

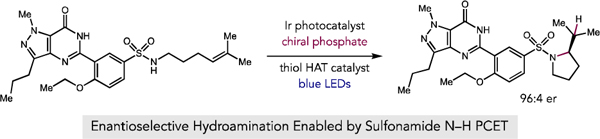
Abstract
An enantioselective, radical-based method for the intramolecular hydroamination of alkenes with sulfonamides is reported. These reactions are proposed to proceed via N-centered radicals formed by proton-coupled electron transfer (PCET) activation of sulfonamide N–H bonds. Non-covalent interactions between the neutral sulfonamidyl radical and a chiral phosphoric acid generated in the PCET event are hypothesized to serve as the basis for asymmetric induction in a subsequent C–N bond forming step, achieving selectivities of up to 98:2 er. These results offer further support for the ability of non-covalent interactions to enforce stereoselectivity in reactions of transient and highly reactive open-shell intermediates.
Graphical Abstract
Non-covalent interactions provide a powerful means to control selectivity in asymmetric transformations, both in nature and in the laboratory.1 However, a lack of clear understanding about how these weak interactions can serve to bind and activate open-shell intermediates has largely precluded their use a control element in the enantioselective reactions of free radical species.2–4 Seeking address this deficit, our group has become interested in the use of proton-coupled electron transfer (PCET) as a platform for developing catalytic asymmetric free radical chemistry. Oxidative PCET—which effects formal bond homolysis through the joint movement of a proton and electron to a Brønsted base and one-electron oxidant, respectively5—generally requires pre-equilibrium hydrogen bonding between the reactive E–H bond of the substrate and the Brønsted base prior to the electron transfer step. In addition to controlling site-selectivity,6 this H–bond interface can remain intact following the PCET event, furnishing a transient hydrogen-bonded complex between the nascent free radical and the conjugate acid of the Brønsted base.7 In principle, this association can then serve as a basis for achieving asymmetric induction in subsequent bond forming steps when chiral bases are employed.
This manner of asymmetric PCET activation has been demonstrated previously in a reductive aza-pinacol cyclization and in the oxidative activation of indoles en route to the formation of pyrolloindolines (Figure 1a).8 In these examples, the neutral ketyl radical and indole radical cation remain associated with a chiral anionic phosphate, wherein the negatively charged acceptor was proposed to increase the strength of the post-PCET hydrogen bonding interaction. We questioned whether this blueprint could be extended further to enable high enantioselectivity in the reactions of neutral free radical intermediates associated with neutral hydrogen bond donors. To evaluate this hypothesis, we chose to investigate enantioselective variants of a recently reported hydroamination of alkenes with sulfonamides.9,10 This transformation proceeds through a key sulfonamidyl radical intermediate generated via PCET activation of the substrate N–H bond by an excited-state Ir(III) oxidant and a dialkyl phosphate base. This electrophilic nitrogen-centered radical then undergoes addition to a pendant olefin to furnish a new C–N bond.11 We hypothesized that if chiral phosphate bases were employed, then a successor H-bonding complex between the chiral phosphoric acid and the neutral sulfonamidyl radical could nucleate a network of non-covalent interactions that would in turn differentiate competing diastereomeric transition states for C–N bond formation (Figure 1b).7 Here, we report the successful realization of this goal, and preliminary mechanistic observations consistent with the design hypothesis presented above (Figure 1c). To put this effort in context, we note that relatively few methods for catalytic asymmetric C–N bond formation with N-radical intermediates have been reported to date, and all involve strong bonding interactions between the substrates and the chiral catalysts. Meggers and MacMillan13 have reported methods where a free N-radical undergoes addition to an alkene acceptor bound to either a chiral Rh-complex or a chiral enamine, respectively, while Zhang and Chemler have developed approaches using metalloradical complexes.14
Figure 1.
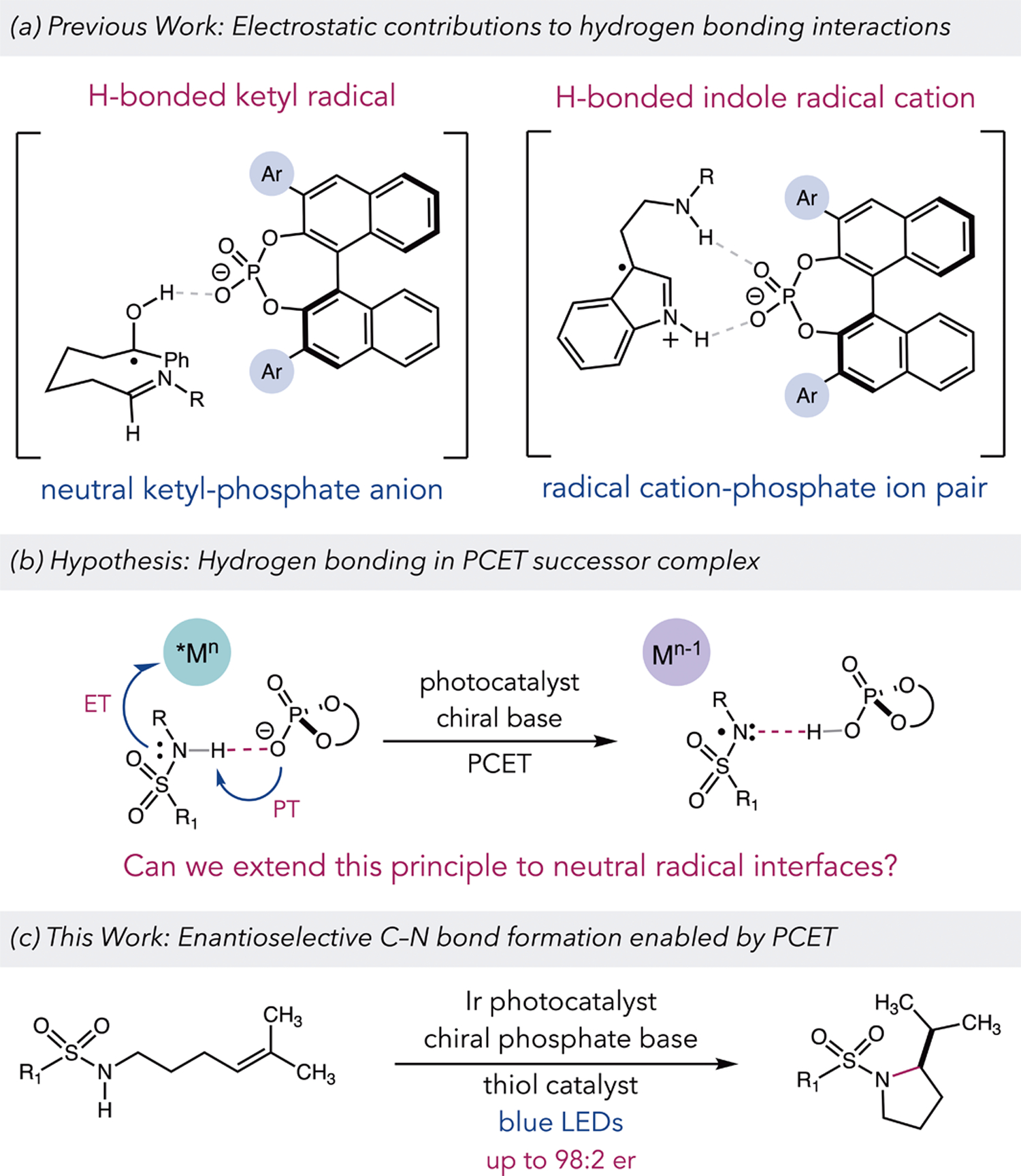
(a) Examples of PCET-enabled enantioselective reactions; (b) Hypothesis for PCET-enabled asymmetric olefin hydroamination. (c) Enantioselective hydroamination with sulfonamides
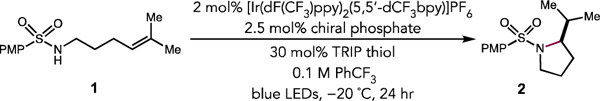
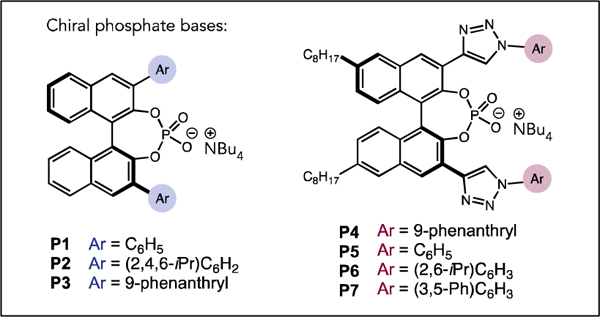
We began by evaluating the cyclization of acyclic 4-methoxyphenyl (PMP) sulfonamide 1 to form pyrrolidine 2 under the previously reported PCET conditions for hydroamination in the presence of a variety of chiral phosphate bases. Phenyl-substituted BINOL phosphate P1 and commercially available TRIP-phosphate P2 provided pyrrolidine product 2 in reasonable yield and low, but measurable levels of enantioselectivity (Table 1, Entries 1 and 2). A modest increase in selectivity and reactivity was observed with 9-phenanthrene substituted P3, affording 2 in 96% yield and 35:65 er (Table 1, Entry 3). Further evaluation of chiral phosphate scaffolds led to an improvement in selectivity with the analogous 1,2,3-triazole containing P4, which provided 2 in 62% yield and 86:14 er (Table 1, Entry 4).15 As this 1,2,3-triazole containing chiral phosphate scaffold provided higher enantioselectivity, we evaluated the effect of substitution pattern on the aryl triazole moiety. The parent phenyl substituted catalyst P5 afforded 2 in 93:7 er. Substituents at the ortho positions (P6) were detrimental to selectivity compared to P5 yielding the product in 87:13 er (Table 1, Entry 5). Of the catalysts examined, the highest selectivities were observed with meta substituents on the arene (See SI, Tables S1 and S2). Of these, phosphate P7 proved optimal, affording the product 2 in 85% yield and 95:5 er (Table 1, Entry 7).
Table 1.
Optimization Studiesa
 | |||
|---|---|---|---|
| Entry | Phosphate | Yield (%) | er |
| 1 | P1 | 54 | 55:45 |
| 2 | P2 | 30 | 52:48 |
| 3 | P3 | 96 | 35:65 |
| 4 | P4 | 62 | 86:14 |
| 5 | P5 | 97 | 93:7 |
| 6 | P6 | 98 | 87:13 |
| 7 | P7 | 85 | 95:5 |
 | |||
| Entry | Change from Entry 7 | Yield (%) | er |
| 8 | room temperature | 93 | 89:11 |
| 9 | thiophrenol H-atom donor | 81 | 94:6 |
| 10 | 10 mol% P7 | 89 | 95:5 |
| 11 | no base | <1 | - |
| 12 | no thiol | <5 | - |
| 13 | no photocatalyst | <1 | - |
| 14 | no light | <1 | - |
Reactions were conducted on a 0.05 mmol scale, and yields determined by NMR analysis relative to an internal standard. Enantioselectivity was determined by HPLC analysis on a chiral stationary phase.
With phosphate P7, we next evaluated the sensitivity of the transformation to variations in other reaction parameters. At room temperature, 2 was formed in 93% yield, but with a lower selectively of 89:11 er (Table 1, entry 8). Further changes to the conditions, such as substituting commercially available thiophenol for TRIP-thiophenol resulted in a similar enantioselectivity of 94:6 er (Table 1, Entry 9). Increasing the catalyst loading of P7 to 10 mol% improved the yield marginally, but afforded no enhancement in selectivity (Table 1, Entry 10). Control reactions excluding base, light, and photocatalyst resulted in no detectable product formation, and only trace product was observed in the absence of the thiol HAT catalyst (Table 1, Entries 11–14).
We then sought to evaluate the reaction scope with respect to both the sulfonamide (products 2–22) and alkene (products 23–27) moieties of the substrate. Uniformly high selectivities were observed upon variation of the para substituent of the sulfonamide arene (2–8). Substituents at the ortho positions (9, 91% yield, 92:8 er & 10, 78% yield, 90:10 er) modestly reduced the observed enantioselectivity, whereas selectivity was retained upon meta substitution (11, 79% yield, 95:5 er). The reaction accommodated benzofuran (12, 92% yield, 96:4 er), thiophene (13, 53% yield, 93:7 er), and thiazole (14, 79% yield, 97:3 er) heterocycles. Benzyl substitution on the sulfonamide led to a modest decrease in selectivity (15, 98% yield, 91:9 er), whereas a longer phenethyl chain was well tolerated (16, 98% yield, 96:4 er). The reaction was also successful for sulfamate ester (17, 87% yield, 92:8 er) and sulfamide (18, 80% yield, 94:6 er) substrates. We further applied this methodology to more complex sulfonamide substrates. Product 19, containing a thioether linked 1,2,4-triazole moiety, was afforded in 84% yield and 87:13 er at room temperature under otherwise standard conditions. Sultiame-derived product 20 was isolated in 89% yield and 96:4 er, and the reaction of a celecoxib-derived sulfonamide provided cyclized product 21 in 83% and 95:5 er. The reaction also tolerated the presence of other hydrogen bonding donor and acceptor functionalities to afford sildenafil derivative 22 in 50% yield and 96:4 er.
The enantioselectivity of the reaction was then probed for a variety of alkene substitution patterns (products 23–27). Cyclohexyl-substituted product 23 was delivered in 98% yield and 94:6 er and protected piperidine-substituted alkene provided the cyclized chiral product 24 in 91% yield and 90:10 er. Cyclobutyl-substituted 25 was afforded in 96% yield and 95:5 er. The conditions also afforded high levels of enantioselectivity for cis and trans disubstituted alkenes. While initial reactivity of these substrates was low at –20 °C, the yields were significantly improved with an increase in reaction temperature to 0 °C and substitution of 15 mol% of TRIP-disulfide for the thiol (See SI, Table S2). Product 26 was afforded in 72% yield and 93:7 er from the cis-disubstituted alkene, while the trans isomer afforded 26 in 93% yield and 95:5 er. A bulkier tert-butyl substituted alkene provided product 27 in 93% yield and 98:2 er from the cis isomer. 27 was also generated in 48% yield and 96:4 er from the corresponding trans isomer of the starting acyclic alkene. Unsymmetrical trisubstituted alkene substrates were also effective with a nerol-derived sulfonamide furnishing 28 in 85% yield as a 1.5:1 mixture of diastereomers, wherein each diastereomer was generated in 96:4 er.16
Seeking to shed light on the enantiodetermining step of this transformation, we conducted an amination experiment where the thiol co-catalyst was removed from solution and several equivalents of methyl vinyl ketone were added (Figure 2). From this reaction carboamination product 29 was isolated in 65% yield and 95:5 er - a nearly identical enantioselectivity to the hydroamination reaction of the same sulfonamide substrate. These results suggest that the common C–N bond forming step is stereoselectivity-determining in both transformations.
Figure 2.
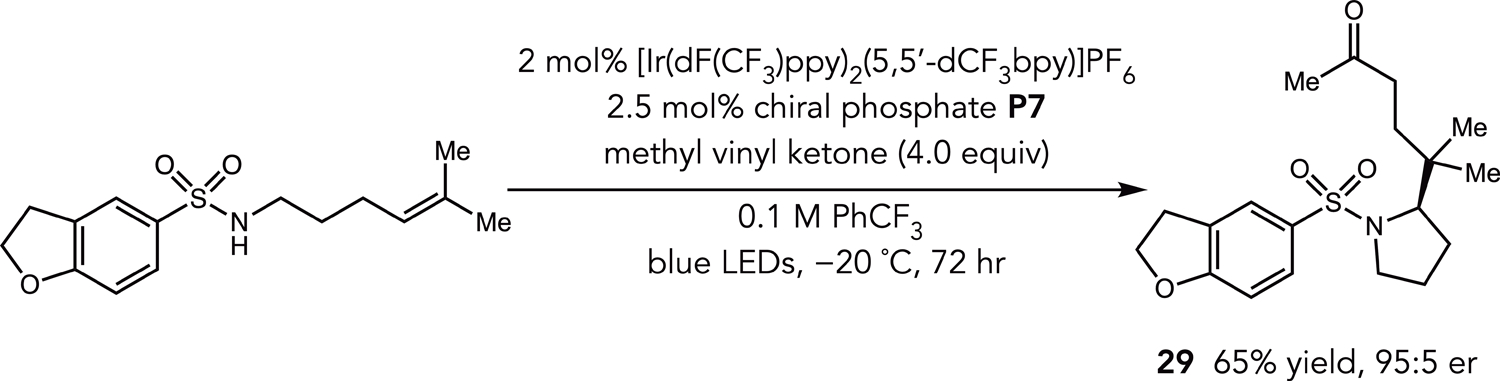
Carboamination Reaction
With this information in hand, we sought to better understand the nature of the interaction between the substrate and the chiral catalyst. While our initial design conjectured a neutral hydrogen bonding interaction resulting from a PCET-generated sulfonamidyl radical, we also considered that an alternative ion-pairing pathway may be responsible for asymmetric induction as previously proposed by Luo and Nicewicz.17 This interaction could arise by an alternative mechanism where the pendant olefin (Ep/2(2-methyl-2-butene) = +1.60 V vs. Fc+/Fc) undergoes an endergonic single electron oxidation by the excited state of the Ir photocatalyst (E1/2 [*Ir(III)/(II)] = +1.30 V vs. Fc+/Fc).18,19 The resulting ion pairing interaction between the alkene radical cation and chiral phosphate anion would then provide a basis for enantioinduction.
In seeking to distinguish between these pathways, we note that previously reported cyclic voltammetry and Stern-Volmer quenching studies were consistent with PCET-activation of sulfonamides under nearly identical conditions using achiral phosphate bases.20 Moreover, in the absence of the phosphate base (Table 1, Entry 11) no cyclized product is detected, and increased loadings of phosphate (Table 1, Entry 10) do not result in higher observed enantioselectivities. These results suggest the absence of a racemic background reaction proceeding through alkene oxidation.21 Furthermore, high reactivity and enantioselectivity were observed in reactions of disubstituted alkenes (products 26–27), for which single electron oxidation is prohibitively endergonic (Ep/2(cyclopentene) = +1.99 V vs. Fc+/Fc) for the Ir complex employed.17
Lastly, an inverse relationship between solvent polarity and enantioselectivity is often been taken as support for an ion pairing mechanism.22 In contrast, we observed that the enantioselectivity of this hydroamination reaction was notably insensitive to solvent dielectric. Nearly identical selectivities are observed for reactions run in either toluene (ε = 2.4, er = 93:7) or acetonitrile (ε = 36.6, er = 94:6), and only a moderate decrease was found for reactions run in highly polar propylene carbonate (ε = 66.2, er = 85:15) (Table 3, Entries 1,5, and 6), though the reactivity was significantly diminsihed.23 This insensitivity to solvent polarity argues strongly against a key role for ionic intermediates in C–N bond forming step.
Table 3.
Enantioselectivity as a Function of Solvent Dielectric
 | ||||
|---|---|---|---|---|
| Entry | Solvent | Yield (%) | er | ε |
| 1 | toluene | 85 | 93:7 | 2.4 |
| 2 | flurobenzene | 77 | 94:6 | 5.5 |
| 3 | tetrahydrofuran | 45 | 94:6 | 7.5 |
| 4 | dichlomethane | 54 | 94:6 | 8.9 |
| 5 | acetonitrile | 15 | 94:6 | 36.6 |
| 6 | N,N-dimethylformamide | trace | 83:17 | 38.3 |
| 7 | propylene carbonate | trace | 83:15 | 66.2 |
Reactions were conducted on a 0.05 mmol scale, and yields determined by NMR analysis relative to an internal standard. Enantioselectivity determined by HPLC analysis on a chiral stationary phase.
In conclusion, we have developed a PCET-based protocol for the asymmetric hydroamination of alkenes with sulfonamides to prepare enantioenriched pyrrolidine products. This method shows high enantioselectivity for a variety of alkene substitution patterns and sulfonamide substrates, including complex drug-derived examples. A variety of observations are consistent with a neutral catalyst-substrate interaction governing selectivity in an enantiodetermining C–N bond forming step. Further work will aim to elucidate the precise interactions underlying the observed selectivity, including the potential role of a post-PCET hydrogen bonding interaction to the nitrogen radical that served as the key design hypothesis in the development of this work. We are optimistic that the results presented here can be extended more broadly to enable the development of other enantioselective reactions of free radical intermediates mediated solely by non-covalent associations.
Supplementary Material
Table 2.
Scope of Enantioselective Amination Reactiona
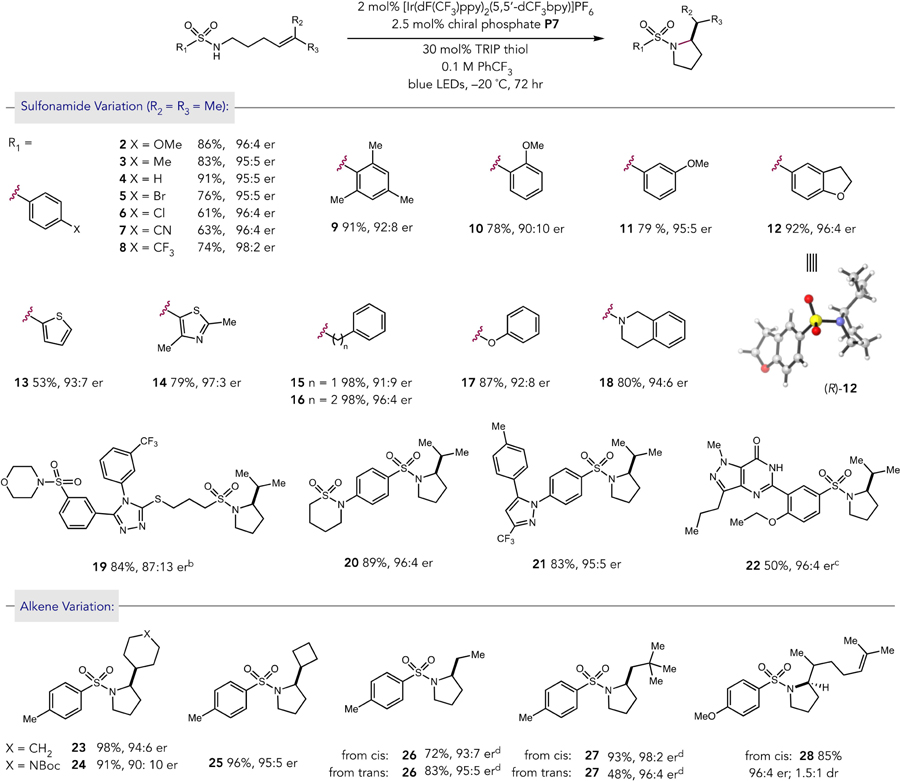 |
Yields and enantioselectivities are for isolated material following chromatography on silica gel and are the average of two experiments. Reactions were conducted on 0.5 mmol scale.
Reaction was run at room temperature.
Reaction was run in dichloromethane.
Reactions were run at 0 °C with substitution of TRIP-disulfide for thiol.
ACKNOWLEDGMENT
Financial support was provided by the NIH (R35 GM134893). Qilei Zhu is thanked for generously providing substrates. J.D. kindly acknowledges the MolDesignS lab for financial support, and also the Research Foundation - Flanders (FWO) for generous funding through Fundamental Research fellowship 11ZY118N and travel grant V416618N.
Footnotes
ASSOCIATED CONTENT
Supporting Information.
The Supporting Information is available free of charge on the ACS Publications website.
Experimental procedures, spectral data, and optical purity determinations (PDF).
Crystal structure of 12 (CIF)
Crystal structure of 14 (CIF)
REFERENCES
- (1).(a) Knowles RR; Jacobsen EN Attractive Noncovalent Interactions in Asymmetric Catalysis: Links Between Enzymes and Small Molecule Catalysts. Proc. Natl. Acad. Sci. U. S. A 2010, 107, 20678–20685. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Davis HJ; Phipps RJ Harnessing non-covalent interactions to exert control over regioselectivity and site-selectivity in catalytic reactions. Chem. Sci 2017, 8, 864–877. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Raynal M; Ballester P; Vidal-Ferran A; van Leeuwen PWNM Supramolecular catalysis. Part 2: artificial enzyme mimics. Chem. Soc. Rev 2014, 43, 1734–1787. [DOI] [PubMed] [Google Scholar]; (d) Taylor MS; Jacobsen EN Asymmetric Catalysis by Chiral Hydrogen-Bond Donors. Angew, Chem. Int. Ed 2006, 45, 1520–1543. [DOI] [PubMed] [Google Scholar]
- (2).(a) For studies of hydrogen bonding to radicals, see: Johnson ER; Salamone M; Bietti M; DiLabio GA Modeling Noncovalent Radical-Molecule Interactions Using Convetional Density-Functional Theory: Beware Erroneous Charge Transfer. J. Phys Chem. A 2014, 117, 947–952. [DOI] [PubMed] [Google Scholar]; (b) Hernández-Soto H; Weinhold F; Francisco JS Radical hydrogen bonding: Origins of stability of radical-molecule complexes. J. Chem. Phys 2007, 127, 164102. [DOI] [PubMed] [Google Scholar]; (c) Johnson ER; DiLabio GA Radicals as Hydrogen Bond Donors and Acceptors. Interdiscip. Sci. Comput. Life Sci 2009, 1, 133–140. [DOI] [PubMed] [Google Scholar]
- (3).(a) For general reviews of enantioselective radical reactions. Studer A; Curran DP Catalysis of Radical Reactions: A Radical Chemistry Perspective. Angew. Chem. Int. Ed 2016, 55, 58–102. [DOI] [PubMed] [Google Scholar]; (b) Sibi MP; Manyem S; Zimmerman J Enantioselective Radical Processes. Chem. Rev 2003, 103, 3263–3295. [DOI] [PubMed] [Google Scholar]; (c) Zhang L; Meggers E Steering Asymmetric Lewis Acid Catalysis Exclusively with Octahedral Metal-Centered Chirality. Acc. Chem. Res 2017, 50, 320–330. [DOI] [PubMed] [Google Scholar]; (d) Yoon TP Photochemical Stereocontrol Using Tandem Photoredox-Chiral Lewis Acid Catalysis. Acc. Chem. Res 2016, 49, 2307–2315. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Silvi M; Melchiorre P Enhancing the potential of enantioselective organocatalysis with light. Nature, 2018, 554, 41–49. [DOI] [PubMed] [Google Scholar]
- (4).(a) For examples of a role for hydrogen bonding in radical reactions: Waldner A; De Mesmaeker A; Hoffmann P; Mindt T; Winkler T α-Sulfinyl Substituted Radicals; 1. Stereoselective Radical Addition Reactions of Cyclic α-Sulfinyl Radicals. Synlett, 1991, 2, 101–104. [Google Scholar]; (b) De Mesmaeker A; Waldner A; Hoffmann P; Mindt T α-Sulfinyl Substituted Radicals; II. Stereoselective Inter- and Intramolecular Addition Reactions of Acyclic α-Sulfinyl Radicals. Synlett, 1993, 11, 871–874. [Google Scholar]; (c) Curran DP; Kuo LH Altering the stereochemistry of Allylation Reactions of Cyclic α-Sulfinyl Radicals with Diarylureas. J. Org. Chem 1994, 59, 3259–3261. [Google Scholar]; (d) Böhm A; Bach T Synthesis of Supramolecular Iridium Catalysts and Their Use in Enantioselective Visible-Light-Induced Reactions. Synlett, 2016, 27, 1056–1060. [Google Scholar]; (e) Bauer A; Westkämper F; Grimme S; Bach T Catalytic enantioselective Reactions Driven by Photoinduced Electron Transfer. Nature 2005, 436, 1139–1140. [DOI] [PubMed] [Google Scholar]; (f) Bach T; Bergmann H; Grosch B; Harms K Highly Enantioselective Intra- and Intermolecular [2 + 2] Photocycloaddition Reactions of 2-Quinolones Mediated by a Chiral Lactam Host: Host−Guest Interactions, Product Configuration, and the Origin of the Stereoselectivity in Solution. J. Am. Chem. Soc 2002, 124, 7982–7990. [DOI] [PubMed] [Google Scholar]; (g) Aechtner T; Dressel M; Bach T Hydrogen Bond Mediated Enantioselectivity of Radical Reactions. Angew. Chem. Int. Ed 2004, 43, 5849–5851. [DOI] [PubMed] [Google Scholar]; (h) Müller C; Bauer A; Bach T Light-Driven Enantioselective Organocatalysis. Angew. Chem., Int. Ed 2009, 48, 6640–6642. [DOI] [PubMed] [Google Scholar]; (i) Sandoval BA; Meichan AJ; Hyster TK Enantioselective Hydrogen Atom Transfer: Discovery of Catalytic Promiscuity in Flavin-Dependent ‘Ene’-Reductases. J. Am. Chem. Soc 2017, 139, 11313–11316. [DOI] [PubMed] [Google Scholar]; (j) Proctor RSJ; Davis HJ Phipps RJ Catalytic enantioselective Minisci-Type addition to heteroarenes. Science, 2018, 360, 419–422. [DOI] [PubMed] [Google Scholar]; (k) Uraguchi D; Kinoshita N; Kizu T; Ooi T Synergistic Catalysis of Ionic Brønsted Acid and Photosensitizer for a Redox Neutral Asymmetric α-Coupling of N-Arylaminomethanes with Aldimines. J. Am. Chem. Soc 2015, 137, 13768–13771. [DOI] [PubMed] [Google Scholar]; (l) Shevchenko GA; Oppelaar B; List B An Unexpected α-Oxidation of Cyclic Ketones with 1,4-Benzoquinone by Enol Catalysis. Angew. Chem. Int. Ed 2018, 57, 10756–10759. [DOI] [PubMed] [Google Scholar]; (m) Cao K; Tan SM; Lee R; Yang S; Jia H; Zhao X; Qiao B; Jiang Z Catalytic Enantioselective Addition of Prochiral Racicals to Vinylpyridines. J. Am. Chem. Soc 2019, 141, 5437–5443. [DOI] [PubMed] [Google Scholar]
- (5).(a) Weinberg DR; Gagliardi CJ; Hull JF; Murphy CF; Kent CA; Westlake BC; Paul A; Ess DH; McCafferty DG; Meyer TJ Proton-Coupled Electron Transfer. Chem. Rev 2012, 112, 4016–4093. [DOI] [PubMed] [Google Scholar]; (b) Mayer JM Proton-Coupled Electron Transfer: A Reaction Chemist’s View. Annu. Rev. Phys. Chem 2004, 55, 363–390. [DOI] [PubMed] [Google Scholar]; (c) Hoffmann N Proton-Coupled Electron Transfer in Photoredox Catalytic Reactions. Eur. J. Org. Chem 2017, 15, 1982–1992. [Google Scholar]; (d) Miller DC; Tarantino KT; Knowles RR Proton-Coupled Electron Transfer in Organic Synthesis: Fundamentals, Applications, and Opportunities. Top. Curr. Chem 2016, 374, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Qiu G; Knowles RR Understanding Chemoselectivity in Proton-Coupled Electron Transfer: A Kinetic Study of Amide and Thiol Activation. J. Am. Chem. Soc 2019, 141, 16574–16578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).(a) Mayer JM; Hrovat DA; Thomas JL; Borden WT Proton-Coupled Electron Transfer versus Hydrogen Atom Transfer in Benzyl/Toluene, Methoxyl/Methanol, and Phenoxyl/Phenol Self-Exchange Reactions. J. Am. Chem. Soc 2002, 124, 11142–11147. [DOI] [PubMed] [Google Scholar]; (b) Mader EA; Mayer JM The Importance of Precursor and Successor Complex Formation in a Bimolecular Proton-Electron Reaction. Inorg. Chem 2010, 49, 3685–3687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).(a) Rono LJ; Yayla HG; Wang DY; Armstrong MF; Knowles RR Enantioselective Photoredox Catalysis Enabled by Proton-Coupled Electron Transfer: Development of an Asymmetric Aza-Pinacol Cyclization. J. Am. Chem. Soc 2013, 135, 17735–17738. [DOI] [PubMed] [Google Scholar]; (b) Gentry EC; Rono LJ; Hale ME; Matsuura R; Knowles RR Enantioselective Synthesis of Pyrroloindolines via Non-Covalent Stabilization of Indole Radical Cations and Applications to the Synthesis of Alkaloid Natural Products. J. Am. Chem. Soc 2018, 140, 3394–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Zhu Q; Graff DE; Knowles RR Intermolecular Anti-Markovnikov Hydroamination of Unactivated Alkenes with Sulfonamides Enabled by Proton-Coupled Electron Transfer. J. Am. Chem. Soc 2018, 140, 741–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).(a) Representative reviews and reports of enantioselective hydroamination reactions: Hannedouche J; Schulz E Asymmetric Hydroamination: A Survey of the Most Recent Developments. Chem. Eur. J 2013, 19, 4972–4985. [DOI] [PubMed] [Google Scholar]; (b) Michon C; Abadie M-A; Medina F; Agbossou-Niedercorn F Recent Metal-Catalyzed Asymmetric Hydroaminations of Alkenes. J. Organometallic Chem 2017, 847, 13–27. [Google Scholar]; (c) Hultzsch KC Transition Metal-Catalyzed Asymmetric Hydroamination of Alkenes (AHA). Adv. Synth. Catal 2005, 347, 367–391. [Google Scholar]; (d) Hii KK Development of Palladium Catalysts for Asymmetric Hydroamination Reactions. Pure Appl. Chem 2006, 78, 341–349. [Google Scholar]; (e) Aillaud I; Collin J; Hannedouche J; Schulz E Asymmetric Hydroamination of Non-Activated Carbon–Carbon Multiple Bonds. Dalton Trans 2007, 44, 5105–5118. [DOI] [PubMed] [Google Scholar]; (f) Yu F; Chen P; Liu G Pd(II)-catalyzed intermolecular enantioselective hydroamination of styrenes. Org. Chem. Front 2015, 2, 819–822. [Google Scholar]; (g) Yang Y; Shi S-L; Niu D; Liu P; Buchwald SL Catalytic Asymmetric Hydroamination of Unactivated Internal Olefins to Aliphatic Amines. Science 2015, 349, 62–66. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Zhou Y; Engl OD; Bandar JS; Chant ED; Buchwald SL CuH-Catalyzed Asymmetric Hydroamidation of Vinylarenes. Angew. Chem. Int. Ed 2018, 57, 6672–6675. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Lutete LM; Kadota I; Yamamoto Y Palladium-Catalyzed Intramolecular Asymmetric Hydroamination of Alkynes. J. Am. Chem. Soc 2004, 126, 1622–1623. [DOI] [PubMed] [Google Scholar]; (j) LaLonde RL; Sherry BD; Kang EJ; Toste FD Gold(I)-Catalyzed Enantioselective Intramolecular Hydroamination of Allenes. J. Am. Chem. Soc 2007, 129, 2452–2453. [DOI] [PubMed] [Google Scholar]; (k) Sevov CS; Zhou JS; Hartwig JF Iridium-Catalyzed Intermolecular Hydroamination of Unactivated Aliphatic Alkenes with Amides and Sulfonamides. J. Am. Chem. Soc 2012, 134, 11960–11963. [DOI] [PubMed] [Google Scholar]; (l) Hannedouche J; Schulz E Hydroamination and Hydroaminoalkylation of Alkenes by Group 3–5 Elements: Recent Developments and Comparison with Late Transition Metals. Organometallics 2018, 37, 4313–4326. [Google Scholar]; (m) Brown AR; Uyeda C; Brotherton CA; Jacobsen EN Enantioselective Thiourea-Catalyzed Intramolecular Cope-Type Hydroamination. J. Am. Chem. Soc 2013, 135, 6747–6749. [DOI] [PMC free article] [PubMed] [Google Scholar]; (n) Hong S; Tian S; Metz MV; Marks TJ C2-Symmetric Bis(oxazolinato)lanthanide Catalysts for Enantioselective Intramolecular Hydroamination/Cyclization. J. Am. Chem. Soc 2003, 125, 14768–14783. [DOI] [PubMed] [Google Scholar]
- (11).Horner JH; Musa OM; Bouvier A; Newcomb M Absolute Kinetics of Amidyl Radical Reactions. J. Am. Chem. Soc 1998, 120, 7738–7748. [Google Scholar]
- (12).(a) Representative reviews on the generation and synthetic utility of nitrogen centered radicals: Zard SZ Recent Progress in the Generation and Use of Nitrogen-Centred Radicals. Chem. Soc. Rev 2008, 37, 1603–1618. [DOI] [PubMed] [Google Scholar]; (b) Xiong T; Zhang Q New Amination Strategies Based on Nitrogen-Centered Radical Chemistry. Chem. Soc. Rev 2016, 45, 3069–3087. [DOI] [PubMed] [Google Scholar]; (c) Chen J-R; Hu X-Q; Lu L-Q; Xiao W-J Visible Light Photoredox-Controlled Reactions of N-Radicals and Radical Ions. Chem. Soc. Rev 2016, 45, 2044–2056. [DOI] [PubMed] [Google Scholar]; (d) Kärkäs MD Photochemical Generation of Nitrogen-Centered Amidyl, Hydrazonyl, and Imidyl Radicals: Methodology Developments and Catalytic Applications. ACS Catal 2017, 7, 4999–5022. [Google Scholar]
- (13).(a) Cecere G; König CM; Alleva JL; MacMillan DWC Enantioselective Direct α-Amination of Aldehydes via a Photoredox Mechanism: A Strategy for Asymmetric Amine Fragment Coupling. J. Am. Chem. Soc 2013, 135, 11521–11524. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Huang X; Webster RD; Harms K; Meggers E Asymmetric Catalysis with Organic Azides and Diazo Compounds Initiated by Photoinduced Electron Transfer. J. Am. Chem. Soc 2016, 138, 12636–12642. [DOI] [PubMed] [Google Scholar]; (c) Zhou Z; Li Y; Han B; Gong L; Meggers E Enantioselective catalytic β-amination through proton-coupled electron transfer followed by stereocontrolled radical–radical coupling. Chem. Sci, 2017, 8, 5757–5763. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Shen X; Harms K; Marsch M; Meggers E A Rhodium Catalyst Superior to Iridium Congeners for Enantioselective Radical Amination Activated by Visible Light Chem. Eur. J 2016, 22, 9102–9105. [DOI] [PubMed] [Google Scholar]
- (14).(a) Lang K; Torker S; Wojtas L; Zhang XP Asymmetric Induction and Enantiodivergence in Catalytic Radical C–H amination via Enantiodifferentiative H-Atom Abstraction and Stereoselective Radical Substitution. J. Am. Chem. Soc 2019, 141, 12388–12396. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Li J; Zhang Z; Wu L; Zhang W; Chen P; Lin Z; Liu G Site-Specific allylic C–H bond functionalization with a copper-bound N-centered radical. Nature 2019, 574, 516–522. [DOI] [PubMed] [Google Scholar]; (c) Turnpenny BW; Hyman KL; Chemler SR Chiral Indoline Synthesis via Enantioselective Intramolecular Copper-Catalyzed Alkene Hydroamination. Organometallics, 2012, 31, 7819–7822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Orlandi M; Toste FD; Sigman MS Multidimensional Correlations in Asymmetric Catalysis through Parameterization of Uncatalyzed Transition States. Angew. Chem. Int. Ed 2017, 56, 14080–14084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16). A terminal olefin and 6-exo cyclization substrate were also tested, but exhibited low reactivity and poor enantioselectivity. See Supporting Information for details.
- (17).(a) Morse PD; Nguyen TM; Cruz CL; Nicewicz DA Enantioselective Counter-Anions in Photoredox Catalysis: The Asymmetric Cation Radical Diels-Alder Reaction. Tetrahedron 2018, 74, 3266–3272. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Yang Z; Li H; Li S; Zhang M-T, Luo S; A chiral ion-pair photoredox organocatalyst: Enantioselective anti-Markovnikov hydroetherification of alkenols. Org. Chem. Front 2017, 4, 1037–1041. [Google Scholar]
- (18).Roth HG; Romero NA; Nicewicz DA Experimental and Calculated Electrochemical Potentials of Common Organic Molecules for Applications to Single Electron Redox Chemistry. Synlett 2016, 27, 714–723. [Google Scholar]
- (19).Yayla HG; Wang H; Tarantino KT; Orbe HS; Knowles RR Catalytic Ring-Opening of Cyclic Alcohols Enabled by PCET Activation of Strong O-H Bonds. J. Am. Chem. Soc 2016, 138, 10794–10797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20). In Stern-Volmer luminescence experiments, quenching is observed only when both base and sulfonamide are present. Results of cyclic voltammetry experiments show an earlier onset potential and increased current response upon increasing the concentration of base. For more information, see reference 7.
- (21).(a) Nguyen TM; Nicewicz DA Anti-Markovnikov hydroamination of Alkenes Catalyzed by an Organic Photoredox System. J. Am. Chem. Soc 2013, 135, 9588–9591. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Nguyen TM; Manohar N; Nicewicz DA Anti-Markovnikov Hydroamination of Alkenes Catalyzed by a Two Component Photoredox System: Direct Access to Phenethylamine Derivatives. Angew. Chem. Int. Ed 2014, 53, 6198–6201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).(a) Loupy A; Tchoubar B Salt Effects in Organic and Organometallic Chemistry, VCH Publishers, Inc.: New York, NY, 1991. [Google Scholar]; (b) Wynberg H; Greijdanus B Solvent effects in homogeneous asymmetric catalysis. J. Chem. Soc., Chem. Commun 1978, 427–428. [Google Scholar]; (c) Brak K; Jacobsen EN Asymmetric Ion-Pairing Catalysis. Angew. Chem. Int. Ed 2013, 52, 534–561. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Bendelsmith AJ; Kim SC; Wasa M; Roche SP; Jacobsen EN Enantioselective Synthesis of α-Allyl Amino Esters via Hydrogen Bond-Donor Catalysis. J. Am. Chem. Soc 2019, 141, 11414–11419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Wohlfarth C Permittivity (Dielectric Constant) of Liquids In CRC Handbook of Chemistry and Physics, 97th edition, Haynes WM; Lide DR; Bruno TJ, Eds.; CRC, Press: Boca Raton, Florida, 2016; 6–199. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.