

Abstract
Bromocriptine mesylate treatment was examined in dogs fed a high fat diet (HFD) for 8 wk. After 4 wk on HFD, daily bromocriptine (Bromo; n = 6) or vehicle (CTR; n = 5) injections were administered. Oral glucose tolerance tests were performed before beginning HFD (OGTT1), 4 wk after HFD began (Bromo only), and after 7.5 wk on HFD (OGTT3). After 8 wk on HFD, clamp studies were performed, with infusion of somatostatin and intraportal replacement of insulin (4× basal) and glucagon (basal). From 0 to 90 min (P1), glucose was infused via peripheral vein to double the hepatic glucose load; and from 90 to 180 min (P2), glucose was infused via the hepatic portal vein at 4 mg·kg−1·min−1, with the HGL maintained at 2× basal. Bromo decreased the OGTT glucose ΔAUC0–30 and ΔAUC0–120 by 62 and 27%, respectively, P < 0.05 for both) without significantly altering the insulin response. Bromo dogs exhibited enhanced net hepatic glucose uptake (NHGU) compared with CTR (~33 and 21% greater, P1 and P2, respectively, P < 0.05). Nonhepatic glucose uptake (non-HGU) was increased ~38% in Bromo in P2 (P < 0.05). Bromo vs. CTR had higher (P < 0.05) rates of glucose infusion (36 and 30%) and non-HGU (~40 and 27%) than CTR during P1 and P2, respectively. In Bromo vs. CTR, hepatic 18:0/16:0 and 16:1/16:0 ratios tended to be elevated in triglycerides and were higher (P < 0.05) in phospholipids, consistent with a beneficial effect of bromocriptine on liver fat accumulation. Thus, bromocriptine treatment improved glucose disposal in a glucose-intolerant model, enhancing both NHGU and non-HGU.
Keywords: dopamine D2 receptor, glucose tolerance, hypercaloric diet, liver
INTRODUCTION
The dopamine D2 agonist bromocriptine mesylate is used in type 2 diabetes management, sometimes in combination with metformin, but mechanisms of action are not clear (29, 92). Bromocriptine mesylate-QR (a unique quick-release formation of micronized bromocriptine) is an oral, centrally acting, sympatholytic dopamine D2 receptor agonist approved for treatment of hyperglycemia in patients with type 2 diabetes (20). Circadian-timed administration of bromocriptine in animal models of insulin resistance and glucose intolerance ameliorates aberrations in hypothalamic mechanisms involved in the regulation of peripheral glucose metabolism (3, 10, 25, 49, 51, 52, 66, 80, 82). In both seasonal and high-fat-fed insulin-resistant and glucose-intolerant rodents, the circadian peak of dopaminergic input activity in the biological clock pacemaker for the body, the superchiasmatic nucleus (SCN), is markedly diminished (50, 53), and this alteration contributes significantly to this metabolic state (9, 51, 53). Direct administration of dopamine to the SCN of high-fat-fed insulin-resistant and glucose-intolerant animals at the time of day that mimics its daily SCN peak in insulin-sensitive animals reverses the insulin-resistant, glucose-intolerant condition (53). Likewise, circadian-timed systemic bromocriptine administration to reestablish the SCN circadian peak in dopamine activity also improves the insulin-resistant, glucose-intolerant state in rodents (11, 14, 25, 52). The pathways by which such central circadian dopaminergic activities improve glucose metabolism include a reduction in elevated sympathetic tone, elevated cortisol secretion, and leptin resistance (3, 11, 25, 53, 81). The SCN controls the daily rhythm (phase and amplitude) of plasma glucose, insulin sensitivity, and β-cell responsiveness to glucose that follow a diurnal pattern in glucose-tolerant animals and humans (19, 39, 40, 45, 74), and there is evidence suggesting this pattern is impaired in type 2 diabetes (71).
In a double-blind study involving obese individuals with type 2 diabetes, 16 wk of bromocriptine-QR as monotherapy or add-on to sulfonylurea was associated with lower HbA1c and fasting glucose levels than placebo treatment (72). Moreover, at the end of treatment, the bromocriptine-QR-treated subjects exhibited a reduction in mean glucose concentrations following an oral glucose tolerance test (OGTT), compared with the increase in glucose concentrations observed in the placebo-treated control subjects despite similar mean plasma insulin concentrations in the two groups during the OGTT. In addition, bromocriptine versus placebo treatment was associated with increased glucose disposal under hyperinsulinemic euglycemic clamp conditions (72).
The liver is a key organ in glucose homeostasis by virtue of its ability to export glucose for the use of other tissues during fasting and to extract glucose to reduce glycemia in times of plenty. Failure of the liver to make the transition from net output to uptake of glucose in the postprandial state is a major contributor to the development of impaired glucose tolerance and hyperglycemia (2, 44). Thus, the question arises whether enhancement of net hepatic glucose uptake (NHGU) played a role in the improvement in glucose tolerance displayed by the subjects receiving bromocriptine. NHGU is difficult to quantify in the human because of the invasiveness of the hepatic portal vein catheterization required. Portal and hepatic vein catheterization is feasible in the dog, and the canine model provides a useful method for examining the liver’s role in glucose disposal (43).
Dopamine receptor agonists inhibit prolactin secretion, and the effectiveness of bromocriptine in suppressing prolactin levels has been suggested to be a surrogate marker of its effectiveness in improving glucose tolerance (13). Thus, for maximum effectiveness, bromocriptine must be given close to the time of morning waking, when prolactin secretion normally peaks. The present studies were carried out to determine whether daily administration of bromocriptine could improve hepatic and/or whole body glucose disposal in a dogs fed a high-fat-diet (HFD). The HFD has previously been demonstrated to have impaired glucose tolerance, as well as blunted hepatic glucose disposal under hyperinsulinemic hyperglycemic clamp conditions (17).
RESEARCH DESIGN AND METHODS
Animal Care, Diet, Timeline, and Surgical Procedures
The protocol was approved by the Vanderbilt University Institutional Animal Care and Use Committee, and the animals were housed and cared for according to Association for Assessment and Accreditation of Laboratory Animal Care International guidelines. Studies were carried out on 11 adult male mongrel dogs acquired from a USDA-approved vendor. The dogs were separated into two groups: placebo control (CTR; n = 5) and treated with bromocriptine mesylate (Bromo; n = 6). They were housed in a facility with a 0600–1800 light cycle and initially received a standard chow and meat diet calculated to be weight maintaining (16).
Timeline.
An OGTT was carried out on all dogs during week 0, before any treatment. The dogs were then started on the HFD, with fat, protein, and carbohydrate providing 52, 22, and 26% of total energy, respectively (17). The diet was fed in amounts that were in excess of energy maintenance needs. The dogs were maintained on the HFD until the end of the study procedures. A record was kept of the amount of food provided each day as well as the amount remaining from the previous day, and the dogs were weighed at least once weekly.
After 4 wk on the diet, the Bromo dogs underwent a second OGTT to assess change in the glycemic excursion and hormonal responses since week 0. Subsequently, each dog received a daily subcutaneous injection of bromocriptine, 15 µg/kg (Bromo group), or vehicle (CTR group; 30% ethanol in sterile water, ~0.4 mL to approximate volume in the Bromo group) within 30 min of the beginning of the light cycle, with the daily injections continuing for 4 wk. The Bromo dose was based on bromocriptine-QR pharmacodynamic studies in humans (82), the effective dose of bromocriptine to inhibit plasma prolactin in dogs, and bromocriptine pharmacokinetic dose response data in dogs (data not shown). The circadian timing of the Bromo injections was set to the onset of daily locomotor activity, which approximates the daily peak in brain dopamine activity associated with daily waking in studied species (50, 53, 59, 82). During the fourth week of Bromo or vehicle treatment, all dogs underwent a final OGTT; 3–4 days after the OGTT, each dog underwent a hyperinsulinemic hyperglycemic clamp experiment.
Surgical preparation.
After 10–12 days of bromocriptine or vehicle treatment, each dog underwent surgery under general anesthesia, as previously described (16), to insert sampling catheters in the femoral artery, hepatic portal vein, and left common hepatic vein; blood flow probes around the portal vein and hepatic artery; and a splenic and a jejunal vein catheter to allow infusion into the portal circulation.
Experimental Design
OGTTs.
On the day before each OGTT, each dog was fed one can of meat at noon to ensure equivalent food intake in all animals. The dogs were then fasted until the following morning. On the morning of each OGTT, the dog was placed in a Pavlov harness, and a catheter was introduced via a peripheral leg vein and threaded into a deep venous location to allow collection of samples for glucose, insulin, glucagon, and C-peptide (16). After a 50-min period of acclimation, a 10-min basal sampling period began, followed by oral administration of the glucose load (0.9 g glucose/kg; Polycose, Abbott Nutrition, dissolved in 60 mL of water). The body weight on the day of the first OGTT was used for calculation of all subsequent glucose doses. At the end of sampling (180 min after administration of the glucose load), the catheter was removed.
Hyperinsulinemic hyperglycemic clamp experiment.
For the hyperinsulinemic hyperglycemic clamp experiment, the fasting conditions were exactly as for the OGTTs. Each experiment consisted of a 100-min equilibration period (−120 to −20 min), a 20-min period of basal sampling (−20 to 0 min), and a 180-min experimental period divided into two subperiods (P1, 0–90 min; P2, 90–180 min). At −120 min, a priming dose of [3-3H]glucose (38 μCi) was administered, and a constant infusion of [3-3H]glucose (0.38 μCi/min) was begun. At time 0 (10:30 AM for all dogs), a constant peripheral venous infusion of somatostatin (0.8 µg·kg−1·min−1) began, and continuous intraportal infusions of insulin (4× basal, 1.2 mU·kg−1·min−1) and glucagon (basal, 0.55 ng·kg−1·min−1) were started. A peripheral infusion of 50% glucose was administered, with the infusion rate adjusted as needed to double the hepatic glucose load. In P2, an intraportal infusion of 20% glucose was administered (4 mg·kg−1·min−1), and the peripheral glucose infusion rate was adjusted as necessary to maintain the hepatic glucose load at the same level as in P1. At the end of the study, each animal was anesthetized with pentobarbital sodium while all infusions continued, liver tissue was freeze-clamped in situ, and the animal was then euthanized.
Analyses
Arterial, portal vein, and hepatic vein plasma glucose, [3H]glucose, glucagon, insulin, and nonesterified fatty acid (NEFA) levels and blood lactate and glycerol concentrations were measured by standard methods (28, 61). Hepatic glycogen concentrations were assessed by the method of Keppler and Decker (41). Hepatic tissue fatty acids were extracted, and individual species were quantified as previously described (15).
Western blotting procedures for hepatic phosphorylated (p)Akt/Akt, pGSK/GSK, and glucokinase (GK) were performed as described previously (24, 75). Actin and glucokinase regulatory protein (GKRP) antibodies were purchased from Santa Cruz Biotechnology, and glycogen synthase (GS) and pAkt (Ser473) antibodies were purchased from Cell Signaling. Dr. Masakazu Shiota (Vanderbilt University School of Medicine) provided the GK antibody. ImageJ software (http://rsb.info.nih.gov/ij/) was used for quantification. Liver samples from four overnight-fasted dogs (maintained on a standard canine diet of meat and chow) that had undergone no experimental intervention were utilized for reference purposes; they are referred to as Basal samples.
Western blotting of muscle tissue and hepatic assays not listed in the previous paragraph were performed as follows. Frozen muscle tissue (kept at −80°C until use) was ground with a mortar and pestle on dry ice and then homogenized in Radio-Immune Precipitation Assay (RIPA) buffer (ThermoFisher, Waltham, MA; cat. no. 89900) supplemented with protease and phosphatase inhibitors (aprotinin, bestatin, E-64, leupeptin, sodium fluoride, sodium orthovanadate, sodium pyrophosphate, β-glycerophosphate, and EDTA) (ThermoFisher, cat. no. 78442). Muscle protein concentration in the homogenized lysate was quantified with Rapid Gold protein assay (ThermoFisher, cat. no. A53226). Proteins were separated on 4–15% Criterion TGX Stain-Free Gel (Bio-Rad, Hercules, CA; cat. no.5678084) at 200 V for 45 min, activated with UV light with Bio-Rad ChemiDoc XRS+, and transferred to low-fluorescence polyvinylidene difluoride (LFPVDF) membrane (Bio-Rad, cat. no.1620263). Proteins were transferred to LFPVDF membrane with Criterion blotter (Bio-Rad, cat. no. 1704070) at 100 V for 40 min. Total protein in each lane was detected with Bio-Rad ChemiDoc XRS+ fluorescence channel and quantified with Bio-Rad ImageLab 6.0.1 software per manufacturer’s instructions using software defaults. Membrane blocking agent was chosen based on primary antibody manufacturer’s instructions: 5% bovine serum albumin for 1 h for Akt, pAkt (Ser473), and pGSK-3α/β antibodies; EveryBlot Blocking Buffer (Bio-Rad cat. no. 12010020) for 5 min followed by 5% nonfat dry milk (Bio-Rad cat. no. 1706404) for 1 h for acyl-CoA dehydrogenase medium chain (ACADM), carbohydrate response element-binding protein (CHREBP), peroxisome proliferator-activated receptor-α (PPARα), thioredoxin-interacting protein (TXNIP), and sirtuin 1 (SIRT1) antibodies. After blocking, membranes were incubated overnight with mild shaking at room temperature in Tris-buffered saline (TBS) buffer (Bio-Rad, cat. no. 1706435) supplemented with 0.1% Tween 20 (Bio-Rad, cat. no. 1706531) (TBST buffer) with the following primary antibodies from Cell Signaling Technology (CST, Beverly, MA), Novus (Centennial, CO), and Abcam (Cambridge, MA): pAkt (Ser473) (CST, cat. no. 9271), diluted 1:1,000; Akt (CST, cat. no. 9272), diluted 1:1,000; ACADM/MCAD (medium-chain acyl-CoA dehydrogenase; Abcam, cat. no. ab110296), diluted to 0.125 µg/m1; CHREBP (Abcam, cat. no. ab92809), diluted 1:1,000; PPARα (Novus, cat. no. NB600-636), diluted 1:1,000; TXNIP (Abcam, cat. no. ab188865), diluted 1:1,000; pGSK-3α/β (Ser21/9) (CST, cat. no.8566), diluted 1:1,000; SIRT1 (Abcam, cat. no. ab12193), diluted 1:2,000; IDE (Abcam, cat. no. ab32216), diluted to 1 µg/mL; insulin-degrading enzyme (IDE; SCBT, cat. no. sc-393887), diluted 1:100; stearoyl-CoA desaturase-1 (SCD1; Proteintech, cat. no. 23393-1-AP), diluted 1:1,000; and elongation of very long-chain fatty acids protein-6 (ELOVL6; Abcam, cat. no. ab69857), diluted to1 µg/mL. Following overnight incubation with primary antibodies, membranes were rinsed five times for 5 min in TBST buffer. Secondary horseradish peroxidase (HRP)-conjugated antibodies were purchased from Bio-Rad (Immun-Star Goat Anti-Rabbit (GAR)-HRP Conjugate; Bio-Rad, cat. no. 1705046, and Immun-Star Goat Anti-Mouse (GAM)-HRP Conjugate; Bio-Rad, cat. no. 1705047), reconstituted in 2 mL of distilled water, diluted 1:20,000 in TBST buffer, incubated with the membranes for 1 h at room temperature, and then rinsed five times for 5 min in TBST. Chemiluminescence of the HRP-conjugated antibody-probed membrane was detected with Clarity Western ECL Substrate (Bio-Rad, cat. no. 1705061), recorded with a Bio-Rad ChemiDoc XRS+ chemiluminescence channel, quantified with Bio-Rad ImageLab 6.0.1 software per manufacturer’s instructions using software defaults, and adjusted by total protein amount in each sample. Chemiluminescence and fluorescence intensity quantification results [expressed in arbitrary units (AU)] were compared only between the samples blotted on the same membrane. Molecular weight of each protein of interest was determined with Precision Plus Protein Kaleidoscope Standards (Bio-Rad, cat. no.1610375) and Glogos II markers (Agilent, Santa Clara, CA) and confirmed to be consistent with the molecular weight of the protein of interest.
GS and phosphorylase activities were determined by the method of Golden et al. (31). The method described by Barzilai and Rossetti (1) was utilized for assessment of GK activity.
Calculations
During the OGTTs, the areas under the curve (AUCs) of the glucose and insulin responses were calculated as the change from their basal concentrations over the first 30 and 120 min after glucose ingestion (ΔAUC0–30min and ΔAUC0–120min, respectively). The 0- to 120-min time period was chosen based on previous reports demonstrating that glucose absorption is largely complete in that time frame (26, 32, 85). The C-peptide response was calculated as the AUC from 0 ng/mL (AUC0–30min and AUC0–120min, respectively) (4, 57). The C-peptide/insulin ratio, an indicator of insulin clearance (4, 38, 57, 86), was calculated for the 0- to 30- and 0- to 120-min periods after glucose ingestion during each OGTT.
The hepatic glucose load entering the liver (HGL) was calculated as follows: arterial blood glucose concentration (GA; this is nonradioactively labeled glucose) × total hepatic blood flow (HBF; arterial + portal vein flow) + portal glucose infusion rate (mg·kg−1·min−1) – gut uptake of glucose (15). Gut uptake of glucose was calculated as portal vein blood flow (PBF) × (GA − GP), where GP indicates the portal vein glucose concentration (62). Net hepatic glucose balance (NHGB) was calculated with direct and indirect methods. The direct calculation was (GH × hepatic vein flow) – [(GA × arterial flow) + (GP × portal vein flow)], where GH is the hepatic vein glucose concentration, and plasma or blood flow rates are utilized as appropriate. The indirect method was HGL minus the hepatic glucose load exiting the liver (where the load exiting is HBF × GH). Net hepatic balance of substrates other than glucose was calculated by the direct method only. Hepatic fractional extraction of glucose was calculated as net hepatic glucose uptake (NHGU)/HGL. Hepatic glucose uptake (HGU; unidirectional) was calculated as the hepatic fractional extraction of [3H]glucose × HGL. The hepatic fractional extraction of [3H]glucose (unitless) was calculated as the hepatic balance of [3H]glucose divided by the hepatic load of [3H]glucose (15). We converted plasma [3H]glucose to blood values by using conversion factors previously described (65). Plasma insulin and glucagon levels entering the hepatic sinusoids were calculated as described by Chu et al. (27). Glycogen synthesis by the direct pathway = hepatic [3H]-labeled glycogen ÷ average inflowing plasma [3H]glucose specific radioactivity (79).
Ratios of selected fatty acid species were utilized as markers of the activities of enzymes involved in hepatic lipid metabolism, based on previous reports (5, 37, 46, 68–70, 83, 84). These include 18:0/16:0 as an index of ELOVL6 activity, 16:1/16:0 as an index of SCD1 activity, and the ratio 16:0/18:2ω6 as an index of de novo lipogenesis.
Statistical Analyses
Data are expressed as means ± SE. Mean values for P1 or P2 are the means for the last hour of each period. Two-way analysis of variance with or without repeated measures design was used for comparison between groups, one-way analysis of variance was used for assessment of changes over time with a single group, and post hoc analysis was performed using the Student-Newman-Keuls multiple comparisons test (SigmaStat; Systat, Richmond, CA). A P value of <0.05 was considered significant. For Western blot data, statistical analyses were performed using Student’s two-tailed t test to determine the treatment difference for Western blot detected levels of all muscle proteins except GSKβ-Ser9, where a one-tailed t test was used, since it was established that the bromocriptine treatment improved insulin-stimulated glucose disposal, glucose tolerance, and muscle activated Akt before testing for any change in GSKβ-Ser9 and a significant increase in its level was anticipated.
RESULTS
Weight Change and Food Intake
The body weights before the study were very similar in the two groups (20.9 ± 1.1 and 21.5 ± 0.5 kg in CTR and Bromo, respectively, P = 0.29). All dogs gained weight during the initial 4 wk of HFD consumption before drug treatment, with the CTR dogs gaining less than the Bromo group (Δ2.5 ± 0.9 and 4.8 ± 0.5 kg, respectively, P < 0.05), despite the fact that there was no difference in the treatment of the two groups during that time period. Body weight declined slightly and to a similar extent during vehicle or bromocriptine treatment (−0.3 ± 0.7 and −0.6 ± 0.6 kg, respectively, P = 0.2 between groups), so that the final weights were also not different between groups. Food intake relative to body weight declined in both groups during the final 4 wk of study compared with the first 4 wk, but the decrease was significantly greater in the Bromo dogs (daily decrease of 28 ± 13 and 73 ± 8 kcal/kg body wt, CTR and Bromo, respectively, P < 0.005). No vomiting was observed in either the Bromo or CTR dogs.
OGTT Data
Glucagon concentrations remained at basal levels during each of the OGTTs and did not differ between groups at any time (see Supplemental Table S1; https://doi.org/10.6084/m9.figshare.11538909).
Week 0.
The glucose, C-peptide, and insulin concentrations during the initial OGTTs were not significantly different in the two groups, as anticipated (Fig. 1, A–L). The fasting plasma glucose values were 104 ± 3 and 108 ± 1 mg/dL (P = 0.09), and the peak plasma glucose concentrations were 147 ± 10 and 143 ± 7 mg/dL (P = 0.33) in CTR and Bromo, respectively. The ΔAUC0–30min and ΔAUC0–120min for the insulin response relative to the glycemic response did not differ between groups (Fig. 1, M and N). Similarly, the C-peptide-to-insulin response ratio was not different in the two groups (Fig. 1, O and P).
Fig. 1.

Oral glucose tolerance test data. Deep venous plasma glucose (A and B), C-peptide (E and F), and insulin (I and J) concentrations during consumption of the chow and meat diet (week 0; ○), after 4 wk of high-ft diet (HFD) but before bromocriptine treatment (week 4; Bromo group only; △), and after 3.5 wk of bromocriptine or placebo treatment (week 8; ●); data are means ± SE. Area under the curve changes from basal (ΔAUCs) during the first 30 and 120 min after glucose ingestion, respectively, are shown for the glucose response in C and D and for the insulin response in K and L; ΔAUCs of the insulin response relative to that of glucose are shown in M and N. AUC0–30min and AUC0–120min, respectively, of the C-peptide response (calculated as change from 0 rather than as change from basal) is shown in G and H; C-peptide AUC0–30min and AUC0–120min/insulin ΔAUC are shown in O and P. Data for each individual dog are shown in C, D, G, H, and K–P with a horizontal line showing the median for the group; controls (CTR) are shown with open symbols and group treated with bromocriptine mesylate (Bromo) with filled symbols; n = 5 CTR and n = 6 Bromo. Responses that are marked with the same letter are not significantly different from one another, whereas those marked only with different letters differ significantly (P < 0.05).
Week 4.
During the second OGTT in the Bromo group, after 4 wk on the HFD but before drug treatment, the basal glucose, insulin, and C-peptide concentrations were not significantly different from those of the initial OGTT (P = 0.10, 0.16, and 0.12, respectively). The ΔAUC0–30min for glucose and insulin also did not differ significantly from the week 0 results. The ΔAUC0–120 for both glucose and insulin had risen over those observed during the OGTT in week 0, however (P < 0.05), and the C-peptide AUC0–30min and AUC0–120min tended to be elevated compared with the week 0 response (P = 0.06). There were no significant differences in the insulin/glucose and the C-peptide/insulin ratios between the first and second OGTTs within the Bromo group, either in AUC0–30min or AUC0–120min.
Week 8.
At the time of the final OGTT, the fasting glucose, insulin, and C-peptide concentrations did not differ significantly from those during week 0 in either group. On the other hand, the ΔAUC0–30min and ΔAUC0–120min, respectively, for plasma glucose were 4.8 and 1.5 times the response during the first OGTT in CTR, but the values in Bromo were only 1.8 and 1.1 times those during the first OGTT (P < 0.05 between groups for both). The C-peptide AUC0–30min and AUC0–120min in the CTR group were 2.6 and 1.4 times as great, respectively, in the final OGTT versus the initial OGTT (P < 0.01), but the responses in the Bromo group did not differ between the first and third OGTTs. Moreover, the C-peptide AUC0–120min declined significantly in the Bromo group during the third versus the second OGTT. In both groups, the ΔAUC0–30min and ΔAUC0–120min of the insulin response increased significantly between the first and the final OGTTs, and there was no significant difference between groups in the insulin response during the final OGTT. The week 8 insulin/glucose ΔAUC0–30min did not increase significantly above the week 0 response in either group and did not differ between groups. The week 8 insulin/glucose ratio ΔAUC0–120min rose significantly above the week 4 value in the Bromo group, although it did not differ from the first OGTT in either group. The C-peptide/insulin ratio did not differ between the first and final OGTTs within the CTR group. However, the ratio decreased (P < 0.05) during the third OGTT compared with the first and second OGTTs within the Bromo group during both the 0- to 30- and the 0- to 120-min time frames. Also, the ratio was lower (P < 0.05) in Bromo versus CTR during the final OGTT in both the 0- to 30- and the 0- to 120-min time periods.
Clamp Study Results
Hormone concentrations.
Arterial plasma insulin concentrations tended to be higher in Bromo than in CTR both under basal conditions and during the clamp (P = 0.08) with the use of ANOVA (Table 1). When the concentrations during each study period (basal, P1, and P2) were compared between groups with t tests, the arterial insulin concentrations were significantly higher in Bromo than in CTR during P1. Similarly, although there were no significant differences in the hepatic sinusoidal concentrations during the clamp with ANOVA (P = 0.32 over the entire clamp period), t tests of the mean responses of the two groups during the basal period and P2 were greater in Bromo than in CTR (P < 0.05). Hepatic insulin clearance in Bromo during the basal period was 1.8-fold that in CTR (P < 0.05), and it was numerically higher during the clamp periods (P = 0.2 between groups during both clamp periods). Conversely, whole body insulin clearance was ~25% greater in CTR than in Bromo during the clamp periods (P < 0.05).
Table 1.
Hormone concentrations during clamp studies
| Basal Period | Clamp Period 1 | Clamp Period 2 | |
|---|---|---|---|
| Arterial insulin (µU/mL) | |||
| CTR | 7 ± 2 | 23 ± 2 | 24 ± 1 |
| Bromo | 10 ± 2 | 30 ± 3* | 27 ± 4 |
| Hepatic sinusoidal insulin (µU/mL) | |||
| CTR | 16 ± 3 | 72 ± 5 | 69 ± 6 |
| Bromo | 19 ± 4* | 75 ± 6 | 82 ± 6* |
| Arterial glucagon (ng/L) | |||
| CTR | 42 ± 4 | 44 ± 6 | 38 ± 3 |
| Bromo | 41 ± 5 | 47 ± 4 | 41 ± 10 |
| Hepatic sinusoidal glucagon (ng/L) | |||
| CTR | 50 ± 7 | 60 ± 4 | 56 ± 5 |
| Bromo | 51 ± 7 | 57 ± 3 | 60 ± 6 |
| Arterial c-peptide (ng/mL) | |||
| CTR | 0.3 ± 0.1 | 0.0 ± 0.0 | 0.0 ± 0.0 |
| Bromo | 0.2 ± 0.1 | 0.0 ± 0.0 | 0.0 ± 0.0 |
Data are means ± SE for 2 (basal) or 4 (clamp periods 1 and 2) sampling times; controls (CTR) n = 5; group treated with bromocriptine mesylate (Bromo) n = 6. There were no significant differences between groups with 2-way ANOVA (P = 0.1 between groups for hepatic sinusoidal insulin), and there were no significant differences between clamp periods 1 and 2 within either group; t tests were subsequently used to compare the insulin data during each of the 3 time periods.
P < 0.05 between groups by t test.
Arterial and hepatic sinusoidal plasma glucagon during the clamp period did not change significantly from basal in either group, and there were no differences between groups at any time (Table 1). Arterial plasma C-peptide concentrations fell to the limits of detection in both groups during the clamp, and the concentrations did not differ in the two groups during the basal period. Nevertheless, the C-peptide/insulin ratio in the basal period was approximately twice as great in CTR as it was in Bromo (2.3 ± 0.5 vs. 1.2 ± 0.2, respectively, P < 0.05).
Glucose parameters.
Fasting (basal period) plasma glucose concentrations were not different between groups (Fig. 2A). During P1, the arterial glucose concentrations were increased to ≈220 mg/dL (P < 0.05 vs. basal period) to double the hepatic glucose load (Fig. 2B). During P2, arterial blood glucose was clamped at ≈200 mg/dL to maintain a doubling of the hepatic glucose load in the presence of intraportal glucose infusion. Total hepatic blood flow during the clamp period was 23 ± 2 and 22 ± 2 mL·kg−1·min−1 in CTR and Bromo, respectively (P = 0.33 between groups).
Fig. 2.

Clamp study data. Arterial plasma glucose (A), hepatic glucose load (HGL; B), hepatic glucose uptake (HGU; C), net hepatic glucose balance (NHGB; D), glucose infusion rate (GIR) (E), and nonhepatic glucose uptake (non-HGU; F) in controls (CTR; ○, open symbols; n = 5) and group treated with bromocriptine mesylate (Bromo; ●, filled symbols; n = 6) during the basal (−20 to 0 min) and experimental periods (0 to 180 min) of hyperinsulinemic hyperglycemic clamps. Data are means ± SE. *P < 0.05 between groups based on ANOVA; symbols mark results of post hoc analysis.
Tracer-determined hepatic glucose uptake (HGU) during the basal period was low and did not differ significantly between groups (0.2 ± 0.1 and 0.0 ± 0.1 mg·kg−1·min−1 in CTR and Bromo, respectively; Fig. 2C). In both groups, hyperinsulinemia and hyperglycemia stimulated a significant increase in HGU within 15 min, with a mean rate of 1.3 ± 0.1 and 1.2 ± 0.3 mg·kg−1·min−1 during the last 30 min (considered the steady-state period) of P1 in CTR and Bromo, respectively (P < 0.05 vs. basal period in both groups, P = 0.32 between groups). HGU demonstrated an additional increase in response to portal glucose delivery (mean of 1.8 ± 0.4 vs. 1.8 ± 0.3 mg·kg−1·min−1 during P2, P = 0.18 between groups).
Both groups exhibited net hepatic glucose output during the basal period (Fig. 2D). The CTR group switched to NHGU within 60 min of the start of the HIHG clamp, with a mean rate of 0.9 ± 0.2 mg·kg−1·min−1 during the last half-hour of P1 and a further increase to 2.6 ± 0.2 mg·kg−1·min−1 during the last 30 min of P2. The Bromo group switched to NHGU within 30 min of the start of P1. The mean rate of NHGU during P1 in the Bromo group was 0.9 ± 0.5 mg·kg−1·min−1, and it increased to 3.3 ± 0.4 mg·kg−1·min−1 during P2, P < 0.05 between groups. The glucose infusion rate (GIR) was greater in Bromo than in CTR (36% greater in P1 and 30% greater in P2, P < 0.05; Fig. 2E). Moreover, non-HGU was significantly greater (~40 and 27% during P1 and P2, respectively) in the Bromo versus the CTR group (P < 0.05 between groups; Fig. 2F).
Net hepatic carbon retention (NHCR), an index of net hepatic glycogen synthesis, did not differ significantly in the two groups (P = 0.19). NHCR in CTR averaged 0.8 ± 0.1 and 2.7 ± 0.2 mg·kg−1·min−1 during the last 30 min of P1 and P2, respectively, and the corresponding rates in Bromo were 1.0 ± 0.5 and 3.4 ± 0.4 mg·kg−1·min−1.
Lactate, alanine, glycerol, and NEFA.
Similar arterial blood lactate concentrations and rates of net hepatic lactate uptake existed in both groups under basal conditions (Table 2). In response to hyperinsulinemia and hyperglycemia, blood lactate increased in both groups, with the increase being significantly greater in Bromo than in CTR. The increase in lactate concentrations was consistent with that following mixed-meal ingestion in both dogs and humans (16, 87). This occurred in conjunction with a switch to net hepatic lactate output in both groups, with the rates in CTR and Bromo being very similar throughout P1 and P2. The arterial concentrations and net hepatic uptake rates of alanine and glycerol did not differ significantly between groups at any time (Table 2). Both groups exhibited a decline in glycerol and NEFA concentrations with the onset of the clamp, with the initial decline in NEFA being greater in Bromo. Net hepatic glycerol and NEFA uptake declined in parallel with their concentrations.
Table 2.
Arterial lactate, alanine, glycerol and NEFA concentrations (µmol/L) and hepatic balance data (µmol·kg−1·min−1) during clamp studies
| Basal Period | Clamp Period 1 |
Clamp Period 2 |
|||||
|---|---|---|---|---|---|---|---|
| 30 min | 60 min | 90 min | 120 min | 150 min | 180 min | ||
| Arterial blood lactate | |||||||
| CTR | 221 ± 12 | 248 ± 29 | 486 ± 60† | 411 ± 46† | 450 ± 27† | 496 ± 25† | 568 ± 65† |
| Bromo | 270 ± 10 | 384 ± 63 | 611 ± 80† | 598 ± 55*† | 664 ± 74*† | 712 ± 69*† | 630 ± 64† |
| Net hepatic lactate balancea | |||||||
| CTR | −3.7 ± 0.6 | −1.0 ± 1.3† | 4.0 ± 1.3† | 3.0 ± 1.5† | 2.8 ± 1.5† | 1.4 ± 1.3† | 0.5 ± 1.4† |
| Bromo | −4.1 ± 0.5 | 0.4 ± 2.2† | 4.7 ± 1.6† | 3.5 ± 1.0† | 1.6 ± 1.1† | 0.2 ± 1.4 | 1.1 ± 1.1† |
| Arterial blood alanine | |||||||
| CTR | 227 ± 34 | 235 ± 25 | 279 ± 27† | 261 ± 24 | 264 ± 25 | 253 ± 21 | 250 ± 21 |
| Bromo | 246 ± 24 | 271 ± 33 | 303 ± 31† | 277 ± 24 | 260 ± 19 | 255 ± 22 | 248 ± 12 |
| Net hepatic alanine uptake | |||||||
| CTR | 1.4 ± 0.3 | 1.2 ± 0.2 | 1.3 ± 0.2 | 1.5 ± 0.2 | 1.5 ± 0.3 | 1.7 ± 0.4 | 1.9 ± 0.3 |
| Bromo | 1.4 ± 0.1 | 1.1 ± 0.1 | 1.1 ± 0.2 | 1.2 ± 0.1 | 1.2 ± 0.1 | 1.5 ± 0.1 | 1.4 ± 0.1 |
| Arterial blood glycerol | |||||||
| CTR | 96 ± 10 | 46 ± 11† | 35 ± 7† | 33 ± 4† | 34 ± 6† | 26 ± 4† | 21 ± 4† |
| Bromo | 105 ± 10 | 53 ± 7† | 46 ± 4† | 47 ± 8† | 46 ± 12† | 44 ± 10† | 33 ± 5† |
| Net hepatic glycerol uptake | |||||||
| CTR | 2.1 ± 0.4 | 0.9 ± 0.2† | 0.7 ± 0.2† | 0.7 ± 0.2† | 0.6 ± 0.2† | 0.6 ± 0.1† | 0.3 ± 0.1† |
| Bromo | 1.7 ± 0.1 | 0.8 ± 0.1† | 0.6 ± 0.1† | 0.6 ± 0.1† | 0.7 ± 0.2† | 0.6 ± 0.1† | 0.5 ± 0.1† |
| Arterial plasma NEFA | |||||||
| CTR | 957 ± 49 | 394 ± 56† | 243 ± 50† | 149 ± 40† | 85 ± 25† | 96 ± 41† | 104 ± 27† |
| Bromo | 917 ± 84 | 249 ± 42*† | 106 ± 40*† | 77 ± 37† | 59 ± 34† | 28 ± 12† | 29 ± 10† |
| Net hepatic NEFA uptake | |||||||
| CTR | 2.7 ± 0.2 | 1.3 ± 0.4† | 0.8 ± 0.3† | 0.4 ± 0.1† | 0.1 ± 0.3† | 0.4 ± 0.3† | 0.4 ± 0.1† |
| Bromo | 3.2 ± 0.3 | 0.6 ± 0.4† | 0.2 ± 0.1† | 0.4 ± 0.2† | 0.2 ± 0.2† | 0.1 ± 0.0† | 0.2 ± 0.8† |
Data are means ± SE. Basal period values are means of 2 sampling times; controls (CTR) n = 5; group treated with bromocriptine mesylate (Bromo) n = 6. aNegative lactate balance data indicate net uptake. All substrates other than lactate exhibited net hepatic uptake only and are shown as positive values.
P < 0.05 between groups with 2 way repeated-measures ANOVA (symbols indicate time points identified as different with post hoc analysis);
P < 0.05 for difference from basal within the same group, using 1-way repeated-measures ANOVA (symbol indicates time points identified as different).
Liver and muscle tissue analyses.
Neither the terminal hepatic glycogen concentrations nor the mass of glycogen synthesized by the direct pathway differed significantly between groups (Fig. 3, A and B). GK activity was similar in the two groups (Fig. 3C). Both groups exhibited significant increases in pAkt and pGSK expression compared with the Basal response (Fig. 3, D and E), but GK expression did not differ between the Basal, CTR, and Bromo groups (Fig. 3F).
Fig. 3.
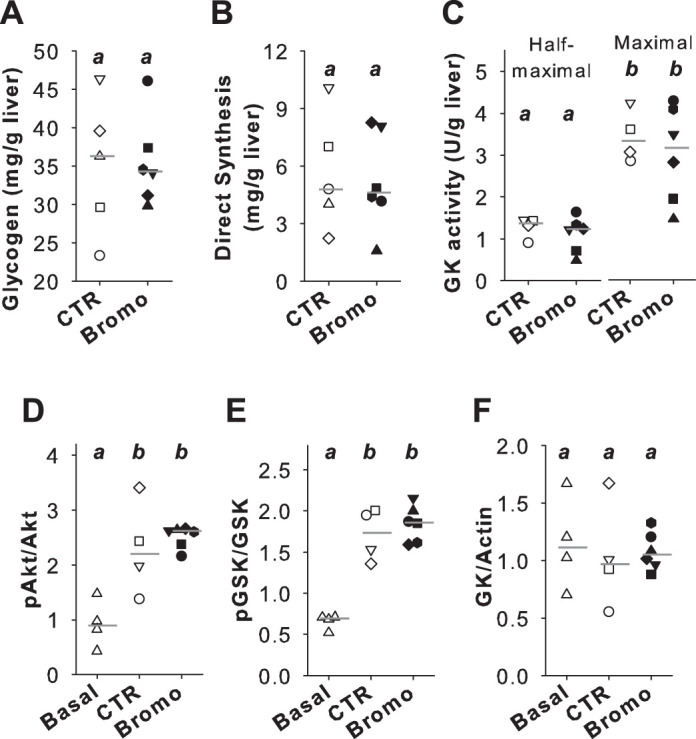
Hepatic tissue analyses. Hepatic glycogen (A), direct glycogen synthesis (B), glucokinase (GK) activity (C), and Western blotting of phosphorylated (p)Akt relative to total Akt (D), pGSK relative to total GSK (E), and GK relative to actin (F). Individual dog data are shown with a unique symbol for each dog in the control (CTR) and group treated with bromocriptine mesylate (Bromo) groups, with a horizontal line indicating the median of the group. For CTR (open symbols), n = 5 in A and B and n = 4 in C–F; n = 6 Bromo (filled symbols). Basal samples in D–F (△) were taken from 4 dogs maintained on a chow and meat diet that were fasted overnight before tissue collection and underwent no clamp study. Raw data for Western blots are shown in Supplemental Figure S1 (https://doi.org/10.6084/m9.figshare.11991351). Where groups are marked with a different letter, they differ significantly (P < 0.05).
In muscle tissue, there were a number of differences in protein expression and phosphorylation between groups. The level of Akt phosphorylated at Ser473 (Fig. 4A) was more than three times as high in Bromo versus CTR (P < 0.05), whereas the amount of total Akt did not differ significantly between groups (P = 0.4; Fig. 4B). Muscle ACADM expression in Bromo was more than twofold that in CTR (P < 0.05; Fig. 4C). Muscle ChREBP level was 30% higher in Bromo versus CTR (P < 0.05; Fig. 4D), and PPARα was 35% higher in Bromo versus CTR (P < 0.05; Fig. 4E). The TXNIP level was reduced by 25% in Bromo versus CTR (P < 0.05; Fig. 4F). The level of muscle GSKβ phosphorylated at Ser9 was 30% higher in Bromo versus CTR (P < 0.05; Fig. 4G). Although the muscle level of SIRT1 showed an upward trend (19% increase) in Bromo versus CTR, this increase did not reach statistical significance (Fig. 4H). IDE was evaluated with two different antibodies (see research design and methods), and both antibodies yielded very similar results. IDE expression did not differ between groups in either muscle or liver (Fig. 4, I and J; data shown are from the Abcam antibody). Hepatic SCD1 and ELOVL-6 expressions were similar in CTR and Bromo (Fig. 4, K and L). In addition, the groups exhibited no differences in expression of the IDE in either muscle or liver (data not shown).There was no significant difference in the total hepatic triglyceride concentrations between groups (0.84 ± 0.23 and 0.75 ± 0.16 µg/mg tissue; CTR and Bromo, respectively, P = 0.34), but the total phospholipid concentration was 27% less in the Bromo group (19.0 ± 1.2 vs. 13.9 ± 1.0 µg/mg liver, CTR vs. Bromo, respectively, P < 0.01). Moreover, there were significant between-group differences in the content of some fatty acid species in both the triglyceride and phospholipid profiles in the liver (Table 3). Overall, 18:1ω9, 16:0, 18:2, 18:0, 20:4, 18:1ω7, and 16:1 accounted for 97–98% of triglyceride composition in both groups (with the contributions of these species arranged in descending order). The first six of these species were also among the most common identified in phospholipids; in addition, 22:5ω3, 22:4ω6, and 20:3ω6 each contributed more than 1% of phospholipid content. The 16:0/18:2 ratio was significantly reduced in both triglycerides and phospholipids in Bromo versus CTR (Table 3). Conversely, the 18:0/16:0 and 16:1/16:0 ratios tended to be elevated in Bromo versus CTR in hepatic triglycerides (P = 0.08 and 0.06, respectively), and they were higher (P < 0.05) in hepatic phospholipids in the Bromo group.
Fig. 4.

Western blot data for control (CTR[open symbols; n = 5) and group treated with bromocriptine mesylate (Bromo; filled symbols; n = 6). Muscle tissue was analyzed for Akt phosphorylated at (p)Ser473 (A), Akt (B), acyl-CoA dehydrogenase medium chain (ACADM; C), carbohydrate response element-binding protein (ChREBP; D), peroxisome proliferator-activated receptor-α (PPARα; E), thioredoxin-interacting protein (TXNIP; F), GSKβ-Ser9 (G), sirtuin 1 (SIRT1; H), and insulin degrading enzyme (IDE; I). Liver tissue analyses included IDE (J), stearoyl-CoA desaturase-1 (SCLD1; K), and elongation of long-chain fatty acids family member 6 (ELOVL6; L). All data were normalized to the mean of the CTR group, which was set equivalent to 1. Each dog is shown with a unique symbol, with the group median marked with a horizontal line. One Bromo dog had an atypical response (>50% difference from the remainder of the group) for both ACADM and PPARa, and its data are excluded from those figures. Raw data for Western blots are shown in Supplemental Fig. S2 (https://doi.org/10.6084/m9.figshare.12066993). *P < 0.05 vs CTR.
Table 3.
Liver triglyceride and phospholipid profiles (%total for each lipid species) and ratios for selected species
| Species | Triglycerides |
Phospholipids |
||||
|---|---|---|---|---|---|---|
| CTR | Bromo | P value | CTR | Bromo | P value | |
| 14:0 | 1.7 ± 0.3 | 1.3 ± 0.1 | 0.079 | 0.2 ± 0.0 | 0.1 ± 0.0 | 0.138 |
| 16:0 | 23.4 ± 0.1 | 19.8 ± 0.9 | 0.002* | 13.6 ± 0.4 | 12.4 ± 0.2 | 0.002* |
| 16:1 | 2.2 ± 0.1 | 2.4 ± 0.3 | 0.303 | 0.5 ± 0.0 | 0.6 ± 0.1 | 0.052 |
| 18:0 | 9.8 ± 0.6 | 10.9 ± 1.3 | 0.245 | 27.7 ± 0.2 | 28.4 ± 0.1 | 0.002* |
| 18:1 ω9 | 33.5 ± 0.6 | 34.5 ± 1.5 | 0.298 | 5.5 ± 0.4 | 5.1 ± 0.1 | 0.111 |
| 18:1 ω7 | 3.6 ± 0.3 | 4.1 ± 0.2 | 0.045* | 1.9 ± 0.1 | 2.2 ± 0.1 | 0.007* |
| 18:2 | 20.7 ± 0.3 | 20.6 ± 0.6 | 0.460 | 15.8 ± 0.4 | 15.6 ± 0.3 | 0.324 |
| 18:3 ω6 | 0.6 ± 0.2 | 0.4 ± 0.2 | 0.291 | 0.1 ± 0.0 | 0.0 ± 0.0 | 0.002* |
| 18:3 ω3 | 0.1 ± 0.2 | 0.3 ± 0.2 | 0.155 | 0.2 ± 0.0 | 0.0 ± 0.0 | 0.006* |
| 20:3 ω6 | 0.0 ± 0.0 | 0.1 ± 0.1 | 0.195 | 1.1 ± 0.3 | 1.2 ± 0.1 | 0.321 |
| 20:4 | 4.4 ± 0.3 | 4.9 ± 0.9 | 0.326 | 26.8 ± 0.5 | 27.5 ± 0.4 | 0.092 |
| 20:5 | 0.1 ± 0.0 | 0.2 ± 0.0 | 0.016* | |||
| 22:4 ω6 | 0.0 ± 0.0 | 0.5 ± 0.1 | 0.012* | 1.8 ± 0.2 | 1.9 ± 0.1 | 0.215 |
| 22:5 ω6 | 0.6 ± 0.0 | 0.6 ± 0.1 | 0.325 | |||
| 22:5 ω3 | 0.0 ± 0.0 | 0.3 ± 0.1 | 0.064 | 2.5 ± 0.1 | 2.3 ± 0.1 | 0.106 |
| 22:6 | 1.7 ± 0.2 | 1.6 ± 0.1 | 0.388 | |||
| Ratios | ||||||
| 16:0/18:2 | 1.1 ± 0.02 | 1.0 ± 0.02 | 0.000003* | 0.5 ± 0.01 | 0.4 ± 0.01 | 0.001* |
| 18:0/16:0 | 0.4 ± 0.03 | 0.6 ± 0.09 | 0.08 | 2.0 ± 0.05 | 2.3 ± 0.05 | 0.001* |
| 16:1/16:0 | 0.09 ± 0.01 | 0.12 ± 0.01 | 0.06 | 0.1 ± 0.01 | 0.2 ± 0.01 | 0.001* |
Data are means ± SE; controls (CTR) n = 5; group treated with bromocriptine mesylate (Bromo) n = 6. Individual species and ratios of selected species were compared between groups with t tests.
P < 0.05 between groups.
DISCUSSION
In keeping with previous findings from our laboratory (17), consumption of the HFD for ~7.5 wk resulted in deterioration of glucose tolerance in all CTR animals despite higher insulin and C-peptide concentrations during the final versus the first OGTT. On the other hand, daily Bromo treatment for ~3.5 wk reduced the impact of the HFD on glucose tolerance such that there was no significant difference in the glycemic response between the first and third OGTTs in that group. The median for insulin ΔAUC0–120min during the second OGTT in the Bromo group was 64% greater than that observed during the first OGTT in that group, a response not significantly different from that of the final OGTT in the CTR group. The CTR group displayed an increase in the C-peptide response and no significant change in the C-peptide/insulin ratio during the final versus the first OGTT, consistent with an increase in insulin secretion playing the major role in the increase in the insulin ΔAUC0–120min. In contrast, the C-peptide AUC0–120min and the C-peptide/insulin ratio were significantly lower in the Bromo group during the final OGTT compared with the 4-wk OGTT, before the start of Bromo treatment. This strongly suggests that a reduction in insulin clearance contributed to the marked increase in the insulin response during the final OGTT in the Bromo dogs. In agreement with this, the C-peptide/insulin ratio during the basal period before the clamp studies was significantly higher in the CTR versus the Bromo group.
Although there was a reduction in food intake during Bromo treatment, it was relatively modest. The reason for the reduction is unknown, although vomiting was not observed, as previously mentioned. In humans, nausea does occur with bromocriptine-QR treatment, but generally in a minority of patients (6, 30, 42, 77). Nausea is usually transient and dose dependent (6, 30), and the relatively low dose utilized in this study may have helped to reduce more apparent side effects.
Although euglycemic clamps are frequently used for assessment of metabolic responses in humans and animal models, they have limited value in examining the liver’s response. The liver does not usually exhibit substantial glucose uptake under euglycemic conditions (23, 33, 76), but it is a major site of postprandial glucose extraction and storage (43, 60), The liver is the primary organ that is able to store glycogen under feeding conditions and release it as glucose for use by other tissues during fasting (58). Under conditions in which three meals a day are consumed, the liver exhibits uptake at least 2/3 of each 24-h period (7). To examine the liver under conditions where glucose uptake and storage would be expected and mimic the normal route of hepatic exposure to glucose under feeding conditions, we utilized hyperinsulinemic hyperglycemic clamp studies with intraportal glucose infusion. Hepatic responses under conditions mimicking feeding are difficult to assess in the human because of the invasiveness of the procedures required, and small-animal models such as rodents often fail to replicate human responses (55, 67). We previously carried out clamp studies under identical experimental conditions to those utilized in the current studies, except that they included dogs fed a normal chow and meat diet; the chow-fed dogs had no consistent weight changes during the 4-wk period before the clamps (17, 18). The chow-fed dogs shifted from net hepatic glucose output to NHGU within 15 min of the start of the clamp period and exhibited NHGU at a rate of ~1.8 mg·kg−1·min−1 during the last 30 min of P1, with a doubling of that rate during the final 30 min of P2. Compared with the chow-fed dogs, NHGU was markedly blunted in the current high-fat-fed CTR dogs (~50 and 25% during P1 and P2, respectively), with a delay in the shift from net hepatic glucose output to NHGU until after the 30-min time point of P1. The blunting of NHGU in the current CTR dogs was consistent with our previous findings in dogs fed the HFD for 4 wk (17). On the other hand, the Bromo dogs switched to NHGU within 30 min of the start of the clamp. Whereas NHGU in CTR and Bromo was very similar during the final 30 min of P1, the mean rate in the Bromo group during the final 30 min of P2 was ~27% greater than that in the CTR dogs. Moreover, the NHGU rate in the Bromo group over the final 30 min of P2 was similar to (~92% of) that observed in our previously reported chow-fed dogs (18), consistent with an improvement in NHGU brought about by bromocriptine treatment.
Previously, we (17) have demonstrated that consumption of the HFD does not significantly alter non-HGU under hyperinsulinemic-hyperglycemic conditions, and in keeping with this, non-HGU in the CTR group was similar to that in our previous chow-fed dogs (17, 18). However, non-HGU was significantly enhanced in Bromo during both clamp periods, suggesting a stimulatory effect of Bromo on muscle glucose uptake. Data from other animal models are consistent with such an impact (27, 78). Muscle composition in the dog and human is similar despite greater muscle mass in proportion to body weight in the dog (22, 56). The muscle of Bromo dogs exhibited increased expression of genes linked to improvement in glucose disposition, including Akt-Ser473, ChREBP, GSK3β-Ser9, and PPARa (47, 63, 64, 73, 89, 90, 94). The decrease in muscle TXNIP in Bromo dogs is also consistent with the enhancement of muscle glucose uptake (88). Thus, the data from our muscle analyses are consistent with the increased insulin-mediated glucose disposal during the hyperinsulinemic hyperglycemic clamp in this group, and bromocriptine’s impact on a number of regulatory pathways appears to contribute to its stimulation of muscle glucose uptake.
The insulin response relative to the glycemic response during the OGTTs did not increase significantly over time in either group, suggesting that Bromo treatment had little impact on β-cell function. Hepatic insulin clearance during the clamp study was enhanced in the Bromo group, whereas whole body clearance was not. The increase in hepatic clearance may reflect an improvement in insulin sensitivity (4, 48, 93).
We noted a number of differences in hepatic fatty acids between the treatment groups. Palmitate (16:0) is an important dietary fatty acid (5, 46), and its concentration was significantly lower in both the hepatic triglycerides and phospholipids of Bromo compared with CTR dogs. It is noteworthy that pork serves as the primary protein and fat source in the HFD utilized in these studies, and pork is a significant source of palmitate (5, 91). This suggests that the greater intake of the HFD, relative to body weight, in the CTR versus Bromo group during the final 4 wk of study might have contributed to the higher hepatic 16:0 concentrations observed in the CTR dogs. However, palmitate is also one of the major products of de novo lipogenesis, and the ratio of 16:0 to 18:2ω6 fatty acids in blood and tissue samples has been utilized as an index of this process (8, 35, 36). The higher 16:0/18:2ω6 ratio in the CTR versus Bromo group (18 and 12% for triglycerides and phospholipids, respectively) is consistent with a suppression of lipogenesis by bromocriptine treatment, and such a suppressive effect of bromocriptine has been reported in rodents (12, 14, 81, 95). Thus, although the mechanism remains undetermined, it appears that bromocriptine intake is associated with a reduction in hepatic palmitate concentrations. Although the metabolic impact of such a reduction over the long term remains undetermined, some data link elevated endogenous palmitate production to increases in oxidative stress, impaired insulin secretion, and other metabolic defects (5, 34, 54). Moreover, reductions in hepatic oxidative stress-responsive and proinflammatory protein transcription factors have been reported in bromocriptine-treated insulin-resistant rodents (25). In regard to other liver lipid findings, the ratio of 18:0 to 16:0 has served as a marker of elongase activity (36, 37). In Bromo compared with CTR dogs, there was a 34% elevation of the 18:0/16:0 ratio in hepatic triglycerides and a 12% elevation in phospholipids. Moreover, the ratio of 16:1 to 16:0 has been utilized as an index of SCD1 enzyme activity, with an increase in the ratio correlating with a decrease in liver fat content in some (69, 83, 84), but not all (46), human studies. Bromocriptine treatment has been associated with a reduction in liver fat in rodent models of fatty liver (21, 25). Thus, the increased 16:1/16:0 ratio (28% greater in both triglycerides and phospholipids), in conjunction with the lower 16:0/18:2ω6 ratio in the Bromo group, is highly intriguing. We observed no difference in hepatic ELOVL6 or SCD1 expression between groups, although bromocriptine treatment of insulin-resistant, hypertensive rats reduced liver SREBP-1 (a transcription factor that regulates the expressions of ELOVL6 and SCD1) protein by 40% (25). Further study of the impact of Bromo and related agents on hepatic lipids appears to be warranted.
In summary, in a glucose-intolerant, high fat-fed dog model, circadian-timed daily bromocriptine treatment for ~3.5 wk improved glucose tolerance relative to control animals without stimulating an increase in insulin secretion or plasma insulin concentrations above control levels. Moreover, it increased glucose disposal during a hyperinsulinemic hyperglycemic clamp both in the presence and inthe absence of the portal glucose signal. There was a significant bromocriptine-related increase in NHGU, but more than 90% of the increase in glucose disposal was associated with the nonhepatic tissues. Chronic bromocriptine treatment appears to have a substantial impact on glucose disposal, particularly in skeletal muscle.
GRANTS
These studies were funded by VeroScience LLC of Tiverton, RI, and S2 Therapeutics of Bristol, TN. Hormone analysis was performed by Vanderbilt’s Hormone Assay and Analytical Services Core, with hepatic lipid assayed in the Lipid Core, and surgical and experimental expertise was provided by Vanderbilt’s Animal Core; all are supported by the Vanderbilt Diabetes Research and Training Center National Institute of Diabetes and Digestive and Kidney Diseases Grant DK-020593. The Hormone Assay Core also receives support from DK-059637. A. D. Cherrington holds the Jacquelyn A. Turner and Dr. Dorothy J. Turner Chair in Diabetes Research.
DISCLOSURES
A. H. Cincotta, M. Ezrokhiare, N. Cominos, and Y. Zhang are employees of VeroScience, and A. H. Cincotta is a shareholder in that company.
AUTHOR CONTRIBUTIONS
A.H.C., M.E., and A.D.C. conceived and designed research; M.C.M., M.S.S., and B.F. performed experiments; M.C.M., L.L.S., A.H.C., M.E., N.C., Y.Z., and A.D.C. analyzed data; M.C.M., L.L.S., A.H.C., and A.D.C. interpreted results of experiments; M.C.M. prepared figures; M.C.M. drafted manuscript; M.C.M., L.L.S., A.H.C., M.E., N.C., Y.Z., and A.D.C. edited and revised manuscript; M.C.M., M.S.S., L.L.S., A.H.C., M.E., N.C., Y.Z., B.F., and A.D.C. approved final version of manuscript.
REFERENCES
- 1.Barzilai N, Rossetti L. Role of glucokinase and glucose-6-phosphatase in the acute and chronic regulation of hepatic glucose fluxes by insulin. J Biol Chem 268: 25019–25025, 1993. [PubMed] [Google Scholar]
- 2.Basu A, Basu R, Shah P, Vella A, Johnson CM, Nair KS, Jensen MD, Schwenk WF, Rizza RA. Effects of type 2 diabetes on the ability of insulin and glucose to regulate splanchnic and muscle glucose metabolism: evidence for a defect in hepatic glucokinase activity. Diabetes 49: 272–283, 2000. doi: 10.2337/diabetes.49.2.272. [DOI] [PubMed] [Google Scholar]
- 3.Bina KG, Cincotta AH. Dopaminergic agonists normalize elevated hypothalamic neuropeptide Y and corticotropin-releasing hormone, body weight gain, and hyperglycemia in ob/ob mice. Neuroendocrinology 71: 68–78, 2000. doi: 10.1159/000054522. [DOI] [PubMed] [Google Scholar]
- 4.Bojsen-Møller KN, Lundsgaard AM, Madsbad S, Kiens B, Holst JJ. Hepatic insulin clearance in regulation of systemic insulin concentrations-role of carbohydrate and energy availability. Diabetes 67: 2129–2136, 2018. doi: 10.2337/db18-0539. [DOI] [PubMed] [Google Scholar]
- 5.Carta G, Murru E, Banni S, Manca C. Palmitic acid: Physiological role, metabolism and nutritional implications. Front Physiol 8: 902, 2017. doi: 10.3389/fphys.2017.00902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chamarthi B, Gaziano JM, Blonde L, Vinik A, Scranton RE, Ezrokhi M, Rutty D, Cincotta AH. Timed bromocriptine-qr therapy reduces progression of cardiovascular disease and dysglycemia in subjects with well-controlled type 2 diabetes mellitus. J Diabetes Res 2015: 1, 2015. doi: 10.1155/2015/157698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cherrington AD. Banting Lecture 1997. Control of glucose uptake and release by the liver in vivo. Diabetes 48: 1198–1214, 1999. doi: 10.2337/diabetes.48.5.1198. [DOI] [PubMed] [Google Scholar]
- 8.Chong MF, Hodson L, Bickerton AS, Roberts R, Neville M, Karpe F, Frayn KN, Fielding BA. Parallel activation of de novo lipogenesis and stearoyl-CoA desaturase activity after 3 d of high-carbohydrate feeding. Am J Clin Nutr 87: 817–823, 2008. doi: 10.1093/ajcn/87.4.817. [DOI] [PubMed] [Google Scholar]
- 9.Cincotta AH, Ezrokhi M, Trubitsyna Y, Luo S. Lesion of dopaminergic affecent neurons communicating with the biological clock induces metabolic syndrome in rats. Diabetes 64: A540, 2015. [Google Scholar]
- 10.Cincotta AH, Luo S, Zhang Y, Liang Y, Bina KG, Jetton TL, Scislowski PW. Chronic infusion of norepinephrine into the VMH of normal rats induces the obese glucose-intolerant state. Am J Physiol Regul Integr Comp Physiol 278: R435–R444, 2000. doi: 10.1152/ajpregu.2000.278.2.R435. [DOI] [PubMed] [Google Scholar]
- 11.Cincotta AH, MacEachern TA, Meier AH. Bromocriptine redirects metabolism and prevents seasonal onset of obese hyperinsulinemic state in Syrian hamsters. Am J Physiol 264: E285–E293, 1993. doi: 10.1152/ajpendo.1993.264.2.E285. [DOI] [PubMed] [Google Scholar]
- 12.Cincotta AH, Meier AH. Prolactin permits the expression of a circadian variation in lipogenic responsiveness to insulin in hepatocytes of the golden hamster (Mesocricetus auratus). J Endocrinol 106: 173–176, 1985. doi: 10.1677/joe.0.1060173. [DOI] [PubMed] [Google Scholar]
- 13.Cincotta AH, Meier AH, Cincotta M Jr. Bromocriptine improves glycaemic control and serum lipid profile in obese Type 2 diabetic subjects: a new approach in the treatment of diabetes. Expert Opin Investig Drugs 8: 1683–1707, 1999. doi: 10.1517/13543784.8.10.1683. [DOI] [PubMed] [Google Scholar]
- 14.Cincotta AH, Schiller BC, Meier AH. Bromocriptine inhibits the seasonally occurring obesity, hyperinsulinemia, insulin resistance, and impaired glucose tolerance in the Syrian hamster, Mesocricetus auratus. Metabolism 40: 639–644, 1991. doi: 10.1016/0026-0495(91)90057-4. [DOI] [PubMed] [Google Scholar]
- 15.Coate KC, Kraft G, Irimia JM, Smith MS, Farmer B, Neal DW, Roach PJ, Shiota M, Cherrington AD. Portal vein glucose entry triggers a coordinated cellular response that potentiates hepatic glucose uptake and storage in normal but not high-fat/high-fructose-fed dogs. Diabetes 62: 392–400, 2013. doi: 10.2337/db12-0417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Coate KC, Kraft G, Lautz M, Smith M, Neal DW, Cherrington AD. A high-fat, high-fructose diet accelerates nutrient absorption and impairs net hepatic glucose uptake in response to a mixed meal in partially pancreatectomized dogs. J Nutr 141: 1643–1651, 2011. doi: 10.3945/jn.111.145359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Coate KC, Kraft G, Moore MC, Smith MS, Ramnanan C, Irimia JM, Roach PJ, Farmer B, Neal DW, Williams P, Cherrington AD. Hepatic glucose uptake and disposition during short-term high-fat vs. high-fructose feeding. Am J Physiol Endocrinol Metab 307: E151–E160, 2014. doi: 10.1152/ajpendo.00083.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Coate KC, Scott M, Farmer B, Moore MC, Smith M, Roop J, Neal DW, Williams P, Cherrington AD. Chronic consumption of a high-fat/high-fructose diet renders the liver incapable of net hepatic glucose uptake. Am J Physiol Endocrinol Metab 299: E887–E898, 2010. doi: 10.1152/ajpendo.00372.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Coomans CP, van den Berg SA, Lucassen EA, Houben T, Pronk AC, van der Spek RD, Kalsbeek A, Biermasz NR, Willems van Dijk K, Romijn JA, Meijer JH. The suprachiasmatic nucleus controls circadian energy metabolism and hepatic insulin sensitivity. Diabetes 62: 1102–1108, 2013. doi: 10.2337/db12-0507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cycloset. [Package insert] Tiverton, RI: VeroScience LLC, 2019. [Google Scholar]
- 21.Davis LM, Pei Z, Trush MA, Cheskin LJ, Contoreggi C, McCullough K, Watkins PA, Moran TH. Bromocriptine reduces steatosis in obese rodent models. J Hepatol 45: 439–444, 2006. doi: 10.1016/j.jhep.2006.03.019. [DOI] [PubMed] [Google Scholar]
- 22.Dries B, Vanwanseele B, Jonkers I, Dingemanse W, Vander Sloten J, Villamonte-Chevalier A, Van der Vekens E, Polis I, Vanderperren K, Van Bree H, Gielen I. Musculotendon excursion potential, tendon slack and muscle fibre length: the interaction of the canine gastrocnemius muscle and tendon. J Anat 233: 460–467, 2018. doi: 10.1111/joa.12845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Edgerton DS, Moore MC, Winnick JJ, Scott M, Farmer B, Naver H, Jeppesen CB, Madsen P, Kjeldsen TB, Nishimura E, Brand CL, Cherrington AD. Changes in glucose and fat metabolism in response to the administration of a hepato-preferential insulin analog. Diabetes 63: 3946–3954, 2014. doi: 10.2337/db14-0266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Edgerton DS, Ramnanan CJ, Grueter CA, Johnson KM, Lautz M, Neal DW, Williams PE, Cherrington AD. Effects of insulin on the metabolic control of hepatic gluconeogenesis in vivo. Diabetes 58: 2766–2775, 2009. doi: 10.2337/db09-0328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ezrokhi M, Luo S, Trubitsyna Y, Cincotta AH. Neuroendocrine and metabolic components of dopamine agonist amelioration of metabolic syndrome in SHR rats. Diabetol Metab Syndr 6: 104, 2014. [Erratum in Diabetol Metab Syndr 7: 61, 2015.] doi: 10.1186/1758-5996-6-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Feizi A, Meamar R, Eslamian M, Amini M, Nasri M, Iraj B. Area under the curve during OGTT in first-degree relatives of diabetic patients as an efficient indicator of future risk of type 2 diabetes and prediabetes. Clin Endocrinol (Oxf) 87: 696–705, 2017. doi: 10.1111/cen.13443. [DOI] [PubMed] [Google Scholar]
- 27.Furigo IC, Suzuki MF, Oliveira JE, Ramos-Lobo AM, Teixeira PDS, Pedroso JA, de Alencar A, Zampieri TT, Buonfiglio DC, Quaresma PGF, Prada PO, Bartolini P, Soares CRJ, Donato J Jr. Suppression of prolactin secretion partially explains the antidiabetic effect of bromocriptine in ob/ob mice. Endocrinology 160: 193–204, 2019. doi: 10.1210/en.2018-00629. [DOI] [PubMed] [Google Scholar]
- 28.Galassetti P, Shiota M, Zinker BA, Wasserman DH, Cherrington AD. A negative arterial-portal venous glucose gradient decreases skeletal muscle glucose uptake. Am J Physiol 275: E101–E111, 1998. doi: 10.1152/ajpendo.1998.275.1.E101. [DOI] [PubMed] [Google Scholar]
- 29.Garber AJ, Abrahamson MJ, Barzilay JI, Blonde L, Bloomgarden ZT, Bush MA, Dagogo-Jack S, DeFronzo RA, Einhorn D, Fonseca VA, Garber JR, Garvey WT, Grunberger G, Handelsman Y, Hirsch IB, Jellinger PS, McGill JB, Mechanick JI, Rosenblit PD, Umpierrez GE. Consensus statement by the American Association of Clinical Endocrinologists and American College of Endocrinology on the Ccomprehensive Type 2 Diabetes Management Algorithm. Endocr Pract 24: 91–120, 2018. doi: 10.4158/CS-2017-0153. [DOI] [PubMed] [Google Scholar]
- 30.Gaziano JM, Cincotta AH, O’Connor CM, Ezrokhi M, Rutty D, Ma ZJ, Scranton RE. Randomized clinical trial of quick-release bromocriptine among patients with type 2 diabetes on overall safety and cardiovascular outcomes. Diabetes Care 33: 1503–1508, 2010. doi: 10.2337/dc09-2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Golden S, Wals PA, Katz J. An improved procedure for the assay of glycogen synthase and phosphorylase in rat liver homogenates. Anal Biochem 77: 436–445, 1977. doi: 10.1016/0003-2697(77)90257-3. [DOI] [PubMed] [Google Scholar]
- 32.Harder-Lauridsen NM, Rosenberg A, Benatti FB, Damm JA, Thomsen C, Mortensen EL, Pedersen BK, Krogh-Madsen R. Ramadan model of intermittent fasting for 28 d had no major effect on body composition, glucose metabolism, or cognitive functions in healthy lean men. Nutrition 37: 92–103, 2017. doi: 10.1016/j.nut.2016.12.015. [DOI] [PubMed] [Google Scholar]
- 33.Honka MJ, Latva-Rasku A, Bucci M, Virtanen KA, Hannukainen JC, Kalliokoski KK, Nuutila P. Insulin-stimulated glucose uptake in skeletal muscle, adipose tissue and liver: a positron emission tomography study. Eur J Endocrinol 178: 523–531, 2018. doi: 10.1530/EJE-17-0882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hoppa MB, Collins S, Ramracheya R, Hodson L, Amisten S, Zhang Q, Johnson P, Ashcroft FM, Rorsman P. Chronic palmitate exposure inhibits insulin secretion by dissociation of Ca(2+) channels from secretory granules. Cell Metab 13: 487, 2011. doi: 10.1016/j.cmet.2011.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hudgins LC, Baday A, Hellerstein MK, Parker TS, Levine DM, Seidman CE, Neese RA, Tremaroli JD, Hirsch J. The effect of dietary carbohydrate on genes for fatty acid synthase and inflammatory cytokines in adipose tissues from lean and obese subjects. J Nutr Biochem 19: 237–245, 2008. doi: 10.1016/j.jnutbio.2007.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hudgins LC, Hellerstein MK, Seidman CE, Neese RA, Tremaroli JD, Hirsch J. Relationship between carbohydrate-induced hypertriglyceridemia and fatty acid synthesis in lean and obese subjects. J Lipid Res 41: 595–604, 2000. [PubMed] [Google Scholar]
- 37.Hudgins LC, Parker TS, Levine DM, Hellerstein MK. A dual sugar challenge test for lipogenic sensitivity to dietary fructose. J Clin Endocrinol Metab 96: 861–868, 2011. doi: 10.1210/jc.2010-2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jung SH, Jung CH, Reaven GM, Kim SH. Adapting to insulin resistance in obesity: role of insulin secretion and clearance. Diabetologia 61: 681–687, 2018. doi: 10.1007/s00125-017-4511-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kalsbeek A, Bruinstroop E, Yi CX, Klieverik LP, La Fleur SE, Fliers E. Hypothalamic control of energy metabolism via the autonomic nervous system. Ann N Y Acad Sci 1212: 114–129, 2010. doi: 10.1111/j.1749-6632.2010.05800.x. [DOI] [PubMed] [Google Scholar]
- 40.Kalsbeek A, Foppen E, Schalij I, Van Heijningen C, van der Vliet J, Fliers E, Buijs RM. Circadian control of the daily plasma glucose rhythm: an interplay of GABA and glutamate. PLoS One 3: e3194, 2008. doi: 10.1371/journal.pone.0003194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Keppler D, Decker K. Glycogen: determination with amyloglycosidase. In: Methods of Enzymatic Analysis (Bergmeyer HU, editor). New York: Verlag Chemie Weinheim, Academic Press, 1974, p. 1127–1131. [Google Scholar]
- 42.Kerr JL, Timpe EM, Petkewicz KA. Bromocriptine mesylate for glycemic management in type 2 diabetes mellitus. Ann Pharmacother 44: 1777–1785, 2010. doi: 10.1345/aph.1P271. [DOI] [PubMed] [Google Scholar]
- 43.Kleinert M, Clemmensen C, Hofmann SM, Moore MC, Renner S, Woods SC, Huypens P, Beckers J, de Angelis MH, Schürmann A, Bakhti M, Klingenspor M, Heiman M, Cherrington AD, Ristow M, Lickert H, Wolf E, Havel PJ, Müller TD, Tschöp MH. Animal models of obesity and diabetes mellitus. Nat Rev Endocrinol 14: 140–162, 2018. doi: 10.1038/nrendo.2017.161. [DOI] [PubMed] [Google Scholar]
- 44.Krssak M, Brehm A, Bernroider E, Anderwald C, Nowotny P, Dalla Man C, Cobelli C, Cline GW, Shulman GI, Waldhäusl W, Roden M. Alterations in postprandial hepatic glycogen metabolism in type 2 diabetes. Diabetes 53: 3048–3056, 2004. doi: 10.2337/diabetes.53.12.3048. [DOI] [PubMed] [Google Scholar]
- 45.Lee A, Ader M, Bray GA, Bergman RN. Diurnal variation in glucose tolerance. Cyclic suppression of insulin action and insulin secretion in normal-weight, but not obese, subjects. Diabetes 41: 750–759, 1992. doi: 10.2337/diab.41.6.750. [DOI] [PubMed] [Google Scholar]
- 46.Lee JJ, Lambert JE, Hovhannisyan Y, Ramos-Roman MA, Trombold JR, Wagner DA, Parks EJ. Palmitoleic acid is elevated in fatty liver disease and reflects hepatic lipogenesis. Am J Clin Nutr 101: 34–43, 2015. doi: 10.3945/ajcn.114.092262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lemberger T, Braissant O, Juge-Aubry C, Keller H, Saladin R, Staels B, Auwerx J, Burger AG, Meier CA, Wahli W. PPAR tissue distribution and interactions with other hormone-signaling pathways. Ann N Y Acad Sci 804, 1 Peroxisomes: 231–251, 1996. doi: 10.1111/j.1749-6632.1996.tb18619.x. [DOI] [PubMed] [Google Scholar]
- 48.Lundsgaard AM, Sjøberg KA, Høeg LD, Jeppesen J, Jordy AB, Serup AK, Fritzen AM, Pilegaard H, Myrmel LS, Madsen L, Wojtaszewski JFP, Richter EA, Kiens B. Opposite regulation of insulin sensitivity by dietary lipid versus carbohydrate excess. Diabetes 66: 2583–2595, 2017. doi: 10.2337/db17-0046. [DOI] [PubMed] [Google Scholar]
- 49.Luo S, Luo J, Cincotta AH. Chronic ventromedial hypothalamic infusion of norepinephrine and serotonin promotes insulin resistance and glucose intolerance. Neuroendocrinology 70: 460–465, 1999. doi: 10.1159/000054508. [DOI] [PubMed] [Google Scholar]
- 50.Luo S, Luo J, Cincotta AH. Suprachiasmatic nuclei monoamine metabolism of glucose tolerant versus intolerant hamsters. Neuroreport 10: 2073–2077, 1999. doi: 10.1097/00001756-199907130-00015. [DOI] [PubMed] [Google Scholar]
- 51.Luo S, Luo J, Meier AH, Cincotta AH. Dopaminergic neurotoxin administration to the area of the suprachiasmatic nuclei induces insulin resistance. Neuroreport 8: 3495–3499, 1997. doi: 10.1097/00001756-199711100-00016. [DOI] [PubMed] [Google Scholar]
- 52.Luo S, Meier AH, Cincotta AH. Bromocriptine reduces obesity, glucose intolerance and extracellular monoamine metabolite levels in the ventromedial hypothalamus of Syrian hamsters. Neuroendocrinology 68: 1–10, 1998. doi: 10.1159/000054344. [DOI] [PubMed] [Google Scholar]
- 53.Luo S, Zhang Y, Ezrokhi M, Li Y, Tsai TH, Cincotta AH. Circadian peak dopaminergic activity response at the biological clock pacemaker (suprachiasmatic nucleus) area mediates the metabolic responsiveness to a high-fat diet. J Neuroendocrinol 30: e12563, 2018. doi: 10.1111/jne.12563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ly LD, Xu S, Choi SK, Ha CM, Thoudam T, Cha SK, Wiederkehr A, Wollheim CB, Lee IK, Park KS. Oxidative stress and calcium dysregulation by palmitate in type 2 diabetes. Exp Mol Med 49: e291, 2017. doi: 10.1038/emm.2016.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Martignoni M, Groothuis GM, de Kanter R. Species differences between mouse, rat, dog, monkey and human CYP-mediated drug metabolism, inhibition and induction. Expert Opin Drug Metab Toxicol 2: 875–894, 2006. doi: 10.1517/17425255.2.6.875. [DOI] [PubMed] [Google Scholar]
- 56.Mathewson MA, Kwan A, Eng CM, Lieber RL, Ward SR. Comparison of rotator cuff muscle architecture between humans and other selected vertebrate species. J Exp Biol 217: 261–273, 2014. doi: 10.1242/jeb.083923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Meier JJ, Holst JJ, Schmidt WE, Nauck MA. Reduction of hepatic insulin clearance after oral glucose ingestion is not mediated by glucagon-like peptide 1 or gastric inhibitory polypeptide in humans. Am J Physiol Endocrinol Metab 293: E849–E856, 2007. doi: 10.1152/ajpendo.00289.2007. [DOI] [PubMed] [Google Scholar]
- 58.Meyer C, Dostou JM, Welle SL, Gerich JE. Role of human liver, kidney, and skeletal muscle in postprandial glucose homeostasis. Am J Physiol Endocrinol Metab 282: E419–E427, 2002. doi: 10.1152/ajpendo.00032.2001. [DOI] [PubMed] [Google Scholar]
- 59.Monti JM, Monti D. The involvement of dopamine in the modulation of sleep and waking. Sleep Med Rev 11: 113–133, 2007. doi: 10.1016/j.smrv.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 60.Moore MC, Coate KC, Winnick JJ, An Z, Cherrington AD. Regulation of hepatic glucose uptake and storage in vivo. Adv Nutr 3: 286–294, 2012. doi: 10.3945/an.112.002089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Moore MC, Rossetti L, Pagliassotti MJ, Monahan M, Venable C, Neal D, Cherrington AD. Neural and pancreatic influences on net hepatic glucose uptake and glycogen synthesis. Am J Physiol Endocrinol Metab 271: E215–E222, 1996. doi: 10.1152/ajpendo.1996.271.2.E215. [DOI] [PubMed] [Google Scholar]
- 62.Myers SR, Biggers DW, Neal DW, Cherrington AD. Intraportal glucose delivery enhances the effects of hepatic glucose load on net hepatic glucose uptake in vivo. J Clin Invest 88: 158–167, 1991. doi: 10.1172/JCI115273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nguyen-Ngo C, Jayabalan N, Salomon C, Lappas M. Molecular pathways disrupted by gestational diabetes mellitus. J Mol Endocrinol 63: R51–R72, 2019. doi: 10.1530/JME-18-0274. [DOI] [PubMed] [Google Scholar]
- 64.Ortega-Prieto P, Postic C. Carbohydrate sensing through the transcription factor ChREBP. Front Genet 10: 472, 2019. doi: 10.3389/fgene.2019.00472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pagliassotti MJ, Holste LC, Moore MC, Neal DW, Cherrington AD. Comparison of the time courses of insulin and the portal signal on hepatic glucose and glycogen metabolism in the conscious dog. J Clin Invest 97: 81–91, 1996. doi: 10.1172/JCI118410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Panda S. Circadian physiology of metabolism. Science 354: 1008–1015, 2016. doi: 10.1126/science.aah4967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Perlman RL. Mouse models of human disease: An evolutionary perspective. Evol Med Public Health 2016: 170–176, 2016. doi: 10.1093/emph/eow014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Peter A, Cegan A, Wagner S, Elcnerova M, Königsrainer A, Königsrainer I, Häring HU, Schleicher ED, Stefan N. Relationships between hepatic stearoyl-CoA desaturase-1 activity and mRNA expression with liver fat content in humans. Am J Physiol Endocrinol Metab 300: E321–E326, 2011. doi: 10.1152/ajpendo.00306.2010. [DOI] [PubMed] [Google Scholar]
- 69.Peter A, Cegan A, Wagner S, Lehmann R, Stefan N, Königsrainer A, Königsrainer I, Häring HU, Schleicher E. Hepatic lipid composition and stearoyl-coenzyme A desaturase 1 mRNA expression can be estimated from plasma VLDL fatty acid ratios. Clin Chem 55: 2113–2120, 2009. doi: 10.1373/clinchem.2009.127274. [DOI] [PubMed] [Google Scholar]
- 70.Peter A, Weigert C, Staiger H, Machicao F, Schick F, Machann J, Stefan N, Thamer C, Häring HU, Schleicher E. Individual stearoyl-coa desaturase 1 expression modulates endoplasmic reticulum stress and inflammation in human myotubes and is associated with skeletal muscle lipid storage and insulin sensitivity in vivo. Diabetes 58: 1757–1765, 2009. doi: 10.2337/db09-0188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Peter R, Dunseath G, Luzio SD, Chudleigh R, Roy Choudhury S, Owens DR. Daytime variability of postprandial glucose tolerance and pancreatic B-cell function using 12-h profiles in persons with Type 2 diabetes. Diabet Med 27: 266–273, 2010. doi: 10.1111/j.1464-5491.2010.02949.x. [DOI] [PubMed] [Google Scholar]
- 72.Pijl H, Ohashi S, Matsuda M, Miyazaki Y, Mahankali A, Kumar V, Pipek R, Iozzo P, Lancaster JL, Cincotta AH, DeFronzo RA. Bromocriptine: a novel approach to the treatment of type 2 diabetes. Diabetes Care 23: 1154–1161, 2000. doi: 10.2337/diacare.23.8.1154. [DOI] [PubMed] [Google Scholar]
- 73.Rahman S, Islam R. Mammalian Sirt1: insights on its biological functions. Cell Commun Signal 9: 11, 2011. doi: 10.1186/1478-811X-9-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rakshit K, Qian J, Colwell CS, Matveyenko AV. The islet circadian clock: entrainment mechanisms, function and role in glucose homeostasis. Diabetes Obes Metab 17, Suppl 1: 115–122, 2015. doi: 10.1111/dom.12523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ramnanan CJ, Edgerton DS, Rivera N, Irimia-Dominguez J, Farmer B, Neal DW, Lautz M, Donahue EP, Meyer CM, Roach PJ, Cherrington AD. Molecular characterization of insulin-mediated suppression of hepatic glucose production in vivo. Diabetes 59: 1302–1311, 2010. doi: 10.2337/db09-1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ramnanan CJ, Kraft G, Smith MS, Farmer B, Neal D, Williams PE, Lautz M, Farmer T, Donahue EP, Cherrington AD, Edgerton DS. Interaction between the central and peripheral effects of insulin in controlling hepatic glucose metabolism in the conscious dog. Diabetes 62: 74–84, 2013. doi: 10.2337/db12-0148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ramteke KB, Ramanand SJ, Ramanand JB, Jain SS, Raparti GT, Patwardhan MH, Murthy M, Ghanghas RG. Evaluation of the efficacy and safety of bromocriptine QR in type 2 diabetes. Indian J Endocrinol Metab 15, Suppl 1: 33–39, 2011. doi: 10.4103/2230-8210.83062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Reda E, Hassaneen S, El-Abhar HS. Novel trajectories of bromocriptine antidiabetic action: leptin-IL-6/JAK2/p-STAT3/SOCS3, p-IR/p-Akt/GLUT4, PPAR-gamma/adiponectin, Nrf2/PARP-1, and GLP-1. Front Pharmacol 9: 771, 2018. doi: 10.3389/fphar.2018.00771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Satake S, Moore MC, Igawa K, Converse M, Farmer B, Neal DW, Cherrington AD. Direct and indirect effects of insulin on glucose uptake and storage by the liver. Diabetes 51: 1663–1671, 2002. doi: 10.2337/diabetes.51.6.1663. [DOI] [PubMed] [Google Scholar]
- 80.Schwartz SS, Zangeneh F. Evidence-based practice use of quick-release bromocriptine across the natural history of type 2 diabetes mellitus. Postgrad Med 128: 828–838, 2016. doi: 10.1080/00325481.2016.1214059. [DOI] [PubMed] [Google Scholar]
- 81.Scislowski PW, Tozzo E, Zhang Y, Phaneuf S, Prevelige R, Cincotta AH. Biochemical mechanisms responsible for the attenuation of diabetic and obese conditions in ob/ob mice treated with dopaminergic agonists. Int J Obes Relat Metab Disord 23: 425–431, 1999. doi: 10.1038/sj.ijo.0800893. [DOI] [PubMed] [Google Scholar]
- 82.Scranton R, Cincotta A. Bromocriptine—unique formulation of a dopamine agonist for the treatment of type 2 diabetes. Expert Opin Pharmacother 11: 269–279, 2010. doi: 10.1517/14656560903501544. [DOI] [PubMed] [Google Scholar]
- 83.Silbernagel G, Kovarova M, Cegan A, Machann J, Schick F, Lehmann R, Häring HU, Stefan N, Schleicher E, Fritsche A, Peter A. High hepatic SCD1 activity is associated with low liver fat content in healthy subjects under a lipogenic diet. J Clin Endocrinol Metab 97: E2288–E2292, 2012. doi: 10.1210/jc.2012-2152. [DOI] [PubMed] [Google Scholar]
- 84.Stefan N, Peter A, Cegan A, Staiger H, Machann J, Schick F, Claussen CD, Fritsche A, Häring HU, Schleicher E. Low hepatic stearoyl-CoA desaturase 1 activity is associated with fatty liver and insulin resistance in obese humans. Diabetologia 51: 648–656, 2008. doi: 10.1007/s00125-008-0938-7. [DOI] [PubMed] [Google Scholar]
- 85.Takahashi K, Nakamura H, Sato H, Matsuda H, Takada K, Tsuji T. Four plasma glucose and insulin responses to a 75 g ogtt in healthy young japanese women. J Diabetes Res 2018: 1, 2018. doi: 10.1155/2018/5742497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tamaki M, Fujitani Y, Hara A, Uchida T, Tamura Y, Takeno K, Kawaguchi M, Watanabe T, Ogihara T, Fukunaka A, Shimizu T, Mita T, Kanazawa A, Imaizumi MO, Abe T, Kiyonari H, Hojyo S, Fukada T, Kawauchi T, Nagamatsu S, Hirano T, Kawamori R, Watada H. The diabetes-susceptible gene SLC30A8/ZnT8 regulates hepatic insulin clearance. J Clin Invest 123: 4513–4524, 2013. doi: 10.1172/JCI68807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Theytaz F, de Giorgi S, Hodson L, Stefanoni N, Rey V, Schneiter P, Giusti V, Tappy L. Metabolic fate of fructose ingested with and without glucose in a mixed meal. Nutrients 6: 2632–2649, 2014. doi: 10.3390/nu6072632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Thielen L, Shalev A. Diabetes pathogenic mechanisms and potential new therapies based upon a novel target called TXNIP. Curr Opin Endocrinol Diabetes Obes 25: 75–80, 2018. doi: 10.1097/MED.0000000000000391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Tsintzas K, Norton L, Chokkalingam K, Nizamani N, Cooper S, Stephens F, Billeter R, Bennett A. Independent and combined effects of acute physiological hyperglycaemia and hyperinsulinaemia on metabolic gene expression in human skeletal muscle. Clin Sci (Lond) 124: 675–686, 2013. doi: 10.1042/CS20120481. [DOI] [PubMed] [Google Scholar]
- 90.Turner MC, Martin NRW, Player DJ, Ferguson RA, Wheeler P, Green CJ, Akam EC, Lewis MP. Characterising hyperinsulinemia-induced insulin resistance in human skeletal muscle cells. J Mol Endocrinol 64: 125–132, 2020. doi: 10.1530/JME-19-0169. [DOI] [PubMed] [Google Scholar]
- 91.van den Brandt PA. Red meat, processed meat, and other dietary protein sources and risk of overall and cause-specific mortality in The Netherlands Cohort Study. Eur J Epidemiol 34: 351–369, 2019. doi: 10.1007/s10654-019-00483-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lopez Vicchi F, Luque GM, Brie B, Nogueira JP, Garcia Tornadu I, Becu-Villalobos D. Dopaminergic drugs in type 2 diabetes and glucose homeostasis. Pharmacol Res 109: 74–80, 2016. doi: 10.1016/j.phrs.2015.12.029. [DOI] [PubMed] [Google Scholar]
- 93.Watada H, Tamura Y. Impaired insulin clearance as a cause rather than a consequence of insulin resistance. J Diabetes Investig 8: 723–725, 2017. doi: 10.1111/jdi.12717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Weigert C, Hennige AM, Brodbeck K, Häring HU, Schleicher ED. Interleukin-6 acts as insulin sensitizer on glycogen synthesis in human skeletal muscle cells by phosphorylation of Ser473 of Akt. Am J Physiol Endocrinol Metab 289: E251–E257, 2005. doi: 10.1152/ajpendo.00448.2004. [DOI] [PubMed] [Google Scholar]
- 95.Zhang Y, Scislowski PW, Prevelige R, Phaneuf S, Cincotta AH. Bromocriptine/SKF38393 treatment ameliorates dyslipidemia in ob/ob mice. Metabolism 48: 1033–1040, 1999. doi: 10.1016/S0026-0495(99)90202-0. [DOI] [PubMed] [Google Scholar]