

Abstract
What happens to DNA replication when it encounters a damaged or nicked DNA template has been under investigation for five decades. Initially it was thought that DNA polymerase, and thus the replication-fork progression, would stall at road blocks. After the discovery of replication-fork helicase and replication re-initiation factors by the 1990s, it became clear that the replisome can “skip” impasses and finish replication with single-stranded gaps and double-strand breaks in the product DNA. But the mechanism for continuous fork progression after encountering roadblocks is entangled with translesion synthesis, replication fork reversal and recombination repair. The recently determined structure of the bacteriophage T7 replisome offers the first glimpse of how helicase, primase, leading-and lagging-strand DNA polymerases are organized around a DNA replication fork. The tightly coupled leading-strand polymerase and lagging-strand helicase provides a scaffold to consolidate data accumulated over the past five decades and offers a fresh perspective on how the replisome may skip lesions and complete discontinuous DNA synthesis. Comparison of the independently evolved bacterial and eukaryotic replisomes suggests that repair of discontinuous DNA synthesis occurs post replication in both.
Keywords: Replisome, Replication fork, Lesion skipping, Helicase reload, Polymerase restart
1. Introduction
The mechanism for DNA replication was immediately obvious when Watson and Crick determined the DNA double-helix structure in 1953 [1]. Shortly afterward, DNA was successfully copied in the test tube with DNA polymerase I (Pol I) purified from E. coli by Arthur Kornberg’s lab [2,3]. In the ensuing two decades, DNA was shown to be susceptible to ultraviolet light (UV) damage, chemical adducts, oxidation and ionization radiation, and lesions caused by these damaging agents were thought to block DNA replication and induce the “SOS” response [4–10]. However, in 1968 Rupp and Howard-Flanders showed that replication is only slowed down but not blocked by unrepaired UV lesions, and in the presence of UV lesions replication products are shorter than usual and their lengths roughly match the distances between lesions [11]. Further experiments showed that replication continued from preexisting forks with the formation of single-stranded gaps [12–14]. The mechanism for such lesion-tolerant and discontinuous replication, however, was unclear at the time because it was assumed that DNA polymerase was responsible for advancing the replication fork and the blocked polymerase would prevent further movement of the fork.
Although by 1979 nearly 20 polypeptides had been identified to form a replisome in E. coli [15], DNA polymerase was the only enzyme known to be blocked by DNA lesions. It was not until 1986 that E. coli DnaB, which had been known to be essential for replication [16,17], was found to have the helicase activity [18]. This led to the recognition that parental DNA duplex unwinding by DnaB helicase has a central role in advancing the replication fork. Contrary to the generally accepted prevailing dogma that the leading strand is always synthesized continuously, Rupp proposed a model in which leading strands can be restarted [19]. Discoveries of replication re-start after skipping a roadblock [20] and primosomal proteins for replication re-initiation in 1990 [21,22] led to inclusion of the helicase and primase in addition to DNA polymerases in the replisome and of their combined roles in dealing with roadblocks [23]. However in the late 1990s, the discovery of a brand-new Y-family of DNA polymerases, which are specialized in bypassing DNA lesions that block normal replicative DNA polymerases [24], and analyses of diverse features of these fascinating DNA polymerases and translesion synthesis (TLS) [25–27] took the front seat in studying effects of lesions on DNA replication. Because DNA translesion synthesis can take place post replication separately from replication-fork progression [28–32], the mechanism by which replisomes circumvent damaged DNA remains unresolved.
In recent years, the finding that the majority of replication forks in human cells can traverse (skip or circumvent) interstrand crosslinks (ICLs) [33], which completely block the progression of DNA polymerase and replication-fork helicase, re-centers our attention on the persistence of the fork progression over impasses and roadblocks. The observation of “fork traverse” in human cells is parallel to the early finding of slowed and fragmented but complete DNA replication in UV-damaged E. coli cells. The continuous fork progression is different from and potentially independent of TLS. Both bacterial and eukaryotic replisomes must have roadside assistance and use built-in mechanisms to reassemble after encounters with roadblocks and continue DNA synthesis, even discontinuously, to finish replication.
Unlike transcription and translation, DNA replication and replisomes independently evolved in bacteria and eukaryotes and the two worlds apparently share no molecular or organizational conservation. In recent years, E. coli and yeast replisomes have been completely reconstituted in vitro [34,35]. The first detailed replisome structure, which is of bacteriophage T7 and the simplest replication machinery known, was reported in 2019 [36]. Aided by the replisome structure, here we integrate existing genetic, biochemical and structural data to review the replisome organization and ways that it can be broken and reassembled to complete replication, which is the first priority in cell survival and imperious over repair of DNA.
2. Structure of the replisome
Every DNA replisome requires four enzymatic activities, DNA unwinding by a hexameric replication-fork helicase, leading- and lagging-strand DNA synthesis by polymerases, and RNA-primer synthesis for each DNA strand by a primase. Other factors, such as proofreading 3′ to 5′ exonuclease (intrinsic or extrinsic to DNA polymerases), single-strand binding protein (e.g. SSB in E. coli and RPA in eukaryotes), processivity factors for DNA polymerases (thioredoxin for T7 Pol, β-sliding clamp in E. coli and PCNA in eukaryotes), and adaptors coordinating helicase with polymerase or primases, enhance the replisomes’ performance and are highly diverse among species [37–41]. Bacteriophage T7 presents the simplest replisome by combining the helicase and primase in one polypeptide chain (gp4) and using the gp5 DNA polymerase for both leading and lagging strand synthesis. E. coli and yeast replisomes are more complicated and involve many accessary subunits. Notably, most accessary subunits are located between the helicase and lagging-strand primase (in bacteria) or primosome (in eukaryotes, including a two-subunit primase and two-subunit DNA Pol α) [41] for the successive and discontinuous synthesis of Okazaki fragments.
Despite the lack of evolutionary conservation and varied complexity, structures of parts of E. coli and yeast replisomes [42–45], and of the complete bacteriophage T7 replisome [36] suggest a similar three-tiered architecture (Fig. 1A). A hexameric replication DNA helicase (Fig. 1B) forms the central tier between the leading-strand DNA polymerase and the lagging-strand primase/primosome. A DNA fork substrate to be replicated is bound either between the leading strand polymerase and lagging-strand helicase in bacterial replisomes or between the leading-strand helicase and lagging-strand primosome in eukaryotes (Fig. 1A) (see details below). A lagging-strand DNA polymerase is naturally located on the primase side. In popular textbook diagrams of a replication fork, helicase, leading and lagging-strand DNA polymerases are depicted to move in the same direction parallel to the downstream parental DNA (Fig. 1C). However, in the structure of T7 replisome, the helicase translocates in a direction perpendicular to the downstream DNA and unwinds the double helix tangentially like unspooling a coil, and the leading- and lagging-strand DNA polymerases are nearly antiparallel as dictated by the opposite polarity of two DNA strands (Fig. 1D) [36]. The T7 lagging-strand polymerase needs to be coupled with the primase only during handover of an RNA primer, and otherwise operates independently of the three-tier core of replisomes. Two lagging-strand polymerases, one actively synthesizing DNA and the other waiting for a primer, can be simultaneously tethered to the T7 primase tier [36].
Fig. 1.
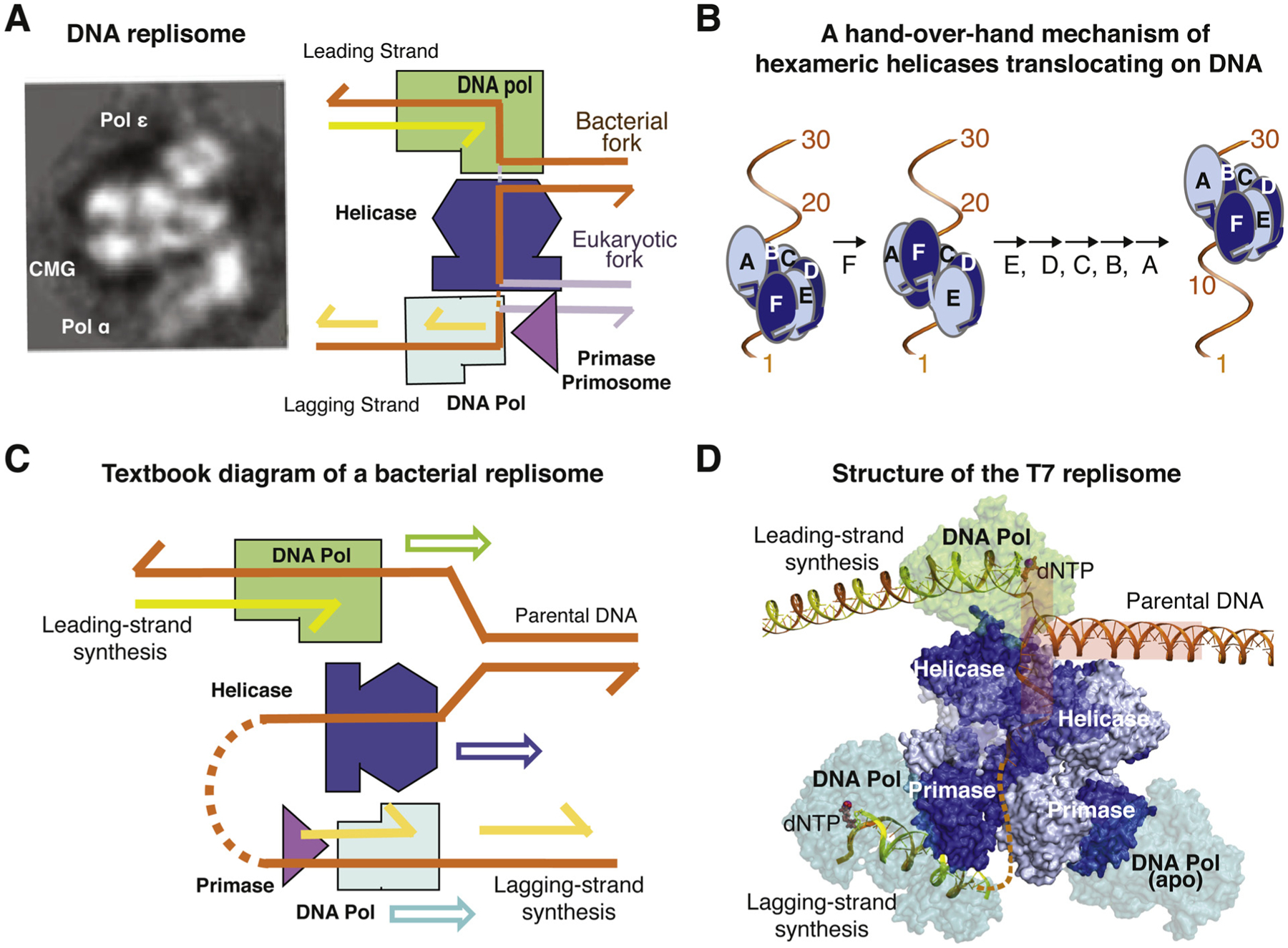
Diagrams of DNA replisomes. (A) Low-resolution cryoEM reconstitution of yeast CMG, Pol ε and primosome complex reveals a three-tier-core structure. Diagrams of the DNA fork substrate in the three-tier-core structure. The conserved DNA part are shown in orange, and the different downstream parental DNA in bacterial and eukaryotic replisome is shown in gold and silver, respectively. (B) A hand-over-hand mechanism of replication-fork helicase translocation along DNA. (C) A textbook diagram of the replisome and directions of the three motor proteins along the DNA fork substrate. (D) Structure of the T7 replisome. The “T” shaped DNA fork is highlighted by a semi-transparent T.
In bacteria, a replication helicase binds the lagging strand and translocates in the 5′ to 3′ direction (Fig. 1A). In the T7 replisome [36], the DNA fork adopts a T shape with the two separated strands 180° to each other forming the top of the “T” and the downstream duplex perpendicular to both strands forming the stem of the “T” (Fig. 1D). The helicase and polymerase each pull one DNA strand tangentially to the DNA helix. The two enzymes not only physically contact each other to stabilize the DNA substrate, but T7 DNA polymerase also physically divides the leading and lagging strand at the single-double strand junction with a separation pin and “feeds” the lagging strand to the helicase for translocation. These structural features substantiate the observations that the two enzymes work concertedly during replication and enhance each other’s DNA unwinding and synthesis activity [46]. In the T7 replisome, the leading-strand template base is but one nucleotide away from the parental DNA duplex, while the helicase on the lagging strand varies from 1 to 3 nt away from the parental duplex. The T7 helicase translocates along ssDNA 2 nt per step by sequentially moving one subunit at a time (5′ to 3′ or 3′ to 5′) by a hand-over-hand mechanism [36]. In every step, the moving “hand” is a single subunit and the hand holding the DNA includes the remaining five subunits (Fig. 1B). Despite the different step sizes, 2 nt advancement per ATP hydrolysis by the helicase and 1 nt incorporation each time by the polymerase, the two enzymes move together in unison, and each is less processive and slower without the other.
In eukaryotes, a replication helicase binds the leading strand and translocates in the 3′ to 5′ direction ahead of the leading-strand polymerase (Fig. 1A) [47]. Although the eukaryotic replication helicase CMG is a heterohexamer MCM2–7, it requires Cdc45 and the tetrameric GINS complex as co-factors [48,49] and moves in the opposite direction of bacterial helicases. Both types of replication-fork helicases form a spiral lock-washer and wrap around 12 nts of a right-handed ssDNA coil (Fig. 1B) [36,45,47]. The motoring ATPase domains of these replication helicases are located at the C-terminal half of the protein, and the N- to C-terminus direction of replication helicases is always aligned with the 5′ to 3′ polarity of the ssDNA independent of the direction of helicase translocation [47,50] (Fig. 1A).
In eukaryotic replisomes, the DNA replication fork is between the N-terminal helicase domain and the primosome, and the motor domains of the CMG helicase subunits maintain the direct interaction with the leading-strand DNA polymerase Pol ε [44,45]. Pol α is unique in eukaryotes and extends the RNA primer with deoxyribonucleotides before DNA Pol δ can take over with the lagging-strand synthesis [35,51] Although Pol α, δ and ε belong to the same B-family of DNA polymerases, Pol α doesn’t have the intrinsic proofreading exonuclease activity [52], and the Pol ε catalytic subunit is twice as large as that of Pol δ due to a gene fusion event [53]. Based on the recent results, Pol ε is a dedicated leading strand polymerase due to the physical association with CMG helicase [35,54]. In contrast, Pol δ is a gap-filling polymerase and synthesizes all Okazaki fragments in the lagging strand [55,56]. In addition, Pol δ makes short DNA patches during repair processes and initial leading-strand synthesis by extending primers from the primosome before handing over to Pol ε [35,57]. In contrast to Pol ε, Pol δ depends on PCNA for its processivity [58].
3. Lesion skipping by bacterial replisomes
Many different types of DNA lesions on both leading and lagging strands can impede the normal replisome progression in bacteria. Different types of lesions and locations, whether on the leading or lagging strand, however, would have different impacts on replication.
3.1. Scenario 3.1 Damage on the leading strand
If the leading-strand polymerase is stalled by a DNA lesion, the helicase is likely to dissociate from the polymerase temporarily and proceed alone inefficiently on the lagging strand. If the lesion is a minor base modification, for example, an oxidized guanine (8-oxoG), the leading-strand polymerase may overcome the roadblock by mis-incorporating dA opposite it [59–61] and catch up and re-associate with the helicase (Fig. 2A). After replication, E. coli uses MutY and MutM glycosylases to eliminate the mismatched dA and 8-oxoG, respectively, by the base excision repair pathway [62].
Fig. 2.
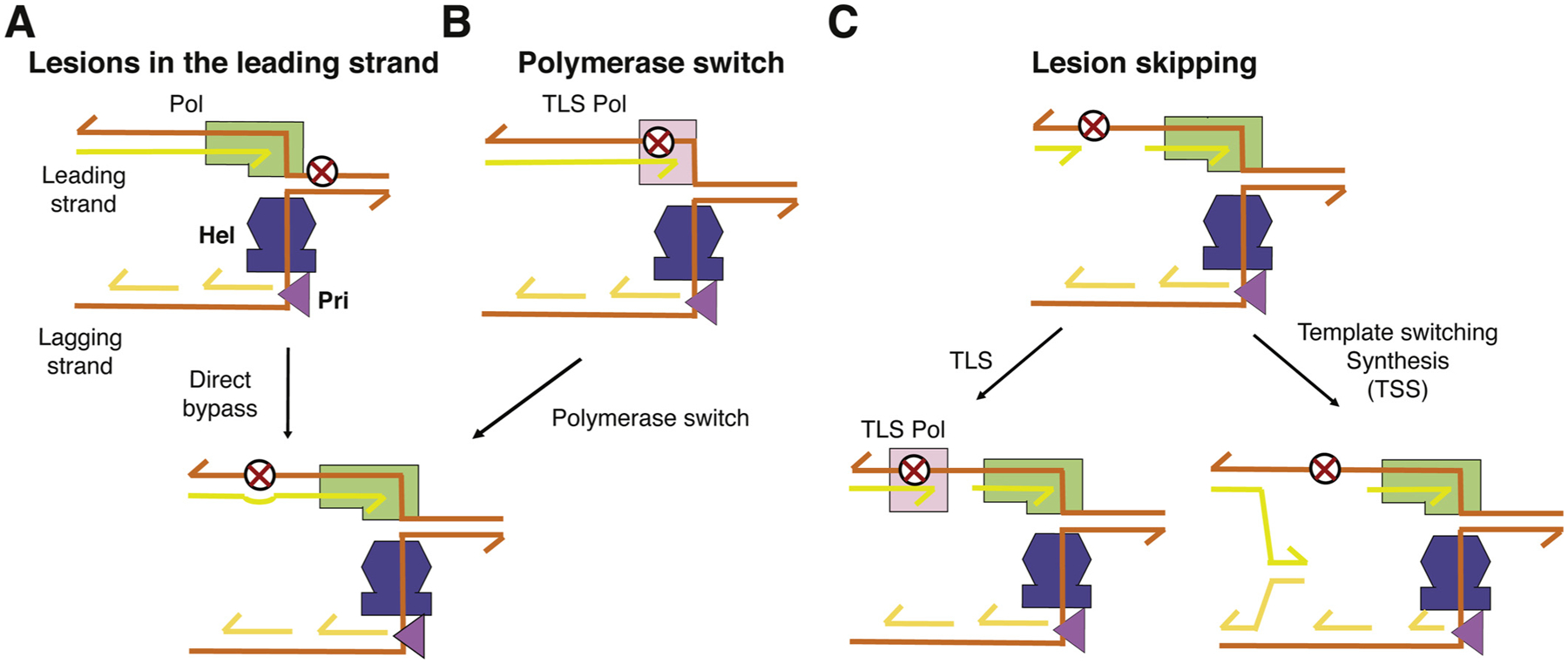
Mechanisms for lesion skipping by bacterial replisome along the leading strand. (A) DNA replicative polymerase bypasses a minor DNA base lesion, e.g. 8-oxoG. (B) Lesion bypass by TLS polymerase, which requires at least two polymerase-switch steps before the polymerase and helicase re-association. (C) When a leading-strand polymerase is completely stalled by a roadblock, e.g. UV-induced pyrimidine dimers, the leading strand DNA synthesis requires a new primer to restart. The incomplete DNA synthesis due to the lesion may be completed by TLS or template-switching synthesis (TSS) post replication.
However, upon encountering a UV induced pyrimidine dimer, a bulky adduct on DNA bases or an abasic site, a replicative polymerase cannot easily continue nucleotide incorporation. If a specialized TLS polymerase, such as E. coli Pol IV or Pol V, is available nearby, it may carry out translesion synthesis on-site and thus allow the replicative polymerase to dissociate and re-associate from the leading strand and continue afterwards [30,63] (Fig. 2B). The difficult lesions, which require a TLS polymerase to bypass but without a TLS polymerase nearby to assist, or a DNA nick, which immediately stops the polymerase, can lead to a long-lasting polymerase-helicase dissociation [64]. The helicase, which continues on the lagging strand, eventually needs to recruit a replacement leading-strand polymerase to re-start DNA synthesis beyond the lesion site (Fig. 2C). The primase, which is a part of the helicase in T7 phage or a dedicated primase (DnaG) in E. coli, must occasionally make primers for the leading-strand synthesis in addition to regularly making primers for the lagging-strand synthesis. Alternatively, RNAs generated by transcription of the leading strand may also be used for replication restart [65]. Re-start of the leading-strand synthesis results in gaps and fragments in DNA product, which is exactly what Rupp and Howard-Flanders observed in E. coli cells upon UV irradiation in 1968 [11]. The gaps between DNA fragments can be filled after either TLS synthesis or DNA synthesis with template switching via homologous recombination, and both occur post replication (Fig. 2D) [28,29,66].
3.2. Scenario 3.2 Damage on the lagging strand
A replication-fork helicase (DnaB in E. coli) will be the first that is exposed to lesions on the lagging strand before primase and DNA polymerase. As replication-fork helicases only interact with the DNA backbone and the diameter of ssDNA bound to the helicases is 22 Å, larger than the B-DNA duplex (20 Å) [36,67], abnormal DNA bases, including bulky adduct and UV pyrimidine dimers, would have little effect on helicase translocation [68]. Damaged bases are likely to be also skipped over during lagging strand synthesis by primase and DNA polymerase due to their discontinuous nature [69] (Fig. 3A). The resulting gaps between Okazaki fragments can be repaired by TLS polymerase or template-switching synthesis TSS mediated and resolved by homologous recombination as can repair of gaps in the leading strand (Fig. 2D). The lesions that can derail the helicase are interstrand crosslinks and DNA nicks. We are not sure what happens to interstrand crosslinks in bacteria, as such lesions block both leading-strand polymerase and lagging-strand helicase.
Fig. 3.
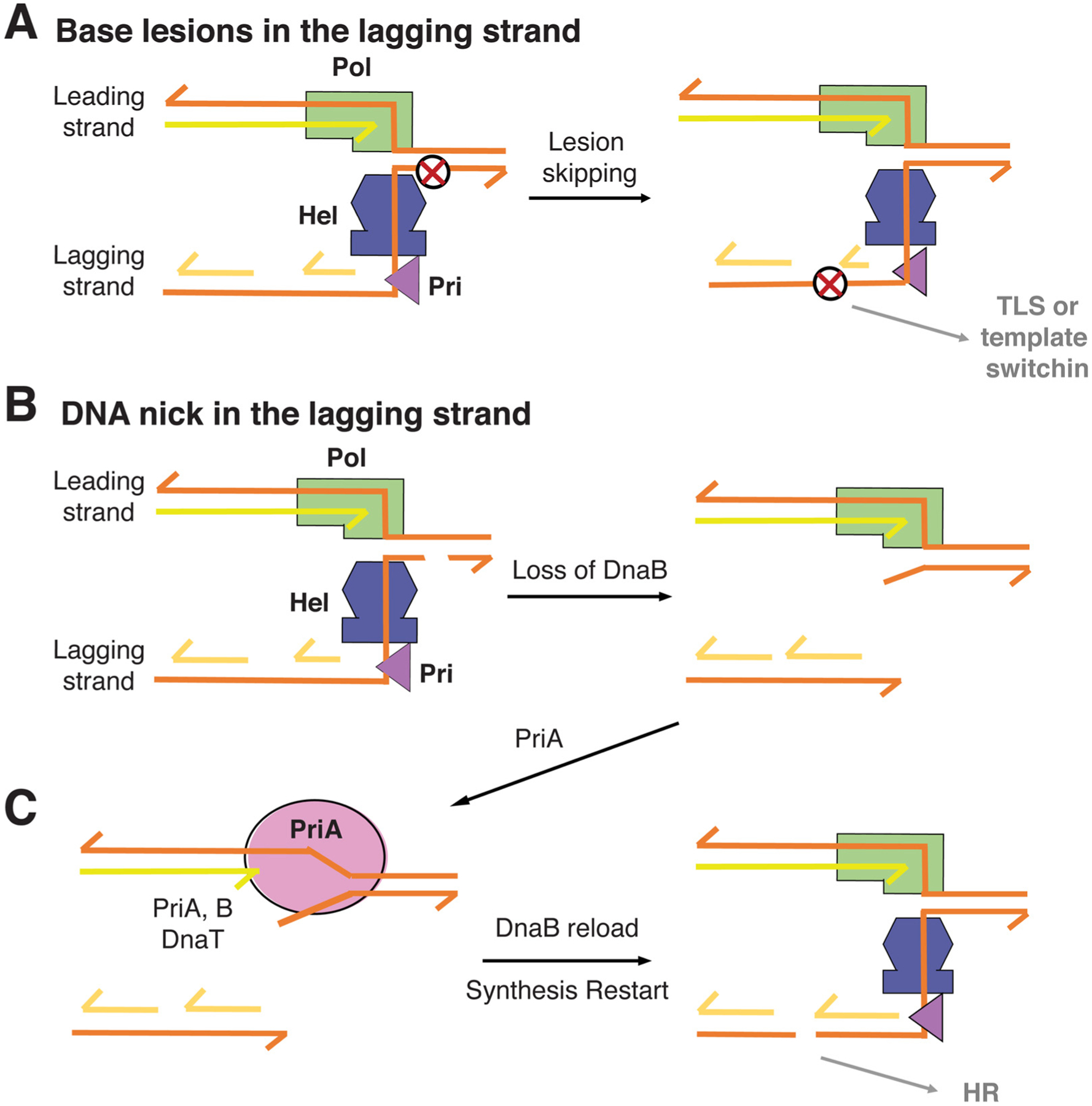
Mechanisms for lesion skipping by bacterial replisome along the lagging strand. (A) Most lagging-strand lesions have no ill effect on the helicase and are skipped by primase and polymerase alike. (B) A nick in the lagging strand leads to helicase dissociation, while the leading strand polymerase continues at a reduced speed. (C) PriA may displace the leading strand DNA polymerase, and together with PriB, PriC and DnaT help to reload DnaB. The double-strand break in the lagging strand depends on homologous recombination for repair.
When encountering nicks, which are prevalent due to spontaneous loss of bases and endonuclease cleavage of abasic sites, the lagging-strand helicase most probably falls off and leaves the DNA polymerase on the leading strand to continue DNA synthesis alone at a reduced speed and processivity. The resulting DNA fork comprises a leading strand polymerase, which carries out strand-displacement synthesis 1-nt away from the ds-ss junction, and a ssDNA tail on the lagging strand (Fig. 3B). Such a DNA structure is perfect for DnaB helicase reloading by PriABC and DnaT in E. coli [70–72]. It is often asumed that PriA helicase, which translocates along DNA in the 3′ to 5′ direction, unwinds the lagging-strand template towards the upstream and away from the replication fork [73,74]. However, the reasons for the loss of DnaB helicase in the first place and the necessity for DnaB reloading were not taken into account for the involvement of PriA. Unwinding of the lagging strand upstream doesn’t seem to aid lesion bypass and cannot help reassembly of a replisome beyond the DNA nick to complete replication. Based on the available data [23,75], we propose that PriA actually unwinds the downstream DNA by binding the leading-strand template in the 3′ to 5′ direction. By preferential binding to the 3′ end at a stalled fork, PriA readily competes off the leading-strand polymerase [76]. With the help of ssDNA-binding PriB and DnaT, PriA can generate a region of ssDNA on the lagging strand for DnaB reloading [72] (Fig. 3C). PriA doesn’t need to unwind the parental strand extensively as the ATPase activity of PriA is not essential for DnaB reloading [77]. Once DnaB is reloaded and PriA has translocated away from the 3′ end, a leading-strand polymerase may regain the fork and replication restarts as depicted in Fig. 2C. Repair of the double-strand break in the lagging strand requires homologous recombination [70].
4. Lesion skipping by eukaryotic replisomes
Eukaryotic replisomes would have an easier time to complete DNA replication than the bacterial counterparts for the following two reasons. Firstly, unlike bacteria, in which there is only one replication origin and also a defined termination site, eukaryotes use many replication origins for replication of a single chromosome and have no defined termination sites [41]. In addition to the early, middle, and late firing origins in Eukaryotes, dormant origins, which are “spare” for emergency, may be used to supplement replication forks, whose progressions are stalled by lesions, and complete replication [78,79]. Secondly, in eukaryotic replisomes the leading strand DNA polymerase and CMG helicase both move along the leading strand (Fig. 1A). In contrast to bacterial replisomes, which have to suffer from lesions on the leading and lagging strand, in eukaryotes lesions on the lagging strand are avoided by primosome and DNA polymerase and are inconsequential for completing replication (Fig. 4A). As synthesis of Okazaki fragments is frequent (~ every 200 nt or so) and priming sites are stochastic [80,81], skipped lesions and resulting gaps on the lagging strand can be dealt with post replication. Double-strand breaks due to nicks in the lagging strand may be repaired by homologous recombination (Fig. 4B) [82,83]. The helicase and leading-strand polymerase of eukaryotic replisomes, however, may disassemble as outlined below.
Fig. 4.
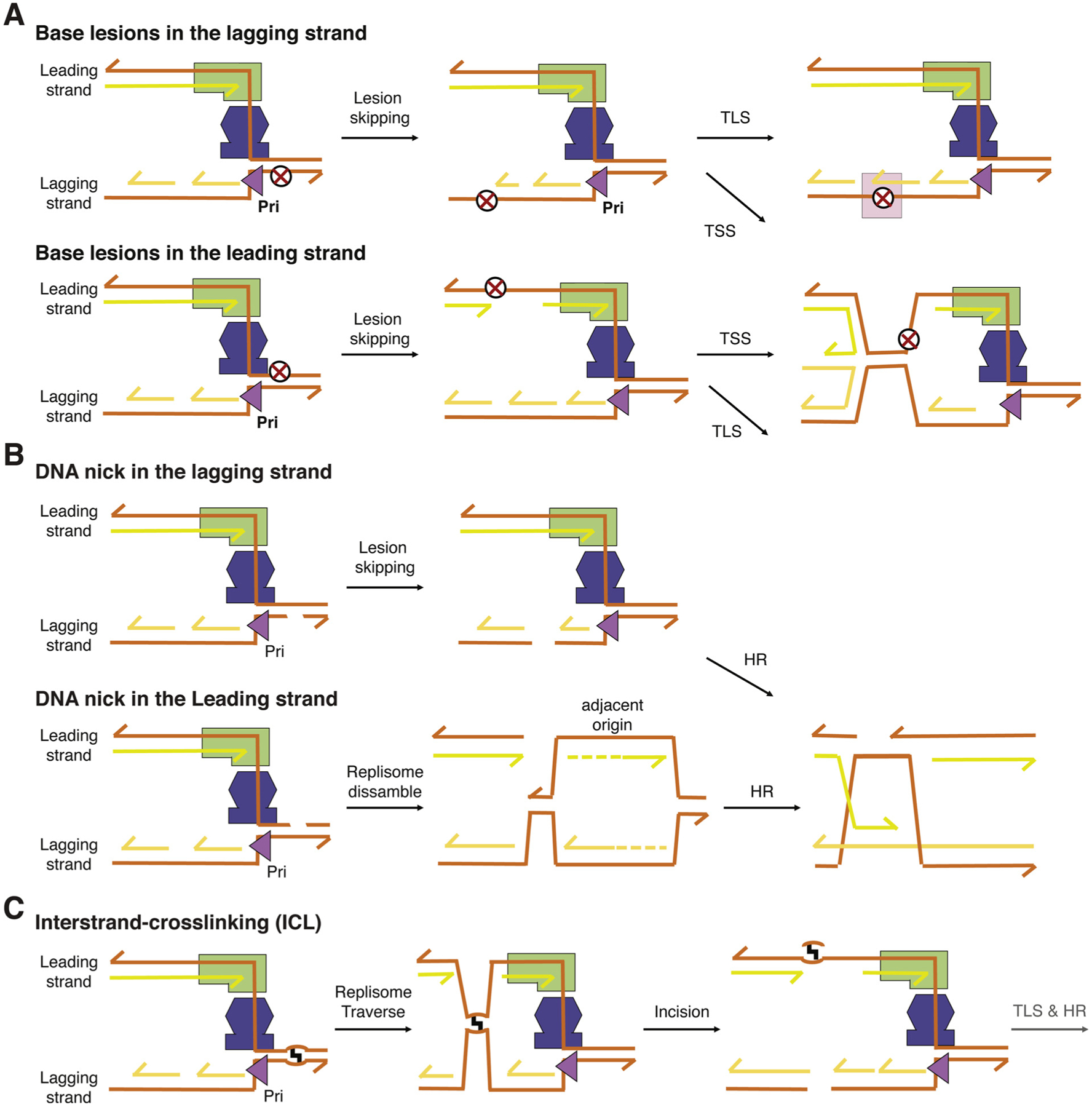
Mechanisms for lesion skipping by eukaryotic replisome. (A) Base lesions in either leading or lagging strands. When a leading-strand polymerase is completely stalled, to continue DNA synthesis requires a new primer to restart and reloading of Pol ε. The gapped DNA synthesis product due to the lesion may be repaired by TLS or template-switching synthesis post replication. (B) Nicks in either template strand lead to double strand breaks after replication. If the leading strand is nicked, replication can be completed only by a fork approaching from the other side as CMG cannot be reloaded. The resulting double-strand breaks in either leading or lagging strands are repaired by homologous recombination (HR). (C) In the presence of an ICL, the majority of replisomes are able to traverse the lesion and continue DNA synthesis on the other side of the ICL. The lesion, gap and break in the replication product are removed by repair pathways including DNA incision, TLS and HR.
4.1. Scenario 4.1 A stalled DNA Pol ε and restart of DNA synthesis in the leading strand
As in bacteria, some DNA lesions may only stall the DNA Pol ε for a short time, and translesion synthesis is carried out either by Pol ε directly or by “onsite” specialized TLS polymerases with quick polymerase switches. Under such circumstances, replication would resume with a short slow down. Like bacteria, eukaryotes have a MutY homolog, MYH, to take out mis-incorporated dA opposite 8-oxoG and MutM homolog OGG1 to remove 8-oxoG [84]. If lesions and particularly a nick cause a prolonged stalling of DNA Pol ε or its dissociation from CMG, DNA synthesis on the leading strand needs to restart (Fig. 4A–B). It is not clear whether eukaryotic primosomes may continue to supply primers for the leading-strand DNA synthesis outside of replication origins. A special DNA primase, PrimPol, is capable of ab initio DNA synthesis and can supply primers for the leading strand restart [85,86]. Skipped lesions and associated DNA gaps can be repaired by TLS or TSS after the replication fork has moved on (Fig. 4A). Based on the replisome structure (Fig. 1A), any single-stranded DNA on the leading strand indicates abnormal replication. The presence of the single-stranded DNA binding protein RPA on the leading strand sends out a signal of replication stress and recruits replication repair proteins [87].
4.2. Scenario 4.2 A stalled CMG and fork traverse
DNA interstrand crosslinks (ICLs) have always been considered absolute blocks to replisomes. In the plasmid replication assay using Xenopus cell extract, a CMG stalls on one side of an ICL, and replication is completed by a second fork coming from the other side [88,89]. While the dual fork model has been very influential, a cell-based DNA fiber analysis of genomic replication tracts revealed an alternative. More than 50% of replication forks restarted DNA synthesis on the side of the ICL distal from the initial encounter and progressed away from the lesion [33]. These results were confirmed by the Lopes group, who observed the replication-restart DNA structures by electron microscopy [90].
FANCM, an ATP motor protein [91], is important for ICL “traverse” and becomes associated with the ICL proximal replisomes that subsequently lose the GINS complex [92]. The structure of yeast CMG reveals that GINS functions like a latch that closes the spiral-shaped MCM2–7 [47]. We speculate that without GINS, Cdc45 and MCM2–7 could pass by an ICL. FANCM may help to remodel CMG or unwind the downstream duplex creating a landing site for MCM2–7. The concept of an elastic replisome is further supported by studies on the encounter of replicative helicases with DNA-protein crosslinks on the leading strand. Both the SV40 T antigen and the CMG can unwind past these structures, demonstrating their “remarkable capacity to overcome obstacles on the translocation strand” [93,94]. Once the bulk of the replisome has skipped (traversed) an ICL, synthesis of the leading strand after reloading of Pol ε will be similar to bypass a base adduct (Fig. 4A). Gaps in the daughter strands at skipped ICLs and the ICLs themselves, may be repaired by DNA incisions followed by TLS and recombination post replication [88] (Fig. 4C).
Despite the resilience of the CMG, persistent fork stalling can occur at ICLs and nicks in the leading strand. Under these conditions, DNA synthesis initiated from a neighboring origin, including a dormant one, can complete replication (Fig. 4B). ICL traverse, bypass of bulky protein-DNA crosslinks, and dual fork replication on either side of an obstruction, all support a “replication imperative”: the requirement to complete S phase as expeditiously as possible.
“Fork reversal” may be triggered by replication hindered by lesions. A four-way junction structure associated with DNA replication supports the “fork reversal” hypothesis [95,96]. Recent work in the Xenopus extract system demonstrates that, after dual fork convergence at the ICL, one of the forks undergoes reversal, a process that requires disassembly of the CMG [97]. These results also indicate that fork reversal and subsequent repair events occur post replication.
5. Fork reversal may occur post replication
The above summary of consequences of various DNA lesions on leading or lagging strand synthesis suggests that DNA replication is imperious over any impediments. Once initiated, it progresses to the bitter end and leaves lesions, gaps and double-strand breaks behind. The difference between bacterial and eukaryotic replisomes shows an increased probability for replication to complete in eukaryotes as replisomes are impervious to damage on a half of each genome (lagging strand). Instead of the reloading a replication-fork helicase (DnaB), eukaryotes employ a “traverse” mechanism to skip over roadblocks and use spare origins to complete replication.
Since 2002, “fork reversal” has been a widely studied phenomenon in replication hindered by lesions. Although a DNA four-way junction structure associated with DNA replication supports the “fork reversal” hypothesis [95,96], whether fork reversal takes place before or after the fork has traversed an ICL lesion has not been determined. Recent reports suggest that fork reversal and fork traverse both occur in 15 min to 1 h after ICL treatment [33,90,92]. Because fork reversal requires complete disassembly of replisomes and cannot lead to fork traverse of an ICL, we suggest that replication continues and fork reversal may occur post replication as all repair processes.
6. Concluding remarks
Eukaryotic replisomes are much more complex and more regulated machineries than bacteriophage T7 and E. coli equivalents. There are likely to be more than one way to get around any lesions. The summary outlined here is a framework to organize different mechanisms for bacteria and eukaryotes to complete DNA at the cost of gapped and fragmented DNA products, which can be repaired after replisomes and replication forks have progressed past lesion sites.
Acknowledgments
We thank K. Marians for critical reading of the manuscript. This research was supported by the National Institute of Diabetes and Digestive and Kidney Diseases to W. Y. (DK036146), the National Institute of Aging to M.S. (AG000746), and the Henry Koerner Center at Yale and the Radiobiology Program of the Yale Cancer Center to W.D.R.
Abbreviations:
- TLS
translesion synthesis
- TSS
template switching synthesis
- ICL
interstrand crosslink
- HR
homologous recombination
Footnotes
This Special Issue is edited by Philip C. Hanawalt.
This article is part of the special issue Cutting-edge Perspectives in Genomic Maintenance VI.
References
- [1].Watson JD, Crick FH, The structure of DNA, Cold Spring Harb. Symp. Quant. Biol 18 (1953) 123–131. [DOI] [PubMed] [Google Scholar]
- [2].Bessman MJ, Kornberg A, Lehman IR, Simms ES, Enzymic synthesis of deoxyribonucleic acid, Biochim. Biophys. Acta 21 (1956) 197–198. [DOI] [PubMed] [Google Scholar]
- [3].Lehman IR, et al. , Enzymatic synthesis of deoxyribonucleic acid. V. Chemical composition of enzymatically synthesized deoxyribonucleic acid, Proc. Natl. Acad. Sci. U. S. A 44 (1958) 1191–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Merz T, Swanson CP, Hemalatha CN, Radiosensitivity and problem of chromosome breakage and rejoining, Brookhaven Symp. Biol 14 (1961) 53–61. [PubMed] [Google Scholar]
- [5].Rupert CS, Photoenzymatic repair of ultraviolet damage in DNA. II. Formation of an enzyme-substrate complex, J. Gen. Physiol 45 (1962) 725–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Rupert CS, Photoenzymatic repair of ultraviolet damage in DNA. I. Kinetics of the reaction, J. Gen. Physiol 45 (1962) 703–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Setlow RB, Setlow JK, Evidence that ultraviolet-induced thymine dimers in DNA cause biological damage, Proc. Natl. Acad. Sci. U. S. A 48 (1962) 1250–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Witkin EM, Mutation and the repair of radiation damage in bacteria, Radiat. Res (Suppl. 6) (1966) 30–53. [PubMed] [Google Scholar]
- [9].Tomasz M, H2O2 generation during the redox cycle of mitomycin C and dna-bound mitomycin C, Chem. Biol. Interact 13 (1976) 89–97. [DOI] [PubMed] [Google Scholar]
- [10].Radman M, SOS repair hypothesis: phenomenology of an inducible DNA repair which is accompanied by mutagenesis, Basic Life Sci. 5A (1975) 355–367. [DOI] [PubMed] [Google Scholar]
- [11].Rupp WD, Howard-Flanders P, Discontinuities in the DNA synthesized in an excision-defective strain of Escherichia coli following ultraviolet irradiation, J. Mol. Biol 31 (1968) 291–304. [DOI] [PubMed] [Google Scholar]
- [12].Iyer VN, Rupp WD, Usefulness of benzoylated naphthoylated DEAE-cellulose to distinguish and fractionate double-stranded DNA bearing different extents of single-stranded regions, Biochim. Biophys. Acta 228 (1971) 117–126. [DOI] [PubMed] [Google Scholar]
- [13].Hewitt R, Gaskins P, Influence of ultraviolet irradiation on chromosome replication in ultraviolet-sensitive bacteria, J. Mol. Biol 62 (1971) 215–221. [DOI] [PubMed] [Google Scholar]
- [14].Rupp WD, Wilde CE 3rd, Reno DL, Howard-Flanders P, Exchanges between DNA strands in ultraviolet-irradiated Escherichia coli, J. Mol. Biol 61 (1971) 25–44. [DOI] [PubMed] [Google Scholar]
- [15].Kornberg A, The enzymatic replication of DNA, CRC Crit. Rev. Biochem 7 (1979) 23–43. [DOI] [PubMed] [Google Scholar]
- [16].Masker WE, Hanawalt PC, Ultraviolet-stimulated DNA synthesis in toluenzied Escherichia coli deficient in DNA polymerase I, Proc. Natl. Acad. Sci. U. S. A 70 (1973) 129–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Olsen WL, Staudenbauer WL, Hofschneider PH, Replication of bacteriophage M13: specificity of the Escherichia coli dnaB function for replication of double-stranded M13 DNA, Proc. Natl. Acad. Sci. U. S. A 69 (1972) 2570–2573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].LeBowitz JH, McMacken R, The Escherichia coli dnaB replication protein is a DNA helicase, J. Biol. Chem 261 (1986) 4738–4748. [PubMed] [Google Scholar]
- [19].Rupp WD, et al. , Neidhart FC (Ed.), Escherichia Coli and Salmonella Cellular and Molecular Biology, ASM Press, Washington DC, 1996, pp. 2277–2294. [Google Scholar]
- [20].Cupido M, Bypass of pyrimidine dimers in DNA of bacteriophage T4 via induction of primer RNA, Mutat. Res 109 (1983) 1–11. [DOI] [PubMed] [Google Scholar]
- [21].Lee EH, Masai H, Allen GC Jr., A. Kornberg, The priA gene encoding the primosomal replicative n’ protein of Escherichia coli, Proc. Natl. Acad. Sci. U. S. A 87 (1990) 4620–4624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Nurse P, DiGate RJ, Zavitz KH, Marians KJ, Molecular cloning and DNA sequence analysis of Escherichia coli priA, the gene encoding the primosomal protein replication factor Y, Proc. Natl. Acad. Sci. U. S. A 87 (1990) 4615–4619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Heller RC, Marians KJ, The disposition of nascent strands at stalled replication forks dictates the pathway of replisome loading during restart, Mol. Cell 17 (2005) 733–743. [DOI] [PubMed] [Google Scholar]
- [24].Ohmori H, et al. , The Y-family of DNA polymerases, Mol. Cell 8 (2001) 7–8. [DOI] [PubMed] [Google Scholar]
- [25].Yang W, An overview of Y-Family DNA polymerases and a case study of human DNA polymerase eta, Biochemistry 53 (2014) 2793–2803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Yang W, Gao Y, Translesion and Repair DNA Polymerases: Diverse Structure and Mechanism, Annu. Rev. Biochem 87 (2018) 239–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Yang W, Woodgate R, What a difference a decade makes: insights into translesion DNA synthesis, Proc. Natl. Acad. Sci. U. S. A 104 (2007) 15591–15598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Henrikus SS, et al. , DNA polymerase IV primarily operates outside of DNA replication forks in Escherichia coli, PLoS Genet. 14 (2018) e1007161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Robinson A, et al. , Regulation of mutagenic DNA polymerase V activation in space and time, PLoS Genet. 11 (2015) e1005482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Indiani C, McInerney P, Georgescu R, Goodman MF, O’Donnell M, A sliding-clamp toolbelt binds high- and low-fidelity DNA polymerases simultaneously, Mol. Cell 19 (2005) 805–815. [DOI] [PubMed] [Google Scholar]
- [31].Hedglin M, Benkovic SJ, Eukaryotic translesion DNA synthesis on the leading and lagging strands: unique detours around the same obstacle, Chem. Rev 117 (2017) 7857–7877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Karras GI, Jentsch S, The RAD6 DNA damage tolerance pathway operates uncoupled from the replication fork and is functional beyond S phase, Cell 141 (2010) 255–267. [DOI] [PubMed] [Google Scholar]
- [33].Huang J, et al. , The DNA translocase FANCM/MHF promotes replication traverse of DNA interstrand crosslinks, Mol. Cell 52 (2013) 434–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Graham JE, Marians KJ, Kowalczykowski SC, Independent and stochastic action of DNA polymerases in the replisome, Cell 169 (2017) 1201–1213 e1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Yeeles JTP, Janska A, Early A, Diffley JFX, How the eukaryotic replisome achieves rapid and efficient DNA replication, Mol. Cell 65 (2017) 105–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Gao Y, et al. , Structures and operating principles of the replisome, Science 363 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Benkovic SJ, Valentine AM, Salinas F, Replisome-mediated DNA replication, Annu. Rev. Biochem 70 (2001) 181–208. [DOI] [PubMed] [Google Scholar]
- [38].Hamdan SM, Richardson CC, Motors, switches, and contacts in the replisome, Annu. Rev. Biochem 78 (2009) 205–243. [DOI] [PubMed] [Google Scholar]
- [39].Reha-Krantz LJ, DNA polymerase proofreading: multiple roles maintain genome stability, Biochim. Biophys. Acta 1804 (2010) 1049–1063. [DOI] [PubMed] [Google Scholar]
- [40].Pellegrini L, Costa A, New insights into the mechanism of DNA duplication by the eukaryotic replisome, Trends Biochem. Sci 41 (2016) 859–871. [DOI] [PubMed] [Google Scholar]
- [41].O’Donnell M, Langston L, Stillman B, Principles and concepts of DNA replication in bacteria, archaea, and eukarya, Cold Spring Harb. Perspect. Biol 5 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Bailey S, Eliason WK, Steitz TA, Structure of hexameric DnaB helicase and its complex with a domain of DnaG primase, Science 318 (2007) 459–463. [DOI] [PubMed] [Google Scholar]
- [43].Fernandez-Leiro R, Conrad J, Scheres SH, Lamers MH, cryo-EM structures of the E. Coli replicative DNA polymerase reveal its dynamic interactions with the DNA sliding clamp, exonuclease and tau, Elife 4 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Sun J, et al. , The architecture of a eukaryotic replisome, Nat. Struct. Mol. Biol 22 (2015) 976–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Goswami P, et al. , Structure of DNA-CMG-Pol epsilon elucidates the roles of the non-catalytic polymerase modules in the eukaryotic replisome, Nat. Commun 9 (2018) 5061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Nandakumar D, Pandey M, Patel SS, Cooperative base pair melting by helicase and polymerase positioned one nucleotide from each other, Elife 4 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Georgescu R, et al. , Structure of eukaryotic CMG helicase at a replication fork and implications to replisome architecture and origin initiation, Proc. Natl. Acad. Sci. U. S. A 114 (2017) E697–E706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Takayama Y, et al. , GINS, a novel multiprotein complex required for chromosomal DNA replication in budding yeast, Genes Dev. 17 (2003) 1153–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Ilves I, Petojevic T, Pesavento JJ, Botchan MR, Activation of the MCM2–7 helicase by association with Cdc45 and GINS proteins, Mol. Cell 37 (2010) 247–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Li H, O’Donnell ME, The eukaryotic CMG helicase at the replication fork: emerging architecture reveals an unexpected mechanism, Bioessays 40 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Copeland WC, Wang TS, Enzymatic characterization of the individual mammalian primase subunits reveals a biphasic mechanism for initiation of DNA replication, J. Biol. Chem 268 (1993) 26179–26189. [PubMed] [Google Scholar]
- [52].Doublie S, Zahn KE, Structural insights into eukaryotic DNA replication, Front. Microbiol 5 (2014) 444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Tahirov TH, Makarova KS, Rogozin IB, Pavlov YI, Koonin EV, Evolution of DNA polymerases: an inactivated polymerase-exonuclease module in Pol epsilon and a chimeric origin of eukaryotic polymerases from two classes of archaeal ancestors, Biol. Direct 4 (2009) 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Pursell ZF, Isoz I, Lundstrom EB, Johansson E, Kunkel TA, Yeast DNA polymerase epsilon participates in leading-strand DNA replication, Science 317 (2007) 127–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Nick McElhinny SA, Gordenin DA, Stith CM, Burgers PM, Kunkel TA, Division of labor at the eukaryotic replication fork, Mol. Cell 30 (2008) 137–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Daigaku Y, et al. , A global profile of replicative polymerase usage, Nat. Struct. Mol. Biol 22 (2015) 192–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Garbacz MA, et al. , Evidence that DNA polymerase delta contributes to initiating leading strand DNA replication in Saccharomyces cerevisiae, Nat. Commun 9 (2018) 858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Hu Z, Perumal SK, Yue H, Benkovic SJ, The human lagging strand DNA polymerase delta holoenzyme is distributive, J. Biol. Chem 287 (2012) 38442–38448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Nghiem Y, Cabrera M, Cupples CG, Miller JH, The mutY gene: a mutator locus in Escherichia coli that generates G.C——T.A transversions, Proc. Natl. Acad. Sci. U. S. A 85 (1988) 2709–2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Hatahet Z, Zhou M, Reha-Krantz LJ, Morrical SW, Wallace SS, In search of a mutational hotspot, Proc. Natl. Acad. Sci. U. S. A 95 (1998) 8556–8561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Sabouri N, Viberg J, Goyal DK, Johansson E, Chabes A, Evidence for lesion bypass by yeast replicative DNA polymerases during DNA damage, Nucleic Acids Res. 36 (2008) 5660–5667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Michaels ML, Cruz C, Grollman AP, Miller JH, Evidence that MutY and MutM combine to prevent mutations by an oxidatively damaged form of guanine in DNA, Proc. Natl. Acad. Sci. U. S. A 89 (1992) 7022–7025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Gabbai CB, Yeeles JT, Marians KJ, Replisome-mediated translesion synthesis and leading strand template lesion skipping are competing bypass mechanisms, J. Biol. Chem 289 (2014) 32811–32823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Yeeles JT, Marians KJ, Dynamics of leading-strand lesion skipping by the replisome, Mol. Cell 52 (2013) 855–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Georgescu RE, Yao NY, O’Donnell M, Single-molecule analysis of the Escherichia coli replisome and use of clamps to bypass replication barriers, FEBS Lett. 584 (2010) 2596–2605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Giannattasio M, et al. , Visualization of recombination-mediated damage bypass by template switching, Nat. Struct. Mol. Biol 21 (2014) 884–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Itsathitphaisarn O, Wing RA, Eliason WK, Wang J, Steitz TA, The hexameric helicase DnaB adopts a nonplanar conformation during translocation, Cell 151 (2012) 267–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Sun B, et al. , T7 replisome directly overcomes DNA damage, Nat. Commun 6 (2015) 10260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].McInerney P, O’Donnell M, Functional uncoupling of twin polymerases: mechanism of polymerase dissociation from a lagging-strand block, J. Biol. Chem 279 (2004) 21543–21551. [DOI] [PubMed] [Google Scholar]
- [70].Michel B, Sandler SJ, Replication restart in Bacteria, J. Bacteriol 199 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Michel B, Sinha AK, Leach DRF, Replication fork breakage and restart in Escherichia coli, Microbiol. Mol. Biol. Rev 82 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Windgassen TA, Wessel SR, Bhattacharyya B, Keck JL, Mechanisms of bacterial DNA replication restart, Nucleic Acids Res. 46 (2018) 504–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Jones JM, Nakai H, Escherichia coli PriA helicase: fork binding orients the helicase to unwind the lagging strand side of arrested replication forks, J. Mol. Biol 312 (2001) 935–947. [DOI] [PubMed] [Google Scholar]
- [74].Windgassen TA, Leroux M, Satyshur KA, Sandler SJ, Keck JL, Structure-specific DNA replication-fork recognition directs helicase and replication restart activities of the PriA helicase, Proc. Natl. Acad. Sci. U. S. A 115 (2018) E9075–E9084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Heller RC, Marians KJ, Non-replicative helicases at the replication fork, DNA Repair (Amst.) 6 (2007) 945–952. [DOI] [PubMed] [Google Scholar]
- [76].Xu L, Marians KJ, PriA mediates DNA replication pathway choice at recombination intermediates, Mol. Cell 11 (2003) 817–826. [DOI] [PubMed] [Google Scholar]
- [77].Zavitz KH, Marians KJ, ATPase-deficient mutants of the Escherichia coli DNA replication protein PriA are capable of catalyzing the assembly of active primosomes, J. Biol. Chem 267 (1992) 6933–6940. [PubMed] [Google Scholar]
- [78].Ge XQ, Jackson DA, Blow JJ, Dormant origins licensed by excess Mcm2–7 are required for human cells to survive replicative stress, Genes Dev. 21 (2007) 3331–3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Ibarra A, Schwob E, Mendez J, Excess MCM proteins protect human cells from replicative stress by licensing backup origins of replication, Proc. Natl. Acad. Sci. U. S. A 105 (2008) 8956–8961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].DePamphilis ML, Wassarman PM, Replication of eukaryotic chromosomes: a close-up of the replication fork, Annu. Rev. Biochem 49 (1980) 627–666. [DOI] [PubMed] [Google Scholar]
- [81].Burgers PMJ, Kunkel TA, Eukaryotic DNA replication fork, Annu. Rev. Biochem 86 (2017) 417–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Maher RL, Branagan AM, Morrical SW, Coordination of DNA replication and recombination activities in the maintenance of genome stability, J. Cell. Biochem 112 (2011) 2672–2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].McVey M, Khodaverdian VY, Meyer D, Cerqueira PG, Heyer WD, Eukaryotic DNA polymerases in homologous recombination, Annu. Rev. Genet 50 (2016) 393–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Nohmi T, Kim SR, Yamada M, Modulation of oxidative mutagenesis and carcinogenesis by polymorphic forms of human DNA repair enzymes, Mutat. Res 591 (2005) 60–73. [DOI] [PubMed] [Google Scholar]
- [85].Mouron S, et al. , Repriming of DNA synthesis at stalled replication forks by human PrimPol, Nat. Struct. Mol. Biol 20 (2013) 1383–1389. [DOI] [PubMed] [Google Scholar]
- [86].Kobayashi K, et al. , Repriming by PrimPol is critical for DNA replication restart downstream of lesions and chain-terminating nucleosides, Cell Cycle 15 (2016) 1997–2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Marechal A, Zou L, RPA-coated single-stranded DNA as a platform for post-translational modifications in the DNA damage response, Cell Res. 25 (2015) 9–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Raschle M, et al. , Mechanism of replication-coupled DNA interstrand crosslink repair, Cell 134 (2008) 969–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Zhang J, et al. , DNA interstrand cross-link repair requires replication-fork convergence, Nat. Struct. Mol. Biol 22 (2015) 242–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Mutreja K, et al. , ATR-mediated global fork slowing and reversal assist fork traverse and prevent chromosomal breakage at DNA interstrand cross-links, Cell Rep. 24 (2018) 2629–2642 e2625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Xue X, Sung P, Zhao X, Functions and regulation of the multitasking FANCM family of DNA motor proteins, Genes Dev. 29 (2015) 1777–1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Huang J, et al. , Remodeling of interstrand crosslink proximal replisomes is dependent on ATR, FANCM, and FANCD2, Cell Rep. 27 (2019) 1794–1808 e1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Yardimci H, et al. , Bypass of a protein barrier by a replicative DNA helicase, Nature 492 (2012) 205–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Sparks JL, et al. , The CMG helicase bypasses DNA-Protein cross-links to facilitate their repair, Cell 176 (2019) 167–181 e121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Sogo JM, Lopes M, Foiani M, Fork reversal and ssDNA accumulation at stalled replication forks owing to checkpoint defects, Science 297 (2002) 599–602. [DOI] [PubMed] [Google Scholar]
- [96].Neelsen KJ, Lopes M, Replication fork reversal in eukaryotes: from dead end to dynamic response, Nat. Rev. Mol. Cell Biol 16 (2015) 207–220. [DOI] [PubMed] [Google Scholar]
- [97].Amunugama R, et al. , Replication fork reversal during DNA interstrand crosslink repair requires CMG unloading, Cell Rep. 23 (2018) 3419–3428. [DOI] [PMC free article] [PubMed] [Google Scholar]