

Abstract
Epigallocatechin gallate (EGCG), a major polyphenol in green tea, exhibits diverse biological activities. Previous studies show that EGCG could effectively suppress HBV gene expression and replication, but the role of EGCG in HBV replication and its underlying mechanisms, especially the signaling pathways involved, remain unclear. In this study we investigated the mechanisms underlying EGCG inhibition on HBV replication with a focus on the signaling pathways. We showed that EGCG (12.5−50 μM) dose-dependently inhibited HBV gene expression and replication in HepG2.2.15 cells. Similar results were observed in HBV mice receiving EGCG (25 mg· kg−1· d−1, ip) for 5 days. In HepG2.2.15 cells, we showed that EGCG (12.5−50 μM) significantly activate ERK1/2 MAPK signaling, slightly activate p38 MAPK and JAK2/STAT3 signaling, while had no significant effect on the activation of JNK MAPK, PI3K/AKT/mTOR and NF-κB signaling. By using specific inhibitors of these signaling pathways, we demonstrated that ERK1/2 signaling pathway, but not other signaling pathways, was involved in EGCG-mediated inhibition of HBV transcription and replication. Furthermore, we showed that EGCG treatment dose-dependently decreased the expression of hepatocyte nuclear factor 4α (HNF4α) both at the mRNA and protein levels, which could be reversed by pretreatment with the ERK1/2 inhibitor PD98059 (20 μM). Moreover, we revealed that EGCG treatment dose-dependently inhibited the activity of HBV core promoter and the following HBV replication. In summary, our results demonstrate that EGCG inhibits HBV gene expression and replication, which involves ERK1/2-mediated downregulation of HNF4α.These data reveal a novel mechanism for EGCG to inhibit HBV gene expression and replication.
Keywords: HBV, EGCG, ERK1/2, HNF4α, core promoter, PD98059, HepG2.2.15 cells, HBV mice
Introduction
Hepatitis B virus (HBV) infection remains a major world health threat, despite the availability of an effective vaccine. Approximately 260 million people are chronically infected with HBV worldwide, and these individuals may develop end-stage liver diseases, such as liver fibrosis and hepatocellular carcinoma [1, 2]. Interferon-α (IFN-α) and nucleos(t)ide analogs (such as lamivudine, entecavir) have been approved for the treatment of HBV infection; however, the relatively lower cure rate, side effects and drug resistance have limited their application [3, 4]. Thus, there is a strong need for novel anti-HBV agents.
Green tea is one of the most widely consumed beverages worldwide. In traditional Chinese medicine, green tea is regarded as a panacea, possessing antidiarrheal, antidotal, and antipyretic properties, indicating anti-infective activity in our modern concept [5, 6]. Epigallocatechin-3-gallate (EGCG) is the main component of green tea and is considered to account for most of its beneficial effects [7, 8]. Recent experimental evidence shows that EGCG displays an inhibitory effect on a variety of viruses [9–14]. We and others have also demonstrated that EGCG could effectively suppress HBV gene expression and replication [15–18]; however, the role of EGCG in HBV replication and its underlying mechanisms, especially the signaling pathways involved, remain to be further explored.
In natural infection, HBV transcription and replication are mainly restricted to the hepatocytes in the livers of humans and a limited number of primates [19, 20]. The reason for HBV tropism is largely unknown. Although it is assumed that infection is limited to the liver because of the tissue-restricted expression of the viral receptor [21, 22], liver-enriched transcription factors, especially hepatocyte nuclear factor 4α (HNF4α), are found to contribute to the hepatocyte-specific tropism of HBV [23]. Accumulating evidence indicates that the regulation of the expression of HNF4α may play an important role in determining HBV transcription and replication [24, 25]. For example, the activation of signaling pathways, such as MAPKs, has been reported to affect the expression level of HNF4α and contribute to the regulation of HBV replication [26].
In the present investigation, we found that EGCG inhibited HBV gene expression and replication both in vitro and in vivo. Furthermore, our data showed that EGCG treatment could activate the p38 MAPK, ERK1/2 MAPK, and JAK2/STAT3 signaling pathways; however, it is the inhibition of ERK1/2, but not other signaling pathways, that contributed to the EGCG-mediated inhibition of HBV gene expression and replication. Furthermore, our data demonstrated that the ERK1/2 signaling pathway was implicated in the EGCG-mediated inhibition of HNF4α and thus contributed to the EGCG-mediated suppression of HBV gene expression and replication.
Materials and methods
Materials
EGCG was purchased from Sigma Chemical (St Louis, MO, USA), MEK1/2 inhibitor, PD98059, p38 inhibitor SB203580, and JAK2/STAT3 inhibitor AG490 were purchased from Calbiochem (La Jolla, CA, USA). Antibodies against p-AKT (#9271), AKT (#9272), p-mTOR (#2971), mTOR (#2972), p-ERK1/2 (#9101), ERK1/2 (#9102), p-p38 (#9211), p38 (#9212), p-JNK (#9251), JNK (#9252), p-JAK2 (#3771), p-STAT3 (#9138), STAT3 (#9139), p-IκBα (#2859), and p-NF-κB-p65 (#3033) were obtained from Cell Signaling Technology (Beverly, MA, USA). Antibodies against HNF4α (BS6888) and GAPDH (AP0063) were obtained from BioWorld Biotechnology (Minneapolis, MN, USA).
Cell culture and transfection
HepG2.2.15 cells were maintained at 37 °C in DMEM containing 10% FBS in a 5% CO2 atmosphere. For transfection, the cells were subjected to electrotransfection using Amaxa Nucleofector Technology (Amaxa GmbH, Koln, Germany).
Animal study
To establish an HBV infection mouse model, ten micrograms of pAAV-HBV1.2 was hydrodynamically injected into the tail veins of C57BL/6 mice in a volume of PBS equivalent to 8% of the mouse body weight as described previously [27]. Four weeks after the injection, serum HBsAg was detected to confirm the successful establishment of the HBV mouse model. For EGCG administration, HBV mice were injected intraperitoneally (ip) with EGCG (25 mg/kg) daily for 5 consecutive days. The mouse sera were then collected and assayed for HBsAg and HBV-DNA, and liver tissues were collected for Western blotting or immunohistochemical staining. All animal experiments were conducted according to the Guide for the Care and Use of Medical Laboratory Animals (Ministry of Health, PR China, 1998) and with the approval of the Ethics Committee of Fudan University (Shanghai, China).
Western blot analysis
Western blot analysis was performed as described previously [15, 28, 29]. Briefly, the proteins were denatured in SDS, resolved by SDS-PAGE, and transferred onto polyvinylidene fluoride membranes. The membranes were blocked for 2 h with nonfat dry milk solution (5% in TBS) containing 0.05% Tween-20 and then sequentially probed with the indicated primary antibodies and peroxidase-conjugated secondary antibodies. The bands were visualized by using SuperSignal chemiluminescent substrate (CWbiotech, Beijing, China).
Quantitative reverse transcription PCR (qRT-PCR)
qRT-PCR was performed as described previously [28, 30]. Briefly, the cells or liver tissues were harvested, and total RNA was isolated with RNAiso Plus (Takara, Dalian, China). cDNA was synthesized from 0.5 μg of RNA using the PrimeScript™ RT Reagent Kit with gDNA Eraser (Perfect Real Time) (Takara). Prior to reverse transcription, gDNA Eraser was used to remove contaminating DNA according to the manufacturer’s instructions. The qRT-PCR analysis of cDNA targets was performed using a Lightcycler 480 and a SYBR Green system (Takara). The primers used in this study are as follows: HBV pgRNA, forward, 5′- TGTTCAAGCCTCCAAGCT-3′ and reverse, 5′-GGAAAGAAGTCAGAAGGCAA-3′; HBV RNA, forward, 5′-GCACTTCGCTTCACCTCTGC-3′ and reverse, 5′-CTCAAGGTCGGTCGTTGACA-3′; HNF4α, forward, 5′-TGTCCCGACAGATCACCTC-3′ and reverse, 5′-CACTCAACGAGAACCAGCAG-3′; GAPDH, forward, 5′-CGGGTGGGAATGTTGAGG-3′ and reverse, 5′-TGGCGGGAGATGTGGGTAC-3′. The levels of pgRNA, HBV RNA or HNF4α were calculated following normalization to GAPDH levels by the comparative ΔΔ threshold cycle method. The specificity of the amplification reactions was confirmed by melt curve analysis. The results are representative of three independent experiments.
Extraction and quantitative analysis of intracellular core particle-associated HBV-DNA
The method for the extraction and analysis of intracellular core particle-associated HBV-DNA in HepG2.2.15 cells or pAAV-HBV1.2-injected mouse livers was described previously [15, 28, 29]. Briefly, HBV genomic DNA was then extracted with a Viral Genome Purification kit (CWbiotech, Beijing, China) following the manufacturer’s protocol and subjected to quantitative PCR (qPCR) using an HBV diagnostic kit (Kehua Biotech, Shanghai, China) according to the manufacturer’s instructions. For Southern hybridization, the intracellular core particle-associated HBV-DNA was extracted and probed with a digoxigenin (DIG)-labeled full-length HBV probe synthesized with a DIG probe synthesis kit (Roche Diagnostics, Mannheim, Germany).
Immunochemical staining
For immunohistochemical staining for HBcAg, paraffin-embedded sections were treated for 10 min at room temperature with 3% hydrogen peroxide and washed with PBS. After the sections were blocked with 5% BSA at 37 °C for 30 min, a rabbit polyclonal antibody against HBcAg (Dako, Glostrup, Denmark) was applied at a 1:500 dilution overnight at 4 °C. The secondary antibody labeled with biotin was incubated with the sections for 30 min, and then avidin-biotin complex reagent was used. Peroxidase staining was developed with 3,3′-diaminobenzidine solution and counterstained with hematoxylin.
Luciferase assay
The plasmids SpLUC, CpLUC, pS(1)pLUC, and XpLUC, each containing one complete HBV genome with the luciferase (LUC) gene governed by the surface, core, preS, or X promoter, respectively, were employed as reporters, as described previously [25, 29]. For the reporter gene assay, the HBV promoter-dependent LUC reporter plasmids were transfected into HepG2.2.15 cells. Twenty-four hours post transfection, the cells were further stimulated with the indicated concentrations of EGCG for 24 h, and the LUC activity in the lysates of transfected cells was determined with a LUC reporter assay system (Promega, Madison, WI, USA). In all transfection assays, pCMV-β-gal was cotransfected to normalize the transfection efficiency.
Statistical analysis
The data are expressed as the mean ± SEM. Comparisons between two groups were performed with a t-test, and multiple groups were compared using one-way analysis of variance. Statistical analysis was performed with the SPSS version 18.0 statistical software package. A value of P < 0.05 was considered statistically significant.
Results
EGCG inhibited HBV gene expression and replication both in vitro and in vivo
To investigate the effect of EGCG on HBV gene expression and replication, we treated HepG2.2.15 cells with increasing concentrations of EGCG for 24 h and then detected the level of HBsAg and HBeAg in culture supernatants by ELISA, the level of HBcAg in cell lysates by Western blotting, the level of HBV transcripts by qRT-PCR, and the level of intracellular core particle-associated HBV-DNA by quantitative real-time PCR and Southern blotting. Our results showed that EGCG treatment markedly decreased the levels of HBeAg (Fig. 1b) and HBcAg (Fig. 1c) but only had a slight effect on the level of HBsAg (Fig. 1a). Accordingly, our data showed that EGCG treatment downregulated the level of HBV transcripts, especially that of pgRNA (Fig. 1d). Further study showed that EGCG could suppress HBV replication in a dose-dependent manner (Fig. 1e, f). To exclude the possibility that the inhibitory effect of EGCG on HBV gene expression and replication was due to its toxic effect on cells, we determined the cell viability of EGCG-treated cells by CCK8 assays. As shown in Fig. 1g, the concentration of EGCG used in the present investigation did not have a significant effect on the survival of HepG2.2.15 cells.
Fig. 1.

EGCG inhibited HBV gene expression and replication in HepG2.2.15 cells. HepG2.2.15 cells were treated with increasing amounts of EGCG (12.5, 25, 50 μM) for 24 h. The levels of HBsAg (a) and HBeAg (b) in culture supernatants were determined by ELISA; the level of HBcAg (c) in the cell lysates was determined by Western blotting; the levels of HBV pgRNA and HBV RNAs (d) were determined by qRT-PCR; the levels of intracellular core particle-associated HBV-DNA (e, f) were determined by quantitative real-time PCR and Southern blotting, respectively; and the cell viability (g) was determined by CCK8 assay. The values are expressed as the means ± SEM of three independent experiments
To further evaluate the inhibitory effect of EGCG on HBV replication, we established an HBV mouse model by the hydrodynamic injection of pAAV1.2 as described previously and then treated HBV mice with EGCG (25 mg/kg every day, ip) for 5 days, followed by the evaluation of the effect of EGCG on HBV gene expression and replication. The results showed that EGCG treatment decreased the levels of HBsAg (Fig. 2a) and strongly downregulated the levels of HBeAg (Fig. 2b) and HBV-DNA (Fig. 2c) in mouse sera. Data from qRT-PCR showed that EGCG treatment decreased the levels of HBV transcripts, especially that of pgRNA, in mouse liver tissues (Fig. 2e). We also evaluated the expression of the HBV core antigen (HBcAg) in hepatocytes by immunohistochemical staining and found that EGCG treatment significantly downregulated the level of HBcAg in mouse liver tissue, as expected (Fig. 2d). Furthermore, our data revealed that EGCG treatment efficiently decreased the core particle-associated HBV-DNA level in mouse liver tissues (Fig. 2f). To exclude the possibility that the EGCG-mediated inhibition of HBV replication was due to its possible toxic effect on hepatocytes, we tested the level of ALT in mouse sera. The results showed that EGCG treatment had little effect on ALT levels (Fig. 2g).
Fig. 2.

The effect of EGCG on HBV gene expression and replication in mice. To evaluate the effect of EGCG on HBV replication, HBV mice were treated with or without EGCG (25 mg/kg every day, ip) for 5 days (n = 10 per group). The serum concentrations of HBsAg (a), HBeAg (b), and HBV-DNA (c) were determined by ELISA and quantitative real-time PCR; the level of HBcAg (d) in liver tissue was determined by immunohistochemical staining; the levels of HBV pgRNA and HBV RNAs (e) in liver tissues were determined by qRT-PCR; the levels of intracellular core particle-associated HBV-DNA (f) in liver tissues were determined by Southern blotting; and the levels of ALT (g) in mouse sera were determined by an ALT/GPT ELISA kit. The values were expressed as the means ± SEM of three independent experiments; *P < 0.05 (unpaired, two-tailed Student’s t tests)
Taken together, our data indicated that EGCG possessed an inhibitory effect on HBV gene expression and replication both in vitro and in vivo.
The ERK1/2 signaling pathway was involved in the EGCG-mediated inhibition of HBV
Reports indicate that EGCG plays a crucial role in regulating signaling pathways [31–34], while HBV replication has a close relationship with the activation of some signaling pathways [28, 35–37]. We therefore paid particular attention to the possible role of signaling pathways in EGCG inhibition of HBV in the present investigation. We treated HepG2.2.15 cells with increasing amounts of EGCG for 24 h and then detected the activation of various signaling pathways by Western blotting. The results showed that EGCG treatment significantly upregulated the levels of phosphorylated ERK1/2 (p-ERK1/2), slightly upregulated the levels of p-p38, p-JAK2 and p-STAT3, and had little effect on the levels of p-JNK, p-AKT, p-mTOR, p-p70S6K, p-IκBα, and p-NF-κB-p65 (Fig. 3a), indicating that EGCG treatment could significantly activate ERK1/2 MAPK signaling and slightly activate p38 MAPK and JAK2/STAT3 signaling, but had no significant effect on the activation of JNK MAPK, PI3K/AKT/mTOR, and NF-κB signaling. To investigate the role of ERK MAPK, p38 MAPK and JAK2/STAT3 signaling in the EGCG-mediated inhibition of HBV, HepG2.2.15 cells were pretreated with ERK inhibitor (PD98059), p38 inhibitor (SB203580) or JAK2/STAT3 inhibitor (AG490) for 1 h, followed by treatment with 25 μM EGCG for another 24 h. Our data showed that PD98059, SB203580, or AG490 could efficiently inhibit the activation of their corresponding signaling pathways (Fig. 3b); however, the inhibition of ERK1/2, but not other signaling pathways, significantly attenuated the EGCG-mediated inhibition of HBV gene expression and replication (Fig. 3c).
Fig. 3.

ERK1/2 signaling pathway was involved in the EGCG-mediated inhibition of HBV. a The effect of EGCG on the activation of the MAPK (p38, ERK, JNK), AKT/mTOR, and JAK2/STAT3 pathways. HepG2.2.15 cells were treated with increasing doses of EGCG (25 μM and 50 μM) for 24 h, and the cells were then subjected to Western blotting with the indicated antibodies. b The effect of pharmacological inhibitors on the activation of p38, ERK MAPK, or JAK2/STAT3 signaling pathways. HepG2.2.15 cells were pretreated with the ERK inhibitor PD98059 (20 μM), the p38 inhibitor 203580 (10 μM) or the JAK2/STAT3 inhibitor AG490 (50 μM) for 1 h and then treated with 25 μM of EGCG for 24 h. The levels of p-p38, p-ERK, p-JAK2, or p-STAT3 were determined by Western blotting. c The role of p38, ERK MAPKs, or JAK2/STAT3 signaling pathways in the EGCG-mediated inhibition of HBV. HepG2.2.15 cells were treated as in b, and the levels of HBeAg or HBV-DNA were determined by ELISA or qPCR, respectively. The values were expressed as the means ± SEM of three independent experiments; *P < 0.05 (unpaired, two-tailed Student’s t tests)
The ERK1/2 signaling pathway contributes to the EGCG-mediated downregulation of HNF4α
Our above data showed that EGCG-activated ERK1/2 signaling, which has been reported to play important roles in the regulation of the liver-enriched factor HNF4α, a key player in HBV gene expression and replication [24, 26, 38]. We thus further investigated the effect of EGCG on the expression of HNF4α. Our data showed that EGCG treatment could downregulate the expression of HNF4α at both the mRNA and protein levels in a dose-dependent manner (Fig. 4a, b). Notably, our data showed that the EGCG-mediated downregulation of HNF4α appeared to correlate with EGCG-activated ERK signaling (Fig. 4c). Further studies showed that the inhibition of ERK signaling by treatment with PD98059 significantly attenuated the EGCG-mediated inhibition of HNF4α at both the mRNA and protein levels (Fig. 4d, e), indicating that the ERK1/2 signaling pathway might play an important role in the EGCG-mediated inhibition of HNF4α.
Fig. 4.

The ERK1/2 signaling pathway was involved in EGCG-mediated inhibition of HNF4α. a The effect of EGCG on the mRNA level of HNF4α in HepG2.2.15 cells. HepG2.2.15 cells were treated with increasing doses of EGCG (25 and 50 μM) for 24 h. The mRNA levels of HNF4α were then determined by quantitative RT-PCR. b The effect of EGCG on the protein level of HNF4α in HepG2.2.15 cells. HepG2.2.15 cells were treated as in a, and the protein levels of HNF4α were then determined by Western blotting. c HepG2.2.15 cells were treated with 50 μM of EGCG for the indicated time periods, and Western blotting was then performed using antibodies against p-ERK1/2, HNF4α and GAPDH. The effect of ERK signaling on the EGCG-mediated inhibition of HNF4α. HepG2.2.15 cells were pretreated with ERK inhibitor PD98059 for 1 h and then treated with 50 μM of EGCG for 24 h, and the mRNA levels (d) and protein levels (e) of HNF4α were determined by qRT-PCR and Western blotting, respectively. The values were expressed as the means ± SEM of three independent experiments; *P < 0.05; (unpaired, two-tailed Student’s t tests)
EGCG inhibited the activity of HBV promoters
HNF4α, as an important transcriptional factor, plays a crucial role in HBV transcriptional activity [20, 25]. We thus further investigated the effect of EGCG on the activity of all four HBV promoters: surface (S), core (C), preS (pS) and X promoters. Our data showed that EGCG treatment significantly suppressed the activity of HBV core and preS promoters but had no significant effect on the activity of S and X promoters (Fig. 5a-d). These data were correlated with the EGCG-mediated effect on HBV gene expression and replication, i.e., EGCG displayed a stronger inhibitory effect on the level of HBeAg and HBV-DNA than on that of HBsAg.
Fig. 5.
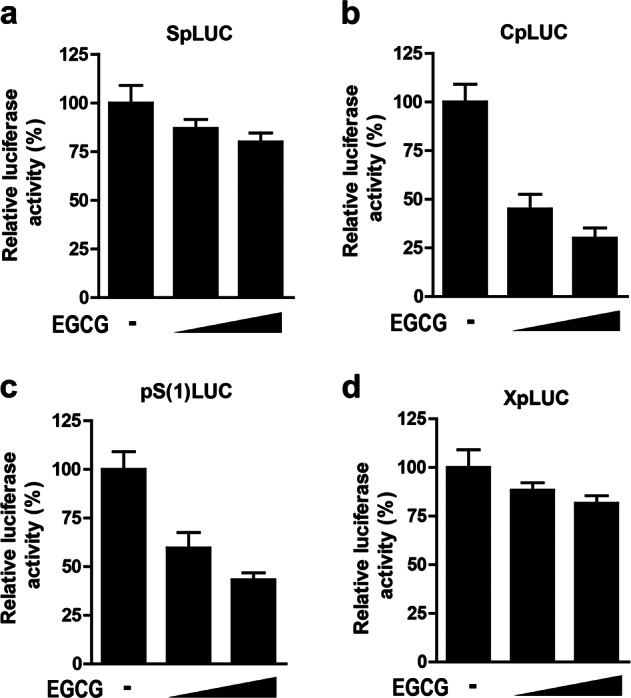
The effect of EGCG on the activity of HBV promoters. HepG2.2.15 cells were transfected with SpLUC (a), CpLUC (b), pS(1) LUC (c) or XpLUC (d), respectively. Twenty-four hours post transfection, the cells were further treated with increasing doses of EGCG (25 µM and 50 µM) for another 24 h. The luciferase activity in cell lysates was then determined. The luciferase activity of EGCG-untreated cells was set to 100%. In all transfection assays, pCMV-β-gal was cotransfected to normalize the transfection efficiency. The values are expressed as the means ± SEM of three independent experiments
ERK1/2-HNF4α contributed to the EGCG-mediated inhibition of HBV core promoter activity and subsequent HBV replication
The above data showed that EGCG showed a strong inhibitory effect on the HBV core promoter, while the core promoter has been demonstrated to play a crucial role in HBV replication. We further investigated the role of the ERK1/2-HNF4α axis in EGCG-mediated inhibition of the HBV core promoter. Our data showed that the ERK1/2 inhibitor PD98059, but not the p38 inhibitor SB203580 or the JAK2/STAT3 inhibitor AG490, significantly attenuated EGCG-mediated inhibition of HBV core promoter activity (Fig. 6a), correlated with their effect on HBeAg and HBV-DNA as shown in Fig. 3c. Furthermore, our data revealed that the ectopic expression of HNF4α significantly reversed the EGCG-mediated inhibition of HBV core promoter activity (Fig. 6b). Furthermore, we determined the effect of HNF4α on the EGCG-mediated inhibition of HBV gene expression and replication. As shown in Fig. 6c, the ectopic expression of HNF4α also significantly reversed the suppressive effect of EGCG on HBeAg and HBV-DNA levels. Taken together, these data indicated that the ERK1/2-HNF4α axis was involved in the EGCG-mediated inhibition of HBV gene expression and replication.
Fig. 6.

ERK1/2-HNF4α axis contributed to EGCG inhibition of HBV core promoter activity. a The effect of ERK signaling on EGCG-mediated inhibition of HBV core promoter activity. The cells were transfected with a core promoter-dependent reporter plasmid (CpLUC). Twenty-four hours post transfection, the cells were treated with EGCG in the absence or presence of the ERK inhibitor PD98059 (20 μM), the p38 inhibitor 203580 (10 μM) or the JAK2/STAT3 inhibitor AG490 (50 μM) for 24 h, followed by the detection of luciferase activity in the cell lysates. b The effect of ectopic HNF4α expression on the EGCG-mediated inhibition of HBV promoter activity. The HNF4α expression plasmid (pHNF4α-HA) was transfected into HepG2.2.15 cells together with CpLUC. Twenty-four hours post transfection, the cells were further stimulated with EGCG for another 24 h, followed by the detection of luciferase activity. Below: The whole cell lysate was subjected to Western blotting using antibodies against HA and GAPDH. c The effect of HNF4α expression on EGCG-mediated inhibition of HBV gene expression and replication. HepG2.2.15 cells were transfected with control empty vector or an increasing dose of pHNF4α for 24 h and then treated with EGCG for another 24 h. HBeAg and HBV-DNA levels were determined by ELISA and qPCR, respectively. The values were expressed as the means ± SEM of three independent experiments; *P < 0.05; (unpaired, two-tailed Student’s t tests)
Discussion
Green tea, one of the most widely consumed beverages worldwide, has been shown to have diverse physiological and pharmacological health benefits, such as anticarcinogenic, anti-inflammatory, and antioxidant activities [5, 39, 40]. In the past several decades, increasing evidence indicates that EGCG, the main constituent of green tea, displays antiviral activities with different modes of action against diverse viruses, including some important human pathogens, such as human immunodeficiency virus, influenza A virus, hepatitis C virus and HBV [10, 12–16]. Our previous data showed that EGCG induced a complete autophagic process, which is unfavorable for HBV replication [15]. However, even after the inhibition of EGCG-induced autophagy by the knockdown of some key regulators of autophagy (such as ATG5 and ATG7), EGCG still retained a significant inhibitory effect on HBV replication [15], indicating that, as a multifunctional effector, EGCG might exert anti-HBV activity through other mechanisms.
In the present investigation, we further confirmed the inhibitory effect of EGCG on HBV gene expression and replication both in vitro and in vivo. The reports indicate that EGCG plays an important role in the regulation of diverse signaling pathways. For example, PKCα and ERK1/2 signaling pathways are involved in EGCG-mediated protection against restraint stress-induced neural injuries in rats [32], and EGCG could attenuate high-fat-and high-fructose-induced cognitive defects by regulating the IRS/AKT and ERK/CREB/BDNF signaling pathways [33]. The activation of several signaling pathways, such as PI3K/AKT/mTOR signaling, has been suggested to play important roles in the regulation of HBV replication [35, 37]. Our previous investigation demonstrated that ROS-JNK signaling was involved in HBV/HBx-induced autophagosome formation and thus contributed to the regulation of HBV replication [28]. In mechanistic studies, we focused on the role of signaling pathways in the EGCG-mediated inhibition of HBV. Our data showed that EGCG treatment could activate the ERK1/2, p38 MAPK, and JAK2/STAT3 signaling pathways but had little effect on other signaling pathways, such as JNK MAPK, PI3K/AKT/mTOR, and NF-κB signaling. However, further studies revealed that the activation of ERK1/2, but not p38 or JAK2/STAT3 signaling, played an important role in the suppression of HBV gene expression and replication by EGCG.
Several reports have indicated that ERK1/2 MAPK may have a close relationship with the regulation of HNF4α [26, 41, 42], while HNF4α is now regarded as an important therapeutic target for controlling HBV replication [24, 43, 44]. Therefore, we investigated the effect of EGCG on the expression of HNF4α. Our data showed that EGCG treatment could downregulate the expression level of HNF4α at both the mRNA and protein levels in a dose-dependent manner, and the effect of EGCG on ERK1/2 signaling was correlated with the EGCG-mediated downregulation of HNF4α. Furthermore, our data demonstrated that the inhibition of ERK1/2 signaling indeed significantly reversed the EGCG-mediated downregulation of HNF4α.
HNF4α, as an important transcriptional factor, plays a crucial role in HBV transcriptional activity. We therefore also investigated the effect of EGCG on the activity of all four HBV promoters: core, S, preS and X promoters. EGCG treatment displayed a strong inhibitory effect on the HBV core promoter, while its effect on the HBV surface promoter was much less significant, which was correlated with the data showing that EGCG had a stronger inhibitory effect on the level of HBeAg and HBV-DNA than that of HBsAg. Furthermore, our data revealed that the inhibition of ERK1/2 activity or HNF4α overexpression significantly reversed the EGCG-mediated inhibition of HBV core promoter activity and the subsequent HBV replication, indicating that the ERK1/2-HNF4α axis contributed to the EGCG-mediated inhibition of HBV gene expression and replication. Similar to our data, Zhao et al. [26] reported that an antimicrobial peptide, Mucroporin-M1, also suppressed HBV replication by activating ERK1/2 signaling and downregulating HNF4α. Together, these data suggest that targeting the ERK1/2-HNF4 axis may be helpful for controlling HBV infection.
In summary, our data demonstrated that EGCG inhibited HBV gene expression and replication both in vitro and in vivo, in which EGCG-activated ERK1/2 signaling played an important role. Further study showed that EGCG-activated ERK1/2 signaling might lead to the downregulation of HNF4α, a key player in HBV gene expression and replication, and the ERK1/2-HNF4α axis was revealed to contribute to the EGCG-mediated inhibition of HBV.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (31470839, 31872731, and 21334001), the Jiangsu Natural Science Youth Fund (No. BK20170209), the China Postdoctoral Research Foundation (No. 180942), the Jiangsu Province Postdoctoral Research Foundation (1701012A) and the Key Talents of Young Medical Science Project in Jiangsu Province (No. QNRC2016169).
Author contributions
ZYW, YQL, ZWG, and XHZ conducted the experiments; ZYW, YQL, MDL, and BG analyzed the data; BG and TCX supervised the project; and BG, ZYW, and TCX wrote the paper.
Competing interests
The authors declare no competing interests.
Contributor Information
Tong-chun Xue, Email: xue.tongchun@zs-hospital.sh.cn.
Bo Gao, Email: gaobofd@126.com.
References
- 1.Tsai KN, Kuo CF, Ou JJ. Mechanisms of hepatitis B virus persistence. Trends Microbiol. 2018;26:33–42. doi: 10.1016/j.tim.2017.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schweitzer A, Horn J, Mikolajczyk RT, Krause G, Ott JJ. Estimations of worldwide prevalence of chronic hepatitis B virus infection: a systematic review of data published between 1965 and 2013. Lancet. 2015;386:1546–55. doi: 10.1016/S0140-6736(15)61412-X. [DOI] [PubMed] [Google Scholar]
- 3.Wong GL, Wong VW, Chan HL. Virus and host testing to manage chronic hepatitis B. Clin Infect Dis. 2016;62(Suppl 4):298–305. doi: 10.1093/cid/ciw024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ward H, Tang L, Poonia B, Kottilil S. Treatment of hepatitis B virus: an update. Future Microbiol. 2016;11:1581–97. doi: 10.2217/fmb-2016-0128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Narotzki B, Reznick AZ, Aizenbud D, Levy Y. Green tea: a promising natural product in oral health. Arch Oral Biol. 2012;57:429–35. doi: 10.1016/j.archoralbio.2011.11.017. [DOI] [PubMed] [Google Scholar]
- 6.Xu J, Xu Z, Zheng W. A review of the antiviral role of green tea catechins. Molecules 2017;22: pii: E1337. [DOI] [PMC free article] [PubMed]
- 7.Xi J, Shen D, Zhao S, Lu B, Li Y, Zhang R. Characterization of polyphenols from green tea leaves using a high hydrostatic pressure extraction. Int J Pharm. 2009;382:139–43. doi: 10.1016/j.ijpharm.2009.08.023. [DOI] [PubMed] [Google Scholar]
- 8.Guo Q, Zhao B, Li M, Shen S, Xin W. Studies on protective mechanisms of four components of green tea polyphenols against lipid peroxidation in synaptosomes. Biochim Biophys Acta. 1996;1304:210–22. doi: 10.1016/S0005-2760(96)00122-1. [DOI] [PubMed] [Google Scholar]
- 9.Reygaert WC. The antimicrobial possibilities of green tea. Front Microbiol. 2014;5:434. doi: 10.3389/fmicb.2014.00434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Calland N, Albecka A, Belouzard S, Wychowski C, Duverlie G, Descamps V, et al. (-)-Epigallocatechin-3-gallate is a new inhibitor of hepatitis C virus entry. Hepatology. 2012;55:720–29. doi: 10.1002/hep.24803. [DOI] [PubMed] [Google Scholar]
- 11.Wang CY, Hour MJ, Lai HC, Chen CH, Chang PJ, Huang SH, et al. Epigallocatechin-3-gallate inhibits the early stages of Japanese encephalitis virus infection. Virus Res. 2018;253:140–46. doi: 10.1016/j.virusres.2018.06.009. [DOI] [PubMed] [Google Scholar]
- 12.Yang ZF, Bai LP, Huang WB, Li XZ, Zhao SS, Zhong NS, et al. Comparison of in vitro antiviral activity of tea polyphenols against influenza A and B viruses and structure-activity relationship analysis. Fitoterapia. 2014;93:47–53. doi: 10.1016/j.fitote.2013.12.011. [DOI] [PubMed] [Google Scholar]
- 13.Steinmann J, Buer J, Pietschmann T, Steinmann E. Anti-infective properties of epigallocatechin-3-gallate (EGCG), a component of green tea. Br J Pharmacol. 2013;168:1059–73. doi: 10.1111/bph.12009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li S, Hattori T, Kodama EN. Epigallocatechin gallate inhibits the HIV reverse transcription step. Antivir Chem Chemother. 2011;21:239–43. doi: 10.3851/IMP1774. [DOI] [PubMed] [Google Scholar]
- 15.Zhong L, Hu J, Shu W, Gao B, Xiong S. Epigallocatechin-3-gallate opposes HBV-induced incomplete autophagy by enhancing lysosomal acidification, which is unfavorable for HBV replication. Cell Death Dis. 2015;6:e1770. doi: 10.1038/cddis.2015.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lai YH, Sun CP, Huang HC, Chen JC, Liu HK, Huang C. Epigallocatechin gallate inhibits hepatitis B virus infection in human liver chimeric mice. BMC Complement Alter Med. 2018;18:248. doi: 10.1186/s12906-018-2316-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang HC, Tao MH, Hung TM, Chen JC, Lin ZJ, Huang C. (-)-Epigallocatechin-3-gallate inhibits entry of hepatitis B virus into hepatocytes. Antivir Res. 2014;111:100–11. doi: 10.1016/j.antiviral.2014.09.009. [DOI] [PubMed] [Google Scholar]
- 18.Xu J, Gu W, Li C, Li X, Xing G, Li Y, et al. Epigallocatechin gallate inhibits hepatitis B virus via farnesoid X receptor alpha. J Nat Med. 2016;70:584–91. doi: 10.1007/s11418-016-0980-6. [DOI] [PubMed] [Google Scholar]
- 19.Trepo C, Chan HL, Lok A. Hepatitis B virus infection. Lancet. 2014;384:2053–63. doi: 10.1016/S0140-6736(14)60220-8. [DOI] [PubMed] [Google Scholar]
- 20.Quasdorff M, Protzer U. Control of hepatitis B virus at the level of transcription. J Viral Hepat. 2010;17:527–36. doi: 10.1111/j.1365-2893.2010.01315.x. [DOI] [PubMed] [Google Scholar]
- 21.Schieck A, Schulze A, Gahler C, Muller T, Haberkorn U, Alexandrov A, et al. Hepatitis B virus hepatotropism is mediated by specific receptor recognition in the liver and not restricted to susceptible hosts. Hepatology. 2013;58:43–53. doi: 10.1002/hep.26211. [DOI] [PubMed] [Google Scholar]
- 22.Yan H, Zhong G, Xu G, He W, Jing Z, Gao Z, et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. Elife. 2012;1:e49. doi: 10.7554/eLife.00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tang H, McLachlan A. Transcriptional regulation of hepatitis B virus by nuclear hormone receptors is a critical determinant of viral tropism. Proc Natl Acad Sci USA. 2001;98:1841–46. doi: 10.1073/pnas.98.4.1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen EQ, Sun H, Feng P, Gong DY, Liu C, Bai L, et al. Study of the expression levels of Hepatocyte nuclear factor 4 alpha and 3 beta in patients with different outcome of HBV infection. Virol J. 2012;9:23. doi: 10.1186/1743-422X-9-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Raney AK, Johnson JL, Palmer CN, McLachlan A. Members of the nuclear receptor superfamily regulate transcription from the hepatitis B virus nucleocapsid promoter. J Virol. 1997;71:1058–71. doi: 10.1128/JVI.71.2.1058-1071.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhao Z, Hong W, Zeng Z, Wu Y, Hu K, Tian X, et al. Mucroporin-M1 inhibits hepatitis B virus replication by activating the mitogen-activated protein kinase (MAPK) pathway and down-regulating HNF4alpha in vitro and in vivo. J Biol Chem. 2012;287:30181–90. doi: 10.1074/jbc.M112.370312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang LR, Wu HL, Chen PJ, Chen DS. An immunocompetent mouse model for the tolerance of human chronic hepatitis B virus infection. Proc Natl Acad Sci USA. 2006;103:17862–67. doi: 10.1073/pnas.0608578103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhong L, Shu W, Dai W, Gao B, Xiong S. Reactive oxygen species-mediated c-Jun NH2-terminal kinase activation contributes to hepatitis B virus X protein-induced autophagy via regulation of the beclin-1/Bcl-2 interaction. J Virol. 2017;91:e00001–17. doi: 10.1128/JVI.00001-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gao B, Duan Z, Xu W, Xiong S. Tripartite motif-containing 22 inhibits the activity of hepatitis B virus core promoter, which is dependent on nuclear-located RING domain. Hepatology. 2009;50:424–33. doi: 10.1002/hep.23011. [DOI] [PubMed] [Google Scholar]
- 30.Sato S, Li K, Kameyama T, Hayashi T, Ishida Y, Murakami S, et al. The RNA sensor RIG-I dually functions as an innate sensor and direct antiviral factor for hepatitis B virus. Immunity. 2015;42:123–32. doi: 10.1016/j.immuni.2014.12.016. [DOI] [PubMed] [Google Scholar]
- 31.Li Y, Ying C, Zuo X, Yi H, Yi W, Meng Y, et al. Green tea polyphenols down-regulate caveolin-1 expression via ERK1/2 and p38MAPK in endothelial cells. J Nutr Biochem. 2009;20:1021–27. doi: 10.1016/j.jnutbio.2008.12.001. [DOI] [PubMed] [Google Scholar]
- 32.Zhao X, Liu F, Jin H, Li R, Wang Y, Zhang W, et al. Involvement of PKCalpha and ERK1/2 signaling pathways in EGCG’s protection against stress-induced neural injuries in Wistar rats. Neuroscience. 2017;346:226–37. doi: 10.1016/j.neuroscience.2017.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mi Y, Qi G, Fan R, Qiao Q, Sun Y, Gao Y, et al. EGCG ameliorates high-fat- and high-fructose-induced cognitive defects by regulating the IRS/AKT and ERK/CREB/BDNF signaling pathways in the CNS. FASEB J. 2017;31:4998–5011. doi: 10.1096/fj.201700400RR. [DOI] [PubMed] [Google Scholar]
- 34.Liu S, Sun Z, Chu P, Li H, Ahsan A, Zhou Z, et al. EGCG protects against homocysteine-induced human umbilical vein endothelial cells apoptosis by modulating mitochondrial-dependent apoptotic signaling and PI3K/Akt/eNOS signaling pathways. Apoptosis. 2017;22:672–80. doi: 10.1007/s10495-017-1360-8. [DOI] [PubMed] [Google Scholar]
- 35.Zhou X, Wang Y, Metselaar HJ, Janssen HL, Peppelenbosch MP, Pan Q. Rapamycin and everolimus facilitate hepatitis E virus replication: revealing a basal defense mechanism of PI3K-PKB-mTOR pathway. J Hepatol. 2014;61:746–54. doi: 10.1016/j.jhep.2014.05.026. [DOI] [PubMed] [Google Scholar]
- 36.Zheng Y, Li J, Johnson DL, Ou JH. Regulation of hepatitis B virus replication by the ras-mitogen-activated protein kinase signaling pathway. J Virol. 2003;77:7707–12. doi: 10.1128/JVI.77.14.7707-7712.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guo H, Zhou T, Jiang D, Cuconati A, Xiao GH, Block TM, et al. Regulation of hepatitis B virus replication by the phosphatidylinositol 3-kinase-akt signal transduction pathway. J Virol. 2007;81:10072–80. doi: 10.1128/JVI.00541-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Veto B, Bojcsuk D, Bacquet C, Kiss J, Sipeki S, Martin L, et al. The transcriptional activity of hepatocyte nuclear factor 4 alpha is inhibited via phosphorylation by ERK1/2. PLoS One. 2017;12:e172020. doi: 10.1371/journal.pone.0172020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tipoe GL, Leung TM, Hung MW, Fung ML. Green tea polyphenols as an anti-oxidant and anti-inflammatory agent for cardiovascular protection. Cardiovasc Hematol Disord Drug Targets. 2007;7:135–44. doi: 10.2174/187152907780830905. [DOI] [PubMed] [Google Scholar]
- 40.Li MJ, Yin YC, Wang J, Jiang YF. Green tea compounds in breast cancer prevention and treatment. World J Clin Oncol. 2014;5:520–28. doi: 10.5306/wjco.v5.i3.520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.de Boussac H, Ratajewski M, Sachrajda I, Koblos G, Tordai A, Pulaski L, et al. The ERK1/2-hepatocyte nuclear factor 4alpha axis regulates human ABCC6 gene expression in hepatocytes. J Biol Chem. 2010;285:22800–08. doi: 10.1074/jbc.M110.105593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hatzis P, Kyrmizi I, Talianidis I. Mitogen-activated protein kinase-mediated disruption of enhancer-promoter communication inhibits hepatocyte nuclear factor 4alpha expression. Mol Cell Biol. 2006;26:7017–29. doi: 10.1128/MCB.00297-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chou YC, Chen ML, Hu CP, Chen YL, Chong CL, Tsai YL, et al. Transforming growth factor-beta1 suppresses hepatitis B virus replication primarily through transcriptional inhibition of pregenomic RNA. Hepatology. 2007;46:672–81. doi: 10.1002/hep.21726. [DOI] [PubMed] [Google Scholar]
- 44.Hosel M, Quasdorff M, Wiegmann K, Webb D, Zedler U, Broxtermann M, et al. Not interferon, but interleukin-6 controls early gene expression in hepatitis B virus infection. Hepatology. 2009;50:1773–82. doi: 10.1002/hep.23226. [DOI] [PubMed] [Google Scholar]