

Abstract
Background
Due to the frequent reassortment and zoonotic potential of influenza A viruses, rapid gain of sequence information is crucial. Alongside established next-generation sequencing protocols, the MinION sequencing device (Oxford Nanopore Technologies) has become a serious competitor for routine whole-genome sequencing. Here, we established a novel, rapid and high-throughput MinION multiplexing workflow based on a universal RT-PCR.
Methods
Twelve representative influenza A virus samples of multiple subtypes were universally amplified in a one-step RT-PCR and subsequently sequenced on the MinION instrument in conjunction with a barcoding library preparation kit from the rapid family and the MinIT performing live base-calling. The identical PCR products were sequenced on an IonTorrent platform and, after final consensus assembly, all data was compared for validation. To prove the practicability of the MinION-MinIT method in human and veterinary diagnostics, we sequenced recent and historical influenza strains for further benchmarking.
Results
The MinION-MinIT combination generated over two million reads for twelve samples in a six-hour sequencing run, from which a total of 72% classified as quality screened, trimmed and mapped influenza reads to produce full genome sequences. Identities between the datasets of > 99.9% were achieved, with 100% coverage of all segments alongside a sufficient confidence and 4492fold mean depth. From RNA extraction to finished sequences, only 14 h were required.
Conclusions
Overall, we developed and validated a novel and rapid multiplex workflow for influenza A virus sequencing. This protocol suits both clinical and academic settings, aiding in real time diagnostics and passive surveillance.
Keywords: Nanopore sequencing, MinION, Influenza A viruses, Full genome sequencing, Next-generation sequencing, Avian influenza viruses, Multiplexing
Background
Next-generation sequencing (NGS) methods, especially second-generation sequencers, have shown their capability of whole-genome sequencing (WGS) over the past decade for a wide spectrum of pathogens including influenza A viruses (IAV) [1, 2]. Due to widespread avian influenza virus (AIV) outbreaks with high mortality among poultry and wild birds in combination with the unceasing risk of zoonosis, avian origin IAV has devastating economic and anthropological impacts [3, 4]. Frequent reassortment events and vast genetic diversity of these viruses show the necessity for fast and accurate WGS [5].
While the characterisation of IAV has greatly benefitted from WGS utilising first- and second-generation sequencers, limiting factors such as high costs, process duration, extensive protocols and large, stationary equipment leave room for improvement [6]. In recent times, the new era of third-generation sequencers has started to fill this gap. Among these, the portable MinION third-generation nanopore sequencing device (Oxford Nanopore Technologies, Oxford, UK; ONT) has developed to become a serious competitor [7], especially in regard to real-time sequencing and multiplex barcoding possibilities [8].
Employing the MinION with a broad range of IAV subtypes of both avian and human origin, we developed and validated a high-throughput sequencing workflow and speedy screening method for unknown IAV samples. During an outbreak situation, this method could dramatically reduce the cost and time for WGS, thus accelerating the response and aiding in disease control.
Methods
Nucleic acid extraction
Twelve egg-grown avian virus isolates (Table 1), four human IAV isolates and two avian swab samples (Additional File 1, Table S1) of different subtypes were collected in the German National Reference Laboratory for Avian Influenza, located at the Friedrich-Loeffler-Institut, Insel Riems, Germany. RNA was extracted using TRIZOL LS (Thermo Fisher Scientific, Waltham, USA) and the QIAamp Viral RNA Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions.
Table 1.
Summary of MinION data from all reference samples sequenced with name, subtype and barcode. Mean coverage, mapped reads and number of nucleotide differences are shown for each individual segment. The consensus identity is calculated for all segments in %
| Barcode | Sample | Subtype | PB2 | PB1 | PA | HA | NA | NP | MP | NS | Sum | Consensus identity % | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | R30–06 | H1N1 | Mean Coverage | 749.9 | 188.3 | 700.9 | 2771.9 | 8756.7 | 7987.3 | 13,551.5 | 11,637.7 | 5793.025 | 99.98528113 |
| Mapped Reads | 8102 | 1131 | 4059 | 8207 | 22,156 | 20,995 | 31,635 | 30,095 | 126,380 | ||||
| Nucleotide Differences | 1 | 1 | 2 | ||||||||||
| 2 | R3111–07 | H2N9 | Mean Coverage | 2250.2 | 1360.5 | 1001 | 4396.1 | 626.7 | 4582.6 | 8212.6 | 7477 | 3738.3375 | 99.99264057 |
| Mapped Reads | 32,171 | 11,283 | 7372 | 11,560 | 2390 | 10,367 | 18,613 | 18,298 | 112,054 | ||||
| Nucleotide Differences | 1 | 1 | |||||||||||
| 3 | R2555–06 | H3N1 | Mean Coverage | 6373.3 | 2200.9 | 2080.6 | 929.1 | 1968 | 3874.8 | 5749.1 | 5016.4 | 3524.025 | 99.98528113 |
| Mapped Reads | 40,737 | 14,924 | 14,166 | 4074 | 6248 | 9628 | 13,859 | 8970 | 112,606 | ||||
| Nucleotide Differences | 1 | 1 | 2 | ||||||||||
| 4 | HAIV-81 | H4N6 | Mean Coverage | 1462 | 3401.4 | 792.2 | 4161.2 | 8039.7 | 11,356.3 | 14,790.1 | 9955.9 | 6744.85 | 99.98528113 |
| Mapped Reads | 18,920 | 20,238 | 6874 | 11,778 | 20,138 | 27,900 | 35,597 | 23,680 | 165,125 | ||||
| Nucleotide Differences | 1 | 1 | 2 | ||||||||||
| 5 | R1612–08 | H5N3 | Mean Coverage | 5033.4 | 3013.7 | 5260.7 | 1441.3 | 3776.2 | 1802.1 | 6314.7 | 5898.6 | 4067.5875 | 99.96320283 |
| Mapped Reads | 35,383 | 19,354 | 31,659 | 8876 | 10,321 | 4821 | 14,220 | 12,908 | 137,542 | ||||
| Nucleotide Differences | 1 | 1 | 2 | 1 | 5 | ||||||||
| 6 | R617–07 | H6N2 | Mean Coverage | 3933.5 | 2889.7 | 2995.6 | 4024 | 3403.6 | 3564.3 | 9820.9 | 5480.4 | 4514 | 99.99264057 |
| Mapped Reads | 29,256 | 21,688 | 23,359 | 13,971 | 9179 | 8292 | 22,581 | 13,515 | 141,841 | ||||
| Nucleotide Differences | 1 | 1 | |||||||||||
| 7 | R11–01 | H7N7 | Mean Coverage | 5558.2 | 1201 | 1696.6 | 3737.4 | 3467.6 | 6623.9 | 7390.9 | 6832.7 | 4563.5375 | 99.98528113 |
| Mapped Reads | 46,729 | 12,888 | 17,912 | 15,132 | 8502 | 16,267 | 18,781 | 16,333 | 152,544 | ||||
| Nucleotide Differences | 1 | 1 | 2 | ||||||||||
| 8 | R249–08 | H9N2 | Mean Coverage | 4445.5 | 3087.6 | 2260.4 | 1729.2 | 2652.5 | 2341.6 | 5403.8 | 6044.2 | 3495.6 | 99.98528113 |
| Mapped Reads | 32,149 | 20,998 | 14,076 | 6316 | 7275 | 6366 | 12,914 | 13,244 | 113,338 | ||||
| Nucleotide Differences | 1 | 1 | 2 | ||||||||||
| 9 | WV1677–03 | H10N4 | Mean Coverage | 4296.7 | 1296.4 | 2896.6 | 3175.4 | 2728 | 3976.2 | 7689.3 | 5656 | 3964.325 | 99.9779217 |
| Mapped Reads | 41,559 | 12,764 | 26,781 | 9445 | 8429 | 10,422 | 17,043 | 13,420 | 139,863 | ||||
| Nucleotide Differences | 2 | 1 | 3 | ||||||||||
| 10 | R2675–06 | H11N6 | Mean Coverage | 924.2 | 912.9 | 1378.1 | 1499.1 | 2260.8 | 4951.9 | 4345.1 | 3944 | 2527.0125 | 99.99264057 |
| Mapped Reads | 14,361 | 12,470 | 13,834 | 5702 | 5402 | 12,233 | 10,004 | 9893 | 83,899 | ||||
| Nucleotide Differences | 1 | 1 | |||||||||||
| 11 | R2613–06 | H13N8 | Mean Coverage | 866.9 | 1040.9 | 1199.3 | 6152 | 5035.8 | 10,790.8 | 19,984.4 | 16,654.6 | 7715.5875 | 99.9779217 |
| Mapped Reads | 9250 | 10,656 | 10,600 | 19,030 | 14,503 | 26,581 | 48,937 | 44,445 | 184,002 | ||||
| Nucleotide Differences | 1 | 1 | 1 | 3 | |||||||||
| 12 | Se-99 | H16N3 | Mean Coverage | 9922 | 426.7 | 599.1 | 115.8 | 2673.5 | 2096 | 5058.4 | 5190.9 | 3260.3 | 99.96320283 |
| Mapped Reads | 3843 | 2695 | 5041 | 393 | 11,992 | 5296 | 12,593 | 11,951 | 53,804 | ||||
| Nucleotide Differences | 1 | 1 | 1 | 2 | 5 |
IAV-End-RT-PCR and purification
RNA was amplified with one pair of influenza-specific primers (forward and reverse) at a 10 pmol/μl concentration [9] using Invitrogen Superscript III One-Step RT-PCR with Platinum Taq (Thermo Fisher Scientific). The IAV-End-RT-PCR included 5 μl RNA template, 1 μl forward and 1 μl reverse primer, 12.5 μl reaction mix, 1 μl SuperScript III RT/Platinum Taq mix and 4.5 μl RNase free water to obtain a total volume of 25 μl. In this protocol, all influenza segments are amplified simultaneously using a one-step RT-PCR and one set of primers adapted to the conserved 3′ and 5′ segment ends. Amplicon length therefore ranges from the smallest non-structural protein segment (866 nt) to the largest polybasic 2 protein segment (2316 nt), in accordance to the individual segment lengths.
Cycling conditions for the respective IAV-End-RT-PCR were conducted as described: An initial primary reverse transcription step of 30 min at 55 °C, then denaturation at 94 °C for 2 min, followed by five cycles of 94 °C for 30 s, 45 °C for 30 s and 68 °C for 3 min, then an additional 30 cycles of 94 °C for 30 s, 57 °C for 30 s and 68 °C for 3 min, and to conclude a final elongation step at 68 °C for 5 min.
After amplification, samples were purified with AMPure XP Magnetic Beads (Beckman Coulter, Fullerton, USA) in an × 0.65 sample volume to bead volume ratio. Quantification was conducted with the NanoDrop™ 1000 Spectrophotometer (Thermo Fisher Scientific).
Sequencing of IAV-End-PCR products – IonTorrent platform
The purified avian RT-PCR amplicons were sequenced on the IonTorrent platform (Thermo Fisher Scientific) as previously described [5, 10]. Before library preparation for the respective platform, the samples were mechanically fragmented to a 500 bp size on a Covaris M220 Ultrasonicator (Covaris Ltd., Brighton, UK). The GeneRead DNA L Core Kit (Qiagen) was subsequently used for library preparation with Xpress Barcode Adapters (Qiagen). After a following size selection and clean-up step with AMPure XP Beads (Beckman Coulter), the final library was quality checked on an Agilent Bioanalyzer 2100 (Agilent Technologies, Böblingen, Germany) and quantized via qPCR with the KAPA Library Quantification Kit (Roche, Mannheim, Germany). Sequencing was conducted on the IonTorrent S5XL (Thermo Fisher Scientific) in combination with the Ion OneTouch 2 System (Thermo Fisher Scientific), encompassing twelve AIV samples per Ion 530 Chip (Thermo Fisher Scientific).
Analysis of IonTorrent sequencing data
The raw data produced was screened for adapter and primer contamination, followed by a quality trimming step. By using the Geneious Software Suite (v11.1.5; Biomatters, Auckland, New Zealand), consensus sequences were generated via a map to reference approach utilising Bowtie2 (v2.3.0; pre-set “Medium Sensitivity”) [11].
Sequencing of IAV-End-PCR products – MinION sequencer
The identical purified IAV RT-PCR amplicons utilised on the IonTorrent were likewise employed for MinION sequencing along with the human strains. Following the manufacturer’s instructions, the Rapid Barcoding Kit (RBK-004, ONT) was applied: This 15 min two-step method includes a transposase for simultaneous cleaving of template DNA in conjunction with attachment of twelve barcodes to the cleaved ends (step 1), followed by pooling of the barcoded samples in the desired ratio and addition of Rapid Sequencing Adapters (step 2; ONT). After library preparation, the pooled samples were loaded onto a FLO-MIN106 R9.4.1 flow cell following the manufacturer’s instructions (ONT). A six-hour run was conducted with standard settings.
Analysis of MinION sequencing data
Real time basecalling was performed with the MinIT and integrated Guppy v3.0.4 software (ONT) to produce fast5 and fastQ files. The automatic real time division into passed and failed reads by the MinIT works as a quality check, removing reads with quality scores < 7. The quality checked reads were demultiplexed and trimmed for adapters and primers using ONT Guppy Barcoding Software v3.1.5 + 781ed575, followed by mappings and a final consensus production in Geneious (v11.1.5; Biomatters) with Bowtie2 (v2.3.0; pre-set “Medium Sensitivity”) [11]. Due to the segmented influenza genome and thus comparable length of ONT and IonTorrent reads, usage of an identical mapping process was possible. MinION data quality was documented with NanoPlot v1.25.0 [12].
Data availability
All sequence data (raw data, assemblies, consensus sequences) were made publicly available in the European Nucleotide Archive (ENA) under project accession PRJEB35098. Data accessions are summarised in Additional File 1, Table S2.
Results
The IAV-End-RT-PCR successfully amplified all samples and segments. In the case of the IonTorrent run, 33 h were necessary to achieve twelve full genomes with 100% coverage (Fig. 1). A sum of 11,499,351 reads were produced after quality check and removal of polyclonal reads with an average of 97% classified influenza reads, evenly distributed between the individual barcodes. Overall, 258,488 reads were without a barcode (2.2%). An average trimmed read length of 276 nt was achieved.
Fig. 1.
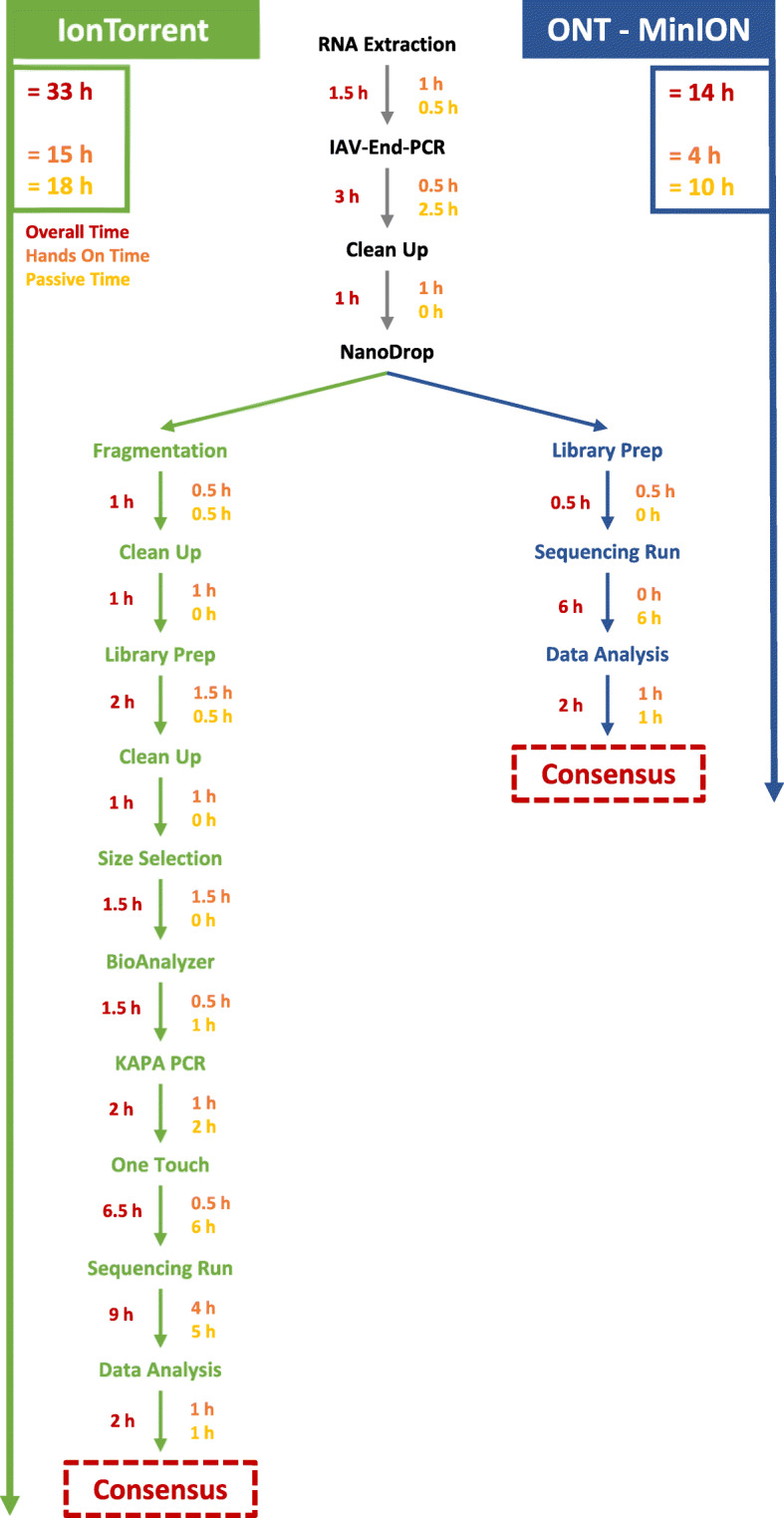
Comparison of IonTorrent S5XL and MinION workflow from RNA extraction to finished consensus sequences for twelve samples. Overall time is split into hands on and passive time needed for each protocol step
For MinION sequencing, starting at the RNA extraction to final consensus sequence, a total of 14 h was necessary to produce twelve complete genomes with 100% coverage (Fig. 1). In total, 2,090,778 reads were generated in the six-hour run, of which 90.43% classified as passed reads. After demultiplexing, a final count of 1,667,946 reads (79.78% of overall reads, 87.01% of passed reads) was available for further analysis leaving 248,963 passed reads (11.91% of overall reads, 12.98% of passed reads) without or unable to be allocated to a barcode. The read distribution between the twelve barcodes was roughly even, with barcodes 10 and 12 producing less than average sequence counts (Additional File 1, Table S1). Read quality was monitored by evaluating read length versus read quality per sample (Additional File 1, Figure S1) with an overall mean read quality reaching from 12.3 to 12.6, typical for MinION data. Read length ranges from 375 to 567 nt with an average of 479 nt, making the data accessible for standard mapping algorithms.
Analyses of the generated full genome sequences revealed 1,522,998 mapped MinION reads (91.30% of all passed and demultiplexed reads) and a consensus identity between the IonTorrent and MinION data of > 99.9%, with a range of one to a maximum of five nucleotide differences throughout the entire genome. Respectively, virtually all deviations were detected in homopolymer regions. Altogether, a mean coverage of 4492 reads was achieved for all MinION data (Table 1).
The additionally MinION sequenced human IAV isolates and avian swab samples passed all quality checks to produce six further complete genomes with similar read quality and high coverage. The quality check process included the primary distinction of passed (quality scores > 7) and failed reads (quality scores < 7) during live basecalling and further evaluation of the read quality by conducting NanoPlot (v1.25.0) to receive, inter alia, mean read quality and read length. Here, the cut-off value for mean read quality was set at ≥10 for further usage of the sequencing data.
Discussion
The proposed MinION workflow allows high-throughput sequencing in real time with a rapid library preparation protocol. Although MinION sequencing of IAV has previously been conducted [13], the novel combination of the IAV-End-RT-PCR with the Rapid Barcoding Kit (ONT) reduces the time for library preparation to a minimum and the analysis of low yield samples is achievable thanks to prior universal amplification. The respective RT-PCR allows for the production of less overall sequencing data due to the high proportion of viral influenza reads and a minimal host share, concurrently saving time and monetary means. The Rapid Barcoding Kit (ONT) also dramatically cuts the hands-on time needed for other NGS platforms, additionally reducing labour amount and, thus, expenditure. By utilising the MinIT (ONT), real time basecalling allows real time analysis, consequently leading to fast results, often crucial in the clinical setting.
Next-generation sequencers can produce immense amounts of data at a moderate cost, yet the application in clinical diagnostics is limited due to capital investment, complexity and time-consuming protocols. Easy, rapid and cost-efficient sequencing on the MinION platform could make sequencing accessible to a wide range of research backgrounds and might change the diagnostic process in the healthcare system. The availability of third-generation sequencers is pushing sequencing in the direction of becoming an integral part of many laboratories. The proposed IAV MinION sequencing protocol could easily be introduced into existing laboratory environments and allow direct, rapid and cost-efficient identification of diverse IAV strains. In addition, this protocol allows high-throughput sequencing of, for example, AIV samples from passive surveillance studies, shown to be of great importance for the molecular epidemiology of the worldwide AIV situation [14, 15].
The suitability of sequencing approaches for field surveillance was demonstrated with swine influenza viruses and the comparison to Illumina sequencing library strategies show comparable results with longer total analysis time and specific hands-on-time, respectively [16]. The use of multiplexing strategies is preferable due to the advantages of barcoding in higher throughput, better cost efficiency and decreased sequencing run time. The use of amplification with universal primers also allows an easier workflow and, in this study, allows better comparability of the achieved results, as the identical PCR products were sequenced on both platforms. PCR-free sequencing approaches for IAV have been described using direct RNA sequencing on the MinION platform to receive the complete coding genome of IAV [7]. This method allows avoidance of a prior PCR, and thus, the concurrent potentially resulting bias. Although this method is of great interest, the samples were high titre isolates and the limit of detection reached Ct values of 17, which lies outside the range of most clinical samples. Albeit the produced consensus sequences shared maximally 98.97% identity to the reference, the complexity and expenditure to achieve these results is not viable in the clinical setting. Multiplexing of direct RNA sequencing has yet to be produced for the MinION platform, additionally raising expenditures. In comparison, the here described MinION workflow allows superior consensus identity levels for low viral load samples at a lower cost and time consumption. In the future, direct RNA sequencing will certainly play a significant role; however, the current technological capabilities will most likely first allow the entry of third-generation sequencers into the clinical setting, in line with the aim of our proposed protocol.
Metagenomic nanopore sequencing has previously been piloted for clinical respiratory IAV samples [17]. The results of this study are promising for the combination of nanopore sequencing and metagenomics. However, although the detection of individual IAV reads was described in samples with Ct values of up to 36, whole genome sequences with the necessary coverage depth were only achieved at much lower Ct levels. Additionally, far larger datasets are needed to attain full genome coverage in comparison to the PCR-based MinION protocol. Deep sequencing and the concomitant possibility of SNP and variant detection is likewise only achievable with greater coverage depth, also attainable with the proposed protocol.
A multitude of varying bioinformatics analysis tools are available for sequencing data produced on ONT platforms, all aiming towards the improvement of the currently standing error profile. Especially in the current worldwide SARS-CoV-2 pandemic, third-generation sequencing platforms with distinct bioinformatic workflows have been implemented to obtain whole genomes [18, 19]. In the proposed protocol, the aim was to concentrate on the laboratory work and keep the implementation as accessible as possible, thus allowing better comparisons of both platforms via the utilisation of the identical annotation workflow. The comparability was shown by the generation of highly identical consensus sequences proving that the higher error rates of individual reads could be compensated by higher coverage.
The error rate of individual reads likewise affects demultiplexing of reads [20]. Misindexed reads are a known problem for nanopore sequencing, with on average 0.056% of total reads assigned to the incorrect barcode. When conducting metagenomic sequencing, a value of 0.056% misindexed reads can immensely influence the final genome construction, as often a fast majority of > 99% of the produced reads derive from the host [21, 22], leaving only few viral reads for analysis. Using the proposed method, high coverage and a large percent of viral influenza reads are expected due to the upstream IAV-End-RT-PCR (here: 91.3% of all passed and demultiplexed MinION reads were identified as influenza reads). Therefore, the fraction of misindexed reads does not affect the final consensus production and is, thus, in this case negligible.
Overall, sample processing of twelve samples can be achieved in 14 h, less than half the time required with the IonTorrent, without the need for large, expensive devices. Remarkable is the very low hands on time needed with the transposase-based rapid library MinION protocol. Although the accuracy of the MinION is known to be lower than other NGS platforms, especially struggling with homopolymer regions [23], adequate coverage leads to almost identical consensus sequences [24], as our data confirms. The MinION proved to be adaptably applicable for not only avian and human isolates, but also for representative clinical swab samples. In addition, swab samples from the recent 2020 outbreak of a novel IAV clade 2.3.4.4b H5N8 reassortant in Germany were successfully sequenced to produce full genome sequences using this method [25], demonstrating the practicality and applicability of the respective workflow.
Ongoing improvements from ONT are expected to advance basecalling accuracy with new technologies, e.g. the R10 flow cells, alongside the development of more accurate direct RNA sequencing kits to avoid PCRs and resulting biases. In addition, library preparation using automatic systems like the VolTRAX V2 (ONT) will allow even less hands-on time and reduce contamination while improving reproducibility.
Conclusions
In summary, we developed and validated a novel rapid multiplex workflow for IAV sequencing using the MinION in combination with a one-step RT-PCR and the Rapid Barcoding Kit (ONT). This protocol is ideal for both clinical and academic settings, aiding in real time diagnostics, applicable to any IAV sample and indispensable for active outbreaks and passive surveillance.
Supplementary information
Acknowledgements
We would like to thank Kathrin Steffen, Patrick Zitzow, Aline Maksimov and Diana Parlow for excellent technical assistance.
Abbreviations
- IAV
Influenza A viruses
- AIV
Avian influenza viruses
- HPAIV
Highly pathogenic avian influenza viruses
- LPAIV
Low pathogenic avian influenza viruses
- ONT
Oxford Nanopore Technologies
- RT-PCR
Reverse transcription polymerase chain reaction
- NGS
Next-generation sequencing
- WGS
Whole-genome sequencing
- ENA
European Nucleotide Archive
- RNA
Ribonucleic acid
- Nt
Nucleotide
Authors’ contributions
JK produced, analysed and curated the sequencing data and was responsible for the first written draft of the manuscript alongside further editing and figure work. TH provided the samples for sequencing, participated in the conceptualisation and methodology and was a major contributor in the editing of the manuscript. MB participated in the conceptualisation, in funding acquisition and supervision and was a major contributor in the editing of the manuscript. AP contributed to the formal data analyses, conceptualisation of the project, funding acquisition and overall supervision alongside playing a major role in the editing process of the manuscript. All authors read and approved the final manuscript.
Funding
This work was in part financed by EU Horizon 2020 program grant agreement “COMPARE” No. 643476, “VEO” No. 874735 and “DELTA-FLU” No. 727922. Open access funding provided by Projekt DEAL.
Availability of data and materials
The datasets generated and analysed in the current study are available in the European Nucleotide Archive (ENA) under project accession PRJEB35098. Data accessions are summarised in Additional File 1, Table S2.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary information accompanies this paper at 10.1186/s12879-020-05367-y.
References
- 1.Bertelli C, Greub G. Rapid bacterial genome sequencing: methods and applications in clinical microbiology. Clin Microbiol Infect. 2013;19:803–813. doi: 10.1111/1469-0691.12217. [DOI] [PubMed] [Google Scholar]
- 2.Vernikos G, Medini D, Riley DR, Tettelin H. Ten years of pan-genome analyses. Curr Opin Microbiol. 2015;23:148–154. doi: 10.1016/j.mib.2014.11.016. [DOI] [PubMed] [Google Scholar]
- 3.Globig A, Staubach C, Sauter-Louis C, Dietze K, Homeier-Bachmann T, Probst C, et al. Highly Pathogenic Avian Influenza H5N8 Clade 2.3.4.4b in Germany in 2016/2017. Front Vet Sci. 2017;4:240. doi: 10.3389/fvets.2017.00240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Paules C, Subbarao K. Influenza. Lancet. 2017;390:697–708. doi: 10.1016/S0140-6736(17)30129-0. [DOI] [PubMed] [Google Scholar]
- 5.Pohlmann A, Starick E, Grund C, Höper D, Strebelow G, Globig A, et al. Swarm incursions of reassortants of highly pathogenic avian influenza virus strains H5N8 and H5N5, clade 2.3.4.4b, Germany, winter 2016/17. Sci Rep. 2018;8:15. doi: 10.1038/s41598-017-16936-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Imai K, Tamura K, Tanigaki T, Takizawa M, Nakayama E, Taniguchi T, et al. Whole genome sequencing of influenza a and B viruses with the MinION sequencer in the clinical setting: a pilot study. Front Microbiol. 2018;9:2748. doi: 10.3389/fmicb.2018.02748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Keller MW, Rambo-Martin BL, Wilson MM, Ridenour CA, Shepard SS, Stark TJ, et al. Direct RNA sequencing of the coding complete influenza a virus genome. Sci Rep. 2018;8:14408. doi: 10.1038/s41598-018-32615-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.de Jesus JG, Giovanetti M, Rodrigues Faria N, LCJ A. Acute Vector-Borne Viral Infection: Zika and MinION Surveillance. Microbiol Spectr. 2019. 10.1128/microbiolspec.AME-0008-2019. [DOI] [PMC free article] [PubMed]
- 9.Hoffmann E, Stech J, Guan Y, Webster RG, Perez DR. Universal primer set for the full-length amplification of all influenza a viruses. Arch Virol. 2001;146:2275–2289. doi: 10.1007/s007050170002. [DOI] [PubMed] [Google Scholar]
- 10.Wylezich C, Papa A, Beer M, Höper D. A versatile sample processing workflow for metagenomic pathogen detection. Sci Rep. 2018;8:13108. doi: 10.1038/s41598-018-31496-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Langmead B, Salzberg SL. Fast gapped-read alignment with bowtie 2. Nat Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.de Coster W, D’Hert S, Schultz DT, Cruts M, van Broeckhoven C. NanoPack: visualizing and processing long-read sequencing data. Bioinformatics. 2018;34:2666–2669. doi: 10.1093/bioinformatics/bty149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang J, Moore NE, Deng Y-M, Eccles DA, Hall RJ. MinION nanopore sequencing of an influenza genome. Front Microbiol. 2015;6:766. doi: 10.3389/fmicb.2015.00766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alarcon P, Brouwer A, Venkatesh D, Duncan D, Dovas CI, Georgiades G, et al. Comparison of 2016-17 and previous epizootics of highly pathogenic avian influenza H5 Guangdong lineage in Europe. Emerg Infect Dis. 2018;24:2270–2283. doi: 10.3201/eid2412.171860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Venkatesh D, Poen MJ, Bestebroer TM, Scheuer RD, Vuong O, Chkhaidze M, et al. Avian influenza viruses in wild birds: virus evolution in a multihost ecosystem. J Virol. 2018. 10.1128/JVI.00433-18. [DOI] [PMC free article] [PubMed]
- 16.Rambo-Martin BL, Keller MW, Wilson MM, Nolting JM, Anderson TK, Vincent AL, et al. Influenza A Virus Field Surveillance at a Swine-Human Interface. mSphere. 2020;5. [DOI] [PMC free article] [PubMed]
- 17.Lewandowski K, Xu Y, Pullan ST, Lumley SF, Foster D, Sanderson N, et al. Metagenomic Nanopore sequencing of influenza virus direct from clinical respiratory samples. J Clin Microbiol. 2019. 10.1128/JCM.00963-19. [DOI] [PMC free article] [PubMed]
- 18.Wang M, Fu A, Hu B, Tong Y, Liu R, Liu Z, et al. Nanopore Targeted Sequencing for the Accurate and Comprehensive Detection of SARS-CoV-2 and Other Respiratory Viruses. Small. 2020:e2002169. 10.1002/smll.202002169. [DOI] [PMC free article] [PubMed]
- 19.Fauver JR, Petrone ME, Hodcroft EB, Shioda K, Ehrlich HY, Watts AG, et al. Coast-to-Coast Spread of SARS-CoV-2 during the Early Epidemic in the United States. Cell. 2020;181:990–996.e5. doi: 10.1016/j.cell.2020.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xu Y, Lewandowski K, Lumley S, Pullan S, Vipond R, Carroll M, et al. Detection of viral pathogens with multiplex Nanopore MinION sequencing: be careful with cross-talk. Front Microbiol. 2018;9:2225. doi: 10.3389/fmicb.2018.02225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gu W, Miller S, Chiu CY. Clinical metagenomic next-generation sequencing for pathogen detection. Annu Rev Pathol. 2019;14:319–338. doi: 10.1146/annurev-pathmechdis-012418-012751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Greninger AL. The challenge of diagnostic metagenomics. Expert Rev Mol Diagn. 2018;18:605–615. doi: 10.1080/14737159.2018.1487292. [DOI] [PubMed] [Google Scholar]
- 23.Forth JH, Forth LF, King J, Groza O, Hübner A, Olesen AS, et al. A deep-sequencing workflow for the fast and efficient generation of high-quality African swine fever virus whole-genome sequences. Viruses. 2019. 10.3390/v11090846. [DOI] [PMC free article] [PubMed]
- 24.Hoenen T. Sequencing of Ebola Virus Genomes Using Nanopore Technology. Bio Protoc. 2016. 10.21769/BioProtoc.1998. [DOI] [PMC free article] [PubMed]
- 25.King J, Schulze C, Engelhardt A, Hlinak A, Lennermann S-L, Rigbers K, et al. Novel HPAIV H5N8 Reassortant (Clade 2.3.4.4b) Detected in Germany. Viruses. 2020. 10.3390/v12030281. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All sequence data (raw data, assemblies, consensus sequences) were made publicly available in the European Nucleotide Archive (ENA) under project accession PRJEB35098. Data accessions are summarised in Additional File 1, Table S2.
The datasets generated and analysed in the current study are available in the European Nucleotide Archive (ENA) under project accession PRJEB35098. Data accessions are summarised in Additional File 1, Table S2.