

Abstract
Many key residues, which mediate the interaction between SARS-CoV2 spike glycoprotein (S protein) and human ACE2 receptor, have been reviewed using the SARS-CoV2 S spike protein with human ACE2 complex. The initial SARS-CoV2 S protein and ACE2 protein complex structure is formed by RBD structure of SARS-CoV2 S protein and ACE2 protein. However, the cryo-EM structure study targeting SARS-Cov S protein with human ACE2 complex has shown that there exist different binding conformations during the binding process facing ACE2 protein. It suggests the interaction between SARS-CoV2 S spike protein complex might have different binding conformations, which request full-length of SARS-CoV2 S protein complex in the structure-functional analysis. In this study, we built a full-length SARS-CoV2 S protein with human ACE2 complex by computational methods. Residues K31, H34, E35 in ACE2 protein were showed both in our full-length model and RBD structure model, which recognized as critical residues in previous studies. Surprisingly, ACE2 residues E564, R559, N556 were only found participating in the interaction of our full-length model, which suggested the full-length model has bigger binding interface. This finding was further supported by the interaction network of full-length model and RBD model. Meanwhile, the method bias was taken into consideration. Eventually, the MM-PBSA results showed the full-length model had a stronger binding free energy (almost 5-fold) than the RBD structure model of SARS-CoV2 S spike protein complex. In computational level, we present a stronger binding model containing a full-length structure of SARS-CoV2 S protein with ACE2 complex.
Keywords: Spike protein, SARS-CoV2, ACE2, MD, MM-PBSA
Graphical abstract
Highlights
-
•
In this study, we built a full-length SARS-Cov-2 S protein with human ACE2 complex by computational methods, which might present the bigger binding info.
-
•
Residues K31, H34, E35 in ACE2 protein were showed as critical residues in previous studies in our full-length model and RBD structure model.
-
•
ACE2 residues E564, R559, N556 were found in the interaction of our full-length model.
-
•
The full-length model had a stronger binding free energy (almost 5-fold) than the RBD structure model.
-
•
In computational level, we present a stronger binding model containing a full-length structure of SARS-CoV-2 S protein with ACE2 complex.
1. Introduction
In early 2020, a novel coronavirus named SARS-CoV2 (COVID-19) has resulted in a global health issue with its high infectious ability and lethal consequence. It has become a continuous threat to the wellbeing of human life which has made this RNA virus to be an emergency subject [[1], [2], [3], [4]]. It is believed that the spike glycoprotein (S protein) on the virion surface mediates receptor recognition and host selectivity [5]. Meanwhile, evidence has shown the SARS-Cov and SARS-CoV2 spike proteins interact with angiotensin-converting enzyme 2 (ACE2) [6,7]. Previously, the study of cryo-EM structure has reviewed the different conformation states of SARS-Cov S protein and further confirmed that the up conformation of receptor-binding domain (RBD) is required for ACE2 binding [8]. This full-length model has shown the binding process of ACE2 protein might request full-length structure of S protein to carry on binding procedure between virus and human. In addition, only the RBD structure of SARS-COVID-2 S protein complex with human ACE2 has been solved by X-ray diffraction 2.68 Å (code: 6VW1) [9]. Based that, several structure studies have located some key residues mediating the spike protein and ACE2 protein and ACE2 residue K31, E35, M82, K353, Q24, D30, Y41, Y83, R357 are multiple showed in these studies [7,[9], [10], [11], [12]]. And the binding pocket of ACE2 could be initially located. However, there still exist a need of full-length model to ensure that all the binding interfaces were under fully consideration Hence, the model containing full-length SARS-CoV2 S protein targeting human ACE2 might present the bigger binding info than RBD complex model.
With computational methods, we built a complex containing full-length SARS-CoV2 S protein with human ACE2, shown in Fig.S1. According to our results, we further predicted the interaction between SARS-CoV2 S protein and human ACE2 might mediate by inside and outside the RBD structure of SARS-COVID-2 S protein together. To avoid the method bias, we used the same method to predict ACE2 protein structure and full-length structure of SARS-Cov S protein complex and key residues were found as well as previous studies. Meanwhile, we calculated the binding free energy of RBD structure and full-length structure of SARS-CoV2 S protein complex to find the better computational model. This study might provide further information about SARS-Cov and SARS-CoV2 targeting human ACE2.
2. Material and methods
2.1. Template and sequence
To carry on docking and MD simulations, the SARS-Cov and ACE2 complex structure, ACE2-bound conformation 1 (code: 6ACG), was used as a template of SARS-CoV2 bound human ACE2 complex [13]. Structure of SARS-CoV2 chimeric receptor-binding domain complexed with its receptor human ACE2 (code: 6VW1) was used as a control group [8]. The SARS-CoV2 sequence used for the modelling process and binding was obtained from NCBI (NC_045512.2) [14]. The algorithm of alignment was ClustalW server (https://www.Genome.Jp/tools-bin/clustalw) and ESPriPt sever (espript. Ibcp. fr/ ESPript/ cgi- bin/ ESPript. cgi) [15].
2.2. Protein modelling
Homology model of SARS-CoV2 was built through server [16]. Several models were built and the quality of them was evaluated. The best model was obtained using the template 6ACG. Then, by using rosetta2019 program, the initial structure of SARS-CoV2 was refined for docking [17].
2.3. Protein-protein docking
The Rosetta program was used for docking to further locate the binding interface [18]. The full-length S protein complexes containing SARS-CoV2 or SARS-Cov with human ACE2 complexes were carried on docking for 30 times. Docking results were evaluated by ref2015 score-function of Rosetta program. And no significant shift was observed in 30 results, thus the best model was obtained for MD simulation.
2.4. MD simulation and binding free energy
Complexes were carried on MD simulations using the GROMACS program [19,20]. The AMBER ff99sb force field was used in MD simulation. And all models were dissolved with TIP3P water models. All periodic boxes were set to ensure that the complex center was at least 0.5 nm away from the wall. The electroneutral system was separately guaranteed by adding the Na+ or CL− separately. After that, all systems were minimized by running the steepest descent method with step 1E-3 ps with 10,000 steps. Then, to achieve equilibration, V-rescale method and Berendsen method were used for 100 ps run. Finally, all well-prepared systems were performed under 300 K and 1 bar. The full-length SARS-CoV2 S protein complex was calculated for 30 ns and so did full-length SARS-Cov S protein complex. And the structure of SARS-CoV2 chimeric receptor-binding domain complexed with its receptor human ACE2 was calculated for 100 ns under the same condition. The two full-length complexes were calculated 30 ns because each system contained 471,596 atoms and 472,054 atoms, while the RBD structure of SARS-CoV2 S protein system contained 86,388 atoms. Full-length models were almost 6-fold bigger than RBD structure complex. The most common structures were analyzed by cluster process which presented the binding state of each complex. Then, the MMPBSA.py package of AMBERTOOLS18 program was used for MM-PBSA calculation [21]. Further, we calculated the interaction network in full-length model and RBD model by using RING server, which was designed to analyze the interaction between ligands and receptor [22]. The algorithm firstly generated the network by a list of residue-residue pairs to present interaction based on distance measurements. Then the algorithm characterized contact info of input structure by using the specific type of interaction.
3. Results
3.1. Homology modelling and docking
The sequence alignment results suggested that the sequence identity, between the full-length SARS-CoV2 S protein and the SARS-Cov S protein (code: 6ACG), was 75.12%, shown in Fig.S2-S3. Thus, the initial model was built by using the template 6ACG. To evaluate and refine the structure, Rosetta program was used in this study. The human ACE2 protein was docking to refined structure, firstly optimized the rigid body of complex, then optimized the ACE2 protein backbone to generate the low-resolution structure. Eventually, the side-chain rotamers were refined to generate the high-resolution structure. The top 10 docking results were listed in Table 1 . The best docking result 2019nCoV_ACE2_Model_2_0024 was used for further MD simulation. The detail of docking was shown in Fig.S4.
Table 1.
The top 10 models from evaluations of full-length SARS-CoV2 S protein with human ACE2. The total_score presented the quality of each complex. I_sc present the interface quality in each complex while pep_sc present the ACE2 quality of each complex.
| Number | Total_score | I_sc | pep_sc | Description |
|---|---|---|---|---|
| 1 | −1880.688 | −22.979 | −963.38 | 2019nCoV_ACE2_Model_2_0024 |
| 2 | −1868.721 | −19.983 | −993.26 | 2019nCoV_ACE2_Model_2_0002 |
| 3 | −1861.189 | −16.55 | −944.202 | 2019nCoV_ACE2_Model_2_0027 |
| 4 | −1826.062 | −14.535 | −955.218 | 2019nCoV_ACE2_Model_2_0012 |
| 5 | −1825.966 | −38.261 | −945.635 | 2019nCoV_ACE2_Model_2_0005 |
| 6 | −1818.623 | −13.703 | −937.926 | 2019nCoV_ACE2_Model_2_0011 |
| 7 | −1813.706 | −28.912 | −930.05 | 2019nCoV_ACE2_Model_2_0010 |
| 8 | −1807.027 | −23.033 | −949.358 | 2019nCoV_ACE2_Model_2_0016 |
| 9 | −1805.228 | −16.303 | −946.937 | 2019nCoV_ACE2_Model_2_0009 |
| 10 | −1792.69 | −24.743 | −921.192 | 2019nCoV_ACE2_Model_2_0028 |
3.2. MD simulation and binding free energy
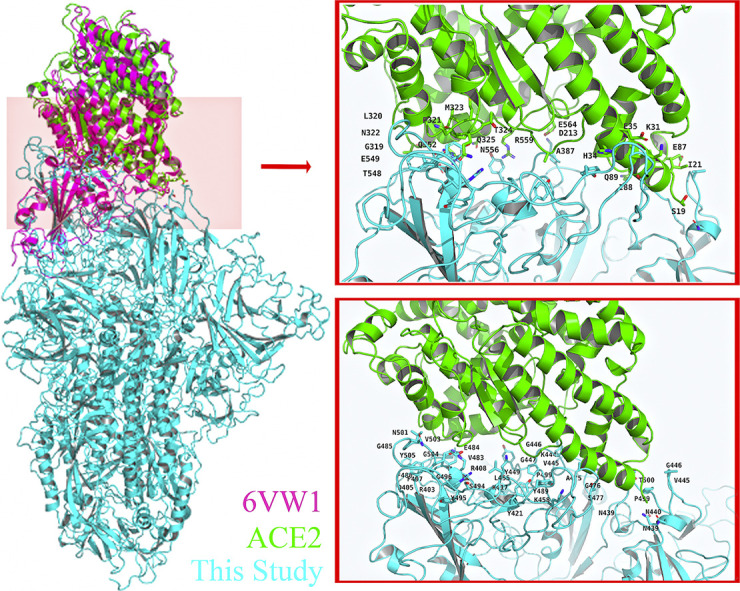
GROMACS2019 program was used to obtain the trajectory. The cluster structure of the MD simulation trajectory was analyzed and used to present the binding free energy between full-length SARS-CoV2 S protein and RBD SARS-CoV2 S protein with ACE2, separately. And MM-PBSA package from AMBERTOOLS-18 program was used to analyze the cluster structure of trajectory. The root-mean-square-deviation (RMSD) showed the systems were stable since 20 ns, showed in Fig. 1 . The 100 ns MD cluster structure reviewed ACE2 residues H34, Y41, Q42, D30, K31, K353, R357, E329, Q325 were involved in the interaction, while the full-length model suggested not only ACE2 residues K31, H34, Q325 but also residues E564, N556, R559 in ACE2 have a participant in the interaction, shown in Fig. 2 . These residues further suggested that there might be more residues involving in the binding pocket, which suggested full-length SARS-CoV2 S protein with ACE2 complex might be a better model with rational computation evidence. At last, with the MM-PBSA package from AMBER, the binding free energy of the full-length model and RBD model were calculated. And the energy results showed that the binding free energy of full-length SARS-CoV2 S protein complex is 449.06 KJ/Mol in GB level and 472.66 KJ/Mol in PB level, while the binding free energy of RBD SARS-CoV2 S protein complex is −1897.53 KJ/Mol in GB level and − 1990.53 KJ/Mol in PB level, shown in Fig. 3 . We further calculated the interaction network in the full-length model and RBD model. The results showed in Fig. 4 . The interaction network of RBD model showed that the S protein residues in 19–355 position were involved in interaction with ACE2 protein. Interestingly, the interaction network of full-length model showed that the S protein residues in 549–559 position plus 31–325 position were involved in interaction with ACE2 protein as well, which had shown a bigger interaction network than RBD model.
Fig. 1.

RMSD results of the backbone of SARS-CoV2 (COVID-19), SARS-Cov and RBD complex 6VW1 were present. Again, two full-length complexes were calculated 30 ns because they were 5-fold bigger than 6VW1 system.
Fig. 2.

ACE2 Residues in full-length SARS-CoV2 S protein (A), full-length SARS-Cov S protein (B) and RBD structure of SARS-Cov 2 (C) were present. S protein residues in full-length SARS-CoV2 S protein (D), full-length SARS-Cov S protein (E) and RBD structure of SARS-Cov 2 (F) were present as well. Human ACE2 protein was marked in green. SARS-CoV2 S protein was marked in cyan and SARS-Cov S protein was marked in orange. In the result of RBD model 6VW1, the 100 ns MD structure reviewed ACE2 residues H34, Y41, Q42, D30, K31, K353, R357, E329, Q325 were involved in the interaction, while the full-length model suggested not only ACE2 residues K31, H34, Q325 but also residue E564, N556, R559 in ACE2 might mediate the interaction. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Fig. 3.

The binding free energy of the full-length model and RBD model were calculated. And the energy results showed that the binding free energy of the full-length SARS-CoV2 S protein complex is 449.06 KJ/Mol in GB level and 472.66 KJ/Mol in PB level, while the binding free energy of RBD SARS-CoV2 S protein complex is −1897.53 KJ/Mol in GB level and − 1990.53 KJ/Mol in PB level.
Fig. 4.

The network analysis results were calculated. The interaction network of RBD model (A) showed that the S protein residues in 30–329 amino acid sequence position complexed with ACE2 protein. And the interaction network of full-length model (B) showed that the S protein residues in 548–559 acid sequence position were interacted with ACE2 protein, which further suggested the interaction network in full-length model was bigger than RBD model.
4. Discussion
Key residues in RBD structure of S protein have been identified in SARS-CoV2 S protein RBD structure with ACE2 complex (code: 6VW1). Residue Q498 in SARS-CoV2 S protein has been found multiple appearances in structure study [11,23]. While, other residues have been mentioned (such as N487, Y489, Y453, G496, Y449, T500, G 502, Y505) in SARS-CoV2 S protein as well [9]. Residues K353 and M82 in human ACE2 have been found as the most common residues when we compared recently studies [7,[9], [10], [11],13]. Meanwhile, some residues Q24, D30, K31, H34, E35, Y41, Y83, R357 have multiple participations in some of ACE2 interface structure studies [7,[9], [10], [11]]. These residues together have provided valuable information to study the mechanism of binding between RBD structure of SARS-CoV2 S protein and ACE2 protein, which emphasized that ACE2 residues around K31, M82, K353 might be critical residues. In this study, we calculated the 100 ns trajectory of RBD structure of SARS-CoV2 S protein (code: 6VW1). And the cluster structure also showed residues Q24, D30, K31, H34, E35, Y41, M82, Y83, K353, R357 were mediating the binding interface, which further confirmed key residues again. However, the binding interface had a shift in the 30 ns cluster structure of full-length model when we compared the full-length structure with RBD structure of SARS-CoV2 S protein complex. And residues K31, H34, E35 were found in our full-length complex model as well, which again reviewed these residues might be critical.
Surprisingly, residues E564, R559, N556 were found appearance in the binding of full-length SARS-CoV2 S protein complex due to the shift interface. These unfamiliar key residues were not showed in mostly previous structure studies but were reviewed by R559S mutation ddG study [12]. We used the same computational process to analysis full-length SARS-CoV S protein with ACE2 complex (code: 6ACG) as well to get rid of method bias. Interestingly, key residues were characterized as residues K31 and K353 in our 30 ns full-length SARS-CoV S protein with ACE2 complex. And this result was similar to previous structure studies [7,[9], [10], [11],13]. This finding suggested the shift of binding interface in our RBD and full-length SARS-CoV2 S protein complex was probably because of the different structure integrity. To further review the different binding affinity between the full-length and RBD structure of SARS-CoV2 S protein targeting ACE2, we calculated the binding free energy between the cluster structure of 30 ns full-length model and the 100 ns RBD model. The results showed the full-length model has a stronger binding free energy (almost 5-fold) than RBD structure model, which further reviewed the importance of structural integrity facing SARS-CoV2 S protein with ACE2 complex. In the computational full-length model, the S protein binding complex of SARS-CoV2 had a bigger interface than SARS-CoV targeting ACE2. This study could provide further information towards SARS-CoV2 S protein with ACE2 complex. As well as the interaction network of full-length model showed that the S protein residues in 548–559 acid sequence position were interacted with ACE2 protein together with residues in 31–325 position, which further suggested the interaction network in full-length model was bigger than RBD model. With our computational results, it seems like the electrostatic residue E564, R559, N556 might have a bigger contribution to interaction because the full-length model (which involving residue E564, R559, N556) is more rational than RBD model (which are not involving residue E564, R559, N556) in computational level.
5. Conclusion
Different binding conformation states of SARS-CoV S protein in cryo-EM structure study targeting human ACE2 complex suggested that SARS-CoV2 S spike protein complex might have different binding conformations targeting same protein. It requested full-length SARS-CoV-2 S spike protein to further present potential binding interfaces. In this study, ACE2 residues K31, H34, E35 were not only multiple showed in our full-length model and RBD structure of SARS-CoV2 S spike protein with human ACE2 complex, as well as other studies reviewed. Surprisingly, ACE2 residues E564, R559, N556 were found participating in the interaction of our full-length model but not in the RBD model. The MM-PBSA results showed the full-length model had a stronger binding free energy (almost 5-fold) than the RBD structure model. These findings further conformed the importance of structural integrity in structure function relationship study targeting SARS-CoV2 S protein with ACE2 complex. Overall, we concluded that the interaction of SARS-CoV2 S protein with ACE2 complex might be mainly mediated by ACE2 residues around K31, H34, E35 besides E564, R559, N556. And in computational level, we present a stronger binding model containing a full-length structure of SARS-CoV2 S protein with human ACE2 complex.
Author statement
Under supervision by Lirui, Lirui performed sample preparation and data analysis. LiHaonan developed mechanics modelling theory of this paper. Chenwanyi contributed language part and in charge of some language revision.
All authors read and contributed to the manuscript.
Declaration of Competing Interest
All authors declare there are no financial/commercial conflicts of interest.
Acknowledgments
This work was supported by colleagues and friends
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.bpc.2020.106472.
Appendix A. Supplementary data
Supplementary material
References
- 1.Cerofolini L. Orientation of immobilized antigens on common surfaces by a simple computational model: exposition of SARS-CoV-2 spike protein RBD epitopes. Biophys. Chem. 2020;265 doi: 10.1016/j.bpc.2020.106441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chakraborty H., Bhattacharjya S. Mechanistic insights of host cell fusion of SARS-CoV-1 and SARS-CoV-2 from atomic resolution structure and membrane dynamics. Biophys. Chem. 2020;265 doi: 10.1016/j.bpc.2020.106438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li W. Delving deep into the structural aspects of a furin cleavage site inserted into the spike protein of SARS-CoV-2: a structural biophysical perspective. Biophys. Chem. 2020;264 doi: 10.1016/j.bpc.2020.106420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rao P. Reckoning a fungal metabolite, pyranonigrin a as a potential Main protease (M(pro)) inhibitor of novel SARS-CoV-2 virus identified using docking and molecular dynamics simulation. Biophys. Chem. 2020;264 doi: 10.1016/j.bpc.2020.106425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Simmons G. Proteolytic activation of the SARS-coronavirus spike protein: cutting enzymes at the cutting edge of antiviral research. Antivir. Res. 2013;100(3):605–614. doi: 10.1016/j.antiviral.2013.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhixin L., Xiuli W., Jian L., Jing Y., Huabing T., Jianyong Z., Qiwei Z., Jiangguo W., Long L. Composition and dicergence of coronavirus spike proteins and host ACE2 receptores predict potential intermediate hosts of SARS-CoV-2. J. Med. Virol. 2020:1–15. doi: 10.1002/jmv.25726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Junwen L. Y.L., Xiaolu J., Leiliang Z., Spike protein recognition of mammalian ACE2 predicts the host range and an optimized ACE2 for SARS-CoV-2 infection. Biochem. Biophys. Res. Commun. 2020:1–5. doi: 10.1016/j.bbrc.2020.03.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Song W. Cryo-EM structure of the SARS coronavirus spike glycoprotein in complex with its host cell receptor ACE2. PLoS Pathog. 2018;14(8) doi: 10.1371/journal.ppat.1007236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jun L., Jinfang Y., Sisi S., Huan Z., Shilong F., Qi Z., Xuanling S., Qisheng W., Linqi Z., Xingquan W. Structure of the SARS-CoV-2 spike receptorbinding domain bound to the ACE2 receptor. Nature. 2020:1–8. doi: 10.1038/s41586-020-2180-5. [DOI] [PubMed] [Google Scholar]
- 10.Ortega J.T. Role of changes in SARS-CoV-2 spike protein in the interaction with the human ACE2 receptor: an in silico analysis. EXCLI J. 2020;19:410–417. doi: 10.17179/excli2020-1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Renhong Y., Yaning L., Lu X., Yingying G., Qiang Z. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science. 2020;367:1444–1448. doi: 10.1126/science.abb2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hussain M. Structural variations in human ACE2 may influence its binding with SARS-CoV-2 spike protein. J. Med. Virol. 2020;92:1580–1586. doi: 10.1002/jmv.25832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shang J. Structural basis of receptor recognition by SARS-CoV-2. Nature. 2020;581:221–224. doi: 10.1038/s41586-020-2179-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang W.E. RT-LAMP for rapid diagnosis of coronavirus SARS-CoV-2. Microb. Biotechnol. 2020;13:950–961. doi: 10.1111/1751-7915.13586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Robert X., Gouet P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014;42 doi: 10.1093/nar/gku316. (Web Server issue): p. W320-4) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Waterhouse A. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 2018;46(W1):W296–W303. doi: 10.1093/nar/gky427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alford R.F. The Rosetta all-atom energy function for macromolecular Modeling and design. J. Chem. Theory Comput. 2017;13(6):3031–3048. doi: 10.1021/acs.jctc.7b00125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tyka M.D. Alternate states of proteins revealed by detailed energy landscape mapping. J. Mol. Biol. 2011;405(2):607–618. doi: 10.1016/j.jmb.2010.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rakhshani H., Dehghanian E., Rahati A. Enhanced GROMACS: toward a better numerical simulation framework. J. Mol. Model. 2019;25(12) doi: 10.1007/s00894-019-4232-z. [DOI] [PubMed] [Google Scholar]
- 20.Makarewicz T., Kazmierkiewicz R. Improvements in GROMACS plugin for PyMOL including implicit solvent simulations and displaying results of PCA analysis. J. Mol. Model. 2016;22(5) doi: 10.1007/s00894-016-2982-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tingjun H., Youyong L., Wei W. Assessing the Performance of the MM/PBSA and MM/GBSA Methods. 1. The Accuracy of Binding Free Energy Calculations Based on Molecular Dynamics Simulations. J. Chem. Inf. Model. 2010;51:69–82. doi: 10.1021/ci100275a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Piovesan D., Minervini G., Tosatto S.C. The RING 2.0 web server for high quality residue interaction networks. Nucleic Acids Res. 2016;44(W1):W367–W374. doi: 10.1093/nar/gkw315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tian X. Potent binding of 2019 novel coronavirus spike protein by a SARS coronavirus-specific human monoclonal antibody. Emerg. Microbes Infect. 2020;9(1):382–385. doi: 10.1080/22221751.2020.1729069. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material