

ABSTRACT
Background
Recent literature suggests that the Western diet's imbalance between high ω-6 (n–6) and low ω-3 (n–3) PUFA intake contributes to fatty liver disease in obese youth.
Objectives
We tested whether 12 wk of a low n–6:n–3 PUFA ratio (4:1) normocaloric diet mitigates fatty liver and whether the patatin-like containing domain phospholipase 3 (PNPLA3) rs738409 variant affects the response.
Methods
In a single-arm unblinded study, obese youth 9–19 y of age with nonalcoholic fatty liver disease were treated with a normocaloric low n–6:n–3 PUFA ratio diet for 12 wk. The primary outcome was change in hepatic fat fraction (HFF%), measured by abdominal MRI. Metabolic parameters included alanine aminotransferase (ALT), lipids, measures of insulin sensitivity, and plasma oxidized linoleic acid metabolites (OXLAMs). Outcomes were also analyzed by PNPLA3 rs738409 genotype. Wilcoxon's signed rank test, the Mann–Whitney U test, and covariance pattern modeling were used.
Results
Twenty obese adolescents (median age: 13.3 y; IQR: 10.5–16.4 y) were enrolled and 17 completed the study. After 12 wk of dietary intervention, HFF% decreased by 25.8% (P = 0.009) despite stable weight. We observed a 34.4% reduction in ALT (P = 0.001), 21.9% reduction in triglycerides (P = 0.046), 3.28% reduction in LDL cholesterol (P = 0.071), and a 26.3% improvement in whole body insulin sensitivity (P = 0.032). The OXLAMs 9-hydroxy-octadecandienoic acid (9-HODE) (P = 0.011), 13-HODE (P = 0.007), and 9-oxo-octadecadienoic acid (9-oxoODE) (P = 0.024) decreased after 12 wk. HFF% declined in both the not-at-risk (CC/CG) and at-risk (GG) PNPLA3 rs738409 genotype groups, with significant (P = 0.016) HFF% reduction in the GG group. Changes in 9-HODE (P = 0.023), 9-oxoODE (P = 0.009), and 13-oxoODE (P = 0.003) differed between the 2 genotype groups over time.
Conclusions
These data suggest that, independently of weight loss, a low n–6:n–3 PUFA diet ameliorates the metabolic phenotype of adolescents with fatty liver disease and that response to this diet is modulated by the PNPLA3 rs738409 genotype.
This trial was registered at clinicaltrials.gov as NCT01556113.
Keywords: childhood obesity, PNPLA3, insulin resistance, nonalcoholic fatty liver disease, metabolic syndrome
Introduction
Nonalcoholic fatty liver disease (NAFLD) is a common complication of childhood obesity and an ominous marker of the metabolic syndrome (1). Indeed, children with NAFLD exhibit insulin resistance, glucose intolerance, hypertriglyceridemia (1), and cardiovascular disease (2, 3).
Although many factors contribute to the development of fatty liver, the composition of the diet plays a pivotal role. Recent literature suggests that the dietary imbalance of high ω-6 (n–6) and low ω-3 (n–3) PUFA intake, characteristic of the Western diet (average ratio 15:1), leads to the development of NAFLD (4). Furthermore, diet is the only source of the essential fatty acids linoleic acid (LA; 18:2n–6) and α-linolenic acid (ALA; 18:3n–3), the respective precursors to n–6 and n–3 PUFAs. Accordingly, it has been shown that individuals with NAFLD have a lower n–3 and higher n–6 PUFA dietary intake than healthy controls, consistent with the abundance of n–6 PUFAs in hepatic long-chain fatty acids (5). The excess hepatic uptake of n–6 PUFAs promotes formation and accumulation of oxidized linoleic acid metabolites (OXLAMs), which are risk factors for fatty liver development in both youth and adults (6–8). Furthermore, our group has recently shown the relation of genetics to dietary PUFA intake and development of NAFLD. In fact, a common variant (rs738409) in the patatin-like containing domain phospholipase 3 gene (PNPLA3) (9) enhances the association between OXLAM concentration and fatty liver disease in obese adolescents (10).
In this quasi-experimental study we aimed to examine whether 12 weeks of a low n–6:n–3 PUFA ratio (4:1) diet affects intrahepatic fat content, assessed by MRI, in obese youth with fatty liver. Moreover, given our previous observation of an interaction between the PNPLA3 rs738409 variant and n–6:n–3 PUFA ratio of dietary intake on fatty liver (11), we also explored whether this variant affected individual responses to the diet.
Methods
Study population
Twenty children and adolescents (aged 9–19 y) were recruited. Eligible participants had a BMI ≥95th percentile for age and gender and hepatic fat fraction (HFF%) measured by liver MRI ≥5.5%. Youth with a liver disease other than NAFLD, diabetes, a food allergy to fish, or who were taking medications affecting lipid, glucose, or hepatic metabolism were excluded. The study (NCT01556113) was approved by the Yale Human Investigation Committee and written consent and assent (for minors) were obtained from all participants. The procedures followed were in accordance with the ethical standards of Yale University's Human Investigations Committee in accordance with the Helsinki Declaration of 1975 as revised in 1983.
Study outcomes
Our primary outcome was change in HFF% from baseline (week 0) to follow-up (week 12) of the diet. Secondary outcomes included changes from week 0 to week 12 in plasma alanine aminotransferase (ALT), lipids (triglycerides, LDL cholesterol), and indexes of glucose metabolism [glucose, insulin, whole body insulin sensitivity index (WBISI)]. Plasma concentrations of n–6 PUFA metabolites [oxidized compounds derived from LA (OXLAM)] and n–3 PUFAs [ALA, EPA (20:5n–3), docosapentaenoic acid (DPA; 22:5n–3), and DHA (22:6n–3)] were assessed for linear trends.
We explored the impact of the PNPLA3 rs738409 variant on HFF%, triglycerides, and ALT from week 0 to week 12, and on linear changes in plasma OXLAM and n–3 PUFA concentrations during the study period. Raw study data are available as a supplemental Excel file.
Procedures
In addition to medical and dietary history, participants underwent screening laboratory evaluation, a standard oral-glucose-tolerance test (OGTT) (12), abdominal MRI, and genotyping for PNPLA3 rs738409 at Yale School of Medicine. Participants then began the 12-wk dietary intervention provided by the study personnel (described below). Participants met weekly with the dietitian and team to receive study food, review compliance through weekly food records, and obtain anthropometric measurements. To monitor the compliance to the diet through biochemical measures, every 4 wk, LA, arachidonic acid (AA; 20:4n–6), and its OXLAM products, 9- and 13-hydroxy-octadecandienoic acid (HODE) and 9- and 13-oxo-octadecadienoic acid (oxoODE), were measured from plasma (13). n–3 PUFAs were also measured from plasma, and included ALA, DHA, EPA, and DPA. The OGTT, MRI, and laboratory tests were repeated at the conclusion of the 12-wk intervention.
Meal plan
For the 12 wk of dietary intervention, study personnel provided all needed dietary intake to participants (not for the entire family). To control for the potential confounding variable of weight loss or gain, the meal plan was designed to be normocaloric to prestudy food intake, as assessed by the study dietitian. Three-day food records (1 weekend day and 2 weekdays) were used to assess prestudy food intake and analyzed using the Nutrition Data Software for Research (NDS-R 2014, University of Minnesota). This program determined usual caloric intake and, consequently, the prescribed caloric amount for the intervention. The intervention diet consisted of a low n–6 to n–3 PUFA ratio of 4:1, and macronutrient content was 50%–55% daily total calories from carbohydrate, 20% from protein, and 25%–30% from fat, with saturated fat comprising 8%–10% of daily total caloric intake and monounsaturated and polyunsaturated fats each comprising 8%–10% of daily total caloric intake.
The diet was rich in n–3 PUFAs (e.g., fish and nuts), and although no supplements were used in this study, some foods (spreads, dressings, and oil) were enriched with n–3 PUFAs. Udo's Oil 3-6-9 Blend (Flora, Inc.) was the staple oil utilized and it provided twice the amount of n–3 compared with n–6 PUFAs. The meal plan was “kid-friendly” with foods such as frozen pizza and burgers included each week; there was also 1 fish-free day. This design was used to represent real-world, long-term food intake and, of course, to increase the likelihood of compliance. Participants were instructed to only consume items provided by the meal plan; a sample meal plan is provided in Supplemental Figure 1.
A parent was present during diet instruction, which continued weekly for the first 4 wk. Participants picked up food twice weekly. Food was measured and packaged for each day by the Metabolic Kitchen of the Yale Center for Clinical Investigation. For the final 8 wk, the participants picked up food weekly, with a supplemental food delivery to participant homes by a commercial grocery delivery service. Compliance was assessed with a simplified daily food record, in which participants checked off boxes for foods consumed, and it was returned to the dietitian weekly (Supplemental Figure 1). Food records were scored accordingly to determine the percentage compliance and a score >80% was deemed “compliant.” Weight was also measured weekly and calories were modified if a participant gained or lost >1.6 kg (∼3 pounds) within a week or fluctuated a total of 2.3 kg (5 pounds) from baseline, for the goal of weight maintenance during the 12-wk dietary intervention.
Physical activity
In addition to controlling for the potential confounding variable of weight loss or gain through caloric adjustments as needed, we instructed all participants to keep their physical activity level stable during the intervention. Specifically, if a child did not normally engage in exercise activities, we asked that they remain inactive. If a child was fairly active, we instructed the participant to also continue with the same level of activity.
Smoking and alcohol
The study dietitian instructed participants to not consume alcohol or smoke during the 12-wk dietary intervention. At the OGTT visits at week 0 and week 12, a smoking and alcohol questionnaire was administered and completed by the child and parent.
Abdominal MRI
Abdominal MRI was performed on a Siemens Sonata 1.5 Tesla system. HFF% was calculated from the liver images obtained using a modified Dixon technique (14, 15). Visceral and subcutaneous abdominal fat fractions were assessed as previously described (16).
Biochemical analyses
Plasma glucose concentrations were measured at bedside using the YSI 2300 Stat Plus Glucose Analyzer (Yellow Springs Instruments). Plasma insulin concentrations were measured using antibody RIA from Millipore Sigma, lipid concentrations were measured with an Auto-Analyzer (model 747-200, Roche Diagnostics), and liver enzymes were measured by using standard automated kinetic enzymatic assays. The OGTT-derived WBISI was calculated to reflect insulin sensitivity in this cohort of children with obesity (17, 18).
Measurements of n–6 and n–3 PUFAs
Lipid extractions and protein hydrolyses were performed using disposable threaded borosilicate glass test tubes with Polytetrafluoroethylene (PTFE)-lined caps. Before use, all glassware tubes, caps, and pipette tips were washed with nitric acid to remove trace transition metals, extensively rinsed with Chelex-treated water containing 1 μM diethylenetriamine pentaacetic acid (DTPA; pH 7.0 in H2O), and then rinsed with pure Chelex-treated water. Plastic tips were further rinsed in methanol and air-dried before use. Test tubes were also baked at 500°C overnight to remove residual potential organics. All plasma samples were divided into aliquots in tubes containing an antioxidant cocktail [DTPA (2 mM final) and butylated hydroxytoluene (500 μM final)] with head space overlaid with argon. Samples were thawed in an ice/water bath immediately before sample handling for LC online electrospray ionization tandem MS analysis (8). Fatty acids and oxidized fatty acids in plasma were extracted as previously described (8). Briefly, plasma (50 μL), internal standard [synthetic 15(S)-HETE-d8], and potassium hydroxide were added to the glass test tubes, overlaid with argon, and sealed. Lipids were hydrolyzed at 60°C under an argon atmosphere for 2 h, and then the released fatty acids were extracted into the hexane layer twice by liquid/liquid extraction. With each extraction, argon was used to purge the head space of the tube before sealing and mixing on a vortex/centrifugation. The combined hexane layers were dried under nitrogen gas and then resuspended in 200 μL 85% methanol:water (vol:vol) (8).
Genotyping
Genomic DNA was extracted from peripheral blood leukocytes from whole blood using the guanidine HCl DNA extraction protocol. Automatic sequencing was used to genotype the PNPLA3 rs738409 variant as previously reported (10). We previously observed that subjects homozygous for the risk minor allele (GG) have a much higher intrahepatic fat content than those with the other 2 genotypes (CC and CG) (19). To analyze the effect of the PNPLA3 rs738409 variant on the changes in HFF% after the 12-wk intervention, we grouped the homozygous major allele and the heterozygous (CC/CG) to compare the percentage changes in HFF% between this group and the group of individuals homozygous for the risk minor allele (GG).
Statistical analysis
Descriptive statistics, such as median (IQR) and n (%), were used to characterize the cohort. Owing to the small sample size and nonnormal distribution of most of the outcomes of interest, which is well documented in the literature, we selected nonparametric tests for the unadjusted analyses. Differences in the distributions of outcomes at week 0 and at week 12 of the intervention were assessed using Wilcoxon's signed rank test. Differences in the change from week 0 to week 12 between the genotype groups were assessed using the Mann–Whitney U test. Differences in prevalence were evaluated using a chi-square test or Fisher's exact test, as appropriate. Mean trajectories of log-transformed OXLAMs and PUFAs over time were modeled using covariance pattern (CP) modeling, an approach for longitudinal or repeated within-person observations, whereby a specific correlation structure is applied to the residuals. Optimal correlation structures were selected among the unstructured, AR1, compound symmetry, and Toeplitz, using the likelihood ratio test (from restricted-maximum likelihood) for nested models and the smallest Akaike Information Criterion (AIC) for nonnested models. Using the time trends in the observed means, we tested up to the second-degree polynomial trend in the CP models for the mean trajectories of outcomes over time by incorporating a linear and a quadratic effect for time. When testing for the effect of genotype on the trajectories, we examined all relevant linear contrasts of the effect of time (linear compared with linear plus quadratic) and the binary genotype variable (GG/CG compared with CC), and reported P values from the most parsimonious models, using the Wald test. All subjects were included in the CP modeling because this approach assumes that an outcome at a given time point is missing at random. This is a reasonable assumption for our data, given that there were only 2 missing observations for week 8 OXLAM and n–3 PUFA measurements and we can use all available data for an outcome in the maximum-likelihood-based estimation of the parameters in a CP model. Statistical significance was established at α of 0.05, and because this was a small feasibility study, we also considered α of 0.10 indicative of a potential statistical trend. Data were summarized using boxplots, as well as line plots of the observed means and SEs of the outcomes of interest. Analyses were implemented using SAS 9.4 (SAS Institute) and figures were generated using GraphPad Prism software 8.2.0.
Results
Participant characteristics
Of the 20 youth who were recruited, 3 (females) withdrew during the first week of the study. Table 1 shows demographic and baseline characteristics of the 17 adolescents who completed the 12-wk study. All of the subjects had fatty liver as defined by an HFF% >5.5%; median HFF% was 14.7% (IQR: 9.10%–22.8%) and median plasma ALT was 33.0 U/L (IQR: 10.0–54.0 U/L). During the study, the mean weight remained stable by study design (Supplemental Figure 2) and there were no statistically significant changes in visceral or subcutaneous abdominal fat, or the visceral/subcutaneous abdominal fat ratio (P > 0.05) (Table 1).
TABLE 1.
Week 0 baseline and week 12 follow-up characteristics of 17 obese male and female youths1
| Week 0 (n = 17) | Week 12 (n = 17) | P value2 | |
|---|---|---|---|
| Age, years | 13.3 [10.5–16.4] | 13.6 [10.8–16.7] | 0.001 |
| Sex (M/F) [n(%)] | 6/11 (35/65) | 6/11 (35/65) | 1.000 |
| Race (C/AA/H) [n(%)] | 7/2/8 (41/12/47) | 7/2/8 (41/12/47) | 1.000 |
| Body composition | |||
| BMI, kg/m2 | 32.5 [26.8–39.1] | 31.6 [26.2–39.0] | 0.685 |
| VAT, cm2 | 73.8 [54.2–98.9] | 69.9 [52.5–101.0] | 0.323 |
| SAT, cm2 | 539 [352–713] | 504 [348–641] | 0.464 |
| VAT/(VAT + SAT) ratio | 0.148 [0.107–0.194] | 0.141 [0.101–0.204] | 0.900 |
| Metabolic measurements | |||
| Glucose tolerance (NGT/IGT/T2D) [n(%)] | 13/4/0 (76/24/0) | 15/1/1 (88/6/6) | 0.344 |
| Fasting glucose,3 mg/dL | 87.5 [80.5–93.4] | 89.0 [83.9–91.0] | 0.773 |
| 2-h glucose, 3 mg/dL | 118 [102–142] | 109 [101–126] | 0.323 |
| Fasting insulin,3 μU/mL | 42.3 [24.0–52.0] | 32.1 [15.6–48.1] | 0.050 |
| WBISI | 1.09 [0.695–1.74] | 1.27 [0.820–2.56] | 0.032 |
Values are median [IQR] or n (%). AA, African American; C, Caucasian; H, Hispanic; IGT, impaired glucose tolerance; NGT, normal glucose tolerance; SAT, subcutaneous adipose tissue; T2D, type 2 diabetes; VAT, visceral adipose tissue; WBISI, whole body insulin sensitivity index.
P values are from Wilcoxon's signed rank test for continuous variables or chi-square test for categorical variables.
Measured from blood plasma.
Dietary compliance
The dietitian obtained completed simplified food records from each participant to assess percentage compliance for each week. Of the 17 participants who completed the dietary intervention, 15 participants were compliant (median compliance for the 12-wk intervention: 92.0%; IQR: 87.2%–94.7%). For measures of plasma OXLAM concentrations, there was a significant progressive decline in 9-HODE, 13-HODE, and 9-oxoODE concentrations in the cohort over the 12-wk intervention (P < 0.05) (Supplemental Figure 3). Although there was no clear trend in the plasma concentrations of n–3 PUFAs in the cohort, there was a significant increase in DHA over the 12-wk intervention (P = 0.002) (Supplemental Figure 4).
Physical activity compliance
No participant reported the initiation of an exercise regimen or an increase in physical activity. Participants were sedentary at week 0 and throughout the duration of the intervention, and no participants engaged in vigorous daily activity.
Smoking and alcohol compliance
Questionnaires on smoking and alcohol usage collected at week 0 and week 12 indicated that participants were not smoking or drinking alcohol at the start of the study, as well as throughout the 12-wk intervention.
Changes in liver-related outcomes
In Figure 1A, we show individual changes in HFF% from before (week 0) to after (week 12) 12 wk of dietary intervention. Median HFF% decreased in the entire group by 25.8% (P = 0.009), from 14.7% (IQR: 9.10%–22.8%) at week 0 to 9.70% (IQR: 2.80%–22.4%) at week 12. Among the 14 participants who reduced HFF% by week 12 of the diet intervention, the median HFF% at week 0 was 12.3% (IQR: 9.00%–18.2%) and the median HFF% at week 12 was 7.15% (IQR: 1.78%–12.1%). Of them, 6 subjects reverted to the normal range, as defined by HFF% <5.5%, in whom the median HFF% at week 0 was 8.90% (IQR: 8.43%–10.6%) and the median HFF% at week 12 was 1.56% (IQR: 0.00%–3.95%). Three subjects showed no change or an increase in HFF% after the diet intervention, in whom the median HFF% at week 0 was 17.8% (IQR: 15.9%–30.9%) and the median HFF% at week 12 was 24.4% (IQR: 20.3%–32.6%). At the end of the 12-wk intervention, a significant drop in median plasma ALT concentrations (P = 0.001) was also observed in the overall cohort (week 0: 33.0 IU/L; IQR: 10.0–54.0 IU/L; week 12: 19.0 IU/L; IQR: 5.00–33.0 IU/L) (Figure 1B).
FIGURE 1.
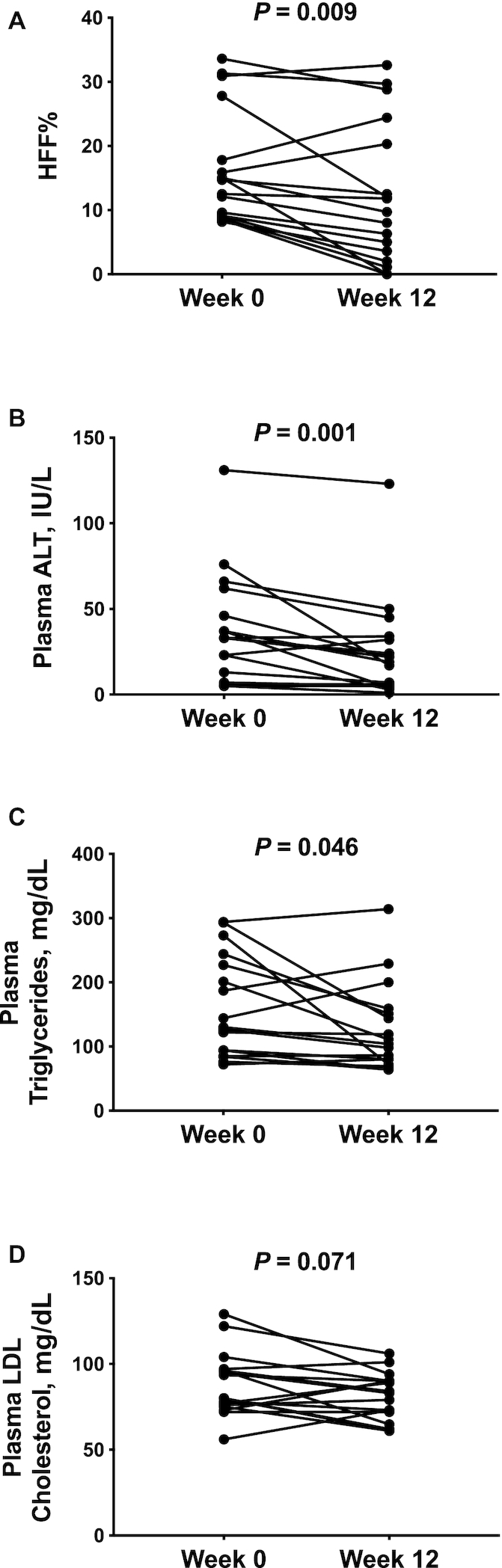
HFF% (A), plasma ALT (B), plasma triglycerides (C), and plasma LDL cholesterol (D) in 17 obese adolescents before (week 0) and after (week 12) a 12-wk low n–6:n–3 PUFA ratio dietary intervention. P values are from Wilcoxon's signed rank test. ALT, alanine aminotransferase; HFF%, hepatic fat fraction.
Changes in glucose- and lipid metabolism–related outcomes
After the intervention, there was a 21.9% reduction of median plasma triglyceride concentrations (week 0: 130 mg/dL; IQR: 89.5–236 mg/dL; week 12: 104 mg/dL; IQR: 76.0–155 mg/dL; P = 0.046) (Figure 1C) and a statistical trend toward a reduction of plasma concentrations of LDL cholesterol (week 0: 80.0 mg/dL; IQR: 75.5–96.5 mg/dL; week 12: 83.0 mg/dL; IQR: 68.4–90.0 mg/dL; P = 0.071) (Figure 1D). The OGTT glycemic responses at week 0 and at week 12 of the intervention were nearly superimposable (P = 0.207) (Figure 2A); however, OGTT plasma insulin concentrations significantly decreased in the cohort at the end of the intervention (P = 0.045) (Figure 2B). Supplemental Figure 5 shows the total AUCs for OGTT plasma glucose and insulin concentrations at week 0 and week 12 for each subject. Of the 4 subjects that presented with impaired glucose tolerance (IGT) at the beginning of the study, 2 subjects reverted to normal glucose tolerance, 1 subject remained with IGT, and 1 subject progressed to type 2 diabetes by the end of the dietary intervention (HFF% at week 0: 15.9%; HFF% at week 12: 20.3%) (Figure 2C, D).
FIGURE 2.
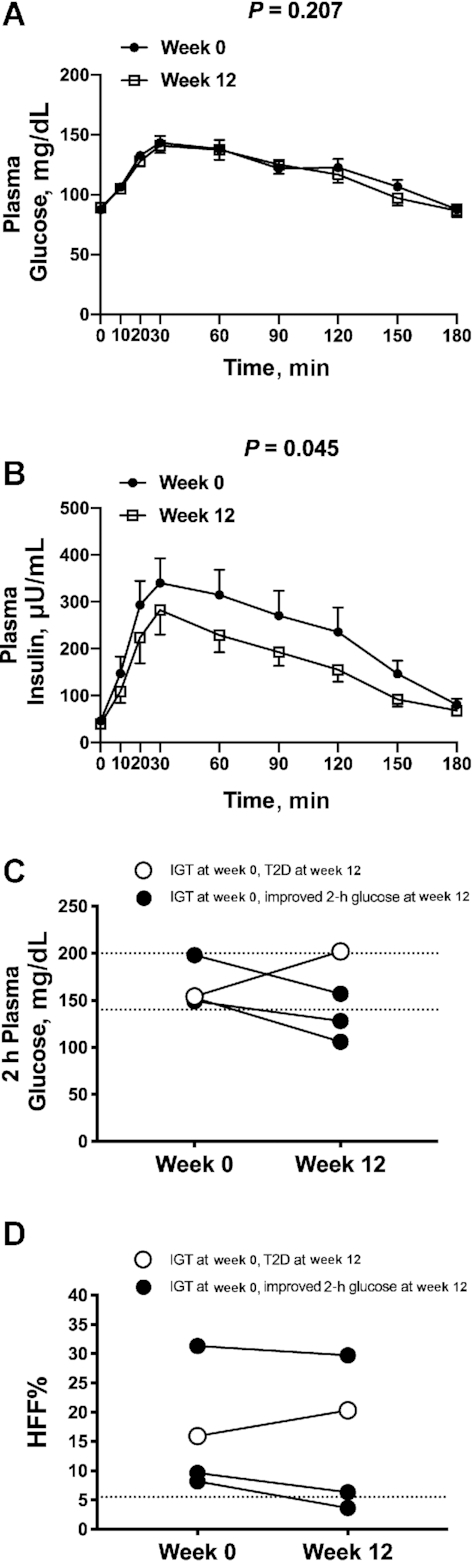
The figure shows changes in glucose and insulin during an OGTT in the entire cohort (A and B) and changes in 2-h glucose and HFF% in the four subjects with IGT (C and D). (A) Plasma glucose and (B) plasma insulin concentrations during an oral-glucose-tolerance test before (week 0) and after (week 12) a 12-wk low n–6:n–3 PUFA ratio dietary intervention. Data are observed mean ± SEM, n = 17. Individual changes in (C) 2-h plasma glucose and (D) HFF% before (week 0) and after (week 12) a 12-wk low n–6:n–3 PUFA ratio dietary intervention for the 4 subjects that presented with IGT at week 0. Of the 4 subjects with IGT at week 0, 3 subjects showed reduced 2-h glucose and HFF% values by week 12 of the diet intervention. The open circle indicates the only subject that progressed to T2D and increased HFF% by week 12 of the intervention. Total AUCs for week 0 and week 12 for (A) plasma glucose and (B) plasma insulin compared with Wilcoxon's signed rank test. HFF%, hepatic fat fraction; IGT, impaired glucose tolerance; T2D, type 2 diabetes.
Changes in n–3 PUFAs and oxidized fatty acids derived from LA (OXLAMs)
Whereas there were no overall changes in the plasma concentrations of LA and AA (P > 0.05) (Supplemental Figure 6), there was a significant progressive decline in plasma concentrations of 9-HODE, 13-HODE, and 9-oxoODE (P < 0.05) and a trend toward a reduction in plasma concentrations of 13-oxoODE (P = 0.053) over the 12-wk intervention (Supplemental Figure 3). For n–3 PUFAs, the plasma concentration of DHA increased significantly (P = 0.002) and there was a trend toward a rise in plasma concentrations of EPA (P = 0.052) over the 12-wk dietary intervention (Supplemental Figure 4).
Changes in liver-related outcomes by PNPLA3 rs738409 genotype
Of the 17 participants, 8 were homozygous for the PNPLA3 rs738409 risk allele (GG), 4 were heterozygous (CG), and 5 subjects were homozygous for the major allele (CC). The CC/CG (3 male, 6 female) and GG (3 male, 5 female) groups had similar age, BMI, and HFF% at week 0 (P > 0.05). Notably, in contrast to the CC/CG group, subjects homozygous for the G allele displayed a significant change in HFF% (P = 0.016) (Figure 3A) and plasma triglycerides (P = 0.016) (Figure 3B) and a trend toward a reduction in plasma ALT (P = 0.055) (Figure 3C) concentrations at the end of the intervention. In Supplemental Figure 7, we show HFF% at week 0 and week 12 stratified by each genotype (CC, CG, GG). Over the 12-wk intervention, OXLAMs decreased in both genotype groups. A significant decline with respect to genotype was found in the OXLAMs 9-HODE (P = 0.023), 9-oxoODE (P = 0.009), and 13-oxoODE (P = 0.003) (Supplemental Figure 8). Plasma concentrations of ALA, EPA, DPA, and DHA (n–3 PUFAs) did not change in a clear pattern during the intervention and were not significantly different among genotypes (P > 0.05) (Supplemental Figure 9). In Supplemental Table 1A, we show week 0 and week 12 characteristics and study outcomes by genotype group and in Supplemental Table 1B, we show mean percentage change from week 0 to week 12 for characteristics and study outcomes by genotype group.
FIGURE 3.
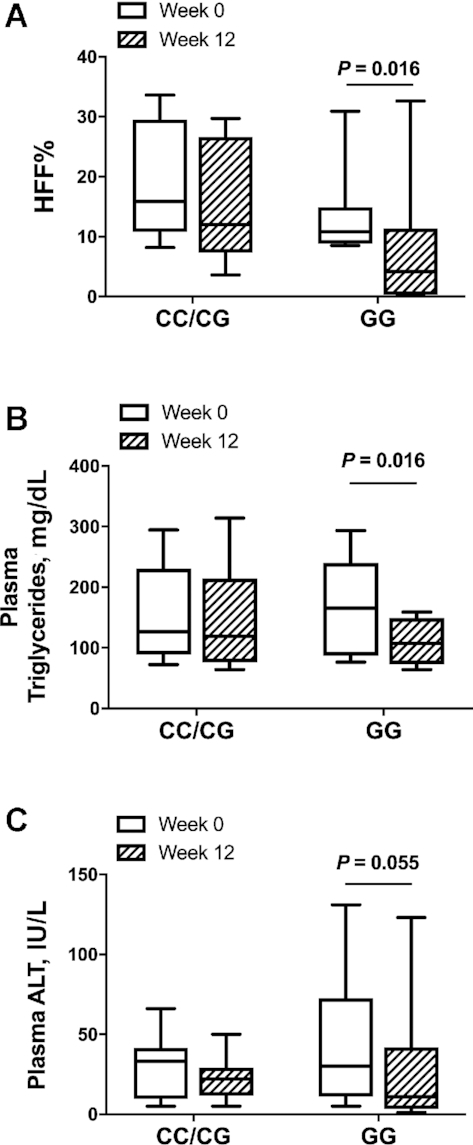
Box plot distribution by patatin-like containing domain phospholipase 3 (PNPLA3) rs738409 variant genotype for HFF% (A), plasma triglycerides (B), and plasma ALT (C) before (week 0) and after (week 12) a 12-wk low n–6:n–3 PUFA ratio dietary intervention. Data are median and IQR, n = 17 (CC/CG: n = 9; GG: n = 8). ALT, alanine aminotransferase; HFF%, hepatic fat fraction.
Discussion
This study has shown, to our knowledge for the first time, that a nonpharmacologic, food-based dietary intervention high in n–3 and low in n–6 PUFA intake improves fatty liver disease in obese adolescents, and restores liver fat content to normal in one-third of participants, in the absence of weight loss. The observed improvement in lipids, lipoprotein concentrations, and insulin sensitivity at the end of the study demonstrates a beneficial impact of a low n–6:n–3 PUFA ratio diet on both diabetes and cardiovascular disease risk factors. Furthermore, patients with the PNPLA3 rs738409 risk genotype tend to have a more robust response to the dietary intervention, suggesting that it may be especially effective in youth homozygous for the PNPLA3 rs738409 risk allele. This study supports the concept that nutritional interventions might be most effective in ameliorating the metabolic phenotype of youth with NAFLD (20), providing additional benefits to dyslipidemia and insulin resistance not present in some drugs for treating NAFLD (21, 22).
Prior work has revealed that adults and children with NAFLD have higher plasma OXLAM concentrations (8, 23). However, the pathophysiology linking a low n–6:n–3 PUFA ratio diet to NAFLD is unknown in youth. The reduction in OXLAMs observed could be the result of the lower substrate supplementation (e.g., n–6 PUFA) that the patients consumed and/or a consequence of a reduction of oxidative stressors. N–3 PUFAs, in fact, act as buffers on free radicals (24), thus high n–3 PUFA concentrations can limit conversion of n–6 PUFAs into downstream metabolites, mitigating the damage that oxidative stress can cause to liver tissue (25).
In addition, prior work has suggested that improved insulin sensitivity may further protect against intrahepatic fat accumulation because improved sensitivity to insulin reduces the flux of free fatty acids from adipose tissue into peripheral circulation and then into the liver (26). Multiple measurements of HFF% and insulin sensitivity could help to further dissect the causative links between improved fatty liver and insulin sensitivity, especially in response to a food-based intervention.
Additional clues to the pathophysiology of hepatic lipid accumulation may be gleaned from the magnitude of the drop in HFF% and in triglycerides in youth with the rs738409 (GG) risk genotype. It has been shown that the interaction between PNPLA3 rs738409 genotype and dietary intake of n–6:n–3 PUFA influences HFF% and ALT concentrations in obese youth (6, 11).
Although early biochemical characterization of the PNPLA3 gene product, adiponutrin, did not show affinity for n–6 PUFAs (27), recent animal data suggest that the physiologic role of adiponutrin is to remodel triglycerides and phospholipids in lipid droplets. Adiponutrin inhibits the lipolytic enzyme adipose triglyceride lipase (ATLG) on lipid droplets in the hepatocyte (28), accommodating the size of lipid droplets in response to feeding (29). Together, these findings support the hypothesis that the mutated protein may indirectly favor accumulation of n–6 PUFA derivatives by affecting lipid droplet remodeling in the hepatocyte. This could explain the interaction observed in clinical studies between the PNPLA3 rs738409 variant and n–6 PUFAs.
However, we acknowledge that this study has some limitations; in fact, the small sample size and the lack of a control intervention represent important drawbacks. Owing to the small sample size, stratification according to genotype could lead to over-interpretation of data. Moreover, because many metabolic parameters were evaluated, there is a risk of making a statistical type I error. Although we did not formally measure physical activity levels, our participants were quite sedentary preintervention and we asked that they keep their activity level consistent to control for possible effects of exercise initiation. If participants engaged in such activity, this could have confounded the effect of reductions seen in HFF%, because exercise can increase fatty acid oxidation and decrease fatty acid synthesis in the liver, even in the absence of weight loss (30).
It remains to be determined whether this nutritional modification is generalizable to a larger population, and whether a paradigm shift away from the Western diet is possible. We envision success in a real-world setting through use of standardized material taught by dietitians in person or via webinar. Improving the feasibility of this nutritional intervention in youth will be possible in some home environments. However, other families may require support to purchase and/or prepare optimal types of foods as well as limit the overwhelming exposure to prepackaged foods in home and school settings.
Another important limitation is the fact that the lipids were measured in the plasma, not in the RBC membrane. In fact, because erythrocytes live for ∼3 mo, a change in erythrocyte lipids might be a more stable indicator of dietary fat composition.
Despite these limitations, the study has several strengths. We meticulously controlled for weight to avoid the confounding factor of weight loss as a mechanism to reduce intrahepatic fat and, indeed, the mean BMI of the cohort did not change significantly from week 0 to week 12 of the intervention. Also, we thoroughly phenotyped the participants using an OGTT to evaluate glucose metabolism, MRI to assess fat distribution, and LC–tandem MS to measure n–6 and n–3 PUFAs for assessing diet compliance. Moreover, the knowledge gained in this study has key implications as we move forward to improve the health of young patients with fatty liver disease at high risk of future complications.
This study has, to our knowledge for the first time, shown improvement in hepatic steatosis and glucose metabolism by a food-based low n–6 and high n–3 PUFA intake dietary intervention. Although this intervention shows feasibility in a small group of obese youth, larger studies are needed, and it would be important to conduct a controlled clinical trial to assess short- and long-term efficacy of a 4:1 n–6 to n–3 PUFA ratio diet on fatty liver disease and its related phenotypes. Moreover, although the mechanism linking OXLAMs, hepatic fat, and insulin resistance remains elusive, our data illustrate the importance of continued investigation on this pathway.
Supplementary Material
Acknowledgments
We are grateful to the patients and their families who agreed to participate in our study perfomed at Yale University and Yale New Haven Hospital, and to the personnel of the Yale Center for Clinical Investigation, Yale Center for Genomic Analysis, Yale Core Lab, and Hospital Research Unit of the Yale New Haven Hospital, including the Metabolic Kitchen. The authors’ responsibilities were as follows––NS and SC: designed the research; MS: designed the diet and provided dietary instruction; BP, JMC, AEF, and CJ: conducted the research; AEF and CJ: provided essential materials; VS, NS, and BTG: analyzed the data and performed the statistical analysis; UE, PLV, GK, MAVN, and SC: provided the subjects; MAVN and NS: wrote the paper; BTG: had primary responsibility for the final content; and all authors: read and approved the final manuscript.
Notes
Supported by American Heart Association grant 13SDG14640038 (to NS), National Institute of Diabetes, Digestive and Kidney grant R01DK114504 (to NS), Juvenile Diabetes Research Foundation grant 2-SRA-2020-884-M-B (to NS), NIH National Center for Advancing Translational Science CTSA grant UL1 TR001863, and Diabetes Research Center at Yale grant P30DK045735.
Author disclosures: The authors report no conflicts of interest.
The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official view of the NIH.
Supplemental Table 1 and Supplemental Figures 1–9 are available from the “Supplementary data” link in the online posting of the article and from the same link in the online table of contents at https://academic.oup.com/jn/.
Abbreviations used: AA, arachidonic acid; ALA, α-linolenic acid; ALT, alanine aminotransferase; CP, covariance pattern; DPA, docosapentaenoic acid; DTPA, diethylenetriamine pentaacetic acid; HFF%, hepatic fat fraction; HODE, hydroxy-octadecandienoic acid; IGT, impaired glucose tolerance; LA, linoleic acid; NAFLD, nonalcoholic fatty liver disease; OGTT, oral-glucose-tolerance test; OXLAM, oxidized linoleic acid metabolite; oxoODE, oxo-octadecadienoic acid; PNPLA3, patatin-like containing domain phospholipase 3; WBISI, whole body insulin sensitivity index.
Contributor Information
Michelle A Van Name, Department of Pediatrics, Yale School of Medicine, New Haven, CT, USA.
Mary Savoye, Department of Pediatrics, Yale School of Medicine, New Haven, CT, USA.
Jennifer M Chick, Department of Pediatrics, Yale School of Medicine, New Haven, CT, USA.
Brittany T Galuppo, Department of Pediatrics, Yale School of Medicine, New Haven, CT, USA.
Ariel E Feldstein, Division of Pediatric Gastrointestinal Diseases, Hepatology, and Nutrition, University of California San Diego, San Diego, CA, USA.
Bridget Pierpont, Department of Pediatrics, Yale School of Medicine, New Haven, CT, USA.
Casey Johnson, Division of Pediatric Gastrointestinal Diseases, Hepatology, and Nutrition, University of California San Diego, San Diego, CA, USA.
Veronika Shabanova, Department of Pediatrics, Yale School of Medicine, New Haven, CT, USA.
Udeme Ekong, Department of Pediatrics, Yale School of Medicine, New Haven, CT, USA.
Pamela L Valentino, Department of Pediatrics, Yale School of Medicine, New Haven, CT, USA.
Grace Kim, Seattle Children's Hospital, Seattle, WA, USA.
Sonia Caprio, Department of Pediatrics, Yale School of Medicine, New Haven, CT, USA.
Nicola Santoro, Department of Pediatrics, Yale School of Medicine, New Haven, CT, USA; Department of Medicine and Health Sciences, “V. Tiberio,” University of Molise, Campobasso, Italy.
References
- 1. Trico' D, Caprio S, Umano GR, Pierpont B, Nouws J, Galderisi A, Kim G, Mata MM, Santoro N. Metabolic features of nonalcoholic fatty liver (NAFL) in obese adolescents: findings from a multiethnic cohort. Hepatology. 2018;68(4):1376–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rutigliano I, Vinci R, De Filippo G, Mancini M, Stoppino L, d'Apolito M, Giardino I, Macarini L, Pettoello Mantovani M, Campanozzi A. Metabolic syndrome, hepatic steatosis, and cardiovascular risk in children. Nutrition. 2017;36:1–7. [DOI] [PubMed] [Google Scholar]
- 3. Pacifico L, Di Martino M, De Merulis A, Bezzi M, Osborn JF, Catalano C, Chiesa C. Left ventricular dysfunction in obese children and adolescents with nonalcoholic fatty liver disease. Hepatology. 2014;59(2):461–70. [DOI] [PubMed] [Google Scholar]
- 4. Toshimitsu K, Matsuura B, Ohkubo I, Niiya T, Furukawa S, Hiasa Y, Kawamura M, Ebihara K, Onji M. Dietary habits and nutrient intake in non-alcoholic steatohepatitis. Nutrition. 2007;23(1):46–52. [DOI] [PubMed] [Google Scholar]
- 5. Parker HM, Johnson NA, Burdon CA, Cohn JS, O'Connor HT, George J. Omega-3 supplementation and non-alcoholic fatty liver disease: a systematic review and meta-analysis. J Hepatol. 2012;56(4):944–51. [DOI] [PubMed] [Google Scholar]
- 6. Santoro N, Caprio S, Giannini C, Kim G, Kursawe R, Pierpont B, Shaw MM, Feldstein AE. Oxidized fatty acids: a potential pathogenic link between fatty liver and type 2 diabetes in obese adolescents?. Antioxid Redox Signal. 2014;20(2):383–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Trico D, Di Sessa A, Caprio S, Chalasani N, Liu W, Liang T, Graf J, Herzog RI, Johnson CD, Umano GR et al. . Oxidized derivatives of linoleic acid in pediatric metabolic syndrome: is their pathogenic role modulated by the genetic background and the gut microbiota?. Antioxid Redox Signal. 2017;30(2):241–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Feldstein AE, Lopez R, Tamimi TA, Yerian L, Chung YM, Berk M, Zhang R, McIntyre TM, Hazen SL. Mass spectrometric profiling of oxidized lipid products in human nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. J Lipid Res. 2010;51(10):3046–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Romeo S, Kozlitina J, Xing C, Pertsemlidis A, Cox D, Pennacchio LA, Boerwinkle E, Cohen JC, Hobbs HH. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet. 2008;40(12):1461–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Santoro N, Kursawe R, D'Adamo E, Dykas DJ, Zhang CK, Bale AE, Cali AM, Narayan D, Shaw MM, Pierpont B et al. . A common variant in the patatin-like phospholipase 3 gene (PNPLA3) is associated with fatty liver disease in obese children and adolescents. Hepatology. 2010;52(4):1281–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Santoro N, Savoye M, Kim G, Marotto K, Shaw MM, Pierpont B, Caprio S. Hepatic fat accumulation is modulated by the interaction between the rs738409 variant in the PNPLA3 gene and the dietary omega6/omega3 PUFA intake. PLoS One. 2012;7(5):e37827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. O'Malley G, Santoro N, Northrup V, D'Adamo E, Shaw M, Eldrich S, Caprio S. High normal fasting glucose level in obese youth: a marker for insulin resistance and beta cell dysregulation. Diabetologia. 2010;53(6):1199–209. [DOI] [PubMed] [Google Scholar]
- 13. Ramsden CE, Ringel A, Feldstein AE, Taha AY, MacIntosh BA, Hibbeln JR, Majchrzak-Hong SF, Faurot KR, Rapoport SI, Cheon Y et al. . Lowering dietary linoleic acid reduces bioactive oxidized linoleic acid metabolites in humans. Prostaglandins Leukot Essent Fatty Acids. 2012;87(4–5):135–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Santoro N, Caprio S, Pierpont B, Van Name M, Savoye M, Parks EJ. Hepatic de novo lipogenesis in obese youth is modulated by a common variant in the GCKR gene. J Clin Endocrinol Metab. 2015;100(8):E1125–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fishbein MH, Gardner KG, Potter CJ, Schmalbrock P, Smith MA. Introduction of fast MR imaging in the assessment of hepatic steatosis. Magn Reson Imaging. 1997;15(3):287–93. [DOI] [PubMed] [Google Scholar]
- 16. Caprio S, Hyman LD, Limb C, McCarthy S, Lange R, Sherwin RS, Shulman G, Tamborlane WV. Central adiposity and its metabolic correlates in obese adolescent girls. Am J Physiol. 1995;269(1 Pt 1):E118–26. [DOI] [PubMed] [Google Scholar]
- 17. Yeckel CW, Weiss R, Dziura J, Taksali SE, Dufour S, Burgert TS, Tamborlane WV, Caprio S. Validation of insulin sensitivity indices from oral glucose tolerance test parameters in obese children and adolescents. J Clin Endocrinol Metab. 2004;89(3):1096–101. [DOI] [PubMed] [Google Scholar]
- 18. Matsuda M, DeFronzo RA. Insulin sensitivity indices obtained from oral glucose tolerance testing: comparison with the euglycemic insulin clamp. Diabetes Care. 1999;22(9):1462–70. [DOI] [PubMed] [Google Scholar]
- 19. Santoro N, Zhang CK, Zhao H, Pakstis AJ, Kim G, Kursawe R, Dykas DJ, Bale AE, Giannini C, Pierpont B et al. . Variant in the glucokinase regulatory protein (GCKR) gene is associated with fatty liver in obese children and adolescents. Hepatology. 2012;55(3):781–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Schwimmer JB, Ugalde-Nicalo P, Welsh JA, Angeles JE, Cordero M, Harlow KE, Alazraki A, Durelle J, Knight-Scott J, Newton KP et al. . Effect of a low free sugar diet vs usual diet on nonalcoholic fatty liver disease in adolescent boys: a randomized clinical trial. JAMA. 2019;321(3):256–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sanyal AJ, Chalasani N, Kowdley KV, McCullough A, Diehl AM, Bass NM, Neuschwander-Tetri BA, Lavine JE, Tonascia J, Unalp A et al. . Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N Engl J Med. 2010;362(18):1675–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Neuschwander-Tetri BA, Loomba R, Sanyal AJ, Lavine JE, Van Natta ML, Abdelmalek MF, Chalasani N, Dasarathy S, Diehl AM, Hameed B et al. . Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo-controlled trial. Lancet. 2015;385(9972):956–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Santoro N, Caprio S, Feldstein AE. Oxidized metabolites of linoleic acid as biomarkers of liver injury in nonalcoholic steatohepatitis. Clin Lipidol. 2013;8(4):411–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Takahashi M, Tsuboyama-Kasaoka N, Nakatani T, Ishii M, Tsutsumi S, Aburatani H, Ezaki O. Fish oil feeding alters liver gene expressions to defend against PPARα activation and ROS production. Am J Physiol Gastrointest Liver Physiol. 2002;282(2):G338–48. [DOI] [PubMed] [Google Scholar]
- 25. Puri P, Wiest MM, Cheung O, Mirshahi F, Sargeant C, Min HK, Contos MJ, Sterling RK, Fuchs M, Zhou H et al. . The plasma lipidomic signature of nonalcoholic steatohepatitis. Hepatology. 2009;50(6):1827–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bush NC, Basu R, Rizza RA, Nair KS, Khosla S, Jensen MD. Insulin-mediated FFA suppression is associated with triglyceridemia and insulin sensitivity independent of adiposity. J Clin Endocrinol Metab. 2012;97(11):4130–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. He S, McPhaul C, Li JZ, Garuti R, Kinch L, Grishin NV, Cohen JC, Hobbs HH. A sequence variation (I148M) in PNPLA3 associated with nonalcoholic fatty liver disease disrupts triglyceride hydrolysis. J Biol Chem. 2010;285(9):6706–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wang Y, Kory N, Cohen JC, Hobbs HH. PNPLA3, CGI-58, and inhibition of hepatic triglyceride hydrolysis in mice. Hepatology. 2019;69(6):2427–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mitsche MA, Hobbs HH, Cohen JC. Patatin-like phospholipase domain-containing protein 3 promotes transfer of essential fatty acids from triglycerides to phospholipids in hepatic lipid droplets. J Biol Chem. 2018;293(18):6958–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Della Pepa G, Vetrani C, Lombardi G, Bozzetto L, Annuzzi G, Rivellese AA. Isocaloric dietary changes and non-alcoholic fatty liver disease in high cardiometabolic risk individuals. Nutrients. 2017;9(10):1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.