kitz P. G.; Novák P.; Berg E. J. Influence of Water Contamination on the SEI Formation in Li-Ion Cells: An Operando EQCM-D Study. ACS Appl. Mater. Interfaces 2020, 12, 15934–15942. 10.1021/acsami.0c01642. [DOI] [PubMed] [Google Scholar]
- Metzger M.; Strehle B.; Solchenbach S.; Gasteiger H. A. Hydrolysis of Ethylene Carbonate with Water and Hydroxide under Battery Operating Conditions. J. Electrochem. Soc. 2016, 163 (7), A1219–A1225. 10.1149/2.0411607jes. [DOI] [Google Scholar]
- Aurbach D.Nonaqueous Electrochemistry, 1st ed.; CRC Press: Boca Raton, FL, 1999. 10.1201/9780367800499 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.