

Abstract
Rare mutations have been proposed to restrict the development of broadly neutralizing antibodies against HIV-1, but this has not been explicitly demonstrated. We hypothesized that such rare mutations might be identified by comparing broadly neutralizing and non-broadly neutralizing branches of an antibody-developmental tree. Because sequences of antibodies isolated from the fusion peptide (FP)-targeting VRC34-antibody lineage suggested it might be suitable for such rare mutation analysis, we carried out next-generation sequencing on B cell transcripts from donor N123, the source of the VRC34 lineage, and functionally and structurally characterized inferred intermediates along broadly neutralizing and poorly neutralizing developmental branches. The broadly neutralizing VRC34.01 branch requires the rare heavy chain mutation Y33P to bind FP, whereas the early bifurcated VRC34.05 branch does not require this rare mutation and evolves less breadth. Our results demonstrate how a required rare mutation can restrict development and shape the maturation of a broad HIV-1-neutralizing antibody lineage.
Keywords: antibody sequencing, antibody maturation, B cell ontogeny, broadly neutralizing antibody, fusion peptide-directed, rare mutations, somatic hypermutation (SHM)
Introduction
Broadly neutralizing antibodies capable of neutralizing a majority of circulating HIV-1 strains can arise from natural infection (Burton and Mascola, 2015; Kwong and Mascola, 2012). Such antibodies, however, often have complex maturation pathways, with unusual features such as extraordinary levels of somatic hypermutation (SHM) (Klein et al., 2013; Scheid et al., 2009; Walker et al., 2009; Wu et al., 2010) or atypically long heavy chain third complementarity-determining regions (CDR H3s) (Borrow and Moody, 2017; Doria-Rose et al., 2014). Next-generation sequencing (NGS) of B cell transcripts has been used to study the development of broadly neutralizing antibodies (Doria-Rose et al., 2014; Landais et al., 2017; Liao et al., 2013; MacLeod et al., 2016; Simonich et al., 2016; Soto et al., 2016; Wu et al., 2015). This has revealed their developmental pathways to be diverse between antibody classes. However, among donors that share a given class of antibody, the developmental pathways often share common characteristics related to the unusual features of the antibody class. For example, CD4-binding site-directed antibodies, such as CH103 (Liao et al., 2013), CH235 (Gao et al., 2014), or VRC01 (Wu et al., 2015), all display extremely long development times to achieve their characteristic extraordinarily high levels of SHM. By contrast, V2-apex targeting antibodies (Bonsignori et al., 2012; Doria-Rose et al., 2014; Walker et al., 2009) have characteristic long CDR H3s that are generated only rarely by recombination, but then have relatively short development times (six months) from lineage appearance to broad neutralization (Doria-Rose et al., 2014).
It has been proposed that low-probability (rare) events that lead to the appearance of observed unusual features might restrict development and shape the maturation of broadly neutralizing antibody lineages (Bonsignori et al., 2017; Hwang et al., 2017; Johnson et al., 2018; Kwong et al., 2017; Sheng et al., 2017; Wiehe et al., 2018). Indeed, mutational reversion of rare mutations to germline residues has been shown to reduce function with the antibody lineages BG520.1, DH270, CH235, and VRC01 (Bonsignori et al., 2017; Wiehe et al., 2018); however, despite this apparent dependence on rare mutations, it has been unclear whether other developmental branches – without the rare mutations – could nonetheless have developed effective neutralization. We hypothesized that rare mutations – including insertions or deletions (Kepler et al., 2014) and amino acid-point substitutions of <0.5% occurrence (see Methods) – that restricted or controlled breadth could be identified by comparing broadly neutralizing and non-broadly neutralizing developmental branches of an HIV-1-directed antibody lineage. Although many developmental trees do not display such differentially neutralizing branches, at least two lineages, VRC34 and PCDN76, did appear to have rare mutations that co-segregated with broadly neutralizing antibodies, but not with poorly neutralizing antibodies (Figure S1).
The most effective member of the VRC34 lineage, VRC34.01, has a 13-residue CDR H3, 15% SHM, and neutralization breadth of 50.5% on our panel of 208-diverse HIV-1 strains (Kong et al., 2016). The structure of VRC34.01 with HIV-1 envelope glycoprotein (Env) trimer reveals recognition of primarily a linear peptide, corresponding to the N-terminal 8 residues of fusion peptide (FP8) at the N-terminus of the gp41 transmembrane subunit, and epitope-based focusing with these N-terminal FP residues appears to be a promising vaccine approach (Cheng et al., 2019; Kong et al., 2019; Xu et al., 2018). Initial isolation has defined the seven lineage members in two separate developmental branches: four of the members, VRC34.01-04, segregate into one branch, and each neutralizes over 30% of a panel of 22 HIV strains; the other three members, VRC34.05-07, segregate into another branch, and each neutralizes at most 1 of the 22 tested strains (Kong et al., 2016).
Here we used next-generation sequencing to delineate thousands of VRC34 lineage sequences for heavy and light chains, and we also used paired sequencing to delineate hundreds of heavy/light pairs. These NGS sequences defined a detailed developmental tree from which we calculated developmental intermediates. We characterized the binding and neutralization of VRC34 lineage members and intermediates with diverse strains as well as with autologous viruses. Structural and functional characterization of developmental intermediates from each branch revealed a rare mutation to be required in the broadly neutralizing branch, but not in the poorly neutralizing branch. Our results illustrate how a rare mutation can both restrict the later development and shape the overall maturation of a broadly neutralizing lineage.
Results
Developmental Pathway for VRC34 Bifurcates Early in Maturation
To understand development of the broadly neutralizing VRC34-lineage, we sought to obtain NGS data from June 2009, the time point from which we previously identified VRC34 lineage members by B cell sorting (Kong et al., 2016). First, we sequenced IgM transcripts from peripheral blood memory B cells and used IgDiscover (Corcoran et al., 2016) to confirm the presence of the previously identified IGHV1-2*02 germline allele used by the VRC34 lineage in donor N123 (Figure S2A). We noticed that VRC34-lineage antibodies have numerous mutations in the FR1 region (Kong et al., 2016), and we thus designed lineage-specific primers to enhance the recovery of VRC34-lineage sequences (Figure S2B). By using a combination of standard and lineage-specific primers, VRC34 lineage-specific sequences were obtained by applying lineage-specific filters to Illumina NGS reads (Figure S2B–S2D). We applied informatics sieving to remove non-lineage sequences and obtained 10336 heavy chain and 5608 light chain reads for the VRC34 lineage, which had 1-22% SHM (nucleotide) in the VH gene and 0-16% in the VL gene (Figure S3A and S3C). Consistent with the initiation of the lineage at a time point several years prior to sampling, very few of the reads assigned to the VRC34 lineage had low SHM: in the heavy chain, only two reads had SHM below 5% (Figure S3A, 2nd panel from right); in the light chain, low-divergence reads clustered in identity-divergence plots in the region of low identity populated by non-lineage antibodies (Figure S3C, 2nd panel from right). After clustering to unique sequences and removal of low-identity reads, we constructed phylogenetic trees for heavy and light chains and inferred developmental intermediates (Figure S3B and S3D). Both trees had a similar shape that bifurcated early: we defined “I(n)-h” or “I(n)-l” for heavy and light intermediates of the VRC34.01-04 branch and “I(n)’-h” and “I(n)’-l” for heavy and light intermediates of the VRC34.05-07 branch.
Having derived separate heavy and light chain trees, we next sought to merge this information into paired heavy-light developmental pathways. We obtained paired heavy-light sequences from PACBIO sequencing and applied filters to obtain 431 full-length heavy-light sequences, which retained heavy-light pairing information (Figure 1A and 1B). We constructed heavy and light chain maximum-likelihood (ML) phylogenetic trees combining inferred-intermediate sequences defined by the Illumina-sequencing and paired heavy and light-chain sequences from PACBIO sequencing (Figures 1C and S3E). The topological information derived from the ML trees enabled inference of the pairing of heavy and light chain intermediates (Figure 1D). Overall, heavy-light chain pairing recapitulated the overall structure of the Illumina-derived trees, with the phylogenetic distances of these heavy and light chain developmental intermediates correlated highly with each other (R2 = 0.9382, P=0.0002).
Figure 1. Phylogenetic Analysis of Paired Antibody Sequences Recapitulates Overall Structure of Illumina-Derived Trees and Permits Pairing of Heavy and Light Chain Developmental Intermediates.
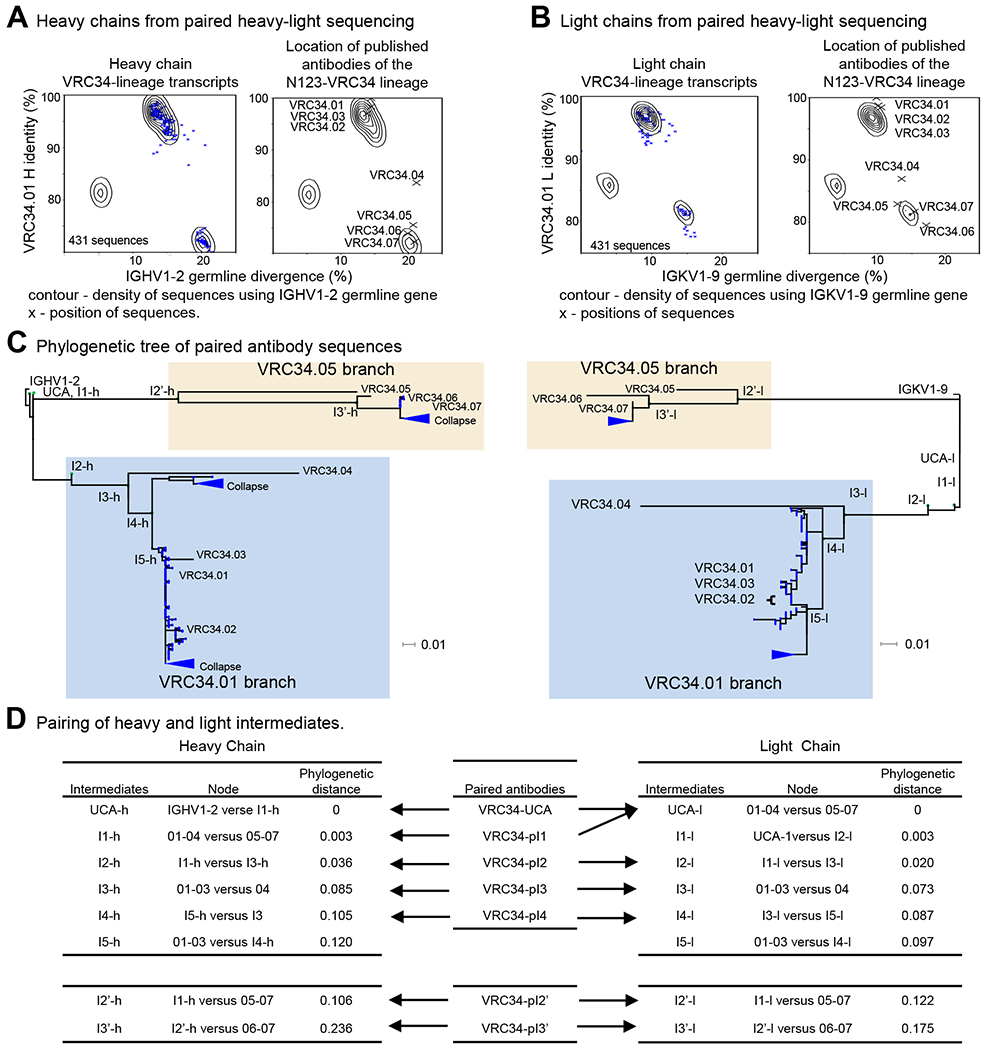
(A) Identity/divergence plots showing selection of N123-VRC34 clonal variants by computational sieving of PACBIO samples. The contour lines were constructed from the paired antibodies using IGHV1-2 germline gene. (Left panel) The blue “x” indicates the results of sieving on V and J genes, CDR H3 length of 13 residues (IMGT numbering), and CDR H3 signature. (Right panel) of VRC34 lineage members are projected on the identity/divergence plot, indicated with “X”. (B) Selection of N123-VRC34 clonal variants by computational sieving of PACBIO samples. The contour lines were constructed from the paired antibodies using IGKV1-9 germline gene. (Left panel) The blue “x” shows the results of sieving on V and J genes, CDR L3 length 9 residues (IMGT numbering), and CDR L3 signature. (Right panel) Projection of N123-VRC34 lineage (“X”) on Identity/divergence plot. (C) Maximum likelihood trees constructed from the paired heavy and light chain sequences. Green dots indicate the location of inferred intermediates obtained from separate heavy and light chain ML trees. The VRC34.01 and VRC34.05 branches are highlighted in light blue and light yellow boxes, respectively. (D) Pairing of heavy and light intermediates.
See also Figures S1, S2 and S3, and Tables S1 and S2.
Sequencing of Autologous Virus from Donor N123 Reveals Non-Canonical FP8 Sequences
Having defined the developmental pathway of the VRC34 lineage in donor N123, we next sought to determine autologous Env sequences to gain insight into their contribution to VRC34 lineage development. We amplified and sequenced approximately 1,000 autologous HIV env genes each from plasma samples taken on December 2008, June 2009 and July 2012 (Figure 2A); these sequences clustered within clade B (Figure 2B). From the earliest time point, we found that 40% Envs had the FP8 sequence AVGIGAAF (residues 512-519). We named this autologous sequence FP8_a1. This sequence differed in only one residue (V518A) from the most prevalent variant of the FP8 sequence in the LANL database, AVGIGAVF (FP8_v1). The second most frequent autologous FP8 sequence (comprising 32% of Env sequences at the earliest time point) was ALGIGAAF (FP8_a2), which differed from FP8_a1 in one residue at position 513 (V513L). The two other highly frequent autologous sequences, AVGFGAAF (FP8_a3) and AAGFGAMF (FP8_a4), accounted for 19% and 8%, respectively, of the sequences from the December 2008 time point. FP8_a4 was the dominant autologous FP8 sequence in June 2009, the time point from which VRC34.01 was identified (Kong et al., 2016), but was not observed in July 2012. At this last time point, FP8_a1 and FP8_a2 became dominant again with frequencies of 54% and 43%, respectively. Overall, we identified four frequent FP8 sequences in donor N123, with FP8_a1 and FP8_a2 dominating in most of the time points analyzed. These dominant FP8 sequences in donor N123 were different from those prevalent in circulating HIV-1 strains in the LANL database.
Figure 2. Sequencing of Autologous Virus Reveals Non-Canonical FP8 Sequences in Donor N123.
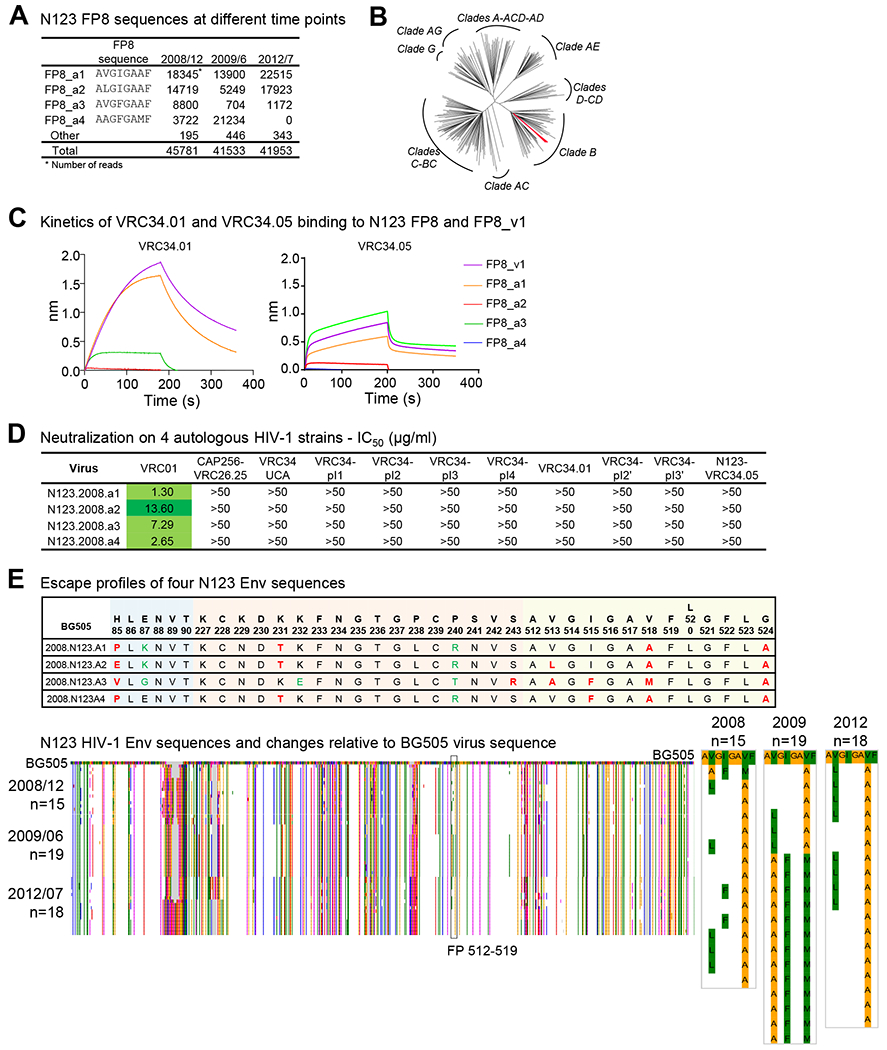
(A) Donor N123 FP8 sequences identified in three time points. (B) Neighbor joining phylogenetic tree of HIV-1 Env sequences from donor N123 and the 208-strain panel. The four Env sequences from donor N123 are colored red. (C) Binding analysis of VRC34.01 (left) and VRC34.05 (right) to FP8_v1 and autologous FP8 sequences. Binding curves were measured with BLI (Octet). (D) Neutralization of VRC34 intermediates on autologous HIV-1 strains. (E) (Upper panel) Escape profiles of N123 Env sequences. Escape mutations (Dingens et al., 2018) were colored in red, and non-escape mutations were shown in green. (Lower left panel) Env sequences from donor N123 aligned with that of BG505. Amino acid variations were colored by Lesk scheme (Lesk, 2002) (Lower right panel) Enlarged sequences from the FP (512-519) region with Lesk coloring.
Having identified autologous FP8 sequences, we characterized their interactions with VRC34 lineage members. We expressed and purified the putative unmutated common ancestor (UCA) and six VRC34 heavy/light-paired developmental intermediates (VRC34-pI1, VRC34-pI2, VRC34-pI3, VRC34-pI4 in the VRC34.01 branch, and VRC34-pI2’ and VRC34-pI3’ in the VRC34.05 branch), along with mature antibodies VRC34.01 and VRC34.05 to evaluate their function. We first assessed the binding of these VRC34 lineage variants with autologous or prevalent FP8 conjugated to KLH (Figure S4A and S4B). The early variants, UCA, VRC34-pI1, VRC34-pI2, and VRC34-pI2’, did not bind the autologous FP or prevalent FP sequences, whereas the late variants, VRC34-pI3, VRC34-pI4, VRC34-pI3’ and mature antibodies VRC34.01 and VRC34.05, bound both autologous FP and prevalent FP sequences.
We also characterized binding kinetics of VRC34.01 and VRC34.05 to peptides FP8_a1 through a4 and FP8_v1 (Figures 2C, S4C and S4D). Notably, VRC34.01 had affinity to FP8_v1 which was higher than its affinity to any of the autologous FP8 peptides. VRC34.05 had higher affinity to FP8_a3 than that to other autologous FP8 peptides, and VRC34.05 binding to FP8_a3 had similar kinetics to its binding to FP8_v1. Thus, FP8_v1 – the most prevalent HIV-1 FP8 sequence – closely approximated the binding behavior of VRC34.01 and VRC34.05 to autologous FP8 peptides.
VRC34 UCA and Early Intermediates Do Not Neutralize HIV
To gain insight into the interaction between VRC34-lineage antibodies and N123 virus, we produced four N123 autologous viruses from December 2008 and tested neutralization by VRC34 antibodies and developmental intermediates. Although autologous FP8 were recognized by late VRC34 lineage members, none of the autologous viruses were neutralized by VRC34-lineage members (Figure 2D and Table S3). To understand this lack of neutralization, we analyzed escape profiles of VRC34.01 from BG505 virus, which we had previously characterized (Dingens et al., 2018), and analyzed contact residues of the four dominant autologous Env sequences (Figure 2E). Mutations at contact residues H85, V518 and G524, along with other escape mutations, defined sites at which the four autologous viruses were incompatible with neutralization by VRC34-lineage antibodies; we interpret this incompatibility to suggest that – at the time points analyzed – the autologous virus had already escaped neutralization by the VRC34 lineage.
As autologous virus was not neutralized by VRC34-lineage members, we assessed their neutralization against 10 heterologous strains (Figure 3A and Table S3). No neutralization was observed for UCA, VRC34-pI1, VRC34-pI2, VRC34-pI2’, VRC34-pI3’ and VRC34.05, whereas VRC34-pI3, VRC34-pI4, and VRC34.01 neutralized 8 or 9 strains. Results from the 10-strain panel indicated that VRC34-pI3 might have better potency and breadth than the broadly neutralizing antibody VRC34.01. We therefore analyzed UCA, VRC34-pI3, VRC34-pI4, VRC34.01, and VRC34.05 on a 208-strain panel (Figure 3B, Table S4). VRC34-pI3 was the broadest and neutralized 119 of the 208-strain panel (57%) with IC50 <50 μg/ml; VRC34-UCA, however, did not neutralize any of the 208 strains, consistent with the inability of VRC34-UCA to bind Env trimer or FP as free peptide. Although mature VRC34.05 bound FP and Env, VRC34.05 only neutralized one strain, BG1168. Thus, substantial SHM was required for the VRC34 lineage to develop neutralization; however, VRC34.05, even with higher SHM of 20.7% (heavy chain nucleotide), did not show broad neutralization. Overall, neutralization was only observed with mature antibodies or late intermediates from the VRC34.01 branch; VRC34 UCA and early intermediates did not neutralize any tested HIV-1 strain.
Figure 3. VRC34 UCA and Early Intermediates Do Not Neutralize HIV.
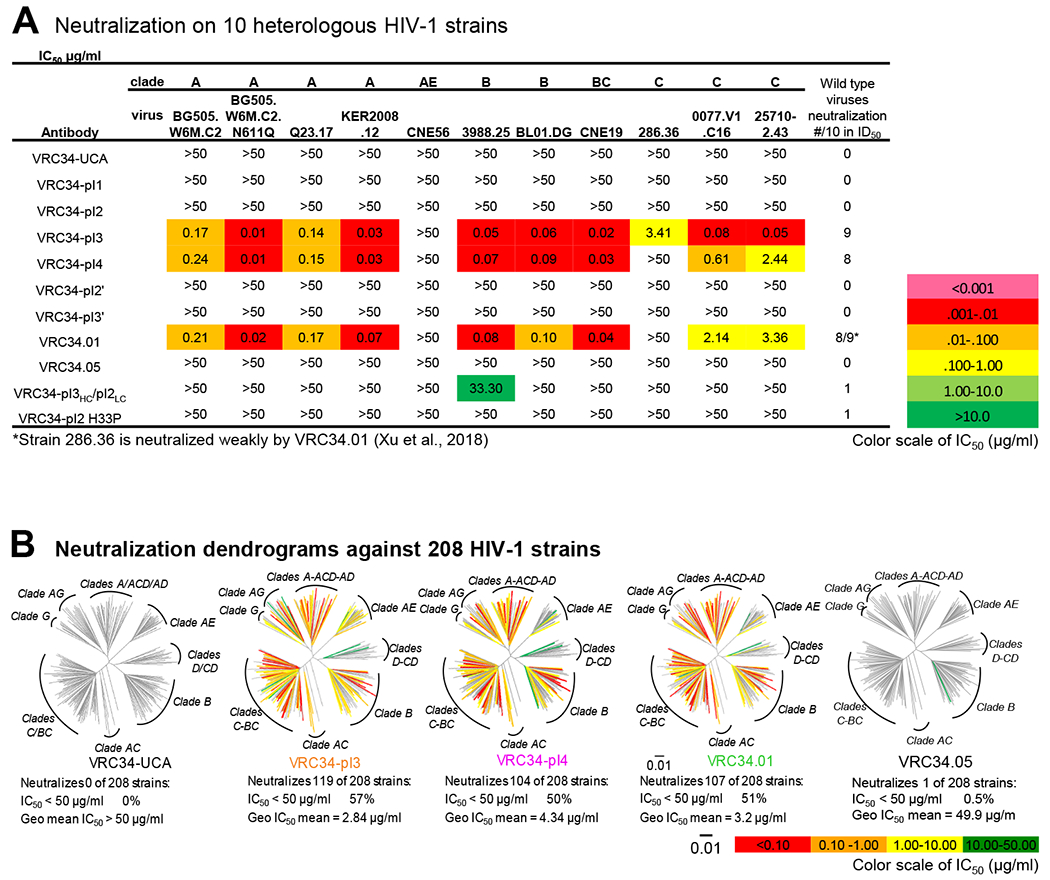
(A) Neutralization of VRC34 intermediates on 10 heterologous HIV-1 strains. (B) Neutralization dendrograms show the diversity of 208 viral strains, with branches colored according to neutralization potency and non-neutralized branches shown in gray.
Analysis of SHM in VRC34 Lineage Development Reveals a Critical Rare Heavy Chain Mutation
To understand the functional importance of rare mutations in VRC34 lineage maturation, we analyzed SHMs of VRC34 lineage variants, their frequencies of occurrence, and their potential contacts with Env (Figure 4A–B). VRC34.01 had 23 and 12 amino acid mutations on heavy and light chain V regions, respectively. VRC34.05 had more SHM than the more potent antibody VRC34.01, with 32 and 20 mutations on the heavy and light chain, respectively. Among these SHMs, six mutations, K23RHC, M48LHC, G56DHC, L82aVHC, Y32ELC, and A50YLC, were shared between VRC34.01 and VRC34.05, with two of them, Y32ELC and A50YLC, being rare mutations as determined using gene-specific substitution profile (Figure 4C and S5A–C) (Sheng et al., 2017). Since these two rare mutations were present in both the broadly neutralizing VRC34.01 branch and the poorly neutralizing VRC34.05 branch, they were insufficient in of themselves for broad neutralization. By contrast, the mutation Y33P was present solely in the broadly neutralizing VRC34.01 branch. This mutation requires two nucleotide changes, Tyr (TAT) to His (CAT) and then to Pro (CCT), with a combined frequency of less than 0.5% occurrence, which meets our definition of a rare mutation. We note that the subsequent mutation P33A (shown as Y33A in the CDRH1 of Figure S5A and P33A in Figure S5C) would also be of low frequency. Thus, analysis of SHM for the VRC34 lineage indicated broad neutralization to segregate with rare mutations at heavy chain residue Y33, a residue which contacts FP in the context of the crystal structure of VRC34.01 in complex with Env trimer (Kong et al., 2016).
Figure 4. VRC34-Lineage Sequences Contain Multiple Rare Mutations.
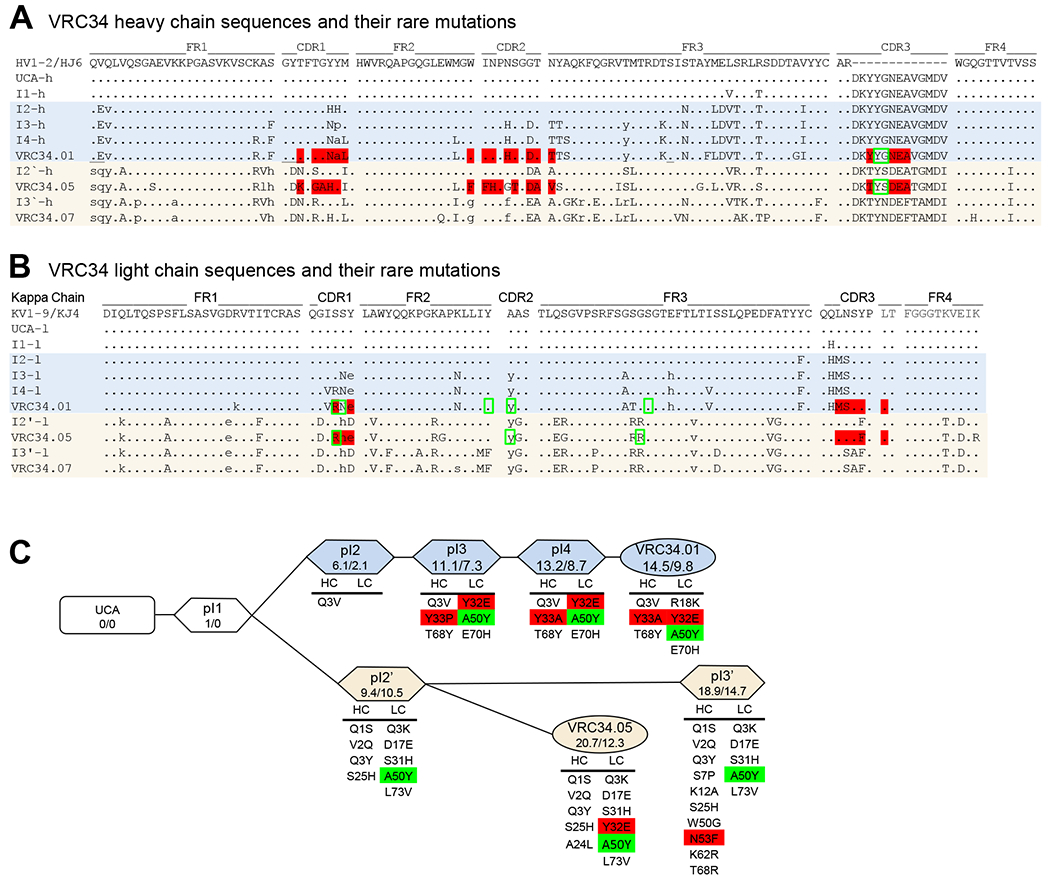
(A and B) Protein sequences of phylogenetically inferred developmental heavy chain (A) and light chain (B) intermediates compared to mature VRC34-lineage members and constituent germline genes. Amino acids that contact FP are displayed in red, and those that contact glycan are labeled in green rectangle. Rare mutations are shown in lowercase. (C) Schematic diagram of VRC34 developmental pathway. Degree of SHM for each VRC34 intermediate is given as % nucleotide mutated (vH/vL) below the name of each antibody variant. Rare mutations are listed under each paired intermediate. Rare mutations that contact FP are highlighted in red, and those that contact glycan are highlighted in green.
See also Figure S5.
Crystal Structures Provide a Molecular Basis for Contribution of Rare Y33P Mutation to FP Recognition
Having identified VRC34-lineage sequences (Figure 1), autologous virus (Figure 2), neutralization by VRC34-lineage antibodies (Figure 3), and the importance of rare mutations at position Y33HC (Figure 4), our next step was to define the details of heavy chain residue 33 interactions in the context of the germline-encoded Y33HC and its somatic variants H33HC, P33HC, and A33HC. We previously found VRC34-lineage antibodies interactions with the most prevalent FP variant (FP8_v1) to provide the closest binding behavior to autologous FP8 peptides (Figure 2C), and thus chose FP8_v1 for binding and structural studies.
First, we mapped the interaction between FP and VRC34-lineage variants by performing Ala-Gly scanning, in which each individual FP residue was mutated to Ala or Gly and binding affinity to the antibody was measured, to determine the individual contribution of each residue on FP to the FP-antibody interaction energy. Because UCA (with Y33HC), pI1 (Y33HC), and pI2 (H33HC) exhibited only weak binding to FP8_v1, no additional binding experiments were pursued; with pI3 (P33HC) and pI4 (A33HC), tight binding was observed, with three FP residues, V513FP, I515FP and G516FP, showing significantly reduced binding upon Ala or Gly substitution. This binding was virtually indistinguishable from that of VRC34.01, indicating that these three amino acids contributed substantially to binding in the VRC34.01 developmental branch (Figure 5A and 5B).
Figure 5. Crystal Structures Reveal the Molecular Basis for Rare Y33P Mutation Requirement.

(A) Development of somatic hypermutations along the maturation pathway of VRC34.01. SHMs of the VRC34 intermediates are mapped on the structure of VRC34.01 shown in ribbon representation. The location of each SHM is indicated with a sphere and colored according to the paired intermediates. (B) Effect of single Ala or Gly mutation on the binding affinity of mutant peptides to VRC34 UCA and intermediates was measured by BLI. Fitted binding curves of the Ala/Gly mutants and parent peptide to VRC34-UCA, VRC34-pI1, VRC34-pI2 demonstrating absence of binding are shown on the left; binding response of individual mutant peptides (alanine (grey bars) and glycine (white bars)) normalized to parent FP for VRC34-pI3, VRC34-pI4 and VRC34.01 are shown on the right. (C) Binding interaction of FP residues with intermediates VRC34-pI2 (left panel), VRC34-pI3 (middle), and VRC34-pI4 (right). VRC34-pI2 was a homology model based on VRC34-pI3. FP is colored red, pI2 mutations are colored blue, pI3 mutations are colored orange, and pI4 mutations are colored purple. (D) Binding kinetics by BLI of chimeric antibodies, VRC34 pI2HC/pI3LC and VRC34 pI3HC/pI2LC, and of single point mutants, pI2 H33P, pI3 P33H and pI3 P33A, with His-tagged FP8_v1 (upper row) and diverse FP-KLH conjugates (lower row). We note that in the lower row, interactions between bivalent antibodies and the multivalent FP-KLH conjugates reduce off rate, leading to very high apparent affinities, which in these BLI measurements “max out” at ~KD=10−12.
Next, we determined co-crystal structures of antigen-binding fragments (Fabs) of VRC34 intermediates pI3 (P33HC) and pI4 (A33HC) in complex with FP8_v1 to provide atomic-level details of their interaction. Crystals of the pI3 and pI4 complexes diffracted to 1.38 and 1.29 Å, respectively (Figures 5C middle and right panel, S6, and Table S5). Both structures showed well-defined electron density for FP residues 512-518, which revealed FP to be captured in a hydrophobic groove, similar to the previously determined VRC 34.01-FP8_v1 complex structure (Kong et al., 2016). Due to their low binding affinity, we were unable to obtain co-crystals for early intermediates with FP8_v1; to obtain insight into their potential interactions, we modeled the structure of the pI2 complex (with H33HC) based on the structure of the pI3 (P33HC) complex with FP8_v1. The modeled structure indicated a significant collision between the pI2 H33HC side chain and the side chain of I515FP (Figure 5C, left panel), thus preventing VRC34-pI2 from binding FP in a conformation similar to that observed in the high affinity complexes between FP and VRC34 lineage variants.
Overall, the sequence and structural analysis revealed that steric accommodation provides a molecular basis for the required rare Y33PHC mutation. In addition to Y33PHC, two rare light chain mutations, Y32ELC and A50YLC were present among many of the mature antibodies in both developmental branches (Figure 4C). To gain insight into the functional impact of these rare light chain mutations, we swapped the heavy and light chains of variants pI2 and pI3 (Figure 5D, left two columns). No binding to diverse FP sequences was observed for the pI2HC/pI3LC swapped variant. In contrast, the pI3HC/pI2LC variant showed binding to three of the four prevalent FP8 sequences (all except FP8_v4), suggesting pI3HC SHMs to be critical for binding FP, and the two light chain rare mutations, Y32ELC and A50YLC, to be not critical.
As SHMs in addition to the rare H33PHC mutation accumulate in the heavy chain between pI2 and pI3, we investigated the degree to which this specific rare mutation contributed to the binding. We prepared a mutant of pI2 with His33 mutated to Pro (pI2 H33PHC) and two pI3 mutants, pI3 P33HHC and pI3 P33AHC (Figure 5D middle and right two columns). pI3 P33AHC bound FP8_v1 with 60 nM affinity (Figure 5D, 2nd from right, upper panel) and also recognized diverse FP in the multivalent context (Figure 5D, 2nd from right, lower panel). pI2 H33PHC had weak binding to FP8_v1 with 4700 nM affinity and recognized diverse FP. In contrast, pI3 P33HHC showed no binding to FP8_v1 or diverse FP-KLH conjugates. Overall, these data indicate the heavy chain mutations: Y33 to H33 to P33 to A33 to be critical for FP binding by antibodies of the VRC34.01 branch.
Poorly Neutralizing VRC34.05 Developmental Branch Does Not Utilize a Y33 Mutation and Evolves Distinct FP Recognition
In contrast to the VRC34.01 branch, the heavy chain germline residue Y33 was retained in the VRC34.05 branch, suggesting an alternative developmental pathway for FP binding. To elucidate this alternative FP-binding, we determined the co-crystal structure of VRC34.05 complexed with FP8_v1 (Table S5). The structure revealed the hydroxy group of Y33HC to form hydrogen bonds with the amide nitrogens of I515FP and G516FP and the carbonyl oxygen of G516FP (Figure 6A). In the structure of VRC34.01 complexed with FP8_v1, the hydroxy group of Y97HC – a different tyrosine from that used by VRC34.05 – formed hydrogen bonds to the same amide nitrogen of I515FP and G516FP, and the carbonyl oxygen of G516FP (Kong et al., 2016); VRC34.01 uses the small side chain of A33HC to make hydrophobic contacts with the side chain of I515FP. Despite these differences, the conformation of the FP8_v1 peptide was similar in the two complexes, and the overall binding mode to FP8 was also similar (RMSD 0.69 Å for FP8 Cα atoms), except for differences at residues 33HC and 97HC (Figure 6B).
Figure 6. The VRC34.05 Developmental Branch Retains Germline 33Tyr and Evolves Distinct FP Recognition.
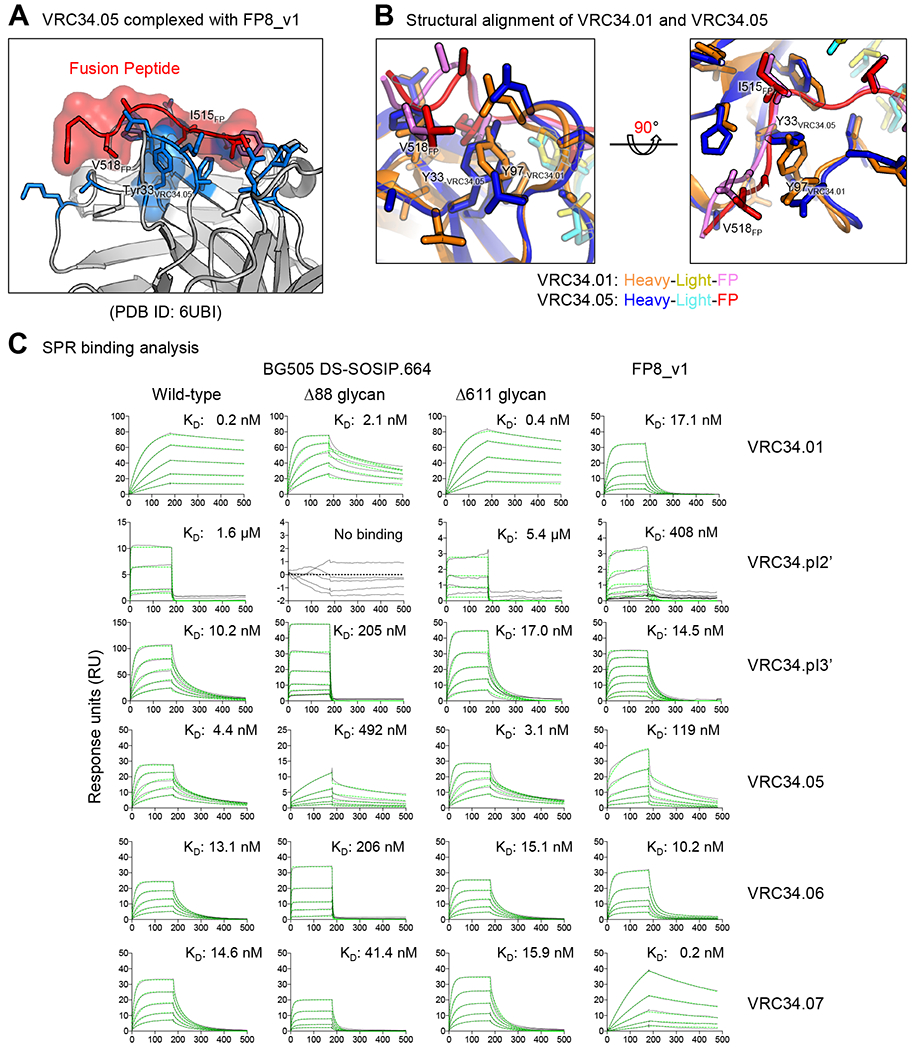
(A and B) Co-crystal structure of VRC34.05-FP8_v1 revealed a different role of residue 33 in FP recognition. VRC34.05 retained germline 33Tyr and used smaller side chain S97 to avoid clash. (C) Binding analysis of VRC34.05-branch variants to wild type and glycan mutants of BG505 DS-SOSIP and FP. The variants had a wide range of affinity. VRC34.05 had large reduction in affinity to ∆88 Env trimer and FP, compared with VRC34.01. Blank-corrected SPR sensorgrams were shown in black, and fitted data using a 1:1 Langmuir model of binding were shown in green dashed lines. Note that y-axis ranges from 5 to 150 RU were used to highlight concentration dependent responses.
To provide insight into the general binding mode of VRC34.05 branch antibodies and differences from the VRC34.01 branch antibodies, we measured binding kinetics for mature VRC34.05-branch members to FP8_v1 and various HIV Env trimers: a prefusion-closed trimer stabilized by disulfide bonds and I to P mutation (DS-SOSIP) (Kwon et al., 2015) as well as two variants, ∆88 and ∆611, in which FP-proximal glycans were removed (Kong et al., 2016; Xu et al., 2018) (Figure 6C and S4; Table S6). VRC34.05 showed 7-fold weaker binding to FP8_v1 than VRC34.01 (119 vs 17 nM affinity, respectively); VRC34.05 bound BG505 trimer with 4 nM affinity, which decreased 100-fold in the absence of glycan N88. VRC34.07 showed very tight binding affinity to FP8_v1 (0.2 nM), but displayed only moderate affinity to Env trimer (15 nM).
Overall, antibodies from both VRC34.01 and VRC34.05 branches had similar on-rate (ka) for binding to BG505 DS-SOSIP, whereas the off-rate (kd) for dissociation from BG505 DS-SOSIP was ~100-fold faster for the VRC34.05 branch antibodies; on the other hand, free FP binding kinetics showed no conserved differences between the two branches – and high variability within the VRC34.05 branch. These observations suggest that the key rare mutation, Y33PHC, in the VRC34.01 branch provides favorable interactions with FP in the trimer context that reduce off-rate for trimer. By contrast, antibodies in the VRC34.05 branch utilize Y97HC instead of Y33PHC to accommodate FP. The consequence of the structural swapping of the roles of tyrosines at 97HC and 33HC as revealed by the SPR measurements (Table S6) is a 100-fold decrease in off-rate in the trimer context, greatly increasing the ability of the VRC34.01 branch to neutralize HIV-1.
Discussion
Definition of the factors controlling the maturation of antibodies to achieve effective virus neutralization is central to understanding how the humoral immune system recognizes and clears viral pathogens. Broadly neutralizing antibodies against HIV arise infrequently, and it has been hypothesized that this is due in part to their requirement for rare mutations to achieve broad neutralization. Relevant to this, Saunders et al. recently showed that immunogens with higher affinity for antibody variants incorporating rare mutations could be used for the targeted selection of these rare mutations (Saunders et al., 2019). However, most broadly neutralizing antibodies against HIV-1 display multiple rare mutations, and the lack of a 1:1 correspondence between rare mutation and function has hindered elucidation of the significance of each rare mutation (Sheng et al., 2016; Wiehe et al., 2018). With the VRC34 lineage, we recognized from sequences of the initial seven antibodies (VRC34.01-.07) (Kong et al., 2016) that the lineage bifurcated into two developmental branches – one poorly neutralizing (VRC34.05-.07) and the other achieving >50% neutralization breadth (VRC34.01-.04). Deep sequencing of N123-B cell transcripts, structural analysis of VRC34 lineage intermediates, and functional mutagenesis studies revealed that a rare mutation, Y33PHC, was required for FP recognition in the broadly neutralizing VRC34.01 developmental branch; the VRC34.05 branch without this critical rare mutation neutralized poorly. Thus, the bifurcated structure of VRC34 lineage development, with co-segregation of rare mutation and function, provided a means to delineate functionally critical rare mutations – and to address the rare mutation requirement in the development of a broadly neutralizing antibody lineage.
Maturation pathways have been defined for a number of broadly neutralizing antibodies. To a first approximation, SHMs occur stochastically, with rare mutations occurring at a frequency defined here as <0.5% occurrence; moreover, to a first approximation, most mutations are functionally neutral. Thus, with ~300 nucleotides (~100 amino acid residues of each variable region), it is expected that some rare mutations will be incorporated as “passengers”, even if they are not positively selected. The difficulty has been in identifying functionally critical versus “passenger” mutations.
In some cases, different rare mutations are present in each of the neutralizing branches, such as has been observed with the VRC01 lineage in donor 45 (Wu et al., 2015), thereby obscuring functional conclusions about rare mutations. In other cases, rare mutations have been studied, but only by reversion (Simonich et al., 2016; Wiehe et al., 2018), making it unclear whether a separate branch could have evolved without the rare mutations (Figure S1 D and E). And in still other cases, function and rare mutations have co-segregated, such as with the PCDN76 lineage, but the localization of these mutations outside of the paratope region have made their functional relevance unclear (Figure S1).
With the VRC34 lineage, rare CDRH1 Y33 mutations occurred in the paratope and interacted directly with FP, and functional data showed these mutations to enhance binding. However, the appearance of Y33 mutations first occurred between I1 and I2 (Y33H) and then between I2 and I3 (H33P), prior to the development of measurable binding or neutralization, which first appears with intermediate I3. Nevertheless, we were able to gain insights into the functional roles of these SHMs using heavy/light chain chimeras and functional mutagenesis. Moreover, we note that all antibodies in the VRC34 lineage that developed broad neutralization derived from the rare mutation Y33P, providing additional evidence for its critical role.
The major implication of this study, however, is not related to utilizing the VRC34 lineage as a vaccine template (which has generally been the focus of studies on rare mutations and lineage development), but is related to the general role of rare mutations in the development of broadly neutralizing antibodies. In general, we show how the requirement for a specific rare mutation (Y33P) controls and shapes the development of neutralization breadth in the VRC34 lineage. Notably, the mechanistic impact of Y33P appears to be a reduction in the off-rate between antibody and Env trimer, yielding the tight Env-trimer affinity required for neutralization. Overall, this paper adds a critical element to our understanding of lineage development – precise delineation of the impact of a required rare mutation on the development of a broadly neutralizing lineage – and thus solidifies our understanding of the role of rare mutations in the development of broadly neutralizing antibodies.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Peter D. Kwong (pdkwong@nih.gov). This study did not generate new unique reagents.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Patient samples
The peripheral blood mononuclear cells (PBMCs) were from a chronically HIV-1 infected donor N123 who was enrolled in the National Institute of Allergy and Infectious Disease (NIAID) (Kong et al., 2016). Human peripheral blood samples were collected after obtaining informed consent and appropriate Institutional Review Board (IRB) approval.
Cell lines
Expi293F cells were from ThermoFisher Scientific Inc (Invitrogen, cat# A14528; RRID: CVCL_D615). TZM-bl cells were from NIH AIDS Reagent Program (www.aidsreagent.org, cat# 8129, RRID:CVCL_B478). The cell lines were used directly from the commercial sources and cultured following manufacturer suggestions as described in Method Details below.
METHOD DETAILS
Individualized IGH germline database
An individualized IGH database was prepared as described previously (Vazquez Bernat et al., 2019). In brief, total RNA was obtained from 10 million Ficol separated PBMC mononuclear cells. 400 ng of total RNA was used in a cDNA synthesis reaction using a primer specific to the IgM constant region and containing a unique molecular identifier. The cDNA was used as a template to produce a PCR amplified IgM library that was subsequently gel purified, indexed, and sequenced using the MiSeq 2 × 300 V3 system. The R1 and R2 reads were merged and analyzed using the IgDiscover program (version 0.10+178.gbf0956e) to identify the germline IGHV, D and J genotype of the subject.
Lineage specific primers
We designed lineage-specific primers to target all the previously reported VRC34 bNAb family variants, as well as any mutations between those known bNAbs and the germline origin FR1 sequences (Table S1). This primer set would amplify any genes encoded by the same heavy and light chain gene families (IGHV1-2, IGKV1-9, and closely related variants); in addition, we also used a 50:50 mix of lineage-specific and all diverse human FR1 primers as well as a set of leader peptide-specific primers for amplification of all genes based on a different priming region (DeKosky et al., 2015; Doria-Rose et al., 2014).
Sequencing of natively paired VH:VL amplicons
Donor N123 was enrolled at the National Institute of Allergy and Infectious Diseases under a clinical protocol. The donor signed written informed consent that was approved by the NIAID Institutional Review Board. This donor was diagnosed with HIV-1 in 2000, was not on antiretroviral treatment, and at the time of PBMC collection had a CD4+ T cell count of 463 cells/μl and a plasma HIV-1 viral load of 4,920 RNA copies/ml. 1.42×106 peripheral B cells were isolated from 25 million PBMCs using a human B cell selection kit (Stemcell Technologies).
We utilized a flow-focusing nozzle to rapidly compartmentalize B cells in single cell emulsion droplets, followed by single B cell lysis inside droplets and single-cell mRNA capture with oligo(dT)-coated magnetic beads (DeKosky et al., 2015; Doria-Rose et al., 2014; McDaniel et al., 2016). Overlap extension RT-PCR was then performed to link heavy and light chains using a Superscript III RT-PCR kit (ThermoFisher Scientific) and either FR1 or leader peptide-targeting primer sets for paired heavy:light amplicon generation (DeKosky et al., 2015; McDaniel et al., 2016). All variable region primers were at a final concentration of 40 nM each, including any lineage-specific primers in the reaction mix (Table S1) (DeKosky et al., 2015; McDaniel et al., 2016). Nested PCR to amplify paired heavy and light chain genes and separate heavy and light chains were performed as described previously (DeKosky et al., 2015; McDaniel et al., 2016). Sequencing of the natively paired antibody repertoire from B cells was performed as previously described (DeKosky et al., 2016). Samples EN60-EN71 comprised full-length, separately amplified VH and VL genes, as described previously (DeKosky et al., 2013; McDaniel et al., 2016). Two samples were also submitted for Pacific Biosciences long-range sequencing (Table S2A), and circular consensus sequences were generated based on standard Pacific Biosciences data analysis protocols. Bioinformatic analysis was performed as previously described (DeKosky et al., 2016). Pacific Biosciences sample data were filtered for a minimum of 10 substring reads and read quality >0.99, and heavy and light chains were identified prior to further analysis.
Autologous plasma HIV env deep sequencing
Virions were concentrated from VRC34 study donor plasma by centrifugation at 21,100 x g for 1.5 h at 4°C. RNA was extracted from virion pellets using QIAmp Viral RNA Mini kits (Qiagen) and immediately reverse transcribed. Reverse transcription reactions included 100 nM primer env3out (TTGCTACTTGTGATTGCTCCATGT) (Salazar-Gonzalez et al., 2008), 100 units SuperScript III Reverse Transcriptase, 500 μM dNTP, 5 mM DTT, 20 units RNAseOUT, and 1× 1st Strand Buffer (all from ThermoFisher Scientific). Reverse transcription reaction conditions were 50 °C for 50 min, 85 °C for 10 min, and 4°C hold. Copy numbers of resulting cDNAs were determined by limiting-dilution PCR using fluorescence-assisted clonal amplification (FCA) (Boritz et al., 2016). An estimated 1000 cDNA templates were then subjected to near-full-length env PCR using forward primer GAGCAGAAGACAGTGGCAATGA and reverse primer CCACTTGCCACCCATBTTATAGCA as described (Laird Smith et al., 2016). PCR-amplified cDNA was prepared for Pacific Biosciences single-molecule real-time (SMRT) sequencing using SMRTbell Template Prep Kit 1.0 and sequenced on a Pacific Biosciences Sequel with a 20 h movie time.
HIV env deep sequence data analysis
Pacific Biosciences CCS reads were generated at 99.9% identity, using the Quiver framework to get consensus sequences. The generated fastq files were clustered by dereplicating reads and keeping a count of read abundance using Collapser command from FASTX-Toolkit (http://hannonlab.cshl.edu/fastx_toolkit). The clustered species were trimmed for primer sequences, and sequence reads under 1000 base pairs in length were discarded. These species were further translated into amino acid sequences, and the dereplication step was repeated to determine unique occurrence of each amino acid sequence. Species with fewer than five reads were discarded at this step to get high confidence subsampled sequences. MAFFT (Katoh et al., 2002) was used to align the sequences, and the alignments were improved manually, with assessment of indels and removal of out-of-frame sequences.
Antibody expression and purification
DNA sequences of antibody heavy and light chain variable regions were codon-optimized and synthesized, and subcloned into pVRC8400 vectors (Kong et al., 2016) by Gene Universal Inc. For antibody Fab production of VRC34 intermediates, a HRV3C protease cleavage site was inserted at the hinge region of IgG heavy chain gene. For protein expression, 0.3 ml of Turbo293 transfection reagent (Speed BioSystems) was mixed with 5.0 ml Opti-MEM medium (Life Technology) and incubated at room temperature for 5 min. Heavy and light chain plasmid DNAs (50 μg each) were mixed with 5.0 ml of Opti-MEM medium in a separate tube, and the mixture added to the Turbo293 Opti-MEM mixture. The transfection mixture was incubated for 15 min at room temperature and then added to 80 ml of Expi293 cells (Life Technology) at 2.5 million cells/ml. The transfected cells were incubated overnight in a shaker incubator at 9% CO2, 37 °C, and 120 rpm. On the second day, 8.0 ml of AbBooster medium (ABI scientific) was added and the incubation continued at 33 °C with the same CO2 and rotation rate for additional 5 days. On day 6 post transfection, supernatants were harvested, filtered, and loaded on a 1.0-ml Protein A column (GE Health Science). Antibodies were eluted with IgG elution buffer (Pierce) and immediately neutralized by addition of one tenth volume of 1M Tris-HCl pH 8.0. The antibody solutions were then dialyzed against PBS. Antibody concentration was adjusted to 1.0 mg/ml and sterile filtered (0.22 μm). To obtain Fab fragments, purified VRC34.01, VRC34.01-UCA, VRC34-pI1-4 and VRC34-pI2’-3’ IgGs were digested with HRV3C overnight at 4 ºC. Purified VRC34.05-07 IgGs were digested with Endoproteinase LysC overnight at room temperature. Fc fragments were removed by passing through a Protein A column. The Fab fragments were further purified through a Superdex 200 column in 5 mM HEPES, pH 7.5, and 150 mM NaCl.
Surface plasmon resonance (SPR)
Binding affinities and kinetics of VRC34 lineage antibodies to wild type and glycan-deleted HIV-1 Env were assessed by SPR on a Biacore S-200 (GE Healthcare) at 25°C in the HBS-EP+ buffer (10 mM HEPES, pH 7.4, 150 mM NaCl, 3 mM EDTA and 0.05% surfactant P-20) as described previously (Kong et al., 2019). Briefly, HIV-1 Env was captured through glycan-reactive 2G12 IgG coupled on the CM5 chip to ~400 RU. Serially diluted Fab of VRC34 lineage antibodies starting at 200 nM were passed through the sample and reference channels for 180 seconds followed by a 300-second dissociation phase at 30 µl/min. The chip surface was regenerated using 3M MgCl2. Blank sensorgrams were obtained with HBS-EP+ buffer in place of antibody Fab solution. Blank-corrected sensorgrams of the concentration series were fitted globally with Biacore S200 evaluation software using a 1:1 Langmuir model of binding.
Binding affinities and kinetics of VRC34 lineage antibodies to FP were assessed similarly in a modified HBS-EP+ buffer containing a reduced EDTA concentration of 50 µM. Ni-NTA chip was first activated by flowing 0.5 mM NiSO4 at 10 µl/min for 60 seconds. His-tagged FP (AVGIGAVFGSGSGSHHHHHHHH) solution in the modified HBS-EP+ buffer at 10 ng/ml was injected into the sample channel at 6 µl/min for 60 seconds. Serial dilutions of Fab of the VRC34 lineage antibodies starting at 50 nM were passed through the sample and reference channels for 180 seconds followed by a 300-second dissociation phase at 30 µl/min. The surface was regenerated by flowing 300 mM imidazole solution for 30 seconds at a flow rate of 50 µl/min. Blank sensorgrams were obtained by injection of the same volume of the modified HBS-EP+ buffer in place of antibody Fab solution. Blank-corrected sensorgrams of the concentration series were fitted globally with Biacore T200 evaluation software (BIAevaluation) using a 1:1 Langmuir model of binding.
Bio-Layer interferometry
A fortéBio Octet Red384 instrument was used to measure binding of VRC34-UCA, pI1, pI2, pI3, pI4, and mature VRC34.01 to FP. Assays were performed at 30°C in tilted black 384-well plates (Geiger Bio-One) in 1×PBS with 5% BSA with agitation set to 1,000 rpm. Anti-human IgG Fc capture biosensors were used to immobilize antibody IgG at 40 μg/ml for 150 sec. Fusion peptides with various lengths and sequences (Figure S4A and S4B) were conjugated to KLH through a bifunctional reagent m-maleimidobenzoyl-N-hydroxysuccinimide ester that links an appended C-terminal cysteine of the peptides to lysine residues of KLH (Xu et al., 2018). Binding of FP-KLH conjugates was measured by dipping immobilized IgG into solutions of FP-KLH at 200 nM. Parallel correction to subtract systematic baseline drift was carried out by subtracting the measurements recorded for a loaded sensor dipped into 1×PBS.
Bioinformatic analysis of antibodyomes
We processed the Illumina and PacBio data using our previously developed bioinformatics pipeline for antibodyome analysis (Joyce et al., 2016). For Illumina paired-end sequencing, we kept the reads with a minimum Q-score of 20 over 50% of the raw reads and merged the paired-end reads using USEARCH (v8.1.1861) (Edgar, 2010) with default parameters. Standalone IgBlast was used to assign V(D)J gene families and alleles to each sequence (Ye et al., 2013). We kept the Ig sequences that do not have stop codon and are productive. For the paired VH:VL obtained from PacBio platform, we discarded the reads with less than 10 substring reads and read quality less than 0.99 and kept the Ig sequences having both heavy and light chain and no stop codon and being productive.
A series of bioinformatic sieves were used to enrich the VRC34 lineage sequences containing the same lineage characteristics for downstream phylogenetic analysis. Both heavy and light chain NGS data sets were sieved to remove sequences that did not share the same germline gene assignments as in the mature VRC34.01, i.e., those that did not use the correct V and J germline assignments (IGHV1-2/IGHJ6 and IGKV1-9/IGKJ4). A third sieve was applied to keep the sequences having the correct length of the CDR3 loop (13 aa for CDR H3 and 9 aa for CDR L3). After these three sieves of germline gene assignments, we applied a sieve of the CDR H3 sequence signature, DK[TY]Y[SGN][NDE], where each pair of the square brackets represents one residue position occupied by one of the amino acids in the brackets, to remove those non-lineage related sequences. This filter retained sequences that contain a full or partial signature for further downstream processing. A similar sequence-signature sieve, Q[HQ][ML][SN][SA][YLF]PL, was applied to the light chain sequences. Because the number of sequences after the signature sieves were still large, sequences were clustered to 97% nt identity (USEARCH) (Edgar, 2010).
Phylogenetic analysis to define intermediates
We used the lineage sequences from Illumina or PacBio to construct heavy chain and light chain phylogenetic trees. After sieving and clustering the VRC34 lineage sequences from Illumina NGS reads, we constructed two maximum-likelihood (ML) trees separately for heavy and light chain sequences. Matured VRC34.01 – VRC34.07 heavy and light chain sequences were added to the VRC34 lineage related heavy and light chain sequences, respectively. Before tree construction, germline-derived heavy and light chain sequences were aligned with ClustalO (v1.2.4) (Sievers et al., 2011) using default parameter and prepared as suggestions in (Cole et al., 2013). The alignments were then provided as input to the construction of a maximum-likelihood phylogenetic tree using RAxML package v.8.2.9 (Stamatakis, 2014), with default parameters. FastML server was used to obtain heavy and light ancestral sequences(Ashkenazy et al., 2012). The outgroup was set as the germline gene and the resulting tree of VRC34 lineage was visualized using Dendroscope (Huson and Scornavacca, 2012).
Env-pseudotyped virus neutralization assay
Antibody neutralization was assessed using the single-round infection assay of TZM-bl cells with a panel of 208 or 10 HIV-1 Env-pseudoviruses (Doria-Rose et al., 2013; Sarzotti-Kelsoe et al., 2014). Serial dilutions of monoclonal antibodies starting at up to 500 ug/ml were mixed with the virus stocks and incubated at 37 °C for 1h. TZM-bl cells (0.5 million/ml) were added and the mixture incubated at 37 °C overnight. Sham medium in place of antibody was used as a control. Infection levels were determined in 2 days with Bright-Glo luciferase assay system (Promega, Madison, WI). Neutralization curves were fitted to a 5-parameter nonlinear regression analysis to obtain IC50, the antibody concentration required to inhibit infection by 50%.
Dendrograms depicting the gp160 protein sequence distances were produced by using ClustalW, Prodist and Neighbor Phylogenetic tree in BioEdit (Hall, 1999) to generate a phylogenetic tree of the HIV-1 gp160 protein sequences of 208 strains used in the neutralization assays. The tree was displayed with Dendroscope (Huson and Scornavacca, 2012) (http://dendroscope.org) and color-coded according to neutralization sensitivity.
Crystallization of antibody-FP8 complexes
FP8_v1 (AVGIGAVF) was dissolved in 100% DMSO at 100 mg/ml concentration, then mixed with Fabs in a 10:1 molar ratio to reach a final protein complex concentration of 10 mg/ml. Fab:FP8 complexes were screened for crystallization using 576 conditions from Hampton screen, Precipitant Synergy screen, and Qiagen Wizard screen with a mosquito robot delivering 100 nl of reservoir solution mixing with 100 nl of protein solution per drop. Crystals of VRC34-pI3 Fab complexed with FP8 were obtained from 0.2 M NaCl and 20% w/v PEG 3350 and grew to dimensions of 300 um x 200 um x 100 um. Crystals of VRC34-pI4 Fab complexed with FP8_v1 were obtained from 0.2 M NH4CH3CO2, 0.1 M Tris pH 8.5, and 25% w/v PEG 3350 and grew to dimensions of 400 um x 400 um x 50 um. Crystals of VRC34.05 Fab complexed with FP8_v1 were obtained from 0.1M HEPES pH 7.0 30%, w/v PEG 6000 and grew to dimensions of 300 um x 300 um x 200 um.
Crystal structure determination
Crystals were cryoprotected in 30% glycerol and flash-frozen in liquid nitrogen. Data were collected at 100 K and a wavelength of 1.00 Å at the SER-CAT beamline ID-22 (Advanced Photon Source, Argonne National Laboratory). Diffraction data were processed with HKL2000 (Otwinowski and Minor, 1997). Structure solution was obtained by molecular replacement with Phaser (McCoy et al., 2007) using homologous Fab:FP complex structure (PDB ID: 5I8E for VRC34.01 Fab and PDB ID: 5I8C for VRC34.01 Fab:FP complex) as search model. Model building was carried out with Coot (Emsley and Cowtan, 2004), followed by refinement with Phenix (Adams et al., 2004). Structure parameter were validated by MOLPROBITY (Davis et al., 2004) during refinement. Ramachandran statistical analysis indicated that the final structures contained no more than 0.2% disallowed residues. Data collection and refinement statistics are shown in Table S5.
Mutation hotspot and rare mutation analysis
Rare mutations were analyzed with Gene-Specific Substitution Profiles (GSSPs), which measure the frequency of amino acid substitutions in each antibody gene from NGS data (Guo et al., 2019; Sheng et al., 2017). To construct GSSPs, we collected and curated transcripts from IgG/IgA antibody repertoires (293 repertoires from 108 donors for heavy chain and 63 repertoires from 16 donors for light chain) (Guo et al., 2019). For each repertoire, curated transcripts were first sorted into groups based on V and J gene usage combinations. Transcripts with 90% CDR3 sequence identity and the same CDR3 length were clustered into clones using USEARCH (Edgar, 2010). We selected one transcript as representative sequence for each clone, representative transcripts from all repertoires were combined to build GSSPs for IGHV1-2 and IGKV1-9 genes (with 19143 and 5923 clones, respectively) using mGSSP in SONAR package (Schramm et al., 2016; Sheng et al., 2017). In GSSPs, rare mutations were defined as 1-substitution frequency > 0.995.
Antibody alanine/glycine scan
Binding of VRC34.01 and intermediates to 16 C-terminal His-tagged FP peptides (residue 512-521), including wildtype and alanine/glycine single mutants, was assessed using a fortéBio Octet HTX instrument (Kong et al., 2019; Xu et al., 2018). Briefly, peptides at 50 µg/ml in PBS were loaded onto Ni-NTA biosensors by binding through their C-terminal His-tag. Typical capture levels were between 1.1 and 1.3 nm, with a variability of < 0.1 nm within a row of eight tips. These peptide-bound biosensors were equilibrated in PBS for 60 s followed by capture of Fabs (250 nM) of VRC34.01, VRC34-pI1-PI4 or an RSV F antibody Motavizumab for 120 s and a subsequent dissociation step in PBS. Data analysis was carried out using Octet software, version 9.0, and normalized responses were plotted using PRISM (GraphPad).
QUANTIFICATION AND STATISTICAL ANALYSIS
Distances of each VRC34 lineage variant to UCA for heavy and light chains were obtained from the paired heavy and light trees to guide the pairing of the heavy and light chain intermediates obtained from Illumina sequencing. Pearson correlation was used to calculate the correlation between heavy and light intermediates.
DATA AND SOFTWARE AVAILABILITY
Coordinates and structural factors for VRC34.05, VRC34.01-pI3 and VRC34.01-pI4 in complex with FP8_v1 are available in Protein Data Bank under accession code 6UBI, 6UCE, 6UCF, respectively. The NGS data used in this study have been deposited in the NCBI BioProject under accession code PRJNA566192.
Supplementary Material
Acknowledgments
We thank S.K. Farney for assistance of visualization of VRC34 somatic mutations, C. Moore, G. Padilla, J. Rathmann, C. Whittaker, A. B. McDermott of the Vaccine Immunology Program for assistance with 208 strain neutralization, J. Stuckey for assistance with figures, and members of the Structural Biology Section and Structural Bioinformatics Core, Vaccine Research Center, for discussions and comments on the manuscript. We thank J. Baalwa, D. Ellenberger, F. Gao, B. Hahn, K. Hong, J. Kim, F. McCutchan, D. Montefiori, L. Morris, J. Overbaugh, E. Sanders-Buell, G. Shaw, R. Swanstrom, M. Thomson, S. Tovanabutra, C. Williamson, and L. Zhang for contributing the HIV-1 envelope plasmids used in our neutralization panel. Support for this work was provided by the Intramural Research Program of the Vaccine Research Center, National Institute of Allergy and Infectious Diseases (NIAID), by NIH grant 1R21AI138024-01A1 to Z.S, and by the Swedish Research Council grant #2017-00968 to M.M.C. and G.K.H. Use of sector 22 (Southeast Region Collaborative Access team) at the Advanced Photon Source was supported by the US Department of Energy, Basic Energy Sciences, Office of Science, under contract number W-31-109-Eng-38.
Footnotes
Declaration of Interests
The authors declare no competing interests.
References
- Adams PD, Gopal K, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Pai RK, Read RJ, Romo TD, et al. (2004). Recent developments in the PHENIX software for automated crystallographic structure determination. J Synchrotron Radiat 11, 53–55. [DOI] [PubMed] [Google Scholar]
- Ashkenazy H, Penn O, Doron-Faigenboim A, Cohen O, Cannarozzi G, Zomer O, and Pupko T (2012). FastML: a web server for probabilistic reconstruction of ancestral sequences. Nucleic Acids Res 40, W580–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonsignori M, Kreider EF, Fera D, Meyerhoff RR, Bradley T, Wiehe K, Alam SM, Aussedat B, Walkowicz WE, Hwang KK, et al. (2017). Staged induction of HIV-1 glycan-dependent broadly neutralizing antibodies. Sci Transl Med 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonsignori M, Montefiori DC, Wu X, Chen X, Hwang KK, Tsao CY, Kozink DM, Parks RJ, Tomaras GD, Crump JA, et al. (2012). Two distinct broadly neutralizing antibody specificities of different clonal lineages in a single HIV-1-infected donor: implications for vaccine design. J Virol 86, 4688–4692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boritz EA, Darko S, Swaszek L, Wolf G, Wells D, Wu X, Henry AR, Laboune F, Hu J, Ambrozak D, et al. (2016). Multiple Origins of Virus Persistence during Natural Control of HIV Infection. Cell 166, 1004–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borrow P, and Moody MA (2017). Immunologic characteristics of HIV-infected individuals who make broadly neutralizing antibodies. Immunol Rev 275, 62–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton DR, and Mascola JR (2015). Antibody responses to envelope glycoproteins in HIV-1 infection. Nat Immunol 16, 571–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng C, Xu K, Kong R, Chuang GY, Corrigan AR, Geng H, Hill KR, Jafari AJ, O’Dell S, Ou L, et al. (2019). Consistent elicitation of cross-clade HIV-neutralizing responses achieved in guinea pigs after fusion peptide priming by repetitive envelope trimer boosting. PLoS One 14, e0215163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole MF, Cox VE, Gratton KL, and Gaucher EA (2013). Reconstructing evolutionary adaptive paths for protein engineering. Methods Mol Biol 978, 115–125. [DOI] [PubMed] [Google Scholar]
- Corcoran MM, Phad GE, Vazquez Bernat N, Stahl-Hennig C, Sumida N, Persson MA, Martin M, and Karlsson Hedestam GB (2016). Production of individualized V gene databases reveals high levels of immunoglobulin genetic diversity. Nat Commun 7, 13642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis IW, Murray LW, Richardson JS, and Richardson DC (2004). MOLPROBITY: structure validation and all-atom contact analysis for nucleic acids and their complexes. Nucleic Acids Res 32, W615–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeKosky BJ, Ippolito GC, Deschner RP, Lavinder JJ, Wine Y, Rawlings BM, Varadarajan N, Giesecke C, Dorner T, Andrews SF, et al. (2013). High-throughput sequencing of the paired human immunoglobulin heavy and light chain repertoire. Nat Biotechnol 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeKosky BJ, Kojima T, Rodin A, Charab W, Ippolito GC, Ellington AD, and Georgiou G (2015). In-depth determination and analysis of the human paired heavy- and light-chain antibody repertoire. Nat Med 21, 86–91. [DOI] [PubMed] [Google Scholar]
- DeKosky BJ, Lungu OI, Park D, Johnson EL, Charab W, Chrysostomou C, Kuroda D, Ellington AD, Ippolito GC, Gray JJ, and Georgiou G (2016). Large-scale sequence and structural comparisons of human naive and antigen-experienced antibody repertoires. Proc Natl Acad Sci U S A 113, E2636–2645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dingens AS, Acharya P, Haddox HK, Rawi R, Xu K, Chuang GY, Wei H, Zhang B, Mascola JR, Carragher B, et al. (2018). Complete functional mapping of infection- and vaccine-elicited antibodies against the fusion peptide of HIV. PLoS Pathog 14, e1007159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doria-Rose N, Bailer R, Louder M, Lin C-L, Turk E, Laub L, Longo N, Connors M, and Mascola JR (2013). High throughput HIV-1 microneutralization assay In Protocol Exchange. [Google Scholar]
- Doria-Rose NA, Schramm CA, Gorman J, Moore PL, Bhiman JN, DeKosky BJ, Ernandes MJ, Georgiev IS, Kim HJ, Pancera M, et al. (2014). Developmental pathway for potent V1V2-directed HIV-neutralizing antibodies. Nature 509, 55–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461. [DOI] [PubMed] [Google Scholar]
- Emsley P, and Cowtan K (2004). Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Gao F, Bonsignori M, Liao HX, Kumar A, Xia SM, Lu X, Cai F, Hwang KK, Song H, Zhou T, et al. (2014). Cooperation of B cell lineages in induction of HIV-1-broadly neutralizing antibodies. Cell 158, 481–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Chen K, Kwong PD, Shapiro L, and Sheng Z (2019). cAb-Rep: A Database of Curated Antibody Repertoires for Exploring antibody diversity and Predicting Antibody Prevalence. Front Immunol 10, 2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall T (1999). BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symposium Series, 95–98.10780396 [Google Scholar]
- Huson DH, and Scornavacca C (2012). Dendroscope 3: an interactive tool for rooted phylogenetic trees and networks. Syst Biol 61, 1061–1067. [DOI] [PubMed] [Google Scholar]
- Hwang JK, Wang C, Du Z, Meyers RM, Kepler TB, Neuberg D, Kwong PD, Mascola JR, Joyce MG, Bonsignori M, et al. (2017). Sequence intrinsic somatic mutation mechanisms contribute to affinity maturation of VRC01-class HIV-1 broadly neutralizing antibodies. Proc Natl Acad Sci U S A 114, 8614–8619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson EL, Doria-Rose NA, Gorman J, Bhiman JN, Schramm CA, Vu AQ, Law WH, Zhang B, Bekker V, Abdool Karim SS, et al. (2018). Sequencing HIV-neutralizing antibody exons and introns reveals detailed aspects of lineage maturation. Nat Commun 9, 4136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joyce MG, Wheatley AK, Thomas PV, Chuang GY, Soto C, Bailer RT, Druz A, Georgiev IS, Gillespie RA, Kanekiyo M, et al. (2016). Vaccine-Induced Antibodies that Neutralize Group 1 and Group 2 Influenza A Viruses. Cell 166, 609–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Misawa K, Kuma K, and Miyata T (2002). MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res 30, 3059–3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kepler TB, Liao HX, Alam SM, Bhaskarabhatla R, Zhang R, Yandava C, Stewart S, Anasti K, Kelsoe G, Parks R, et al. (2014). Immunoglobulin gene insertions and deletions in the affinity maturation of HIV-1 broadly reactive neutralizing antibodies. Cell Host Microbe 16, 304–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein F, Diskin R, Scheid JF, Gaebler C, Mouquet H, Georgiev IS, Pancera M, Zhou T, Incesu RB, Fu BZ, et al. (2013). Somatic mutations of the immunoglobulin framework are generally required for broad and potent HIV-1 neutralization. Cell 153, 126–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong R, Duan H, Sheng Z, Xu K, Acharya P, Chen X, Cheng C, Dingens AS, Gorman J, Sastry M, et al. (2019). Antibody Lineages with Vaccine-Induced Antigen-Binding Hotspots Develop Broad HIV Neutralization. Cell 178, 567–584 e519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong R, Xu K, Zhou T, Acharya P, Lemmin T, Liu K, Ozorowski G, Soto C, Taft JD, Bailer RT, et al. (2016). Fusion peptide of HIV-1 as a site of vulnerability to neutralizing antibody. Science 352, 828–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon YD, Pancera M, Acharya P, Georgiev IS, Crooks ET, Gorman J, Joyce MG, Guttman M, Ma X, Narpala S, et al. (2015). Crystal structure, conformational fixation and entry-related interactions of mature ligand-free HIV-1 Env. Nat Struct Mol Biol 22, 522–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwong PD, Chuang GY, DeKosky BJ, Gindin T, Georgiev IS, Lemmin T, Schramm CA, Sheng Z, Soto C, Yang AS, et al. (2017). Antibodyomics: bioinformatics technologies for understanding B-cell immunity to HIV-1. Immunol Rev 275, 108–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwong PD, and Mascola JR (2012). Human antibodies that neutralize HIV-1: identification, structures, and B cell ontogenies. Immunity 37, 412–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laird Smith M, Murrell B, Eren K, Ignacio C, Landais E, Weaver S, Phung P, Ludka C, Hepler L, Caballero G, et al. (2016). Rapid Sequencing of Complete env Genes from Primary HIV-1 Samples. Virus Evol 2, vew018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landais E, Murrell B, Briney B, Murrell S, Rantalainen K, Berndsen ZT, Ramos A, Wickramasinghe L, Smith ML, Eren K, et al. (2017). HIV Envelope Glycoform Heterogeneity and Localized Diversity Govern the Initiation and Maturation of a V2 Apex Broadly Neutralizing Antibody Lineage. Immunity 47, 990–1003 e1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesk AM (2002). Introduction to bioinformatics (Oxford ; New York: Oxford University Press; ). [Google Scholar]
- Liao HX, Lynch R, Zhou T, Gao F, Alam SM, Boyd SD, Fire AZ, Roskin KM, Schramm CA, Zhang Z, et al. (2013). Co-evolution of a broadly neutralizing HIV-1 antibody and founder virus. Nature 496, 469–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLeod DT, Choi NM, Briney B, Garces F, Ver LS, Landais E, Murrell B, Wrin T, Kilembe W, Liang CH, et al. (2016). Early Antibody Lineage Diversification and Independent Limb Maturation Lead to Broad HIV-1 Neutralization Targeting the Env High-Mannose Patch. Immunity 44, 1215–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, and Read RJ (2007). Phaser crystallographic software. J Appl Crystallogr 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDaniel JR, DeKosky BJ, Tanno H, Ellington AD, and Georgiou G (2016). Ultra-high-throughput sequencing of the immune receptor repertoire from millions of lymphocytes. Nat Protoc 11, 429–442. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, and Minor W (1997). [20] Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 276, 307–326. [DOI] [PubMed] [Google Scholar]
- Salazar-Gonzalez JF, Bailes E, Pham KT, Salazar MG, Guffey MB, Keele BF, Derdeyn CA, Farmer P, Hunter E, Allen S, et al. (2008). Deciphering human immunodeficiency virus type 1 transmission and early envelope diversification by single-genome amplification and sequencing. J Virol 82, 3952–3970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarzotti-Kelsoe M, Bailer RT, Turk E, Lin CL, Bilska M, Greene KM, Gao H, Todd CA, Ozaki DA, Seaman MS, et al. (2014). Optimization and validation of the TZM-bl assay for standardized assessments of neutralizing antibodies against HIV-1. J Immunol Methods 409, 131–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders KO, Wiehe K, Tian M, Acharya P, Bradley T, Alam SM, Go EP, Scearce R, Sutherland L, Henderson R, et al. (2019). Targeted selection of HIV-specific antibody mutations by engineering B cell maturation. Science 366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheid JF, Mouquet H, Feldhahn N, Seaman MS, Velinzon K, Pietzsch J, Ott RG, Anthony RM, Zebroski H, Hurley A, et al. (2009). Broad diversity of neutralizing antibodies isolated from memory B cells in HIV-infected individuals. Nature 458, 636–640. [DOI] [PubMed] [Google Scholar]
- Schramm CA, Sheng Z, Zhang Z, Mascola JR, Kwong PD, and Shapiro L (2016). SONAR: A High-Throughput Pipeline for Inferring Antibody Ontogenies from Longitudinal Sequencing of B Cell Transcripts. Front Immunol 7, 372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng Z, Schramm CA, Connors M, Morris L, Mascola JR, Kwong PD, and Shapiro L (2016). Effects of Darwinian Selection and Mutability on Rate of Broadly Neutralizing Antibody Evolution during HIV-1 Infection. PLoS Comput Biol 12, e1004940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng Z, Schramm CA, Kong R, Program NCS, Mullikin JC, Mascola JR, Kwong PD, and Shapiro L (2017). Gene-Specific Substitution Profiles Describe the Types and Frequencies of Amino Acid Changes during Antibody Somatic Hypermutation. Front Immunol 8, 537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Soding J, et al. (2011). Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7, 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonich CA, Williams KL, Verkerke HP, Williams JA, Nduati R, Lee KK, and Overbaugh J (2016). HIV-1 Neutralizing Antibodies with Limited Hypermutation from an Infant. Cell 166, 77–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soto C, Ofek G, Joyce MG, Zhang B, McKee K, Longo NS, Yang Y, Huang J, Parks R, Eudailey J, et al. (2016). Developmental Pathway of the MPER-Directed HIV-1-Neutralizing Antibody 10E8. PLoS One 11, e0157409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazquez Bernat N, Corcoran M, Hardt U, Kaduk M, Phad GE, Martin M, and Karlsson Hedestam GB (2019). High-Quality Library Preparation for NGS-Based Immunoglobulin Germline Gene Inference and Repertoire Expression Analysis. Front Immunol 10, 660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker LM, Phogat SK, Chan-Hui PY, Wagner D, Phung P, Goss JL, Wrin T, Simek MD, Fling S, Mitcham JL, et al. (2009). Broad and potent neutralizing antibodies from an African donor reveal a new HIV-1 vaccine target. Science 326, 285–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiehe K, Bradley T, Meyerhoff RR, Hart C, Williams WB, Easterhoff D, Faison WJ, Kepler TB, Saunders KO, Alam SM, et al. (2018). Functional Relevance of Improbable Antibody Mutations for HIV Broadly Neutralizing Antibody Development. Cell Host Microbe 23, 759–765 e756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Yang ZY, Li Y, Hogerkorp CM, Schief WR, Seaman MS, Zhou T, Schmidt SD, Wu L, Xu L, et al. (2010). Rational design of envelope identifies broadly neutralizing human monoclonal antibodies to HIV-1. Science 329, 856–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Zhang Z, Schramm CA, Joyce MG, Kwon YD, Zhou T, Sheng Z, Zhang B, O’Dell S, McKee K, et al. (2015). Maturation and Diversity of the VRC01-Antibody Lineage over 15 Years of Chronic HIV-1 Infection. Cell 161, 470–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu K, Acharya P, Kong R, Cheng C, Chuang GY, Liu K, Louder MK, O’Dell S, Rawi R, Sastry M, et al. (2018). Epitope-based vaccine design yields fusion peptide-directed antibodies that neutralize diverse strains of HIV-1. Nat Med 24, 857–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye J, Ma N, Madden TL, and Ostell JM (2013). IgBLAST: an immunoglobulin variable domain sequence analysis tool. Nucleic Acids Res 41, W34–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Coordinates and structural factors for VRC34.05, VRC34.01-pI3 and VRC34.01-pI4 in complex with FP8_v1 are available in Protein Data Bank under accession code 6UBI, 6UCE, 6UCF, respectively. The NGS data used in this study have been deposited in the NCBI BioProject under accession code PRJNA566192.