

Abstract
The cAMP-responsive element binding protein (CREB) binding protein (CBP) and adenoviral E1A-binding protein (P300) are two closely related multifunctional transcriptional coactivators. Both proteins contain a bromodomain (BrD) adjacent to the histone acetyl transferase (HAT) catalytic domain, which serves as a promising drug target for cancers and immune system disorders. Several potent and selective small-molecule inhibitors targeting CBP BrD have been reported, but thus far small-molecule inhibitors targeting BrD outside of the BrD and extraterminal domain (BET) family are especially lacking. Here, we established and optimized a TR-FRET-based high-throughput screening platform for the CBP BrD and acetylated H4 peptide. Through an HTS assay against an in-house chemical library containing 20 000 compounds, compound DC_CP20 was discovered as a novel CBP BrD inhibitor with an IC50 value of 744.3 nM. This compound bound to CBP BrD with a KD value of 4.01 μM in the surface plasmon resonance assay. Molecular modeling revealed that DC_CP20 occupied the Kac-binding region firmly through hydrogen bonding with the conserved residue N1168. At the celluslar level, DC_CP20 dose-dependently inhibited the proliferation of human leukemia MV4-11 cells with an IC50 value of 19.2 μM and markedly downregulated the expression of the c-Myc in the cells. Taken together, the discovery of CBP BrD inhibitor DC_CP20 provides a novel chemical scaffold for further medicinal chemistry optimization and a potential chemical probe for CBP-related biological function research. In addition, this inhibitor may serve as a promising therapeutic strategy for MLL leukemia by targeting CBP BrD protein.
Keywords: CBP bromodomain, small-molecule inhibitor, high-throughput screening, TR-FRET, molecular modeling, human leukemia MV4-11 cells
Introduction
The transcriptional coactivator CBP [also known as cAMP-responsive element binding protein (CREB) binding protein] and its homolog P300 (adenoviral E1A-binding protein of 300 kDa) are two closely related histone acetyl transferases (HATs) [1, 2] that contain an HAT catalytic domain and a bromodomain [3]. The HAT domain functions as an acetyl-lysine “writer” by acetylating histone tails and other nuclear proteins, while the bromodomain (BrD) functions as an acetyl-lysine “reader” by identification of lysine acetylation state [4, 5]. Numerous studies have demonstrated that CBP plays an important role in various biochemical processes, including cell-type-specific gene transcription, cellular growth control, cell-cycle progression, DNA repair, maintenance of the differentiated state, and embryonic development [6]. The dysregulation of CBP is functionally associated with many diseases, such as inflammation and cancer [5, 7–9].
CBP/P300 bromodomains (BrD) are α-helical modules of ~110 residues that recognize the ε-N-acetylation of lysine residues, which is a family of evolutionarily conserved protein interaction modules that were discovered in the early 1990s in the brahma gene from Drosophila melanogaster [10, 11]. The bromodomain of CBP is a member of the non-BET bromodomain family, and many studies have shown that the bromodomain of CBP regulates Myc [12–14], an oncogene widely expressed in diverse cancers, especially in lymphoid malignancies and myeloma [15]. In addition, the success of BET (bromodomain and extraterminal) inhibitors has stimulated intensive research enthusiasm against other BrD proteins, including the CBP bromodomain. In addition, CBP BrD is a promising and profound drug target for therapeutic intervention, and several potent and selective small-molecule inhibitors targeting CBP BrD have been reported in the literature [11, 15–25]. The first potent inhibitors for CBP BrD were acetyl-lysine mimetic moieties based on 5- and 6-isoxazolylbenzimidazoles [26], which were later optimized as SGC-CBP30 [21]. Compound MS7972 was reported to inhibit the CBP-p53 interaction at 50 μM [21]. Other CBP BrD probes, such as I-CBP112, PF-CBP1, and CPI-637, displayed good selectivity for CBP BrD over other bromodomains and showed good cellular activity. Recently, CellCentric disclosed the development of CCS1477, an orally available bromodomain inhibitor with a single-digit nanomolar affinity for CBP/p300, which is planned to begin phase I clinical trials in late-stage prostate cancer [27]. All of these inhibitors support the high-potential chemical probes for further CBP-related biological function research and promote the development of therapeutic agents beyond the BET bromodomain family. Since the role of CBP in various pathways is elusive, there is still a pressing need for novel chemotypes of CBP BrD inhibitors with prominent pharmacokinetic and physicochemical properties.
Here, we report the discovery and evaluation of a CBP BrD inhibitor through an optimized TR-FRET (time-resolved fluorescence energy transfer)-based high-throughput screening assay against 20 000 compounds with different chemical scaffolds. The leading compound was discovered and named DC_CP20, and its IC50 value against the binding of CBP BrD with acetylated lysine was 744.3 nM. In addition, molecular docking studies proposed that DC_CP20 bound inside the Kac-binding pocket in a competitive manner. Furthermore, at the cellular level, the compound DC_CP20 showed effective inhibition of the proliferation of human leukemia MV4-11 cells with minimal effects on normal cells, and this molecule also downregulated the expression of the downstream oncogene Myc. These results demonstrated that this novel CBP BrD inhibitor was promising as a drug candidate for further optimization and development in therapies for CBP-related cancers.
Materials and methods
Protein expression and purification
The DNA fragment encoding the bromodomain of human CBP (residues 1082–1197) was codon optimized for Escherichia coli and subcloned into the pGEX 6p-1 vector (GE Healthcare, Chicago, IL, USA) containing a glutathione S-transferase tag. The CBP BrD protein was overexpressed in E. coli BL21 (DE3) cells in the LB medium at 37 °C for 4–6 h until the OD600 reached 0.6–0.8, then 0.4 mM IPTG (isopropyl-1-thio-D-galactopyranoside) (Sangon Biotech, Shanghai, China, Cat#A600168) was added, and the culture was incubated overnight at 16 °C, followed by harvesting by centrifugation. Cell pellets were resuspended and sonicated in precooled lysis buffer A (20 mM HEPES, pH 7.4, 150 mM NaCl). The supernatant was loaded onto a GST affinity column (GE Healthcare, Chicago, IL, USA) after centrifugation at 18 000 r/min for 40 min at 4 °C. The recombinant proteins were eluted with buffer B (20 mM HEPES, pH 7.4, 150 mM NaCl, 20 mM L-glutathione reduced), then concentrated and further purified by gel-filtration chromatography using a Superdex 75 10/300GL column (GE Healthcare, Chicago, IL, USA) in 20 mM HEPES, pH 7.4, 150 mM NaCl, and 1 mM TCEP. Fractions containing protein were pooled, concentrated, and stored at −80 °C for biochemical characterization.
Time-resolved fluorescence energy transfer (TR-FRET) high-throughput screening assay
TR-FRET technology was used to screen in-house chemical libraries for small-molecule inhibitors of CBP BrD. The final volume of the reaction was 40 μL. The compounds were diluted in CPD buffer with 20 mM HEPES, pH 7.4, and 150 mM NaCl and then transferred to white 384-well plates (PerkinElmer, Waltham, MA, USA, Cat#6007299) and incubated with 10 nM GST-CBP BrD in assay buffer [20 mM HEPES, pH 7.4, 150 mM NaCl, 0.1% bovine serum albumin (w/v), 0.01% Triton X-100 (v/v)] at room temperature for 30 min. After incubating with 100 nM H4 substrate peptide [N-C: SGRG-K(Ac)-GG-K(Ac)-GLG-K(Ac)-GGA-K(Ac)-RHRKVGG-K(biotin)] (ChinaPeptides, Suzhou, China) for 30 min, TR-FRET fluorophores, MAb anti-GST-Eu cryptate donor fluorophores and MAb anti-GST-XL665 acceptor fluorophores (Cisbio, Codolet, France, Cat#61GSTKLB and 61GSTXLB), were diluted in assay buffer. Subsequently, 10 μL of the fluorophores was added to each well of the plates and incubated at room temperature for 90 min. Finally, the signals were measured by an EnVision Multilabel plate reader (PerkinElmer; mirror LANCE/DELFIA Dual/Bias, Eu ex 337 nm, Eu em 615 nm; XL665 em 665/10 nm) and analyzed with GraphPad Prism 7.0 (GraphPad Software Inc., La Jolla, CA, USA).
Z′ factor and S/B calculation
The Z′ factor quantifies the suitability of the high-throughput screening assay, and is calculated according to the following equation: Z′ = 1–3(σn + σp) /|(μn−μp)|
The means and standard deviations of the positive (p) and negative (n) controls are denoted as μp, σp and μp, σn, respectively [28]. The negative controls were performed in different proportions of DMSO, and 2 μM SGC-CBP30 was used as the positive control and was added to each well of the same plate. The S/B value is the ratio of the average of the negative controls and the average of the positive controls.
Molecular modeling
Protein preparation and docking-grid generation
The apo crystal structure of CBP BrD was downloaded from the Protein Data Bank (PDB Code: 4OUF). Specifically, all of the solvent molecules, except conservative water (residue number: 1313), were removed, and only the A chain was retained. Subsequently, the remaining protein structure was prepared using the Protein Preparation Wizard module (Schrödinger, LLC, New York, NY, USA, 2017) in Maestro (Maestro, version 11.1; Schrödinger, LLC: New York, NY, USA, 2017) with standard Glide protocols [29], including assigning bond orders, adding hydrogens, creating disulfide bonds, and generating het states using Epik at pH 7.4 ± 2.0. Then, H-bond optimization and restrained minimization were performed with the OPLS3 force field and the default value for RMSD of 0.30 Å for converging heavy atoms. The receptor grid was defined as a closed box centered at crucial residues, including N1168, R1173, and Y1125, and other settings were set to default values.
Ligand preparation for docking
The 3D coordinates of the ligands in the compound candidates list were generated using LigPrep. The possible ionization of these ligands was generated at pH 7.4 ± 2.0. Desalting and tautomers were generated with the OPLS3 force field.
Glide docking procedure
Docking was performed using Maestro with the standard precision (SP) mode or extra precision (XP) mode. The ligands were sampled in a flexible manner, adding Epik state penalties to the docking score and rewarding intramolecular hydrogen bonds. After Glide docking, the poses scored lower than −5.0 were picked up.
Induced fit docking procedure
Docking was performed using Maestro with the induced fit docking procedure. The ligands were sampled in a flexible manner, adding Epik state penalties to the docking score and rewarding intramolecular hydrogen bonds. Extended sampling was used as a docking protocol to generate up to 80 poses using automatic docking settings. The crucial amino acids N1168, Y1125, and R1173 were selected as the docking box center, and residues within 5 Å of ligand poses were refined. Finally, the poses scored lower than −6.0 were picked up.
Surface plasmon resonance (SPR)-binding assay
The SPR-binding assays were performed on a Biacore T200 instrument (GE Healthcare, Chicago, IL, USA) at 25 °C, as previously described [30, 31]. CBP BrD protein was covalently immobilized on a CM5 chip using a standard amine-coupling procedure in 10 mM sodium acetate (pH 5.0). The chip was first equilibrated in HBS buffer (20 mM HEPES (pH 7.4), 150 mM NaCl, 0.05% (v/v) surfactant P20, and 0.2% (v/v) DMSO) overnight. The compounds were serially diluted with HBS buffer and injected for 120 s (contact phase), followed by 240 s (dissociation phase). The KD values of the tested compounds were determined by Biacore T200 evaluation software (GE Healthcare, Chicago, IL, USA).
Cell-proliferation assays
MV4-11, HUV-EC-C, and MRC-5 cell lines were purchased from ATCC. All cell lines were maintained in the RPMI-1640 medium or DMEM (Life Technologies, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum and 1% Pen/Strep (Life Technologies, Carlsbad, CA, USA). MV4-11 cells were seeded onto 96-well plates at a density of 5 × 103 mL−1 in a volume of 50 μL and incubated for 30 min. HUV-EC-C and MRC-5 cells were seeded onto 96-well plates at a density of 3 × 103 mL−1 in a volume of 50 μL and incubated for 12 h. Then, 50 μL of different concentrations of compounds or DMSO control was added to the plate and incubated for 3 days. Cell Titer-Glo luminescent assays (Promega, Madison, WI, USA) were used to determine the fraction of viable cells according to the manufacturer’s instructions. The plates were read on a multilabel reader (Envision, PerkinElmer, Waltham, MA, USA). All treatments were performed in triplicate. All data were analyzed with GraphPad Prism 7.0 (GraphPad Software Inc., La Jolla, CA, USA).
Western blot analysis
The cells were seeded onto six-well plates at a density of 1 × 106 mL−1 and treated with different concentrations of compounds or DMSO control for 6 h. The cell lysates were prepared using 1 × SDS cell lysis buffer and boiled for 20 min. The total cell lysates were separated by 12% SDS-polyacrylamide gels and transferred to nitrocellulose membranes. The blots were blocked with blocking buffer (5% nonfat milk in TBST) for 30−60 min at room temperature and incubated with anti-cMYC (Cell Signaling Technology, Danvers, MA, USA, Cat#5605S) and anti-GAPDH (Sangon Biotech, Shanghai, China, Cat#D110016) primary antibodies overnight at 4 °C. Then, the blots were probed with HRP-conjugated anti-rabbit (Sangon Biotech, Shanghai, China, Cat#D110058) or anti-mouse (Sangon Biotech, Shanghai, China, Cat#D110087) IgG secondary antibodies for 1 h at room temperature. Following another three washes, the bands were detected with a Amersham Imager 600 imaging system (GE Healthcare, Chicago, IL, USA) using ECL substrate (Share-Bio, Shanghai, China, Cat#sb-wb011).
Statistical analyses
All the data are presented as the mean ± the standard error of the mean (SEM) which were from at least three independent experiments. All the statistical analyses were performed using GraphPad Prism 7.0 (GraphPad Software Inc., La Jolla, CA, USA).
Results
Establishment of a high-throughput screening (HTS) assay targeting CBP bromodomain
Currently, HTS has matured to become an integral part of pharmaceutical research and a cornerstone in the expansion of biomedical knowledge [32]. The TR-FRET technique is a reliable and efficient method for detecting enzyme activity and protein–protein interactions. In this report, we developed a high-throughput screening assay based on the TR-FRET technique, which was applied to screen in-house chemical libraries containing ~20,000 diverse compound collections. In the TR-FRET assay, the signal value became stable when the two fluorophores carrying CBP BrD and substrate peptides were close to each other. In contrast, the signal would decrease if the compound blocks the interaction (Fig. 1a). To determine the optimal HTS assay conditions, different concentrations of CBP BrD were titrated ranging from 1.56 nM to 100 nM with excess H4 substrate to determine the optimum protein concentration for the following screening experiments. The titration was then reversed with different concentrations of peptides in the presence of a fixed amount of CBP BrD protein and determined the apparent KD values between CBP BrD and acetylated substrate peptide H4 (Fig. 1b, c). Considering the sensitivity and robustness of the HTS assay, the relatively low concentrations (20 nM CBP BrD and 100 nM biotinylated H4 peptide) that provided reliable signals were selected for future applications.
Fig. 1.

The establishment and evaluation of the TR-FRET assay. a The schematic diagram of the TR-FRET assay. b TR-FRET saturation-binding curves with increasing concentrations of GST-CBP BrD. c TR-FRET saturation-binding curves with increasing concentrations of lysine-acetylated H4 peptide. d Evaluation of Z′ factors for the high-throughput screening assay in the absence or presence of 0.25% DMSO. e IC50 determination for SGC-CBP30 against CBP BrD. All experiments were performed at least three times. The data were analyzed in GraphPad Prism 7.0
Since most of the compounds used DMSO as a solvent and DMSO can bind to the CBP bromodomain (PDB code 3P1E), DMSO tolerance is a vital factor influencing the stability of high-throughput screens. Various concentration gradients of DMSO (0.25%, 0.5%, 1%, 2%, and 4%) were used for testing the DMSO tolerance and stability of the HTS assay (Fig. 1d; Supplementary Fig. S1). When the percentage of DMSO was 0.25%, the signal-to-background ratio (S/B) was 25.10, but there was almost no window in the readings when the ratio increased to 4%. In addition, the Z′ factor is commonly used as an indicator of high-throughput screening assay performance, and an assay with a Z′ factor of less than 0.5 is not recommended for HTS [28]. To test whether the TR-FRET assay was suitable as a high-throughput screening assay, we calculated the Z′ factors at different DMSO concentrations (0.25%, 0.5%, 1%, 2%, and 4%), and the results indicated that increased DMSO concentrations led to lower Z′ factors. The assay had an excellent Z′ factor of 0.87 with 0.25% DMSO. To increase the robustness and accuracy of the assay, we performed the screening assay under a 0.25% DMSO level. The feasibility of this HTS assay was further verified by testing the inhibitory activity of the positive compound SGC-CBP30; the test values were consistent with those in previous reports [15] (Fig. 1e).
Discovery of compound DC_CP20 through TR-FRET-based high-throughput screening
We applied the well-optimized HTS method to screen in-house chemical libraries containing ~20 000 different chemicals (Fig. 2a). A primary screening was performed at a single concentration (50 μM), and the compounds displaying greater than 70% inhibition were selected for further evaluation. After primary TR-FRET-based high-throughput screening and second-round validation, we generated seven chemical scaffolds containing 26 candidate compounds with significant dose-dependent inhibitory activities for CBP BrD, and the inhibition rate of these compounds at concentrations of 25 μM and 50 μM is shown in Fig. 2b. Among these candidate inhibitors, DC_CP20, with a novel chemical scaffold of 3-phenyl- [1,2,4] triazolo [3,4-b] [1,3,4] thiadiazole for CBP BrD, showed optimal inhibitory activity (IC50 = 744.3 nM) (Fig. 2c, d). Thus, DC_CP20 was chosen as the representative compound for further in-depth characterization.
Fig. 2.

The discovery of compound DC_CP20 and hits validation. a Flowchart of hierarchical high-throughput screening for CBP BrD inhibitors. b Inhibitory activity of top 26 compounds from high-throughput screening determined by TR-FRET assay. c Structure of compound DC_CP20. d IC50 determination for compound DC_CP20 against CBP BrD by TR-FRET assay. e SPR-binding data for the interactions of DC_CP20 with CBP BrD. The compound was prepared at concentration of 0.173, 0.390, 0.585, 0.878, 1.975, 2.963, 4.444, and 6.667 μM, respectively
Binging assay based on SPR
As the SPR assay is one of the most useful biophysical methods for hit validation, we conducted SPR assays to further validate the interaction of CBP BrD with DC_CP20 and to determine their binding affinity. As shown in Fig. 2e, DC_CP20 directly bound to CBP BrD with an equilibrium dissociation constant (KD) of 4.01 μM, which was consistent with its IC50 value of 744.3 nM. Taken together, DC_CP20 can bind to CBP BrD and then inhibit CBP BrD activity in vitro.
Binding mode and structure–activity relationships (SARs) analysis
To further understand the molecular mechanism of the inhibitory properties of DC_CP20 toward CBP BrD, a putative binding mode was docked by Glide by using the SP mode of Maestro (Schrödinger LLC 2015, USA). As shown in Fig. 3a, b, DC_CP20 stably occupied the hydrophobic pocket core in CBP BrD, and the dimethylamino group interacted with the side chain of the N1168 residue through a hydrogen bond. Significantly, the residue of N1168 is a conserved Kac-binding site for CBP BrD [33–35]. In addition, the N1 of the [1,2,4] triazolo [3,4-b] [1,3,4] thiadiazole moiety was also involved in the water hydrogen-bonding bridge with Y1125 and M1133. Moreover, some nonpolar interactions, such as hydrophobic interactions, were also formed between CBP BrD and DC_CP20. The chemical scaffold 3-phenyl- [1,2,4] triazolo [3,4-b] [1,3,4] thiadiazole is sandwiched between L1120 and the gatekeeper (V1174), while the phenyl group makes van der Waals interactions with I1122, L1120, and P1117.
Fig. 3.

Putative binding mode between DC_CP20 and CBP BrD. a A close up view of the interaction between CBP BrD and DC_CP20. The ligand and interacting residues are shown as sticks; water molecules are shown as spheres; hydrogen bonds are indicated by yellow dotted lines. b The Kac-binding region of CBP BrD occupied by DC_CP20
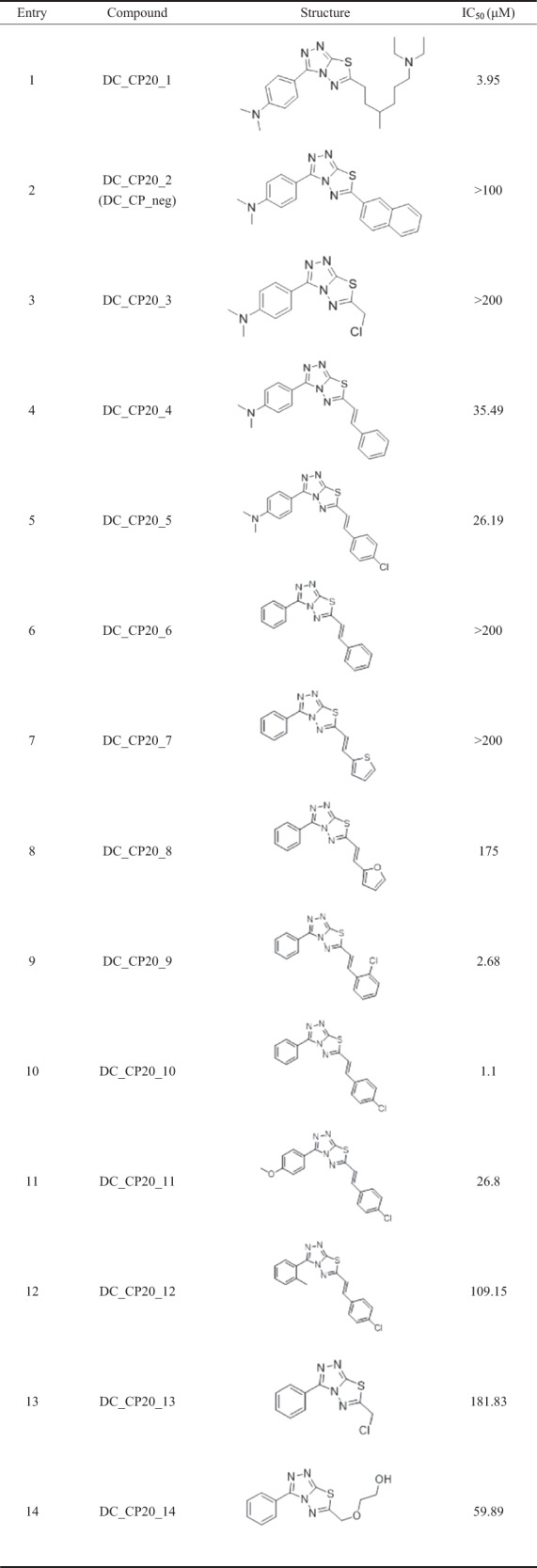
To further explore the relationship between the chemical structures and their anti-CBP BrD activity, we detected the inhibitory activity of various derivatives of the chemical scaffold 3-phenyl- [1,2,4] triazolo [3,4-b] [1,3,4] thiadiazole (Table 1). First, the results showed that the dimethylamino group or the methoxy group in the para position of the benzene ring weakened the inhibitory activity, probably because of the steric hindrance and electrostatic interactions of compounds DC_CP20_01 and DC_CP20_11. Second, when (E)-1-chloro-4-(prop-1-en-1-yl) phenyl became a side chain of the scaffold, the inhibitory activity of compounds DC_CP20_10 and DC_CP20_05 was slightly reduced. Moreover, the determined IC50 of compound DC_CP03 and compound DC_CP13 suggested that the inhibitory activity would become weak if the side chain was replaced by small groups, such as an ethyl group. Similarly, the large side chain would impair the inhibitory activity probably because of its steric hindrance effects. For example, the IC50 value of the negative compound DC_CP_neg was 100 μM (Supplementary Fig. S2), which was more than 100 times the IC50 value of compound DC_CP20. Collectively, the docking results and SAR analysis demonstrated the binding mechanism of the lead compound. In addition, the above insights should help guide further design and discovery of the more potent CBP BrD inhibitors derived from compound DC_CP20.
Table 1.
The chemical structure and inhibition activities of DC_CP20 derivatives
Evaluation of compound DC_CP20 at the cellular level
Recent studies have indicated that CBP/p300 are involved in recurrent leukemia-associated chromosomal translocations and are key regulators of cell growth. These studies also supported consideration of p300 and CBP as therapeutic targets in AML (acute myeloid leukemia) [15]. Therefore, to further evaluate the inhibitory activity of DC_CP20 at the cellular level, we investigated its effects on the proliferation of MV4-11 in vitro using a cell Titer-Glo luminescent assay. As expected, DC_CP20 showed potent antiproliferative activity against tested leukemia cell lines in a dose-dependent manner, with IC50 values of 19.2 μM, while the negative compound DC_CP_neg presented minimal effect on cell growth (Fig. 4a, b). In addition, DC_CP20 showed minimal effect on the proliferation of normal cells, including the umbilical vascular endothelium cells HUV-EC-C and normal lung tissue cell line MRC5 (Fig. 4c, d). Previous genetic studies have demonstrated that CBP BrD regulates the expression of Myc in diverse human cancers [13, 14]. To evaluate the effects of DC_CP20 on the expression of Myc protein, gradient concentrations of DC_CP20 were applied to evaluate protein levels. As shown in Fig. 4e, DC_CP20 downregulated the expression of Myc in MV4-11 cells. Collectively, these results demonstrated the high-target engagement of the identified CBP BrD inhibitor in MV4-11 cells.
Fig. 4.

Characterization of hit compounds at cellular level. (a) Cell growth inhibition curves of compound DC_CP20 for the MV4-11 cell lines at 72 h. (b) Cell growth histogram of negative compound DC_CP_neg for the MV4-11 cell lines at 72 h. (c) Cell growth histogram of compound DC_CP20 for the HUV-EC-C normal cell lines and (d) MRC5 normal cell lines at 72 h. (e) Treatment with DC_CP20 for 6 h dose-dependently inhibited the protein abundance of c-Myc in MV4-11 cells
Discussion
The CBP bromodomain is a non-BET bromodomain that is crucial for diverse cellular processes [36] and has emerged as an attractive target for drug discovery. Thus, there is an urgent demand to develop novel chemical scaffolds for CBP BrD.
In this study, we developed a stable and reproducible TR-FRET-based HTS assay targeting CBP BrD to identify small molecules that block the interaction between CBP BrD and its substrate peptide H4 (K5ac, K8ac, K12ac, and K16ac). The high Z′ factors (>0.7) at different DMSO concentrations demonstrated the robustness and reliability of this TR-FRET-based high-throughput screening assay. Using this platform, we carried out a pilot screen against an in-house chemical library, leading to the identification of DC_CP20, which showed a favorable IC50 value of 744.3 nM in vitro. The binding affinity was validated by SPR with a KD value of 4.01 μM. The KD value measured by the SPR method was consistent with the IC50 value detected by the TR-FRET assay. Further molecular docking results demonstrated that DC_CP20 occupied the Kac-binding region, which accounted for its inhibitory activity in vitro and provided an atomic-resolution interpretation of structure–activity relationship. A series of derivatives of DC_CP20 were tested for the inhibitory activity of CBP BrD, which provided a preliminary structure–activity relationship that can be evaluated for further chemical modification. At the cellular level, DC_CP20 could inhibit the proliferation of MV4-11 cell lines with an IC50 of 19.2 μM, and this compound showed no effect on normal cells. In addition, treatment with DC_CP20 significantly downregulated the expression of Myc at a concentration of 12.5 μM. DC_CP20 exerted differential activities between the TR-FRET assay and the cell Titer-Glo luminescent assay, an effect that is probably due to the weak permeability of our molecules in cells, suggesting that these compounds exhibited less strong potency at the cellular level.
In conclusion, the TR-FRET-based high-throughput screening assay established here may be a reliable platform that could promote the drug discovery process for CBP BrD. In addition, this technique may be a potential strategy to identify more potent compounds by combining this platform with a rational drug molecular design and further optimization of the chemical structure. In addition, the novel scaffold of the CBP BrD inhibitor was promising as a drug candidate after further optimization and development in the therapeutic intervention of CBP-related cancers.
Supplementary information
Acknowledgements
We thank the staff members of the Large-scale Protein Preparation System at the National Facility for Protein Science in Shanghai (NFPS), Zhangjiang Lab, China for providing technical support and assistance in data collection and analysis. We gratefully acknowledge financial support from the National Natural Science Foundation of China (91853205 and 81625022 to CL, 81430084 to DHD); KC Wong Education Foundation to CL, Chinese Academy of Sciences (XDA12020353 to CL and CASIMM0120184015 to HZ) and China Postdoctoral Science Foundation (2017M621571 to LYY), Science and Technology Commission of Shanghai Municipality (18431907100 and 19XD1404700 to CL), and National Science & Technology Major Project of China (2018ZX09711002 to DHD).
Author contributions
CL generated the original hypothesis and designed the study; FCZ performed the protein purification, compound screening, molecular docking and other biochemical assays. ZYS performed the cell proliferation assay and Western blot assay. FCZ and ZYS wrote the paper. CL reviewed the paper. CPL, LPL and YZ performed the protein expression and purification. DZ, JW and YTC assisted to finish the HTS. SHX, HJ, TL and PX assisted to finish SPR assay. LYY, DHD and HZ read and contributed to the final manuscript.
Footnotes
These authors contributed equally: Feng-cai Zhang, Zhong-ya Sun
Contributor Information
Chuan-peng Liu, Email: liucp74@hotmail.com.
Cheng Luo, Email: cluo@simm.ac.cn.
Supplementary information
The online version of this article (10.1038/s41401-019-0256-2) contains supplementary material, which is available to authorized users.
References
- 1.Bannister AJ, Kouzarides T. The CBP co-activator is a histone acetyltransferase. Nature. 1996;384:641–3. doi: 10.1038/384641a0. [DOI] [PubMed] [Google Scholar]
- 2.Raisner R, Kharbanda S, Jin L, Jeng E, Chan E, Merchant M, et al. Enhancer activity requires CBP/P300 bromodomain-dependent histone H3K27 acetylation. Cell Rep. 2018;24:1722–9. doi: 10.1016/j.celrep.2018.07.041. [DOI] [PubMed] [Google Scholar]
- 3.Das C, Roy S, Namjoshi S, Malarkey CS, Jones DNM, Kutateladze TG, et al. Binding of the histone chaperone ASF1 to the CBP bromodomain promotes histone acetylation. Proc Natl Acad Sci USA. 2014;111:E1072–E81. doi: 10.1073/pnas.1319122111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weinert BT, Narita T, Satpathy S, Srinivasan B, Hansen BK, Scholz C, et al. Time-resolved analysis reveals rapid dynamics and broad scope of the CBP/p300 acetylome. Cell. 2018;174:231–44 e12. doi: 10.1016/j.cell.2018.04.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Iyer NG, Ozdag H, Caldas C. P300/CBP and cancer. Oncogene. 2004;23:4225–31. doi: 10.1038/sj.onc.1207118. [DOI] [PubMed] [Google Scholar]
- 6.Dancy BM, Cole PA. Protein lysine acetylation by p300/CBP. Chem Rev. 2015;115:2419–52. doi: 10.1021/cr500452k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bedford DC, Brindle PK. Is histone acetylation the most important physiological function for CBP andp300? Aging (Albany NY) 2012;4:247–55. doi: 10.18632/aging.100453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Theodoulou NH, Tomkinson NC, Prinjha RK, Humphreys PG. Clinical progress and pharmacology of small molecule bromodomain inhibitors. Curr Opin Chem Biol. 2016;33:58–66. doi: 10.1016/j.cbpa.2016.05.028. [DOI] [PubMed] [Google Scholar]
- 9.Giles RH, Peters DJ, Breuning MH. Conjunction dysfunction: CBP/p300 in human disease. Trends Genet. 1998;14:178–83. doi: 10.1016/S0168-9525(98)01438-3. [DOI] [PubMed] [Google Scholar]
- 10.Filippakopoulos P, Knapp S. Targeting bromodomains: epigenetic readers of lysine acetylation. Nat Rev Drug Disco. 2014;13:337–56. doi: 10.1038/nrd4286. [DOI] [PubMed] [Google Scholar]
- 11.Xu M, Unzue A, Dong J, Spiliotopoulos D, Nevado C, Caflisch A. Discovery of CREBBP bromodomain inhibitors by high-throughput docking and hit optimization guided by molecular dynamics. J Med Chem. 2016;59:1340–9. doi: 10.1021/acs.jmedchem.5b00171. [DOI] [PubMed] [Google Scholar]
- 12.Conery AR, Centore RC, Neiss A, Keller PJ, Joshi S, Spillane KL, et al. Bromodomain inhibition of the transcriptional coactivators CBP/EP300 as a therapeutic strategy to target the IRF4 network in multiple myeloma. Elife. 2016;5:e19432. doi: 10.7554/eLife.19432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vervoorts J, Luscher-Firzlaff JM, Rottmann S, Lilischkis R, Walsemann G, Dohmann K, et al. Stimulation of c-MYC transcriptional activity and acetylation by recruitment of the cofactor CBP. EMBO Rep. 2003;4:484–90. doi: 10.1038/sj.embor.embor821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Faiola F, Liu XH, Lo SY, Pan SQ, Zhang KL, Lymar E, et al. Dual regulation of c-Myc by p300 via acetylation-dependent control of Myc protein turnover and coactivation of Myc-induced transcription. Mol Cell Biol. 2005;25:10220–34. doi: 10.1128/MCB.25.23.10220-10234.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Crawford TD, Romero FA, Lai KW, Tsui V, Taylor AM, de Leon Boenig G, et al. Discovery of a potent and selective in vivo probe (GNE-272) for the bromodomains of CBP/EP300. J Med Chem. 2016;59:10549–63. doi: 10.1021/acs.jmedchem.6b01022. [DOI] [PubMed] [Google Scholar]
- 16.Spiliotopoulos D, Zhu J, Wamhoff EC, Deerain N, Marchand JR, Aretz J, et al. Virtual screen to NMR (VS2NMR): Discovery of fragment hits for the CBP bromodomain. Bioorg Med Chem Lett. 2017;27:2472–8. doi: 10.1016/j.bmcl.2017.04.001. [DOI] [PubMed] [Google Scholar]
- 17.Jin L, Garcia J, Chan E, de la Cruz C, Segal E, Merchant M, et al. Therapeutic targeting of the CBP/p300 bromodomain blocks the growth of castration-resistant prostate cancer. Cancer Res. 2017;77:5564–75. doi: 10.1158/0008-5472.CAN-17-0314. [DOI] [PubMed] [Google Scholar]
- 18.Picaud S, Fedorov O, Thanasopoulou A, Leonards K, Jones K, Meier J, et al. Generation of a selective small molecule inhibitor of the CBP/p300 bromodomain for leukemia therapy. Cancer Res. 2015;75:5106–19. doi: 10.1158/0008-5472.CAN-15-0236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chekler EL, Pellegrino JA, Lanz TA, Denny RA, Flick AC, Coe J, et al. Transcriptional profiling of a selective CREB binding protein bromodomain inhibitor highlights therapeutic opportunities. Chem Biol. 2015;22:1588–96. doi: 10.1016/j.chembiol.2015.10.013. [DOI] [PubMed] [Google Scholar]
- 20.Sachchidanand, Resnick-Silverman L, Yan S, Mutjaba S, Liu WJ, Zeng L, et al. Target structure-based discovery of small molecules that block human p53 and CREB binding protein association. Chem Biol. 2006;13:81–90. doi: 10.1016/j.chembiol.2005.10.014. [DOI] [PubMed] [Google Scholar]
- 21.Xiang Q, Wang C, Zhang Y, Xue X, Song M, Zhang C, et al. Discovery and optimization of 1-(1H-indol-1-yl)ethanone derivatives as CBP/EP300 bromodomain inhibitors for the treatment of castration-resistant prostate cancer. Eur J Med Chem. 2018;147:238–52. doi: 10.1016/j.ejmech.2018.01.087. [DOI] [PubMed] [Google Scholar]
- 22.Hay DA, Fedorov O, Martin S, Singleton DC, Tallant C, Wells C, et al. Discovery and optimization of small-molecule ligands for the CBP/p300 bromodomains. J Am Chem Soc. 2014;136:9308–19. doi: 10.1021/ja412434f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bronner SM, Murray J, Romero FA, Lai KW, Tsui V, Cyr P, et al. A unique approach to design potent and selective cyclic adenosine monophosphate response element binding protein, binding protein (CBP) inhibitors. J Med Chem. 2017;60:10151–71. doi: 10.1021/acs.jmedchem.7b01372. [DOI] [PubMed] [Google Scholar]
- 24.Popp TA, Tallant C, Rogers C, Fedorov O, Brennan PE, Muller S, et al. Development of selective CBP/P300 benzoxazepine bromodomain inhibitors. J Med Chem. 2016;59:8889–912. doi: 10.1021/acs.jmedchem.6b00774. [DOI] [PubMed] [Google Scholar]
- 25.Romero FA, Taylor AM, Crawford TD, Tsui V, Cote A, Magnuson S. Disrupting acetyl-lysine recognition: Progress in the development of bromodomain inhibitors. J Med Chem. 2016;59:1271–98. doi: 10.1021/acs.jmedchem.5b01514. [DOI] [PubMed] [Google Scholar]
- 26.Hay D, Fedorov O, Filippakopoulos P, Martin S, Philpott M, Picaud S, et al. The design and synthesis of 5-and 6-isoxazolylbenzimidazoles as selective inhibitors of the BET bromodomains. Medchemcomm. 2013;4:140–4. doi: 10.1039/C2MD20189E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Breen ME, Mapp AK. Modulating the masters: chemical tools to dissect CBP and p300 function. Curr Opin Chem Biol. 2018;45:195–203. doi: 10.1016/j.cbpa.2018.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang JH, Chung TDY, Oldenburg KR. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J Biomol Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 29.Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, et al. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem. 2004;47:1739–49. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 30.Lu T, Hu JC, Lu WC, Han J, Ding H, Jiang H, et al. Identification of small molecule inhibitors targeting the SMARCA2 bromodomain from a high-throughput screening assay. Acta Pharm Sin. 2018;39:1544–52. doi: 10.1038/aps.2017.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen S, Wang Y, Zhou W, Li S, Peng J, Shi Z, et al. Identifying novel selective non-nucleoside DNA methyltransferase 1 inhibitors through docking-based virtual screening. J Med Chem. 2014;57:9028–41. doi: 10.1021/jm501134e. [DOI] [PubMed] [Google Scholar]
- 32.Macarron R, Banks MN, Bojanic D, Burns DJ, Cirovic DA, Garyantes T, et al. Impact of high-throughput screening in biomedical research. Nat Rev Drug Disco. 2011;10:188–95. doi: 10.1038/nrd3368. [DOI] [PubMed] [Google Scholar]
- 33.Plotnikov AN, Yang S, Zhou TJ, Rusinova E, Frasca A, Zhou MM. Structural insights into acetylated-histone H4 recognition by the bromodomain-PHD finger module of human transcriptional coactivator CBP. Structure. 2014;22:353–60. doi: 10.1016/j.str.2013.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zeng L, Zhang Q, Gerona-Navarro G, Moshkina N, Zhou MM. Structural basis of site-specific histone recognition by the bromodomains of human coactivators PCAF and CBP/p300. Structure. 2008;16:643–52. doi: 10.1016/j.str.2008.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hugle M, Lucas X, Ostrovskyi D, Regenass P, Gerhardt S, Einsle O, et al. Beyond the BET family: Targeting CBP/p300 with 4-acyl pyrroles. Angew Chem Int Ed Engl. 2017;56:12476–80. doi: 10.1002/anie.201705516. [DOI] [PubMed] [Google Scholar]
- 36.Goodman RH, Smolik S. CBP/p300 in cell growth, transformation, and development. Genes Dev. 2000;14:1553–77. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.